 Remi N. Ketchum1*
Remi N. Ketchum1* Edward G. Smith2
Edward G. Smith2 Grace O. Vaughan2
Grace O. Vaughan2 Britney L. Phippen1Dain McParland2Noura Al-Mansoori2
Britney L. Phippen1Dain McParland2Noura Al-Mansoori2 Tyler J. Carrier1
Tyler J. Carrier1 John A. Burt2
John A. Burt2 Adam M. Reitzel1
Adam M. Reitzel1- 1Department of Biological Sciences, University of North Carolina at Charlotte, Charlotte, NC, United States
- 2Center for Genomics and Systems Biology, New York University Abu Dhabi, Abu Dhabi, United Arab Emirates
The microbial assemblages of marine organisms play fundamental biological roles in their eukaryotic hosts. Studies aimed at characterizing this diversity have increased over the last decade and with the availability of high-throughput sequencing, we are now able to characterize bacteria that were non-culturable and, therefore, went undetected. With the number of marine microbiome studies growing rapidly, it is increasingly important to develop a set of “best practices” in order to accurately represent the bacterial communities present, and correct for biases. To address this, we sampled the gut communities of the pan-tropical echinoid Echinometra mathaei from two environmentally distinct populations along the Arabian Peninsula. We used three common DNA extraction procedures and compared inferred bacterial diversity from each method through 16S ribosomal RNA (rRNA) gene amplicon sequencing. Our results show that the addition of a bead-beating and lysozyme step more effectively capture traditionally difficult to lyse taxa, such as gram-positive bacteria. Further, DNA extraction method plays an important role in estimates of Shannon diversity, with diversity indices significantly higher in both sites combined when a lysozyme and bead beating step was used. Finally, we conducted a literature synthesis to highlight the current diversity of approaches used to characterize the microbiome of marine invertebrates and found that the inclusion of a lysozyme treatment is uncommon (2% of surveyed studies), despite the importance of this step in recovery of rare OTUs as shown in our study.
Introduction
Bacteria associate with animals and can play a significant role in their development, immunity, metabolism, and physiology (Mcfall-Ngai et al., 2013; Bordenstein and Theis, 2015; Theis et al., 2016). In marine environments, the functional role of the microbiome has been associated with diverse processes including cycling of essential nutrients, the passage of trace minerals, production of secondary metabolites, and vitamin synthesis (reviewed by Bourne et al., 2016; Webster and Reusch, 2017). There has been an increasing amount of research focused on characterizing microbial community dynamics and shifts, and understanding how the functional role of the microbiome may co-vary with changes in the environment (Kohl and Carey, 2016; Carrier and Reitzel, 2017). For example, specific bacterial communities were found to be tightly correlated with thermal tolerance in a coral host (Ziegler et al., 2017), expression levels of innate immunity genes in the three spine stickleback (Small et al., 2017), and geographic and depth gradients in a sponge species (Reveillaud et al., 2014).
The Earth Microbiome Project (EMP) resulted in a set of recommended approaches for characterization of microbial taxa based on sequence analysis. While sponge microbial research has recently adopted the EMP protocol (Thomas et al., 2016; Moitinho-Silva et al., 2017), studies of associated bacterial communities of other marine invertebrate taxa currently lack a standardized approach. Sequence-based methods for describing associated microbes are sensitive to methodological steps and these differences may limit the ability to compare between studies or species. It is well established in human and environmental samples that bias can be introduced at each methodological step and significant research has gone into elucidating the role that methodology plays on interpretation of bacterial communities (McOrist et al., 2002; Yuan et al., 2012; Albertsen et al., 2015; De Bruin and Birnboim, 2016; Pollock et al., 2018). The variation in methodology that currently exists can be confounding in many ways, including differences in inferred microbial communities due to methods (species differ in efficacy for DNA extraction), information differences (different regions of 16S rRNA or metagenomics), and technical variation (PCR replicates and purifications). However, the majority of this research has been conducted on the human microbiome, activated sludge, and soil (Miller et al., 1999; McOrist et al., 2002; Vanysacker et al., 2010; Yuan et al., 2012). The role of methodology in the microbial communities of marine organisms remains comparatively understudied.
While the impact of each step in a sequence-based analysis of the microbiome is likely important, extraction, and amplification of nucleic acids is central in characterizing a mixed bacterial community. DNA extractions should, as accurately as possible, characterize the bacterial diversity present. For example, methods for DNA extraction in corals influence the DNA yield and inferred microbial community (Weber et al., 2017). Exclusion of particular steps (e.g., mechanical and/or chemical lysis) (Lesser and Walker, 1992) in extraction protocols may result in unsampled portions of the microbial community due to difficult to lyse bacteria (e.g., gram-positive bacteria or endospores), which may play important functional roles in their hosts. Extraction protocols that result in a greater portion of the bacterial community being represented are important for identifying common, core, and rare associated microbes.
Echinoderms are dominant members of benthic marine habitats throughout the world's oceans. Over the last few decades, research using histological approaches has reported subcuticular bacteria in all classes of echinoderms (e.g., Holland and Nealson, 1978; Kelly et al., 1995). The localization and apparent specificity of these bacteria has suggested that echinoderms select particular bacteria for these associations, but any specific functions have only rarely been described (Walker and Lesser, 1989; Lesser and Walker, 1992). Efforts to culture echinoderm-associated bacteria have been either unsuccessful or difficult to repeat (discussed in Kelly et al., 1995). Recent studies using sequence-based approaches have reported diverse microbial communities that differ between species and developmental stage for asteroids (Galac et al., 2016) and echinoids (Carrier and Reitzel, 2018). Shifts in the microbial community associated with echinoid spines was reported for adults cultured under different temperature and pH conditions (Webster et al., 2016). Microbial communities of adult digestive systems of holothuroids (Gao et al., 2014), and echinoids (Meziti et al., 2007; Nelson et al., 2010; Hakim et al., 2015, 2016) have identified specific microbial communities located in different regions of the digestive system. Together, these previous studies suggest that echinoderms associate with unique microbial communities that may shift in response to different environmental conditions.
Here, we determined the microbial community in the hind-gut of Echinometra mathaei, a keystone urchin species (Uthicke et al., 2013), from two reef populations along the Arabian Peninsula. One population, inside the Persian/Arabian Gulf (PAG), Dhabiya, experiences extreme environmental conditions, while the other, inside the Gulf of Oman (GO), Al Aqah, experiences cooler thermal maxima and a more narrow range of temperatures (Smith et al., 2017). Sampling from these two sites allowed us to determine the effect of DNA extraction method in conspecifics across two contrasting environments in this species' contiguous geographic range. We use three different nucleic acid extraction techniques to compare bacterial communities and elucidate specific bacterial taxa that may be missed depending on the methods used. We then compare our approach with a literature survey of the methodologies currently used in studies with other marine invertebrates in order to place our results in the context of the broader community studying these microbial associations.
Materials and Methods
Sample Collections
Adult Echinometra mathaei were sampled in November 2016 from two reef sites: Al Aqah (in the Gulf of Oman, GO) and Dhabiya (in the Persian/Arabian Gulf, PAG, see Figure 1). The sampling sites were chosen because they experience vastly different environmental conditions and they were as geographically distant (~270 miles apart) as was possible given our sampling capabilities. Fifteen individuals were taken from each reef and placed in a 100 L cooler filled with ~40 L of seawater until tissue extractions (within 1–2 h). For gut extractions, individual urchins were then cut in half with a sterile knife, and a fragment of intestine located closest to the anus, and its contents, were removed with forceps and placed in RNAlater (Ambion). After one hour (to allow RNAlater to infiltrate the tissue), tubes were placed in a −20°C freezer.
Figure 1. Map of Arabian Peninsula indicating the location of the two sampling sites.
DNA Extraction
Prior to extraction, each sample tube was homogenized using a vortex (3x at 15 s) and mixed with a wide bore 100 μL pipette tip to ensure that each solution was uniform. This initial step reduced or eliminated bias that could occur simply through extraction of different sections of the sample. An aliquot of ~20 μL of the original sample (gut tissue and contents) was placed in three separate tubes, one for each different extraction method. Each method used the DNeasy Blood & Tissue Kit (QIAGEN). All extraction materials were autoclaved, UV sterilized, and DNA extractions and PCRs were performed under a PCR hood.
Method 1. The first extraction method used only the DNA extraction kit and followed the manufacturer's protocols for extracting animal tissue, with slight modification. We added 40 μL of proteinase K and 220 μL of AL buffer (instead of the recommended 20 and 200 μL, respectively) as recommended by the User-Developed Protocol for DNeasy Blood and Tissue Kit when samples are preserved in RNAlater. These modifications were kept consistent throughout each of the other two extraction methods.
Method 2. In addition to the kit described above, we added a bead beating step after addition of Buffer ATL and before placing sample on a heat block. We added 0.5 mL of 0.5 mm zirconia-silica beads (Fisher Scientific) and used the Bead Beater (BioSpec Products) for 40 s at 2,400-3,800 strokes/min and repeated this a total of three times.
Method 3. In addition to the kit and the bead beating step, Method 3 included the addition of 180 μL enzymatic lysis buffer; 20 mM TrisCL (pH 8), 2 mM sodium EDTA, 1.2% Triton, 20 mg/ml lysozyme (as described in DNeasy Blood and Tissue Manual). The buffer was added in place of Buffer ATL, incubated at 37°C and followed by the bead beating step (the extraction then proceeded as per the guidelines in the QIAGEN manual).
Samples that did not contain sufficient DNA (< 2ng/μL) were either extracted again or concentrated using an ethanol precipitation. All samples were then quantified using a Qubit dsDNA High Sensitivity Assay Kit on the Qubit® 2.0 Fluorometer and visualized using a 2% agarose gel. Prior to PCR amplification, all samples were normalized to a concentration of 1 ng/μL.
PCR Amplification and Sequencing
PCRs were performed in triplicate 25 μL reactions and pooled per individual sample. The 16S rRNA V3/V4 region was amplified using the universal V3/V4 PCR primers (forward: 5′- CTACGGGNGGCWGCAG-3′; reverse: 5′-GACTACHVGGGTATCTAATCC-3′) (Klindworth et al., 2013). Each PCR contained 5 ng of DNA, 1.5 μL of forward primer (final concentration of 10 mM), 1.5 μL of reverse primer (final concentration of 10 mM), 12 μL of KAPA Biosciences HiFi HotStart Ready Mix, and 5 μL of water for a final volume of 25 μL. PCR cycling conditions were 95°C for 3 min, followed by 20 cycles of 95°C for 30 s, 50°C for 30 s, and 72 for 30 s, with a final extension time of 5 min at 72°C. Successful amplification was visualized at 35 cycles on a 2% agarose gel.
MiSeq indexing adaptors were added according to the Illumina 16S Metagenomic Sequencing Library Preparation protocol and an AxyPrep Mag™ PCR Clean-up Kit (Axygen Biosciences, Corning). 16S rRNA gene amplicon libraries were sequenced along with a 30% PhiX control at the University of North Carolina Charlotte sequencing facility on the Illumina MiSeq platform using 2x300 bp paired-end reads. As an extra quality control measure for potential contamination in the reagents, we sequenced different types of no template controls: two blanks that were processed through the DNA extraction column to identify potential contaminants from the kit, as well as sequencing one no template control that was only a water sample added to the original PCR reaction to identify potential contaminants in other parts of the procedure.
Sequence Data Processing
We merged the paired-end reads using PEAR v.0.9.10 with default settings, and used Trimmomatic v.0.36 for moderate quality trimming (threshold quality = 20; minimum length = 50) (Bolger et al., 2014; Zhang et al., 2014b). Chimeras were identified using UCHIME and removed using the filter_fasta.py command in QIIME v.1.9.1 (Caporaso et al., 2010; Edgar et al., 2011). All subsequent analysis was performed in QIIME v.1.9.1. Merged, trimmed, non-chimeric sequences were clustered into Operational Taxonomic Units (OTUs) at 97% sequence similarity using open-reference picking with the UCLUST algorithm. OTUs with fewer than 10 sequence reads were removed. One DNA sample from the PAG resulted in few sequence reads (maximum was 2,432 reads which was lower than the blanks) regardless of DNA extraction method and was therefore removed from later analyses, resulting in a total of 87 samples. We also filtered out any OTUs that were present in a higher abundance within the three blank samples than within our biological samples. This resulted in deletion of 228 OTUs comprising 11,916 sequence reads from all three blank samples combined and deletion of 28,101 sequence reads from all 87 samples combined. This was less than 0.8% of the total reads resulting from the biological samples (see Supplemental Table 1 for complete list of removed OTUs). We also removed chloroplast sequences as they are assumed to be brought into the gut through ingestion. Taxonomy was assigned to the remaining OTUs through comparison with the Greengenes database 13_5. OTU counts were rarefied to 17,903 sequence reads (the lowest number of sequences over all samples included in the analysis) using the single_rarefaction.py command and the rarefied OTU table was used in downstream analysis.
16S rRNA Microbial Community Analysis
Taxonomically annotated sequences were used to create bacterial community composition stacked plots at the family level to compare between methods and sites. Venn diagrams were generated using Venny 2.1.0 (Oliveros, 2007) to compare the number of shared and unique OTUs between methods and sites. PCoA plots were generated using Euclidean distance and Shannon and Simpson diversity indices were calculated and processed in QIIME. A Kruskal-Wallis test was used to identify OTUs whose abundance was significantly different between methods. A non-parametric, two sample t-test was run using QIIME to test significance between alpha diversity indices for all pairwise comparisons between methods using the default number of Monte Carlo permutations (999) and the Bonferroni correction method. Non-metric multidimensional scaling (MDS) was used to visualize differences between methods in Dhabiya, PAG. Variations between methodologies from both collection sites were examined using an analysis of similarity (ANOSIM), a robust method which does not require normally distributed data or balanced replicates between groupings (Voss et al., 2007). Non-metric MDS ordinations and ANOSIM were conducted using Primer 7 (Clarke, 1993).
Literature Search
We searched the Web of Science (access dates 20/02/2018 - 24/02/2018) for publications reporting empirical research on the associated bacterial community for marine invertebrates. Our intention for these searches was not to be exhaustive but to identify and compare methodological approaches primarily related to DNA extraction from tissues. Literature searches were completed using two common “topics” search terms, “marine” and “microbio*,” and a third specific search term for four groups of animals, “coral,” “sponge,” “echino*,” and “mollus*.” We replaced the search term “microbio*” with “bacteria,” which resulted in considerably more articles (e.g., 4.6X more for “coral”), but our preliminary comparisons suggested many were largely unrelated to our area of interest and thus we determined using “microbio*” was a more effective search term. Results for these four searches first included publications over all of the years in the database (1900–2017) to compare publication trends (Supplemental Figure 1). To restrict our search for a more in-depth literature comparison, we then reviewed all publications in these results for the years 2015–2017. We removed publications that were not relevant, typically because they were not specific to the taxonomic group (e.g., water samples), did not study microbes from live organisms (e.g., paleontology or non-living structures like shells), or did not use next generation or clone-library sequence-based methodology (e.g., culture-based studies). For the pruned list of publications, we categorized the methodology of each approach based on DNA extraction method, PCR strategy, and sequencing technology. A number of publications summarized their methods when using a commercial kit using phrases like “following manufacturer's protocol” or similar. For these, we categorized the methods using the default steps for DNA extraction without additional steps recommended elsewhere in the manual.
Results
Bacterial Community Composition Between Methods and Sites
Sequencing of the V3/V4 region of the 16S rRNA resulted in a total of 6,554,465 reads from 93 samples: 90 E. mathaei samples (15 unique samples from Dhabiya (PAG), 15 unique samples from Al Aqah (GO); 30 total samples extracted using three different methods) and three blanks. DNA extraction Method 1 used the DNeasy Blood and Tissue Kit (QIAGEN), Method 2 added a bead beating step to this kit protocol, and Method 3 added the bead beating step and lysozyme to the kit protocol. After quality filtering, our pipeline retained 87 total samples with 3,469,193 sequence reads. To evaluate differences in the bacterial community composition, sequences were categorized taxonomically to the family level, where possible. We found family level taxonomic assignment to be the most informative, as genus and species level classification was often unsuccessful and resulted in many unknown strains. Samples from both geographic sites and all three methods were composed of similar principal bacterial families, although varying in levels of abundance (Figure 2).
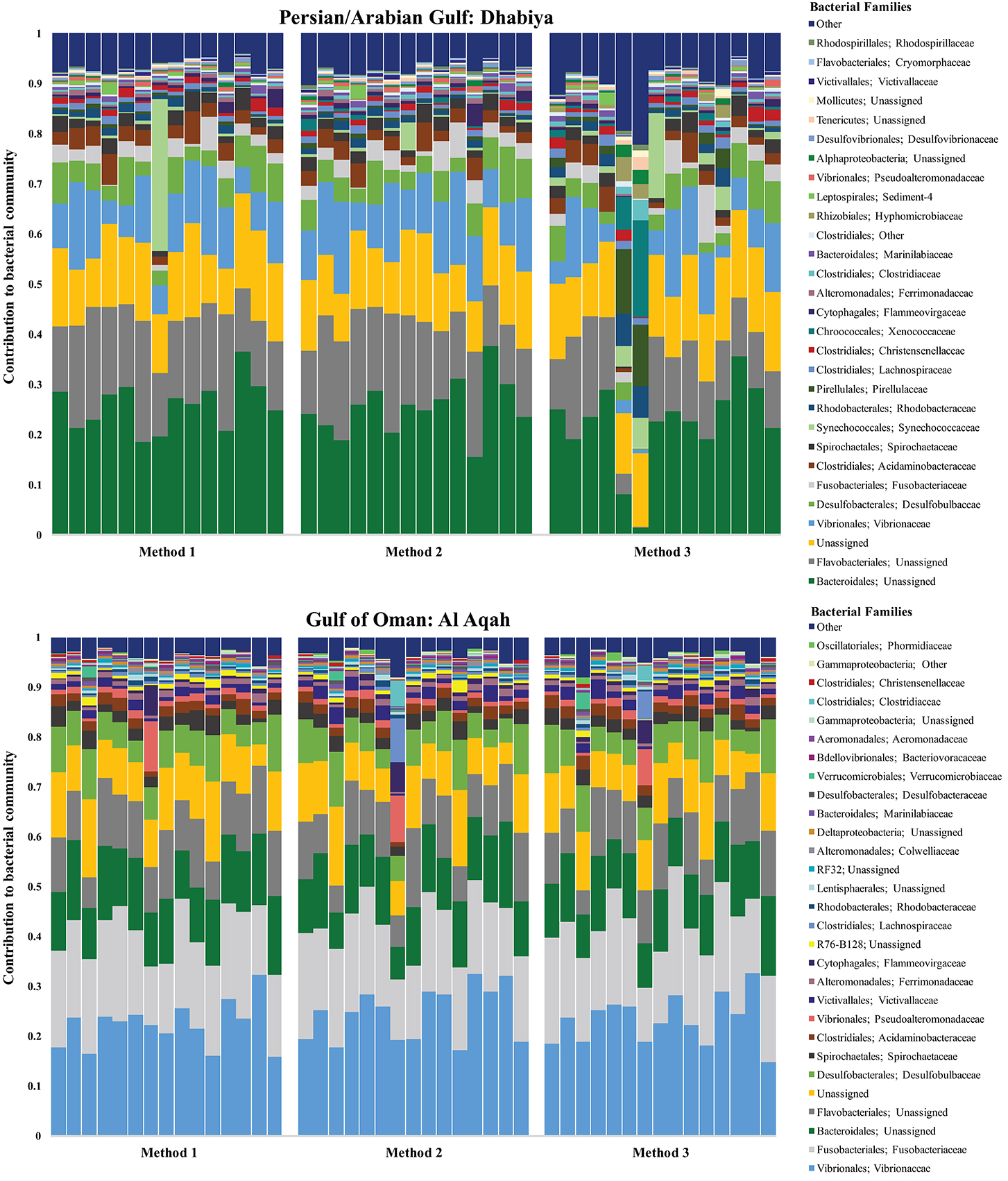
Figure 2. Bacterial community composition of gut contents from Echinometra mathaei from two populations; Persian/Arabian Gulf (on top), Gulf of Oman (on bottom). This taxonomy stacked column plot is characterized to the family level, where possible. Each color represents one of the 29 most abundant families and the remaining taxa are grouped into the category “other.” Samples are grouped by method.
For example, almost all 87 samples contained Bacteriodales (range: ~2–38%, average number of reads: 3,203, standard deviation: 1442.4), Flavobacteriales (~5–24%, 2,269, 850.8), Desulfobulbaceae (~1–11%, 1,076, 449.3), and Fusobacteriaceae (~1–26%, 2,078, 1620.8), with a similar proportion of reads classifying as unassigned (~4-19%, 2,191, 609.7). Individuals from the PAG had a total of 304 OTUs while those from the GO had 271, with the PAG containing 50 unique OTUs, and the GO containing 17 (Figure 3). The location where samples were collected explained 63.7% of the variation in the samples (PC1 vs. PC2) (Figure 4). For this reason, we separated our data based on location in order to describe the differences driven solely by methodology. Method 3 contains 11 unique OTUs while both Method 1 and 2 contain only 4. Further, Method 2 and 3 share more OTUs (n = 23) than Method 1 and 2 (n = 7) or Method 1 and 3 (n = 8).
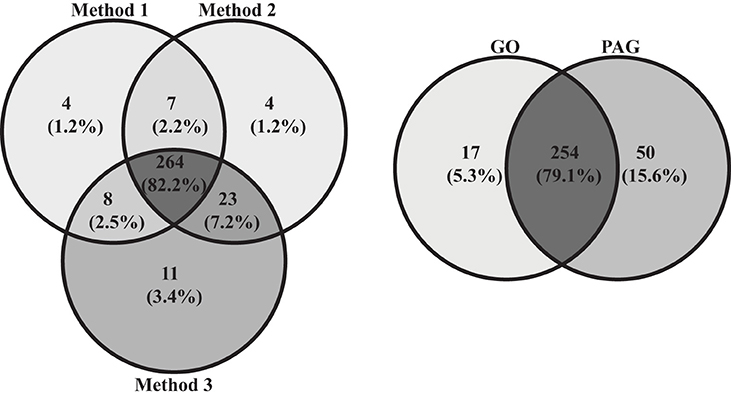
Figure 3. Venn diagrams comparing number of OTUs within and between the PAG and the GO, as well as within and between the three different methods.
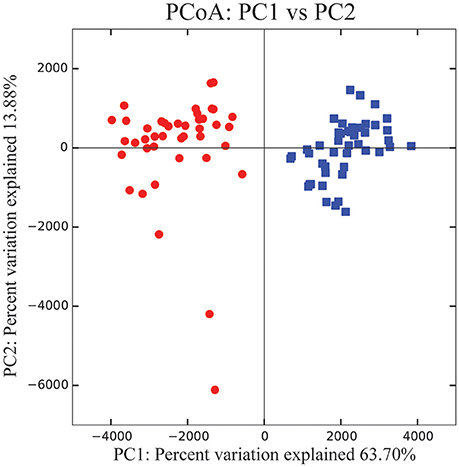
Figure 4. PCoA plot showing PC1 vs. PC2 using Euclidean distance (Bray-Curtis distance yielded nearly identical results). Red circles indicate samples collected from the PAG, while blue squares indicate samples collected from the GO.
Inside the PAG, there were 13 bacterial families that differed significantly in abundance between the methodologies used (Table 1). For every bacterial family whose abundance differed significantly between methods, Methods 2 and 3 contained a higher number of mean sequence reads than Method 1 for all but one bacteria (Acidimicrobiales; C111) (Table 2). Notably, five out of 13 of these taxa were gram-positive bacteria, groups known to have thick cell walls: Gaiellales, Turicibacteraceae, Staphylococcaceae, Pseudonocardiaceae, and Acidimicrobiales. Moreover, two of these 13 taxa were cyanobacteria; Xenococcaceae, and Pseudanabaenaceae, with differential representation between methods. The most significant difference (Kruskal–Wallis, P < 0.001) between methods was Pseudomonadaceae. Lastly, the remaining five bacterial taxa consisted of Hyphomicrobiaceae, Amoebophilaceae, Rhodospirillales, Ellin6529, and Methylobacteriaceae. Inside the GO, there were four significant differences between methods; two cyanobacteria (Cyanobacteriaceae and Xenococcaceae), one gram-positive bacteria (C111) and Pseudomonadaceae.
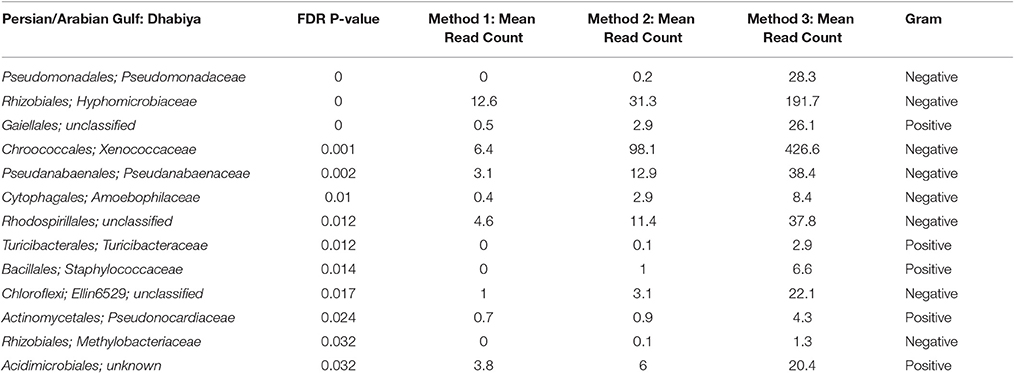
Table 1. All bacterial families that are significantly different between the three methods in the Persian/Arabian Gulf.
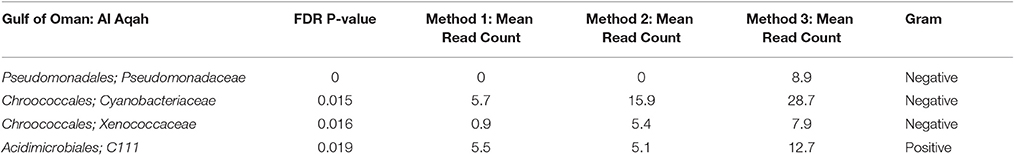
Table 2. All bacterial families that are significantly different between the three methods in the Gulf of Oman.
Shannon diversity was higher inside the PAG than the GO, independent of methodology (Figure 5). Shannon diversity for both sites combined was significantly higher in Method 3 than Method 1 (non-parametric t-test, P = 0.018). Simpson diversity showed a similar trend to Shannon diversity when comparing methodologies, although this difference was not significant (non-parametric t-test, P = 0.123) (Supplemental Figure 2). Multivariate statistics revealed an overall significant community difference between Methods 1 and 3 in both sites combined (ANOSIM, P = 0.011, R = 0.084). Although the differences between Methods 1 and 2 (ANOSIM, P = 0.421, R = 0.001) and Method 2 and 3 (ANOSIM, P = 0.122, R = 0.031) were not statistically significant, the non-metric MDS plot showed near uniform directionality between methods in the PAG (Figure 6). This was indicative of consistent changes in the abundance of certain taxa (see Tables 1, 2) between methods that were driving the vectors connecting individuals in a consistent direction relative to the other two methods. Further, the effect that method had on community composition was heavily determined by individual (as seen in Figures 5, 6), with some individuals displaying a greater variation in vector length between methods. This suggests that these individuals contain a higher abundance of difficult to lyse bacteria.
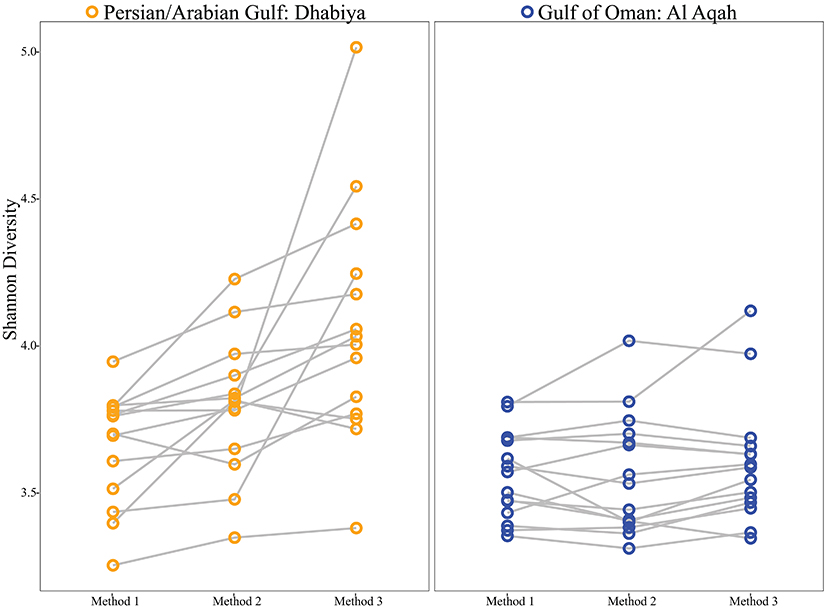
Figure 5. Shannon Diversity indices of Persian/Arabian Gulf samples (left) and Gulf of Oman samples (right). Each node corresponds to an individual sample and each line connects one individual's tissue subjected to the three DNA extraction methods.
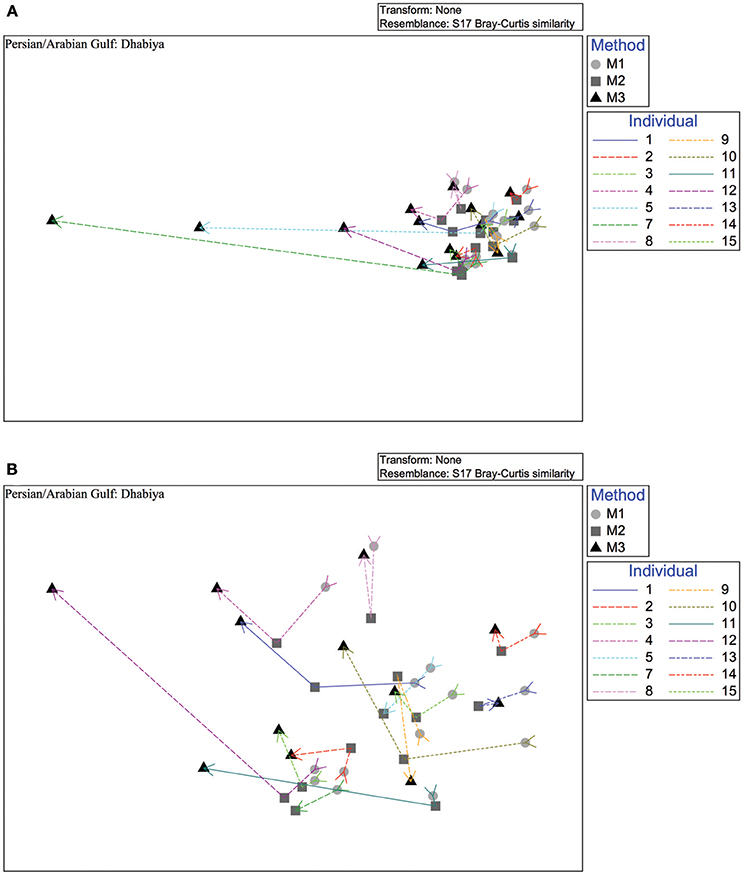
Figure 6. Non-metric multidimensional scaling plot based on Bray-Curtis similarities of the microbial method comparison data depicting Method 1 (circle), Method 2 (square), and Method 3 (triangle). Lines connect individual urchin samples. The upper plot (A) consists of all samples within the Persian/Arabian Gulf site, Dhabiya, while the lower plot (B) does not contain the two outliers (Sample ID: DH7, DH5) for a more in-depth view.
Literature Analysis
Our comparison in methodologies for characterizing E. mathaei's microbiome indicated that bead beating and lysozyme significantly influenced the community composition of microbiota. Therefore, we surveyed the literature to assess the commonality of these approaches in studies of marine invertebrate microbiomes and to better understand these results relative to the broader research community. Our literature search for manuscripts using our search terms (see Materials and Methods) at Web of Science resulted in 129 articles for corals, 167 for sponges, 14 for echinoderms, and 50 for molluscs, beginning in the early to mid-1990s (Supplemental Figure 1).
In general, publications increased over time and were generally highest in most recent years. The review of articles from 2015 to 2017 returned from our search resulted in substantially fewer that were relevant for a comparison of methodologies for sequence-based microbiome comparisons in these groups of marine invertebrates (15 of 67 for coral, 36 of 91 for sponge, 2 of 15 for mollusc*, 2 of 5 for echino*). From these 55 retained publications, comparisons of DNA extraction approach (53 total, two studies lacked sufficient details; Supplemental Table 2) resulted in 20 different commercial kits used in 44 studies and 9 studies used non-kit extractions [e.g., CTAB procedure as in (Fiore et al., 2013), and bead-beating method (Taylor et al., 2004)].
We then categorized the methods using commercial kits depending on if there was a column-based purification step, bead vortexing, or beating step, a lysozyme digest, or a combination. Of the 53 studies, 44 used a column-based step, 29 used bead vortexing or beating, and only one specifically listed a lysozyme step. Next, we assessed the sequencing techniques and genetic region for comparing microbial taxa (Supplemental Table 3). A majority of studies utilized high throughput sequencing (32 Illumina MiSeq or HiSeq, 16 Roche pyrosequencing, 1 Ion Torrent, 1 PacBio), with the remaining five using either Sanger-sequenced clone libraries or gel-based analysis (e.g., DGGE). While metagenomic methods are increasing in frequency, almost all studies (49 of 55) we identified in our search used 16S rRNA for comparisons of microbial communities. The specific variable region(s) of 16S rRNA that were used varied across studies. Lastly, only two studies (Marcelino and Verbruggen, 2016; Nakagawa et al., 2017) explicitly stated in their methods that negative control samples were used in their analysis.
Discussion
In the past, microbial community studies were restricted to culture-based approaches that isolated microorganisms as pure cultures and performed downstream analysis on these cultures. This is a limiting approach as most bacteria in natural environments cannot be cultured (Su et al., 2015). One of the major advances in modern sequencing technologies is the ability to use phylogenetic markers, such as the conserved 16S rRNA gene, to identify bacterial members of the microbiome that are non-culturable or rare (Lagkouvardos et al., 2016). However, using molecular methods that exclude certain bacteria, for example those with robust cell walls not lysed using traditional approaches, will underestimate microbial diversity and limit our interpretation of bacterial communities. It is vital that the microbial research community develops a set of best practices that most accurately represent the bacterial communities present (Miller et al., 1999; Yuan et al., 2012; Albertsen et al., 2015; Larsen et al., 2015; Burbach et al., 2016). Our goal in this study was to examine how DNA extraction approaches may influence the characterization of microbiome diversity through an empirical study of a sea urchin collected from different geographic areas and how these methods vary through literature-based comparisons.
Here we show that DNA extraction method plays an important role in estimates of Shannon diversity for bacterial communities associated with gut samples from the urchin E. mathaei in a site- and individual-specific manner. Within the GO, method did not result in considerable differences in Shannon diversity metrics, although there were specific families of bacteria that are found in significantly different proportions between methods. However, in the PAG, we observed the maximum Shannon diversity in Method 3, and this was significantly higher than diversity estimates from Method 1. These site-specific effects could be a result of methodology playing a more important role in identifying a more diverse community, as the PAG has higher Shannon indices than the GO, regardless of method. Further, we found the role methodology has on bacterial communities is specific to the individual. In other words, there are particular individuals that demonstrate larger community shifts when using Method 3. This variation could be a result of these individuals containing more difficult to lyse bacteria than others. Although Simpson diversity indices are generally higher using Method 3 for PAG samples, the difference between methods was not significant. This is not surprising, as Simpson diversity is heavily weighted by dominant OTUs (Hill et al., 2003), and it is the rare OTUs that are driving the differences between methods (Supplemental Figure 2). These results indicate that it may be challenging to compare microbial communities between studies that used different methods and highlights the importance of using the most robust method possible, especially when studying an uncharacterized system. As shown in our literature analysis, where DNA extraction techniques vary substantially, these biases could be influential in community characterization.
Of the bacterial taxa that were better captured with Method 3, there were five gram-positive bacteria in the PAG and one in the GO. These results were not surprising as gram-positive bacteria have a thick peptidoglycan layer in their cell wall that makes them more difficult to lyse than most gram-negative bacteria. Lysozymes are effective at hydrolyzing the glycosidic linkages in the peptidoglycan layer of the cell walls (Mehta et al., 2015). Gram-positive bacteria are common in many different environments and play crucial ecological roles. Actinobacteria, for example, are secondary metabolite producers and play an important role in organic matter turnover and the carbon cycle (Zhang et al., 2014a). There was also an increased abundance of two cyanobacteria and Pseudomonadaceae after lysozyme treatment in both sampling sites as well. Cyanobacteria have a resilient cell wall that is thicker and has a more highly crosslinked peptidoglycan layer than many gram-negative bacteria (Mehta et al., 2015). Although Pseudomonadaceae is gram-negative, Pseudomonas aeruginosa (the most well-described species within the Pseudomonadaceae family) is difficult to lyse without EDTA and lysozyme (Eagon and Carson, 1965). Further, we saw a general trend of increasing mean sequence reads from Method 1 to 3, with Method 3 consistently outperforming the other two methods (Tables 1, 2). It is also important to note that there were no OTUs that were represented significantly higher in Method 1 or 2 than Method 3. These results emphasize that although bead beating does help lyse hardy bacteria, it does not do so as effectively as when lysozyme and bead beating are combined. Microbial studies that are not using bead beating and lysozyme may be missing key microbial players.
The significantly different bacterial families that we present here typically comprised <0.2% of an individual's microbial community extracted using Method 3, although Xenococcaceae did contribute ~2.8% of the individual's community. While these families are not the most abundant taxa in our dataset, they may play important ecological or physiological roles. For example, several studies have found that rare species are disproportionately active compared to their abundant counterparts and therefore, should not be overlooked (Dimitriu et al., 2010; Debroas et al., 2015; Jousset et al., 2017). In one study, similar sized communities with a more diverse rare microbiome were shown to have higher respiration than communities with a smaller overall rare microbiome (Dimitriu et al., 2010). It is also important to note that although some of these taxa may be present in relatively low abundances in this dataset, other communities may be composed of larger abundances of difficult to lyse bacteria and therefore, method may play a more influential role. There is no way to assess the relative abundances of difficult to lyse bacteria a priori. Therefore, choosing the most rigorous methodology is recommended as a default choice to ensure accurate bacterial community representation, as well as allow for cross comparisons between studies.
Method-based biases can limit comparisons of microbial communities between studies that used different methods, thereby decreasing the potential to synthesize across groups of organisms. Many animal species or phylogenetic groups may have specific protocols for DNA extraction (e.g., inclusion of CTAB for molluscs and corals) to increase yield of nucleic acids for downstream analysis. Similarly, individual bacterial species have different optimal lysis conditions. Our survey of recently published studies on the microbial communities of marine invertebrates showed that while most studies used a commercial kit for DNA extraction, the methodological steps varied. Specifically, the inclusion of bead vortexing or beating was part of about 50% of studies and a lysozyme step was found in less than 5% of studies, which could result in reduced representation of particular groups of bacteria. The impact of these variations in the kit or the protocol remains sparsely reported in the literature for animal-associated bacteria of marine invertebrates (Weber et al., 2017). Moreover, many of these kits contain multiple protocols for DNA extraction depending on the type of tissue or organismal group. Future reporting by authors on which specific protocol was used would be beneficial for later utility in meta-analyses of the field.
In addition to rigorous methodology, microbial studies also need to account for bacterial contamination. Contamination can be introduced throughout the experimental process, from collection to sample processing. A significant amount of research has investigated the potential for contamination from DNA extraction kits and our data support the need to account for this contamination (Salter et al., 2014; Weiss et al., 2014; Glassing et al., 2016). With three blank samples (two of which were run through a DNA extraction column), we had roughly 14,000 total sequence reads, highlighting the prominence of contaminants. It is possible that these contaminants were also introduced at different steps of the extraction methodology (e.g., pipette tips or sample tubes). However, the blanks that were run through the extraction column had >60% more sequence reads and more OTUs (400 compared to 138) than the blank that was only used during the PCR step. Although removal of all the OTUs that are present in blank and biological samples may remove some biological signal (if contamination is a result of the biological sample contaminating the blanks), it is important to consider the ramifications of not accounting for the presence of contaminants. Indeed, in our review of the literature, we only identified two studies that definitely used a blank in their study. It is also important to note that each extraction kit may contain a different set of contaminants (the “kitome”), making it crucial to run and account for blanks in parallel with the biological samples (Salter et al., 2014; Weiss et al., 2014; Glassing et al., 2016). Our review of publications over the last three years showed that 20 different commercial kits were used to extract DNA. This potential for bias from these kits and variation between lots of the same kit will likely add a number of rare OTUs to the characterization of the microbial community. These bacteria would not only increase the derived diversity of the microbiome but could also result in shared, rare microbes between studies of species that happen to use the same kits. Thus, although kit-based DNA extractions may increase repeatability of methods, studies that do not account for the potential of contaminating DNA from particular bacteria will include bacteria not specific to the biological system under study.
In summary, we show that DNA extraction methods that incorporate a bead beating and lysozyme step more accurately characterize this bacterial community. While we observe significant differences in community composition between DNA extraction methods, this is only one step in a multi-step procedure. Sample collection, primer pair selection, PCR conditions, and sequencing approach each have their own propensity for introducing bias that also merit further study to determine to what extent each step effects the inferred microbial diversity. Further, we describe the current state of marine microbial methodology in invertebrates and highlight the importance of developing a standardized DNA extraction protocol for marine microbial community analysis. Finally, it is important that studies select rigorous extraction methods to most accurately sample the community and increase consistency between studies.
Ethics Statement
The samples collected were invertebrates and therefore did not require ethical committee approval. Collections were permitted by the Environmental Agency in Abu Dhabi.
Author Contributions
RK designed the study, collected samples, and performed all lab work. ES, BP, and AR aided in designing the study. JB provided funding and staff to support field collections and assisted with data analysis and writing. ES, GV, DM, and NA-M aided in sample collections. All authors contributed ideas throughout the process, read, edited, and approved the final manuscript.
Funding
RK was supported by a NSF Graduate Research Fellowship (GRF) and a Lerner-Gray Marine Research Grant. TC was also supported by a NSF GRF. AR was supported by Human Frontier Science Program Award RGY0079/2016.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2018.00255/full#supplementary-material
Supplemental Figure 1. Bar chart showing growth in number of publications from 1992 to 2017 in the four invertebrates; coral (in blue), sponge (in orange), mollusc*(in gray), and echino*(in yellow).
Supplemental Figure 2. Simpson Diversity indices of Gulf of Oman samples (right) and Persian/Arabian Gulf samples (left). Each node corresponds to an individual sample and each line connects one individual's tissue subjected to the three DNA extraction methods.
Supplemental Table 1. Complete list of all OTUs deleted from our analysis due to their presence in blank sequences. Table contains taxonomic assignment and mean number of sequence reads in each method and blank sample.
Supplemental Table 2. Meta-data for marine microbial publication data synthesis including; taxonomic group, kit used, extraction method, and publication information.
Supplemental Table 3. Meta-data for marine microbial publication data synthesis including; taxonomic group, sequencing method, and region sequenced.
Acknowledgments
We thank Karen Lopez (UNC Charlotte) for her technical assistance with library preparation and sequencing for the sea urchin samples. We also thank New York University Abu Dhabi for supportive funding and the Environmental Agency – Abu Dhabi for permits.
References
Albertsen, M., Karst, S. M., Ziegler, A. S., Kirkegaard, R. H., and Nielsen, P. H. (2015). Back to Basics – the influence of DNA extraction and primer choice on phylogenetic analysis of activated sludge communities. PLoS ONE 10:e0132783. doi: 10.1371/journal.pone.0132783
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bordenstein, S. R., and Theis, K. R. (2015). Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PLoS Biol. 13:e1002226. doi: 10.1371/journal.pbio.1002226
Bourne, D. G., Morrow, K. M., and Webster, N. S. (2016). Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Annu. Rev. Microbiol. 70, 317–340. doi: 10.1146/annurev-micro-102215-095440
Burbach, K., Seifert, J., Pieper, D. H., and Camarinha-Silva, A. (2016). Evaluation of DNA extraction kits and phylogenetic diversity of the porcine gastrointestinal tract based on Illumina sequencing of two hypervariable regions. Microbiologyopen 5, 70–82. doi: 10.1002/mbo3.312
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335. doi: 10.1038/nmeth.f.303
Carrier, T. J., and Reitzel, A. M. (2017). The hologenome across environments and the implications of a host-associated microbial repertoire. Front. Microbiol. 8:802. doi: 10.3389/fmicb.2017.00802
Carrier, T. J., and Reitzel, A. M. (2018). Convergent shifts in host-associated microbial communities across environmentally elicited phenotypes. Nat. Commun. 9:952. doi: 10.1038/s41467-018-03383-w
Clarke, K. R. (1993). Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18, 117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x
Debroas, D., Hugoni, M., and Domaizon, I. (2015). Evidence for an active rare biosphere within freshwater protists community. Mol. Ecol. 24, 1236–1247. doi: 10.1111/mec.13116
De Bruin, O. M., and Birnboim, H. C. (2016). A method for assessing efficiency of bacterial cell disruption and DNA release. BMC Microbiol. 16:197. doi: 10.1186/s12866-016-0815-3
Dimitriu, P. A., Lee, D., and Grayston, S. J. (2010). An evaluation of the functional significance of peat microorganisms using a reciprocal transplant approach. Soil Biol. Biochem. 42, 65–71. doi: 10.1016/j.soilbio.2009.10.001
Eagon, R. G., and Carson, K. J. (1965). Lysis of cell walls and intact cells of Pseudomonas aeruginosa by ethylenediamine tetraacetic acid and by lysozyme. Can. J. Microbiol. 11, 193–201. doi: 10.1139/m65-025
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fiore, C. L., Jarett, J. K., and Lesser, M. P. (2013). Symbiotic prokaryotic communities from different populations of the giant barrel sponge, Xestospongia muta. Microbiologyopen 2, 938–952. doi: 10.1002/mbo3.135
Galac, M. R., Bosch, I., and Janies, D. A. (2016). Bacterial communities of oceanic sea star (Asteroidea: Echinodermata) larvae. Mar. Biol. 163:162. doi: 10.1007/s00227-016-2938-3
Gao, F., Li, F., Tan, J., Yan, J., and Sun, H. (2014). Bacterial community composition in the gut content and ambient sediment of sea cucumber Apostichopus japonicus revealed by 16S rRNA gene pyrosequencing. PLoS ONE 9:e100092. doi: 10.1371/journal.pone.0100092
Glassing, A., Dowd, S. E., Galandiuk, S., Davis, B., and Chiodini, R. J. (2016). Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 8:24. doi: 10.1186/s13099-016-0103-7
Hakim, J. A., Koo, H., Dennis, L. N., Kumar, R., Ptacek, T., Morrow, C. D., et al. (2015). An abundance of Epsilonproteobacteria revealed in the gut microbiome of the laboratory cultured sea urchin, Lytechinus variegatus. Front. Microbiol. 6:1047. doi: 10.3389/fmicb.2015.01047
Hakim, J. A., Koo, H., Kumar, R., Lefkowitz, E. J., Morrow, C. D., Powell, M. L., et al. (2016). The gut microbiome of the sea urchin, Lytechinus variegatus, from its natural habitat demonstrates selective attributes of microbial taxa and predictive metabolic profiles. FEMS Microbiol. Ecol. 92:fiw146–fiw146. doi: 10.1093/femsec/fiw146
Hill, T. C., Walsh, K. A., Harris, J. A., and Moffett, B. F. (2003). Using ecological diversity measures with bacterial communities. FEMS Microbiol. Ecol. 43, 1–11. doi: 10.1111/j.1574-6941.2003.tb01040.x
Holland, N. D., and Nealson, K. H. (1978). The fine structure of the echinoderm cuticle and the subcuticular bacteria of echinoderms. Acta Zool. 59, 169–185. doi: 10.1111/j.1463-6395.1978.tb01032.x
Jousset, A., Bienhold, C., Chatzinotas, A., Gallien, L., Gobet, A., Kurm, V., et al. (2017). Where less may be more: how the rare biosphere pulls ecosystems strings. ISME J. 11:853. doi: 10.1038/ismej.2016.174
Kelly, M. S., Barker, M. F., Mckenzie, J. D., and Powell, J. (1995). The incidence and morphology of subcuticular bacteria in the echinoderm fauna of New Zealand. Biol. Bull. 189, 91–105. doi: 10.2307/1542459
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Kohl, K. D., and Carey, H. V. (2016). A place for host–microbe symbiosis in the comparative physiologist's toolbox. J. Exp. Biol. 219, 3496–3504. doi: 10.1242/jeb.136325
Lagkouvardos, I., Joseph, D., Kapfhammer, M., Giritli, S., Horn, M., Haller, D., et al. (2016). IMNGS: A comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci. Rep. 6:33721. doi: 10.1038/srep33721
Larsen, A. M., Mohammed, H. H., and Arias, C. R. (2015). Comparison of DNA extraction protocols for the analysis of gut microbiota in fishes. FEMS Microbiol. Lett. 362:fnu031. doi: 10.1093/femsle/fnu031
Lesser, M. P., and Walker, C. W. (1992). Comparative study of the uptake of dissolved amino acid in sympatric brittle stars with and without endosymbiotic bacteria. Comp. Biochem. Physiol. Part B 101, 217–223. doi: 10.1016/0305-0491(92)90182-Q
Marcelino, V. R., and Verbruggen, H. (2016). Multi-marker metabarcoding of coral skeletons reveals a rich microbiome and diverse evolutionary origins of endolithic algae. Sci. Rep. 6:31508. doi: 10.1038/srep31508
Mcfall-Ngai, M., Hadfield, M. G., Bosch, T. C., Carey, H. V., Domazet-Lošo, T., Douglas, A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Nat. Acad. Sci. U.S.A. 110, 3229–3236. doi: 10.1073/pnas.1218525110
Mcorist, A. L., Jackson, M., and Bird, A. R. (2002). A comparison of five methods for extraction of bacterial DNA from human faecal samples. J. Microbiol. Methods 50, 131–139. doi: 10.1016/S0167-7012(02)00018-0
Mehta, K. K., Evitt, N. H., and Swartz, J. R. (2015). Chemical lysis of cyanobacteria. J. Biol. Eng. 9:10. doi: 10.1186/s13036-015-0007-y
Meziti, A., Kormas, K. A., Pancucci-Papadopoulou, M. A., and Thessalou-Legaki, M. (2007). Bacterial phylotypes associated with the digestive tract of the sea urchin Paracentrotus lividus and the Ascidian microcosmus sp. Russ. J. Mar. Biol. 33, 84–91. doi: 10.1134/S1063074007020022
Miller, D. N., Bryant, J. E., Madsen, E. L., and Ghiorse, W. C. (1999). Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl. Environ. Microbiol. 65, 4715–4724.
Moitinho-Silva, L., Nielsen, S., Amir, A., Gonzalez, A., Ackermann, G. L., Cerrano, C., et al. (2017). The sponge microbiome project. Gigascience 6, 1–7. doi: 10.1093/gigascience/gix077
Nakagawa, S., Saito, H., Tame, A., Hirai, M., Yamaguchi, H., Sunata, T., et al. (2017). Microbiota in the coelomic fluid of two common coastal starfish species and characterization of an abundant Helicobacter-related taxon. Sci. Rep. 7:8764. doi: 10.1038/s41598-017-09355-2
Nelson, L., Blair, B., Murdock, C., Meade, M., Watts, S., and Lawrence, A. L. (2010). Molecular analysis of gut microflora in captive-raised sea urchins (Lytechinus variegatus). J. World Aquac. Soc. 41, 807–815. doi: 10.1111/j.1749-7345.2010.00423.x
Oliveros, J. C. (2007). VENNY: An Interactive Tool for Comparing Lists With Venn Diagrams. Available online at: http://bioinfogp.cnb.csic.es/tools/venny/index.html
Pollock, J., Glendinning, L., Wisedchanwet, T., and Watson, M. (2018). The madness of microbiome: attempting to find consensus “best practice” for 16S microbiome studies. Appl. Environ. Microbiol. 84:e02627–17. doi: 10.1128/AEM.02627-17
Reveillaud, J., Maignien, L., Eren, A. M., Huber, J. A., Apprill, A., Sogin, M. L., et al. (2014). Host-specificity among abundant and rare taxa in the sponge microbiome. ISME J. 8, 1198–1209. doi: 10.1038/ismej.2013.227
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12:87. doi: 10.1186/s12915-014-0087-z
Small, C. M., Milligan-Myhre, K., Bassham, S., Guillemin, K., and Cresko, W. A. (2017). Host genotype and microbiota contribute asymmetrically to transcriptional variation in the threespine stickleback gut. Genome Biol. Evol. 9, 504–520. doi: 10.1093/gbe/evx014
Smith, E. G., Hume, B. C. C., Delaney, P., Wiedenmann, J., and Burt, J. A. (2017). Genetic structure of coral-Symbiodinium symbioses on the world's warmest reefs. PLoS ONE 12:e0180169. doi: 10.1371/journal.pone.0180169
Su, X., Sun, F., Wang, Y., Hashmi, M. Z., Guo, L., Ding, L., et al. (2015). Identification, characterization and molecular analysis of the viable but nonculturable Rhodococcus biphenylivorans. Sci. Rep. 5:18590. doi: 10.1038/srep18590
Taylor, M. W., Schupp, P. J., Dahllöf, I., Kjelleberg, S., and Steinberg, P. D. (2004). Host specificity in marine sponge-associated bacteria, and potential implications for marine microbial diversity. Environ. Microbiol. 6, 121–130. doi: 10.1046/j.1462-2920.2003.00545.x
Theis, K. R., Dheilly, N. M., Klassen, J. L., Brucker, R. M., Baines, J. F., Bosch, T. C. G., et al. (2016). Getting the hologenome concept right: An eco-evolutionary framework for hosts and their microbiomes. mSystems 1:e00028–16. doi: 10.1128/mSystems.00028-16
Thomas, T., Moitinho-Silva, L., Lurgi, M., Björk, J. R., Easson, C., Astudillo-García, C., et al. (2016). Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 7:11870. doi: 10.1038/ncomms11870
Uthicke, S., Soars, N., Foo, S., and Byrne, M. (2013). Effects of elevated pCO2 and the effect of parent acclimation on development in the tropical Pacific sea urchin Echinometra mathaei. Mar. Biol. 160, 1913–1926. doi: 10.1007/s00227-012-2023-5
Vanysacker, L., Declerck, S. A., Hellemans, B., De Meester, L., Vankelecom, I., and Declerck, P. (2010). Bacterial community analysis of activated sludge: an evaluation of four commonly used DNA extraction methods. Appl. Microbiol. Biotechnol. 88, 299–307. doi: 10.1007/s00253-010-2770-5
Voss, J. D., Mills, D. K., Myers, J. L., Remily, E. R., and Richardson, L. L. (2007). Black band disease microbial community variation on corals in three regions of the wider Caribbean. Microb. Ecol. 54, 730–739. doi: 10.1007/s00248-007-9234-1
Walker, C. W., and Lesser, M. P. (1989). Nutrition and development of brooded embryos in the brittlestar Amphipholis squamata: do endosymbiotic bacteria play a role? Mar. Biol. 103, 519–530. doi: 10.1007/BF00399584
Weber, L., Deforce, E., and Apprill, A. (2017). Optimization of DNA extraction for advancing coral microbiota investigations. Microbiome 5:18. doi: 10.1186/s40168-017-0229-y
Webster, N. S., Negri, A. P., Bott,é, E. S., Laffy, P. W., Flores, F., Noonan, S., et al. (2016). Host-associated coral reef microbes respond to the cumulative pressures of ocean warming and ocean acidification. Sci. Rep. 6:19324. doi: 10.1038/srep19324
Webster, N. S., and Reusch, T. B. H. (2017). Microbial contributions to the persistence of coral reefs. ISME J. 11:2167. doi: 10.1038/ismej.2017.66
Weiss, S., Amir, A., Hyde, E. R., Metcalf, J. L., Song, S. J., and Knight, R. (2014). Tracking down the sources of experimental contamination in microbiome studies. Genome Biol. 15:564. doi: 10.1186/s13059-014-0564-2
Yuan, S., Cohen, D. B., Ravel, J., Abdo, Z., and Forney, L. J. (2012). Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLoS ONE 7:e33865. doi: 10.1371/journal.pone.0033865
Zhang, H., Ding, W., He, X., Yu, H., Fan, J., and Liu, D. (2014a). Influence of 20–Year Organic and inorganic fertilization on organic carbon accumulation and microbial community structure of aggregates in an intensively cultivated sandy loam soil. PLoS ONE 9:e92733. doi: 10.1371/journal.pone.0092733
Zhang, J., Kobert, K., Flouri, T., and Stamatakis, A. (2014b). PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620. doi: 10.1093/bioinformatics/btt593
Keywords: sea urchin, microbiome, Arabian Peninsula, 16S rRNA, lysozyme, bead-beating
Citation: Ketchum RN, Smith EG, Vaughan GO, Phippen BL, McParland D, Al-Mansoori N, Carrier TJ, Burt JA and Reitzel AM (2018) DNA Extraction Method Plays a Significant Role When Defining Bacterial Community Composition in the Marine Invertebrate Echinometra mathaei. Front. Mar. Sci. 5:255. doi: 10.3389/fmars.2018.00255
Received: 28 March 2018; Accepted: 06 July 2018;
Published: 31 July 2018.
Edited by:
Nicola Segata, University of Trento, ItalyReviewed by:
Punyasloke Bhadury, Indian Institute of Science Education and Research Kolkata, IndiaKonstantinos Ar. Kormas, University of Thessaly, Greece
Copyright © 2018 Ketchum, Smith, Vaughan, Phippen, McParland, Al-Mansoori, Carrier, Burt and Reitzel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Remi N. Ketchum, cmtldGNodTFAdW5jYy5lZHU=