 Thomas M. Blattmann1,2*
Thomas M. Blattmann1,2* Daniel B. Montluçon2
Daniel B. Montluçon2 Negar Haghipour2,3
Negar Haghipour2,3 Naoto F. Ishikawa1,2
Naoto F. Ishikawa1,2 Timothy I. Eglinton2
Timothy I. Eglinton2- 1Biogeochemistry Program, Research Institute for Marine Resources Utilization, Japan Agency for Marine-Earth Science and Technology, Yokosuka, Japan
- 2Geological Institute, Department of Earth Sciences, ETH Zurich, Zurich, Switzerland
- 3Laboratory of Beam Physics, Department of Physics, ETH Zurich, Zurich, Switzerland
The “building blocks of life” are found nearly ubiquitously in the environment in the form of proteins, peptides, and single amino acids. To shed light on amino acid sources, cycling, and preservation in sedimentary environments, we present a method using high-pressure liquid chromatography to separate and isolate underivatized amino acids extracted from sediments to conduct compound-specific radiocarbon analysis. This method consists of three main steps including (1) amino acid extraction by hydrofluoric and hydrochloric acids followed by desalting, (2) liquid chromatographic isolation and purification using two complementary column chemistries, and (3) post-purification and measurement by accelerator mass spectrometry. The resulting blank of this procedure is estimated to contain 2.2 ± 1.3 μgC with 0.25 ± 0.09 Fm.
Introduction
Compound-specific radiocarbon in environmental samples has seen an explosion in interest for various biomolecules including steroids (Eglinton et al., 1996, 1997; Griffith et al., 2012), lipids (Eglinton et al., 1996, 1997; Smittenberg et al., 2004; Shah et al., 2008; Druffel et al., 2010; Birkholz et al., 2013; Wakeham and McNichol, 2014), lignin (Feng et al., 2013, 2017), pollutants (Giger et al., 1984; Griffith et al., 2012), pigments (Kusch et al., 2010), biomineral occluded compounds (Ingalls et al., 2004), and pyrogenic carbon (Coppola et al., 2018). Amino acids are among the most abundant and ubiquitous organic molecules in the environment and, to date, their stable carbon and nitrogen isotopic information has been investigated for both ecological (Ohkouchi et al., 2017) and sedimentary studies (Keil and Fogel, 2001). Previous investigations in distal deep-sea sediments on the radiocarbon content of the amino acid compound class reflects its primary productivity source (Wang et al., 1996, 1998; Hwang et al., 2005). However, the information contained in their amino acid-specific radiocarbon content has hitherto remained sparingly investigated. For amino acids, procedures for analyzing their compound-specific radiocarbon content have been developed for dissolved organic matter (Quan, 2005), collagen (Tripp et al., 2006), and for animal tissue (Ishikawa et al., 2018). Motivated by the prospect of exploring new research frontiers in biogeochemistry including food webs and organic matter-mineral interactions (Blattmann and Ishikawa, Unpublished), we report a method to conduct compound-specific radiocarbon on amino acids extracted from sediments.
Materials and Methods
The methodology for sedimentary amino acid-specific radiocarbon isotope analysis introduced here is subdivided into three main steps: (1) amino acid extraction, (2) amino acid isolation, and (3) final amino acid purification and radiocarbon analysis. An overview of the procedure is provided in Figure 1. This section also contains discussion and rationale concerning method choice, which is specific to and modifiable for other studies as a function of their specific scientific objectives and sample matrix.
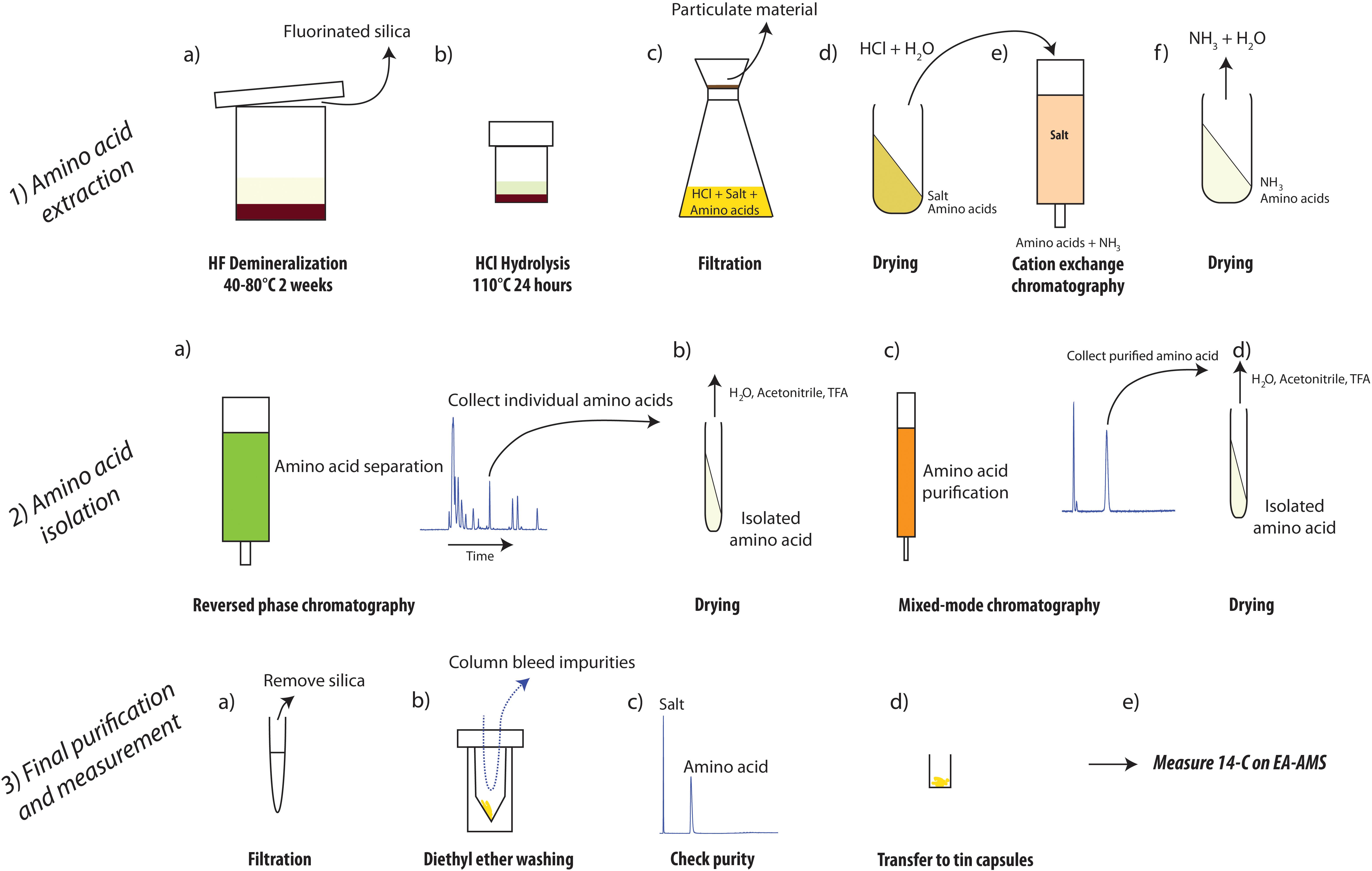
Figure 1. Overview of designed procedure recommended for extraction and preparation of amino acids from sediments for measurement of radiocarbon isotopic composition. Step 3b was not carried out in this study but is detailed in Ishikawa et al. (2018).
Blanks, Standard Amino Acid Mixture, and Test Samples
The five samples discussed here include a (I) procedural blank without added compounds (hitherto sample “I”) covering the entire procedure (see Figure 1), (II) a standard amino acid mixture with known concentrations and amino acid-specific radiocarbon isotopic compositions which was (a) subject to acid hydrolysis (Sample “IIa”; Steps 1a–2a), and (b) a second set which was only processed by liquid chromatographic separation and isolation (Sample “IIb”; from step 2a onward), and (III) two natural sediments (Sample “III”; Steps 1a–2a). This study reports chromatograms from samples listed under I, II, and III and the radiocarbon concentrations recovered from samples listed under I and IIb.
During protein hydrolysis by HCl, which frees amino acids by cleaving peptide bonds, tryptophan, cysteine, and methionine are partially degraded (Otter, 2012) and asparagine and glutamine are deaminated to aspartic acid and glutamic acid, respectively (Hunt, 1985). For these reasons, asparagine, cysteine, and glutamine were not included in the standard amino acid mixture. The standard amino acid mixture consisted of alanine, arginine, aspartic acid, glutamic acid, glycine, histidine, hydroxyproline, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine, and valine. The concentrations and consensus radiocarbon concentrations of the amino acids ultimately investigated in this study are reported in Table 1.
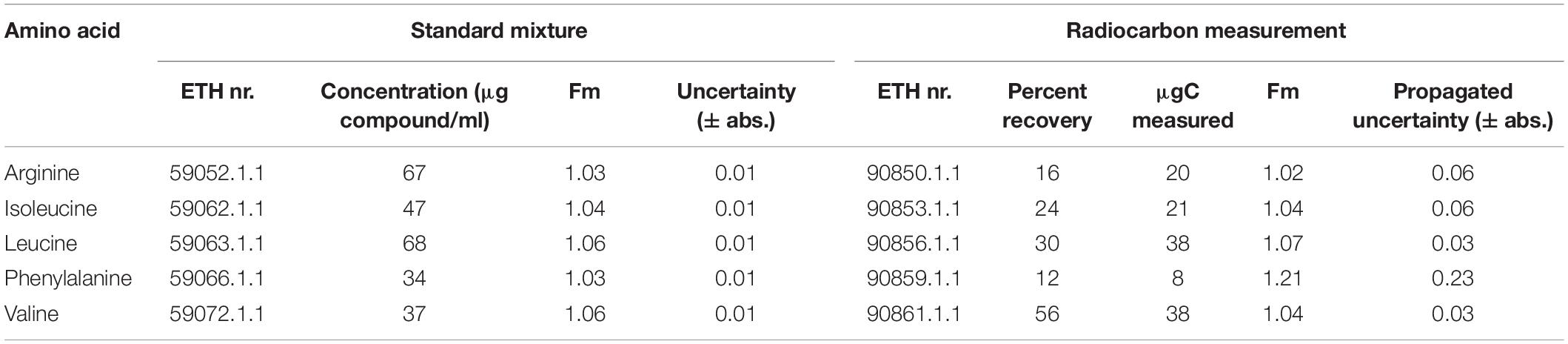
Table 1. Analyzed amino acids with consensus radiocarbon values from unprocessed reference materials and their respective constant contamination-corrected radiocarbon concentrations measured on processed standards (IIb) based on 23 injections of 80 μl of standard mixture.
The two riverbed sediments test samples (Sample “III”) were from the termini of large southeast Asian rivers collected in 2016 with one sample from the mouth of the Pearl River in southern China, hitherto referred to as sample “PO3,” and one from the mouth of the Cagayan River from the Philippines, hitherto referred to as sample “LZ31TB.” Upon sampling, samples were kept cool and frozen as soon as possible until they were freeze-dried. The sediments were dry sieved to < 200 μm, which was chosen over wet sieving to not risk sorptive exchanges. From each sample, approximately 30 g of dry sample material was then weighed in for analysis.
Amino Acid Extraction
Rationale
In geosciences, hydrolysis of proteins in dissolved organic matter, particulate organic matter, and sediments is usually carried out using HCl of different concentrations (6–12 M HCl), temperature (100–150°C), and time (70 min–24 h) (c.f. Keil et al., 1998; McCarthy et al., 2004; Lang et al., 2013; Priotto and Lara, 2013). Additional variants of amino acid extraction include vapor phase HCl treatment of seawater dissolved organic matter (Keil and Kirchman, 1991) and combinations of HF and HCl treatments and non-hydrolytic amino acid recovery techniques from marine sediments (Nunn and Keil, 2005, 2006). However, yields of amino acids extracted from mineral matrices are increased (variable but in some cases by >300%) by treatment of sample material with HF prior to HCl hydrolysis (Stevenson and Cheng, 1970; Cheng, 1975; Cheng et al., 1975), as some amino acids are strongly associated with silicate minerals, precluding their extraction with HCl hydrolysis alone (Cheng, 1975; Stevenson and Cheng, 1970; c.f. Blattmann and Ishikawa, Unpublished). Therefore, following the primary research objectives to investigate organic matter-mineral interactions set forth by Blattmann (2018), sequential HF and HCl extraction/hydrolysis was pursued here.
Procedure Description
Extraction
Samples were introduced in 300 ml Teflon beakers (Savillex) and were spiked with 700 μl norleucine standard (841 μg/ml) as internal reference. Slowly, ≈280 ml 38–40% HF (Merck 1.000329.1000) was added to each beaker to allow controlled release of carbonate-bound CO2. Beakers were heated on hotplates with heat conducting racks (Analab) at 40°C for 2 days under acid reflux conditions. After 2 days, samples were agitated, and pelleted sample material was crushed with a metallic spatula to promote further demineralization and the temperature was increased to 50°C to begin drying. Samples were dried over the course of 35 days. Upon reaching complete dryness, which was assessed visually, the demineralized sample mass was transferred to 60 ml Savillex Teflon hydrolysis vessels. 30 ml of 6 M HCl (prepared from 37% AnalaR NORMAPUR Reag. Ph. Eur. 20252.290), was added to vessel and the headspace was purged with nitrogen gas and then sealed. After 16 h buried in a sand bath at 110°C (± 5°C), the samples were cooled and transferred to glass centrifuge tubes where the hydrolysates were washed three times with MilliQ water to separate them from the solid residue. The supernatants were collected and dried on a solvent evaporator system (Büchi Multi-vapor P12 unit coupled to Büchi V-700 vacuum pump and Büchi F-105 recirculating chiller) with NaOH pellets in the solvent trap of the vertical condenser to reduce HCl vapor exposure to the membrane pump. Dried hydrolysates were then dissolved in MilliQ water and passed through a 47 mm diameter 0.2 μm pore size polyethersulfone membrane filter (Sterlitech) and then dried again on the solvent evaporator system.
Desalting
Using cation exchange resin, inorganic salt was removed following modified methods (Degens and Bajor, 1960; Cheng, 1975; Takano et al., 2010; Rubino et al., 2014). A column was packed with a bedload volume of 60 ml of 200–400 mesh Dowex 50W X8 cation exchange resin (Fluka 44519-100g). Both ends were capped with glass wool (11 μm diameter) and mobile phases were passed through the resin with a Teflon-tape wrapped stopper and tubing introducing nitrogen gas at 2–4 ml/s. The NH3 solution was prepared from a 25% stock solution (VWR AnalaR NORMAPUR 1133.1000), the NaOH solution was prepared from pellets (Reg. Ph. Eur. VWR 28244.295), the HCl was prepared from a 37% stock solution (37% AnalaR NORMAPUR Reag. Ph. Eur. 20252.290), and H2SO4 solution was prepared from a 95.0–98.0% stock solution (Sigma-Aldrich 258105-500ML). The resin was conditioned for desalting following Takano et al. (2010): (1) 180 ml of 1M HCl, (2) 180 ml of water, (3) 180 ml of 1M NaOH, (4) 180 ml of water, (5) 180 ml of HCl, and (6) 180 ml of water. Once conditioned, 10 ml of sample solution (redissolved in 80 ml of 0.1 M HCl) was transferred to the column. The column was washed with MilliQ water until eluent approached near neutral (pH ≈ 6) as monitored using pH strips (typically required 500–1500 ml for sediment samples). Amino acids were then eluted with 240 ml 10% NH3. The desalted amino acid fraction was then dried on the solvent evaporator system. Following sediment samples, pronounced darkening of the cation exchange resin was observed. These strongly retained substances could only be eluted with 0.5 M oxalic acid solution modified after the recommendation by Rubino et al. (2014). Approximately 3–6 bed loads sufficed in removing these retained impurities by acting as a complexing agent and eluting these unknown cations in a flocculent brine. The column was then regenerated using the sequence recommended by Takano et al. (2010). Despite this, some discoloration became apparent after several runs, which appeared not to affect column performance. However, in between samples, enhanced column regeneration was carried out by rinsing with 0.5 M H2SO4 (Small et al., 1975) and allowing the cation exchange beads to rest for a few hours in this mobile phase. During this treatment, the resin filled itself with gaseous voids and returned to its factory-original golden hue. The gas that evolves from the reaction indicates oxidation of organic substances retained on the cation exchange resin.
Chromatographic Separation
Rationale
This section describes the chromatographic separation of underivatized amino acids. In the case of adding a derivative to an amino acid, the carbon isotopic influence of the added carbon needs to be subtracted from the indigenous amino acid carbon to calculate the true radiocarbon isotopic composition. In the case of derivatization (e.g., ethyl ester) the resulting propagated error would quickly balloon to unacceptably large values for radiocarbon isotopic composition apparent from calculations analogous to stable carbon isotopic composition corrections (Chikaraishi and Ohkouchi, 2010). Therefore, only underivatized amino acid preparation with offline carbon isotopic determination was considered during the design of this method (c.f. Hare et al., 1991; Broek et al., 2013; Takano et al., 2015).
The two liquid chromatographic isolation steps following the desalting procedure are necessary given the presence of other compounds in fractions isolated by the first separation. The same conclusion was reached previously in complex environmental matrices (high molecular weight dissolved organic matter), which contained impurities including, among others, monosaccharides and deoxy sugars (Quan, 2005). In contrast to Quan (2005), who used a cation exchange resin after reversed phase chromatography, a mixed mode cation exchange-reversed phase column chemistry was used here.
Procedure Description
For all separations and detection runs, an Agilent 1260 high-performance liquid chromatograph (HPLC) coupled to a charged aerosol detector (CAD) (ESA Corona CAD and Thermo Fisher Scientific Corona CAD Veo) was used. For the liquid chromatographic separation of amino acids, two columns were used using MilliQ water, acetonitrile (Fisher Chemical Optima LC/MS grade A955-212), and trifluoroacetic acid (for HPLC ≥ 99.0% Sigma-Aldrich 302031-100ML) in the mobile phases. In a first step, a reversed-phase CAPCELL PAK C18 MG column (preparative scale, 20 mm × 250 mm, particle size 5 μm, Shiseido, Tokyo, Japan) performed the separations shown in Figure 2. This column achieved baseline separation of phenylalanine, isoleucine, leucine, valine, proline, and arginine from the remaining amino acids. In a second step, phenylalanine, isoleucine, leucine, valine, and arginine were purified using a mixed mode ion-exchange reversed-phase Primesep A column (analytical scale, 4.6 mm × 50 mm, particle size 5 μm, SIELC Technologies, IL, United States). The sample material was dissolved in solvent matching the chemistry of the mobile phase at the time of sample injection of the next respective chromatographic run. For collection of chromatographic isolates in both steps, an Agilent fraction collector cooled to approximately 15°C with 40 round-bottom glass vials (20 ml each) was employed. Following the steps outlined in Figure 1, the chromatographic isolates from both steps were dried on the solvent evaporator system described previously. Following Ishikawa et al. (2018), the mobile phases used for the preparative scale CAPCELL C18 column were MilliQ water with 0.1% (v/v) TFA (solvent A), and acetonitrile with 0.1% (v/v) TFA (solvent B). To protect the column, a Supelco ColumnSaver (0.5 μm filter; 55214-U) and C18 phase guard column (Phenomenex Kinetex C18 5 μm) were used. Each sample injection was set at 80 μl, the column compartment was set at a constant 30°C, and the solvent gradient for each run was linearly programmed as follows: 0 min (A: 100%, B: 0%) to 30 min (A: 90%, B: 10%), to 80 min (A: 80%, B: 20%), to 100 min (A: 80%, B: 20%). The column is then flushed with 100% B for 20 min followed by equilibration with 100% A for 60 min for the next injection at a constant flow rate of 2 ml/min. See Figure 2 for chromatograms and see Supplementary Table 1 for sample collection time windows.
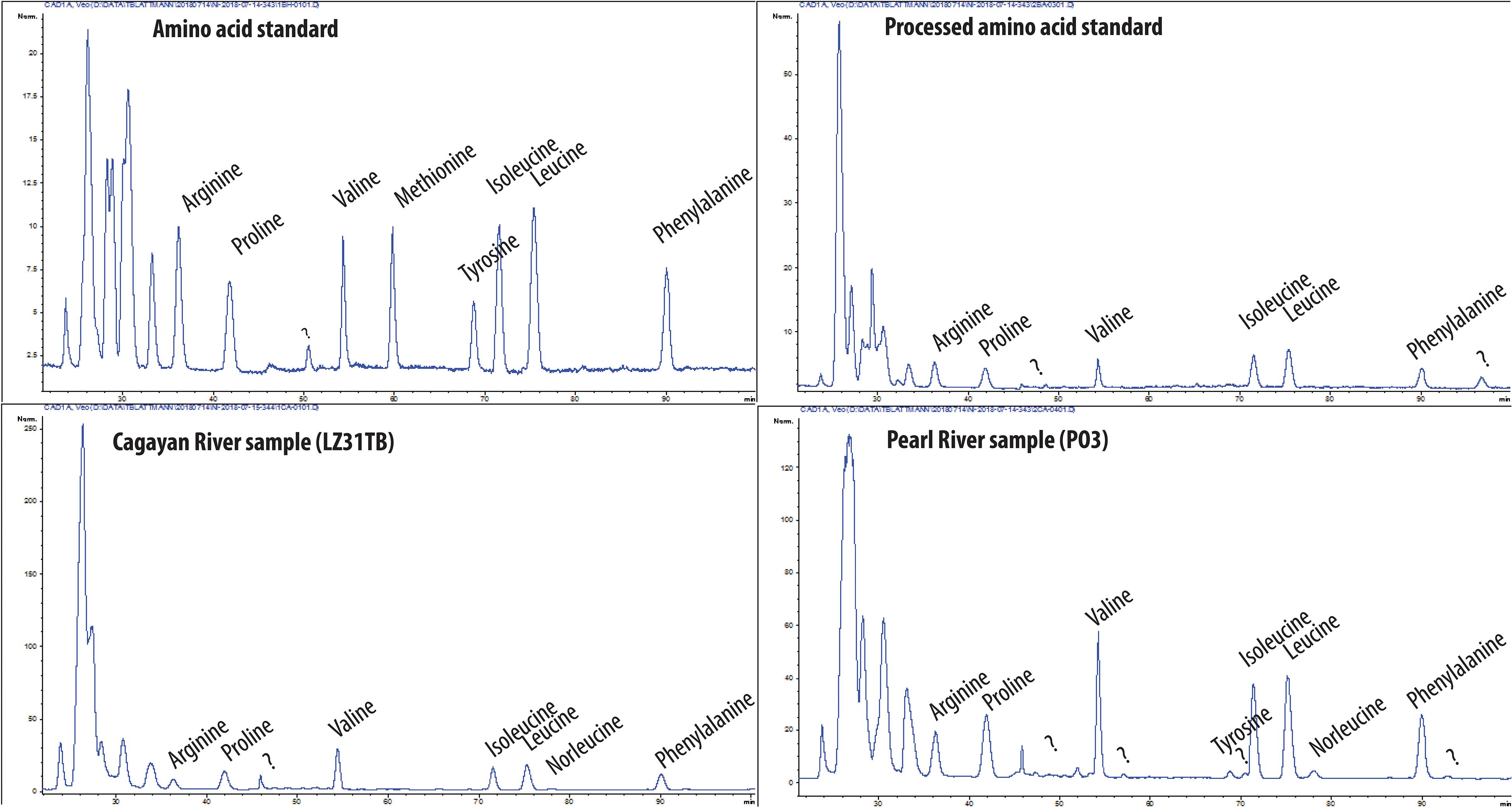
Figure 2. Chromatograms showing CAD traces using the preparative scale CAPCELL C18 column. Top left: unprocessed 18 amino acid standard; top right: 18 amino acid standard which has been subject to the acid hydrolysis and desalting procedure; bottom left: Cagayan River sample LZ31TB; bottom right: Pearl River sample PO3.
The mobile phases used for the analytical scale Primesep A column were MilliQ water with 0.05% (v/v) TFA (solvent A), and acetonitrile with 0.05% (v/v) TFA (solvent B). Here, amino acids were purified with a constant flow rate of 1.5 ml/min under isocratic conditions (A: 95%, B: 5%) for phenylalanine, isoleucine, leucine, and valine, After each injection, the column was flushed with 100% B for 5 min (linearly increasing the flow from 1.5 to 2 ml/min) followed by equilibration (A: 95%, B: 5%) for the next injection for 15 min at 1.5 ml/min. The column compartment was set at a constant 20°C. For arginine, a different isocratic mixture was used (A: 65%, B: 35%) and the flow rate was set at a constant 2 ml/min with a flushing time of 5 min with 100% B followed by equilibration for 15 min (A: 65%, B: 35%). See Supplementary Table 1 for sample collection time windows.
Throughout the isolation and purification steps, the CAD was removed from the column outflow and the eluent was directed to the fraction collector while a diode array detector (carefully monitoring the 200–214 nm wavelength region) was used to monitor elution times of amino acids to quality control the separation performance.
Purification and Measurement
Purified amino acid isolates were dried on the solvent evaporator system and dissolved in ≈500 μl of 0.1 M HCl. Following procedures recommended in Ishikawa et al. (2018), these were then introduced into Eppendorf tubes with internal filters (NANOSEP MF GHP.45 μm hydrophilic polypropylene membrane) to remove any precipitates. Prior to use, the Eppendorf nanosep tubes were leached and centrifuged (Eppendorf Centrifuge 5418) with 0.1 M HCl twice. Sample material was then transferred and dried in V-bottom combusted (450°C) glass vials (BGB 080400-XL-ML 0.9 ml) under a gentle stream of N2 with a heating block set at 40°C. Upon dryness, 1 ml of MilliQ water was added, and the amino acids were dissolved over several hours at 40°C. To check the purity of the amino acids, a small aliquot (1–2 μl) was injected on an analytical-scale reversed-phase column, either Phenomenex Kinetex core shell C18 (4.6 mm × 100 mm, particle size 5 μm) or Shiseido C18 CAPCELL PAK C18 MG column (4.6 mm × 250 mm, particle size 5 μm), monitoring eluent with CAD. Samples that showed a single peak after the initial injection peak were deemed pure and were prepared for radiocarbon analysis (see Supplementary Table 1).
Isolated amino acids and blanks, denoted IIb and I, respectively, were introduced into tin capsules (Elementar 2.88 × 6 × 0.1 mm; product number 03 951 620). Prior to use, tin capsules were rinsed with dichloromethane three times over a glass fiber filter (Whatman GF/F) held on a combusted all-glass vacuum filtration unit, reducing extraneous carbon from the capsules surfaces (Ogawa et al., 2010; Ruff et al., 2010). Dried capsules were then set on a hotplate at 95°C and sample solution was added by syringe in the desired amounts. Tin capsules were wrapped and radiocarbon isotopic composition was measured on an elemental analyzer coupled to a MICADAS accelerator mass spectrometer (EA-AMS) in a setup described elsewhere (Synal et al., 2007; McIntyre et al., 2017).
Discussion
The procedural blank revealed no CAD-detectable extraneous compounds during the liquid chromatographic retention times relevant for the collection of amino acids. However, non-volatile contaminants were introduced during the prior procedure (HF demineralization, HCl hydrolysis, filtering, and desalting). The retention time of non-volatile contaminants (presumably from solvent residues) are consistent with the presence of inorganic salts (see Supplementary Figure 1; Ishikawa et al., 2018). Additionally, the elution of a substance at 33–36 min (only detected by ultraviolet absorption) is possibly attributable to column bleed from the cation exchange resin (divinylbenzene and styrene). In the second liquid chromatographic separation by HPLC using the PrimesepA column, the blank for collected CAPCELL fractions corresponding to the amino acid retention times of valine, isoleucine, leucine, and phenylalanine (I) reveal the absence of co-eluting substances in the relevant time windows. In the case of the CAPCELL blank corresponding to the amino acid retention times of arginine, the CAD detector revealed the presence of non-volatile eluents on the PrimesepA column (I), which however elute approximately 2 min prior to the elution of arginine, thereby eliminating this unknown substance from the final product during fraction collection in the defined time window.
The chromatograms of procedural blank I (see Supplementary Figures 1–6) demonstrate that extraneous amino acids were not a contamination issue, which is a concern owing to their ubiquity (skin flakes, dust, etc.) and possible amino acid contaminants in acids used for hydrolysis (Wolman and Miller, 1971). Each step adds and removes different forms of contamination (Ishikawa et al., 2018) and assuming here that the history of sample processing prior to step 2a (Figure 1) is irrelevant due to removal of contaminants inherited from previous steps, the blank correction applied here is represented by the blank observed by EA-AMS from procedural blank I. In the case of amino acids collected under isocratic 95-5-0.05 water-acetonitrile-TFA conditions (v/v) for the purification of isoleucine, leucine, phenylalanine, and valine, the blanks (n = 4), which were determined by direct measurement of their mass and radiocarbon isotopic composition using EA-AMS, averaged 0.23 ± 0.08 Fm and 2.3 ± 1.5 μgC (see Supplementary Table 2). In the case of arginine collected under isocratic 65-35-0.05 water-acetonitrile-TFA conditions, a larger blank may be expected due to increased column bleed under mobile phase conditions with a higher proportion of organic solvent (Bour et al., 2016; Ishikawa et al., 2018). However, for the arginine equivalent time window, a blank of 1.7 μgC with 0.32 ± 0.01 Fm was quantified by direct EA-AMS measurement (see Supplementary Table 2), appearing slightly smaller than the average blank size mentioned previously, but within uncertainty of the previous blank. For correcting the processed standards, a constant contamination of 2.2 ± 1.3 μgC with 0.25 ± 0.09 Fm (average of all five direct blank measurements) was applied which bins together the blanks isolated under elution time windows corresponding to those of the reported amino acids and purified with the PrimesepA column under isocratic 95-5-0.05 and 65-35-0.05 water-acetonitrile-TFA conditions. This simplified assignment of constant contamination has large uncertainty compared to more rigorously constrained contamination case studies (Haghipour et al., 2019), yet, using Equations 1 and 2 [following Wacker and Christl (2012); see Supplementary Material], corrects all standards (IIb) within error of their respective consensus radiocarbon isotopic compositions (Table 1). Nonetheless, this blank size is consistent with observations made by Ishikawa et al. (2018) who quantify the total blank as approximately 2.4 μgC for samples without “post-purification” (filtration and rinsing with diethyl ether; see procedure 3b in Figure 1). Ishikawa et al. (2018) argue that extraneous carbon in the form of column bleed is effectively removed with the diethyl ether rinsing approach, which based on the experiences gained here, is a position which is further recommended here. Overall, these results, together with the chromatographic baseline separation and purity of amino acids (arginine, valine, leucine, isoleucine, and phenylalanine) recovered after demineralization, hydrolysis, and desalting from standard (IIa) and sample material (III) (see Figure 2 and Supplementary Figures 7–20), demonstrate the suitability of this approach for conducting amino acid-specific radiocarbon measurements on complex environmental matrices.
Summary and Outlook
Here, we demonstrate the successful extraction and liquid chromatographic separation of underivatized amino acids from sediments. Isolated amino acids from a standard reference mixture were measured for their radiocarbon isotopic composition by EA-AMS. Blanks and procedural amino acid standards provide preliminary constraints on contamination input, which brings processed amino acid standards within error of their consensus values. In order to improve the quality of the measurements, diethyl ether rinsing of amino acids should be carried out to remove impurities derived from column bleed as recommended by Ishikawa et al. (2018). The results of the measurements of sedimentary amino acids together with refined blank assessments will be reported in forthcoming publications.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
TB, DM, NH, and NI conducted and supported the laboratory work and measurements. TB, DM, and TE designed the study. All authors contributed to the discussion and writing.
Funding
This work was supported by the ETH Zurich (ETH Research Grant 41 14-1) and JAMSTEC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Fernando Siringan and Erin Tinacba (Marine Science Institute, University of the Philippines), Meixun Zhao and Pengfei Hou (Ocean University of China, Qingdao), and Andrea Winter (ETH Zurich) are thanked for their fieldwork and logistical support. Cameron McIntyre and Lukas Wacker (Laboratory of Ion Beam Physics, ETH Zurich) are thanked for radiocarbon measurements and support. This work benefited from discussions with Yoshinori Takano (JAMSTEC). We thank the reviewers RS and ZL for their careful reviews of this work. We are indebted to the Hanse-Wissenschaftskolleg and the Geochemical Society for providing funding for the conference in which this work was presented and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) project number 422798570.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.00174/full#supplementary-material
References
Birkholz, A., Smittenberg, R. H., Hajdas, I., Wacker, L., and Bernasconi, S. M. (2013). Isolation and compound specific radiocarbon dating of terrigenous branched glycerol dialkyl glycerol tetraethers (brGDGTs). Organ. Geochem. 60, 9–19. doi: 10.1016/j.orggeochem.2013.04.008
Blattmann, T. M. (2018). Topics on Radiocarbon Geochemistry and Organic Matter-Mineral Interactions. Ph.D. thesis, ETH Zürich, Zürich. doi: 10.3929/ethz-b-000313397
Bour, A. L., Walker, B. D., Broek, T. A. B., and McCarthy, M. D. (2016). Radiocarbon analysis of individual amino acids: carbon blank quantification for a small-sample high-pressure liquid chromatography purification method. Analy. Chem. 88, 3521–3528. doi: 10.1021/acs.analchem.5b03619
Broek, T. A. B., Walker, B. D., Andreasen, D. H., and McCarthy, M. D. (2013). High-precision measurement of phenylalanine δ15N values for environmental samples: a new approach coupling high-pressure liquid chromatography purification and elemental analyzer isotope ratio mass spectrometry. Rapid Commun. Mass Spectr. 27, 2327–2337. doi: 10.1002/rcm.6695
Cheng, C. N. (1975). Extracting and desalting amino acids from soils and sediments: evaluation of methods. Soil Biol. Biochem. 7, 319–322. doi: 10.1016/0038-0717(75)90074-7
Cheng, C. N., Shufeldt, R. C., and Stevenson, F. J. (1975). Amino acid analysis of soils and sediments: extraction and desalting. Soil Biol. Biochem. 7, 143–151. doi: 10.1016/0038-0717(75)90012-7
Chikaraishi, Y., and Ohkouchi, N. (2010). “An improved method for precise determination of carbon isotopic composition of amino acids,” in Earth, Life, and Isotopes, eds N. Ohkouchi, I. Tayasu, and K. Koba (Kyoto: Kyoto University Press), 355–366.
Coppola, A. I., Wiedemeier, D. B., Galy, V., Haghipour, N., Hanke, U. M., Nascimento, G. S., et al. (2018). Global-scale evidence for the refractory nature of riverine black carbon. Nat. Geosci. 11, 584–588. doi: 10.1038/s41561-018-0159-8
Degens, E. T., and Bajor, M. (1960). Die Verteilung von Aminosäuren in bituminösen Sedimenten und ihre Bedeutung f r die Kohlen- und Erdölgeologie. Glückauf 96, 1525–1534.
Druffel, E. R. M., Zhang, D., Xu, X., Ziolkowski, L. A., Southon, J. R., dos Santos, G. M., et al. (2010). Compound-specific radiocarbon analyses of phospholipid fatty acids and n-alkanes in ocean sediments. Radiocarbon 52, 1215–1223. doi: 10.1017/s0033822200046294
Eglinton, T. I., Aluwihare, L. I., Bauer, J. E., Druffel, E. R. M., and McNichol, A. P. (1996). Gas chromatographic isolation of individual compounds from complex matrices for radiocarbon dating. Anal. Chem. 68, 904–912. doi: 10.1021/ac9508513
Eglinton, T. I., Benitez-Nelson, B. C., Pearson, A., McNichol, A. P., Bauer, J. E., and Druffel, E. R. M. (1997). Variability in radiocarbon ages of individual organic compounds from marine sediments. Science 277, 796–799. doi: 10.1126/science.277.5327.796
Feng, X., Benitez-Nelson, B. C., Montluçon, D. B., Prahl, F. G., McNichol, A. P., Xu, L., et al. (2013). 14C and 13C characteristics of higher plant biomarkers in Washington margin surface sediments. Geochim. Cosmochim. Acta 105, 14–30. doi: 10.1016/j.gca.2012.11.034
Feng, X., Vonk, J. E., Griffin, C., Zimov, N., Montluçon, D. B., Wacker, L., et al. (2017). 14C variation of dissolved lignin in arctic river systems. ACS Earth Space Chem. 1, 334–344. doi: 10.1021/acsearthspacechem.7b00055
Giger, W., Sturm, M., Sturm, H., Schaffner, C., Bonani, G., Balzer, R., et al. (1984). 14C/12C-ratios in organic matter and hydrocarbons extracted from dated lake sediments: nuclear instruments and methods in physics research section B. Beam Interact. Mater. Atoms 5, 394–397. doi: 10.1016/0168-583x(84)90548-2
Griffith, D. R., Wacker, L., Gschwend, P. M., and Eglinton, T. I. (2012). Carbon isotopic (13C and 14C) composition of synthetic estrogens and progestogens. Rapid Commun. Mass Spectr. 26, 2619–2626. doi: 10.1002/rcm.6385
Haghipour, N., Ausin, B., Usman, M. O., Ishikawa, N., Wacker, L., Welte, C., et al. (2019). Compound-specific radiocarbon analysis by elemental analyzer–accelerator mass spectrometry: precision and limitations. Anal. Chem. 91, 2042–2049. doi: 10.1021/acs.analchem.8b04491
Hare, P. E., Fogel, M. L., Stafford, T. W. Jr., Mitchell, A. D., and Hoering, T. C. (1991). The isotopic composition of carbon and nitrogen in individual amino acids isolated from modern and fossil proteins. J. Archaeol. Sci. 18, 277–292. doi: 10.1016/0305-4403(91)90066-x
Hunt, S. (1985). “Degradation of amino acids accompanying in vitro protein hydrolysis,” in Chemistry and Biochemistry of the Amino Acids, ed. G. C. Barrett (Dordrecht: Springer), 376–398. doi: 10.1007/978-94-009-4832-7_12
Hwang, J., Druffel, E. R. M., and Komada, T. (2005). Transport of organic carbon from the California coast to the slope region: a study of Δ14C and δ13C signatures of organic compound classes. Glob. Biogeochem. Cycles 19, 1–9. doi: 10.1029/2004GB002422
Ingalls, A. E., Anderson, R. F., and Pearson, A. (2004). Radiocarbon dating of diatom-bound organic compounds. Mar. Chem. 92, 91–105. doi: 10.1016/j.marchem.2004.06.019
Ishikawa, N. F., Itahashi, Y., Blattmann, T. M., Takano, Y., Ogawa, N. O., Yamane, M., et al. (2018). Improved method for isolation and purification of underivatized amino acids for radiocarbon analysis. Anal. Chem. 90, 12035–12041. doi: 10.1021/acs.analchem.8b02693
Keil, R. G., and Fogel, M. L. (2001). Reworking of amino acid in marine sediments: stable carbon isotopic composition of amino acids in sediments along the Washington coast. Limnol. Oceanogr. 46, 14–23. doi: 10.4319/lo.2001.46.1.0014
Keil, R. G., and Kirchman, D. L. (1991). Dissolved combined amino acids in marine waters as determined by a vapor-phase hydrolysis method. Mar. Chem. 33, 243–259. doi: 10.1016/0304-4203(91)90070-d
Keil, R. G., Tsamakis, E., Giddings, J. C., and Hedges, J. I. (1998). Biochemical distributions (amino acids, neutral sugars, and lignin phenols) among size-classes of modern marine sediments from the Washington coast. Geochim. Cosmochim. Acta 62, 1347–1364. doi: 10.1016/s0016-7037(98)00080-5
Kusch, S., Kashiyama, Y., Ogawa, N. O., Altabet, M., Butzin, M., Friedrich, J., et al. (2010). Implications for chloro- and pheopigment synthesis and preservation from combined compound-specific δ13C, δ15N, and Δ14C analysis. Biogeosciences 7, 4105–4118. doi: 10.5194/bg-7-4105-2010
Lang, S. Q., Früh-Green, G. L., Bernasconi, S. M., and Butterfield, D. A. (2013). Sources of organic nitrogen at the serpentinite-hosted Lost City hydrothermal field. Geobiology 11, 154–169. doi: 10.1111/gbi.12026
McCarthy, M. D., Benner, R., Lee, C., Hedges, J. I., and Fogel, M. L. (2004). Amino acid carbon isotopic fractionation patterns in oceanic dissolved organic matter: an unaltered photoautotrophic source for dissolved organic nitrogen in the ocean? Mar. Chem. 92, 123–134. doi: 10.1016/j.marchem.2004.06.021
McIntyre, C. P., Wacker, L., Haghipour, N., Blattmann, T. M., Fahrni, S., Usman, M., et al. (2017). Online 13C and 14C gas measurements by EA-IRMS–AMS at ETH Zürich. Radiocarbon 59, 893–903. doi: 10.1017/rdc.2016.68
Nunn, B. L., and Keil, R. G. (2005). Size distribution and amino acid chemistry of base-extractable proteins from Washington coast sediments. Biogeochemistry 75, 177–200. doi: 10.1007/s10533-004-6546-9
Nunn, B. L., and Keil, R. G. (2006). A comparison of non-hydrolytic methods for extracting amino acids and proteins from coastal marine sediments. Mar. Chem. 98, 31–42. doi: 10.1016/j.marchem.2005.06.005
Ogawa, N. O., Nagata, T., Kitazato, H., and Ohkouchi, N. (2010). “Ultra-sensitive elemental analyzer/isotope ratio mass spectrometer for stable nitrogen and carbon isotope analyses,” in Earth, Life, and Isotopes, eds N. Ohkouchi, I. Tayasu, and K. Koba (Kyoto: Kyoto University Press), 339–353.
Ohkouchi, N., Chikaraishi, Y., Close, H. G., Fry, B., Larsen, T., Madigan, D. J., et al. (2017). Advances in the application of amino acid nitrogen isotopic analysis in ecological and biogeochemical studies. Organ. Geochem. 113, 150–174. doi: 10.1016/j.orggeochem.2017.07.009
Otter, D. E. (2012). Standardised methods for amino acid analysis of food. Br. J. Nutr. 108, S230–S237. doi: 10.1017/S0007114512002486
Priotto, S., and Lara, R. J. (2013). On the optimization of hydrolysis conditions for simultaneous determination of amino acids and amino sugars in marine sediments. J. Mar. Biol. Oceanogr. 2, 1–5.
Quan, T. M. (2005). Chemical Characterization of Dissolved Organic Matter (DOM) in Seawater: Structure, Cycling, and the Role of Biology. Ph.D. thesis, Massachusetts Institute of Technology, Woods Hole Oceanographic Institution. doi: 10.1575/1912/1569
Rubino, M., Milin, S., D’Onofrio, A., Signoret, P., Hatté, C., and Balesdent, J. (2014). Measurement of δ13C values of soil amino acids by GC–C–IRMS using trimethylsilylation: a critical assessment. Isotopes Environ. Health Stud. 50, 516–530. doi: 10.1080/10256016.2014.959444
Ruff, M., Fahrni, S., Gäggeler, H. W., Hajdas, I., Suter, M., Synal, H. A., et al. (2010). On-line radiocarbon measurements of small samples using elemental analyzer and MICADAS gas ion source. Radiocarbon 52, 1645–1656. doi: 10.1017/s003382220005637x
Shah, S. R., Mollenhauer, G., Ohkouchi, N., Eglinton, T. I., and Pearson, A. (2008). Origins of archaeal tetraether lipids in sediments: insights from radiocarbon analysis. Geochim. Cosmochim. Acta 72, 4577–4594. doi: 10.1016/j.gca.2008.06.021
Small, H., Stevens, T. S., and Bauman, W. C. (1975). Novel ion exchange chromatographic method using conductimetric detection. Anal. Chem. 47, 1801–1809. doi: 10.1021/ac60361a017
Smittenberg, R. H., Hopmans, E. C., Schouten, S., Hayes, J. M., Eglinton, T. I., and Sinninghe Damsté, J. S. (2004). Compound-specific radiocarbon dating of the varved holocene sedimentary record of saanich Inlet, Canada. Paleoceanography 19, 1–16. doi: 10.1029/2003PA000927
Stevenson, F. J., and Cheng, C. N. (1970). Amino acids in sediments: recovery by acid hydrolysis and quantitative estimation by a colorimetric procedure. Geochim. Cosmochim. Acta 34, 77–88. doi: 10.1016/0016-7037(70)90152-3
Synal, H.-A., Stocker, M., and Suter, M. (2007). MICADAS: a new compact radiocarbon AMS system: nuclear instruments and methods in physics research section B. Beam Interact. Mater. Atoms 259, 7–13. doi: 10.1016/j.nimb.2007.01.138
Takano, Y., Chikaraishi, Y., and Ohkouchi, N. (2015). Isolation of underivatized amino acids by ion-pair high performance liquid chromatography for precise measurement of nitrogen isotopic composition of amino acids: development of comprehensive LC×GC/C/IRMS method. Intern. J. Mass Spectr. 379, 16–25. doi: 10.1016/j.ijms.2014.11.012
Takano, Y., Kashiyama, Y., Ogawa, N. O., Chikaraishi, Y., and Ohkouchi, N. (2010). Isolation and desalting with cation-exchange chromatography for compound-specific nitrogen isotope analysis of amino acids: application to biogeochemical samples. Rapid Commun. Mass Spectr. 24, 2317–2323. doi: 10.1002/rcm.4651
Tripp, J. A., McCullagh, J. S. O., and Hedges, R. E. M. (2006). Preparative separation of underivatized amino acids for compound-specific stable isotope analysis and radiocarbon dating of hydrolyzed bone collagen. J. Separat. Sci. 29, 41–48. doi: 10.1002/jssc.200500247
Wacker, L., and Christl, M. (2012). Data reduction for small radiocarbon samples: error propagation using the model of constant contamination. Lab. Ion Beam Phys. Ann. Rep. 2011:36.
Wakeham, S. G., and McNichol, A. P. (2014). Transfer of organic carbon through marine water columns to sediments - insights from stable and radiocarbon isotopes of lipid biomarkers. Biogeosciences 11, 6895–6914. doi: 10.5194/bg-11-6895-2014
Wang, X.-C., Druffel, E. R. M., Griffin, S., Lee, C., and Kashgarian, M. (1998). Radiocarbon studies of organic compound classes in plankton and sediment of the northeastern Pacific Ocean. Geochim. Cosmochim. Acta 62, 1365–1378. doi: 10.1016/s0016-7037(98)00074-x
Wang, X.-C., Druffel, E. R. M., and Lee, C. (1996). Radiocarbon in organic compound classes in particulate organic matter and sediment in the deep northeast Pacific Ocean. Geophys. Res. Lett. 23, 3583–3586. doi: 10.1029/96gl03423
Keywords: 14C, compound-specific radiocarbon analysis, demineralization, hydrolysis, desalting, protein, peptide, purification
Citation: Blattmann TM, Montluçon DB, Haghipour N, Ishikawa NF and Eglinton TI (2020) Liquid Chromatographic Isolation of Individual Amino Acids Extracted From Sediments for Radiocarbon Analysis. Front. Mar. Sci. 7:174. doi: 10.3389/fmars.2020.00174
Received: 29 October 2019; Accepted: 05 March 2020;
Published: 20 March 2020.
Edited by:
Cindy Lee, Stony Brook University, United StatesReviewed by:
Zhanfei Liu, University of Texas at Austin, United StatesRienk H. Smittenberg, Stockholm University, Sweden
Copyright © 2020 Blattmann, Montluçon, Haghipour, Ishikawa and Eglinton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas M. Blattmann, YmxhdHRtYW5udEBqYW1zdGVjLmdvLmpw