 Zhan-Fei Wei
Zhan-Fei Wei Wen-Li Li1†
Wen-Li Li1† Jun Li
Jun Li Li-Sheng He
Li-Sheng He Yong Wang
Yong Wang- 1Institute of Deep-Sea Science and Engineering, Chinese Academy of Sciences, Sanya, China
- 2College of Earth Sciences, University of Chinese Academy of Sciences, Beijing, China
To obtain an accurate picture of microbial processes is essential for in-depth understanding of the dynamics of microbial communities and metabolic potentials. In this study, an apparatus for automatic multiple in situ nucleic acid collections (MISNAC) loaded and controlled by a lander was designed and applied for in situ microbial filtration, cell lysis and nucleic acid collection at different time points during three cruises in the South China Sea. The MISNAC apparatus completed 390 L in situ water filtration and collected a total of 1,176 ng DNA with six working units in four deployments at ∼1,000 m depth. Microbial community structures in water samples obtained by the MISNAC and in situ microbial filtration and fixation (ISMIFF) apparatus, respectively, were compared by analyzing sequences of 16S rRNA amplicons. The result showed that these communities were largely consistent, regardless of the differences in the communities in the samples from different cruises and time points. In addition, the transcriptomes of the samples collected by the MISANC and a Niskin bottle dramatically differed in abundance and diversity of 16S rRNA gene reads, indicating that more in situ RNA molecules were preserved by the MISNAC, compared to the Niskin. The application of the MISNAC apparatus paved the way to time-series changes of deep-sea microorganisms and their response to various environmental factors.
Introduction
The oceans, as the habitats for a vast variety of organisms, are still mysterious for us. It is remarkable that diverse biological communities playing vital roles in biogeochemical cycling were identified at the different layers of the oceans (Azam et al., 1983). Deep-sea planktonic microorganisms can survive in even tens of thousands of meters below the sea surface, with a striking feature of dark, high-pressure environment (Eloe et al., 2011; Nunoura et al., 2016). The deep waters in oceans are dramatically dynamic in terms of various environmental factors and geographical patterns, which have driven diversification of ecosystems in the tremendous body of waters in the oceans (Urbach et al., 2007). Sampling activities at the different depths and sites in the oceans have been conducted, but the collected samples still could not meet the requirement to capture the subtle changes of the deep-sea ecosystems. Because the majority of microorganisms in the deep-sea environment are not yet culturable, the ‘dark matter’ microbes in the deep waters were mostly undetectable. Metabolisms and in situ activities of the deep-sea microbes are largely illusive due to lack of high-quality nucleic acids for metatranscriptomic and metagenomic studies. Decoding the genomes of different microbial ecotypes is also challenging without long genomic reads from the third generation sequencers, which may be resolved by extraction of high-quality DNA/RNA using deep-sea in situ sampling facilities.
Marine microbiological studies mainly relied on water sampling equipment such as Niskin bottles, by which water samples from different layers of the ocean are collected from the deep ocean and brought to shipboard for filtration and subsequent experiments. For the deep-sea samples, it may take several hours to bring them up for further processing. Therefore, changes in pressure, light density, temperature and redox condition may affect the morphology, metabolism, and community structure of the microbes in the deep water samples (Estrada et al., 2009; Feike et al., 2012; Marietou and Bartlett, 2014). Particularly, for the microbial inhabitants in hydrothermal vents and anoxic zones, the drastic environmental differences will result in a different gene expression profile (Breier et al., 2014; Olins et al., 2017). Previous studies have compared the metatranscriptomes of the microbes in suboxic waters collected by Niskin bottles and in situ filtration apparatus in the central Baltic Sea and the Mediterranean Sea, which showed that sample handling, pressure, and other changes during the sample recovery might lead to physiological changes of the microbes and degradation of labile messenger RNA (Feike et al., 2012; Edgcomb et al., 2016). For sampling from the hadal trench (>6000 m depth), the microbial communities of water samples collected by Niskin bottles differed from those in situ filtered and preserved by an in situ microbial filtration and fixation (ISMIFF) apparatus (Wang et al., 2019a). To resolve the low biomass problem, the ISMIFF could filter a large volume of waters (up to 600 L) and fix the microbes in situ (Wang et al., 2019a). Although the microbes had been fixed, the degradation of nucleic acids and proteins continued. Without high-quality DNA and RNA, metagenomics and metatranscriptomics studies will be hindered. To solve this problem, nucleic acids of the deep-sea microbes should be released and attached to polymer columns or magnetic beans at the deep-sea sampling site. Furthermore, microbial samples could be extracted for better preservation and subsequent molecular work with deep-sea in situ microfluidic techniques. The deep-sea environmental sample processor (D-ESP) were designed and used to in situ finish a variety of molecular assays such as quantitative PCR in the deep-sea zone (Scholin et al., 2009; Ussler et al., 2013). More importantly, with the ability of multiple microbial filtrations and nucleic acid extractions, the variations of microbial communities and metabolic activities at a series of time points could be detected. In this study, on the basis of the ISMIFF, we designed an apparatus for multiple in situ nucleic acid collections (MISNAC), which is loaded and controlled by a lander named ‘Phoenix.’ The MISNAC apparatus was able to automatically finish in situ enrichment of microorganisms, cells lysis and nucleic acid collections at different time points at bottom sites of the South China Sea (SCS).
Materials and Methods
MISNAC Apparatus Structure and Workflow
The MISNAC apparatus was in size of 90 cm × 90 cm × 40 cm, and was weighted 76 kg in air and 45 kg in sea water. The apparatus could be carried and controlled by a lander named “Phoenix” that had two 6 kW⋅h Li-ion batteries and might supply 24 V power for the MISNAC. The working depth of the MISNAC was up to 3000 m. The apparatus mainly consists of four subsystems: a chamber supporting and transition subsystem, a pipe docking subsystem, a flow control subsystem and a nucleic acid collection subsystem (Figure 1A). The chamber supporting and transition subsystem is composed of 10 filtration chambers, a supporting turntable with a transition gear, a motor and an angle sensor to monitor the switch of the filtration chambers. The pipe docking subsystem mainly includes a lead screw, a guide rail, a sealing joint linked to the chamber, a motor and a sensor. The flow control subsystem consisting of valves, pumps and a pressure sensor controls the inlets of seawaters, cell lysis buffer, and ethanol into a polyether tube at different steps. The nucleic acid collection subsystem is composed of two containers for lysate and ethanol, respectively, and polymer columns (Tiangen, Beijing, China) filled up with silica resin for nucleic acid attachment.
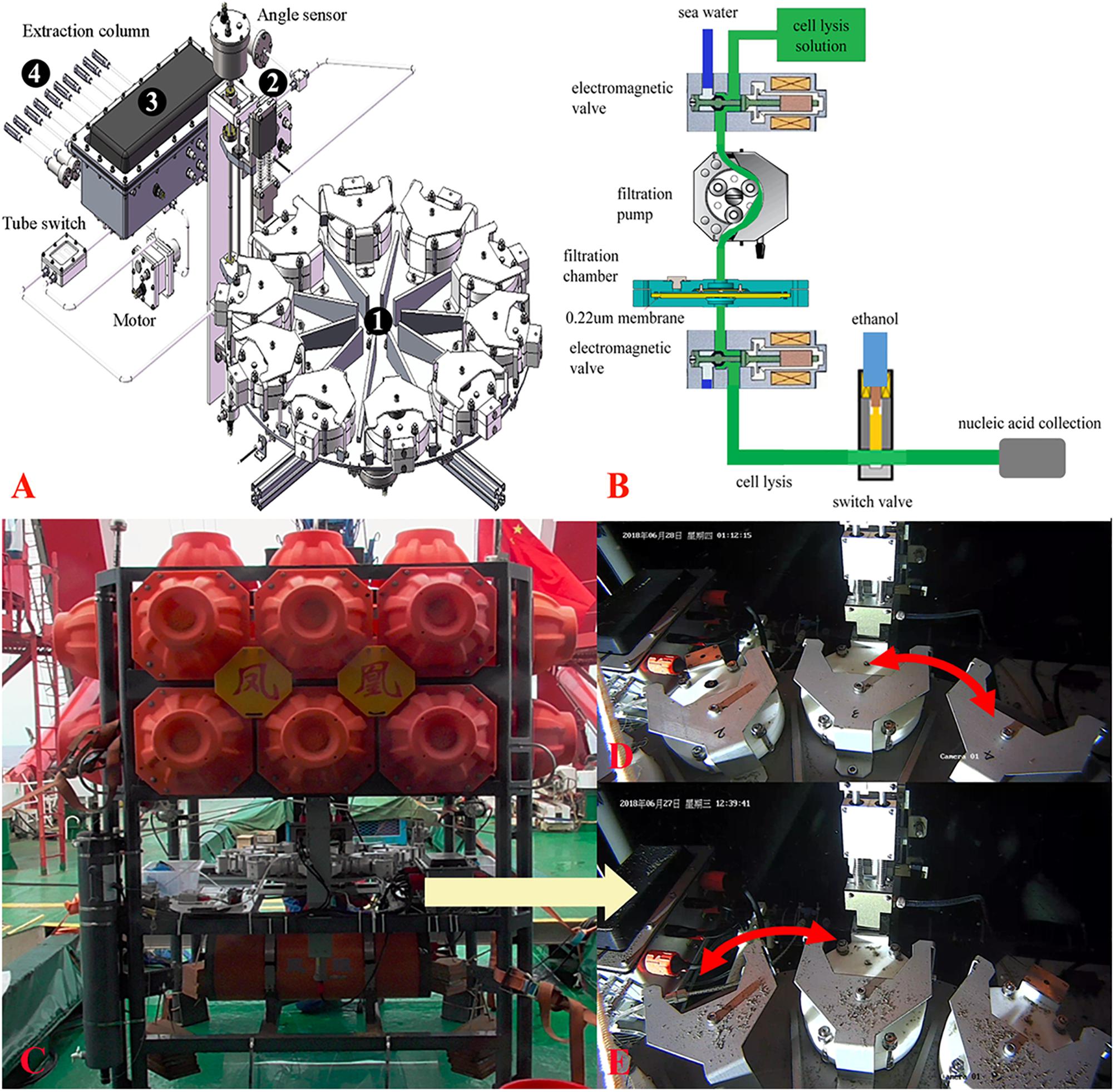
Figure 1. Schematic structure of multiple in situ nucleic acid collections (MISNAC) from deep-sea zone. The four parts of MISNAC: a chamber supporting and transition subsystem (1), a pipe docking subsystem (2), a flow control subsystem (3) and a nucleic acid collection subsystem (4) are labeled on the diagram (A). The workflow of nucleic acid extraction was demonstrated in (B). The MISNAC apparatus was equipped on a lander named ‘Phoenix’ (C). The switch of filtration chambers of the MISNAC apparatus was demonstrated by the photographs taken by a camera on the lander (D,E).
The workflow of the MISNAC apparatus includes several steps described below (Figure 1B). First, seawater was filtered in situ after the movement of the pipe docking system that had connected the filtration pipe. A maximal speed was set to ∼500 ml/min. Second, about 200 ml cell lysis buffer (Tiangen, Beijing, China) was injected into the 100-ml filtration chamber to replace the remaining seawater. The lysate would be maintained in the chamber for ∼30 min for complete lysis of the microbial cells on the membrane. The microbial lysate was then flowed out of the filtration chamber, and mixed with equivalent dosage of 90% ethanol that was being pumped into the pipe from another container. Third, the polymer columns (Tiangen, Beijing, China) in the nucleic acid collection subsystem at the end of the pipe would collect the precipitated nucleic acids in the microbial lysate. The MISNAC apparatus has nine working units and one rinsing unit for cleaning of the pipe. At the ends of the pipe outlets, there are one-way valves to stop inflow of seawater. Thereby, after one working unit is completed, the whole pipeline will be rinsed automatically. The chamber supporting subsystem was then moved to the next working unit and the pipe docking subsystem connected the pipe again for the next filtration. The nucleic acids from the subsequent working units would be retained to a new polymer column switched by the flow control subsystem. Therefore, the MISNAC could collect nine sets of microbial nucleic acids in one lander deployment.
In situ Automatic Workflow for Multiple Nucleic Acid Extraction
The MISNAC apparatus was tested during three cruises in June–July, 2018 and September, 2019, at ∼1,000 m depth in the SCS (Supplementary Figure S1). Before the MISNAC apparatus was deployed with the lander (Figure 1C), the chambers and pipelines were cleaned with 75% ethanol and distilled deionized water to avoid contamination. A 0.22 μm polycarbonate membrane (Millipore, Bedford, MA, United States) with 142 mm diameter was placed into the filtration chambers, and then the pipe and chambers were filled with distilled deionized water. The MISNAC apparatus began to filter the water samples 1 h after landing of the lander on the seafloor, which could avoid uptake of the sedimentary particles disturbed by the lander into the chambers. The following actions were launched by the commands programed in the control unit of the lander according to the workflow described above (Figures 1D,E). Moreover, the working units without cell lysis and nucleic acid collection steps were regarded as ISMIFF control groups although DNA fixation buffer was not used (Wang et al., 2019a). The lander carried the conductivity, temperature, and depth (CTD) (Sea-Bird, Bellevue, WA, United States) instrument and Mini-Pro CO2 (Pro-Oceanus, Bridgewater, NS, Canada) sensor to monitor the changes of deep-sea environments factors.
During the cruises in June–July, 2018, upon onboard of the lander, the nucleic acids attached in the columns were washed with distilled deionized water by centrifuge at 4,000 rpm, and stored at -20°C onboard. In shipborne laboratory, the quality and quantity of nucleic acids were estimated by Qubit 2.0 Fluorometer (Life, United States) immediately. The polycarbonate membranes from the ISMIFF units were transferred and stored at -80°C. Afterward, the polycarbonate membranes for ISMIFF samples were sliced into small pieces by a sterilized scissor and then subjected to total DNA extraction using the MO BIO Power Soil DNA Isolation Kit (MoBio, Carlsbad, CA, United States) in the laboratory according to the manufacturer’s protocol. The quality and quantity of the extracted DNA were estimated using Qubit 2.0 Fluorometer (Life, United States).
During the cruise in September, 2019, we changed the polymer columns (Tiangen, Beijing, China) that were more specific for RNA collection. The RNA molecules were washed off from polymer columns along with DNA using DNase/RNase-Free deionized water (Tiangen, Beijing, China) and stored at -80°C immediately after the lander returned on board. The quality and quantity of RNA were estimated by Qubit 2.0 Fluorometer (Life, United States) in shipborne laboratory. In addition, 10 L water sample was collected during the cruise using a Niskin bottle. The water sample was then filtered using 0.22 μm polycarbonate membrane (Millipore, Bedford, MA, United States) and immediately stored at -80°C as a control. In the laboratory, the membrane was sliced into small pieces and lysed using the lysis buffer (Tiangen, Beijing, China). The RNA/DNA collected by the polymer columns (Tiangen, Beijing, China) was washed with DNase/RNase-Free deionized water (Tiangen, Beijing, China).
High-Throughput Sequencing and Analysis of Amplicon Reads of 16S rRNA Genes
RNA fragments were removed from the nucleic acids obtained by the MISNAC units and ISMIFF using RNaseA (TaKaRa, Dalian, China) with a final concentration of 0.1 ng/μl and incubated for 10 min at 37°C. The V3–V4 region of 16S rRNA genes was amplified using a pair of universal primers: forward primer 341F (5′-CCTAYGGGRBGCASCAG-3′) and reverse primer 802R (5′-TACNVGGGTATCTAATCC-3′) (Claesson et al., 2010; Klindworth et al., 2012) with a 6-nucleotide barcode. The 6-nucleotide barcodes added to the 5′ end of the universal primers were used to distinguish different samples. The PCR reaction system contained 1 ng DNA as template, 20 pmol of each forward primer 341F and reverse primer 802R, 1 μl HS DNA Polymerase, 4 μl dNTP Mixture and 10 μl 5 × PrimerSTAR® Buffer (TaKaRa, Dalian, China) in a 50 μl total reaction volume. Moreover, the PCR reaction procedure comprised an initial denaturation at 98°C for 10 s, followed by 28 cycles consisting of denaturation at 98°C for 10 s, annealing at 50°C for 15 s, and extension at 72°C for 30 s and a final 5 min elongation at 72°C with a thermal cycler (Bio-Rad, United States). The PCR products of partial 16S rRNA genes were purified using the Cycle-Pure Kit (Omega, Norcross, GA, United States) to degrade excess primers and nucleotides. Illumina library for the equally-pooled 16S rRNA gene amplicons was prepared by the TruSeq® Nano DNA LT Kit (Illumina, San Diego, CA, United States) and sequenced on an Illumina Miseq platform (Illumina, San Diego, CA, United States).
The raw sequencing data of 16S rRNA gene amplicons were trimmed using the NGS QC Toolkit (v2.3.3) (Patel and Mukesh, 2012), and then the paired-end reads were assembled using PEAR (v0.9.5) (Zhang et al., 2014). Afterward, the assembled reads were analyzed by QIIME pipeline (Caporaso et al., 2010). Based on the barcodes, the 16S rRNA reads were assigned to different water samples. The separated reads with the similarity threshold of 97% were clustered to the operational taxonomic units (OTUs) using UCLUST (Edgar, 2010), and the longest read of each OTU was used as the representative read for further taxonomic classification analysis with the SILVA 132 database by PyNAST and Ribosomal Database Project (RDP) classifier version 2.2 (Qiong et al., 2007; Gregory Caporaso et al., 2010). The taxa at phyla and order levels were used for microbial community structure and diversity analyses with exclusion of the OTUs assigned to chloroplasts, mitochondria and eukaryotes. Moreover, the taxa at order level were used for principal component analysis (PCA) analysis.
Identification and Classification of 16S rRNA Genes in Metatranscriptomes
DNA fragments were removed from the nucleic acid extraction obtained by the MISNAC and Niskin using the TURBO DNA-freeTM Kit (Ambion, Carlsbad, CA, United States). The cDNA was synthesized from RNA for library preparation using the Ovation RNA-Seq System V2 Kit (Qiagen, Hildon, Germany). The cDNA libraries were constructed by the TruSeq Nano DNA LT Kit (Illumina, San Diego, CA, United States) and sequenced on an Illumina Miseq platform (Illumina, San Diego, CA, United States). The MISNAC and Niskin samples were processed with two replicates. The raw data of metatranscriptomes were trimmed by the Fastp to remove the low quality reads (Q20 over 80% of the reads) and adaptors (Chen et al., 2018). The quality-filtered reads were merged using PEAR (v0.9.5) (Zhang et al., 2014). The 16S rRNA fragments > 150 bp were extracted from the paired-end merged reads with rRNA_HMM (Huang et al., 2012) and were used to explore diversity and structure of microbial communities using the QIIME (Ramiro et al., 2014).
Results and Discussion
To examine the in situ nucleic acid collection abilities of the MISNAC apparatus in the deep-sea zone, three research cruises were conducted at ∼1,000 m depth in the SCS in June–July, 2018 and September, 2019 (Supplementary Figure S1). During the two cruises between June and July of 2018, the MISNAC apparatus loaded by the ‘Phoenix’ lander completed the microbial sampling from 390 L waters by four filtration units in four deployments (Figure 1 and Table 1). In the first cruise, four MISNAC and three ISMIFF units were finished, which resulted in a total of 1,182 ng DNA (Table 1). In the second cruise, two MISNAC and five ISMIFF units were used for collection of 1,904 ng DNA. Although the MISNAC apparatus could complete the nucleic acid collection of nine water filtrations in one launch, only up to three of MISNAC units were used in this study in one launch due to time limitation in the cruises. Moreover, the other working units without cell lysis and nucleic acid collection steps were regarded as ISMIFF controls. In this study, the poor efficiency of nucleic acid collection by the polymer columns at the low temperature probably resulted in low DNA recovery, which has been verified in the laboratory.
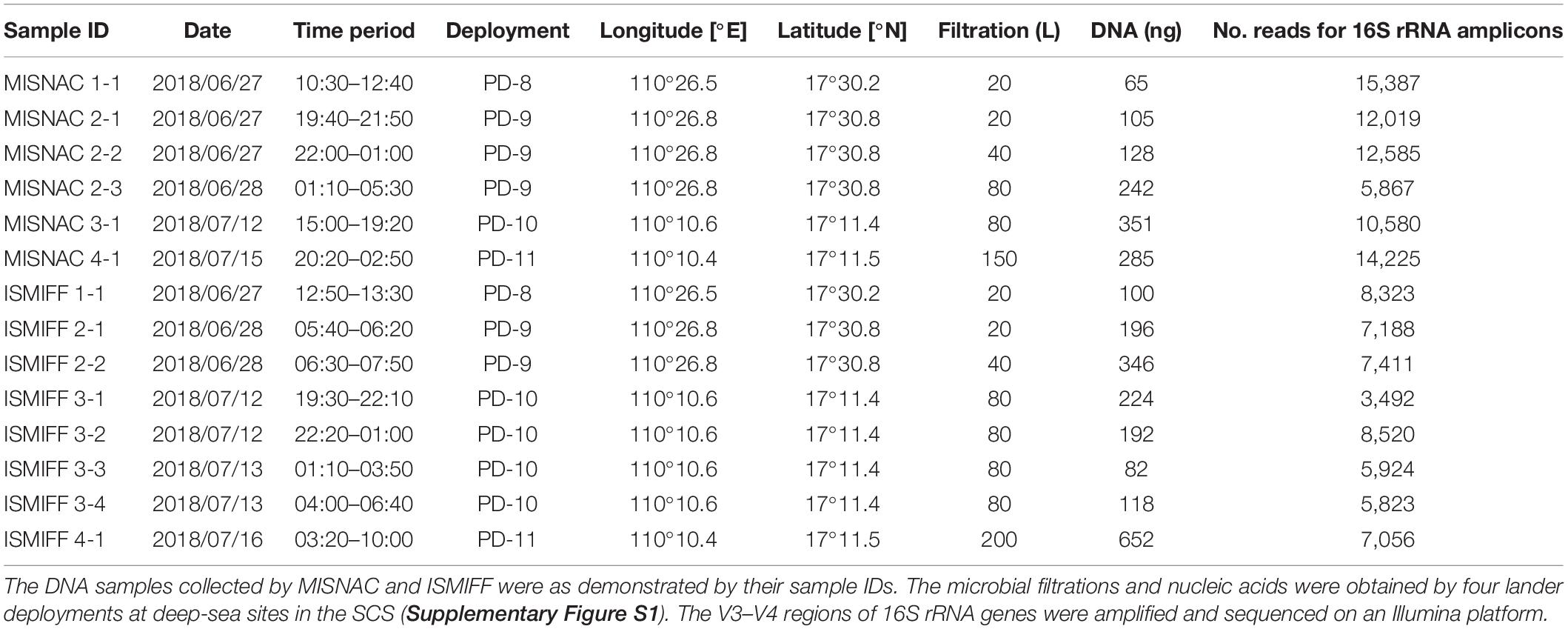
Table 1. DNA samples for sequencing of 16S rRNA gene amplicons.
The temperature, dissolved oxygen, salinity, and CO2 were measured at different time points and sites (Figure 2). The oxygen decreased from the sea surface to the bottom and was higher in PD-8 than those measures at the benthic sites of other deployments. In PD-9, we observed a stepwise decrease of dissolved oxygen from 5.12 to 2.10 ml/L after the lander was sitting on the seafloor. The average CO2 concentrations of PD-8 and PD-9 were 893.32 ± 8.54 and 959.61 ± 7.75 ppm, respectively, which exhibited a striking difference between the two deployments at the same site. For PD-10, we observed a steady decrease of CO2 concentration at ∼1,000 m depth, while a reverse trend was shown at PD-11 site (Figures 2B,C). The salinity at the seafloor fluctuated slightly in hours with an average of 34.25 ± 0.12 PSU (Figure 2).
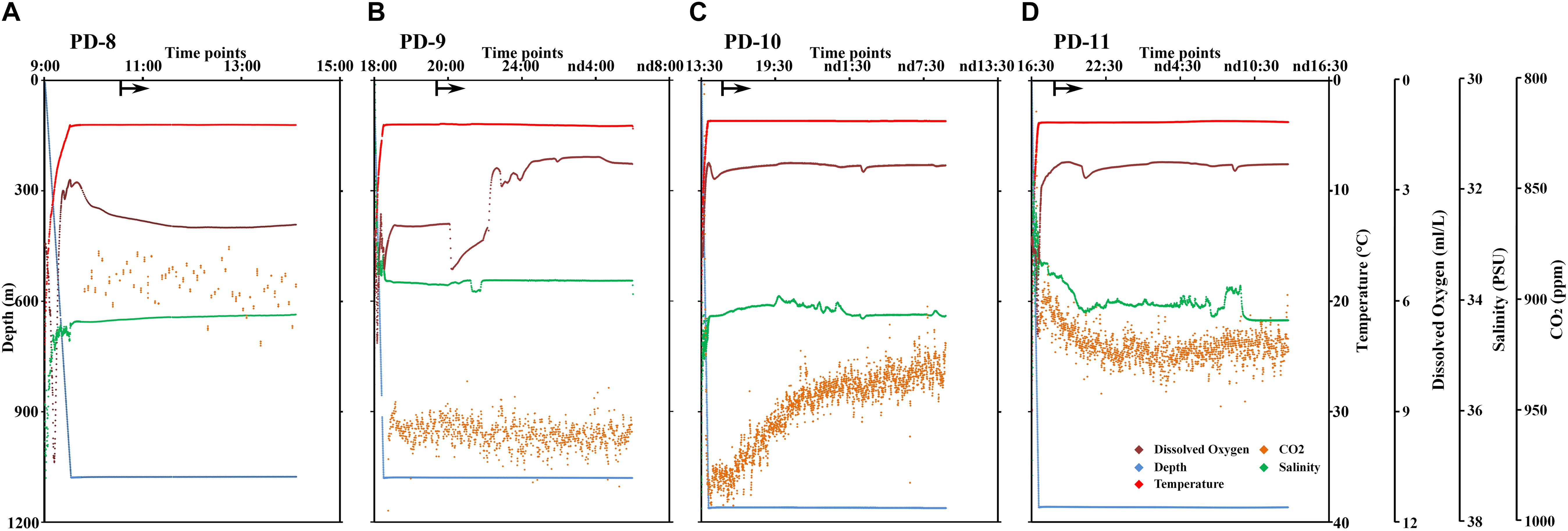
Figure 2. Profile of depth, temperature, dissolved oxygen concentration, salinity, and CO2 concentration along with the time points of lander deployment in the SCS. The data of physical environments were obtained in PD-8 (A), PD-9 (B), PD-10 (C), and PD-11 (D). The locations of four deployments were shown in Table 1 and Supplementary Figure S1. The arrows represent the time points at which the MISNAC apparatus was initiated. nd denotes next day.
The 14 MISNAC and ISMIFF samples were used for sequencing of 16S rRNA amplicons, followed by classification of the reads to reveal the microbial communities in the samples obtained by different methods at different time points (Figure 3 and Supplementary Figure S2). In total, 124,400 16S rRNA reads were obtained (at least 3,492 16S rRNA amplicon reads for each of the samples) (Table 1). We plotted the rarefaction curves of the observed OTUs using the sequences of the 16S rRNA gene amplicons (Figure 4A). All the curves reached a plateau, suggesting that the sequencing depth allowed to capture almost all the species in the samples. We also estimated the diversities of the microbial communities using the Shannon index (Figure 4B). Our statistics result also showed that the microbial communities of the MISNAC and ISMIFF samples were not significantly different in the same cruise (U-test, p > 0.05).
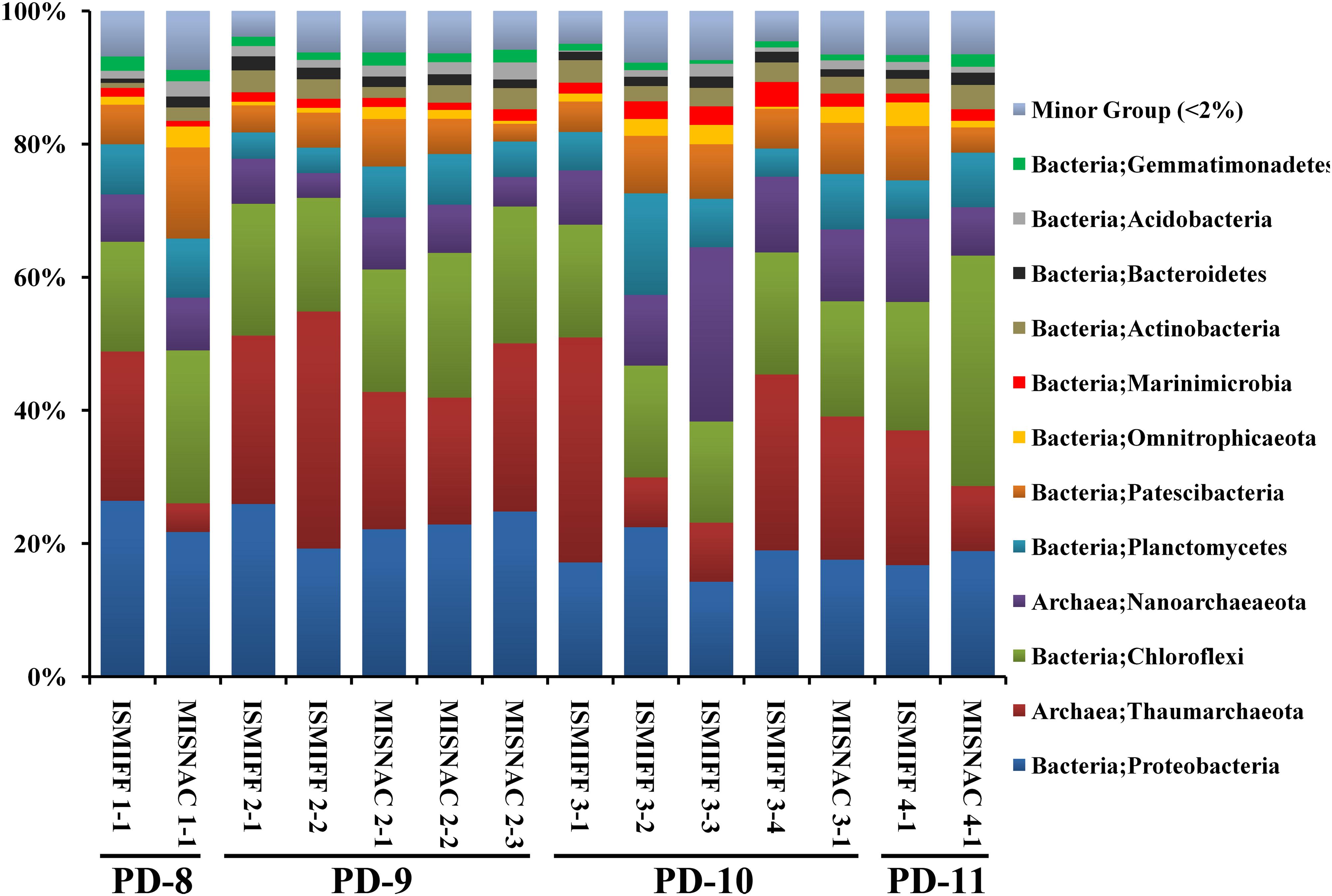
Figure 3. Microbial community structures at phylum level. The microbial samples in waters were collected at two ∼1,000 m depth sites by two methods: in situ filtration on membranes (ISMIFF); multiple in situ nucleic acid extraction (MISNAC). The information of samples was described in Table 1.
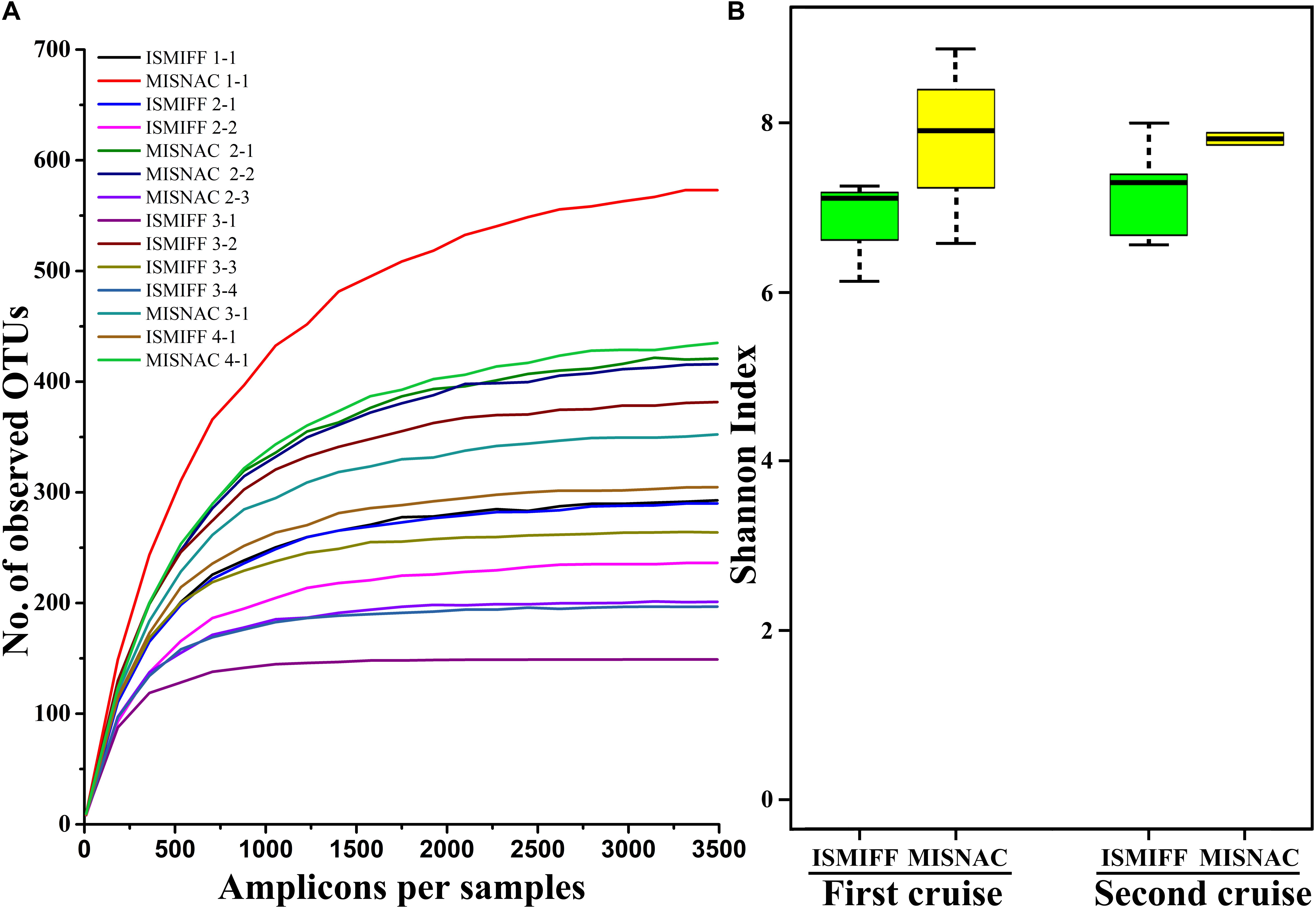
Figure 4. Rarefaction curve of observed OTUs (A) and Shannon index (B). The OTUs and Shannon index were calculated at a 3% dissimilarity using amplicon reads of 16S rRNA genes. The details of the samples are referred to Table 1.
Analysis of the sequencing data of the 16S rRNA gene amplicons showed that 12 phyla were dominant (>2%) in the samples: Proteobacteria, Thaumarchaeota, Chloroflexi, Nanoarchaeaeota, Planctomycetes, Patescibacteria, Omnitrophicaeota, Marinimicrobia, Actinobacteria, Bacteroidetes, Acidobacteria, and Gemmatinonadetes (Figure 3), which were totaled to be 93.7 ± 1.3% of the communities. The dominant phyla were further sorted into 21 major orders (>2%) (Supplementary Figure S2). At both phylum and order levels, the dominant taxa were not significantly different between the communities in the MISNAC and ISMIFF samples (one-way ANOVA, p > 0.05). The presence of some minor groups affiliated with Chloroflexi and Planctomycetes in the MISNAC samples might explain their higher biodiversity, compared with the ISMIFF (Figure 4B). Nevertheless, we could still observe slight differences in microbial communities between the samples obtained by the same method (Figure 3). This might reflect the daily variations of microbial communities in the deep-sea since the waters were filtered at different time points (Table 1). The PCA analysis was performed using the percentages of the orders in the different water samples (Figure 5). The samples from the same cruises tend to be clustered together, while those collected by different methods in each cruise could not be separated clearly. This again suggests that the slight community structural differences were caused by environmental variations such as CO2 flux at the sampling locations and times. This hypothesis will be examined in future by using all the nine sampling units of the MISNAC to obtain nucleic acids for detection of microbial community variations in continuous time periods.
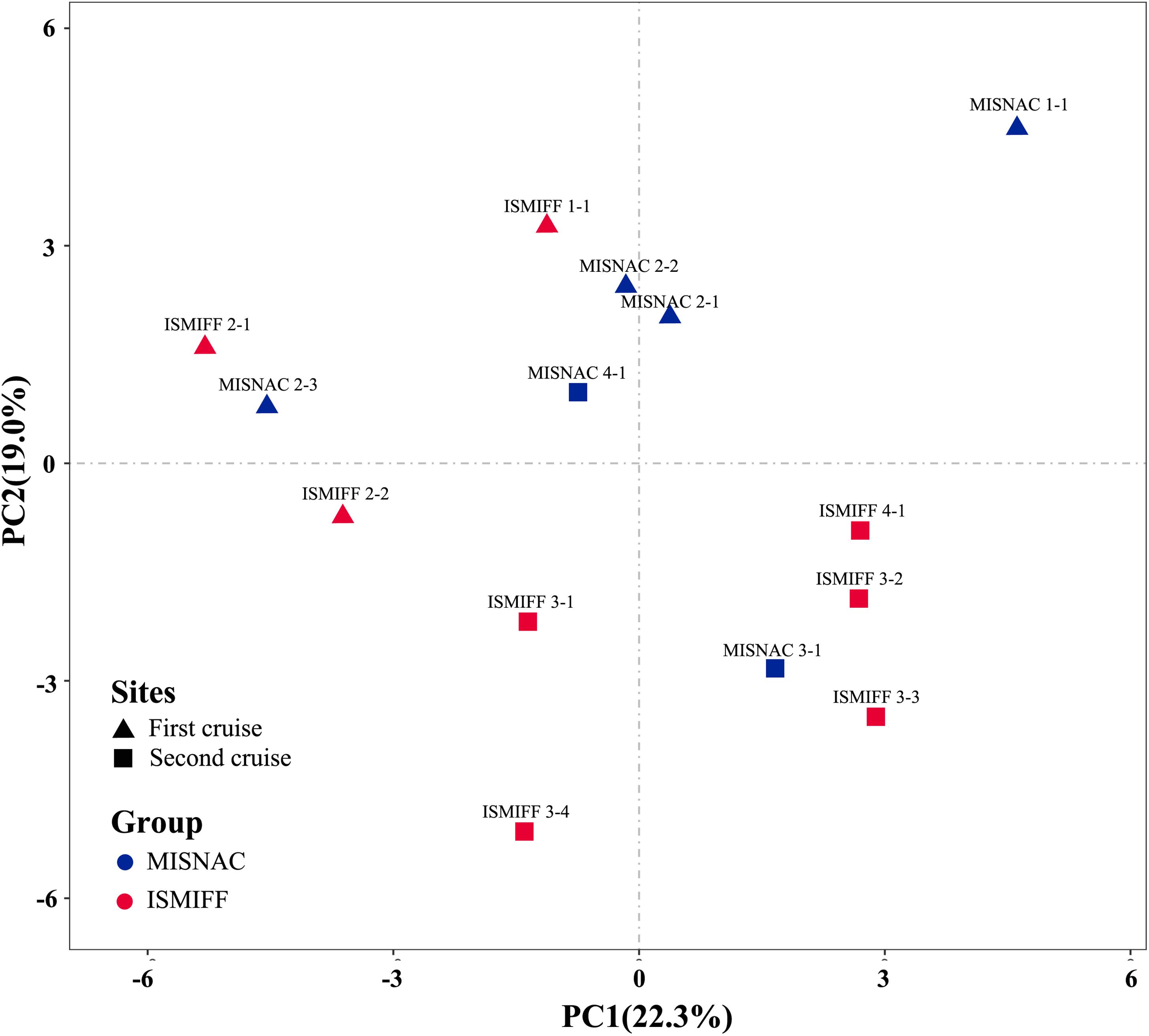
Figure 5. PCA analysis of microbial communities in the samples collected by different methods. The samples were as described in Table 1.
The prevalence of Proteobacteria in epipelagic layer and deep-sea subsurface sediments of the SCS had been reported (Tao et al., 2008; Yi et al., 2018). This is also the fact for the Chloroflexi that were almost ubiquitous in the global deep oceans (Morris et al., 2004; Varela et al., 2010). However, the percentages of Firmicutes in our ISMIFF and MISNAC samples were much less than those recently reported (Yi et al., 2018; Zhang et al., 2019), which might be due to different sampling locations, depths and times. Archaeal 16S rRNA gene sequences were affiliated with two dominant archaeal phyla Thaumarchaeota and Nanoarchaeaeota. Thaumarchaeota represented by Nitrosopumilus was one of the dominant species widely distributed in marine waters including the SCS (Hu et al., 2011; Zhang et al., 2019), which was critical for maintaining both carbon and nitrogen cycles in the waters and sediments (Wang et al., 2019b). In addition, the high abundance of Nanoarchaeaeota was also noticed in the sampling sites, which was not detected in previous studies (Tao et al., 2008; Yi et al., 2018; Zhang et al., 2019). This might be due to the collection of bottom water samples in this study. The spreading of Nanoarchaeaeota was also observed in hadal bottom waters and sediments (Cui et al., 2019; Gao et al., 2019).
Moreover, as the largest tropical marginal sea adjacent to the Pacific Ocean, the SCS was proposed to be vital in the global carbon cycle (Chen et al., 2001; Dai et al., 2009). The dissolved organic matter (DOM) in the deep-sea from the SCS might originate from surface photosynthesis of phytoplankton and terrestrial organic compounds, which might follow day–night rhythm. Therefore, the relative abundance of heterotropic microorganisms might have diurnal periodic changes, which perhaps explains the variations in the microbial community structures of our samples obtained with the same apparatus at different time points. However, the small number of time points for the current study was not sufficient to deduce the periodic changes of the deep-sea communities in day–night cycle.
Recently, we demonstrated the capacity of the MISNAC for RNA collection. During the third cruise, we obtained in situ RNA by the MISNAC with lander deployment PD-12 (Supplementary Figure S1). The sequencing information of metatranscriptomes was shown in Supplementary Table S1. The 16S rRNA gene reads extracted from metatranscriptomes were taxonomically sorted into Proteobacteria, Actinobacteria, Marinimicrobia, Planctomycetes, Chloroflexi, Verrucomicrobia, Thaumarchaeota and Euryarchaeota, indicating that they were the most active microbes in the sampling site (Figure 6A). Using the Niskin sample obtained in the third cruise, we also obtained the metatranscriptome with a replicate for a comparison. The relative abundances of 16S rRNA reads assigned to Euzebyales and Solirubrobacterales increased and those for Vibrionales and SAR11 decreased in the Niskin metatranscriptomes, compared with the MISNAC, which might be caused by the environmental changes such as pressure, temperature, and light during sampling by Niskin bottle and subsequent treatments onboard (Figure 6A). Moreover, the MISNAC metatranscriptomes contained notably more observed OTUs in 16S rRNA fragments, compared to the Niskin counterparts, as suggested by the rarefaction curves (Figure 6B). The result indicated that the in situ RNA of the microbes were better preserved by the MISNAC apparatus, while a large fraction of RNA molecules were degraded in the cells during the sampling by the Niskin bottle. The answer to the factors that raised the transcriptomic differences between the MISNAC and Niskin samples will be provided by future analyses using the frequency of functional genes in the transcriptomes.
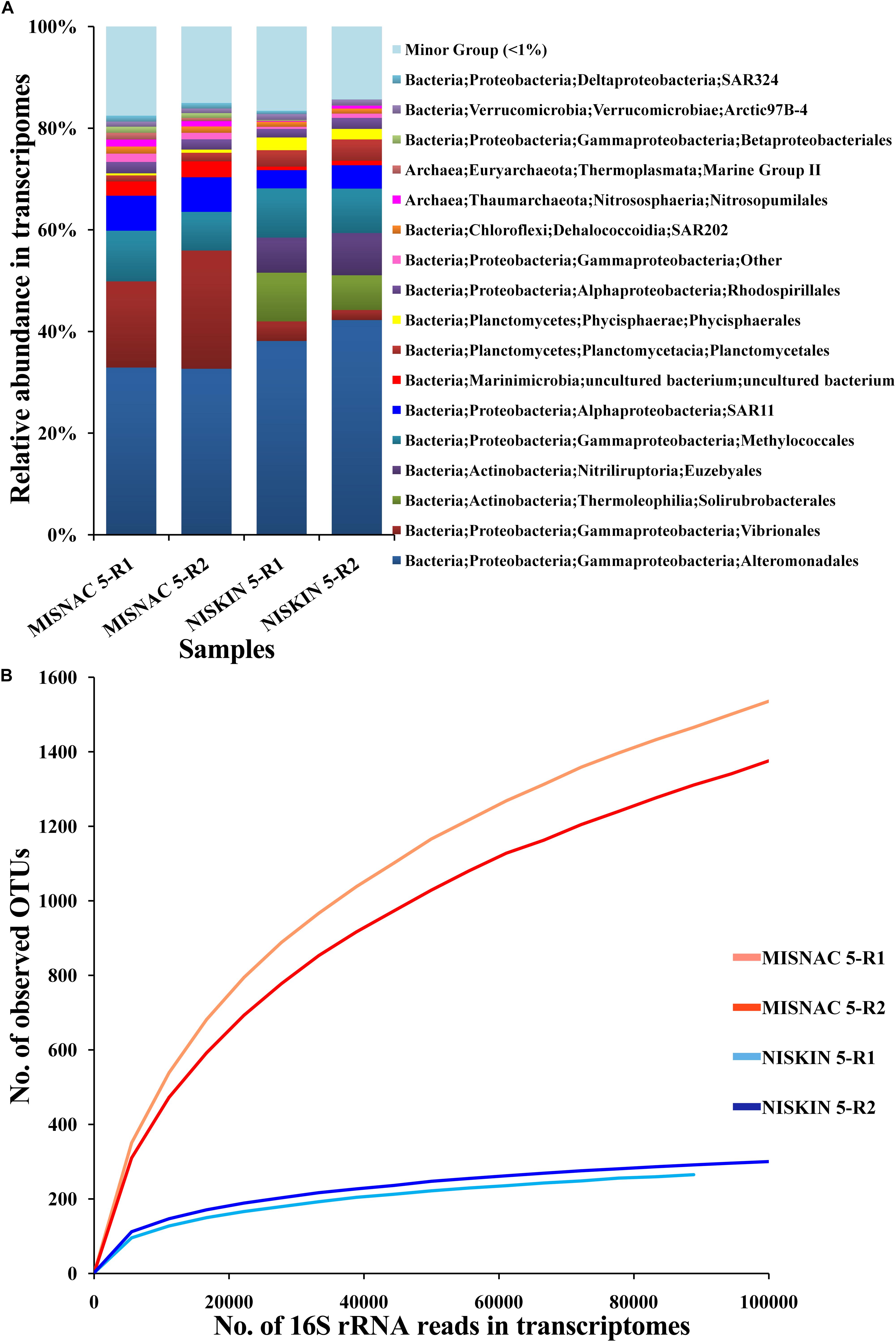
Figure 6. Classification of 16S rRNA transcripts at order level (A) and rarefaction curve of observed OTUs (B) in metatranscriptomes. The microbial samples in waters were collected at ∼1,000 m depth site during the third cruise PD-12 by two methods: multiple in situ nucleic acid extraction (MISNAC) and Niskin bottle (NISKIN). The 16S rRNA reads extracted from the metatranscriptomes were classified to reveal the active microbes in the samples. The number of observed OTUs was calculated at a 3% dissimilarity. The details of the samples are referred to Supplementary Table S1.
The application of MISNAC apparatus in nucleic acid extraction at ∼1,000 m depth in the SCS may facilitate the designation of autonomous in situ laboratories in the deep oceans. To the best of our knowledge, the MISNAC is the first deep-sea apparatus that can independently accomplish a series of tasks using the associated multiple working units, and will provide nucleic acids for subsequent molecular work and cell lysis for in situ analysis of cell components in the microbial cytoplasma. The D-ESP already has a capacity to perform quantitative PCR of functional genes using in situ extracted DNA but it could work at one time point in each deployment (Ussler et al., 2013).
Conclusion
The results in this study suggested that autonomous extraction of nucleic acids with multiple working units was feasible in deep oceans. With the MISNAC for automatic in situ samplings, in situ enrichment of the autotrophic microbes with 13C-labeled CO2 will allow us to uncover and quantify the carbon fixation processes by deep-sea organisms in near future. The largely consistency of microbial community structures between the MISNAC and ISMIFF samples indicates that the autonomous apparatuses could be applied for microbial sample collections in other extreme aquatic environments. Continuous sample collection by MISNAC apparatus will be applied to further explore diel variations of microbial community structure and to capture quick responses of functional genes in deep sea environments in future.
Data Availability Statement
The amplicon reads of 16S rRNA genes used for analysis of microbial communities were submitted to NCBI with the accession number: SRR9333628.
Author Contributions
Z-FW, W-LL, and YW designed the study. Z-FW, W-LL, JL, JC, and Y-ZX developed, prepared, and operated the MISNAC during the study. Z-FW, W-LL, JL, Y-ZX, and L-SH prepared and deployed the ‘Phoenix.’ Z-FW performed the data analysis with W-LL. Z-FW and YW drafted the manuscript. All authors contributed to the results and discussions and to the final version of the manuscript.
Funding
This study was supported by the National Key Research and Development Program of China (2016YFC0302501).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.00081/full#supplementary-material
References
Azam, F., Fenchel, T., Field, J., Gray, J., Meyerreil, L., and Thingstad, F. (1983). The ecological role of water-column microbes in the sea. Mar. Ecol. Prog. Ser. 10, 257–263. doi: 10.3354/meps010257
Breier, J. A., Sheik, C. S., Gomez-Ibanez, D., Sayre-McCord, R. T., Sanger, R., Rauch, C., et al. (2014). A large volume particulate and water multi-sampler with in situ preservation for microbial and biogeochemical studies. Deep Sea Res. Part I Oceanogr. Res. Pap. 94, 195–206. doi: 10.1016/j.dsr.2014.08.008
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336.
Chen, C. T. A., Wang, S. L., Wang, B. J., and Pai, S. C. (2001). Nutrient budgets for the South China Sea basin. Mar. Chem. 75, 281–300. doi: 10.1016/s0304-4203(01)00041-x
Chen, S., Yanqing, Z., Yaru, C., and Jia, G. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Claesson, M. J., Qiong, W., Orla, O. S., Rachel, G. D., Cole, J. R., Ross, R. P., et al. (2010). Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 38:e200. doi: 10.1093/nar/gkq873
Cui, G., Li, J., Gao, Z., and Wang, Y. (2019). Spatial variations of microbial communities in abyssal and hadal sediments across the Challenger Deep. PeerJ 17:e6961. doi: 10.7717/peerj.6961
Dai, M., Meng, F., Tang, T., Kao, S. J., Lin, J., Chen, J., et al. (2009). Excess total organic carbon in the intermediate water of the South China Sea and its export to the North Pacific. Geochem. Geophys. Geosyst. 10:Q12002. doi: 10.1029/2009gc002752
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgcomb, V. P., Taylor, C., Pachiadaki, M. G., Honjo, S., Engstrom, I., and Yakimov, M. (2016). Comparison of Niskin vs. in situ approaches for analysis of gene expression in deep Mediterranean Sea water samples. Deep Sea Res. Part II Top. Stud. Oceanogr. 129, 213–222. doi: 10.1016/j.dsr2.2014.10.020
Eloe, E. A., Shulse, C. N., Fadrosh, D. W., Williamson, S. J., Allen, E. E., and Bartlett, D. H. (2011). Compositional differences in particle-associated and free-living microbial assemblages from an extreme deep-ocean environment. Environ. Microbiol. Rep. 3, 449–458. doi: 10.1111/j.1758-2229.2010.00223.x
Estrada, M., Bayer-Giraldi, M., Felipe, J., Marrasé, C., Sala, M. M., and Vidal, M. (2009). Light and nutrient effects on microbial communities collected during spring and summer in the Beaufort Sea. Aquat. Microb. Ecol. 54, 217–231. doi: 10.3354/ame01268
Feike, J., Jürgens, K., Hollibaugh, J. T., Krüger, S., Jost, G., and Labrenz, M. (2012). Measuring unbiased metatranscriptomics in suboxic waters of the central Baltic Sea using a new in situ fixation system. ISME J. 6, 461–470. doi: 10.1038/ismej.2011.94
Gao, Z., Huang, J. M., Cui, G. J., Li, W. L., Li, J., Wei, Z. F., et al. (2019). In situ meta−omic insights into the community compositions and ecological roles of hadal microbes in the Mariana Trench. Environ. Microbiol. 11, 4092–4108. doi: 10.1111/1462-2920.14759
Gregory Caporaso, J., Bittinger, K., Bushman, F. D., DeSantis, T. Z., Andersen, G. L., and Knight, R. (2010). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267. doi: 10.1093/bioinformatics/btp636
Hu, A., Jiao, N., and Zhang, C. L. (2011). Community structure and function of planktonic Crenarchaeota: changes with depth in the South China Sea. Microb. Ecol. 62, 549–563. doi: 10.1007/s00248-011-9866-z
Huang, Y., Gilna, P., and Li, W. (2012). Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 25, 1338–1340. doi: 10.1093/bioinformatics/btp161
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2012). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Marietou, A., and Bartlett, D. H. (2014). Effects of high hydrostatic pressure on coastal bacterial community abundance and diversity. Appl. Environ. Microbiol. 80, 5992–6003. doi: 10.1128/AEM.02109-14
Morris, R. M., Rappe, M. S., Urbach, E., Connon, S. A., and Giovannoni, S. J. (2004). Prevalence of the Chloroflexi-related SAR202 bacterioplankton cluster throughout the mesopelagic zone and deep ocean. Appl. Environ. Microbiol. 70, 2836–2842. doi: 10.1128/aem.70.5.2836-2842.2004
Nunoura, T., Takaki, Y., Hirai, M., Shimamura, S., Makabe, A., Koide, O., et al. (2016). Hadal biosphere: insight into the microbial ecosystem in the deepest ocean on earth [Ecology]. Proc. Natl. Acad. Sci. U.S.A. 112, 1230–1236. doi: 10.1073/pnas.1421816112
Olins, H. C., Rogers, D. R., Preston, C., Ussler, W., Pargett, D., Jensen, S., et al. (2017). Co-registered geochemistry and metatranscriptomics reveal unexpected distributions of microbial activity within a hydrothermal vent field. Front. Microbiol. 8:1042. doi: 10.3389/fmicb.2017.01042
Patel, R. K., and Mukesh, J. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619
Qiong, W., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/aem.00062-07
Ramiro, L., Shinichi, S., Guillem, S., Cornejo-Castillo, F. M., Isabel, F., Hugo, S., et al. (2014). Metagenomic 16S rDNA Illumina tags are a powerful alternative to amplicon sequencing to explore diversity and structure of microbial communities. Environ. Microbiol. 16, 2659–2671. doi: 10.1111/1462-2920.12250
Scholin, C., Doucette, G., Jensen, S., Roman, B., Pargett, D., and Marin, R. III, et al. (2009). Remote detection of marine microbes, small invertebrates, harmful algae, and biotoxins using the environmental sample processor (ESP). Oceanography 22, 158–167. doi: 10.5670/oceanog.2009.46
Tao, L., Peng, W., and Wang, P. (2008). Microbial diversity in surface sediments of the Xisha Trough, the South China Sea. Acta Ecol. Sin. 28, 1166–1173. doi: 10.1016/s1872-2032(08)60036-0
Urbach, E., Vergin, K. L., Larson, G. L., and Giovannoni, S. J. (2007). Bacterioplankton communities of Crater Lake, OR: dynamic changes with euphotic zone food web structure and stable deep water populations. Hydrobiologia 574, 161–177. doi: 10.1007/978-1-4020-5824-0_10
Ussler, W., Preston, C., Tavormina, P., Pargett, D., Jensen, S., Roman, B., et al. (2013). Autonomous application of quantitative PCR in the deep sea: in situ surveys of aerobic methanotrophs using the deep-sea environmental sample processor. Environ. Sci. Technol. 47, 9339–9346. doi: 10.1021/es4023199
Varela, M. M., Vanaken, H. M., and Herndl, G. J. (2010). Abundance and activity of Chloroflexi-type SAR202 bacterioplankton in the meso- and bathypelagic waters of the (sub)tropical Atlantic. Environ. Microbiol. 10, 1903–1911. doi: 10.1111/j.1462-2920.2008.01627.x
Wang, Y., Gao, Z. M., Li, J., He, L. S., Cui, G. J., Li, W. L., et al. (2019a). Hadal water sampling by in situ microbial filtration and fixation (ISMIFF) apparatus. Deep Sea Res. Part I Oceanogr. Res. Pap. 144, 132–137. doi: 10.1016/j.dsr.2019.01.009
Wang, Y., Huang, J.-M., Cui, G.-J., Nunoura, T., Takaki, Y., Li, W.-L., et al. (2019b). Genomics insights into ecotype formation of ammonia-oxidizing archaea in the deep ocean. Environ. Microbiol. 21, 716–729. doi: 10.1111/1462-2920.14518
Yi, L., Lin Lin, S., Mei Ling, S., Hai Nan, S., Xi Ying, Z., Bin Bin, X., et al. (2018). Vertical and horizontal biogeographic patterns and major factors affecting bacterial communities in the open South China Sea. Sci. Rep. 8:8800. doi: 10.1038/s41598-018-27191-w
Zhang, J., Kobert, K., Flouri, T., and Stamatakis, A. (2014). PEAR: a fast and accurate illumina paired-end reAd mergeR. Bioinformatics 30, 614–620. doi: 10.1093/bioinformatics/btt593
Keywords: in situ filtration, DNA extraction, cell lysis, deep-sea, microbial community
Citation: Wei Z-F, Li W-L, Li J, Chen J, Xin Y-Z, He L-S and Wang Y (2020) Multiple in situ Nucleic Acid Collections (MISNAC) From Deep-Sea Waters. Front. Mar. Sci. 7:81. doi: 10.3389/fmars.2020.00081
Received: 09 August 2019; Accepted: 31 January 2020;
Published: 18 February 2020.
Edited by:
Pei-Yuan Qian, Hong Kong University of Science and Technology, Hong KongReviewed by:
Yu Zhang, Shanghai Jiao Tong University, ChinaYongyu Zhang, Qingdao Institute of Bioenergy and Bioprocess Technology (CAS), China
Copyright © 2020 Wei, Li, Li, Chen, Xin, He and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Wang, d2FuZ3lAaWRzc2UuYWMuY24=
†These authors have contributed equally to this work