 Oded Shamriz
Oded Shamriz Yuval Tal1†
Yuval Tal1†- 1Allergy and Clinical Immunology Unit, Hadassah-Hebrew University Medical Center, Jerusalem, Israel
- 2The Lautenberg Center for Immunology and Cancer Research, Institute of Medical Research Israel-Canada, Hebrew University-Hadassah Medical School, Jerusalem, Israel
- 3Pediatrics Department A, Soroka University Medical Center and Faculty of Health Sciences, Ben-Gurion University of the Negev, Beer Sheva, Israel
Children with chronic mucocutaneous candidiasis (CMC) experience recurrent infections with Candida spp. Moreover, immune dysregulation in the early life of these patients induces various autoimmune diseases and affects normal growth and development. The adaptive and innate immune system components play a significant role in anti-fungal response. This response is mediated through IL-17 production by T helper cells. Inborn errors in IL-17-mediated pathways or Candida spp. sensing molecules are known to cause CMC. In this review, we describe underlying immune mechanisms of monogenic primary immune deficiency disorders known to cause CMC. We will explore insights into current management of these patients and novel available therapies.
Introduction
Children with chronic mucocutaneous candidiasis (CMC) experience recurrent infections with Candida spp. Infections can be mucosal or invasive, and isolated or associated with other infections. CMC can involve the vagina, esophagus, skin, and other organs. Moreover, severe immune dysregulation in the early life of these patients induces various autoimmune diseases and affects normal growth and development. Medical care is complex and usually warrants a combination of systemic anti-fungal and immunosuppressive agents (1–3).
Advances in genetic tests in the recent decade have expanded our knowledge of underlying immune mechanisms in CMC, elucidating an increasing number of newly defined primary immune-deficiency disorders (4). An in-depth characterization of the impaired immune pathways associated with CMC is critical in order to offer treatment tailored to the individual patient.
In this review, we describe monogenic primary immune-deficiency disorders known to cause CMC. Based on insights into underlying immune mechanisms, we explore different targeted therapies currently available or under development for these patients.
Immune Mechanisms Underlying Monogenic Chronic Mucocutaneous Candidiasis
The discovery of monogenic causes for CMC has enabled us to expand our knowledge of fundamental immune mechanisms (Figure 1).
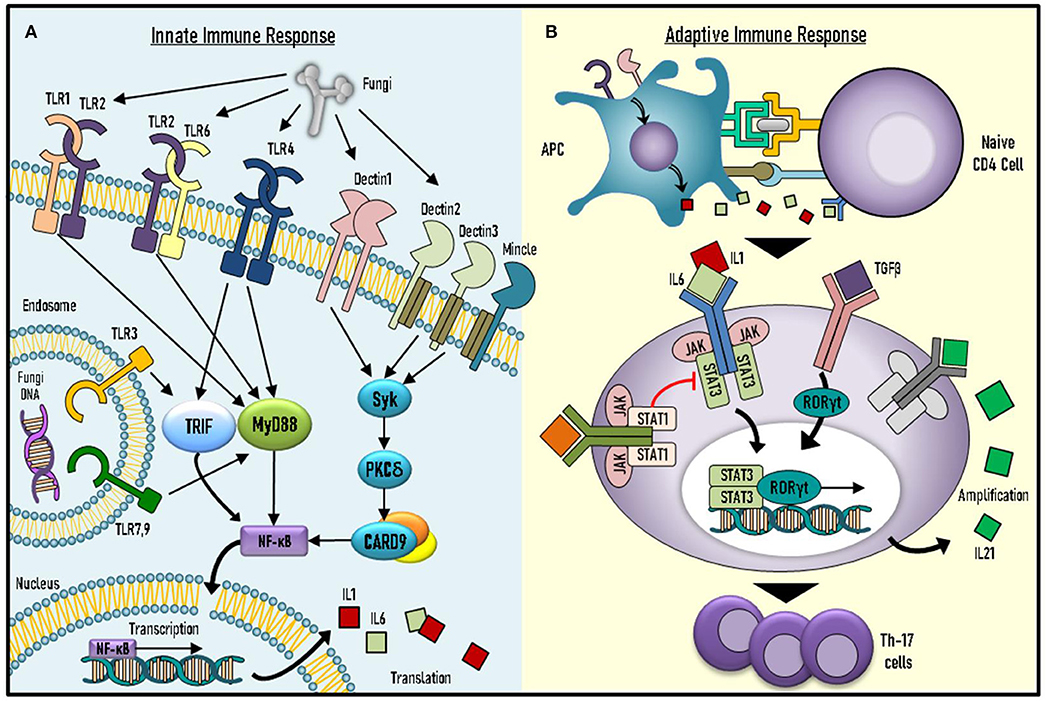
Figure 1. Underlying mechanisms of immune responses against Candida spp. (A) Candida spp. recognition and initial immune response involve key molecules of the innate system. (B) Adaptive response against Candida spp. includes activation and differentiation of naïve CD4+ T cells into effector T helper 17 (Th17) cells. TLR, toll-like receptor; PKC-δ, Protein kinase C-δ; MYD-88, Myeloid differentiation primary response 88; NFκB, Nuclear factor kappa-light-chain-enhancer of activated B cells; IL-Interleukin; APC, Antigen presenting cell; TGF-β, Transforming growth factor beta (TGF-β); JAK-Janus Kinase; STAT, Signal transducer and activator of transcription; RORγT, RAR-related orphan receptor gamma.
Immunity against Candida spp. consists of innate and adaptive responses. The innate response involves recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs) found in different cells of the innate immune system, such as monocytes and natural killer (NK) cells (5). Various PRRs are known to induce pro- and anti-inflammatory cytokine production in response to PAMP ligand binding. These PRRs include toll-like receptors (TLRs) 2, 3, 4, 6, and 9, as well as other receptors, such as dectin 1–3 (5). PAMP ligand binding to Dectin-1 leads to signal transduction via adaptor-molecule caspase activation and recruitment domain-containing 9 (CARD9) (5).
The adaptive immune system components also play a significant role in anti-fungal response. This includes pathways mediated by interleukin (IL)-17 and IL-22, which are produced by Th17 cells (6). Indeed, defective fungal sensing by the innate system, as well as abnormalities in IL-17-mediated pathways can induce CMC (Table 1). Impairments in the adaptive response can be further subdivided into decreased IL-17 cytokine production, impaired IL-17-mediated intracellular signaling or increased peripheral neutralization by anti-IL-17 autoantibodies.
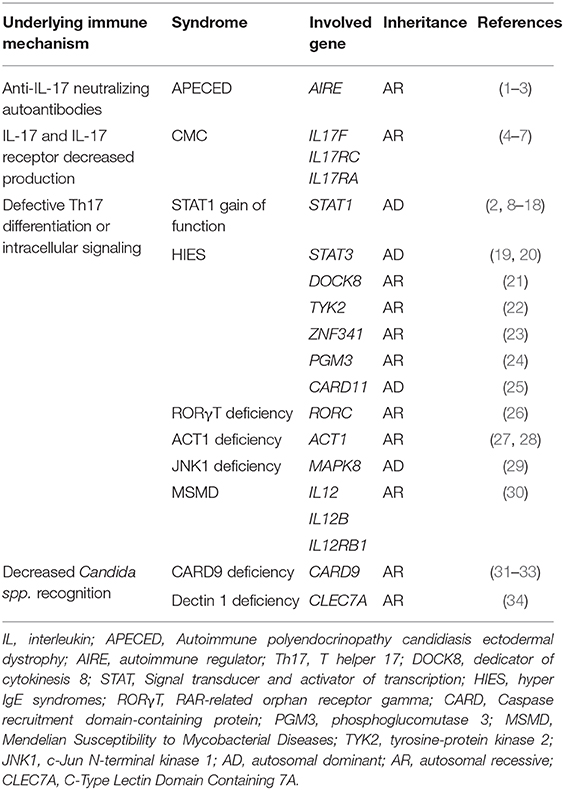
Table 1. Reported genes associated with chronic mucocutaneous candidiasis.
Production of Neutralizing Autoantibodies Against IL-17 and IL-22
T cell development in the thymus includes clonal deletion of self-reactive T cells. This is achieved by the introduction of self-antigens to naive T cells by medullary thymic epithelial (mTEC) and dendritic cells. mTECs express autoimmune regulator (AIRE), an important facilitator of self-antigen gene expression (7).
AIRE deficiency is characterized by loss of self-tolerance and the presence of autoreactive T cells and multiple severe autoimmune diseases. AIRE loss-of-function (LOF) induces autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). APECED is characterized by a classical triad of CMC, hypoparathyroidism, and Addison's disease (8), but other systems can be affected by autoimmunity in APECED, which can induce type 1 diabetes, hypothyroidism, hypogonadism, vitiligo, and various other autoimmune diseases (8). CMC in APECED patients is explained by decreased IL-17 and IL-22 cytokine serum levels, with corresponding increased titers of anti-IL-17 and anti-IL-22 neutralizing autoantibodies (9, 10). Thus, anti-IL-17/22 autoantibody production in APECED demonstrates the important association between immune dysregulation and CMC susceptibility.
Inborn Errors in IL-17 Production or IL-17 Receptor Surface Expression
IL-17R-mediated signaling has been shown in murine models to be essential in the immune response against Candida spp. (11, 12). In 2011, a single patient was reported to have a homozygous c.850C>T mutation in IL-17RA that caused reduced surface expression of IL-17RA on peripheral blood mononuclear cells (PBMC), reduced lymphocyte response to IL-17A/F stimuli, and increased susceptibility to Candida spp. infections (13). Two other patients with CMC had impaired IL-17F cytokine production due to IL-17F gene mutation (13, 14). Since then, several cohorts of CMC patients with IL-17R deficiencies have been reported, including 21 patients from 12 unrelated families with IL-17RA deficiency (15) and three patients with IL-17RC deficiency (16).
Defective Th17 Differentiation or Intracellular Signaling
Antigen-presenting cells produce IL-6, IL1-β, and IL-23, as well as activate transforming growth factor (TGF)-β in response to fungal infections. These cytokines bind to naïve CD4+ T cells and trigger STAT3 followed by RAR-related orphan receptor (ROR)γT-mediated transcription. RORγT enhances production of IL-17A, IL-17F, and IL-21 by lymphocytes, through which they differentiate into Th17 cells. In turn, IL-21 further self-amplifies Th17-mediated immune responses (17, 18).
STAT1 Gain of Function
Inborn errors in any of the key players in Th17 differentiation can result in CMC. STAT1 is such a key component as was demonstrated from the study of autosomal dominant (AD) gain-of-function (GOF) mutations. STAT1 mutations are probably the most common cause of monogenic CMC. These patients present with a wide clinical spectrum of immune dysregulation and increased susceptibility to bacterial, viral and fungal infections (19). Delayed dephosphorylation of STAT1 in these patients impairs the function of IL-6 and IL-21, thus decreasing STAT3-dependent differentiation of naïve CD4+ T cells into Th17 cells (20). Of note, a recent report suggests that some STAT1 GOF mutations may cause STAT1 levels to be high, although phosphorylation is normal (21). Disease severity appears to vary according to the mutation. For example, patients with the T385M mutation are somewhat phenotypically different from others. The T385M clinical spectrum consists of chronic candidiasis, recurrent severe invasive infections with bacterial pathogens, severe viral infections such as cytomegalovirus and John Cunningham virus and, last but not least, severe autoimmune phenomena reminiscent of a combined immunodeficiency disease. These patients show progressive loss of T and B cell function (22).
Hyper IgE Syndromes
Another striking example of impaired Th17 differentiation is STAT3 LOF mutations known to cause autosomal dominant hyper immunoglobulin E syndrome (AD-HIES). These patients have severe eczema, skin abscesses, staphylococcal infections, and decreased or absent Th17 cells, resulting in increased susceptibility to Candida infections (23, 24). Markedly increased IgE levels and eosinophilia are indicative of immune dysregulation in these patients (25). STAT3 LOF patients are distinctive by their non-immunologic features, which include dysmorphic facial features, retained primary teeth, vascular aneurysms, scoliosis, osteoporosis, and other musculoskeletal manifestations (26).
Autosomal recessive (AR) HIES is caused by mutations in dedicator of cytokinesis (DOCK) 8, ZNF341, and tyrosine kinase (TYK)2. DOCK8 plays an important role in T cell activation and proliferation via its role in T cell cytoskeleton and actin reorganization. DOCK8 mutation results in abnormal Th17 polarization and function (27). Clinical manifestations include an immune dysregulation phenotype consisting of allergic disorders, such as atopic dermatitis and food allergies, as well as increased susceptibility to staphylococcal, sino-pulmonary and viral infections (26).
Other gene mutations causing AR-HIES have been reported in ZNF341. This factor regulates the transcription of STAT3, therefore patients with ZNF341 deficiencies are clinically similar to HIES with STAT3 LOF. They are reported to have low levels of STAT3, reduced numbers of Th17 cells, and high risk for CMC (28). TYK2, a JAK family member, is critical for normal IL-12 and type I IFN expression. Mutation of TYK2 can also cause AR-HIES. A patient with a homozygous TYK2 mutation was reported to have increased susceptibility to viral infections due to an impaired IFN-mediated response, and increased risk for fungal infections most probably due to defective IL-12/IL-23-mediated responses (29).
In addition, we should mention phosphoglucomutase (PGM)3 and CARD11 deficiencies, both reported in some studies to induce CMC and HIES. PGM3 deficiency is an AR-HIES disorder characterized by glycosylation defects that have multi-systemic manifestations including a neurodegenerative course. Sassi et al. reported occurrence of CMC in four out of nine patients (30), whereas Zhang et al. and Stray-Pederson et al. did not describe such findings (31, 32). LOF mutations in CARD11 were associated with severe atopy and immune dysregulation (33). In both disorders, it appears that Th17 cells are present, rather than absent. Therefore, the defect is probably functional and in the context of global T cell defects.
IL-12/IL-12 Receptor Pathway
Inborn errors in IL-12-mediated pathways are known to play a major cause for Mendelian susceptibility to mycobacterial disease (MSMD), increasing the risk for mycobacterial and viral infections. Interestingly, impaired defective IL-12 or IL-12R may underlie abnormal IL-23-mediated signaling, thus also exposing these patients to risk of developing CMC (13). Defective IL-23- and IL-12-mediated pathways were previously reported in patients with IL-23R and IL-12Rβ2 deficiencies, respectively. Impaired signaling in these patients induced MSMD; however no CMC was observed (34).
RORC, ACT1, and MAPK8 Mutations
STAT3 induces RORγT transcription, which leads to Th17 differentiation. AR mutations in RORγT have been demonstrated to decrease Th17 cell counts and result in CMC. Interestingly, these patients also presented with increased susceptibility to mycobacterial infections due to impaired interferon (IFN)-γ-mediated immunity, which also requires RORγT (35).
Regarding the IL-17-mediated pathway, one should also remember other proteins downstream. ACT1 is an intracellular adaptor protein in the IL-17-mediated signaling pathway. Several human mutations in ACT1 are known to impair Th17 function and induce CMC (36, 37). Staphylococcus aureus blepharitis (37) and recurrent pneumonia (36) were also noted in these patients, who display characteristics of primary immune deficiency with dysregulation.
Finally, we should also mention mutations in MAPK8. AD MAPK8 mutations resulting in c-Jun N-terminal kinase 1 (JNK1) deficiencies were previously reported to induce CMC. Impaired Th17 differentiation and decreased responses to IL-17A and IL-17F stimuli were shown. Interestingly, JNK1-deficient patients with CMC were also found to have a novel connective tissue disease, thus distinguishing mutant MAPK8 from other monogenic inducers (38).
Decreased Recognition of Candida Infections
The innate response against Candida spp. is complex. Recognition of fungal PAMPs by PRR is critical for Candida spp. sensing, as is the Dectin-1–Syk–CARD9 signaling pathway. Biallelic mutations in CARD9 are reported to induce CMC and general increased susceptibility to fungal infections (39–45). In comparison with IL-17-associated inborn errors, CARD9 deficiency is thought to induce a more severe and invasive candidiasis, affecting various tissues including even the central nervous system (CNS) (46).
Dectin-1 deficiency has also been shown to induce reduced recognition of β-glucans with increased susceptibility to Candida spp. infections. However, an important feature of this disorder is the lack of susceptibility to other infections, which defines it as an isolated CMC (47). Impairment of the Dectin-1–Syk–CARD9 pathway also affects the differentiation of CD4+ naïve T cells into Th17 cells, thereby interfering with the adaptive immune response to Candida spp. (6). Indeed, Tyr238X mutation in dectin-1 was previously described to cause CMC and onychomycosis phenotypes, as well as decreased IL-17 levels. However, phagocytosis and killing of Candida spp. in these patients were intact (47). Although dectin-1 deficiency is not included in International Union of Immunological Societies (IUIS) 2019 classification (4), the Tyr238X mutation can be found in gnomAD1.
Current Management of Monogenic Chronic Mucocutaneous Candidiasis
Current management of CMC consists mainly of prophylactic anti-fungal agents, such as fluconazole (1). However, other therapeutic modalities are currently available. Granulocyte-macrophage colony-stimulating factor (GM-CSF) production by PBMC is suggested to be reduced in CARD9-deficient patients. A patient with a hypomorphic CARD9 mutation presenting with CNS candidiasis was found to achieve clinical remission after GM-CSF administration (46), and GM-CSF has been found to be effective in other patients with CARD9 deficiency (48).
Histone deacetylase (HDAC) inhibitors were also examined in the management of CMC, especially in patients with STAT1 GOF mutations. Inhibition of histone acetylation is thought to affect the adaptive and innate immune systems. Indeed, HDAC inhibitors were found to rescue STAT3-mediated pathways in STAT1 GOF patients (49). Moreover, in-vitro treatment with HDAC inhibitors resulted in increased IL-22 production in response to Candida spp. (49).
Hematopoietic stem cell transplantations (HSCT) have some efficacy in CMC. For example, in two patients with CARD9 deficiency, HSCT from haploidentical and fully matched donors was successful, although a second HSCT was required in the first patient. Complete clinical resolution of fungal infections was noted in both patients (45). There are reports of successful HSCT in STAT1 GOF patients as well, with complete resolution of immune dysregulation and rescue of Th17 differentiation and function (50). However, the results of HSCT in STAT1 GOF are generally not favorable, with high rates of secondary graft failure (51).
Targeted immunotherapies for CMC-inducing inborn errors are therefore warranted. Ruxolitinib, a Janus kinase (JAK)1/2 inhibitor, is reportedly effective in STAT1 GOF. Ruxolitinib treatment of a STAT1 GOF child presenting with a clinical picture of CMC and autoimmune cytopenia was shown to directly intervene with the impaired immune pathways. It improved Th17 differentiation, decreased Th1-mediated responses, and attenuated CMC and immune dysregulation (52). Another study found that ruxolitinib in STAT1 GOF patients can rescue NK cell maturation. Moreover, it was effective in restoring perforin expression on NK cells, thus rescuing NK cytotoxic function (53). Other reports of children with STAT1 GOF mutations have confirmed the efficacy and safety of ruxolitinib in this disorder (54–56).
Conclusions
Current advances in next-generation sequencing have revealed various monogenic inducers of CMC. Understanding the impaired immune pathways involved in CMC is critical in the management of these patients. CMC is strongly associated with immune dysregulation and autoimmunity in early childhood. Therefore, a joint collaboration between immunologists, endocrinologists, and infectious disease and other specialists is needed in order to offer a personally tailored, effective, treatment to these patients.
Author Contributions
OS study design, review of the literature, and manuscript writing. YT and AN study supervision and manuscript revisions. AT immune consultation and manuscript revision. All authors agree to be accountable for the content of the work.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
1. Pappas PG, Kauffman CA, Andes DR, Clancy CJ, Marr KA, Ostrosky-Zeichner L, et al. Clinical practice guideline for the management of candidiasis: 2016 update by the infectious diseases society of america. Clin Infect Dis. (2016) 62:e1–50. doi: 10.1093/cid/civ1194
2. Carey B, Lambourne J, Porter S, Hodgson T. Chronic mucocutaneous candidiasis due to gain-of-function mutation in STAT1. Oral Dis. (2019) 25:684–92. doi: 10.1111/odi.12881
3. Okada S. CMCD: chronic mucocutaneous candidiasis disease. Nihon Rinsho Meneki Gakkai Kaishi. (2017) 40:109–17. doi: 10.2177/jsci.40.109
4. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the iuis phenotypical classification. J Clin Immunol. (2020) 40:66–81. doi: 10.1007/s10875-020-00758-x
5. Netea MG, Leo Joosten JW. van der Meer JWM, Kullberg BJ, van de Veerdonk LF. Immune defence against Candida fungal infections. Nat Rev Immunol. (2015) 15:630–42. doi: 10.1038/nri3897
6. Okada S, Puel A, Casanova JL, Kobayashi M. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin Transl Immunol. (2016) 5:e114. doi: 10.1038/cti.2016.71
7. Proekt I, Miller CN, Lionakis MS, Anderson MS. Insights into immune tolerance from AIRE deficiency. Curr Opin Immunol. (2017) 49:71–8. doi: 10.1016/j.coi.2017.10.003
8. Constantine GM, Lionakis MS. Lessons from primary immunodeficiencies: autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol Rev. (2019) 287:103–20. doi: 10.1111/imr.12714
9. Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med. (2010) 207:299–308. doi: 10.1084/jem.20091669
10. Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med. (2010) 207:291–7. doi: 10.1084/jem.20091983
11. Conti HR, Bruno VM, Childs EE, Daugherty S, Hunter JP, Mengesha BG, et al. IL-17 receptor signaling in oral epithelial cells is critical for protection against oropharyngeal candidiasis. Cell Host Microbe. (2016) 20:606–17. doi: 10.1016/j.chom.2016.10.001
12. Conti HR, Gaffen SL. IL-17-mediated immunity to the opportunistic fungal pathogen candida albicans. J Immunol. (2015) 195:780–8. doi: 10.4049/jimmunol.1500909
13. Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. (2011) 332:65–8. doi: 10.1126/science.1200439
14. Bader O, Weig MS, Gross U, Schon MP, Mempel M, Buhl T. Photo quiz. A. 32-year-old man with ulcerative mucositis, skin lesions, nail dystrophy. Chronic mucocutaneous candidiasis by multidrug-resistant Candida albicans. Clin Infect Dis. (2012) 54:972. doi: 10.1093/cid/cir958
15. Levy R, Okada S, Beziat V, Moriya K, Liu C, Chai LY, et al. Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc Natl Acad Sci USA. (2016) 113:E8277–E285. doi: 10.1073/pnas.1618300114
16. Veverka KK, Feldman SR. Chronic mucocutaneous candidiasis: what can we conclude about IL-17 antagonism? J Dermatolog Treat. (2018) 29:475–80. doi: 10.1080/09546634.2017.1398396
17. Zhang S. The role of transforming growth factor beta in T helper 17 differentiation. Immunology, (2018) 155:24–35. doi: 10.1111/imm.12938
18. Puel A, Picard C, Cypowyj S, Lilic D, Abel L, Casanova JL. Inborn errors of mucocutaneous immunity to Candida albicans in humans: a role for IL-17 cytokines? Curr Opin Immunol. (2010) 22:467–74. doi: 10.1016/j.coi.2010.06.009
19. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. (2016) 127:3154–64. doi: 10.1182/blood-2015-11-679902
20. Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. (2011) 208:1635–48. doi: 10.1084/jem.20110958
21. Zimmerman O, Olbrich P, Freeman AF, Rosen LB, Uzel G, Zerbe CS, et al. STAT1 gain-of-function mutations cause high total STAT1 levels with normal dephosphorylation. Front Immunol. (2019) 10:1433. doi: 10.3389/fimmu.2019.01433
22. Sharfe N, Nahum A, Newell A, Dadi H, Ngan B, Pereira SL, et al. Fatal combined immunodeficiency associated with heterozygous mutation in STAT1. J Allergy Clin Immunol. (2014) 133:807–17. doi: 10.1016/j.jaci.2013.09.032
23. Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. (2008) 452:773–6. doi: 10.1038/nature06764
24. de Beaucoudrey Puel AL, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med. (2008) 205:1543–50. doi: 10.1084/jem.20080321
25. Gernez Y, Freeman AF, Holland SM, Garabedian E, Patel NC, Puck JM, et al. Autosomal dominant hyper-IgE syndrome in the USIDNET registry. J Allergy Clin Immunol Pract. (2018) 6:996–1001. doi: 10.1016/j.jaip.2017.06.041
26. Bergerson JRE, Freeman AF. An update on syndromes with a hyper-ige phenotype. Immunol Allergy Clin North Am. (2019) 39:49–61. doi: 10.1016/j.iac.2018.08.007
27. Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. (2009) 124:1289–302.e4. doi: 10.1016/j.jaci.2009.10.038
28. Beziat V, Li J, Lin JX, Ma CS, Li P, Bousfiha A, et al. A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci Immunol. (2018) 3:eaat4956. doi: 10.1126/sciimmunol.aat4956
29. Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. (2006) 25:745–55. doi: 10.1016/j.immuni.2006.09.009
30. Sassi A, Lazaroski S, Wu G, Haslam SM, Fliegauf M, Mellouli F, et al. Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. J Allergy Clin Immunol. (2014) 133:1410–9. doi: 10.1016/j.jaci.2014.02.025
31. Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, neurocognitive impairment. J Allergy Clin Immunol. (2014) 133:1400–9. doi: 10.1016/j.jaci.2014.02.013
32. Stray-Pedersen A, Backe PH, Sorte HS, Morkrid L, Chokshi NY, Erichsen HC, et al. PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am J Hum Genet. (2014) 95:96–107. doi: 10.1016/j.ajhg.2014.05.007
33. Dorjbal B, Stinson JR, Ma CA, Weinreich MA, Miraghazadeh B, Hartberger JM, et al. Hypomorphic caspase activation and recruitment domain 11 (CARD11) mutations associated with diverse immunologic phenotypes with or without atopic disease. J Allergy Clin Immunol. (2019) 143:1482–95. doi: 10.1016/j.jaci.2018.08.013
34. Martinez-Barricarte R, Markle JG, Ma CS, Deenick EK, Ramirez-Alejo N, Mele F, et al. Human IFN-gamma immunity to mycobacteria is governed by both IL-12 and IL-23. Sci Immunol. (2018) 3:aau6759. doi: 10.1126/sciimmunol.aau6759
35. Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, et al. IMMUNODEFICIENCIES. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science. (2015) 349:606–13. doi: 10.1126/science.aaa4282
36. Bhattad S, Dinakar C, Pinnamaraju H, Ganapathy A, Mannan A. Chronic mucocutaneous candidiasis in an adolescent boy due to a novel mutation in TRAF3IP2. J Clin Immunol. (2019) 39:596–9. doi: 10.1007/s10875-019-00664-x
37. Boisson B, Wang C, Pedergnana V, Wu L, Cypowyj S, Rybojad M, et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. (2013) 39:676–86. doi: 10.1016/j.immuni.2013.09.002
38. Li J, Ritelli M, Ma CS, Rao G, Habib T, Corvilain E, et al. Chronic mucocutaneous candidiasis and connective tissue disorder in humans with impaired JNK1-dependent responses to IL-17A/F and TGF-beta. Sci Immunol. (2019) 4:aax7965. doi: 10.1126/sciimmunol.aax7965
39. Glocker EO, Hennigs A, Nabavi M, Schaffer AA, Woellner C, Salzer U, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. (2009) 361:1727–35. doi: 10.1056/NEJMoa0810719
40. Du B, Shen N, Hu J, Tao Y, Mo X, Cao Q. Complete clinical remission of invasive Candida infection with CARD9 deficiency after G-CSF treatment. Comp Immunol Microbiol Infect Dis. (2020) 70:101417. doi: 10.1016/j.cimid.2020.101417
41. Wang X, Wang A, Li R, Yu J. Cutaneous mucormycosis caused by mucor irregularis in a patient with CARD9 deficiency. Br J Dermatol. (2019) 180:213–214. doi: 10.1111/bjd.17144
42. Perez L, Messina F, Negroni R, Arechavala A, Bustamante J, Oleastro M, et al. Inherited CARD9 deficiency in a patient with both exophiala spinifera and aspergillus nomius severe infections. J Clin Immunol. (2020) 40:359–66. doi: 10.1007/s10875-019-00740-2
43. Corvilain E, Casanova JL, Puel A. Inherited CARD9 deficiency: invasive disease caused by ascomycete fungi in previously healthy children and adults. J Clin Immunol. (2018) 38:656–93. doi: 10.1007/s10875-018-0539-2
44. Quan C, Li X, Shi RF, Zhao XQ, Xu H, Wang B, et al. Recurrent fungal infections in a Chinese patient with CARD9 deficiency and a review of 48 cases. Br J Dermatol. (2019) 180:1221–5. doi: 10.1111/bjd.17092
45. Queiroz-Telles F, Mercier T, Maertens J, Sola CBS, Bonfim C, Lortholary O, et al. Successful allogenic stem cell transplantation in patients with inherited card9 deficiency. J Clin Immunol. (2019) 39:462–9. doi: 10.1007/s10875-019-00662-z
46. Gavino C, Cotter A, Lichtenstein D, Lejtenyi D, Fortin C, Legault C, et al. CARD9 deficiency and spontaneous central nervous system candidiasis: complete clinical remission with GM-CSF therapy. Clin Infect Dis. (2014) 59:81–4. doi: 10.1093/cid/ciu215
47. Ferwerda B, Ferwerda G, Plantinga TS, Willment JA, van Spriel AB, Venselaar H, et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med. (2009) 361:1760–7. doi: 10.1056/NEJMoa0901053
48. Drewniak A, Gazendam RP, Tool AT, van Houdt M, Jansen MH, van Hamme JL, et al. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood. (2013) 121:2385–92. doi: 10.1182/blood-2012-08-450551
49. Rosler B, Wang X, Keating ST, Joosten LAB, Netea MG, van de Veerdonk LF. HDAC inhibitors modulate innate immune responses to micro-organisms relevant to chronic mucocutaneous candidiasis. Clin Exp Immunol. (2018) 194:205–19. doi: 10.1111/cei.13192
50. Kiykim A, Charbonnier LM, Akcay A, Karakoc-Aydiner E, Ozen A, Ozturk G, et al. Hematopoietic stem cell transplantation in patients with heterozygous stat1 gain-of-function mutation. J Clin Immunol. (2019) 39:37–44. doi: 10.1007/s10875-018-0575-y
51. Leiding JW, Okada S, Hagin D, Abinun M, Shcherbina A, Balashov DN, et al. Hematopoietic stem cell transplantation in patients with gain-of-function signal transducer and activator of transcription. 1 mutations. J Allergy Clin Immunol. (2018):141:704–17.e5. doi: 10.1016/j.jaci.2017.03.049
52. Weinacht KG, Charbonnier LM, Alroqi F, Plant A, Qiao Q, Wu H, et al. Ruxolitinib reverses dysregulated T helper cell responses and controls autoimmunity caused by a novel signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J Allergy Clin Immunol. (2017) 139:1629–40.e2. doi: 10.1016/j.jaci.2016.11.022
53. Vargas-Hernandez A, Mace EM, Zimmerman O, Zerbe CS, Freeman AF, Rosenzweig S, et al. Ruxolitinib partially reverses functional natural killer cell deficiency in patients with signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations. J Allergy Clin Immunol. (2018) 141:2142–55.e5. doi: 10.1101/157271
54. Al Shehri T, Gilmour K, Gothe F, Loughlin S, Bibi S, Rowan AD, et al. Novel gain-of-function mutation in stat1 sumoylation site leads to CMC/CID phenotype responsive to ruxolitinib. J Clin Immunol. (2019) 39:776–85. doi: 10.1007/s10875-019-00687-4
55. Mossner R, Diering N, Bader O, Forkel S, Overbeck T, Gross U, et al. Ruxolitinib induces interleukin 17 and ameliorates chronic mucocutaneous candidiasis caused by STAT1 gain-of-function mutation. Clin Infect Dis. (2016) 62:951–3. doi: 10.1093/cid/ciw020
Keywords: CMC, chronic mucocutaneous candidiasis, immune dysregulation, primary immune deficiency, autoimmunity
Citation: Shamriz O, Tal Y, Talmon A and Nahum A (2020) Chronic Mucocutaneous Candidiasis in Early Life: Insights Into Immune Mechanisms and Novel Targeted Therapies. Front. Immunol. 11:593289. doi: 10.3389/fimmu.2020.593289
Received: 10 August 2020; Accepted: 08 September 2020;
Published: 16 October 2020.
Edited by:
Ravi Misra, University of Rochester, United StatesCopyright © 2020 Shamriz, Tal, Talmon and Nahum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oded Shamriz, b2RlZHMmI3gwMDA0MDtoYWRhc3NhaC5vcmcuaWw=
†These authors have contributed equally to this work and share first authorship