 Nichole Flynn
Nichole Flynn Naweed I. Syed
Naweed I. Syed- Hotchkiss Brain Institute and Alberta Children’s Hospital Research Institute, Department of Cell Biology and Anatomy, University of Calgary, Calgary, AB, Canada
The mechanism of postsynaptic neurotransmitter receptor clustering has been best described at the neuromuscular junction (NMJ), where pockets of nicotinic acetylcholine receptors (nAChRs) in muscle fiber are redistributed to the synaptic site upon motor neuron innervation. This process of receptor localization is facilitated by agrin signaling, and the stability of the resulting nAChR clusters depends upon the activation of Src family kinases (SFKs). In contrast to the NMJ, however, the cellular signaling mechanisms orchestrating the clustering of nAChRs in the central nervous system (CNS) remain poorly defined. Furthermore, our understanding of the role of SFKs in the CNS is also limited. Here, we provide evidence that SFK activation is required for synapse formation between pairs of identified neurons isolated from the CNS of Lymnaea stagnalis. Furthermore, we suggest that SFKs are involved in the functional redistribution of nAChRs to the synaptic contact sites in isolated axons. To the best of our knowledge, this study is the first to demonstrate a role for SFKs in the clustering of nAChRs in central neurons, suggesting that the mechanisms of receptor clustering between the peripheral nervous system (PNS) and CNS are likely conserved.
Introduction
Historically, the model synapse used to study synaptogenesis is the neuromuscular junction (NMJ), as it is large and experimentally accessible in most preparations (Madhavan and Peng, 2005; Darabid et al., 2014). The cardinal feature of NMJ development is the redistribution of postsynaptic nicotinic acetylcholine receptors (nAChRs) to the site of nerve-muscle contact, following innervation of the muscle by motor neurons (Mejat et al., 2003; Darabid et al., 2014). The process of nAChR clustering is initiated by agrin, a proteoglycan predominantly released by neurons and localized in the basal lamina (Yang and Nelson, 2004; Ghazanfari et al., 2011; Darabid et al., 2014). Src family kinases (SFKs), notably Src and Fyn, are activated by agrin (and other trophic factors), where they then bind to and phosphorylate nAChR β and δ subunits, fostering the linkage between nAChRs and the cytoskeleton (Yang and Nelson, 2004; Ghazanfari et al., 2011; Darabid et al., 2014).
In contrast to the NMJ, little is known about the clustering of cholinergic receptors in the central nervous system (CNS) (Yang and Nelson, 2004; Colon-Ramos, 2009). Furthermore, the precise role of SFKs in the CNS remains largely unexplored (Salter, 1998). Src is one of nine family members, five of which are expressed in the CNS: (1) Fyn, (2) Lck, (3) Lyn, (4) Src, and (5) Yes (Salter, 1998; Kalia and Salter, 2003). SFKs have been found to upregulate N-methyl-D-aspartate receptor function (Yu et al., 1997; Ohnishi et al., 2011), mediating the induction of long-term potentiation in the hippocampus and spatial learning in rodents (Grant et al., 1992; Lu et al., 1998; Salter, 1998; Zhao et al., 2000). However, the role of SFKs in central synapse formation remains to be explored.
Work from our lab has shown that synapses between identified Lymnaea neurons can form between the isolated axons of left pedal dorsal 1 (LPeD1, postsynaptic) neurons and visceral dorsal 4 (VD4, presynaptic) somata, but require an extrinsic source of trophic factors provided by ganglion-conditioned media (CM) (Meems et al., 2003; Xu et al., 2014). It appears that there is a limited pool of nAChRs present in isolated LPeD1 axons (cell body removed) and that contact with the presynaptic soma (in the presence of CM) redirects the nAChRs to the contact site between the isolated cell body and axon (Xu et al., 2014). As such, the Lymnaea soma–axon model of synapse formation provides a unique opportunity to study the redistribution of neurotransmitter receptors from extrasynaptic compartments to the synaptic contact site during synaptogenesis.
Here, we demonstrate that SFKs are required for central synapse formation between pairs of individually identified Lymnaea neurons. We find that SFK kinase activation is required for the neurotrophic factor-induced clustering of nAChRs at the contact site of soma–axon synapses. Furthermore, we show that this phenomenon is specific to contact with certain target cells only and is required for the formation of excitatory, but not inhibitory synapses.
Materials and Methods
Animals and Cell Culture
Freshwater snails (Lymnaea stagnalis) were raised in well-aerated aquaria at room temperature (20–22°C) on a diet of romaine lettuce. Six- to eight-week-old (10–15 mm shell length) animals were used for the isolation of individual neurons, whereas 2- to 4-month-old snails (20–30 mm shell length) were used to make Lymnaea ganglion (brain)-conditioned media (CM). Owing to the fact that Lymnaea are hermaphroditic, distinctions between male and female subjects do not apply.
The cell isolation protocol is well-established and has been previously described in detail (Syed et al., 1990; Ridgway et al., 1991). Briefly, snails were deshelled and anesthetized in a 10% solution of Listerine® in normal Lymnaea saline (in mM: 51.3 NaCl, 1.7 KCl, 4.0 CaCl2, and 1.5 MgCl2) buffered to pH 7.9 with HEPES. The central ring ganglia (CNS of Lymnaea) were then removed under sterile conditions and enzymatically treated with trypsin (2 mg/ml; T9201; Sigma-Aldrich) for 20–22 min followed by a trypsin inhibitor (2 mg/ml; T9003; Sigma-Aldrich) for 15 min. Both the trypsin and trypsin inhibitor were dissolved in defined medium (DM; L-15; Life Technologies; special order). The ganglia were then pinned down to a Sylgard® dish in high osmolarity DM (750 μl of 1M glucose to added 20 ml DM; raises normal osmolarity of the DM from 130–145 to 180–195 mOsm). After removing the outer and inner sheath protecting the ganglia, visually identified neurons were isolated by applying gentle suction through a fire-polished glass pipette that had been treated with Sigmacote®. The neurons were then transferred to a poly-L-lysine-coated culture dish and plated in CM. To create soma–soma pairs, cell bodies were placed side by side in culture, in direct contact with one another. To create soma–axon pairs, postsynaptic LPeD1 neurons with long axons intact were cultured first (with their axons laid straight). Isolated VD4 somata were then placed in contact with the axon, as close as possible to the middle. The cell pairs were then left to settle and adhere to the dish for ∼30 min. The LPeD1 somata were then carefully severed from the axon, using a sharp glass pipette (1.5 mm internal diameter; World Precision Instruments), and removed from the culture dish via gentle suction.
Conditioned media was prepared by incubating 12 central ring ganglia (previously washed in antibiotic saline containing 50 μg/ml gentamycin) in 6.5 ml of DM (serum-free 50% L-15 medium with added inorganic salts in mM: 40 NaCl, 1.7 KCl, 4.1 CaCl2, 1.5 MgCl2, and 20 μM gentamicin; buffered to pH 7.9 with HEPES) in Sigmacote® -treated Petri dishes. The dishes were then placed in a humidified incubator at room temperature for 3 days to make 1 × CM. The CM was frozen at −20°C if not used immediately. The ganglia were then rewashed several times in antibiotic saline and incubated in fresh DM for another 4 days (2 × CM). 2 × CM was used in all experiments.
To test synapse formation between VD4–LPeD1 and VD4–left pedal E (LPeE) pairs, neurons were cultured overnight (14–18 h) in CM or CM + Src kinase inhibitor I (SrcI; 5 μM; 567805; EMD Millipore) and recorded from the next day. SrcI was added 1 h post-plating, before the onset of synaptogenesis. To test the effects of SrcI on synaptic transmission in VD4–LPeD1 pairs, neurons were plated in CM overnight and tested for synapse formation the next day. After confirming the presence of excitatory synapses, 5 μM SrcI was added to the culture dishes and incubated at room temperature for 1–1.5 h. The cell pairs were then recorded from (intracellularly) a second time to compare synaptic strength and membrane properties to untreated conditions.
Electrophysiology
Conventional intracellular recording techniques were used to monitor single cell and synaptic physiology. Glass microelectrodes (1.5 mm internal diameter; World Precision Instruments) were pulled using a vertical pipette puller (Model 700C, David Kopf Instruments) and filled with a saturated solution of K2SO4. Only electrodes with resistances between 20 and 50 MΩ were used. Cells were visualized under an inverted microscope (Axiovert 135; Zeiss), and Narashige micromanipulators were used to impale the cells. The amplified electrical signals (Neuron Data Instrument) were relayed through a digitizer (Digidata 1440A; MDS) and recorded on Axoscope 10.2 (MDS).
To functionally test whether the nAChRs expressed on LPeD1 neurons were excitatory (appropriate—as seen in vivo) or inhibitory (inappropriate), microelectrodes were pulled as stated above. The tip was then enlarged under a microforge, to a size of < 1 μm, and the microelectrode was filled with acetylcholine (ACh; 10–6 M dissolved in DM; A6625; Sigma Aldrich). The ACh-filled microelectrodes were then placed in close proximity to the cells but at a distance to avoid mechanical stimulation (∼60 μm; determined using vehicle control pulses of DM only). ACh was then pressure applied onto the cell bodies (400 ms pulses; 10 psi) using a PV800 pneumatic pump while recording from the cells intracellularly. To determine if excitatory or inhibitory receptors were functionally present, all cells were consistently held at a membrane potential between −55 and −60 mV (above the reversal potential for chloride). A response to a fixed pulse of ACh was deemed “excitatory” if the cell depolarized, usually leading to a train of action potentials. The response was deemed “inhibitory” instead if the cell became hyperpolarized when held at −55 or −60 mV, or if the cell was firing tonically before the pulse of ACh, a hyperpolarization and cessation of firing was induced. Furthermore, the response to ACh was seen to reverse at −70 mV in cells expressing inhibitory nAChRs, but did not in those expressing excitatory receptors.
To test synapse formation, pre- and postsynaptic neurons were impaled simultaneously. A synapse was confirmed to have formed if single-action potentials induced in the presynaptic neuron, VD4, induced 1-for-1 excitatory postsynaptic potentials (EPSPs) or inhibitory postsynaptic potentials (IPSPs) in the respective postsynaptic neurons, LPeD1 and LPeE. At this stage, all cells were consistently held at a membrane potential of −100 mV, to accurately compare postsynaptic potential (PSP) amplitude in one culture condition versus another. To further characterize whether the synapses formed where excitatory or inhibitory, postsynaptic neurons were brought up to a membrane potential of −60 mV (above the reversal potential for chloride). A train of action potentials was then induced in VD4, which either triggered a depolarization (most often leading to spiking) in LPeD1 (forms an excitatory synapse with VD4), or a hyperpolarization and cessation of firing in LPeE (forms an inhibitory synapses with VD4; response reversed at −70 mV, the reversal potential for chloride).
To functionally determine where nAChRs were localized, ACh-filled microelectrodes (10–6 M) were positioned at either the synaptic contact site or extrasynaptic sites on the postsynaptic cell, LPeD1 (two were picked on either end of the axon). ACh was then applied to each site, while injecting the appropriate amount of current to hold LPeD1 at −50 mV, to quantify the response to ACh. Data were analyzed to quantify the number of action potentials elicited by the application of ACh at contact versus distal (extrasynaptic) sites. For the distal sites, where an attempt was made to measure the response to ACh at two extrasynaptic sites, the mean number of action potentials was statistically analyzed. Control experiments were conducted (not shown) by adding a dye (Fast Green FCF; F-7258; Sigma Aldrich) to the ACh solution to ensure minimal spread. In a subset of these experiments, microperfusion was achieved using a suction electrode placed in opposition to the puffing electrode to create a very thin, focally applied stream of ACh. Use of the suction electrode did not affect the results, and so for simplicity, it was not used for the remainder of the study. Inclusion of the dye confirmed that the spread of ACh was minimal using 400 ms pulses, applied at a pressure of 4 psi (even in the absence of the suction electrode).
To test synaptic transmission and short-term plasticity, current was injected into VD4 to trigger postsynaptic responses in LPeD1 or LPeE. As previously described by Luk et al. (2011), square pulses of current were injected into VD4 to trigger a tetanus comprised of at least seven action potentials by holding the neuron in a depolarized state. PSP amplitudes were then compared before and following the tetanus.
Statistical Analysis
The percentages of cells that expressed excitatory nAChRs and the percentage of cell pairs that formed synapses were compared using Pearson’s chi-squared test. If a general effect was discovered with an omnibus chi-squared test (p < 0.05), individual treatments were compared to one another with post hoc chi-squared tests to analyze significance. Synaptic transmission and action potential threshold for VD4–LPeD1 pairs were analyzed using a paired samples t test. Synaptic strength between VD4–LPeE neurons was compared using a multivariate analysis of variance (ANOVA), with the culture treatment as the fixed factor and the mean EPSP amplitude and short-term potentiation as the dependent variables. Data for the nAChR clustering experiment were analyzed using a mixed-model ANOVA, with culture conditions as between group variable and synaptic site as the within group variable. All of the above statistical analyses were performed using SPSS © Statistics 21 for Windows.
Results
SFKs Are Required for Soma–Soma Synapse Formation
We have previously shown that excitatory synapse formation between soma–soma pairs of individually identified L. stagnalis neurons requires an extrinsic source of neurotrophic factors (supplied by CM). More specifically, excitatory synapse formation between the presynaptic neuron VD4 and the postsynaptic neuron LPeD1 relies upon activity-induced calcium influx in LPeD1, activation of the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) cascade, and expression of the transcription factor, Lymnaea menin, which are required for the expression of excitatory nAChRs (Xu et al., 2009; Flynn et al., 2014). Without neurotrophic factors, the nAChR phenotype remains inhibitory by “default” (Xu et al., 2009). We have identified nAChR subunits in Lymnaea that are either cation or chloride selective (van Nierop et al., 2005, 2006); however, the growth factor-triggered events that lead to the clustering of excitatory nAChRs and subsequent synapse formation remain unknown.
To test if Src family kinases (SFKs) are required for excitatory synapse formation (Figure 1), individual LPeD1 neurons were plated alongside single VD4 neurons in a soma–soma configuration (Figure 1B) in CM (control) or CM + SrcI (5 μM). SrcI is a selective, cell permeable inhibitor of SFKs, capable of inhibiting both Src and Lck. After 14–18 h of pairing, intracellular recordings were made from both VD4 and LPeD1 neurons to determine if an excitatory synapse had formed (as would be seen in vivo). In CM, 92% of cell pairs formed excitatory synapses (n = 25; Figure 1A). Of the remaining pairs, 4% formed inappropriate (not seen in vivo) inhibitory synapses, and 4% failed to synapse at all (Figure 1C). In contrast, 0% of VD4–LPeD1 soma–soma pairs formed excitatory synapses in the presence of SrcI (n = 19; Figure 1Ai), which was significantly less than the percentage of pairs that formed excitatory synapses in CM [χ2(1) = 36.625, p < 0.001; Figure 1C]. The majority of pairs (84%) failed to form synapses altogether, while a smaller portion (16%) formed inappropriate inhibitory synapses (Figure 1C). It thus appears that SFKs are required for synapse formation between VD4–LPeD1 pairs.
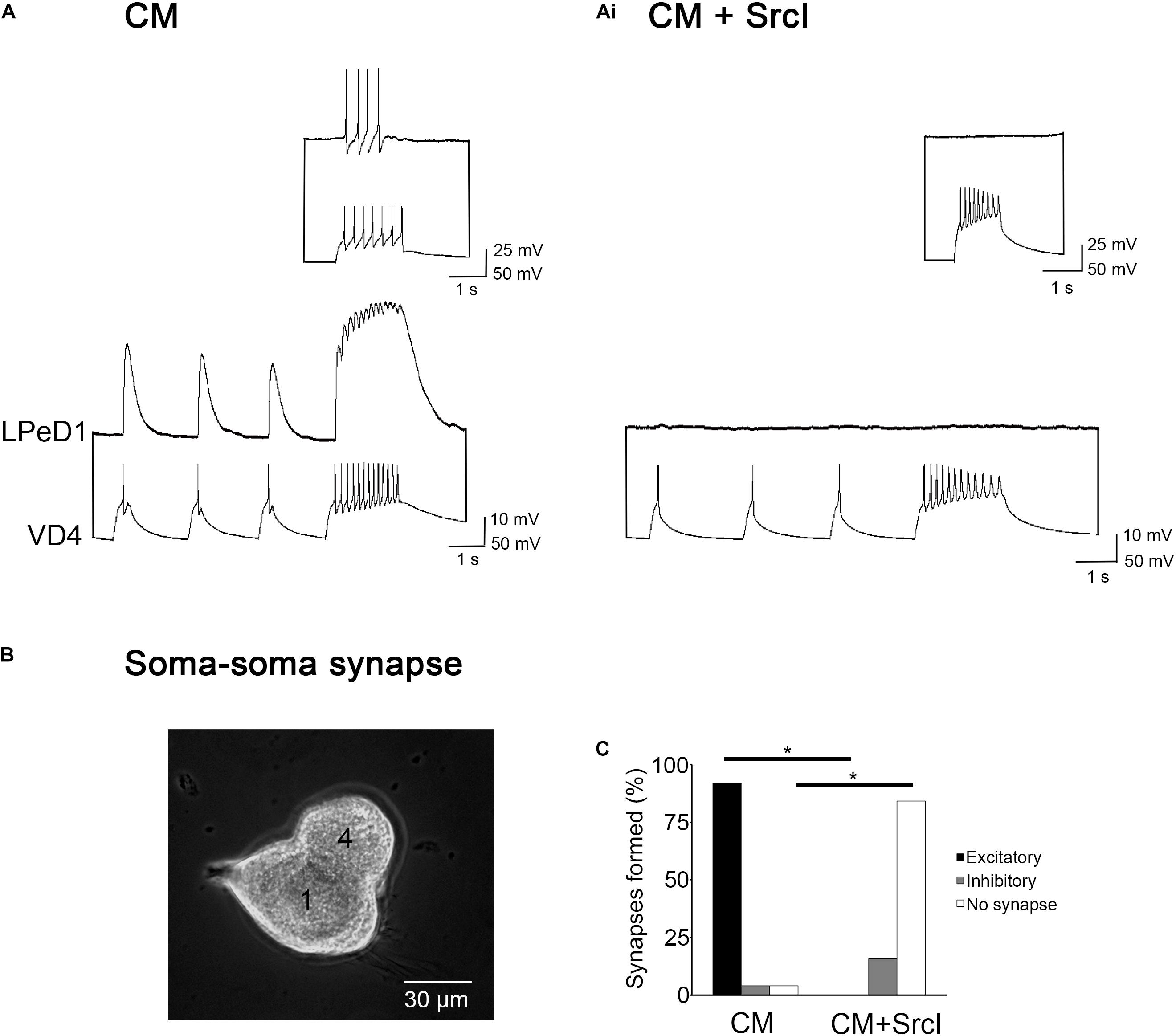
Figure 1. Src family kinases (SFKs) are required for soma–soma synapse formation between visceral dorsal 4 (VD4) and left pedal dorsal 1 (LPeD1) neurons. (A) Individual VD4 and LPeD1 neurons were paired in a soma–soma configuration overnight in conditioned media (CM). Both cells were impaled with sharp intracellular electrodes the next day and held at –100 mV for consistency. Action potentials triggered in VD4 induced 1-for-1 excitatory postsynaptic potentials (EPSPs) in LPeD1 (n = 25). Inset: a train of action potentials triggered in VD4 induced spiking in LPeD1 when held at –60 mV, confirming the presence of an excitatory synapse. (Ai) The SFK inhibitor, SrcI (5 μM), prevented synapse formation (n = 19). Action potentials triggered in VD4 did not induce a response in LPeD1 at –100 mV, or near threshold (–60 mV; inset). (B) Experimental preparation. Phase contrast image of a soma–soma synapse imaged at 20 × magnification. 4 = VD4. 1 = LPeD1. (C) Summary of the percentage of soma–soma pairs that formed synapses, with a significance of p ≤ 0.001, as determined using Pearson’s chi-squared test.
SFK Inhibition Does Not Affect the Postsynaptic Expression of nAChRs
As mentioned above, we have demonstrated that CM serves as an extrinsic source of neurotrophic factors required for the postsynaptic expression of excitatory nAChRs in Lymnaea neurons (Xu et al., 2009; Flynn et al., 2014). To determine if the SrcI-induced perturbation of synapse formation was due to a change in the functional expression of postsynaptic nAChRs, single LPeD1 neurons were plated in CM or CM + SrcI (5 μM) overnight, for 14–18 h (Figure 2). The next day, intracellular recordings were made from these neurons while pressure-applying (puffing) ACh (10–6 M) onto their somata (Figure 2A). In CM, 84% of LPeD1 neurons expressed appropriate (as normally seen in vivo), excitatory receptors (n = 18; Figure 2B). Similarly, neurons treated with SrcI overnight also expressed excitatory receptors [75%; n = 12; Figure 2Bi; χ2(1) = 1.000, p = 0.317]. These data suggest that inhibition of SFKs with SrcI does not perturb synapse formation by affecting the expression profile of nAChRs.
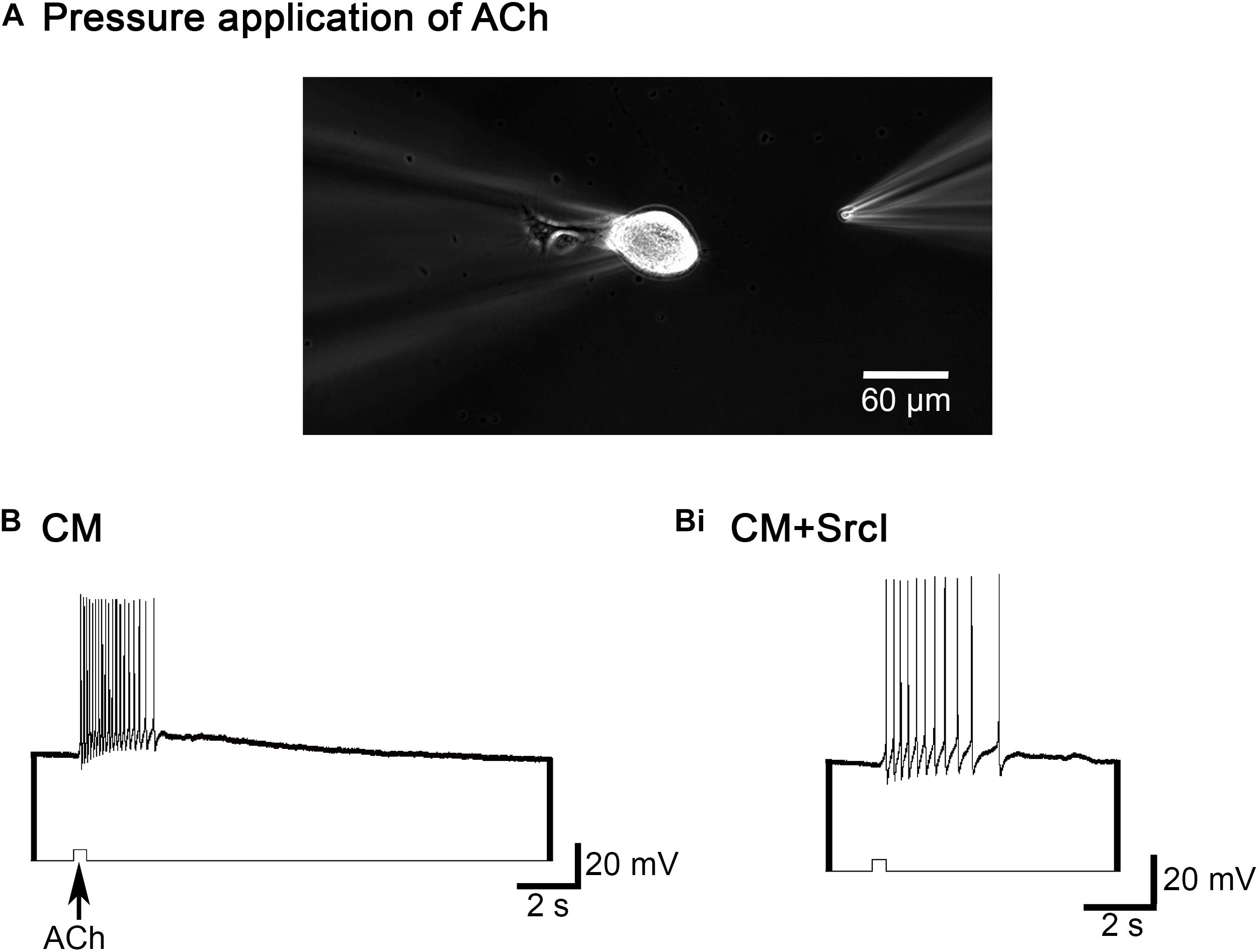
Figure 2. SrcI does not affect the response of left pedal dorsal 1 (LPeD1) (postsynaptic) to acetylcholine (ACh). (A) Experimental preparation. Phase contrast image of a single LPeD1 neuron impaled by a sharp intracellular electrode at 10 × magnification. Another electrode with a tip diameter of 1–5 μm was used to pressure apply ACh onto the cell body of LPeD1. (B) Single LPeD1 neurons were cultured in conditioned media (CM) and impaled with intracellular electrodes. Exogenous application of ACh (10–6 M) triggered an excitatory response in the postsynaptic neuron, LPeD1 (n = 18). (Bi) Exogenous application of ACh also induced an excitatory response in LPeD1 neurons treated with SrcI (n = 12; 5 μM).
SFK Inhibition Does Not Block Synaptic Transmission or Short-Term Potentiation
Src family kinases are expressed both pre- and postsynaptically (Salter, 1998; Kalia and Salter, 2003), where they interact with and regulate both ligand- and voltage-gated ion channels (Thomas and Brugge, 1997; Groveman et al., 2012). As such, we wanted to determine if treatment of VD4–LPeD1 pairs with SrcI perturbed the process of synapse formation by functionally affecting the presynaptic cell, VD4 (Figure 3). To investigate this, synaptic transmission and short-term potentiation (STP) was compared in cell pairs before and following treatment with SrcI (Figures 3A,Ai). VD4 and LPeD1 neuron were soma–soma paired overnight (14–18 h) in CM. The next day, control intracellular recordings were made to confirm that excitatory synapses had indeed formed. SrcI (5 μM) was then added to the media for 1–1.5 h. After incubation with SrcI, the synapses were tested again, to compare the synaptic transmission between treated and untreated conditions. The mean EPSP amplitude of control synapses was 12.3 ± 1.6 mV (n = 16), which was not significantly different from the EPSP amplitude of those same pairs following treatment with SrcI (10.6 ± 0.9 mV; Figure 3B; t-test, p = 0.275), suggesting that synaptic transmission is not the mechanism by which SrcI perturbs synapse formation in VD4–LPeD1 pairs.
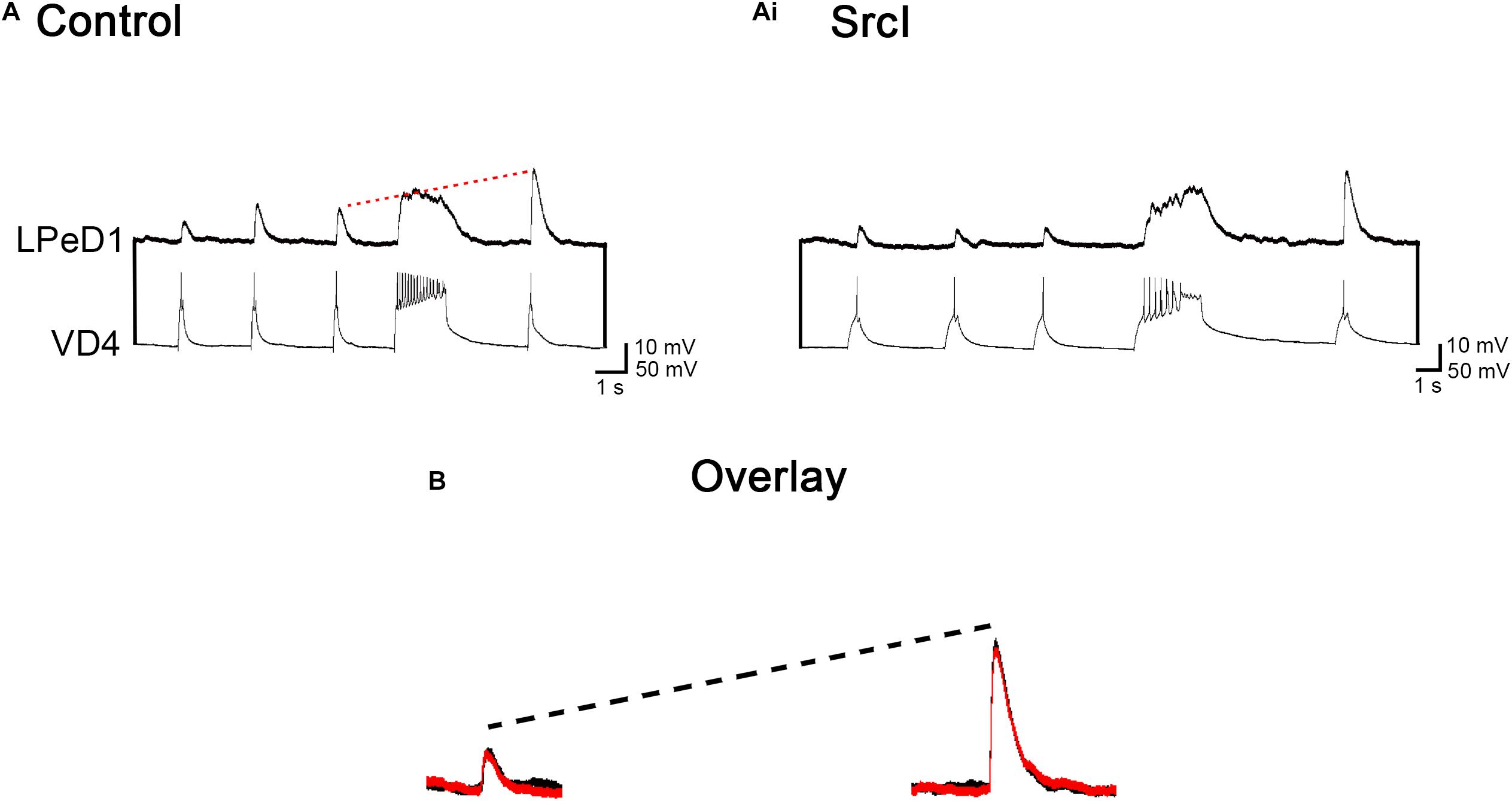
Figure 3. Treatment of visceral dorsal 4 (VD4)–left pedal dorsal 1 (LPeD1) soma–soma pairs with SrcI does not affect synaptic transmission or short-term potentiation (STP). (A) VD4–LPeD1 pairs were cultured in a soma–soma configuration overnight in conditioned media (CM). Both cells were impaled with intracellular electrodes the next day, to confirm that excitatory synapses had formed. Pre- and post-tetanus EPSP amplitudes were measured in LPeD1 (held at –100 mV for consistency). The red dotted line illustrates that post-tetanus EPSPs were potentiated, on average, by 207%, compared to their pretetanus counterparts (n = 16). (Ai) Following control recordings, VD4–LPeD1 pairs were incubated in 5 μM Src for 1–1.5 h. Pre- and post-tetanus EPSP amplitudes were then measured in LPeD1. Post-tetanus EPSPs were potentiated, on average, by 175% (n = 16). (B) Overlay of pretetanus EPSPs (left) and potentiated post-tetanus EPSPs (right) in control and SrcI-treated cell pairs. Black = control. Red = SrcI. Neither synaptic transmission (p = 0.275) nor STP (p = 0.086) were affected by treatment with SrcI, as determined using a paired samples t-test (n = 16).
Synaptic plasticity was also tested in these cell pairs to determine if the presynaptic cell was affected by treatment with SrcI. We have previously shown that a unique form of STP exists in Lymnaea synapses that is use, but not time dependent. This form of STP requires presynaptic calcium and calcium/calmodulin-dependent protein kinase II (CaMKII) (Luk et al., 2011). To produce STP in VD4–LPeD1 synapses, a tetanus was induced in VD4. A post-tetanus triggered action potential, then generated an EPSP in LPeD1 that was significantly larger compared to pretetanus EPSPs. Specifically, STP was compared in VD4–LPeD1 pairs, before and following treatment with 5 μM SrcI (Figures 3A,Ai). There was no significant difference between the amount of potentiation produced in control versus SrcI-treated conditions (n = 16; Figure 3B; t test, p = 0.086). Specifically, post-tetanus EPSPs were potentiated by 207 ± 15% before the addition to SrcI. Following treatment with SrcI, post-tetanus EPSPs were still potentiated by 175 ± 19% (Figure 3B), suggesting that STP (a presynaptic phenomenon) is not affected by SFK inhibition. To ensure that pharmacological treatment was not affecting the general health and/or membrane excitability of LPeD1, input resistance and action potential threshold were compared in cell pairs before and after exposure to SrcI. Both the input resistance and action potential threshold of LPeD1 remained unchanged (data not shown). Overall, synaptic transmission and STP were not impacted by SFK inhibition, suggesting that this cannot explain the SrcI-induced perturbation of synapse formation. Similarly, it could be said that the general health of LPeD1 remained unchanged by treatment with SrcI, based on the fact that the input resistance and action potential threshold of this cell were unchanged.
The Effect of SFK Inhibition on Synaptogenesis Is Cell Pair Specific
Since synaptic transmission and STP were unaffected by treatment with SrcI, it seemed likely that the predominant role of SFKs in synapse formation may involve the postsynaptic cell. To investigate this further, the same presynaptic neuron as used in the above experiments (VD4) was soma–soma paired with a different, identified postsynaptic neuron, left pedal E (LPeE). Like VD4–LPeD1 cell pairs, VD4–LPeE synapses are cholinergic. The difference is that VD4–LPeE pairs form inhibitory, rather than excitatory synapses. VD4–LPeE pairs were cultured in CM or CM + SrcI (5 μM) overnight. After 14–16 h, both neurons were recorded intracellularly to determine (1) if synapses formed in the presence of SrcI, (2) if synaptic strength was affected by SFK inhibition, and (3) if STP was affected by treatment with SrcI (Figure 4). Culturing VD4–LPeE pairs in the presence of SrcI had no effect on the incidence of synapse formation [n = 11; χ2(1) = 1.236, p = 0.266], synaptic strength (n = 9; F1,13 = 0.387, p = 0.545), or STP (n = 9; F1,13 = 0.393, p = 0.542). More specifically, the incidence of synapse formation between VD4–LPeE pairs was 100% in CM and 82% in CM + SrcI (Figures 4A,Ai). To compare synaptic strength between inhibitory VD4–LPeE pairs formed in CM versus CM + SrcI, the postsynaptic cell (LPeE) was held at −100 mV for consistency (below the reversal potential for chloride). As such, IPSPs triggered by action potentials generated in VD4 were reversed in polarity (reverse IPSPs or rIPSPs). The mean amplitude of rIPSPs generated in LPeE cells cultured in CM was 15.8 ± 4.4 and 12.9 ± 2.4 mV for those cultured in CM + SrcI (Figures 4A,B). These rIPSPs were potentiated by 226 ± 32% in CM culture conditions and 196 ± 33% in SrcI-treated VD4–LPeE pairs (Figures 4A,B). Therefore, SrcI had no effect on inhibitory, cholinergic synapse formation between pairs of VD4 and LPeE neurons, suggesting that the SrcI-induced perturbation of synapse formation in excitatory, cholinergic VD4–LPeD1 pairs was specific to the postsynaptic cell LPeD1.
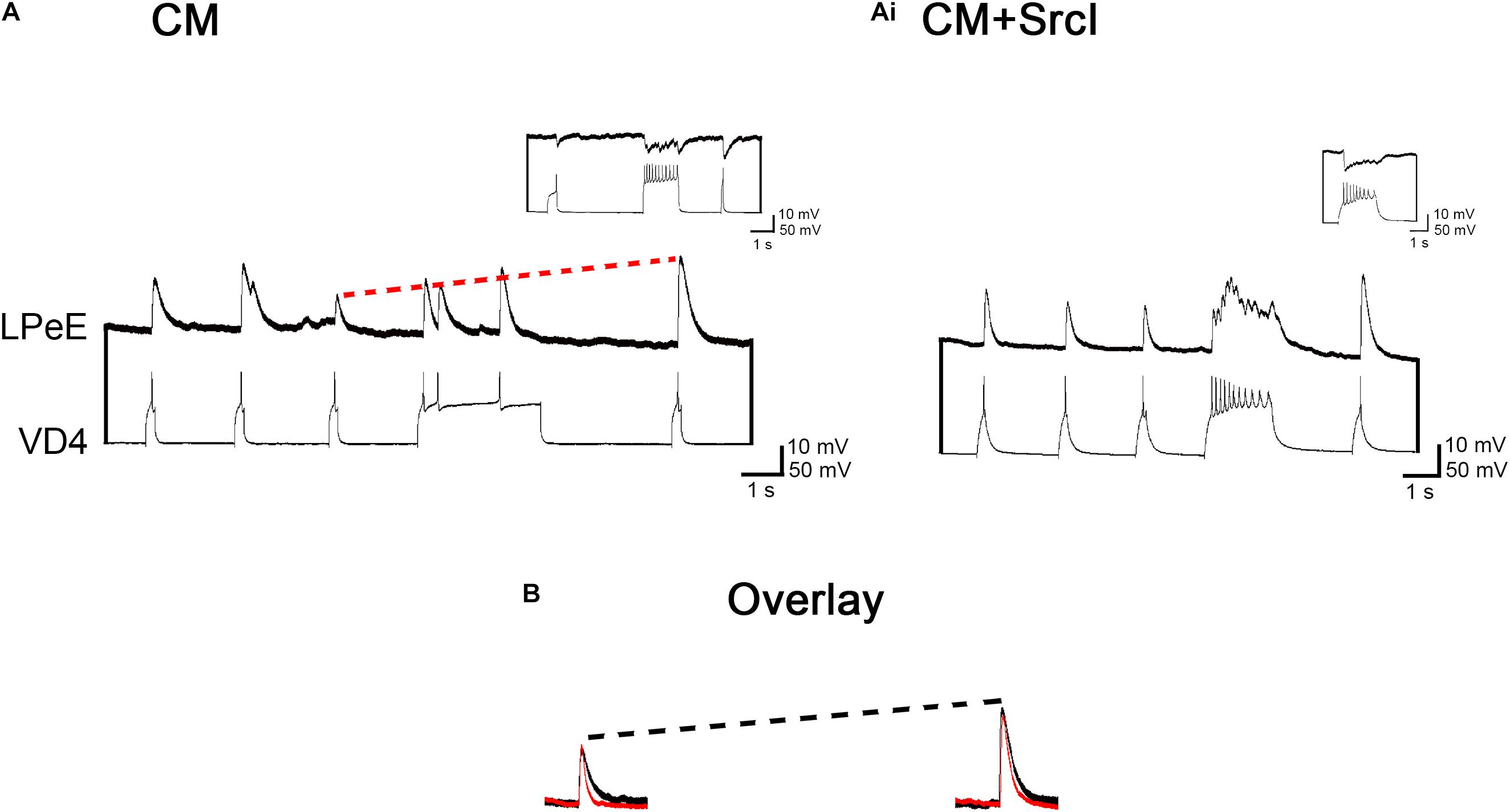
Figure 4. Treatment of visceral dorsal 4 (VD4)–left pedal E (LPeE) soma–soma pairs with SrcI does not affect synapse formation, synaptic strength, or short-term potentiation (STP). (A) VD4–left pedal E (LPeE) pairs were cultured in a soma–soma configuration overnight in conditioned media (CM). Both cells were impaled with intracellular electrodes the next day to confirm that inhibitory synapses had formed. Pre- and post-tetanus reverse inhibitory postsynaptic potential (rIPSP) amplitudes were measured in LPeE (held at –100 mV for consistency). The red dotted line illustrates that post-tetanus rIPSPs were potentiated, on average, by 226%, compared to their pretetanus counterparts. Inset: action potentials triggered in VD4 induced an inhibitory response in LPeE when held at –60 mV (above the reversal potential for Cl–), confirming the presence of an inhibitory synapse. (Ai) A separate group of VD4–LPeE pairs were cultured in CM + SrcI (5 μM) overnight. Pre- and post-tetanus rIPSP amplitudes were measured the next day. Post-tetanus rIPSPs were potentiated, on average, by 196%. Inset: action potentials triggered in VD4 induced an inhibitory response in LPeE when held at –60 mV (above the reversal potential for Cl–), confirming the presence of an inhibitory synapse. (B) Overlay of pretetanus rIPSPs (left) and potentiated post-tetanus rIPSPs (right) in control and SrcI-treated cell pairs. Black = control. Red = SrcI. Neither synaptic strength (p = 0.545) nor STP (p = 0.542) were affected by treatment with SrcI, as determined using a multivariate ANOVA (CM: n = 6; SrcI: n = 9).
SFKs Are Required for Synapses to Form Between VD4 Somata and Isolated LPeD1 Axons
Based on the data presented above, it appears that SFKs are required for excitatory synapse formation between soma–soma paired VD4 and LPeD1 neurons. Furthermore, it is likely that SFK inhibition is affecting the postsynaptic neuron LPeD1 specifically, without perturbing the functional expression of nAChRs in this cell. We thus postulated that SFKs are likely required for the clustering of nAChRs at the synaptic contact site, an important part of postsynaptic differentiation and synaptogenesis.
We have previously demonstrated that synapses form reliably between VD4 somata and isolated LPeD1 axons. This model has proven useful in studying the functional distribution of nAChRs due to the fact that there appears to be a limited pool of nAChRs present in isolated axons (cell bodies removed) (Xu et al., 2014). In particular, we have found that an extrinsic source of trophic factors (CM) is required for the clustering of nAChRs at the synaptic contact site, as demonstrated functionally via intracellular recordings (Meems et al., 2003; Xu et al., 2014).
To test the hypothesis that SFKs are involved in the clustering of postsynaptic nAChRs (Figure 5), VD4 somata and isolated LPeD1 axons (somata removed) were cultured together in CM or CM + SrcI (5 μM) overnight (Figure 5A). The next day (14–16 h later), intracellular recordings were performed to first determine if SFK inhibition perturbed synapse formation in soma–axon pairs as was observed for soma–soma pairs (Figure 6). As was expected, SrcI prevented synapse formation in VD4–LPeD1 soma–axon pairs [Figure 6B; χ2(2) = 38.351, p < 0.001]. The majority of soma–axon pairs treated with SrcI overnight failed to form synapses altogether [n = 19; Figure 6Ai, χ2(1) = 30.915, p < 0.001], as compared to those cultured in CM alone, which formed proper, excitatory synapses 94% of the time [n = 31; Figure 6A, χ2(1) = 38.257, p < 0.001]. SFKs are thus required for synapse formation between VD4 and LPeD1 neurons paired in both soma–soma and soma–axon configuration.

Figure 5. Protocol used to investigate the functional clustering of nicotinic acetylcholine receptors (nAChRs) in visceral dorsal 4 (VD4)–left pedal dorsal 1 (LPeD1) soma–axon pairs. (A) Experimental preparation. Phase contrast image of a VD4–LPeD1 soma–axon pair (10 × magnification). (B) Diagrammatic representation of the electrophysiological protocol used to investigate the clustering of nAChRs in LPeD1 axons. LPeD1 axons were impaled with an intracellular electrode to record the response to acetylcholine (ACh) (10–6 M), which was simultaneously applied at either extrasynaptic (distal) or synaptic contact sites. (C) Phase contrast image of ACh being pressure applied to one of the distal sites (white arrow) on an LPeD1 axon (20 × magnification). (Ci) Phase contrast image of ACh being pressure applied to the synaptic contact site (white arrow) between the VD4 soma and LPeD1 axon (20 × magnification).
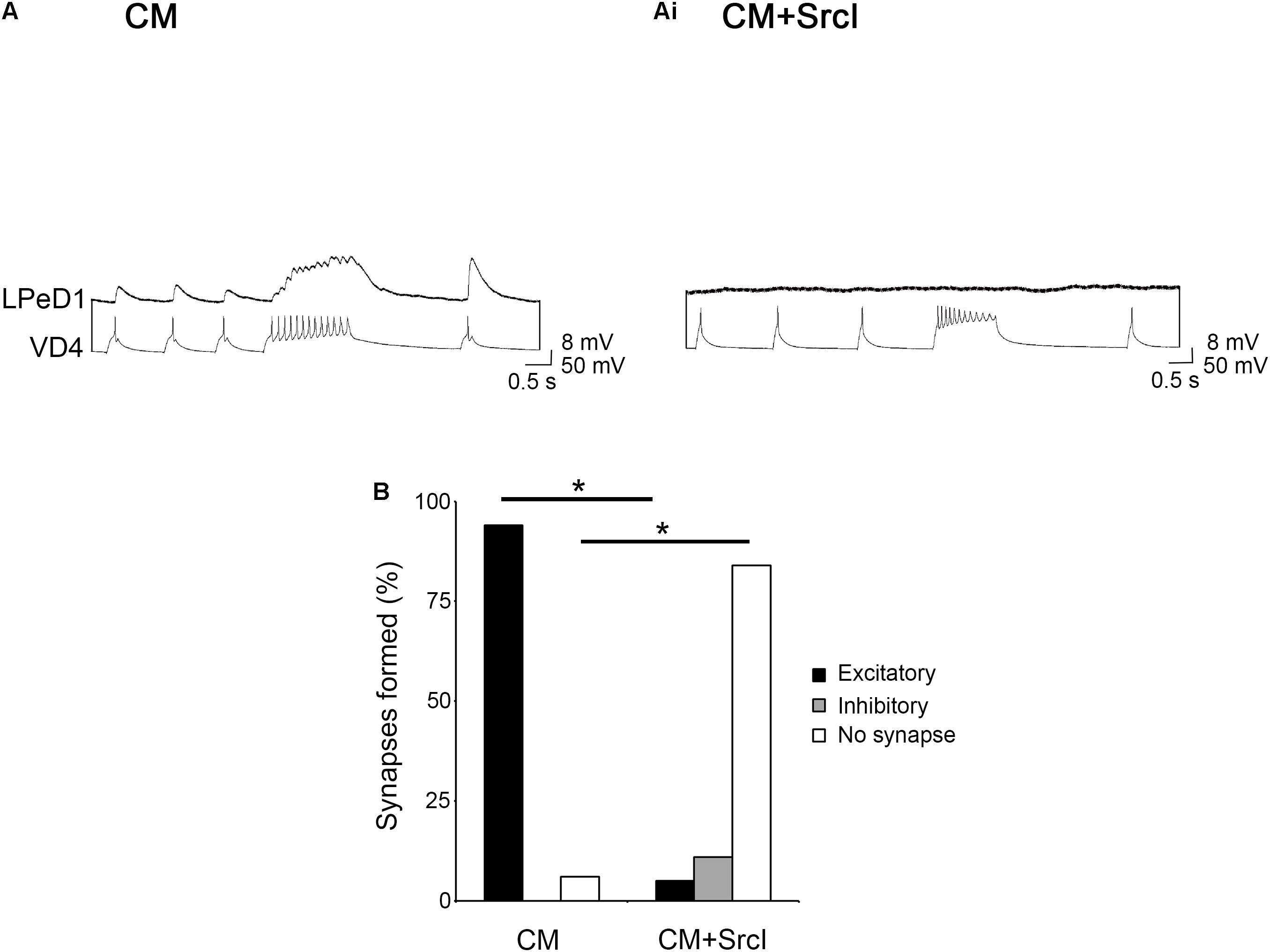
Figure 6. Src family kinases (SFKs) are required for synapse formation between visceral dorsal 4 (VD4) and left pedal dorsal 1 (LPeD1) neurons paired in a soma–axon configuration. (A) Individual VD4 somata and LPeD1 axons (somata removed) were paired overnight in conditioned media (CM). Both cells were impaled with sharp intracellular electrodes the next day and held at –100 mV for consistency. Action potentials triggered in VD4 induced 1-for-1 EPSPs in LPeD1 (n = 31). (Ai) A separate group of soma–axon pairs were cultured in CM + SrcI (5 μM) overnight (n = 19). SrcI perturbed synapse formation in these pairs, as was demonstrated by the lack of response in LPeD1 to action potentials triggered in VD4. (B) Summary of the percentage of soma–soma pairs that formed synapses, with a significance of p ≤ 0.001, as determined using Pearson’s chi-squared test.
SFKs Are Required for the Functional Clustering of nAChRs
The functional clustering of nAChRs was compared in VD4–LPeD1 soma–axon pairs cultured in the presence or absence of SrcI (5 μM). To test this, ACh (10–6 M) was pressure applied to either the synaptic contact site or extrasynaptic sites on either side of the VD4 somata (see Figures 5B–Ci for a visual depiction of this model). The response of LPeD1 to ACh application at these various sites was then analyzed according to the number of action potentials elicited by the puff of ACh, with a larger response (more action potentials) interpreted as a site of higher nAChR concentration (Figure 7). By performing a mixed model ANOVA, it was determined that there was a significant main effect of both puffing site (contact vs. extrasynaptic; F1,21 = 4.835, p = 0.039) and treatment (F1,21 = 9.395, p = 0.006), as well as a significant interaction between the two variables (F1,21 = 9.635, p = 0.005). Specifically, for soma–axon pairs cultured in CM (n = 13), the number of action potentials elicited by pressure application of ACh at the contact site (Figure 7B) was higher, on average, than the number of action potentials induced at extrasynaptic sites (Figure 7Bi). For soma–axon pairs cultured in the presence of SrcI, however, there were no differences in the number of action potentials induced by a puff of ACh at contact or extrasynaptic sites (n = 10; Figures 7Bii,Biii). Finally, more action potentials (6 ± 1) were induced at the synaptic contact site in CM versus CM + SrcI (1 ± 0) (Figure 7C). These data thus suggest that, functionally, there appears to be a higher concentration of nAChRs at the synaptic contact site in soma–axon pairs cultured in CM. Furthermore, it appears that SFKs are required for this redistribution of nAChRs toward the contact site between presynaptic VD4 and postsynaptic LPeD1.
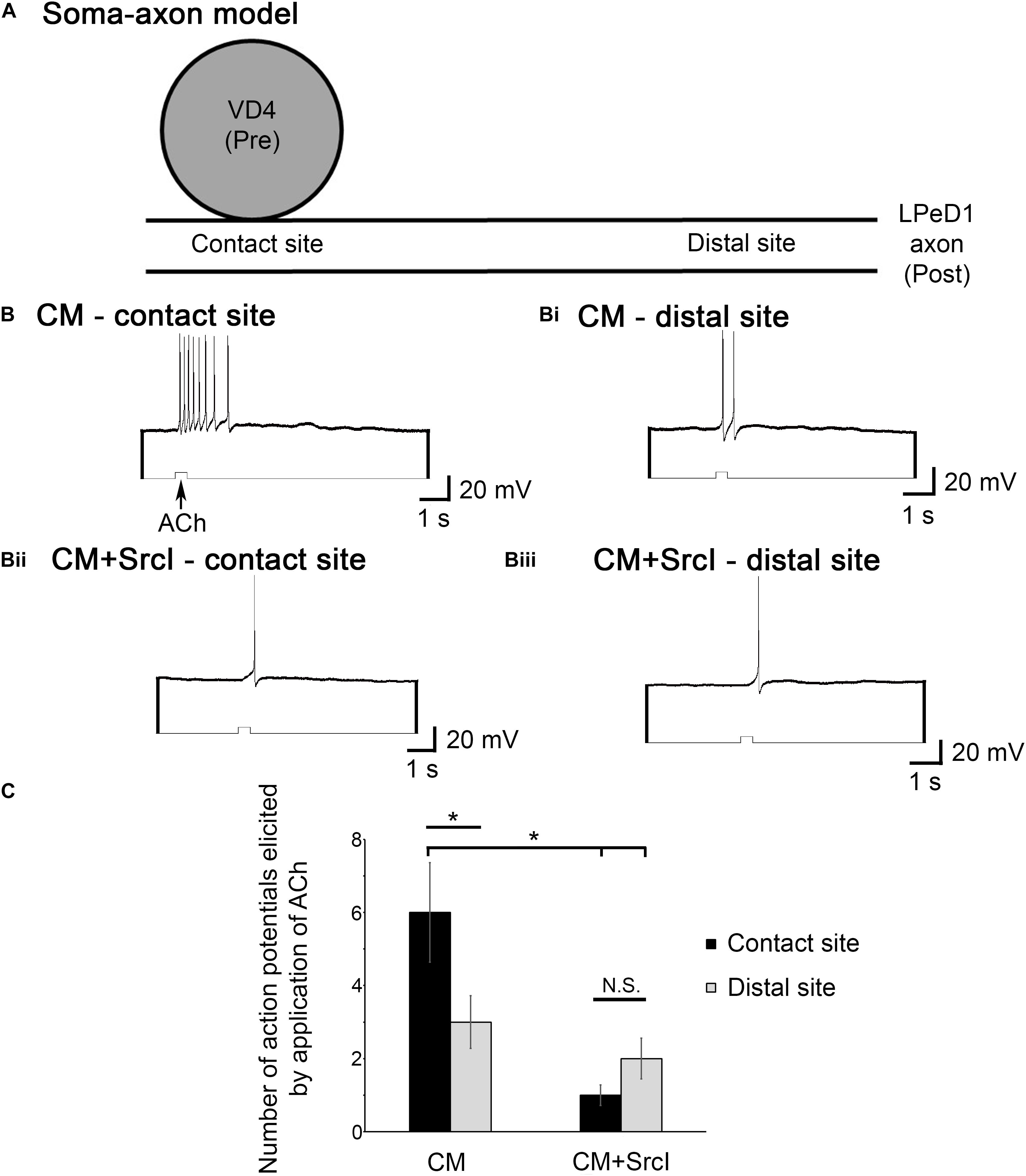
Figure 7. Src family kinases (SFKs) are required for the functional, postsynaptic clustering of nicotinic acetylcholine receptors (nAChRs) at the synaptic contact site. (A) Diagrammatic representation of the soma–axon preparation, outlining synaptic contact sites versus extrasynaptic (distal) sites of acetylcholine (ACh) application. (B–Biii) Individual visceral dorsal 4 (VD4) somata and left pedal dorsal 1 (LPeD1) axons (somata removed) were paired overnight in ganglion-conditioned media (CM) or CM + SrcI (5 μM). The next day, after confirming that synapses had formed, the LPeD1 axon was impaled with a sharp intracellular electrode and held at –100 mV for consistency. ACh was pressure-applied at both contact (B,Bii) and distal sites (Bi,Biii; distal), to compare the response of LPeD1 to ACh according to the number of action potentials elicited by the puff of ACh. (C) Summary of the number of action potentials elicited by puffs of ACh at contact versus distal sites. The number of action potentials elicited by puffs of ACh at the contact site was higher on average in CM (n = 13) than those elicited at the distal sites. For soma–axon pairs cultured in the presence of SrcI (n = 10), there was no differences in the number of action potentials induced by a puff of ACh at contact or distal sites (N.S., not significant). Significantly more action potentials were induced at the synaptic contact site in CM versus CM + SrcI. Significance values were determined using a mixed model ANOVA (p < 0.05). Error bars represent SEM.
Discussion
Most of what we understand about synaptogenesis has been derived from studies of the NMJ (Madhavan and Peng, 2005; Darabid et al., 2014), the cardinal feature of which is the redistribution of postsynaptic nAChRs from extrasynaptic to synaptic sites (Darabid et al., 2014). Innervation of muscle by motor neurons induces the formation of high-density nAChR clusters at nerve contact sites, a process that ensures a rapid and robust postsynaptic response to nerve-released ACh (Mejat et al., 2003; Madhavan and Peng, 2005; Darabid et al., 2014). The stability of these postsynaptic nAChR clusters at the NMJ requires activation of SFKs (Huh and Fuhrer, 2002; Sadasivam et al., 2005). In contrast, little is known about the clustering of nAChRs in the CNS (Yang and Nelson, 2004; Colon-Ramos, 2009), even though these receptors are expressed in most areas of the brain and are key players in the executive function of the prefrontal cortex, for example (dos Santos Coura and Granon, 2012).
In this study, we provided functional evidence of the mechanisms of nAChR clustering in axons individually isolated from the CNS of L. stagnalis. In the presence of an extrinsic source of neurotrophic factors (CM), nAChRs cluster at the synaptic contact site between VD4 somata (presynaptic) and LPeD1 axons (postsynaptic) (Meems et al., 2003; Xu et al., 2014). Here, we show that this process requires SFK activation and is limited to excitatory synapse formation between VD4 and LPeD1 neurons, but not inhibitory synapse formation between VD4 and LPeE neurons. Taking this into consideration, as well as the fact that neither synaptic transmission nor STP are affected by SFK inhibition, we suggest that SFK activation is a requirement in the postsynaptic cell specifically, reminiscent of what is seen at the NMJ.
Two SFKs, Src1 and Src2, have been identified in the sea slug, Aplysia (Wu et al., 2008). In contrast, Src is one of the nine SFKs in vertebrates, five of which are expressed in the CNS (Salter, 1998; Kalia and Salter, 2003). It is thus relatively easier to elucidate the roles of SFKs in the nervous system of mollusks, due to less functional redundancy and their simple, well-characterized nervous systems. In Aplysia for instance, Src1 and Src2 have been shown to associate with both the plasma membrane and cytoskeleton of growth cones, placing them in a prime position to influence synaptogenesis (Wu et al., 2008). At the mouse NMJ, glial-derived neurotrophic factor increases the lateral movement and insertion of nAChRs into muscle membrane, without affecting receptor synthesis, a process that requires Src (Yang and Nelson, 2004). Based on the results presented here, we hypothesize that SFKs stabilize nAChRs in clusters at the synaptic contact site. Using isolated axons with their cell bodies removed, there is a limited pool of nAChRs to be drawn from Xu et al. (2014). We predict that these receptors are redirected to the synaptic contact site, where they then become stabilized/linked to the cytoskeleton by neurotrophic factor-activated SFKs. We fully recognize, however, that an intracellular pool of nAChRs could be drawn from as well and that a distinction remains to be made concerning the involvement of SFKs in the initiation versus stabilization of nAChR clusters. We are also aware that further studies will be needed to delineate the mechanisms by which nAChRs are trafficked and whether their clustering is a result of translocation from a cytoplasmic compartment or due to lateral movement across the membrane. Importantly, future studies will need to focus on devising methods to visualize to nAChRs in Lymnaea, something that has so far proven challenging using conventional methods.
Mechanisms of postsynaptic differentiation, including the clustering of neurotransmitter receptors, have been well-established at the NMJ (Huh and Fuhrer, 2002). What remains to be established, however, is whether similar mechanisms and molecular players orchestrate synaptogenesis in the CNS. Here, we show that SFKs may play a similar role in the clustering of nAChRs in the CNS, as has been demonstrated at the NMJ. Even though Src is the prototypical non-receptor kinase, very little is known about its role in the CNS. Loss of Fyn (an SFK) in mice has been linked to neural dysfunction, including deficits in long-term potentiation (LTP), hippocampal morphology, myelination, and spatial learning (Grant et al., 1992; Thomas and Brugge, 1997; Groveman et al., 2012). As such, it is important that we gain a better understanding of the role of SFKs in the nervous system, as it could lead to novel insights into both healthy and pathological brain function.
Data Availability Statement
The datasets generated for this study can be found in the figshare repository (doi: 10.6084/m9.figshare.7806074).
Ethics Statement
This research was conducted using the lower invertebrate, Lymnaea stagnalis, and is thus exempt from any ethical review process. As such, all experiments are in accordance with the guidelines set forth by the Canadian Council on Animal Care.
Author Contributions
NF: conceptualization, data curation, formal analysis, investigation, methodology, visualization, and writing of the original draft. NS: conceptualization, funding acquisition, methodology, resources, supervision, and writing (review and editing).
Funding
This work was supported by the Canadian Institute of Health Research (CIHR) (64030-60-28350-10015088-00000). NF received studentships from Alberta Innovates—Health Solutions (AIHS) and the National Sciences and Engineering Research Council of Canada (NSERC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Wali Zaidi for excellent technical support. This manuscript was previously published as part of a Ph.D. thesis entitled “Postsynaptic mechanisms of trophic factor-induced excitatory synapse formation in Lymnaea stagnalis,” by NF (Flynn, 2015). This dissertation has been published online in the University of Calgary’s master and doctoral theses archive (The Vault). NF has retained all copyrights for the thesis.
References
Colon-Ramos, D. A. (2009). Synapse formation in developing neural circuits. Curr. Top. Dev. Biol. 87, 53–79. doi: 10.1016/S0070-2153(09)01202-1202
Darabid, H., Perez-Gonzalez, A. P., and Robitaille, R. (2014). Neuromuscular synaptogenesis: coordinating partners with multiple functions. Nat. Rev. Neurosci. 15, 703–718. doi: 10.1038/nrn3821
dos Santos Coura, R., and Granon, S. (2012). Prefrontal neuromodulation by nicotinic receptors for cognitive processes. Psychopharmacology 221, 1–18. doi: 10.1007/s00213-011-2596-2596
Flynn, N. (2015). Postsynaptic Mechanisms of Trophic Factor-Induced Excitatory Synapse Formation in Lymnaea Stagnalis. PhD dissertation, University of Calgary, Calgary.
Flynn, N., Getz, A., Visser, F., Janes, T. A., and Syed, N. I. (2014). Menin: a tumor suppressor that mediates postsynaptic receptor expression and synaptogenesis between central neurons of Lymnaea stagnalis. PLoS One 9:e111103. doi: 10.1371/journal.pone.0111103
Ghazanfari, N., Fernandez, K. J., Murata, Y., Morsch, M., Ngo, S. T., Reddel, S. W., et al. (2011). Muscle specific kinase: organiser of synaptic membrane domains. Int. J. Biochem. Cell Biol. 43, 295–298. doi: 10.1016/j.biocel.2010.10.008
Grant, S. G., O’Dell, T. J., Karl, K. A., Stein, P. L., Soriano, P., and Kandel, E. R. (1992). Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science 258, 1903–1910. doi: 10.1126/science.1361685
Groveman, B. R., Feng, S., Fang, X. Q., Pflueger, M., Lin, S. X., Bienkiewicz, E. A., et al. (2012). The regulation of N-methyl-D-aspartate receptors by Src kinase. FEBS J. 279, 20–28. doi: 10.1111/j.1742-4658.2011.08413.x
Huh, K. H., and Fuhrer, C. (2002). Clustering of nicotinic acetylcholine receptors: from the neuromuscular junction to interneuronal synapses. Mol Neurobiol 25, 79–112.
Kalia, L. V., and Salter, M. W. (2003). Interactions between Src family protein tyrosine kinases and PSD-95. Neuropharmacology 45, 720–728. doi: 10.1016/s0028-3908(03)00313-7
Lu, Y. M., Roder, J. C., Davidow, J., and Salter, M. W. (1998). Src activation in the induction of long-term potentiation in CA1 hippocampal neurons. Science 279, 1363–1367.
Luk, C. C., Naruo, H., Prince, D., Hassan, A., Doran, S. A., Goldberg, J. I., et al. (2011). A novel form of presynaptic CaMKII-dependent short-term potentiation between Lymnaea neurons. Eur J. Neurosci. 34, 569–577. doi: 10.1111/j.1460-9568.2011.07784.x
Madhavan, R., and Peng, H. B. (2005). Molecular regulation of postsynaptic differentiation at the neuromuscular junction. IUBMB Life 57, 719–730. doi: 10.1080/15216540500338739
Meems, R., Munno, D., van Minnen, J., and Syed, N. I. (2003). Synapse formation between isolated axons requires presynaptic soma and redistribution of postsynaptic AChRs. J. Neurophysiol. 89, 2611–2619. doi: 10.1152/jn.00898.2002
Mejat, A., Ravel-Chapuis, A., Vandromme, M., and Schaeffer, L. (2003). Synapse-specific gene expression at the neuromuscular junction. Ann. N. Y Acad. Sci. 998, 53–65. doi: 10.1196/annals.1254.008
Ohnishi, H., Murata, Y., Okazawa, H., and Matozaki, T. (2011). Src family kinases: modulators of neurotransmitter receptor function and behavior. Trends Neurosci. 34, 629–637. doi: 10.1016/j.tins.2011.09.005
Ridgway, R. L., Syed, N. I., Lukowiak, K., and Bulloch, A. G. (1991). Nerve growth factor (NGF) induces sprouting of specific neurons of the snail, Lymnaea stagnalis. J. Neurobiol. 22, 377–390. doi: 10.1002/neu.480220406
Sadasivam, G., Willmann, R., Lin, S., Erb-Vogtli, S., Kong, X. C., Ruegg, M. A., et al. (2005). Src-family kinases stabilize the neuromuscular synapse in vivo via protein interactions, phosphorylation, and cytoskeletal linkage of acetylcholine receptors. J. Neurosci. 25, 10479–10493. doi: 10.1523/JNEUROSCI.2103-05.2005
Salter, M. W. (1998). Src, N-methyl-D-aspartate (n.d.) receptors, and synaptic plasticity. Biochem. Pharmacol. 56, 789–798. doi: 10.1016/s0006-2952(98)00124-5
Syed, N. I., Bulloch, A. G., and Lukowiak, K. (1990). In vitro reconstruction of the respiratory central pattern generator of the mollusk Lymnaea. Science 250, 282–285. doi: 10.1126/science.2218532
Thomas, S. M., and Brugge, J. S. (1997). Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 13, 513–609. doi: 10.1146/annurev.cellbio.13.1.513
van Nierop, P., Bertrand, S., Munno, D. W., Gouwenberg, Y., Spafford, J. D., Syed, N. I., et al. (2006). Identification and functional expression of a family of nicotinic acetylcholine receptor subunits in the central nervous system of the mollusc Lymnaea stagnalis. J. Biol. Chem. 281, 1680–1691. doi: 10.1074/jbc.M508571200
van Nierop, P., Keramidas, A., Bertrand, S., Gouwenberg, Y., Bertrand, D., and Smit, A. B. (2005). Identification of molluscan nicotinic acetylcholine receptor (nAChR) subunits involved in formation of cation- and anion-selective nAChRs. J. Neurosci. 25, 10617–10626. doi: 10.1523/JNEUROSCI.2015-05.2005
Wu, B., Decourt, B., Zabidi, M. A., Wuethrich, L. T., Kim, W. H., Zhou, Z., et al. (2008). Microtubule-mediated Src tyrosine kinase trafficking in neuronal growth cones. Mol. Biol. Cell 19, 4611–4627. doi: 10.1091/mbc.E08-06-0603
Xu, F., Hennessy, D. A., Lee, T. K., and Syed, N. I. (2009). Trophic factor-induced intracellular calcium oscillations are required for the expression of postsynaptic acetylcholine receptors during synapse formation between Lymnaea neurons. J. Neurosci. 29, 2167–2176. doi: 10.1523/JNEUROSCI.4682-08.2009
Xu, F., Luk, C. C., Wiersma-Meems, R., Baehre, K., Herman, C., Zaidi, W., et al. (2014). Neuronal somata and extrasomal compartments play distinct roles during synapse formation between Lymnaea neurons. J. Neurosci. 34, 11304–11315. doi: 10.1523/JNEUROSCI.1651-14.2014
Yang, L. X., and Nelson, P. G. (2004). Glia cell line-derived neurotrophic factor regulates the distribution of acetylcholine receptors in mouse primary skeletal muscle cells. Neuroscience 128, 497–509. doi: 10.1016/j.neuroscience.2004.06.067
Yu, X. M., Askalan, R., Keil, G. J. II, and Salter, M. W. (1997). NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science 275, 674–678. doi: 10.1126/science.275.5300.674
Zhao, W., Cavallaro, S., Gusev, P., and Alkon, D. L. (2000). Nonreceptor tyrosine protein kinase pp60c-src in spatial learning: synapse-specific changes in its gene expression, tyrosine phosphorylation, and protein-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 97, 8098–8103. doi: 10.1073/pnas.97.14.8098
Keywords: synapse, acetylcholine, postsynaptic, Lymnaea stagnalis, synaptogenesis, neurotrophic factors, Src, nicotinic acetylcholine receptor
Citation: Flynn N and Syed NI (2020) Src Family Kinases Play a Role in the Functional Clustering of Central Postsynaptic Nicotinic Acetylcholine Receptors. Front. Mar. Sci. 7:8. doi: 10.3389/fmars.2020.00008
Received: 27 November 2019; Accepted: 09 January 2020;
Published: 19 February 2020.
Edited by:
William Winlow, University of Naples Federico II, ItalyReviewed by:
Rhanor Gillette, University of Illinois at Urbana–Champaign, United StatesVarvara Dyakonova, Koltzov Institute of Developmental Biology (RAS), Russia
Copyright © 2020 Flynn and Syed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naweed I. Syed, nisyed@ucalgary.ca