 Susan Hartzell1
Susan Hartzell1 Sofia Bin1
Sofia Bin1 Chiara Cantarelli2
Chiara Cantarelli2 Meredith Haverly1Joaquin Manrique3Andrea Angeletti4
Meredith Haverly1Joaquin Manrique3Andrea Angeletti4 Gaetano La Manna5Barbara Murphy1
Gaetano La Manna5Barbara Murphy1 Weijia Zhang1Josh Levitsky6Lorenzo Gallon7
Weijia Zhang1Josh Levitsky6Lorenzo Gallon7 Samuel Mon-Wei Yu1
Samuel Mon-Wei Yu1 Paolo Cravedi1*
Paolo Cravedi1*- 1Division of Nephrology, Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 2UO Nefrologia, Azienda Ospedaliera-Universitaria di Parma, Parma, Italy
- 3Nephrology Service, Complejo Hospitalario de Navarra, Pamplona, Spain
- 4Division of Nephrology, Dialysis, Transplantation, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Giannina Gaslini, Genoa, Italy
- 5Department of Experimental Diagnostic and Specialty Medicine, University of Bologna Sant'Orsola- Malpighi Hospital, Bologna, Italy
- 6Division of Gastroenterology, Department of Medicine, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 7Division of Nephrology, Department of Medicine, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
Individuals with kidney failure are at increased risk of cardiovascular events, as well as infections and malignancies, but the associated immunological abnormalities are unclear. We hypothesized that the uremic milieu triggers a chronic inflammatory state that, while accelerating atherosclerosis, promotes T cell exhaustion, impairing effective clearance of pathogens and tumor cells. Clinical and demographic data were collected from 78 patients with chronic kidney disease (CKD) (n = 42) or end-stage kidney disease (ESKD) (n = 36) and from 18 healthy controls (HC). Serum cytokines were analyzed by Luminex. Immunophenotype of T cells was performed by flow cytometry on peripheral blood mononuclear cells. ESKD patients had significantly higher serum levels of IFN-γ, TNF-α, sCD40L, GM-CSF, IL-4, IL-8, MCP-1, and MIP-1β than CKD and HC. After mitogen stimulation, both CD4+ and CD8+ T cells in ESKD group demonstrated a pro-inflammatory phenotype with increased IFN-γ and TNF-α, whereas both CKD and ESKD patients had higher IL-2 levels. CKD and ESKD were associated with increased frequency of exhausted CD4+ T cells (CD4+KLRG1+PD1+CD57−) and CD8+ T cells (CD8+KLRG1+PD1+CD57−), as well as anergic CD4+ T cells (CD4+KLRG1−PD1+CD57−) and CD8+ T cells (CD8+KLRG1−PD1+CD57−). Although total percentage of follicular helper T cell (TFH) was similar amongst groups, ESKD had reduced frequency of TFH1 (CCR6−CXCR3+CXCR5+PD1+CD4+CD8−), but increased TFH2 (CCR6−CXCR3−CXCR5+PD1+CD4+CD8−), and plasmablasts (CD3−CD56−CD19+CD27highCD38highCD138−). In conclusion, kidney failure is associated with pro-inflammatory markers, exhausted T cell phenotype, and upregulated TFH2, especially in ESKD. These immunological changes may account, at least in part, for the increased cardiovascular risk in these patients and their susceptibility to infections and malignancies.
Introduction
Despite significant improved survival in patients with chronic kidney disease (CKD) and end-stage kidney disease (ESKD) in the past decades, the all-cause mortality among these patients has remained exceedingly higher when compared to general population (1, 2). In particular, cardiovascular diseases, infections, and malignancies represent the leading causes of the mortality independent of concomitant comorbidities, such as diabetes or hypertension, suggesting a unique and systemic pathobiology in kidney failure (3–5).
In response to pathogen exposure or tissue damage, the body generates physiological and necessary inflammation to restore homeostasis. However, prolonged inflammatory signals alter T cell functions, leading to a state of exhaustion (6). Exhausted T cells, both CD4+ and CD8+, are characterized by the loss of their effector functions and the expression of inhibitory receptors such as PD-1 and KLRG1. Co-expression of these inhibitory receptors can be of additive effects (6). Functionally, exhausted T cells have impaired cytokine production upon stimulation, but retain some cytotoxicity and degranulation capability (7). Exhausted T cells might be reversed back to fully functional T cells; however, these functional changes become irreversible if stimulus such as inflammation sustains, and eventually lead to cell death (6, 8). Clinically, T cell exhaustion has been associated with unresolved infection and drug-resistant tumors, and emerging immunotherapy such as anti-PD-1 was designed to reverse functional deficits of exhausted T cells (7, 9). In addition, a single-cell analysis also suggested the existence of T cell exhaustion in the development of atherosclerosis (10). Therefore, it is possible that significantly increased risks of cardiovascular death, infection, and malignancy in kidney failure are mediated by exhausted T cells, as a markedly increased T cell exhaustion was seen in a pediatric CKD cohort (11). Follicular helper T cells (TFH), a subset of CD4+ T cells expressing CXC-chemokine receptor 5 (CXCR5), was initially discovered in the germinal centers (GC) of human tonsillar tissues (12). To produce high affinity antibodies against pathogens, B cell requires the assistance from TFH to complete its maturation and class switch recombination inside the GC. Work related to TFH in the kidneys has largely focused on kidney transplantations, and upregulated TFH has been implicated in less favorable outcomes especially due to increased humoral rejections (13). On the other hand, TFH are also linked to atherosclerosis, autoimmune diseases and some lymphoid tumors, suggesting their potential pathogenic role in promoting chronic inflammation (12, 14).
To date, data on T cell exhaustion or TFH in adult CKD and ESKD remains scarce and incomplete. We hypothesized that an association between impaired renal function and exhausted T cell as well as follicular helper T cells, might explain the increased risk of infection, cancer, and cardiovascular disease among CKD and ESKD patients (15–20), and performed a comprehensive immunophenotyping on T cells.
Materials and Methods
Study Population
We included 42 CKD patients (stage 1–5 or with proteinuria) seen at Complejo Hospitalario de Navarra, Pamplona, Spain (Biobank Navarrabiomed; n = 32), or S. Orsola-Malpighi Hospital, University of Bologna, Bologna, Italy (n = 10), 36 ESKD patients who received hemodialysis for > 3 months at the Mount Sinai Kidney Center (n = 21), Northwestern Hospital in Chicago (n = 13), or Complejo Hospitalario de Navarra, Pamplona, Spain (Biobank Navarrabiomed; n = 2), and 18 age-matched healthy controls (HC). All individuals were enrolled from November 2017 to December 2019 (Table 1). Exclusion criteria were pregnancy, recent (<3 months) infectious episode requiring hospitalization, history of kidney transplant, use of immunosuppressive medications at the time of enrollment, inability to give consent, active malignancy. Samples and data from patients included in this study were processed following standard operating procedures with the appropriate approval of the Ethics and Scientific Committees of the participating centers.
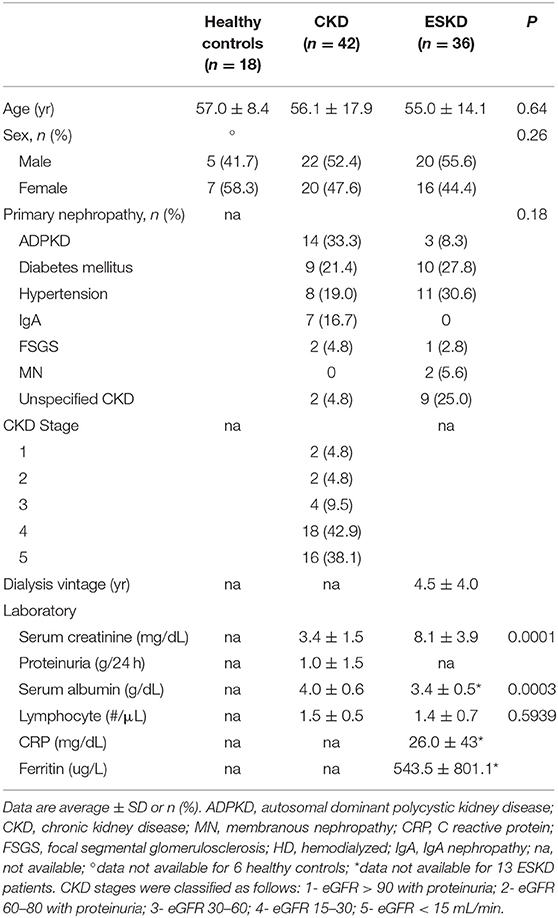
Table 1. Patients’ characteristics.
Luminex
We quantified plasma levels of CD40L, G-CSF/CSF-3, GM-CSF, IFN-γ, IL-1β, IL-10, IL-12p70, IL-13, IL-17A, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, MCP-1, MIP-1β, TNF-α, and TNF-β by Luminex® (xMAP® Technology). We created a multiplex panel combining two commercially available simplex kits: Human Custom Procarta Plex-19 plex (Invitrogen by Thermo Fisher Scientific, Cat. No. PPX-19-MXRWE2G) and Human High Sensitivity T Cell (Merck Millipore, Cat. No. HSTCMAG-28SK). The methodological details including assay protocol, standards and sensitivity are available at the manufacturer's website (http://www.thermofisher.com and http://www.merckmillipore.com, respectively). All samples were measured undiluted and in duplicates. The chemo/cytokine standards were assayed in the same way as patient samples. The data were collected using a xPONENT® software (Luminex, Austin, TX, USA).
Flow Cytometry Analysis
PBMC were isolated from peripheral blood by Ficoll gradient and frozen for batched analysis. We designed six multicolor flow cytometry panels to quantify 60 T cell subsets along with two B cell subsets, and calculated the CD4+/CD8+ T cell ratio (Supplementary Table 1). The following fluorochrome-conjugated anti-human antibodies were used from BD Biosciences (San Jose, CA): CD3-FITC, CD3-PerCP-Cy5.5, CD4-APC, CD4-APC-Cy7, CD8-BV510, CD45RO-FITC, CD45RA-APC, CD45RA-APC-Cy7, CCR4-PE, CD27-PE, CD28-BV421, CD138-BV421, CCR6-BV421, CXCR3-PE, CCR7-A700, IL-17-BV786, IFN-γ-PE-Cy7, iso IgG1k-FITC, iso IgG1k-PE-Cy7, iso IgG2bk-APC, iso IgG1k-APC-Cy7, and iso IgG1k-BV510; from Biolegend (San Diego, CA): CD127-FITC, CD27-APC, CD57-PerCp-Cy5.5, CD19-BV510, PD-1-APC-Cy7, CXCR5-FITC, and TNFα-FITC; from eBioscience (San Diego, CA): CD4-PE-Cy7, and IL-2-PE; from Miltenyi Biotec (San Diego, CA): CD25-APC and KLRG1-PE; from Beckman Coulter (Brea, CA): CD38-PE-Cy7.
Intracellular Cytokine Staining
We performed intracellular staining for IL-17, IL-2, IFN-γ, and TNF-α with extracellular markers for CD4+ and CD8+ after 5 h stimulation by mixture of PMA (Fisher, Waltham, MA), ionomycin (Fisher) and Golgi Plug (Millipore Sigma, Burlington, MA) at 37°C. We fixed and permeabilized the cells using Intracellular Fixation and Permeabilization Buffer Set (eBioscience), according to the manufacturer's instructions. Data were acquired (> 1 × 106 events) on a 3-laser FACSLyric flow cytometer (BD Biosciences) and analyzed with FlowJo® software. Intracellular cytokines were recorded as percentages of CD4+ and CD8+ T cells.
Statistical Analyses
Results were presented as mean and standard deviation or standard error unless stated otherwise. Comparison of continuous variables between groups was performed by Kruskal–Wallis test, unpaired t-test, and categorical variables by two-sided chi-square, where applicable. P < 0.05 was considered as statistically significant. No correction was made for multiple testing. Statistical analysis was performed using GraphpadPrism® version 8.4.2 software package (Graphpad Software Inc., San Diego, CA).
Results
Patients
Patient characteristics are presented in Table 1. The overall age of our cohort was 57.4 ± 15.7 years with no difference among the groups (57.0 ± 8.4 vs. 56.1 ± 17.9 vs. 55.0 ± 14.1 years for HC, CKD, and ESKD, respectively, p = 0.64). In CKD cohort, the most common cause of CKD was autosomal-dominant polycystic kidney disease (ADPKD) (n = 14, 33.3%), followed by diabetic kidney disease (DKD) (n = 9, 21.4%), hypertension (n = 8, 19.0%), IgA nephropathy (n = 7, 16.7%), focal segmental glomerulosclerosis (FSGS) (n = 2, 4.8%), and unspecified CKD (n = 2, 4.8%). In ESKD cohort, the most common cause of ESKD was hypertension (n = 11, 30.6%), followed by DKD (n = 10, 27.8%), unspecified CKD (n = 9, 25.0%), ADPKD (n = 3, 8.3%), membranous nephropathy (MN) (n = 2, 5.6%), and FSGS (n = 1, 2.8%).
Serum Cytokines
To start testing the inflammatory status of patients with kidney failure, we measured inflammatory cytokines: soluble CD40 ligand (sCD40-L), granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), interferon-gamma (IFN-γ), interleukin-1β (IL-1β), IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12p70, IL-13, IL-17A, macrophage inflammatory protein 1-beta (MIP-1β), monocyte chemoattractant protein-1 (MCP-1), tissue necrosis factor-alpha (TNF-α), and tissue necrosis factor-beta (TNF-β) in three study cohorts. Most cytokines were undetectable or extremely low in HC and CKD patients, while most ESKD patients had detectable or high levels. In particular, ESKD patients had signifcantly higher levels of IFN-γ, TNF-α, sCD40L, GM-CSF, IL-4, IL-8, MCP-1, and MIP-1β than CKD patients (Figure 1; Supplementary Figure 1). Altogether, these data confirm and extend previous evidence that kidney failure is associated with a pro-inflammatory state (15, 16).
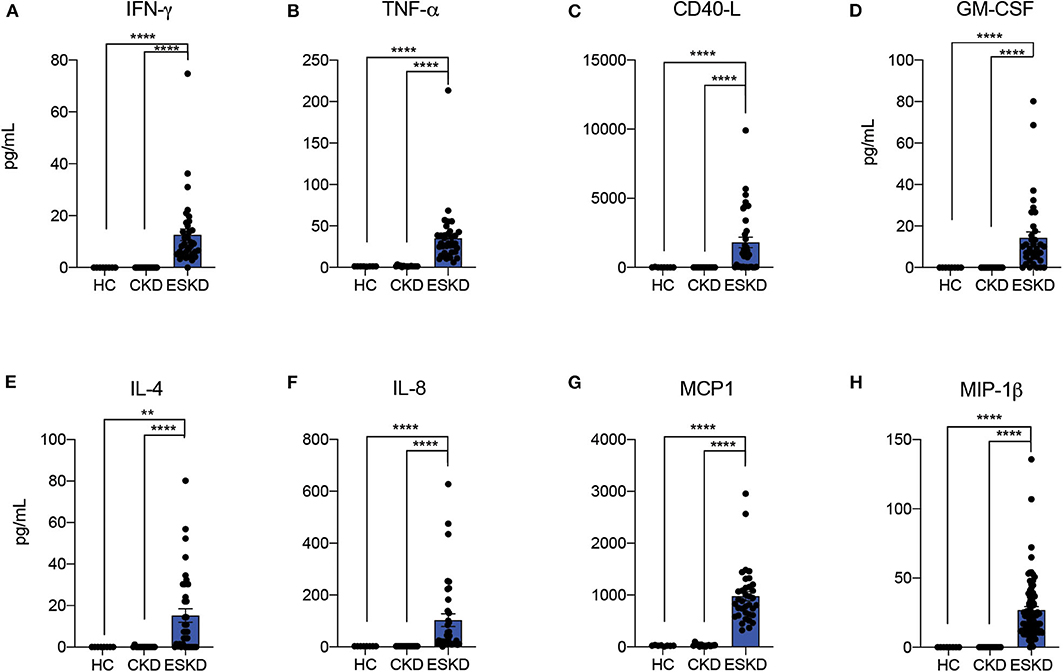
Figure 1. Serum cytokine analysis by Luminex. Data are represented as mean and standard error of the mean (SEM). Each dot represents an individual value. **P < 0.01; ****P < 0.0001. Kruskal–Wallis test.
Naïve, Effector, and Memory CD4+ T Cells
Consistent with prior reports (17–20), we observed fewer CD3+ T cells in CKD (39.5 ± 13.8%) and ESKD (30.3 ± 12.8%) when compared to HC (48.4 ± 10.1%) (Figure 2A). In total T cells, there was no difference in the percentage of total CD4+ T cells among the three groups. Between CKD and ESKD, ESKD group had a significantly lower percentage of total CD8+ T cells with a higher CD4+/CD8+ ratio (Figures 2B–D). Absolute numbers of CD4+ and CD8+ T cells of CKD and ESKD groups demonstrated similar findings to the percentage difference (Supplementary Figure 2), although we did not have absolute numbers in healthy controls during the collection process.
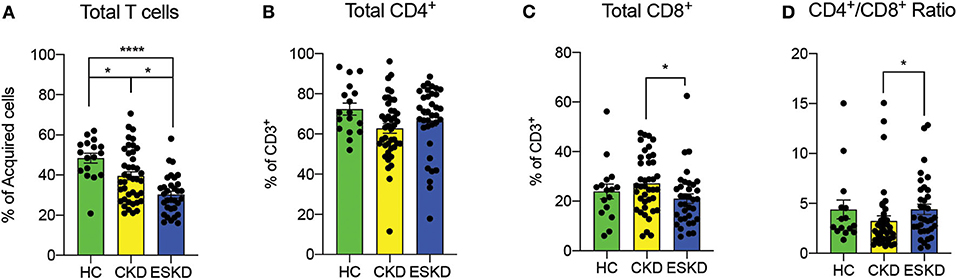
Figure 2. Total T cells, CD4+ and CD8+ cells and CD4+/CD8+ ratio in healthy control, CKD and ESKD patients. (A) CD3+ percentage of acquired cells; (B,C) CD4+ and CD8+ cells percentage of CD3+ T cells, and (D) CD4+/CD8+ ratio. Data are represented as mean and standard error of the mean (SEM). Each dot represents an individual value. *P < 0.05; ****P < 0.0001. Kruskal–Wallis test.
CKD patients had the lowest frequency of naïve CD4+ T cells (CD45RA+CD45RO−CD27+CD28+), while ESKD patients had similar levels to HC (Figure 3A). Both frequencies of effector CD4+ T cells (CD45RA−CD45RO+CD27−CD28−) and memory CD4+ T cells (CD45RA−CD45RO+CD27+CD28+) were remarkably reduced in hemodialysis patients (Figures 3B,C). We next examined the intracellular levels of IFN-γ, IL-2, and TNF-α in CD4+ T cells, as a proxy of their function. We found that CD4+ T cells from CKD or ESKD patients demonstrated a more pro-inflammatory phenotype with increasing IFN-γ, IL-2, and TNF-α after mitogen stimulation, while IL-17 did not significantly differ across the three groups (Figures 3D–G).
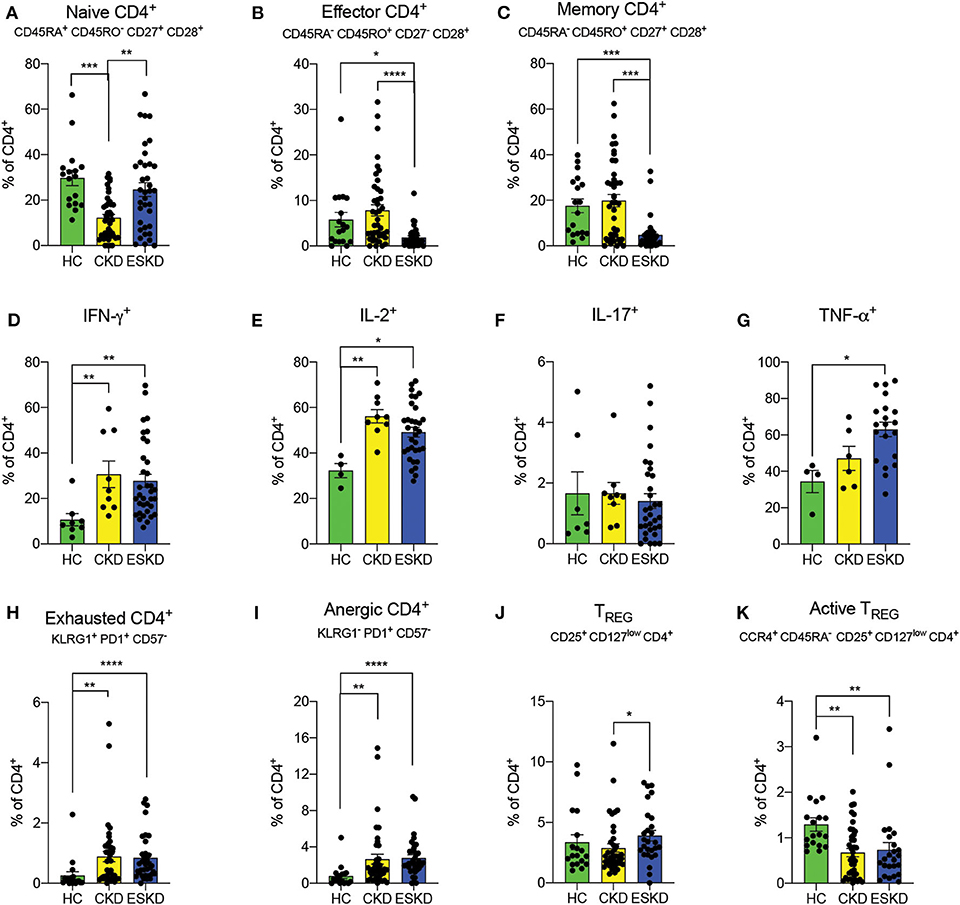
Figure 3. CD4+ T cell subsets in the three study groups. (A–C) Naïve, effector and memory CD4+ T cells. (D–G) IFN- γ, IL-2, IL-17, and TNF-α staining on CD4+ T cells. (H,I) Exhausted and anergic CD4+ T cells. (J,K) Total and active Treg. Data are represented as mean and standard error of the mean (SEM). Each dot represents an individual value. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.000.1. Kruskal–Wallis test. Gating strategies are shown in Supplementary Figures 3, 4.
Dysfunctional and Regulatory CD4+ T Cells
Chronic inflammation has been associated with the emergence of T cell dysfunction (6). To test the hypothesis that a similar phenomenon occurs in patients with kidney failure, we measured phenotypically exhausted and anergic T cells. We found a significant increase of exhausted CD4+ (KLRG1+PD1+CD57−) and anergic T cells (KLRG1−PD1+CD57−) in both CKD and ESKD patients. (Figures 3H,I). Importantly, exhausted CD4+ T cells showed no cytokine production (Supplementary Figure 4).
Previous reports showed reduced levels of regulatory T cells (Treg) (21), a cell subset of CD4+ T cells essential for immune tolerance and eliminating chronic inflammation (22). Herein, we found a markedly decreased frequency of active Treg cells (CCR4+CD45RA−CD25+CD127low−CD4+CD8−CD3+) in both CKD and ESKD groups, whereas the percentage of total Treg (CD25+CD127lowCD4+CD8−CD3+) in ESKD group was statistically higher than CKD but of no difference compared to HC (Figures 3J,K).
Naïve, Memory, Effector, and Dysfunctional CD8+ T Cells
Similar to CD4+ T cells, we observed a decreased frequency of naïve CD8+ T cells in CKD patients, while that of ESKD patients was close to HC (HC: 19.2 ± 11.1% vs. CKD: 10.8 ± 9.1% vs. ESKD: 14.83 ± 11.0%) (Figure 4A). ESKD patients had the lowest effector and memory CD8+ T cells among the groups (Figures 4B,C). In intracellular cytokine analyses, IFN-γ and TNF-α were significantly elevated after mitogen stimulation in ESKD. We also noticed a non-significant trend toward higher IL-2 levels in CKD and ESKD groups, but no difference in IL-17 across the groups (Figures 4D–G). Similar to CD4+ T cells, we found significantly more exhausted and anergic CD8+ T cells in ESKD patients (Figures 4H,I).
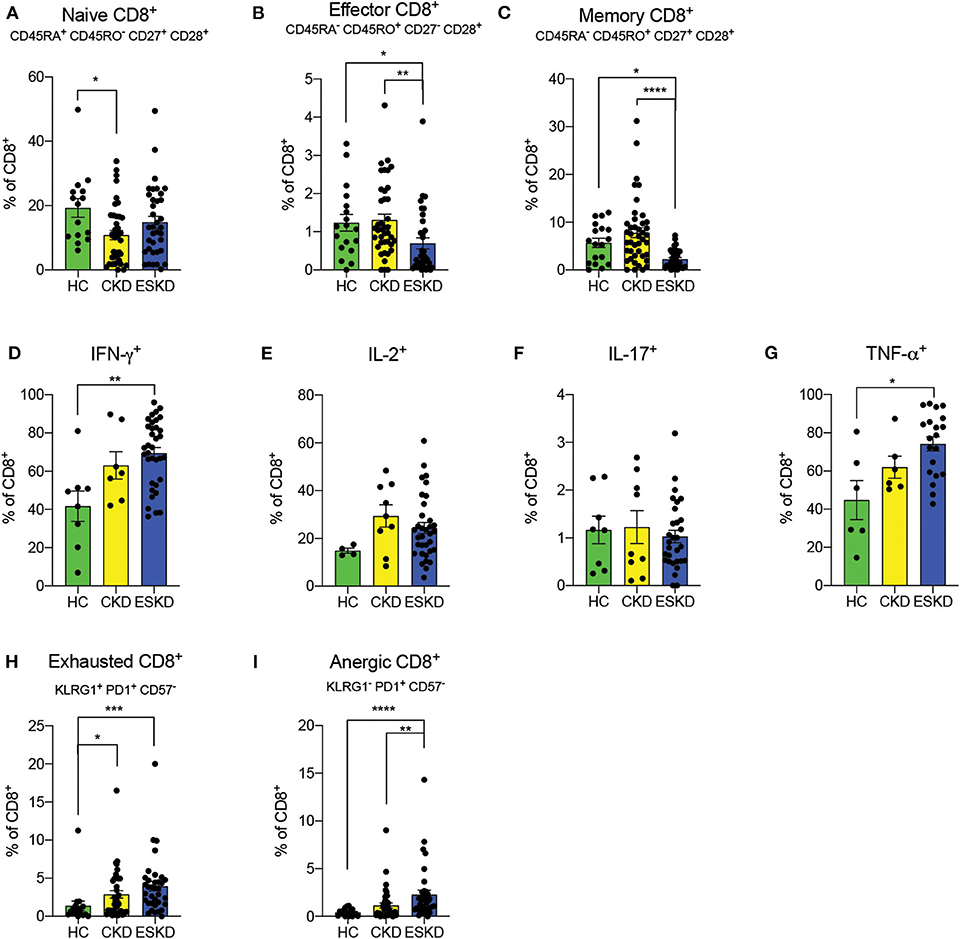
Figure 4. CD8+ T cell subsets in the three study groups. (A–C) Naïve, effector, and memory CD8+ T cells. (D–G) IFN-γ, IL-2, IL-17, and TNF-α staining on CD8+ T cells. (H,I) Exhausted and anergic CD8+ T cells. Data are represented as mean and standard error of the mean (SEM). Each dot represents an individual value. *P < 0.05; ****P < 0.000.1. Kruskal–Wallis test. Gating strategy is shown in Supplementary Figure 3.
Follicular Helper T Cells
We did not observe differences in total TFH (CXCR5+PD1+CD4+CD8−) amongst our cohorts (Figure 5A). ESKD patients had a remarkably lower frequency of TFH1 (CCR6−CXCR3+CXCR5+PD1+CD4+CD8−) cells but more TFH2 (CCR6−CXCR3−CXCR5+PD1+CD4+CD8−) and TFH17 cells (CCR6+CXCR3−CXCR5+PD1+CD4+CD8−) cells (albeit not significant; Figures 5B–D) among three cohorts. TFH1 has a pivotal role in response to viral infections (23, 24). By contrast, TFH2 have been suggested to provide help to B cells within GC, where they convert into plasma blasts and plasma cells and subsequently antibody secreting cells (25). Intriguingly, we also found a significant increase of circulating plasmablasts in ESKD group (Figure 5E) but not plasma cells (Figure 5F), suggestive of a partially promoted B cells maturation process.
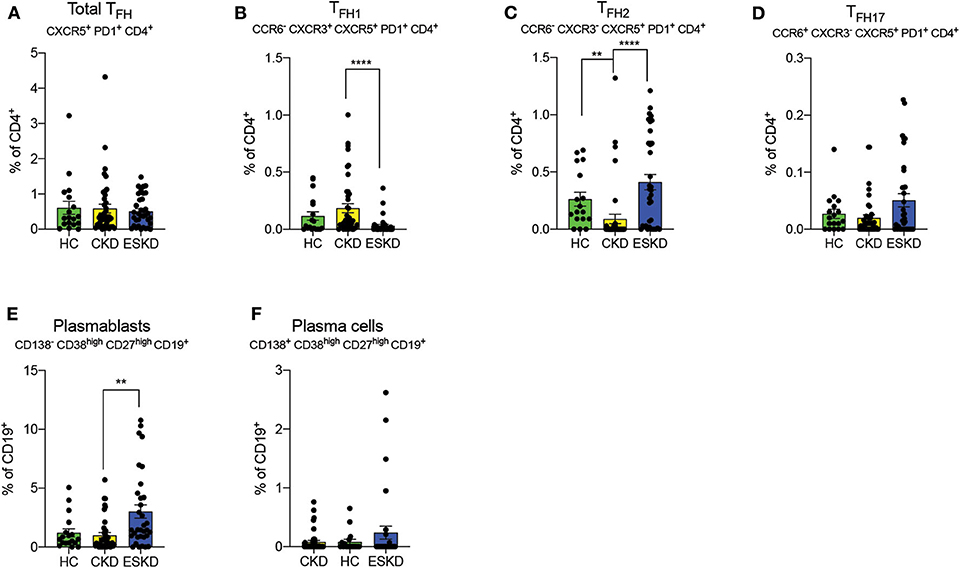
Figure 5. Follicular helper T cell subsets, plasmablasts, and plasma cells. (A–D) Total, TFH1, TFH2 and TFH17. (E,F) Plasmablasts and plasma cells on CD19+ B cells. Each dot represents an individual value. *P < 0.05; **P < 0.01; ****P < 0.000.1. Kruskal–Wallis test.
Discussion
Our study shows that kidney failure is associated with a pro-inflammatory state, characterized by increased T cell production of pro-inflammatory cytokines and high circulating levels of TFH2 and plasmablasts. These alterations may lead to a progressive dysfunction of immune cells, as supported by increased exhausted CD4+ and CD8+ T cells. Altogether, these findings were suggestive of persistent and active pro-inflammatory T cells in both CKD and ESKD patients.
Chronic inflammation can be triggered by reciprocal activation of innate and adaptive immunity (26). In our serum cytokine analysis, we identified a significant pro-inflammatory phenotype characterized by upregulated sCD40L, GM-CSF, IL-4, IL-8, MCP-1, and MIP-1β in the ESKD cohort, apart from the prototypical cytokines such as IFN-γ and TNF-α. CD40L is a costimulatory molecule predominantly expressed on activated CD4+ T cells. Activation of CD40-CD40L axis leads to activation of B cells and cytokine secretion, which in turn activate T cells as positive feedback (27). Increased sCD40L has been associated with chronic HIV infection (28), cancers (29), and pulmonary arterial hypertension (30), and could potentially serve as a biomarker of disease activities. Intriguingly, upregulated circulating sCD40L also enhanced expression and release of sCD40L in platelets (31), supporting the clinical observation of hypercoagulability in ESKD patients. IL-4 is produced by follicular helper T cells and has been shown to regulate B cell maturation in GC (32), which is consistent with our findings in upregulated TFH2 and increased plasmablasts. In addition, IL-4 (33) and MIP-1β (34) are known to polarize macrophage into M1 macrophage, a subtype of macrophage robustly producing cytokines and chemokines such as IL-1β and TNF-α. Although we did not include macrophage into our analysis, a prior review (35) has delineated the pro-inflammatory role of macrophages in kidney failure patients. Of note, the conventional intermittent hemodialysis membranes do not remove middle molecules (500 Da-15 kDa), and therefore the higher levels of pro-inflammatory cytokines were not due to poor clearance compared with others of no statistical significance. Thus, our results support a chronically activated inflammatory state in ESKD.
Regarding T cells, first we noted a similar finding of worsening lymphopenia in patients with CKD and ESKD. However, we did not observe a decreased or inverted ratio of CD4+/CD8+ T cells as previously reported by others (11, 19). This discrepancy might be explained by the unknown status of CMV infection of our patients, as Crépin et al. (18) reported that inverted ratio was only found in CMV-seropositive patients in their cohort. Lymphopenia in CKD/ESKD patients, especially naïve CD4+ and CD8+ T cells and T central memory, was previously attributed to decreased thymic output, increased apoptosis or shortening of telomere length (36). We also observed reduced naïve CD4+ and CD8+ T cells in CKD but not in ESKD cohort. Of note, we strictly defined naïve T cells as CD45RA+CD45RO−CD27+CD28+ in our study rather than CD45RO−CCR7+, which might represent a different population when compared to others (37). It is unclear whether the mobilization of naïve T cells in our ESKD group was mediated by intermittent removal of uremic toxins by hemodialysis or other factors.
Our data demonstrate an association between ESKD and reduced effector CD4+ and CD8+ T cells. Whether this reduction is due to recruitment of these cells to the peripheral tissues or reflects an absolute decline goes beyond the scope of the current study. However, after mitogen stimulation, T cells from ESKD patients demonstrated a pro-inflammatory phenotype with an increase in IFN-γ, IL-2 and TNF-α, whereas CKD patients only had a higher level of IL-2. Altogether, these findings were suggestive of persistent and active pro-inflammatory T cells especially in ESKD patients.
Intriguingly, we found that both CKD and ESKD patients had higher frequencies of exhausted and anergic CD4+ and CD8+ T cells. T cell receptor (TCR) signaling is essential for T cell differentiation into effector cells as well as appropriate clonal expansion (38) and defective TCR signaling have been described in elderly individuals (39) and chronic inflammatory diseases (40). In our study, phenotypically exhausted T cells were not capable of effectively producing cytokines. Therefore, it is tempting to speculate that, upon chronic stimulation (possibly driven by uremic toxins, amongst other stimuli), T cells become dysfunctional, leading to increased risk of infections and malignancies (1).
Despite the inconsistent results of Treg frequency in ESKD patients when compared to HC (17, 21, 41, 42), impaired ex vivo Treg proliferation and suppressive capacity has been shown in patients with ESKD (21). Treg counteracts effector T cells to promote self-tolerance and reduce alloimmunogenicity. Forkhead box P3 (FOXP3), a pivotal transcription factor in Treg homeostasis, is degraded in chronic inflammation by pro-inflammatory cytokines such as TNF-α (43). Thus, downregulated Treg can result in excessive inflammatory responses and aberrant autoimmune diseases. In our cohort, we observed a slight increase in the total frequency of Treg in ESKD patients but a significant decrease of active Treg (CCR4+CD45RA−CD25+CD127low−CD4+CD8−CD3+) in both CKD and ESKD patients. This supports the hypothesis that the chronic inflammatory milieu in ESKD impairs Treg function, instead of affecting their number. A similar finding of upregulation of total Treg was considered to be a reactive response in a cohort ANCA-associated vasculitis (AAV) patients (44).
Upregulation of TFH cells has been associated with chronic inflammation in cardiovascular (45, 46) and autoimmune diseases (47). In our cohort, we observed TFH1, a possibly protective subtype against viral infection, clearly diminished in ESKD. On the contrary, the presence of pro-inflammatory TFH2 and TFH17 was associated with systemic lupus erythematous (SLE) (48) and rheumatoid arthritis (49), respectively, and TFH2 was remarkably increased in our ESKD cohort. To date, there was only one small cohort of ESKD patients, where Assing et al. (50) reported a decreased frequency of peripheral TFH from apoptosis. The authors also suggested the reduced TFH could explain antibody deficiency in ESKD from a disturbed interaction between TFH and B cells in GC. However, the authors did not further classify the subtypes of TFH in their analysis. Indeed, B cells lymphopenia has been shown in patients with CKD or ESKD due to B cell apoptosis and poor B cell differentiation (51, 52), but the mechanism remains poorly understood especially in the context of TFH and B cell interactions. In line with TFH findings, we also observed a higher percentage of circulating plasmablasts, suggesting an increase of B cell maturation within GC. Surprisingly, the upregulated plasmablasts did not result in a similar increase of plasma cells in ESKD patients. After entering circulation from secondary lymphoid tissue, plasmablasts further differentiate into plasma cells, the predominant antibody-secreting cells, and later transition into long-lived plasma cells. This process requires necessary survival factors and environment (bone marrow or spleen) to maintain circulating plasma cells in the periphery (53). For instance, deletion of B cell maturation antigen (BCMA), a B-cell activating factor (BAFF) receptor, was shown to affect the survival of long-lived plasma cells in a murine model (54). Downregulated BAFF receptor on transitional B cells has been implicated in deficient B cell differentiation in ESKD patients (51) but whether it also occurs in plasma cells requires further studies.
Our study has certain limitations. The numbers of our patients were relatively small and the cross-sectional nature of the study prevents testing the association between immunological abnormalities and cardiovascular outcomes, incidence of malignancies, or infectious complications. We did not have information of CMV infection status of our cohort, which could partly contribute to T cell exhaustion. In conclusion, we showed that kidney failure is associated with a pro-inflammatory state that may be responsible for the exhausted phenotype of T cells and the skewed upregulation of TFH2, especially in ESKD. These immunological changes may be, at least in part, responsible for their increased cardiovascular risk and the augmented susceptibility to infections and malignancies.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by IRB-20-03454, Mount Sinai Hospital. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
PC designed the study. SH, SB, CC, and MH performed all the experiments. JM, AA, BM, WZ, and LG provided patient samples. SH, JL, SM-WY, GLM, and PC helped with data interpretation and wrote the initial draft of the paper. All authors approved the paper.
Funding
This study has been supported by an investigator-initiated grant from the Renal Research Institute. We want to particularly acknowledge the patients and the Biobank Navarrabiomed (PT17/0015/0007) integrated in the Spanish National Biobanks Network for their collaboration.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.583702/full#supplementary-material
References
1. United States Renal Data System. USRDS annual data report: Epidemiology of kidney disease in the United States. (2019). Available online at: https://www.usrds.org/2019/view/USRDS_2019_ES_final.pdf (accessed August 10, 2020).
2. Foster BJ, Mitsnefes MM, Dahhou M, Zhang X, Laskin BL. Changes in excess mortality from end stage renal disease in the United States from 1995 to 2013. Clin J Am Soc Nephrol. (2018) 13:91–9. doi: 10.2215/CJN.04330417
3. Cozzolino M, Mangano M, Stucchi A, Ciceri P, Conte F, Galassi A. Cardiovascular disease in dialysis patients. Nephrol Dial Transplant. (2018) 33(Suppl. 3):iii28–34. doi: 10.1093/ndt/gfy174
4. Wong G, Hayen A, Chapman JR, Webster AC, Wang JJ, Mitchell P, et al. Association of CKD and cancer risk in older people. J Am Soc Nephrol. (2009) 20:1341–50. doi: 10.1681/ASN.2008090998
5. Lowrance WT, Ordonez J, Udaltsova N, Russo P, Go AS. CKD and the risk of incident cancer. J Am Soc Nephrol. (2014) 25:2327–34. doi: 10.1681/ASN.2013060604
6. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
7. Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology. (2010) 129:474–81. doi: 10.1111/j.1365-2567.2010.03255.x
8. Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining 'T cell exhaustion'. Nat Rev Immunol. (2019) 19:665–74. doi: 10.1038/s41577-019-0221-9
9. Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. (2015) 6:e1792. doi: 10.1038/cddis.2015.162
10. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
11. George RP, Mehta AK, Perez SD, Winterberg P, Cheeseman J, Johnson B, et al. Premature T cell senescence in pediatric CKD. J Am Soc Nephrol. (2017) 28:359–67. doi: 10.1681/ASN.2016010053
12. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol. (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
13. Macedo C, Hadi K, Walters J, Elinoff B, Marrari M, Zeevi A, et al. Impact of induction therapy on circulating T follicular helper cells and subsequent donor-specific antibody formation after kidney transplant. Kidney Int Rep. (2019) 4:455–69. doi: 10.1016/j.ekir.2018.11.020
14. Crotty S. T follicular helper cell biology: a decade of discovery and diseases. Immunity. (2019) 50:1132–48. doi: 10.1016/j.immuni.2019.04.011
15. Perlman AS, Chevalier JM, Wilkinson P, Liu H, Parker T, Levine DM, et al. Serum inflammatory and immune mediators are elevated in early stage diabetic nephropathy. Ann Clin Lab Sci. (2015) 45:256–63.
16. Yang J, Fang P, Yu D, Zhang L, Zhang D, Jiang X, et al. Chronic kidney disease induces inflammatory CD40+ monocyte differentiation via homocysteine elevation and DNA hypomethylation. Circul Res. (2016) 119:1226–41. doi: 10.1161/CIRCRESAHA.116.308750
17. Freitas GRR, da Luz Fernandes M, Agena F, Jaluul O, Silva SC, Lemos FBC, et al. Aging and end stage renal disease cause a decrease in absolute circulating lymphocyte counts with a shift to a memory profile and diverge in Treg population. Aging Dis. (2019) 10:49–61. doi: 10.14336/AD.2018.0318
18. Crepin T, Legendre M, Carron C, Vachey C, Courivaud C, Rebibou JM, et al. Uraemia-induced immune senescence and clinical outcomes in chronic kidney disease patients. Nephrol Dial Transplant. (2020) 35:624–32. doi: 10.1093/ndt/gfy276
19. Yoon JW, Gollapudi S, Pahl MV, Vaziri ND. Naive and central memory T-cell lymphopenia in end-stage renal disease. Kidney Int. (2006) 70:371–6. doi: 10.1038/sj.ki.5001550
20. Betjes MG, Langerak AW, van der Spek A, de Wit EA, Litjens NH. Premature aging of circulating T cells in patients with end-stage renal disease. Kidney Int. (2011) 80:208–17. doi: 10.1038/ki.2011.110
21. Hendrikx TK, van Gurp EA, Mol WM, Schoordijk W, Sewgobind VD, Ijzermans JN, et al. End-stage renal failure and regulatory activities of CD4+CD25bright+FoxP3+ T-cells. Nephrol Dial Transplant. (2009) 24:1969–78. doi: 10.1093/ndt/gfp005
22. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
23. Baiyegunhi O, Ndlovu B, Ogunshola F, Ismail N, Walker BD, Ndung'u T, et al. Frequencies of circulating Th1-biased T follicular helper cells in acute HIV-1 infection correlate with the development of HIV-specific antibody responses and lower set point viral load. J Virol. (2018) 92:e00659–18. doi: 10.1128/JVI.00659-18
24. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. (2014) 41:529–42. doi: 10.1016/j.immuni.2014.10.004
25. Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. (2011) 34:108–21. doi: 10.1016/j.immuni.2010.12.012
26. Cronkite DA, Strutt TM. The regulation of inflammation by innate and adaptive lymphocytes. J Immunol Res. (2018) 2018:1467538. doi: 10.1155/2018/1467538
27. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. (2009) 229:152–72. doi: 10.1111/j.1600-065X.2009.00782.x
28. Sipsas NV, Sfikakis PP, Kontos A, Kordossis T. Levels of soluble CD40 ligand (CD154) in serum are increased in human immunodeficiency virus type 1-infected patients and correlate with CD4(+) T-cell counts. Clin Diagn Lab Immunol. (2002) 9:558–61. doi: 10.1128/CDLI.9.3.558-561.2002
29. Huang J, Jochems C, Talaie T, Anderson A, Jales A, Tsang KY, et al. Elevated serum soluble CD40 ligand in cancer patients may play an immunosuppressive role. Blood. (2012) 120:3030–8. doi: 10.1182/blood-2012-05-427799
30. Damas JK, Otterdal K, Yndestad A, Aass H, Solum NO, Froland SS, et al. Soluble CD40 ligand in pulmonary arterial hypertension: possible pathogenic role of the interaction between platelets and endothelial cells. Circulation. (2004) 110:999–1005. doi: 10.1161/01.CIR.0000139859.68513.FC
31. Danese S, Katz JA, Saibeni S, Papa A, Gasbarrini A, Vecchi M, et al. Activated platelets are the source of elevated levels of soluble CD40 ligand in the circulation of inflammatory bowel disease patients. Gut. (2003) 52:1435–41. doi: 10.1136/gut.52.10.1435
32. Belanger S, Crotty S. Dances with cytokines, featuring TFH cells, IL-21, IL-4 and B cells. Nat Immunol. (2016) 17:1135–6. doi: 10.1038/ni.3561
33. Luzina IG, Keegan AD, Heller NM, Rook GA, Shea-Donohue T, Atamas SP. Regulation of inflammation by interleukin-4: a review of “alternatives”. J Leukoc Biol. (2012) 92:753–64. doi: 10.1189/jlb.0412214
34. Dorner BG, Scheffold A, Rolph MS, Huser MB, Kaufmann SH, Radbruch A, et al. MIP-1alpha, MIP-1beta, RANTES, and ATAC/lymphotactin function together with IFN-gamma as type 1 cytokines. Proc Natl Acad Sci USA. (2002) 99:6181–6. doi: 10.1073/pnas.092141999
35. Guiteras R, Flaquer M, Cruzado JM. Macrophage in chronic kidney disease. Clin Kidney J. (2016) 9:765–71. doi: 10.1093/ckj/sfw096
36. Winterberg PD, Ford ML. The effect of chronic kidney disease on T cell alloimmunity. Curr Opin Organ Transplant. (2017) 22:22–8. doi: 10.1097/MOT.0000000000000375
37. Larbi A, Fulop T. From “truly naive” to “exhausted senescent” T cells: when markers predict functionality. Cytometry A. (2014) 85:25–35. doi: 10.1002/cyto.a.22351
38. Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. (2002) 2:251–62. doi: 10.1038/nri778
39. Li G, Yu M, Lee WW, Tsang M, Krishnan E, Weyand CM, et al. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat Med. (2012) 18:1518–24. doi: 10.1038/nm.2963
40. Vallejo AN, Weyand CM, Goronzy JJ. T-cell senescence: a culprit of immune abnormalities in chronic inflammation and persistent infection. Trends Mol Med. (2004) 10:119–24. doi: 10.1016/j.molmed.2004.01.002
41. Afzali B, Edozie FC, Fazekasova H, Scotta C, Mitchell PJ, Canavan JB, et al. Comparison of regulatory T cells in hemodialysis patients and healthy controls: implications for cell therapy in transplantation. Clin J Am Soc Nephrol. (2013) 8:1396–405. doi: 10.2215/CJN.12931212
42. Mederacke YS, Vondran FW, Kollrich S, Schulde E, Schmitt R, Manns MP, et al. Transient increase of activated regulatory T cells early after kidney transplantation. Sci Rep. (2019) 9:1021. doi: 10.1038/s41598-018-37218-x
43. Gao Y, Tang J, Chen W, Li Q, Nie J, Lin F, et al. Inflammation negatively regulates FOXP3 and regulatory T-cell function via DBC1. Proc Natl Acad Sci USA. (2015) 112:E3246–54. doi: 10.1073/pnas.1421463112
44. Wang Y, Zhang S, Zhang N, Feng M, Liang Z, Zhao X, et al. Reduced activated regulatory T cells and imbalance of Th17/activated Treg cells marks renal involvement in ANCA-associated vasculitis. Mol Immunol. (2020) 118:19–29. doi: 10.1016/j.molimm.2019.11.010
45. Nus M, Sage AP, Lu Y, Masters L, Lam BYH, Newland S, et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat Med. (2017) 23:601–10. doi: 10.1038/nm.4315
46. Gaddis DE, Padgett LE, Wu R, McSkimming C, Romines V, Taylor AM, et al. Apolipoprotein AI prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat Commun. (2018) 9:1095. doi: 10.1038/s41467-018-03493-5
47. Qin L, Waseem TC, Sahoo A, Bieerkehazhi S, Zhou H, Galkina EV, et al. Insights Into the molecular mechanisms of T follicular helper-mediated immunity and pathology. Front Immunol. (2018) 9:1884. doi: 10.3389/fimmu.2018.01884
48. Kim CJ, Lee CG, Jung JY, Ghosh A, Hasan SN, Hwang SM, et al. the transcription factor ets1 suppresses T follicular helper type 2 cell differentiation to halt the onset of systemic lupus erythematosus. Immunity. (2018) 49:1034–48.e8. doi: 10.1016/j.immuni.2018.10.012
49. Arroyo-Villa I, Bautista-Caro MB, Balsa A, Aguado-Acin P, Bonilla-Hernan MG, Plasencia C, et al. Constitutively altered frequencies of circulating follicullar helper T cell counterparts and their subsets in rheumatoid arthritis. Arthritis Res Ther. (2014) 16:500. doi: 10.1186/s13075-014-0500-6
50. Assing K, Nielsen C, Jakobsen M, Scholze A, Nybo M, Soerensen G, et al. Evidence of perturbed germinal center dynamics, but preserved antibody diversity, in end-stage renal disease. Immun Inflamm Dis. (2016) 4:225–34. doi: 10.1002/iid3.108
51. Pahl MV, Gollapudi S, Sepassi L, Gollapudi P, Elahimehr R, Vaziri ND. Effect of end-stage renal disease on B-lymphocyte subpopulations, IL-7, BAFF and BAFF receptor expression. Nephrol Dial Transplant. (2010) 25:205–12. doi: 10.1093/ndt/gfp397
52. Fernandez-Fresnedo G, Ramos MA, Gonzalez-Pardo MC, de Francisco AL, Lopez-Hoyos M, Arias M. B lymphopenia in uremia is related to an accelerated in vitro apoptosis and dysregulation of Bcl-2. Nephrol Dial Transplant. (2000) 15:502–10. doi: 10.1093/ndt/15.4.502
53. Yu YH, Lin KI. Factors that regulate the generation of antibody-secreting plasma cells. Adv Immunol. (2016) 131:61–99. doi: 10.1016/bs.ai.2016.03.001
Keywords: exhaustion, ESKD, dialysis (ESKD), T cell, treg, immune phenotype
Citation: Hartzell S, Bin S, Cantarelli C, Haverly M, Manrique J, Angeletti A, Manna GL, Murphy B, Zhang W, Levitsky J, Gallon L, Yu SM-W and Cravedi P (2020) Kidney Failure Associates With T Cell Exhaustion and Imbalanced Follicular Helper T Cells. Front. Immunol. 11:583702. doi: 10.3389/fimmu.2020.583702
Received: 15 July 2020; Accepted: 31 August 2020;
Published: 29 September 2020.
Edited by:
Masato Kubo, Tokyo University of Science, JapanReviewed by:
Ankit Saxena, National Institutes of Health (NIH), United StatesTakanori Sasaki, Tokyo University of Science, Japan
Copyright © 2020 Hartzell, Bin, Cantarelli, Haverly, Manrique, Angeletti, Manna, Murphy, Zhang, Levitsky, Gallon, Yu and Cravedi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Cravedi, cGFvbG8uY3JhdmVkaUBtc3NtLmVkdQ==