 Kuipeng Xu
Kuipeng Xu Xinzi Yu
Xinzi Yu Xianghai Tang
Xianghai Tang Fanna Kong
Fanna Kong Yunxiang Mao
Yunxiang Mao- 1Key Laboratory of Marine Genetics and Breeding (OUC), Ministry of Education, Qingdao, China
- 2College of Marine Life Sciences, Ocean University of China, Qingdao, China
- 3Laboratory for Marine Biology and Biotechnology, Pilot National Laboratory for Marine Science and Technology, Qingdao, China
- 4Key Laboratory of Utilization and Conservation for Tropical Marine Bioresources, Hainan Tropical Ocean University, Ministry of Education, Sanya, China
- 5College of Horticulture, Qingdao Agricultural University, Qingdao, China
Pyropia yezoensis is an economically important marine algae crop that, due to its large economic value, has generated considerable interest in the development of breeding programs to improve its production rates. Here, we sequenced the complete mitochondrial and plastid genomes of the P. yezoensis strain RZ-58 using the PacBio RS II sequencing technology. The mitochondrial genome (mtDNA) is 41,692 bp in size with an overall guanine–cytosine (GC) content of 32.72%, and the plastid genome (ptDNA) is 191,977 bp with a GC content of 33.09%. The complete mitochondrial and plastid genomes of 53 individuals from three geographical populations were then resequenced using the next-generation sequencing (NGS) technology to characterize their molecular features. When compared, the plastid genomes displayed similar genomic lengths and conserved gene synteny. However, mitochondrial genomes were quite different in length, which was mainly due to the different patterns of intron distributions. Single-nucleotide polymorphisms (SNPs) were examined to evaluate the genetic diversity of different geographical populations. High diversity was observed across the whole collection with moderate genetic variation between populations. In total, there were 463 and 366 high-quality SNPs detected in the mtDNA and ptDNA, respectively. The Qingdao wild group has the highest diversity with a mean pi of 0.00348 for mtDNA and 0.000388 for ptDNA, while the Yantai group had the lowest diversity. Cluster-based grouping and principal component analysis revealed three subpopulations in the whole collections. However, a genetic break of organellar DNA was observed in populations at sympatric localities, which was inferred as the result of historic biogeographic events. Our findings provide important information to guide marker-assisted selective breeding of Chinese P. yezoensis in the future.
Introduction
Pyropia yezoensis (Ueda), M. S. Hwang and H. G. Choi, is an economically important marine crop that is cultivated on a large scale in China, Japan, and Korea. According to the Food and Agriculture Organization (FAO)’s statistics, ∼2.06 million tons in fresh weight of cultivated Pyropia was produced in 2016 with a commercial value of over 0.95 billion US dollars1. P. yezoensis is typically distributed in the intertidal zone of coastal areas. The thallus constantly suffers from abrupt temperature changes and repeated desiccation and rehydration due to the changing tides. It makes this alga an ideal research model for investigations into the mechanisms that underlay stress tolerance in intertidal seaweeds (Sun et al., 2015). Currently, most of the cultivated P. yezoensis are collected from wild populations, and their genetic variation and patterns of admixture are not well characterized. Traditional selective breeding is extremely time-consuming for improving the genetics of new cultivars and could further reduce the genetic diversity of the cultivated Pyropia stock. Considering its economic and ecological importance, it is necessary to understand the genetic characteristics of this seaweed using modern molecular tools to facilitate breeding operations. To date, there have been several studies that used molecular markers to characterize the biodiversity of Pyropia spp. and P. yezoensis, but most of these relied on data with low density such as amplified fragment length polymorphism (AFLP), restriction fragment length polymorphism (RFLP), inter-simple sequence repeat (ISSR), or simple sequence repeat (SSR) approaches (Niwa et al., 2009; He et al., 2010; Xie et al., 2013; Bi et al., 2014; Cao et al., 2018). As a result, these previously generated data may not be sufficient to provide accurate assessments of recent demographic bottlenecks or for identifying populations with low genetic diversity (Ba et al., 2017). Therefore, the use of high-density genetic markers is needed to improve our understanding of the genetic characteristics of P. yezoensis to conserve germplasm and develop sustainable improvements to breeding strategies.
Single-nucleotide polymorphism (SNP) markers have been widely utilized in recent years due to their high abundance in the genome and the improved resolution they provide for populations (Carvalho et al., 2017; Edea et al., 2017; Fischer et al., 2017). With the development of next-generation sequencing (NGS), it is increasingly feasible to develop and genotype SNP markers across whole genomes rather than targeting specific regions. The identification of genomic SNPs provides an opportunity to apply these data to genome-based association studies in the future. Recent studies have utilized whole mitochondrial or plastid genome approaches to detect SNPs in plants, which showed that organellar SNP markers are a powerful genetic resource for germplasm differentiation, purification, and population genetics (Mcpherson et al., 2013; Sabir et al., 2014; Wei et al., 2016; Shen et al., 2017). The Pyropia species is characterized by a high proportion of organellar DNA and a small organellar genome size, which makes sequencing the organellar genomes affordable and convenient without the need for prior isolation or amplification (Wang et al., 2013; Kong et al., 2014; Xu et al., 2018a).
The third-generation sequencing platform PacBio RS II uses the single-molecule real-time (SMRT) technology to produce long and unbiased sequences that ensure the assembly of complex repeat structures and adenine–thymine (AT)/guanine–cytosine (GC)-rich regions, which is often difficult to do using short reads (Eid et al., 2009; Roberts et al., 2013). This technology has been successfully applied to a wide range of experiments including whole-genome sequencing, targeted sequencing, RNA sequencing, and epigenetics (Filichkin et al., 2018; Sun et al., 2018; Suzuki et al., 2018; Zhou et al., 2018). PacBio has been successfully used in many mitochondrial and chloroplast genomic projects (Chen et al., 2015; Xiang et al., 2016; Peccoud et al., 2017; Kovar et al., 2018). However, errors in reference genomes, which could be carried over by reference-based assembly approaches, misled subsequent studies of mutation detection and molecular evolution (Sloan et al., 2018). Therefore, a highly accurate assembly of organellar genomes would be a useful resource for any future investigations of plant organelle genetics.
Breed characterization requires a basic knowledge of genetic variation that can be effectively measured within and between populations. As a result, this study was designed (1) to sequence the mitochondrial and plastid genomes of P. yezoensis RZ-58 using the PacBio RS II sequencing platform and the 53 wild accessions from China using the Illumina sequencing platform; (2) to detect organelle polymorphisms at the intraspecific and intra-individual level; and (3) to analyze the degree of genetic diversity with a large representation of Chinese P. yezoensis using SNPs in the organellar genome. These genetic data are important factors for the adequate and efficient conservation of core germplasm and could serve as the theoretical basis for future breeding strategies.
Materials and Methods
Materials Sampling
A laboratory-cultured genetically pure line of P. yezoensis RZ-58 was used in the experiments. The original thallus was collected from the nori farm in Putian, Fujian Province, China. At the laboratory, individual somatic cells were enzymatically isolated from the thallus and the allelically homozygous sporophytes (conchocelis) were obtained after spontaneous haploid doubling. The gametophytes from the homozygous sporophytes are homogenous and haploid. The gametophytes were then cultured in bubbling sterilized seawater with Provasoli’s enrichment solution (PES) medium at 8 ± 1°C with a light concentration of 50 μmol photons m–2s–1 and a 12:12 light: dark (L: D) photoperiod. The wild P. yezoensis were collected from three geographical populations: Qingdao (N36°03′29″, E120°18′56″), Yantai (N37°49′35″, E120°44′40″), and Weihai (N37°27′53″, E122°08′27″) of the Shandong Province, China, during March 2016. The samples were randomly collected from the intertidal zone at intervals of at least 5 m to prevent the collection of asexual reproductive ramets. A total of 53 haploid thalli with morphological diversity were selected, 19 from Qingdao, 19 from Yantai, and 15 from Weihai, for the genetic analysis of the different groups. The collected samples were thoroughly cleaned with sterile seawater and frozen at −20°C.
DNA Extraction, Genome Assembly, and Annotation
Total DNA was extracted from the frozen thallus material in accordance with previously reported methods (Wattier et al., 2000). The SMRT sequencing library was constructed using the PacBio® Template Prep Kit with an insert size of 20 kb according to the manufacturer’s instructions. The genome was sequenced using the PacBio RS II platform (Pacific Biosciences, Menlo Park, CA, United States). The PacBio raw reads (polymerase reads) were filtered using the following parameters: removal of the adaptor sequences, minimum polymerase read quality = 0.80, and minimum polymerase read length = 1,000 bp. BLASR was used to extract the organellar sequences using the reference genome (NC_017837 and KC517072) (Chaisson and Tesler, 2012). The extracted sequences were assembled using the RS_HGAP_Assembly 0.3 protocol, and Quiver was run to polish the accurate consensus in the SMRT Analysis v2.3.0 using default parameters (Chin et al., 2013). The circular genomes were then validated by mapping the Illumina HiSeq data and the PCR-based sequencing. We constructed 500-bp paired-end libraries according to the instructions of the NEBNext® UltraTM II DNA Library Prep Kit (NEB, Beijing, China), and the libraries were sequenced on an Illumina Genome Analyzer. The adapters and low-quality reads were removed using the NGS QC Toolkit (Patel and Jain, 2012). Organellar genomes were assembled using the NOVOPlasty software, which utilized a seed-and-extend algorithm that starts from a related or distant single seed sequence (Dierckxsens et al., 2017). We selected the rbcL and atp8 genes as the seeds to assemble the plastid DNA (ptDNA) and mitochondrial DNA (mtDNA), respectively. The genomes were annotated using the online tool ORFfinder2 and aligned using BLASTX and BLASTN searches at the National Center for Biotechnology Information (NCBI) website3. Multiple genomes were aligned and visualized using the mVISTA tool (Mayor et al., 2000). For the molecular identification, the rbcL genes from the wild collections of P. yezoensis and the 82 species of Pyropia (Supplementary Table S1) were downloaded from GenBank and used to construct the phylogenetic tree. The sequences were aligned using the MAFFT version 5 program (Katoh et al., 2005) and trimmed using trimAl with the option “automated1” (Capella-Gutiérrez et al., 2009). The neighbor-joining (NJ) phylogenetic tree was created using MEGA7 with a 1,000-bootstrap replicate (Kumar et al., 2016).
Read Mapping and SNP Genotyping
After the quality controls were applied, the clean reads were mapped to the reference genome using BWA (Li and Durbin, 2009). The reference genomes were indexed, and the command “aln” was used to find the suffix array coordinates of good matches for each read. The best alignments were then converted into the SAM format using the “sampe” command. The alignment results were further corrected using the Picard package with the following two commands: “AddOrReplaceReadGroups” was used to replace all read groups in the INPUT file with a new read group and to assign all of the reads to this read group in the OUTPUT BAM; and “FixMateInformation” was used to ensure that all of the mate-pair information was synchronized between each read and its mate pair4. Potential PCR duplicates were removed using the SAMtools command “rmdup” (Li et al., 2009).
After alignment, we performed SNP calling on a population scale using a Bayesian approach based on the SAMtools program. The command “mpileup” was used with the parameters “- q 1 - C 50 - S - D - m 2 - F 0.002 - u.” The subsequent analysis filtered the raw SNPs using VCFtools (Danecek et al., 2011) and GATK (McKenna et al., 2010) with the following criteria: (1) coverage depth ≥8; (2) mapping quality ≥30; (3) the distance of adjacent SNPs ≥5 bp; (4) only SNPs that occurred in more than 50% of the individuals were retained; and (5) all SNPs with a minor allele frequency (MAF) ≥0.1 were removed. We then used the high-quality SNPs to infer genetic analysis.
Statistical Analysis
Nucleotide diversity (pi), Tajima’s D, transition/transversion ratio (Ts/Tv) and population divergence (FST value) were calculated using VCFtools with a sliding window of 1,000 bp in length and a 500-bp step size (Danecek et al., 2011). Tajima’s D test statistic was calculated using DnaSP based on the differences between the number of segregating sites and the average number of nucleotide differences (Librado and Rozas, 2009). The phylogenetic tree was inferred using TASSEL under the p-distances model using SNPs in a population scale (Bradbury et al., 2007). Principal component analysis (PCA) was performed using the EIGENSOFT software (Price et al., 2006). The organelle genetic groups was further verified with structural classes identified from population structure analysis algorithms using Structure 2.3.4 (Pritchard et al., 2000), which used a Bayesian approach to identify subpopulations with distinct allelic frequencies and places the individuals into K clusters. The number of populations (K) was set from 2 to 7 and repeated 10 times with a burn-in period of 100,000 iterations and 200,000 Markov chain Monte Carlo (MCMC) repeats. The K optimum was evaluated using Structure Harvester (Earl and Vonholdt, 2012).
Results
Structural Variation of P. yezoensis Organellar Genomes
To assemble the high-quality reference organellar genomes, 712 Mb of PacBio® subread bases (a single pass of the template without the adapter sequences) and a total of 92,569 reads were generated with a mean read length of 7,690 bp and an N50 size of 9,898 bp. The finished mitochondrial genome of P. yezoensis is 41,692 bp in size with an overall GC content of 32.72% (Figure 1A). The plastid genome is 191,977 bp with a GC content of 33.09% (Figure 1B). We confirmed that all of the differences identified in our assembly were strongly supported by mapping with the highly accurate Illumina short reads.
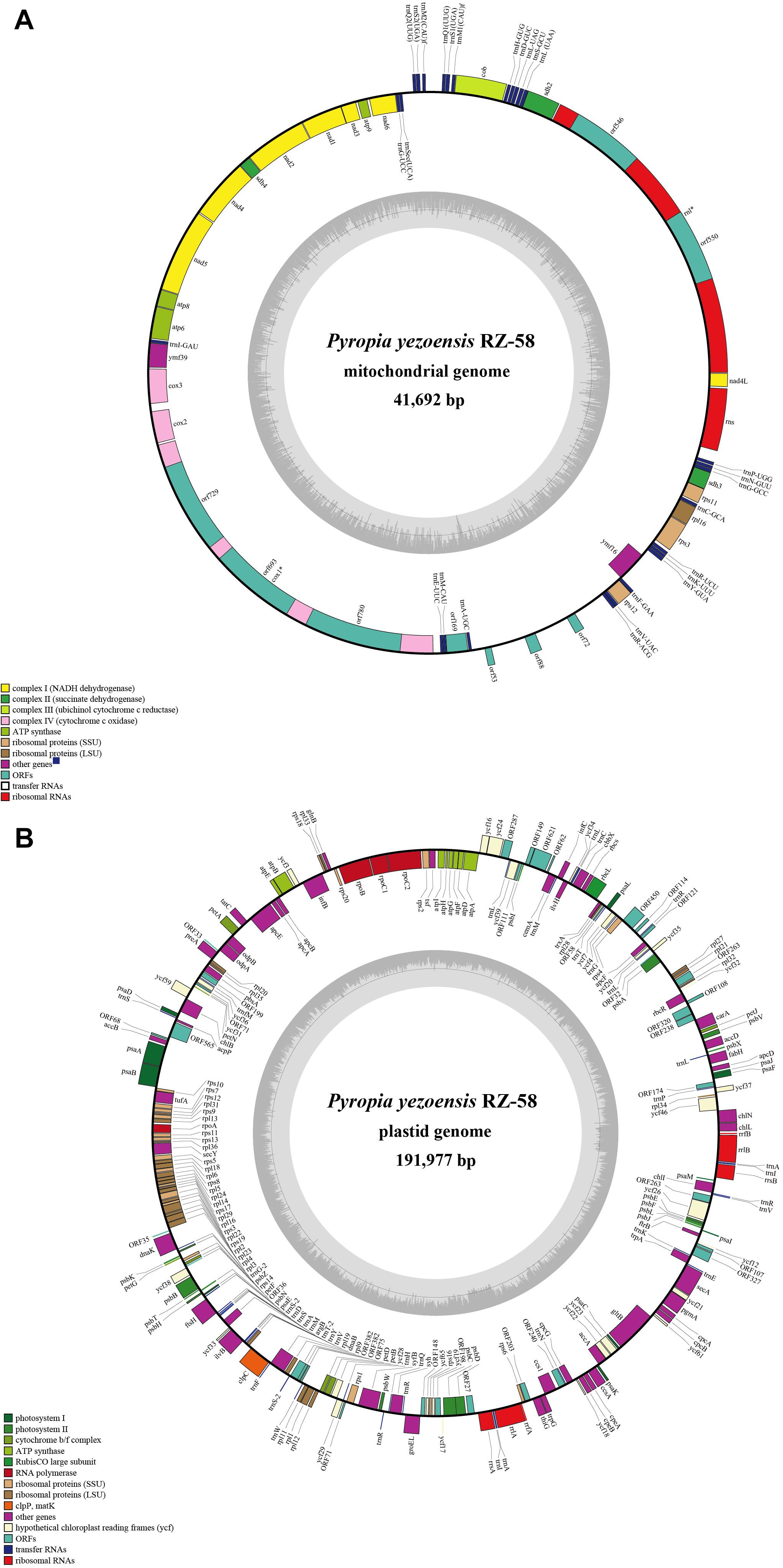
Figure 1. Genome maps of the Pyropia yezoensis mitochondrion (A) and plastid (B). Genes shown outside the outer circle are transcribed clockwise, and those inside are transcribed counterclockwise. Genes that belong to different functional groups are color coded. The dashed area in the inner circle indicates the guanine–cytosine (GC) content of the organellar genome.
We then resequenced 53 accessions of wild P. yezoensis that were collected from Qingdao, Weihai and the Yantai coast of China. After removing low quality reads and enrichment, we obtained ∼4 Gb of organelle sequence bases with coverage that ranged from 260× to 370×. For the molecular identification, a phylogenetic tree that used the plastid rbcL gene showed that all the samples formed a well-supported clade together with the previously identified P. yezoensis (Supplementary Figure S1). The plastid genomes of P. yezoensis were similar in length (191,885 to 192,002 bp) and GC content (33.07 to 33.09%) and had highly conserved gene synteny (Figure 2B and Supplementary Table S2). Sequence differences mainly resulted from tandem repeat polymorphisms that were located in intergenic regions. The repeat motifs ranged in length from 1 to 20 base pairs and the repetitions ranged from 1 to 2. For example, there was a (TTTA)n SSR polymorphism between the chlN and ycf46 genes. A (TAGTTAGTAGTTTAAATAGA)n minisatellite polymorphism was also found between the ORF287 and ycf24 genes.
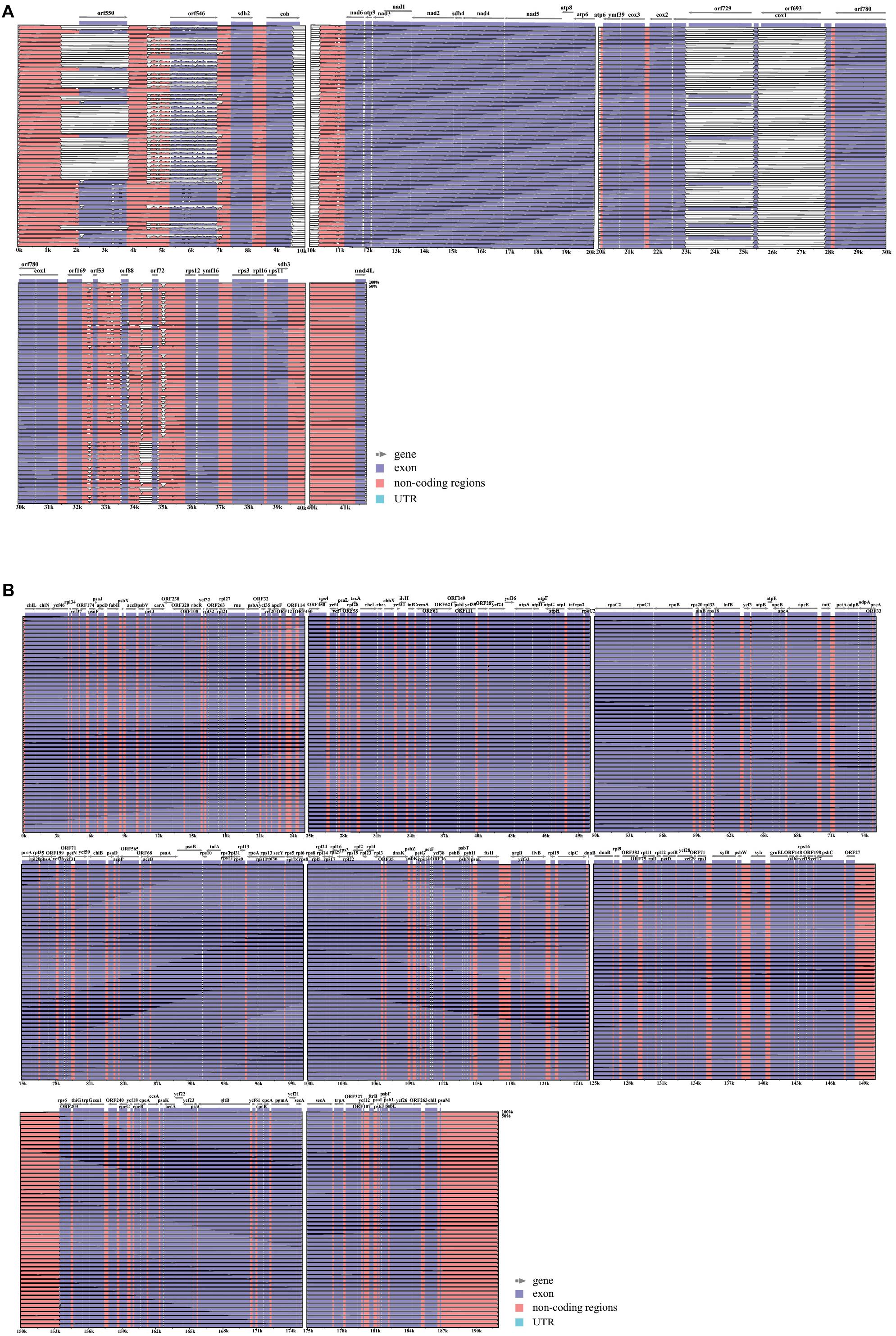
Figure 2. Visualization of the alignment of the organellar genome sequences. (A) VISTA-based identity plots showing sequence identity among the 53 sequenced mitochondrial genomes; (B) VISTA-based identity plots showing sequence identity among the 53 sequenced plastid genomes with Pyropia yezoensis RZ-58 as a reference. Each genome region from the lower boundary to upper boundary indicates the sequence identity ranging from 50% to 100%.
The mitochondrial genome lengths within the populations of P. yezoensis were significantly different, ranging from 34,031 to 40,758 bp (Figure 2A). The GC content ranged from 32.45 to 32.79% (Supplementary Table S2). The observed differences were mainly caused by different numbers of mitochondrial group II introns in the large subunit ribosomal RNA (rnl) gene and the cytochrome c oxidase subunit 1 (cox1) gene. Recent studies have shown that in the Pyropia species, the rnl gene is composed of four coding regions and the cox1 gene is composed of five coding regions (Hwang et al., 2013). According to the presence of the intron, the 53 individuals displayed five structural patterns of the rnl gene and three of the cox1 gene (Figure 3). The combined structural variation of the rnl and cox1 genes exhibited six genotypes in total (one genotype of RZ-58 and five genotypes of the wild collections) among the whole collections (Supplementary Table S2). Only one individual sample, QD23 (Qingdao23), had an intron between each exon of the rnl gene. The others had one or two absent introns (Figure 3A). All 53 individuals and the reference RZ-58 lacked an intron between exon 1 and exon 2 in the cox1 gene (Figure 3B), which was present in the mtDNA of Pyropia haitanensis (NC_017751) and Pyropia tenera (NC_021475). Moreover, each intron contained one full-length intronic ORF. These ORFs encode reverse transcriptases that function in intron mobility and as maturases in RNA splicing. Furthermore, according to the different intronic structural patterns in the population, we annotated the exon–intron boundaries of the rnl and cox1 genes from P. yezoensis mtDNA. The splice junctions of introns 1 and 2 of the rnl gene are a GA/AT and CG/AA motif, respectively (here, we defined the junction start/end as the intronic start/end bases). The splice junctions of introns 2, 3, and 4 of the cox1 gene are a CA/CC, GT/AT, and GT/AC motif, respectively, from the 5’ end to the 3’ end (Supplementary Figure S2).
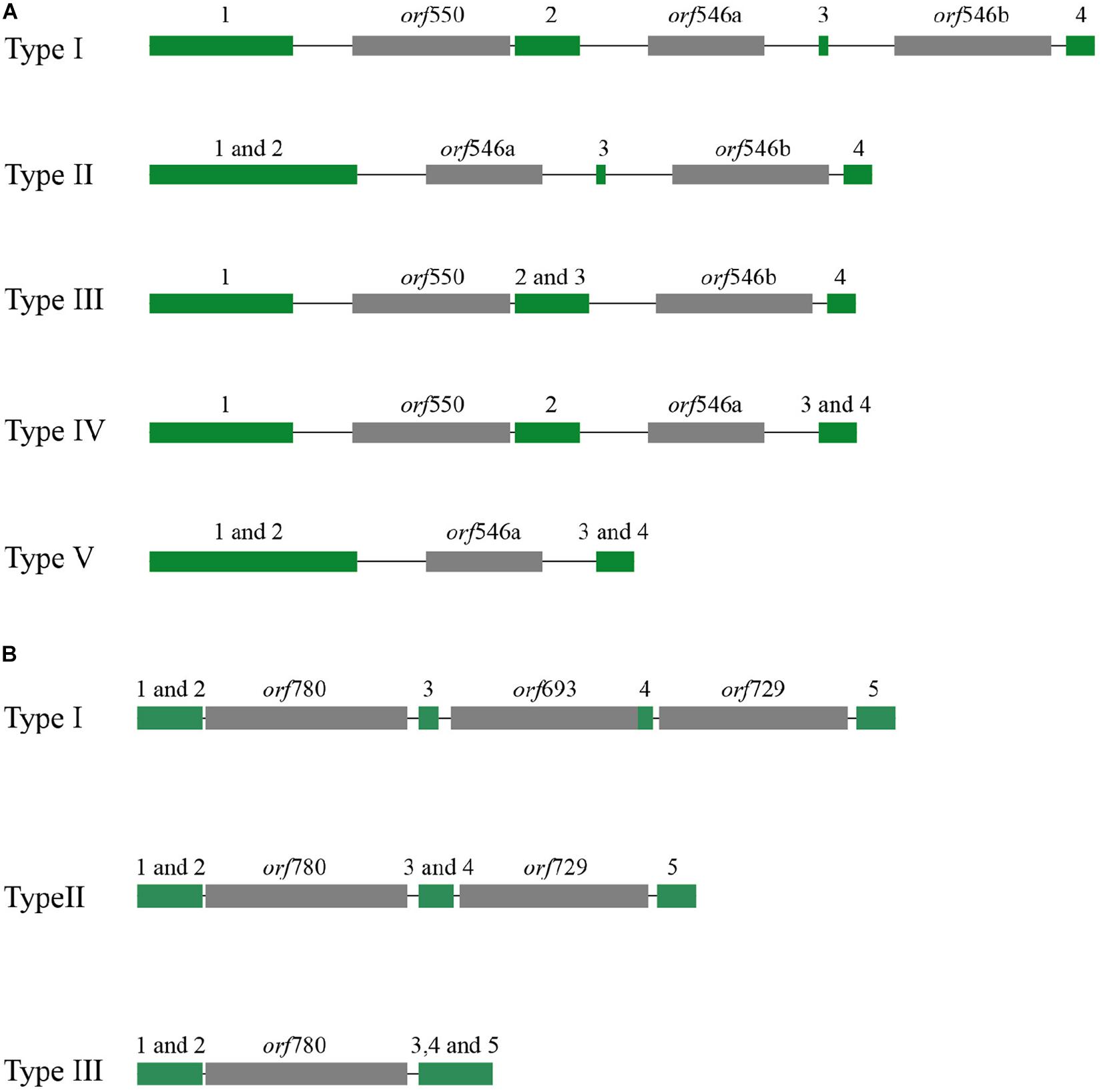
Figure 3. Different types of distributions of group II introns: (A) rnl gene and (B) cox1 gene. The exon of the rnl gene was composed of four coding regions (green block), and cox1 was composed of five regions (numbers over the green block); the intronic open reading frame (ORF) was colored gray.
To further investigate whether there was organellar heteroplasmy within individual P. yezoensis samples, we aligned the NGS data of each individual to their corresponding assembled genomes. The least supported reads of a genotype at each locus was set to four. The aligned results indicated that there was no supporting evidence from these data for heteroplasmy in mtDNA or ptDNA, although four heterozygous loci were found in the ptDNA of all individuals. These two loci were in two direct non-identical repeat regions, which could lead to the misalignment. Validation was then performed using intensive PCR-based sequencing of the repeat regions, which indicated that there were no heterozygous loci. Therefore, we concluded that the P. yezoensis organellar genomes do not display heteroplasmy.
Evaluation of Genetic Diversity Based on Organellar Genomes
Using the mtDNA and ptDNA of P. yezoensis RZ-58 as references, we identified 849 variants, including 829 SNPs and 20 indels across the whole collection (Table 1). There were 463 SNPs and 10 indels detected in the mtDNA with a variation density of 11.35 per 1 kb. After the location of variation across the genome was determined, 364 variants were found within genic regions (Table 1). The different allele types indicated that C/T and A/G were the major forms of base changes with an overall Ts/Tv (transition/transversion) ratio of 4.08. There were 366 SNPs and 10 indels detected in the ptDNA with a variation density of 1.96 per 1 kb. Variation in the plastid genome consisted of 282 variants in genic regions and 94 in intergenic spacers (Table 1). Similarly, C/T and A/G were again the major allele types with an overall Ts/Tv ratio of 2.56. The Qingdao populations held the highest number of SNPs in their mtDNA and the least number of SNPs in their ptDNA (Table 1).

Table 1. Summary of the total SNPs and indels detected in Pyropia yezoensis organellar genomes.
The nucleotide diversity (pi) of the wild Chinese P. yezoensis was calculated at a mean pi of 0.00361 (mtDNA) and 0.000303 (ptDNA) for the whole collection (Table 2). These results indicated that the mutation rate of the mitochondrial genome is greater than that of the plastid genome. A similar trend has been observed in other species of red algae (Smith et al., 2012; Xu et al., 2018b). Among the subgroups, the wild Qingdao P. yezoensis had the highest diversity (0.00348 for mtDNA and 0.000388 for ptDNA) while Yantai had the lowest (0.00222 for mtDNA and 0.000291 for ptDNA).
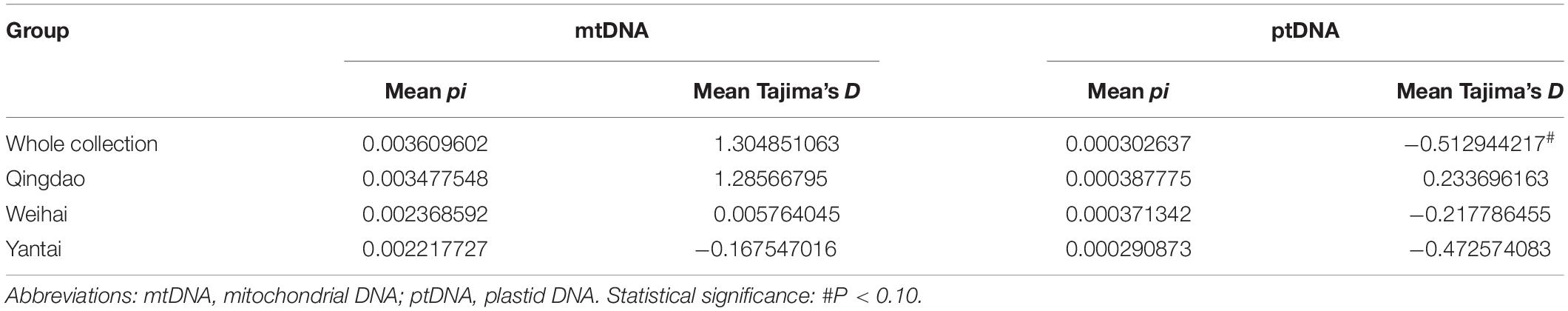
Table 2. Average pi and Tajima’s D value of the whole collection and each group.
The FST values across the three populations were estimated using the organellar SNP data (Supplementary Figure S3). The FST for all pairs of the populations were different and ranged from 0.016 to 0.272 for the mtDNA and 0.047 to 0.087 for the ptDNA. The FST was largest between the wild Qingdao and Yantai populations with average values of 0.272 for mtDNA and 0.087 for ptDNA, which indicates a high degree of genetic isolation between these two groups. The wild Weihai and Yantai populations had the lowest FST values (0.0128 for mtDNA and 0.0436 for ptDNA), which could be attributed to a common ancestry or admixture between the populations.
The Tajima’s D value of the organellar genome was also examined to detect balancing selection. The results identified a location fluctuation in 1 kb bins with positive, negative, and zero values across the genome. The Tajima’s D value for the mtDNA indicated that the wild Qingdao P. yezoensis had more positive positions, which indicated a decrease in its population size and/or balancing selection. In contrast, the Yantai groups had more negative positions, which indicated an expansion in population size and/or purifying selection (Supplementary Figure S3). The Tajima’s D value of the ptDNA also shows more positive positions in the Qingdao group and more negative positions in the Yantai group (Supplementary Figure S3). The closer mean of the Tajima’s D value to 0 indicated weak selection acting on the Weihai group (Table 2). However, Tajima’s D test of each population or whole collection was not found to be significant (p > 0.05).
Cluster Analysis
To address the different evolutionary rates of mtDNA and ptDNA, we combined the mitochondrial and plastid SNPs to conduct the cluster analysis. A total of three sub-groups were identified from the phylogenetic tree (Figure 4A). The dendrogram showed that individuals were mixed among the different geographical populations without obvious regional features. The PCA used the whole SNP dataset and was conducted in EIGENSOFT (Figure 4B). The three main groups were inferred from the first two principal components (PCs), which explained more than 51% of the variation. The PCA plot produced similar results, confirming reliability of the clustering results. We also found that the whole collection clustered according to the different haplotypes of intron structures. Cluster 1 was composed of the R011C0100 haplotype (R = rnl, C = cox1, 0 = intron absent, 1 = intron present). Cluster 2 was composed of the R110C0101 haplotype and cluster 3 was composed of the R110C0101, R111C0100, and R010C0100 haplotypes. Individuals from each haplotype group were present within the same sampling areas at sympatric localities, indicating a genetic break of wild P. yezoensis in the absence of geographic barriers. A Bayesian method based on the MCMC algorithm implemented in STRUCTURE was also performed for identification of genetically different groups. We identified the major substructure groups using an optimal K-value of 3 that was based on the highest ΔK (Supplementary Figure S4), corresponding to the groups identified in wild P. yezoensis. This analysis also revealed there was no significant regional geographic structure in the genetic variation within the groups (Supplementary Figure S5).
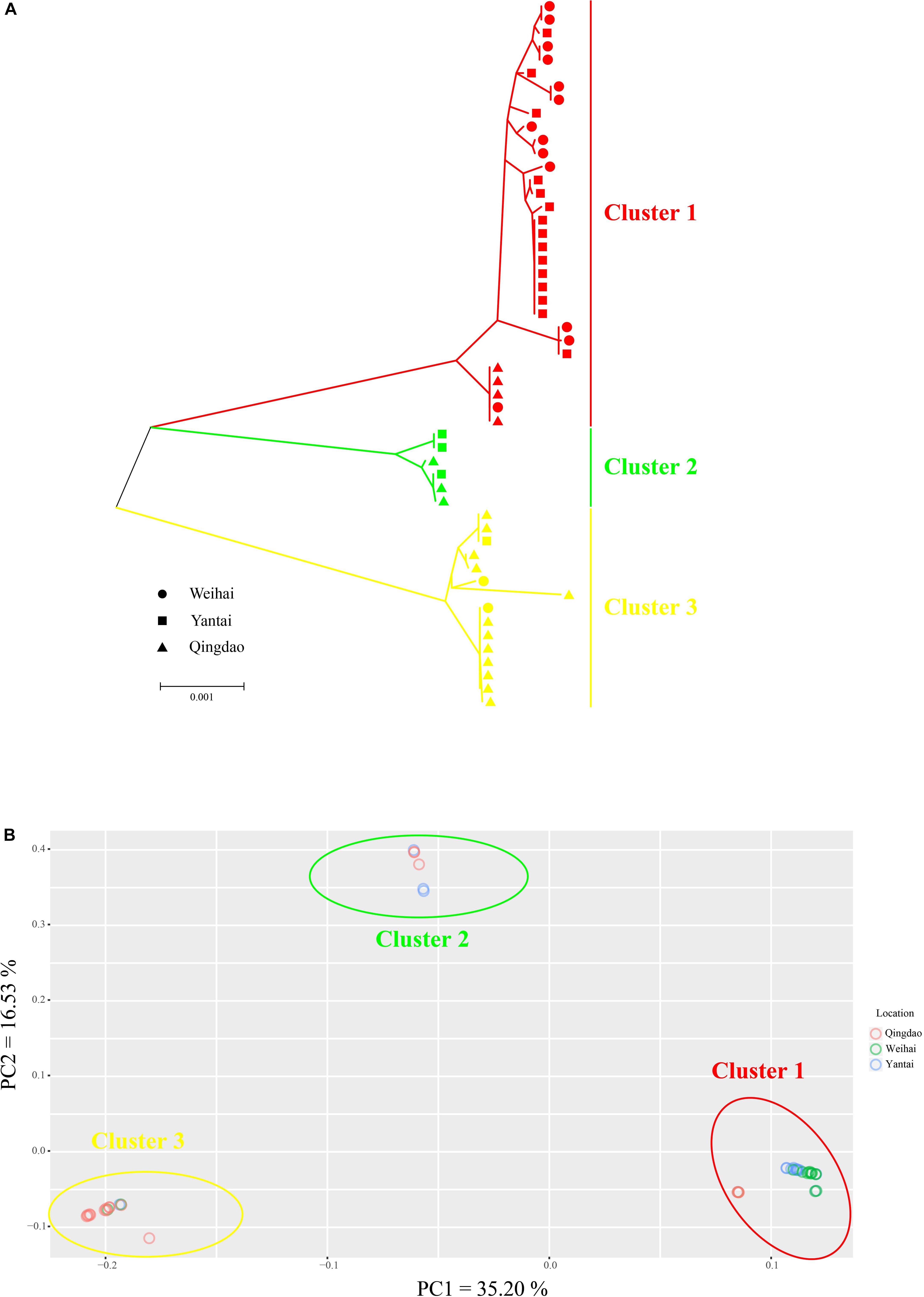
Figure 4. Phylogenetic analysis and principal component analysis (PCA) of the collections. (A) A tanglegram of 53 individuals in three populations of Pyropia yezoensis derived from the neighbor-joining (NJ) method. (B) PCA of all the accessions. The three circles signify the three main groups that were inferred from PCA.
Discussion
Polymorphisms at the Intraspecific and Intraindividual Levels
Variation in the size of P. yezoensis mtDNA was largely due to the different number of mitochondrial group II introns in the rnl and cox1 genes. The differing number of introns was previously reported on in red algae mtDNA at the interspecific level (Hwang et al., 2013; Xu et al., 2018a). In this study, we detected intraspecific variation in intron structure across the 53 Chinese P. yezoensis strains. The combined structural variation of the rnl and cox1 genes resulted in six haplotypes. Recently, Hwang et al. (2018) reported on the genetic features of gene structure in the mitochondrial rnl and cox1 genes of P. yezoensis and found 12 haplotypes from 27 strains from Korea and only one haplotype from 13 strains from Japan. In theory, the combination of the rnl and cox1 genes of P. yezoensis could produce 128 haplotypes. The high intraspecific genetic variation in exon and intron structures could be the more appropriate molecular markers for the identification of species or subspecies. The mitochondrial cox1 gene is considered to be the most suitable DNA barcode for animal and algal species due to its remarkable ability to identify species boundaries (Hebert et al., 2003; Saunders, 2005; Lavinia et al., 2006; Mcdevit and Saunders, 2010). However, the cox1 region is difficult to amplify and sequence compared to other commonly used barcodes for red algae (Sherwood et al., 2010; Du et al., 2015). Results indicate that the failed amplification of cox1 could be caused by the presence or absence of introns. Therefore, we recommend the use of primers that are designed for each exonic region to amplify the cox1 gene.
Heteroplasmy is the presence of DNA from more than one type of organelle in an organism and has been reported in many animals and plants but not yet in this group of red algae (Yamato and Newton, 1999; Chinnery et al., 2000; Steel et al., 2000; Coyer et al., 2004; Frey et al., 2005). However, some of the earlier reports of heteroplasmy may be due to organelle–nuclear DNA transfer (Martin, 2003; Cullis et al., 2009). For instance, the application of NGS followed by read mapping analysis provided no evidence of chloroplast heteroplasmy in sugarcane (Hoang et al., 2015). When assessing heteroplasmy, it is necessary to exclude transferred DNA. In this study, we compared the organelle and nuclear genomes of P. yezoensis in advance, and no transferred DNA fragments could be found (data were not shown). We then re-aligned the NGS data to each of the organellar genomes and found no evidence of heteroplasmy in the mtDNA or the ptDNA. Heteroplasmy could arise from spontaneous mutations or biparental inheritance (Chat et al., 2002; Kmiec et al., 2006). However, a previous study suggested that organelles of P. yezoensis are inherited uniparentally from the female parent (Choi et al., 2010). Moreover, organelle mutations are expected to be purified by genetic drift toward homoplasmy if the mutation is harmful or the two genotypes differ in their replication speed (Hill et al., 2014; Stephan et al., 2015). Therefore, it is plausible that maternal inheritance, an absence of organelle–nuclear DNA transfer and genetic drift could contribute to the homoplasmy observed in the P. yezoensis organellar genomes.
Development of Organellar Genome SNP Markers for P. yezoensis: High Throughput, High Accuracy, and Low Cost
Currently, the complete nuclear genome sequence of Pyropia is unavailable, which is a major constraint for population genetics research for this group. Therefore, information on the polymorphic DNA in organellar genomes can be a useful tool for genetic and evolutionary studies due to the non-recombinant, uniparentally inherited, and effectively haploid genomes (Zeng et al., 2012; Preston et al., 2014). Traditional methods of DNA restriction mapping that use hybridization-based RFLP or SSR have several drawbacks, including low resolution, high labor intensity, and a requirement for large amounts of isolated DNA (Byrne et al., 1993; Wu et al., 2008; Yotsukura et al., 2016). Some of the amplified polymorphic bands were from microorganismal DNA rather than the host DNA due to the number of bacteria or microalgae attached to the surface of the algae. The non-target DNAs from other attached organisms are potentially confounding agents for AFLP DNA fingerprinting. When a reference genome is available, the contaminated DNA can be identified and avoided. Some investigators have predicted that whole-genome analysis will replace other molecular methods such as the AFLP technique. This study represents the first identification of organellar genome SNPs for the analysis of genetic diversity of the Chinese wild P. yezoensis. Compared with existing techniques, this method requires a minimal amount of DNA without any non-target DNA and avoids the laborious experimental procedures that are associated with chloroplast DNA (cpDNA) and mtDNA purification. The sequencing reads of the organellar DNA accounted for 50–90% of the total reads. Considering the small size of the organellar genomes, we could pool the sequencing libraries by adding unique indices. As a result, 200 Mb of sequence bases per individual is enough to achieve a read depth of 400 × for the organellar genomes. Sequencing with higher genomic coverage could decrease the number of missing values and thus obtain even more accurate results. In the present study, we identified a considerable number of SNP markers for the commercially important seaweed P. yezoensis, which led to the identification of 829 SNPs and 20 indels across the diverse collection of specimens. The number of polymorphisms is much larger than the SSRs that were used in previous studies (Kong et al., 2009; Zhang et al., 2009; Hu et al., 2017). Our results demonstrate the high throughput, accurate discovery of organellar DNA polymorphisms, and the low costs associated with this sequencing strategy.
Genetic Diversity in Geographic Populations of P. yezoensis
A broad range of molecular markers is an important genetic resource for breeding applications, which include germplasm conservation, hybridization breeding, quantitative trait loci identification, association mapping, and genomic domestication. All of the above efforts require prior information on genetic diversity (Rao and Hodgkin, 2002; Zhu et al., 2008; Rauf et al., 2010). In this study, applying SNP markers to the whole collection and the geographic populations identified relatively high levels of genetic diversity. A P. yezoensis population that was sampled from five regions along the coast of Shandong Province also showed high levels of genetic diversity as measured by AFLP (Cao et al., 2018). Yang et al. (2003) analyzed genetic variation in 11 lines of P. yezoensis using AFLP and concluded that samples from China shared these features of high genetic diversity. Twelve SSR loci were also used to analyze the genetic diversity of P. yezoensis using wild populations from Qingdao, Yantai, and Weihai (Wei, 2012). These previous results indicated the wild Yantai P. yezoensis had the highest genetic diversity, which conflicts with our finding that the Qingdao population exhibited the highest diversity. The differences in these reported results could be explained by the use of different molecular markers but could also be attributed to decreasing diversity caused by the expansion of cultivated fields in the Yantai coast area during recent years, which overlaps with the habitat of wild P. yezoensis. The genetic diversity of plants can be affected by both genetic and environmental factors (Nevo, 2001). While wild P. yezoensis maintains a relatively high level of genetic diversity, its habitat on natural rocks has been gradually destroyed by coastal pollution and human activities. In order to implement long-term breeding strategies, we first need to make efforts to protect existing wild resources, establish core germplasm banks, and update our breeding and cultivation techniques.
Genetic Break of Organellar DNA at Sympatric Localities
A well-supported organellar DNA break was observed in different geographic populations. Some land plants also display high levels of admixture within their analyzed populations (Aranzana et al., 2010; Barać et al., 2017), which could be attributed to a complex breeding history. We speculate that the presence of a genetic organellar DNA break in the absence of a geographic barrier is the result of historic biogeographic events. Previous studies showed that divergent mtDNA emerges in the absence of structured geographic barriers (Miller et al., 2006). The organellar DNA break might be caused by the sundering of a historically continuous distribution of P. yezoensis due to climate change and the retreat to the coast of Shandong Province, which was facilitated by the winter–spring sea currents in the East China sea. Another contributing factor could be the attractiveness of wild P. yezoensis as a source of food for humans and animals, which led to the dispersal of conchospores or monospores to distinct areas. This phenomenon is often observed for some fruit crops (Mariette et al., 2010; Cao et al., 2012). The genetic break of organellar DNA could lead to the inference that a cryptic species complex within P. yezoensis was reproductively isolated, which was previously reported for the Porphyra/Pyropia complex based on the phylogenetic analysis (Niwa et al., 2009). However, if the observed organellar DNA break reflects reproductive isolation between the haplotype groups, then the break pattern should also be reflected in the nuclear genome of the contact-zone individuals. Organellar and nuclear DNA can differ in their relative responses to historic and recent evolutionary processes due to their distinct modes of inheritance. Discordance between cytoplasmic and nuclear DNA patterns has been previously observed in the wild populations of some species (Egger et al., 2007; Yang and Kenagy, 2009). Therefore, genetic information from nuclear DNA should be further investigated to clarify the relative likelihoods of recent gene flow or the retention of ancestral polymorphisms.
Data Availability Statement
The complete P. yezoensis RZ-58 mitochondrial and plastid genomes are available for download via GenBank under the accession numbers MK695879 and MK695880, respectively. The raw sequencing data for each individual have been deposited into the NCBI Sequence Read Archive under the BioProject: PRJNA550333.
Author Contributions
KX and YM designed the study. KX, XT, and XY performed the experiments. KX, XY, and FK performed the analysis. KX wrote the manuscript. All authors have read and approved the final version of the manuscript.
Funding
This work was financially supported by the Marine S&T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao) (2018SDKJ0302-5), the National Key R&D Program of China (2018YFD0900106), the MOA Modern Agricultural Talents Support Project and National Infrastructure of Fishery Germplasm Resources (2018DKA30470), the Fundamental Research Funds for the Central Universities (201562018, 201762016).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2019.00756/full#supplementary-material
FIGURE S1 | Neighbor-Joining tree for the rbcL sequences of Pyropia. For each node, the bootstrap values are indicated and only values >50 are shown.
FIGURE S2 | Gene structure and annotation of the exon-intron boundaries of the rnl and cox1 genes. “ | ” represents the exon/intron boundary. REF.v1 represents the public mtDNA (NC_017837) and REF.v2 represents the newly assembled mtDNA from this study.
FIGURE S3 | Organellar genome nucleotide diversity (pi), genetic distance (FST), and Tajima’s D test. (A) pi and FST value of the three groups. The circle size indicates the pi value. The FST values between each of the paired groups were represented by the distance between them. The number inside the triangle is FST as calculated by mtDNA and the number outside the triangle is FST as calculated by ptDNA. (B,C) Tajima’s D value for the overall mtDNA (B) and ptDNA (C) in 1 kb bins. The sorted values were plotted for each group.
FIGURE S4 | Delta K (ΔK) graph generated by Structure Harvester.
FIGURE S5 | Population structure analysis of the collections. (A) Population structure clustering using STRUCTURE with K from 2 to 7, sorted by populations. (B) Population structure clustering with an optimal K-value of 3, sorted by a Q matrix.
TABLE S1 | Taxon and GenBank accession numbers of the rbcL genes used in the phylogenetic analyses.
TABLE S2 | Characteristics of the organellar genomes for 53 individuals of P. yezoensis analyzed in this study.
Footnotes
- ^ http://www.fao.org/fishery/factsheets/en
- ^ http://www.ncbi.nlm.nih.gov/projects/gorf/
- ^ http://blast.ncbi.nlm.nih.gov/
- ^ https://github.com/broadinstitute/picard
References
Aranzana, M. J., Abbassi, E.-K., Howad, W., and Arús, P. (2010). Genetic variation, population structure and linkage disequilibrium in peach commercial varieties. BMC Genet. 11:69. doi: 10.1186/1471-2156-11-6
Ba, H., Jia, B., Wang, G., Yang, Y., Kedem, G., and Li, C. (2017). Genome-wide SNP discovery and analysis of genetic diversity in farmed sika deer (Cervus nippon) in Northeast China using double-digest restriction site-associated DNA sequencing. G3 Genes Genomes Genetics 7, 3169–3176. doi: 10.1534/g3.117.300082
Barać, G., Ognjanov, V., Vidaković, D. O., Dorić, D., Ljubojević, M., Dulić, J., et al. (2017). Genetic diversity and population structure of European ground cherry (Prunus fruticosa Pall.) using SSR markers. Sci. Hortic. 224, 374–383. doi: 10.1016/j.scienta.2017.06.060
Bi, Y.-H., Wu, Y.-Y., and Zhou, Z.-G. (2014). Genetic diversity of wild population of Pyropia haitanensis based on SSR analysis. Biochem. Syst. Ecol. 54, 307–312. doi: 10.1016/j.bse.2014.02.010
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., and Buckler, E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Byrne, M., Moran, G. F., and Tibbits, W. N. (1993). Restriction map and maternal inheritance of chloroplast DNA in Eucalyptus nitens. J. Hered. 84, 218–220. doi: 10.1093/oxfordjournals.jhered.a111322
Cao, K., Wang, L., Zhu, G., Fang, W., Chen, C., and Luo, J. (2012). Genetic diversity, linkage disequilibrium, and association mapping analyses of peach (Prunus persica) landraces in China. Tree Genet. Genomes 8, 975–990. doi: 10.1007/s11295-012-0477-8
Cao, Y., Wang, W. J., Liu, F. L., Liang, Z. R., Sun, X. T., Li, X. L., et al. (2018). AFLP fingerprints of Pyropia yezoensis (Bangiales, Rhodophyta) populations revealed the important effect of farming protocols on genetic diversity. Bot. Mar. 61:141. doi: 10.1515/bot-2017-0073
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in largescale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Carvalho, M., Muñoz-Amatriaín, M., Castro, I., Lino-Neto, T., Matos, M., Egea-Cortines, M., et al. (2017). Genetic diversity and structure of Iberian Peninsula cowpeas compared to world-wide cowpea accessions using high density SNP markers. BMC Genomics 18:891. doi: 10.1186/s12864-017-4295-0
Chaisson, M. J., and Tesler, G. (2012). Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR):application and theory. BMC Bioinformatics 13:238. doi: 10.1186/1471-2105-13-238
Chat, J., Decroocq, S., Decroocq, V., and Petit, R. J. (2002). A case of chloroplast heteroplasmy in kiwifruit (Actinidia deliciosa) that is not transmitted during sexual reproduction. J. Hered. 93, 293–300. doi: 10.1093/jhered/93.4.293
Chen, X., Li, Q., Li, Y., Qian, J., and Han, J. (2015). Chloroplast genome of Aconitum barbatum var. puberulum (Ranunculaceae) derived from CCS reads using the PacBio RS platform. Front. Plant Sci. 6:42. doi: 10.3389/fpls.2015.00042
Chin, C.-S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Chinnery, P. F., Thorburn, D. R., Samuels, D. C., White, S. L., Dahl, H. H. M., Turnbull, D. M., et al. (2000). The inheritance of mitochondrial DNA heteroplasmy:random drift, selection or both? Trends Genetics 16, 500–505. doi: 10.1016/S0168-9525(00)02120-X
Choi, S. J., Park, E. J., Endo, H., Kitade, Y., and Saga, N. (2010). Inheritance pattern of chloroplast and mitochondrial genomes in artificial hybrids of Porphyra yezoensis (Rhodophyta). Fish. Sci. 74, 822–829. doi: 10.1111/j.1444-2906.2008.01594.x
Coyer, J., Hoarau, G., Stam, W., and Olsen, J. (2004). Geographically specific heteroplasmy of mitochondrial DNA in the seaweed, Fucus serratus (Heterokontophyta:Phaeophyceae, Fucales). Mol. Ecol. 13, 1323–1326. doi: 10.1111/j.1365-294X.2004.02128.x
Cullis, C. A., Vorster, B. J., Van Der Vyver, C., and Kunert, K. J. (2009). Transfer of genetic material between the chloroplast and nucleus:how is it related to stress in plants? Ann. Bot. 103, 625–633. doi: 10.1093/aob/mcn173
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., Depristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty:de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18. doi: 10.1093/nar/gkw955
Du, G., Wu, F., Guo, H., Xue, H., and Mao, Y. (2015). DNA barcode assessment of Ceramiales (Rhodophyta) in the intertidal zone of the northwestern Yellow Sea. Chin. J. Oceanol. Limnol. 33, 685–695. doi: 10.1007/s00343-015-4088-8
Earl, D. A., and Vonholdt, B. M. (2012). STRUCTURE HARVESTER:a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genetics Resour. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Edea, Z., Dessie, T., Dadi, H., Do, K.-T., and Kim, K.-S. (2017). Genetic diversity and population structure of Ethiopian sheep populations revealed by high-density SNP markers. Front. Genetics 8:218. doi: 10.3389/fgene.2017.00218
Egger, B., Koblmüller, S., Sturmbauer, C., and Sefc, K. M. (2007). Nuclear and mitochondrial data reveal different evolutionary processes in the Lake Tanganyika cichlid genus Tropheus. BMC Evol. Biol. 7:137. doi: 10.1186/1471-2148-7-137
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138. doi: 10.1126/science.1162986
Filichkin, S. A., Hamilton, M., Dharmawardhana, P. D., Singh, S. K., Sullivan, C., Ben-Hur, A., et al. (2018). Abiotic stresses modulate landscape of poplar transcriptome via alternative splicing, differential intron retention, and isoform ratio switching. Front. Plant Sci. 9:5. doi: 10.3389/fpls.2018.00005
Fischer, M. C., Rellstab, C., Leuzinger, M., Roumet, M., Gugerli, F., Shimizu, K. K., et al. (2017). Estimating genomic diversity and population differentiation–an empirical comparison of microsatellite and SNP variation in Arabidopsis halleri. BMC Genomics 18:69. doi: 10.1186/s12864-016-3459-7
Frey, J. E., Frey, B., and Forcioli, D. (2005). Quantitative assessment of heteroplasmy levels in Senecio vulgaris chloroplast DNA. Genetica 123, 255–261. doi: 10.1007/s10709-004-3711-y
He, J.-Y., He, L.-W., Zhang, X., Pan, G.-H., Xu, P., Zhu, J.-Y., et al. (2010). Analysis of genetic diversity of Porphyra yezoensis using AFLP. Oceanol. Limnol. Sin. 4:512. doi: 10.3724/SP.J.1238.2010.00512
Hebert, P. D. N., Cywinska, A., Ball, S. L., and Dewaard, J. R. (2003). Biological identifications through DNA barcodes. Proc. Biol. Sci. 270, 313–321. doi: 10.1098/rspb.2002.2218
Hill, J. H., Zhe, C., and Hong, X. (2014). Selective propagation of functional mitochondrial DNA during oogenesis restricts the transmission of a deleterious mitochondrial variant. Nat. Genet. 46, 389–392. doi: 10.1038/ng.2920
Hoang, N. V., Furtado, A., Mcqualter, R. B., and Henry, R. J. (2015). Next generation sequencing of total DNA from sugarcane provides no evidence for chloroplast heteroplasmy. New Negat. Plant Sci. 1-2, 33–45. doi: 10.1016/j.neps.2015.10.001
Hu, Y., Sui, Z., Zhou, W., Wang, J., Chang, L., Guo, W., et al. (2017). Development of genomic simple sequence repeat markers and genetic diversity analysis of Gracilariopsis lemaneiformis (Rhodophyta). J. Appl. Phycol. 30, 707–716. doi: 10.1007/s10811-017-1237-9
Hwang, I. K., Kim, S.-O., Hwang, M. S., Park, E.-J., Ha, D.-S., and Lee, S.-R. (2018). Intraspecific variation of gene structure in the mitochondrial large subunit ribosomal RNA and cytochrome c oxidase subunit 1 of Pyropia yezoensis (Bangiales, Rhodophyta). ALGAE 33, 49–54. doi: 10.4490/algae.2018.33.2.20
Hwang, M. S., Kim, S.-O., Ha, D.-S., Lee, J. E., and Lee, S.-R. (2013). Complete sequence and genetic features of the mitochondrial genome of Pyropia tenera (Rhodophyta). Plant Biotechnol. Rep. 7, 435–443. doi: 10.1007/s11816-013-0281-4
Katoh, K., Kuma, K. I., Toh, H., and Miyata, T. (2005). MAFFT version 5: improvement in accuracy of multiple sequencealignment. Nucleic Acids Res. 33, 511–518. doi: 10.1093/nar/gki198
Kmiec, B., Woloszynska, M., and Janska, H. (2006). Heteroplasmy as a common state of mitochondrial genetic information in plants and animals. Curr. Genet. 50, 149–159. doi: 10.1007/s00294-006-0082-1
Kong, F., Mao, Y., Yang, H., Qu, H., Yan, X., and Wang, L. (2009). Genetic analysis of Porphyra yezoensis using microsatellite markers. Plant Mol. Biol. Rep. 27, 496–502. doi: 10.1007/s11105-009-0101-8
Kong, F., Sun, P., Cao, M., Wang, L., and Mao, Y. (2014). Complete mitochondrial genome of Pyropia yezoensis:reasserting the revision of genus Porphyra. Mitochondrial DNA 25, 335–336. doi: 10.3109/19401736.2013.803538
Kovar, L., Nageswara-Rao, M., Ortega-Rodriguez, S., Dugas, D. V., Straub, S., Cronn, R., et al. (2018). PacBio-based mitochondrial genome assembly of Leucaena trichandra (Leguminosae) and an intrageneric assessment of mitochondrial RNA editing. Genome Biol. Evol. 10, 2501–2517. doi: 10.1093/gbe/evy179
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7:molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lavinia, R., Stephen, J. R., Gary, L. B., and Juliet, B. (2006). Assessing the use of the mitochondrial cox1 marker for use in DNA barcoding of red algae (Rhodophyta). Am. J. Bot. 93, 1101–1108. doi: 10.3732/ajb.93.8.1101
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Mariette, S., Tavaud, M., Arunyawat, U., Capdeville, G., Millan, M., and Salin, F. (2010). Population structure and genetic bottleneck in sweet cherry estimated with SSRs and the gametophytic self-incompatibility locus. BMC Genet. 11:77. doi: 10.1186/1471-2156-11-77
Martin, W. (2003). Gene transfer from organelles to the nucleus: frequent and in big chunks. Proc. Natl. Acad. Sci. U.S.A. 100, 8612–8614. doi: 10.1073/pnas.1633606100
Mayor, C., Brudno, M., Schwartz, J. R., Poliakov, A., Rubin, E. M., Frazer, K. A., et al. (2000). VISTA:visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16, 1046–1047. doi: 10.1093/bioinformatics/16.11.1046
Mcdevit, D. C., and Saunders, G. W. (2010). A DNA barcode examination of the Laminariaceae (Phaeophyceae) in Canada reveals novel biogeographical and evolutionary insights. Phycologia 49, 235–248. doi: 10.2216/ph09-36.1
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis Toolkit:a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110.20
Mcpherson, H., Merwe, M. V. D., Delaney, S. K., Edwards, M. A., Henry, R. J., Mcintosh, E., et al. (2013). Capturing chloroplast variation for molecular ecology studies:a simple next generation sequencing approach applied to a rainforest tree. BMC Ecol. 13:8. doi: 10.1186/1472-6785-13-8
Miller, M. P., Bellinger, M. R., Forsman, E. D., and Haig, S. M. (2006). Effects of historical climate change, habitat connectivity, and vicariance on genetic structure and diversity across the range of the red tree vole (Phenacomys longicaudus) in the Pacific Northwestern United States. Mol. Ecol. 15, 145–159. doi: 10.1111/j.1365-294X.2005.02765.x
Nevo, E. (2001). Evolution of genome-phenome diversity under environmental stress. Proc. Natl. Acad. Sci. U.S.A. 98, 6233–6240. doi: 10.1073/pnas.101109298
Niwa, K., Iida, S., Kato, A., Kawai, H., Kikuchi, N., Kobiyama, A., et al. (2009). Genetic diversity and introgression in two cultivated species (Porphyra yezoensis and Porphyra tenera) and closely related wild species of Porphyra (Bangiales. Rhodophyta). J. Phycol. 45, 493–502. doi: 10.1111/j.1529-8817.2009.00661.x
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit:a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619
Peccoud, J., Chebbi, M. A., Cormier, A., Moumen, B., Gilbert, C., Marcadé, I., et al. (2017). Untangling heteroplasmy, structure, and evolution of an atypical mitochondrial genome by PacBio sequencing. Genetics 207, 269–280. doi: 10.1534/genetics.117.203380
Preston, M. D., Campino, S., Assefa, S. A., Echeverry, D. F., Ocholla, H., Amambuangwa, A., et al. (2014). A barcode of organellar genome polymorphisms identifies the geographic origin of Plasmodium falciparum strains. Nat. Commun. 5:4052. doi: 10.1038/ncomms5052
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., and Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909. doi: 10.1038/ng1847
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Rao, V. R., and Hodgkin, T. (2002). Genetic diversity and conservation and utilization of plant genetic resources. Plant Cell Tissue Organ Cult. 68, 1–19. doi: 10.1023/A:1013359015812
Rauf, S., Da Silva, J. T., Khan, A. A., and Naveed, A. (2010). Consequences of plant breeding on genetic diversity. Int. J. Plant Breed. 4, 1–21.
Roberts, R. J., Carneiro, M. O., and Schatz, M. C. (2013). The advantages of SMRT sequencing. Genome Biol. 14:405. doi: 10.1186/gb-2013-14-7-405
Sabir, J. S. M., Arasappan, D., Bahieldin, A., Aboaba, S., Bafeel, S., Zari, T. A., et al. (2014). Whole mitochondrial and plastid genome SNP analysis of nine date Palm cultivars reveals plastid heteroplasmy and close phylogenetic relationships among cultivars. PLoS One 9:e94158. doi: 10.1371/journal.pone.0094158
Saunders, G. W. (2005). Applying DNA barcoding to red macroalgae:a preliminary appraisal holds promise for future applications. Philos. Trans. R. Soc. Lond. 360, 1879–1888. doi: 10.1098/rstb.2005.1719
Shen, C. M., Hu, L., Yang, C. H., Yin, C. Y., Li, Z. D., Meng, H. T., et al. (2017). Genetic polymorphisms of 54 mitochondrial DNA SNP loci in Chinese Xibe ethnic minority group. Sci. Rep. 7:44407. doi: 10.1038/srep44407
Sherwood, A. R., Sauvage, T., Kurihara, A., Conklin, K. Y., and Presting, G. G. (2010). A comparative analysis of COI, LSU and UPA marker data for the Hawaiian florideophyte Rhodophyta: implications for DNA barcoding of red algae. Cryptogam. Algol. 31, 451–465. doi: 10.1134/S1021443710060178
Sloan, D. B., Wu, Z., and Sharbrough, J. (2018). Correction of persistent errors in Arabidopsis reference mitochondrial genomes. Plant Cell 30, 525–527. doi: 10.1105/tpc.18.00024
Smith, D. R., Hua, J., Lee, R. W., and Keeling, P. J. (2012). Relative rates of evolution among the three genetic compartments of the red alga Porphyra differ from those of green plants and do not correlate with genome architecture. Mol. Phylogenet. Evol. 65, 339–344. doi: 10.1016/j.ympev.2012.06.017
Steel, D. J., Trewick, S. A., and Wallis, G. P. (2000). Heteroplasmy of mitochondrial DNA in the ophiuroid Astrobrachion constrictum. J. Hered. 91, 146–149. doi: 10.1093/jhered/91.2.146
Stephan, G., Johanna, S., and Ralph, B. (2015). Why are most organelle genomes transmitted maternally Why are most organelle genomes transmitted maternally? Bioessays 37, 80–94. doi: 10.1002/bies.201400110
Sun, P., Mao, Y., Li, G., Cao, M., Kong, F., Wang, L., et al. (2015). Comparative transcriptome profiling of Pyropia yezoensis (Ueda) MS Hwang & HG Choi in response to temperature stresses. BMC Genomics 16:463. doi: 10.1186/s12864-015-1586-1
Sun, S., Zhou, Y., Chen, J., Shi, J., Zhao, H., Zhao, H., et al. (2018). Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat. Genet. 50:1289. doi: 10.1038/s41588-018-0182-0
Suzuki, S., Ranade, S., Osaki, K., Ito, S., Shigenari, A., Ohnuki, Y., et al. (2018). Reference grade characterization of polymorphisms in full-length HLA class I and II genes with short-read sequencing on the Ion PGM system and long-reads generated by single molecule, real-time sequencing on the PacBio platform. Front. Immunol. 9:2294. doi: 10.3389/fimmu.2018.02294
Wang, L., Mao, Y., Kong, F., Li, G., Ma, F., Zhang, B., et al. (2013). Complete sequence and analysis of plastid genomes of two economically important red algae: Pyropia haitanensis and Pyropia yezoensis. PLoS One 8:e6590. doi: 10.1371/journal.pone.0065902
Wattier, R. A., Prodohl, P. A., and Maggs, C. A. (2000). DNA isolation protocol for red seaweed (Rhodophyta). Plant Mol. Biol. Rep. 18, 275–281. doi: 10.1007/bf02823999
Wei, L. (2012). Genetic Diversity Analysis of Porphyra yezoensis and Porphyra haitanensis with Simple Sequence Repeat. Msc Thesis, Ocean University of China, China.
Wei, T., Kim, T. S., and Park, Y. J. (2016). Rice chloroplast genome variation architecture and phylogenetic dissection in diverse oryza species assessed by whole-genome resequencing. Rice 9:57. doi: 10.1186/s12284-016-0129-y
Wu, F., Zhang, Z., Dai, H., Zhang, Y., and Chang, L. (2008). Genetic relationship of some Crataegus spp. (Hawthorn) revealed by chloroplast DNA PCR-RFLP. J. Biotechnol. 136:S103. doi: 10.1016/j.jbiotec.2008.07.235
Xiang, B., Li, X., Qian, J., Wang, L., Ma, L., Tian, X., et al. (2016). The complete chloroplast genome sequence of the medicinal plant Swertia mussotii using the PacBio RS II platform. Molecules 21:1029. doi: 10.3390/molecules21081029
Xie, C., Li, B., Xu, Y., Ji, D., and Chen, C. (2013). Characterization of the global transcriptome for Pyropia haitanensis (Bangiales, Rhodophyta) and development of cSSR markers. BMC Genomics 14:107. doi: 10.1186/1471-2164-14-107
Xu, K., Tang, X., Bi, G., Cao, M., Wang, L., and Mao, Y. (2018a). The first complete organellar genomes of an Antarctic red alga, Pyropia endiviifolia: insights into its genome architecture and phylogenetic position within genus Pyropia (Bangiales, Rhodophyta). J. Oceanol. Limnol. 36, 1315–1328. doi: 10.1007/s00343-018-7088-7
Xu, K., Tang, X., Wang, L., Yu, X., Sun, P., and Mao, Y. (2018b). Divergence time, historical biogeography and evolutionary rate estimation of the order Bangiales (Rhodophyta) inferred from multilocus data. J. Oceanol. Limnol. 36, 870–881. doi: 10.1007/s00343-018-7054-4
Yamato, K., and Newton, K. (1999). Heteroplasmy and homoplasmy for maize mitochondrial mutants: a rare homoplasmic nad4 deletion mutant plant. Heredity 90, 369–373. doi: 10.1093/jhered/90.3.369
Yang, D. S., and Kenagy, G. J. (2009). Nuclear and mitochondrial DNA reveal contrasting evolutionary processes in populations of deer mice (Peromyscus maniculatus). Mol. Ecol. 18, 5115–5125. doi: 10.1111/j.1365-294X.2009.04399.x
Yang, R., Liu, B. Q., Luo, Q. J., Wang, Y. J., and Bao, J. M. (2003). Genetic variation of Porphyra yezoensis by using AFLP1. Acta Oceanol. Sin. 22, 453–457. doi: 10.1029/2002JC001507
Yotsukura, N., Maeda, T., Abe, T., Nakaoka, M., and Kawai, T. (2016). Genetic differences among varieties of Saccharina japonica in northern Japan as determined by AFLP and SSR analyses. J. Appl. Phycol. 28, 1–13. doi: 10.1007/s10811-016-0807-6
Zeng, C.-L., Wang, G.-Y., Wang, J.-B., Yan, G.-X., Chen, B.-Y., Xu, K., et al. (2012). High-throughput discovery of chloroplast and mitochondrial DNA polymorphisms in Brassicaceae Species by ORG-EcoTILLING. PLoS One 7:e47284. doi: 10.1371/journal.pone.0047284
Zhang, P., Zhang, Y., Wang, T. G., and Yan, X. H. (2009). Phylogenetic relationship of the lines of Porphyra haitanensis (Rhodophyta,Bangials) determined by microsatellite DNA markers. J. Fish. Sci. China 16, 842–849. doi: 10.1016/S1874-8651(10)60073-7
Zhou, C., Wang, C., Liu, H., Zhou, Q., Liu, Q., Guo, Y., et al. (2018). Identification and analysis of adenine N 6-methylation sites in the rice genome. Nat. Plants 4:554. doi: 10.1038/s41477-018-0214-x
Keywords: organellar genome, intraspecific variation, organelle heteroplasmy, single-nucleotide polymorphisms, genetic diversity, Pyropia yezoensis
Citation: Xu K, Yu X, Tang X, Kong F and Mao Y (2019) Organellar Genome Variation and Genetic Diversity of Chinese Pyropia yezoensis. Front. Mar. Sci. 6:756. doi: 10.3389/fmars.2019.00756
Received: 11 July 2019; Accepted: 21 November 2019;
Published: 12 December 2019.
Edited by:
Zhijun Dong, Yantai Institute of Coastal Zone Research (CAS), ChinaReviewed by:
Zi-Min Hu, Institute of Oceanology, Chinese Academy of Sciences, ChinaJuan Andrés López, University of Alaska Fairbanks, United States
Copyright © 2019 Xu, Yu, Tang, Kong and Mao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunxiang Mao, eXhtYW9Ab3VjLmVkdS5jbg==; eXhtYW9AaG50b3UuZWR1LmNu