 June Young Lee1†
June Young Lee1† Emaan Madany1†Najwa El Kadi1Sumaarg Pandya1
Emaan Madany1†Najwa El Kadi1Sumaarg Pandya1 Kessandra Ng1
Kessandra Ng1 Michifumi Yamashita1
Michifumi Yamashita1 Caroline A. Jefferies2
Caroline A. Jefferies2 David R. Gibb1,3*
David R. Gibb1,3*- 1Department of Pathology and Laboratory Medicine, Cedars-Sinai Medical Center, Los Angeles, CA, United States
- 2Department of Internal Medicine, Division of Rheumatology, Cedars-Sinai Medical Center, Los Angeles, CA, United States
- 3Division of Transfusion Medicine, Cedars-Sinai Medical Center, Los Angeles, CA, United States
Red blood cell (RBC) transfusion exposes recipients to hundreds of unmatched minor RBC antigens. This exposure can lead to production of alloantibodies that promote clinically significant hemolytic events. Multiple studies have reported an increased frequency of RBC alloimmunization in patients with autoimmunity. However, cellular and molecular mechanisms that underlie autoimmunity-induced alloimmunization have not been reported. Patients with systemic lupus erythematosus (SLE) have a high frequency of alloimmunization and express a type 1 interferon (IFNα/β) gene signature. Thus, we utilized the pristane-induced lupus mouse model to test the hypothesis that inflammation in lupus promotes RBC alloimmunization, and to examine the potential role of IFNα/β. Intraperitoneal injection of pristane, a hydrocarbon oil, led to autoantibody production, glomerulonephritis, and pulmonary hemorrhage in wild type (WT) mice. Pristane treatment significantly induced serum IFNα and expression of multiple interferon-stimulated genes (ISGs) in peripheral blood and peritoneal fluid cells, including inflammatory macrophages. Following transfusion with allogeneic RBCs expressing the KEL glycoprotein, pristane-treated WT mice produced significantly elevated levels of anti-KEL IgM and anti-KEL IgG, compared to untreated mice. Pristane induced comparable levels of inflammatory cells and cytokines in mice lacking the IFNα/β receptor (IFNAR1–/–) or the IFNα/β-inducing transcriptions factors (IRF3/7–/–), compared to WT mice. However, pristane-treated IFNAR1–/– and IRF3/7–/– mice failed to produce ISGs and produced significantly lower levels of transfusion-induced anti-KEL IgG, compared to WT mice. Thus, pristane induction of a lupus-like phenotype promoted alloimmunization to the KEL RBC antigen in an IFNα/β-dependent manner. To our knowledge, this is the first examination of molecular mechanisms contributing to RBC alloimmunization in a model of autoimmunity. These results warrant further investigation of the role of IFNα/β in alloimmunization to other RBC antigens and the contribution of the IFNα/β gene signature to the elevated frequency of alloimmunization in patients with SLE.
Introduction
Red blood cell (RBC) transfusion exposes recipients to hundreds of non-ABO RBC antigens that are not routinely matched between donors and recipients. This exposure can lead to production of anti-RBC alloantibodies that promote clinically significant hemolytic events, including potentially fatal hemolytic transfusion reactions, hemolytic disease of the newborn, and rejection of renal allografts expressing allo-antigens also expressed on RBCs (1–4). Additionally, RBC alloimmunization against multiple RBC antigens can prohibit the availability of compatible RBC units, leading to anemia-associated morbidity and mortality (5, 6). Identifying factors that promote RBC alloantibody responses would allow for identification of at-risk patients who may benefit from interventions that inhibit alloimmunization.
One such factor is the state of inflammation in the transfused recipient. In 1995, Ramsey and Smietana reported that women with autoimmune disease have a high incidence of RBC alloimmunization (7). Subsequent studies showed increased alloimmunization in patients with various autoimmune diseases, including inflammatory bowel disease, rheumatoid arthritis, autoimmune hepatitis, and systemic lupus erythematosus (SLE) (8–10). For example, while 3–5% of all transfused patients form RBC alloantibodies, (11, 12) more than 20% of transfused patients with SLE produce such alloantibodies, representing the second highest incidence, compared to other studied disease populations (8). However, the cellular and molecular mechanisms underlying the elevated incidence in patients with SLE or other chronic autoimmune diseases, including the role of inflammatory pathways, have not been investigated.
A pathway shown to promote autoantibody production and disease severity in SLE is type 1 interferon (IFNα/β) production and signaling. While IFNα/β was discovered for its critical role in anti-viral immunity (13), more recent studies have implicated IFNα/β in the pathogenesis of autoimmune diseases, including rheumatoid arthritis, myositis, Sjögren’s syndrome (SS), systemic sclerosis and SLE (14–18). Approximately two-thirds of adult patients, and nearly all children, with SLE express an IFNα/β gene signature, defined as the expression of multiple interferon-stimulated genes (ISGs) (19–23). In addition, more than 50% of SLE-associated genetic variants have been linked to the IFNα/β pathway (24).
While the contribution of an autoimmune gene signature to RBC alloimmunization has not been examined in humans or animal models, treatment of murine transfusion recipients with specific inflammatory stimuli has been shown to induce or enhance RBC alloimmune responses (11, 25, 26). Co-transfusion with CpG DNA or pre-treatment with polyinosinic:polycytidylic acid [poly(I:C)], a mimetic of viral double stranded RNA, has been shown to induce alloimmunization to a human RBC antigen expressed on mouse RBCs (25–27). Accordingly, polyoma and influenza viral infections in mice enhance the alloimmune response to transfused RBCs (28, 29). Prior studies have also shown that transfusion of pro-inflammatory stored RBCs induces alloimmunization (30–32). Arneja et al. demonstrated that IL-6 is critical for T follicular helper cell promotion of RBC alloimmune responses to stored RBCs (33). Finally, it is noteworthy that not all inflammatory stimuli promote alloimmunization, as gram-negative bacteria and lipopolysaccharide have been shown to inhibit alloimmunization (34, 35). Thus, activation of specific inflammatory pathways in certain conditions can promote or inhibit RBC alloimmunization.
Compared to viral infection and acute stimuli, inflammatory pathways in SLE and other autoimmune diseases differ in strength and chronicity. In patients with chronic autoimmunity, the role of IFNα/β and other cytokine-mediated inflammatory pathways in RBC alloantibody responses is poorly understood. Here, given the increased frequency of alloimmunization in patients with SLE, we examine the degree to which inflammation in a lupus mouse model influences RBC alloimmunization. Use of the pristane-induced lupus model, which exhibits a strong IFNα/β signature (36), also allows examination of the role of IFNα/β in alloimmunization in an autoimmune model.
Materials and Methods
Mice
C57BL/6 and IFNAR1–/– mice were purchased from Jackson Laboratories (Bar Harbor, ME, United States). IFNAR1–/–, IRF3–/–, IRF7–/–, and K1 RBC transgenic mice were previously described (37–39). K1 mice express the KEL glycoprotein, containing the KEL1 antigen, on RBCs. Appropriate gene-deficient mice were bred to produce IRF3/7–/– double knockout mice. All mice were 8–12 weeks of age and had been backcrossed to the C57BL/6 background for more than 8 generations. All pristane-treated mice were female mice injected intraperitoneally with one dose of 0.5 mL pristane (2,6,10,14-tetramethylpentadecane). All animal protocols were approved by the Cedars-Sinai Institutional Animal Care and Use Committee.
RBC Transfusion
Peripheral blood of K1 mice was collected in 12% Citrate Phosphate Dextrose Adenine (CPDA-1, Jorgensen Labs, Melville, NY, United States) by retro-orbital bleeding and leuko-reduced with a Pall (East Hills, NY, United States) syringe filter. Recipient mice were transfused i.v. with 75 μL of leuko-reduced packed RBCs, which is the approximate mouse equivalent of 1 unit of human RBCs.
Clearance Assay
For post-transfusion recovery experiments, mice previously transfused with K1 RBCs received a second transfusion of fluorescently labeled K1 and C57BL/6 RBCs 35 days after the first transfusion. K1 and C57BL/6 RBCs were labeled with DiI (1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindocarbocyanine Perchlorate) and DiO (3,3’-Dioctadecyloxacarbocyanine Perchlorate) lipophilic dyes (Life Technologies, Camarillo, CA, United States), respectively, according to manufacturer’s instructions. A 2:1 ratio of K1 to C57BL/6 RBCs was mixed and transfused into previously transfused recipients and K1 transgenic mice, which served as a negative control. Mice were bled 10 min, 1, 2, and 4 days after transfusion, and the ratio of K1 to C57BL/6 RBCs remaining in circulation was measured by flow cytometry. The percentage of K1 RBCs remaining in circulation, compared to C57BL/6 RBCs remaining, was plotted as post-transfusion recovery.
Measurement of Anti-KEL Alloantibodies
Serum anti-KEL IgM, IgG, IgG1, IgG2b, IgG2c, and IgG3 were measured by flow cytometric crossmatch. Anti-KEL IgM was measured 5 days after transfusion, and anti-KEL IgG was measured 7, 14, 21, and 28 days after transfusion as previously described (40). Briefly, serum from transfused mice was incubated with K1 or C57BL/6 RBCs and subsequently stained for RBC-bound IgM or IgG (goat anti-mouse IgM FITC or goat anti-mouse IgG APC). Secondary antibodies for IgG subsets included goat anti-mouse IgG1 PE, goat anti-mouse IgG2c APC, goat anti-mouse IgG2b FITC, and goat-anti mouse IgG3 BV421 (Jackson ImmunoResearch, West Grove, PA, United States). The adjusted MFI was calculated by subtracting the reactivity of serum with syngeneic C57BL/6 RBCs from the reactivity of serum with K1 RBCs. Graphed anti-KEL IgG data represents the peak antibody response, 21–28 days following transfusion. Flow cytometry of RBCs was performed using a SONY SA 3800 (San Jose, CA, United States) or a Cytek Northern Lights 3000 spectral analyzer (Fremont, CA, United States) and analyzed using FlowJo software (Tree Star, Ashland, OR, United States).
Flow Cytometric Analysis of Leukocytes
Single cell suspensions of peripheral blood leukocytes and splenocytes were analyzed following RBC lysis. Peritoneal fluid cells were collected by flushing the peritoneal cavity with media. Spleens were minced with a razor blade prior to cell filtration with a 70 μM nylon mesh. Suspensions were incubated with Fc receptor block, TruStain FcX, from Biolegend (San Diego, CA, United States) and stained with fluorescently conjugated antibodies specific for cell surface markers, including CD11c (clone N418), B220 (RA3-6B2), F4/80 (BM8), CD11b (M1/70), Ly6C (HK1.4), Ly6G (1A8), Siglec-1 (3D6.112), TCRβ (H57-597), and I-A/I-E (MHC II, M5/114.15.2) from Biolegend. Zombie-Red and Zombie-NIR (Biolegend) were used to exclude dead cells. Cells were acquired with a Cytek Northern Lights 3000 or a LSRII flow cytometer (Becton Dickinson, San Jose, CA, United States) and analyzed using FlowJo.
Quantitative PCR
RNA was isolated from peritoneal fluid and peripheral blood leukocytes using the Qiagen RNeasy mini-kit (Hilden, Germany) and converted to cDNA with the Maxima H Minus cDNA Synthesis Master (Thermo Fisher Scientific, Waltham, MA, United States). The amount of GADPH, Mx1, ISG15, and IRF7 cDNA was measured by a QuantStudio 5 Real-Time PCR System using PowerUp SYBR Green master mix (Thermo Fisher Scientific). Primer sequences are listed in Supplementary Table 1. Thermo Fisher Scientific Connect software was used to determine the relative expression of target genes, compared to GAPDH.
ELISAs, Creatinine, and Hematocrit
Serum autoantibodies and urine albumin were measured by ELISA using the mouse anti-SSA (RO-52) ELISA kit (Signosis, Inc., Santa Clara, CA, United States), the mouse anti-dsDNA IgG ELISA Kit (Alpha Diagnostic International, San Antonio, TX, United States), and the Albuwell M ELISA kit (Ethos Biosciences Inc., Philadelphia, PA, United States). Urine creatinine was measured using a Creatinine Liquicolor kit (Stanbio Laboratory, Boerne, TX, United States). Hematocrit was measured using the HemaTrue® Veterinary Hematology Analyzer (Heska, Loveland, CA, United States).
Histology
Lungs and kidneys were fixed in 10% buffered formalin (Medical Chemical Corporation, Torrance, CA, United States). Slides were cut from paraffin-embedded blocks by the Cedars-Sinai histology lab and stained with hematoxylin and eosin for lungs and periodic acid-Schiff for kidneys. Lung and renal histology slides were scored by a renal pathologist (M.Y.).
Cytokine Measurement
Serum was collected by retro-orbital bleeding and centrifugation, and cytokine measurement and analysis were performed with the LEGENDplex mouse anti-virus response panel multiplex assay (Biolegend) according to manufacturer’s instructions.
Statistics
Statistical analysis was performed using Graph Pad Prism software (San Diego, CA, United States). Statistical significance between two groups was determined using an unpaired Student’s t-test or Mann Whitney U test for parametric and non-parametric data, respectively. Significance between three groups of non-parametric data was determined using a Kruskal-Wallis test with a Dunn’s post-test. Anti-KEL antibody levels and cytokine data are non-parametric. Data bars represent the mean. Error bars represent the standard error of the mean. White circles indicate data from individual mice.
Results
Pristane-Induced Autoimmune Pathology
We utilized the previously described pristane-induced lupus model (41) to assess the effect of lupus-like inflammation on RBC alloantibody responses. Pristane is a hydrocarbon oil that, when injected intraperitoneally, leads to a lupus-like phenotype by a toll-like receptor 7 (TLR7)-dependent mechanism (42). In accordance with prior studies using C57BL/6 mice, the mortality caused by pristane treatment was 0–20% for all experiments (data not shown) (43). Compared to untreated wild type (WT) mice, pristane-treated WT mice produced elevated levels of lupus-associated autoantibodies, including anti-Sjögren’s syndrome related antigen A (SSA) and anti-dsDNA IgG (Supplementary Figures 1A,B). Eight months following pristane treatment of WT C57BL/6 mice, glomeruli exhibited mild mesangial expansion and hypercellularity (Supplementary Figures 1C,D), as previously reported (44). However, pristane-induced glomerular changes did not consistently lead to a significant increase in the urine albumin:creatinine ratio, compared to untreated WT mice (Supplementary Figure 1E). Also consistent with prior studies in C57BL/6 WT mice, pristane-treated mice developed diffuse pulmonary hemorrhage (Supplementary Figures 1F–H) and anemia (Supplementary Figure 1I) two weeks following pristane treatment (44, 45).
We examined the degree to which pristane induced anti-RBC autoantibodies by performing direct antiglobulin tests (DATs), which detect RBC-bound IgG. Two weeks after pristane treatment, none of the WT mice had positive DATs (data not shown). After 8 months, the results were highly variable. On occasion, pristane-treated WT mice had positive DATs (Supplementary Figure 2A), but collectively, there was no significant difference in the percent of RBCs with bound IgG, between pristane-treated and untreated WT mice (Supplementary Figure 2B).
Induction of Anti-KEL Alloimmunization in Pristane-Induced Lupus Mice
To examine RBC alloimmunization in pristane-induced lupus mice, we utilized the previously described mouse transfusion model, in which RBCs from transgenic mice expressing the KEL glycoprotein (K1 RBCs) are transfused into allogeneic recipients (37). Leuko-reduced K1 RBCs were transfused into WT mice treated without or with pristane 2, 14, or 48 days prior to transfusion, and anti-KEL IgM and IgG antibody levels were measured by flow cytometric crossmatch. Consistent with prior studies (29, 37), transfused untreated mice produced low levels of anti-KEL IgM, and produced nearly undetectable levels of anti-KEL IgG. Transfusion 2 days after pristane treatment did not induce elevated alloantibody production, compared to untreated mice. However, recipients treated 14 or 48 days prior to transfusion produced significantly higher levels of anti-KEL IgM and anti-KEL IgG, compared to untreated WT mice (Figures 1A–C). There was no significant difference in anti-KEL IgG produced in mice treated 14 or 48 days before transfusion (Figure 1B and Supplementary Figure 3). Thus, further analysis was completed in mice treated with pristane 14 days prior to transfusion, unless otherwise specified. Anti-KEL IgG in pristane-induced lupus mice included all IgG subclasses made in C57BL/6 mice (Figure 1D).
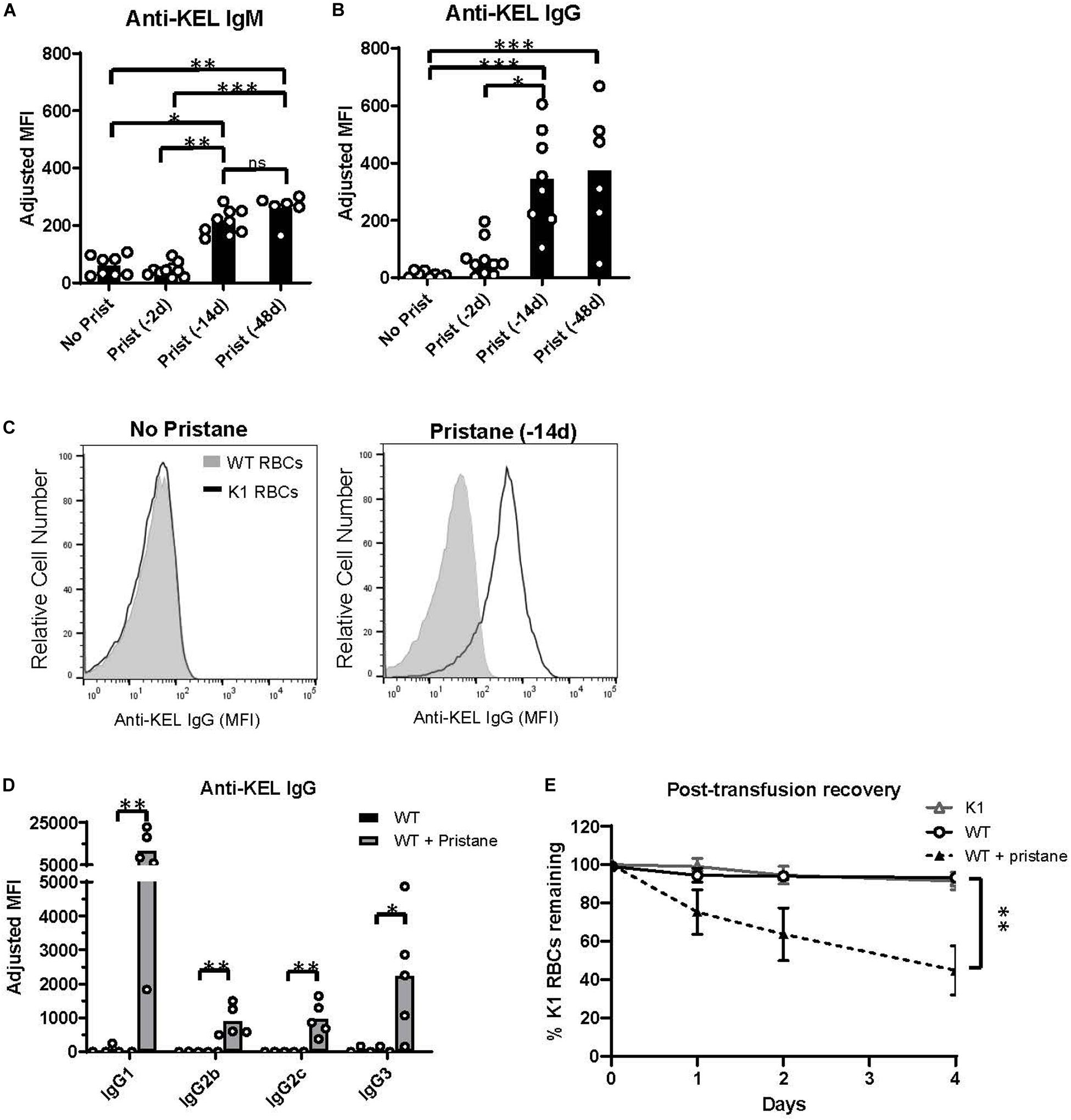
Figure 1. Pristane induces anti-KEL alloantibodies. Recipient WT mice were transfused with K1 RBCs. (A,B) Serum anti-KEL IgM and IgG in untreated mice and mice treated with pristane 2, 14, or 48 days prior to transfusion, measured by flow cytometric crossmatch. The adjusted MFI was calculated by subtracting the MFI of serum incubated with WT RBCs from the MFI of serum incubated with K1 RBCs, shown in (C). (C) Representative histograms of flow cytometric crossmatch, in which post-transfusion serum was incubated with K1 or WT RBCs. (D) Anti-KEL IgG subtypes in mice treated with or without pristane 14 days prior to transfusion. (A–D) Anti-KEL IgM was measured 4–5 days after transfusion. Anti-KEL IgG and subtypes represent the peak IgG response 21–28 days after transfusion. (E) WT mice treated with or without pristane 14 days prior to transfusion were re-transfused with a 2:1 mixture of labeled K1 and WT RBCs 35 days after the first transfusion. Control K1 recipients are negative controls. Ratios of recovered K1 and WT RBCs are calculated as percent of K1 RBCs remaining 1, 2, and 4 days after transfusion. (A–E) Representative of 3 independent experiments, 5–10 mice per group. *p < 0.05, **p < 0.01, ***p < 0.001 by Mann-Whitney U test (D) or Kruskal-Wallis test with a Dunn’s post-test (A,B,E).
To test the clinical significance of anti-KEL antibodies in pristane-induced lupus mice, we examined the ability of anti-KEL antibodies to clear K1 RBCs from circulation. Previously transfused pristane-treated and untreated mice received a second transfusion of a mixture of fluorescently labeled K1 RBCs (DiI+) and WT RBCs (DiO+) 35 days after the first transfusion. K1 mice were also transfused to serve as a negative control. The ratio of K1 RBCs to WT RBCs remaining in circulation was measured by flow cytometry. Compared to K1 and untreated WT recipients, pristane-treated mice preferentially cleared K1 RBCS within 4 days following the second transfusion (Figure 1E).
Pristane-Induced Inflammation in the Peri-Transfusion Period
Prior studies using K1 mice indicate that inflammation at the time of transfusion influences alloimmunization (29, 37). Thus, given the elevated alloantibody responses and clearance in pristane-induced lupus mice, we examined markers of inflammation at the time of transfusion. While there were no significant differences in the number of B and T cells between groups, Ly6G+ neutrophils and Ly6G– Ly6C+ monocytes were significantly elevated in the peripheral blood of pristane-treated WT mice (Figures 2A–C). Additionally, levels of spleen neutrophils, monocytes, CD11b+ F4/80+ macrophages, and CD11chi MHCII+ conventional dendritic cells were elevated in pristane-treated mice (Supplementary Figure 4).
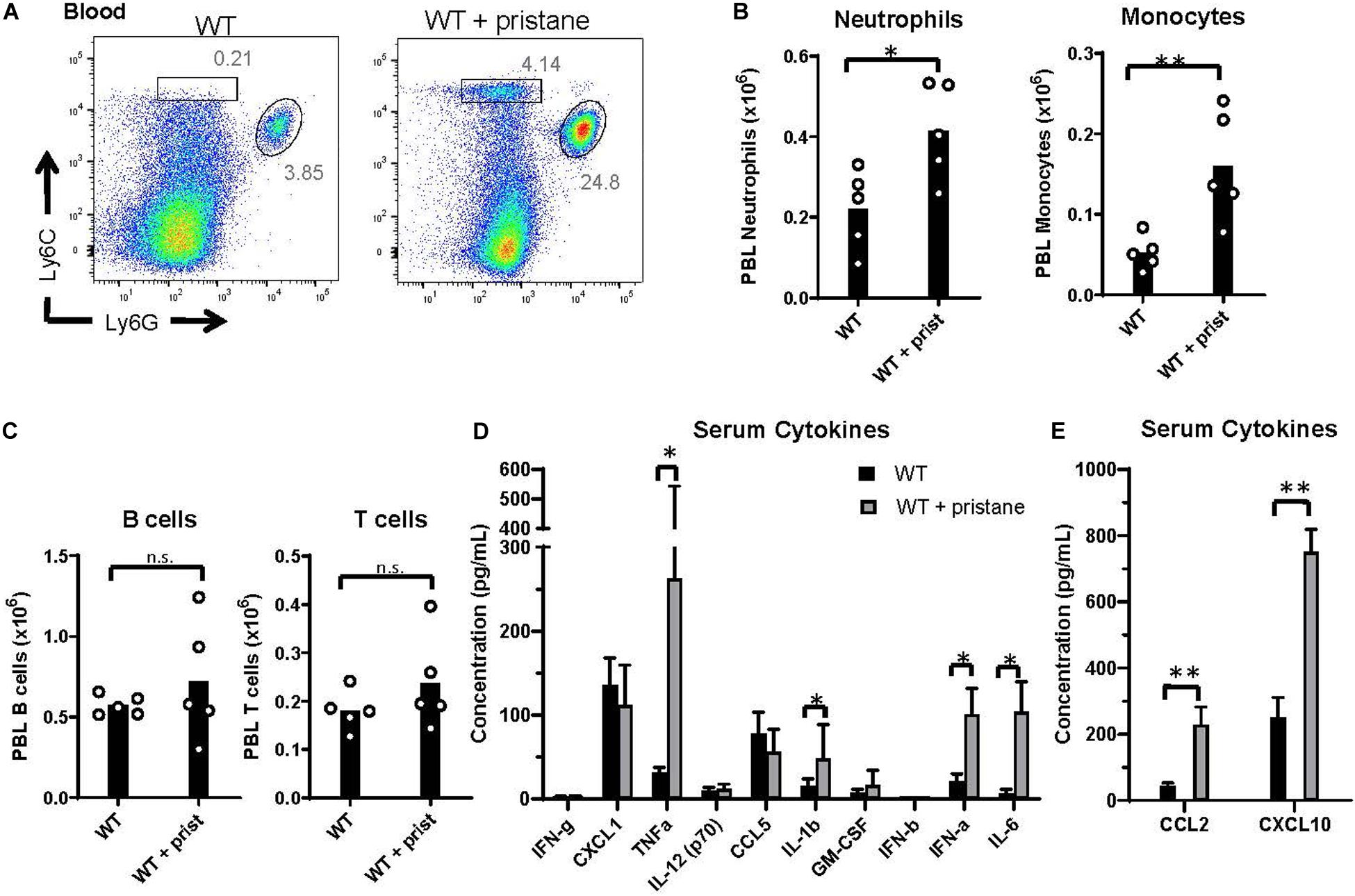
Figure 2. Pristane-induced inflammation in peripheral blood. WT mice were treated with or without pristane 14 days prior to analysis. (A) Representative flow cytometric analysis of Ly6C+ monocytes and Ly6G+ neutrophils gated on live cells. Numbers on plots indicate percentage within drawn gates. (B) Quantification of neutrophils and monocytes gated in (A). (C) Quantification of B220+ B cells and TCRβ+ T cells. (A–C) Cells are gated on Zombie negative live cells. (D,E) Serum cytokine levels measured by multiplex array. Representative of 3 independent experiments with 5 mice per group. *p < 0.05, **p < 0.01, by student’s t-test (B,C) or Mann-Whitney U test (D,E).
Serum cytokine analysis showed that TNFα, IL-1b, IL-6, and IFNα were significantly elevated in pristane-treated mice, compared to untreated mice, in 3 of 3 experiments (Figure 2D). IFNβ was significantly elevated in 1 of 3 experiments (data not shown). In addition, the interferon stimulated genes (ISGs), CCL2 and CXCL10, were elevated in serum of pristane-treated mice (Figure 2E). To determine the chronicity of pristane-induced cytokine production, we quantified serum cytokine and ISG levels in mice treated with or without pristane 2 days, 14 days, or 8 months prior to analysis. Two days after pristane treatment, TNFα, IL-1b, IL-6, IFNα, CCL2, and CXCL10 were not elevated compared to untreated mice. In addition, there was no significant difference in cytokine or ISG levels between mice treated 14 days or 8 months prior (Supplementary Figure 5). This indicates that cytokines and ISGs are chronically produced from 2 weeks to 8 months after pristane treatment. It is also notable that cytokine and ISG production significantly elevated 14 days, but not 2 days, after pristane treatment coincided with transfusion-induced anti-KEL IgM and IgG production.
Prior studies have shown that pristane induces chronic inflammation in the peritoneum (41). Thus, peritoneal fluid was evaluated at the time of transfusion, and CCL2 and CXCL10 were found to be significantly elevated in pristane-treated mice (Figure 3A). CCL2 and CXCL10 are chemokines that recruit inflammatory macrophages, monocytes, and neutrophils to sites of inflammation. These cell subsets were elevated in the peritoneal fluid of pristane-treated mice (Figure 3B). Consistent with other models of peritoneal inflammation (46), pristane treatment led to a reduction of CD11b+ F4/80hi resident macrophages, present in untreated mice, and an increase in inflammatory CD11b+ F4/80int inflammatory macrophages (Figure 3C). Given the elevated levels of IFNα and ISGs in peripheral blood, ISG expression was measured in peritoneal fluid cells. Siglec-1 was previously shown to be upregulated on monocytes following IFNα/β signaling (47). Accordingly, Ly6C+ monocytes and CD11b+ F4/80int inflammatory macrophages expressed elevated levels of Siglec-1 (Figures 3C–F). In addition, collectively, peritoneal fluid cells expressed elevated levels of other ISGs, including Mx1, ISG15, and TLR7 compared to untreated mice (Figure 3G).
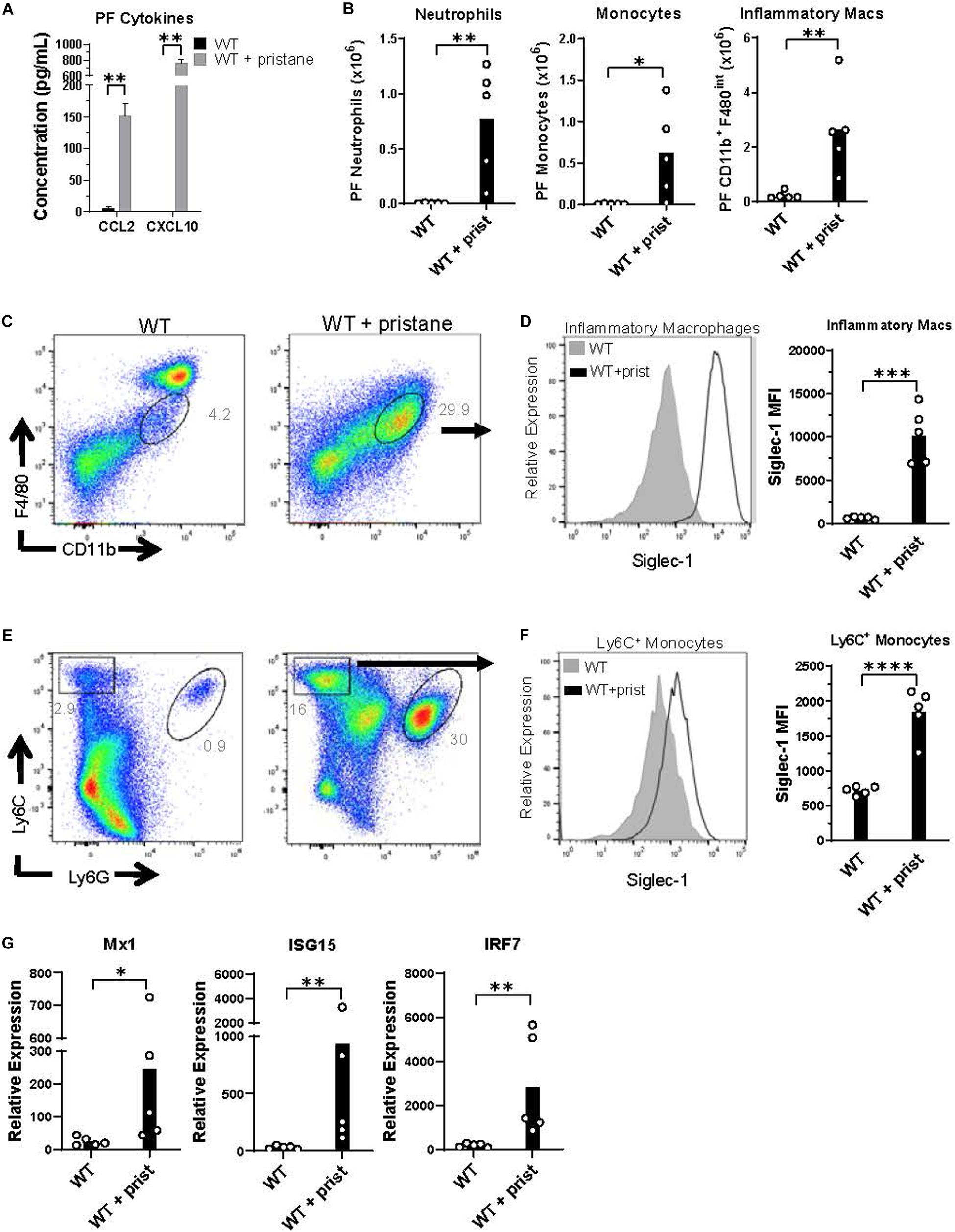
Figure 3. Inflammatory cells in peritoneal fluid express ISGs at the time of transfusion. WT mice were treated with or without pristane 14 days prior to analysis. (A) CCL2 and CXCL10 levels in peritoneal lavage fluid measured by multiplex array. (B) Quantification of peritoneal fluid neutrophils, monocytes, and inflammatory macrophages gated in (C,E). (C,E) Representative flow cytometric analysis of CD11b+F4/80int inflammatory macrophages (gated on live non-lymphocytes, Zombie– TCRβ– B220–), Ly6C+ monocytes and Ly6G+ neutrophils in peritoneal fluid, gated on Zombie negative live cells. (D,F) Representative histograms (left) and quantification (right) of Siglec-1 expression by CD11b+F4/80int inflammatory macrophages and Ly6C+ monocytes gated in (C,E). (G) Relative expression of Mx1, ISG15, and IRF7, compared to GAPDH, by all peritoneal fluid cells, measured by quantitative real-time PCR. Representative of 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by student’s t-test (B,D,F) or Mann-Whitney U test (A,G).
IFNα/β-Independent Inflammatory Cell and Cytokine Responses
Given the pristane-induced production of IFNα and ISGs, we examined inflammation in mice lacking the receptor for IFNα/β (IFNAR1–/–) and mice lacking the transcription factors, interferon response factor (IRF) 3 and IRF7, required for IFNα/β production (IRF3/7–/–). As expected, compared to pristane-treated WT mice, pristane-treated IRF3/7–/– mice produced significantly lower levels of IFNα, whereas pristane-treated IFNAR1–/– and WT mice made comparable levels (Figure 4A). Serum levels of the ISG, CXCL10, were significantly reduced in pristane-treated IRF3/7–/– mice and trended to a non-significant reduction in pristane-treated IFNAR1–/– mice, compared to treated WT mice (Figure 4B). Siglec-1 expression by CD11b+ F4/80int inflammatory macrophages was also reduced in pristane-treated IRF3/7–/– and IFNAR1–/– mice, compared to WT treated mice (Figures 4C–E). In addition, levels of Mx1, ISG15, and IRF7 were significantly reduced in total peritoneal fluid cells of IFNAR1–/– and IRF3/7–/– treated mice, compared to WT treated controls (Figure 4F). A complete absence of IRF7 mRNA confirmed its genetic deficiency in IRF3/7–/– mice.
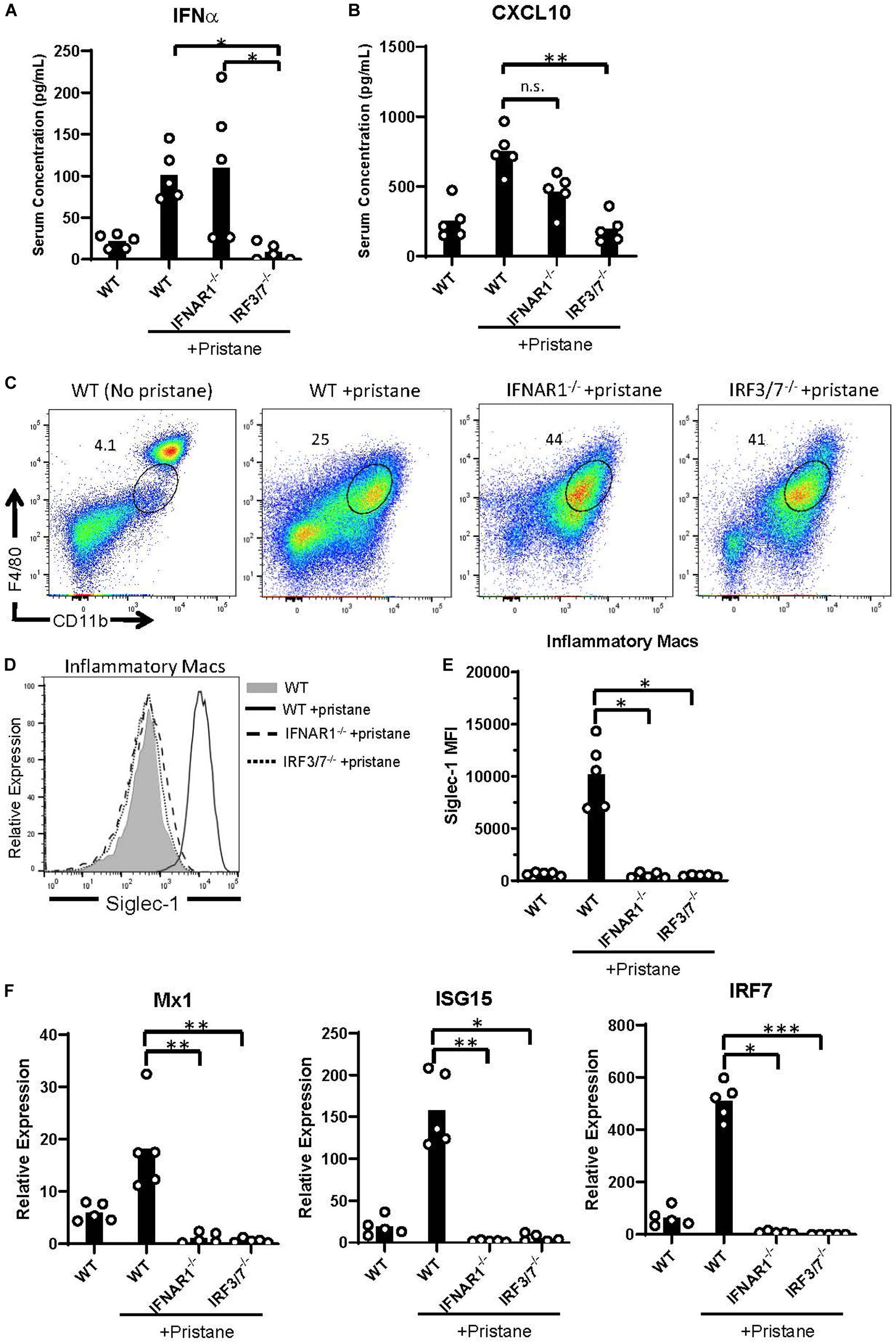
Figure 4. Abrogated ISG expression in pristane-treated IFNAR1– /– and IRF3/7– /– mice. WT, IFNAR1– /–, and IRF3/7– /– mice were treated with pristane 14 days prior to analysis. Untreated WT mice were included as controls. (A,B) Serum IFNα and CXCL10 levels measured by multiplex array. (C) Representative flow cytometric plots of CD11b+ F4/80int inflammatory macrophages, gated on live non-lymphocytes (Zombie– TCRβ– B220–) cells. Numbers on plots indicate percent of cells within drawn gates. (D) Histogram overlays and (E) quantification of Siglec-1 expression by inflammatory macrophages gated in (C). (F) Relative expression of Mx1, ISG15, and IRF7, compared to GAPDH, by all peritoneal fluid cells, measured by quantitative real-time PCR. Representative of 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 by Kruskal-Wallis test with a Dunn’s post-test.
Despite the reduction in IFNα/β and ISG production in IFNAR1–/– and IRF3/7–/– treated mice, peritoneal infiltration by monocytes, neutrophils, and inflammatory macrophages was comparable in pristane-treated IFNAR1–/–, IRF3/7–/– and WT mice (Figures 5A, 4C). Compared to untreated WT mice, pristane also resulted in significantly elevated levels of spleen CD11b+ F4/80+ macrophages in IFNAR1–/– and IRF3/7–/– mice, increased levels of spleen Ly6C+ monocytes and Ly6G+ neutrophils in IRF3/7–/– mice, and trends toward increased levels in IFNAR1–/– mice (Supplementary Figure 6).
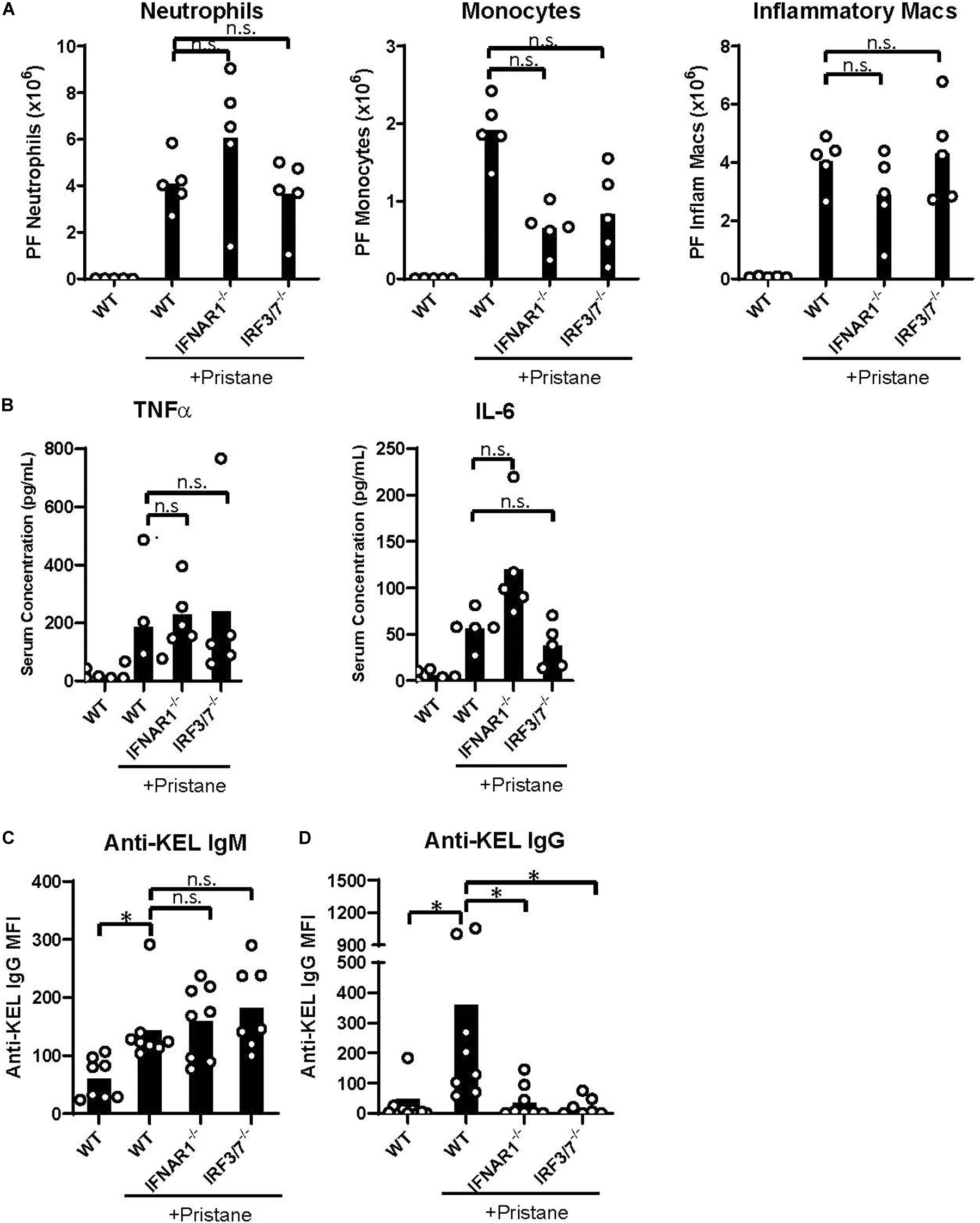
Figure 5. IFNα/β production and signaling promote K1 RBC alloimmunization. WT, IFNAR1–/–, and IRF3/7–/– mice were treated with pristane 14 days prior to analysis (A,B) or transfusion (C,D). Untreated WT mice were included as controls. (A) Quantification of Ly6G+ neutrophils, Ly6C+ monocytes, and CD11b+F4/80int inflammatory macrophages in peritoneal fluid; gated on Zombie-negative live cells. Inflammatory macrophages were also gated on TCRβ–B220– non-lymphocytes. (B) TNFα and IL-6 cytokine levels measured by multiplex array. Representative of 3 independent experiments with 5 mice per group. (C,D) Fourteen days after pristane-treatment, WT, IFNAR1–/–, and IRF3/7–/– mice were transfused with K1 RBCs. (C) Anti-KEL IgM was measured 4–5 days after transfusion. (D) Anti-KEL IgG data represent the peak IgG response 21–28 days after transfusion. 7–8 mice per group. Representative of 3 independent experiments. ∗p < 0.05 by Kruskal-Wallis test with a Dunn’s post-test.
Additionally, all pristane-treated groups produced comparable levels of non-IFNα/β cytokines, including TNFα and IL-6 (Figure 5B). Thus, pristane-induced peritoneal and spleen inflammation is promoted by IFNα/β-independent mechanisms.
IFNα/β-Dependent Anti-KEL RBC Alloimmunization
Finally, given the elevated production of anti-KEL antibodies following K1 RBC transfusion, we examined the role of IFNα/β production and IFNAR1 signaling in K1 RBC alloimmunization. Following transfusion of K1 RBCs, all pristane-treated mice produced anti-KEL IgM at comparable levels (Figure 5C). However, despite the comparable levels of inflammation in all pristane-treated groups, IFNAR1–/– and IRF3/7–/– mice produced significantly lower anti-KEL IgG antibodies, compared to WT treated controls (Figure 5D). Thus, IFNα/β production and signaling promote K1 RBC alloimmunization in pristane-induced lupus mice.
Discussion
Multiple studies have reported an increased frequency of RBC alloimmunization in patients with autoimmune diseases, including SLE (7–10). However, cellular and molecular mechanisms that underlie autoimmunity-induced alloimmunization have not been reported. Here, we report that IFNα/β production and signaling promote RBC alloimmunization in a lupus mouse model. To our knowledge, this is the first examination of molecular mechanisms contributing to RBC alloimmunization in a model of autoimmunity.
Two-thirds of patients with SLE express an IFNα/β gene signature that correlates with autoantibody production and disease severity. Thus, we utilized the well described pristane-induced lupus model, in which IFNα/β production and signaling have been shown to promote autoantibody production and subsequent renal pathology (41). While transient inflammation induced by viral stimuli has previously been shown to promote RBC alloimmunization (27–29), it was unclear whether chronic inflammation in autoimmune models would also induce alloimmunization. Further, in addition to IFNα, pristane induces production of numerous NFκB-induced cytokines, including IL-6, IL-1b, and TNFα, which contribute to other manifestations of lupus. For example, neutrophil invasion of the bone marrow produces TNFα, which perturbs RBC progenitor development, resulting in anemia (45). Thus, it was unclear which pristane-induced inflammatory pathways induced RBC alloimmunization.
While results reported here do not rule out a contributory role for other cytokine pathways, diminished transfusion-induced anti-KEL IgG production in pristane-treated IFNAR1–/– and IRF3/7–/– mice indicates a significant role for IFNα/β production and IFNAR1 signaling in K1 RBC alloimmunization. Of note, a lack of IFNα/β production or signaling did not impact markers of inflammation, including inflammatory cell infiltration in the peritoneal cavity and the spleen, nor did it impact the production of NFκB-induced inflammatory cytokines. Thus, the lack of anti-KEL IgG production in IFNAR1–/– and IRF3/7–/– mice was not due to reduced IFNα/β-independent inflammation.
Pristane-induced production and expansion of inflammatory cells, including neutrophils, monocytes and macrophages, likely contributed to RBC alloimmunization. These cells can phagocytose RBCs, transport antigen, and promote antigen presentation to T cells. In addition, Ly6C+ monocytes in the peritoneum are the primary producers of IFNα/β (48). Inflammatory macrophages in the peritoneum of pristane-treated mice expressed high levels of the ISG, Siglec-1, which was first identified as an erythrocyte binding receptor (49). It was later found to be induced by IFNα/β in patients with systemic sclerosis (47). Siglec-1+ macrophages are located in areas exposed to body fluids, including the peritoneum and the marginal zone of the spleen. They have been shown to capture and transfer antigen to dendritic cells and B cells for antigen presentation (50). Thus, although not tested here, Siglec-1+ macrophages may contribute to RBC alloantibody production.
In this transfusion model, anti-KEL IgG is clinically significant, in that it clears KEL-expressing RBCs and can induce hemolytic events, including hemolytic transfusion reactions (40). In addition to anti-KEL IgG, pristane also enhanced anti-KEL IgM production following K1 RBC transfusion. Notably, in the pristane model, enhancement of anti-KEL IgM was also present in IFNAR1–/– and IRF3/7–/– mice. Thus, IFNα/β is dispensable for anti-KEL IgM, but not anti-KEL IgG production. This observation indicates that IFNα/β may promote a critical process in antibody class switching. IFNα/β has been shown to promote differentiation of germinal center and antibody-producing plasma cells (51, 52). However, it also activates antigen presentation by dendritic cells, and can activate T cells directly (53). Thus, further studies are needed to determine the mechanism of IFNα/β-induced IgG class switching during RBC alloimmunization.
This report builds upon prior data implicating the IFNα/β pathway in RBC alloimmunization following acute viral infection or poly(I:C) treatment. Poly(I:C), injected 3 hr prior to transfusion, induces profoundly high transient levels of serum IFNα that diminish within 24 hr of injection (37). Consequently, K1 RBC transfusion 24 hr before or after poly(I:C) treatment does not induce anti-KEL IgG (37). In contrast, in the current study, pristane treatment resulted in chronic inflammation, IFNα production, and ISG expression that promoted RBC alloimmunization 2 to 6 weeks after pristane treatment. Pristane is thought to induce IFNα/β production by causing the infiltration of inflammatory cells that phagocytose pristane, undergo cell death, and release RNA that binds and activates TLR7 in endosomes (41). In contrast, poly(I:C) is a TLR3 agonist (54) and has been shown to bind the cytosolic receptor melanoma differentiation-associated protein 5 (MDA5), leading to activation of mitochondrial antiviral-signaling protein (MAVS) prior to IFNα/β production, which promotes RBC alloantibody production (37). Activation of disparate pathways may underly differences in quantity and chronicity of IFNα/β production. While TLR7 has been shown to be required for pristane-induced autoantibody production, other pathways warranting further investigation may promote IFNα/β-induced RBC alloimmunization.
It is noteworthy that autoantibody production induced by pristane occurs months after treatment. However, inflammation and IFNAR1 signaling within two weeks of treatment are required for later autoantibody production (41); and data presented here indicate that IFNα/β-mediated inflammation is required for RBC alloantibody production in this model. Future studies are needed to determine whether autoantibody production and autoantibody-mediated pathology promote RBC alloimmunization by IFNα/β-independent mechanisms. Such studies will have the challenge of separating the role of autoimmunity-induced alloimmunization and IFNα/β-induced alloimmunization, as autoantibody production is abrogated in IRF3/7–/– and IFNAR1–/– mice (41). Thus, examination of RBC alloimmunization in other lupus models with less dependence on IFNα/β may be needed.
In summary, we report that inflammation in a lupus mouse model promotes alloimmunization to the KEL RBC antigen in an IFNα/β-dependent manner. These results warrant further investigation of the role of IFNα/β in alloimmunization to other RBC antigens and of the contribution of the IFNα/β gene signature to the increased frequency of alloimmunization in patients with SLE. If these findings extend to human studies, SLE patients with an IFNα/β gene signature may benefit from personalized transfusion protocols.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the Cedars-Sinai Institutional Animal Care and Use Committee.
Author Contributions
EM, JL, NE, CJ, MY, and DRG completed the project design and data analysis. DRG, EM, JL, NE, KN, and SP completed the experiments. MY completed the pathological analysis. DRG wrote the initial draft of the manuscript. All authors contributed to edits of the manuscript.
Funding
This work was funded by NIH/NHLBI (5 K08 HL141446), the American Society of Hematology Scholar Award (5283) and the National Blood Foundation (R13672) to DRG.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SP declared a past co-authorship with one of the authors DRG to the handling editor.
Acknowledgments
The authors thank Jeanne E. Hendrickson, MD of Yale School of Medicine for her continued mentorship of DRG and edits of this manuscript. They thank Chance John Luckey, MD Ph.D. of the University of Virginia School of Medicine for edits. They also thank Robert W. Baloh, MD of Cedars-Sinai Medical Center for IFNAR1–/– mice.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.584254/full#supplementary-material
References
1. Holt S, Donaldson H, Hazlehurst G, Varghese Z, Contreras M, Kingdon E, et al. Acute transplant rejection induced by blood transfusion reaction to the Kidd blood group system. Nephrol Dial Transplant. (2004) 19:2403–6. doi: 10.1093/ndt/gfh333
2. Lerut E, Van Damme B, Noizat-Pirenne F, Emonds MP, Rouger P, Vanrenterghem Y, et al. Duffy and Kidd blood group antigens: minor histocompatibility antigens involved in renal allograft rejection? Transfusion. (2007) 47:28–40. doi: 10.1111/j.1537-2995.2007.01060.x
3. U. D. O. H. A. H. S. (FDA). Fatalities Reported to the FDA Following Blood Collection and Transfusion: Annual Summary for Fiscal Year 2014. (2014). Avaialable online at: http://www.fda.gov/downloads/biologicsbloodvaccines/safetyavailability/reportaproblem/transfusiondonationfatalities/ucm459461.pdf (accessed October 22, 2019).
4. Moise KJ Jr., Argoti PS. Management and prevention of red cell alloimmunization in pregnancy: a systematic review. Obstet Gynecol. (2012) 120:1132–9. doi: 10.1097/AOG.0b013e31826d7dc1
5. Nickel RS, Hendrickson JE, Fasano RM, Meyer EK, Winkler AM, Yee MM, et al. Impact of red blood cell alloimmunization on sickle cell disease mortality: a case series. Transfusion. (2016) 56:107–14. doi: 10.1111/trf.13379
6. Telen MJ, Afenyi-Annan A, Garrett ME, Combs MR, Orringer EP, Ashley-Koch AE. Alloimmunization in sickle cell disease: changing antibody specificities and association with chronic pain and decreased survival. Transfusion. (2015) 55(Pt 2):1378–87. doi: 10.1111/trf.12940
7. Ramsey G, Smietana SJ. Multiple or uncommon red cell alloantibodies in women: association with autoimmune disease. Transfusion. (1995) 35:582–6.
8. Karafin MS, Westlake M, Hauser RG, Tormey CA, Norris PJ, Roubinian NH, et al. Risk factors for red blood cell alloimmunization in the recipient epidemiology and donor evaluation study (REDS-III) database. Br J Haematol. (2018) 181:672–81. doi: 10.1111/bjh.15182
9. Papay P, Hackner K, Vogelsang H, Novacek G, Primas C, Reinisch W, et al. High risk of transfusion-induced alloimmunization of patients with inflammatory bowel disease. Am J Med. (2012) 125:717.e1–8. doi: 10.1016/j.amjmed.2011.11.028
10. Ryder AB, Hendrickson JE, Tormey CA. Chronic inflammatory autoimmune disorders are a risk factor for red blood cell alloimmunization. Br J Haematol. (2015) 174:483–5. doi: 10.1111/bjh.13781
11. Ryder AB, Zimring JC, Hendrickson JE. Factors influencing RBC alloimmunization: lessons learned from murine models. Transfus Med Hemother. (2014) 41:406–19. doi: 10.1159/000368995
12. Yazdanbakhsh K, Ware RE, Noizat-Pirenne F. Red blood cell alloimmunization in sickle cell disease: pathophysiology, risk factors, and transfusion management. Blood. (2012) 120:528–37. doi: 10.1182/blood-2011-11-327361
13. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. (1957) 147:258–67. doi: 10.1098/rspb.1957.0048
14. Olsen N, Sokka T, Seehorn CL, Kraft B, Maas K, Moore J, et al. A gene expression signature for recent onset rheumatoid arthritis in peripheral blood mononuclear cells. Ann Rheum Dis. (2004) 63:1387–92. doi: 10.1136/ard.2003.017194
15. Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis. (2011) 70:2029–36. doi: 10.1136/ard.2011.150326
16. Bave U, Nordmark G, Lovgren T, Ronnelid J, Cajander S, Eloranta ML, et al. Activation of the type I interferon system in primary Sjogren’s syndrome: a possible etiopathogenic mechanism. Arthritis Rheum. (2005) 52:1185–95. doi: 10.1002/art.20998
17. Baechler EC, Bauer JW, Slattery CA, Ortmann WA, Espe KJ, Novitzke J, et al. An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol Med. (2007) 13:59–68. doi: 10.2119/2006-00085.Baechler
18. Assassi S, Mayes MD, Arnett FC, Gourh P, Agarwal SK, McNearney TA, et al. Systemic sclerosis and lupus: points in an interferon-mediated continuum. Arthritis Rheum. (2010) 62:589–98. doi: 10.1002/art.27224
19. Han GM, Chen SL, Shen N, Ye S, Bao CD, Gu YY. Analysis of gene expression profiles in human systemic lupus erythematosus using oligonucleotide microarray. Genes Immun. (2003) 4:177–86. doi: 10.1038/sj.gene.6363966
20. Crow MK, Kirou KA, Wohlgemuth J. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity. (2003) 36:481–90. doi: 10.1080/08916930310001625952
21. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. (2003) 197:711–23. doi: 10.1084/jem.20021553
22. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. (2003) 100:2610–5. doi: 10.1073/pnas.0337679100
23. Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. (2005) 52:1491–503. doi: 10.1002/art.21031
24. Bronson PG, Chaivorapol C, Ortmann W, Behrens TW, Graham RR. The genetics of type I interferon in systemic lupus erythematosus. Curr Opin Immunol. (2012) 24:530–7. doi: 10.1016/j.coi.2012.07.008
25. Bao W, Yu J, Heck S, Yazdanbakhsh K. Regulatory T-cell status in red cell alloimmunized responder and nonresponder mice. Blood. (2009) 113:5624–7. doi: 10.1182/blood-2008-12-193748
26. Yu J, Heck S, Yazdanbakhsh K. Prevention of red cell alloimmunization by CD25 regulatory T cells in mouse models. Am J Hematol. (2007) 82:691–6.
27. Hendrickson JE, Desmarets M, Deshpande SS, Chadwick TE, Hillyer CD, Roback JD, et al. Recipient inflammation affects the frequency and magnitude of immunization to transfused red blood cells. Transfusion. (2006) 46:1526–36. doi: 10.1111/j.1537-2995.2006.00946.x
28. Hudson KE, Lin E, Hendrickson JE, Lukacher AE, Zimring JC. Regulation of primary alloantibody response through antecedent exposure to a microbial T-cell epitope. Blood. (2010) 115:3989–96. doi: 10.1182/blood-2009-08-238568
29. Liu D, Gibb DR, Escamilla-Rivera V, Liu J, Santhanakrishnan M, Shi Z, et al. Type 1 IFN signaling critically regulates influenza-induced alloimmunization to transfused KEL RBCs in a murine model. Transfusion. (2019) 59:3243–52. doi: 10.1111/trf.15482
30. Calabro S, Gallman A, Gowthaman U, Liu D, Chen P, Liu J, et al. Bridging channel dendritic cells induce immunity to transfused red blood cells. J Exp Med. (2016) 213:887–96. doi: 10.1084/jem.20151720
31. Gibb DR, Calabro S, Liu D, Tormey CA, Spitalnik SL, Zimring JC, et al. The Nlrp3 inflammasome does not regulate alloimmunization to transfused red blood cells in mice. EBioMedicine. (2016) 9:77–86. doi: 10.1016/j.ebiom.2016.06.008
32. Hendrickson JE, Hod EA, Spitalnik SL, Hillyer CD, Zimring JC. Storage of murine red blood cells enhances alloantibody responses to an erythroid-specific model antigen. Transfusion. (2010) 50:642–8. doi: 10.1111/j.1537-2995.2009.02481.x
33. Arneja A, Salazar JE, Jiang W, Hendrickson JE, Zimring JC, Luckey CJ. Interleukin-6 receptor-alpha signaling drives anti-RBC alloantibody production and T-follicular helper cell differentiation in a murine model of red blood cell alloimmunization. Haematologica. (2016) 101:e440–4. doi: 10.3324/haematol.2016.149278
34. Evers D, van der Bom JG, Tijmensen J, Middelburg RA, de Haas M, Zalpuri S, et al. Red cell alloimmunisation in patients with different types of infections. Br J Haematol. (2016) 175:956–66. doi: 10.1111/bjh.14307
35. Hendrickson JE, Roback JD, Hillyer CD, Easley KA, Zimring JC. Discrete Toll-like receptor agonists have differential effects on alloimmunization to transfused red blood cells. Transfusion. (2008) 48:1869–77. doi: 10.1111/j.1537-2995.2008.01801.x
36. Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. (2006) 25:383–92. doi: 10.1016/j.immuni.2006.08.010
37. Gibb DR, Liu J, Natarajan P, Santhanakrishnan M, Madrid DJ, Eisenbarth SC, et al. Type I IFN is necessary and sufficient for inflammation-induced red blood cell alloimmunization in mice. J Immunol. (2017) 199:1041–50. doi: 10.4049/jimmunol.1700401
38. Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, et al. Functional role of type I and type II interferons in antiviral defense. Science. (1994) 264:1918–21.
39. Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. (2000) 13:539–48.
40. Stowell SR, Girard-Pierce KR, Smith NH, Henry KL, Arthur CM, Zimring JC, et al. Transfusion of murine red blood cells expressing the human KEL glycoprotein induces clinically significant alloantibodies. Transfusion. (2014) 54:179–89. doi: 10.1111/trf.12217
41. Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. (2009) 30:455–64. doi: 10.1016/j.it.2009.06.003
42. Lee PY, Kumagai Y, Li Y, Takeuchi O, Yoshida H, Weinstein J, et al. TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. J Exp Med. (2008) 205:2995–3006. doi: 10.1084/jem.20080462
43. Satoh M, Weintraub JP, Yoshida H, Shaheen VM, Richards HB, Shaw M, et al. Fas and Fas ligand mutations inhibit autoantibody production in pristane-induced lupus. J Immunol. (2000) 165:1036–43. doi: 10.4049/jimmunol.165.2.1036
44. Chowdhary VR, Grande JP, Luthra HS, David CS. Characterization of haemorrhagic pulmonary capillaritis: another manifestation of Pristane-induced lupus. Rheumatology (Oxford). (2007) 46:1405–10. doi: 10.1093/rheumatology/kem117
45. Zhuang H, Han S, Xu Y, Li Y, Wang H, Yang LJ, et al. Toll-like receptor 7-stimulated tumor necrosis factor alpha causes bone marrow damage in systemic lupus erythematosus. Arthritis Rheumatol. (2014) 66:140–51. doi: 10.1002/art.38189
46. Hermida MDR, Malta R, de Santos SM, Dos-Santos WLC. Selecting the right gate to identify relevant cells for your assay: a study of thioglycollate-elicited peritoneal exudate cells in mice. BMC Res Notes. (2017) 10:695. doi: 10.1186/s13104-017-3019-5
47. York MR, Nagai T, Mangini AJ, Lemaire R, van Seventer JM, Lafyatis R. A macrophage marker, Siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and toll-like receptor agonists. Arthritis Rheum. (2007) 56:1010–20. doi: 10.1002/art.22382
48. Lee PY, Weinstein JS, Nacionales DC, Scumpia PO, Li Y, Butfiloski E, et al. A novel type I IFN-producing cell subset in murine lupus. J Immunol. (2008) 180:5101–8. doi: 10.4049/jimmunol.180.7.5101
49. Crocker PR, Gordon S. Mouse macrophage hemagglutinin (sheep erythrocyte receptor) with specificity for sialylated glycoconjugates characterized by a monoclonal antibody. J Exp Med. (1989) 169:1333–46. doi: 10.1084/jem.169.4.1333
50. Martinez-Pomares L, Gordon S. CD169+ macrophages at the crossroads of antigen presentation. Trends Immunol. (2012) 33:66–70. doi: 10.1016/j.it.2011.11.001
51. Coro ES, Chang WL, Baumgarth N. Type I IFN receptor signals directly stimulate local B cells early following influenza virus infection. J Immunol. (2006) 176:4343–51. doi: 10.4049/jimmunol.176.7.4343
52. Gibb DR, Liu J, Santhanakrishnan M, Natarajan P, Madrid DJ, Patel S, et al. B cells require Type 1 interferon to produce alloantibodies to transfused KEL-expressing red blood cells in mice. Transfusion. (2017) 57:2595–608. doi: 10.1111/trf.14288
53. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
Keywords: RBC alloimmunization, type 1 interferons, lupus, transfusion, autoimmunity
Citation: Lee JY, Madany E, El Kadi N, Pandya S, Ng K, Yamashita M, Jefferies CA and Gibb DR (2020) Type 1 Interferon Gene Signature Promotes RBC Alloimmunization in a Lupus Mouse Model. Front. Immunol. 11:584254. doi: 10.3389/fimmu.2020.584254
Received: 16 July 2020; Accepted: 03 September 2020;
Published: 25 September 2020.
Edited by:
Geraldo Aleixo Passos, University of São Paulo, BrazilReviewed by:
Steven L. Spitalnik, Columbia University, United StatesKrystalyn E. Hudson, Bloodworks Northwest Research Institute, United States
Seema Patel, Emory University, United States
Copyright © 2020 Lee, Madany, El Kadi, Pandya, Ng, Yamashita, Jefferies and Gibb. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David R. Gibb, ZGF2aWQuZ2liYkBjc2hzLm9yZw==
†These authors have contributed equally to this work