 Elyse Y. Bissonnette
Elyse Y. Bissonnette Jean-François Lauzon-Joset
Jean-François Lauzon-Joset Jason S. Debley
Jason S. Debley Steven F. Ziegler
Steven F. Ziegler- 1Centre de Recherche de l’Institut Universitaire de Cardiologie et de Pneumologie de Québec, Department of Medicine, Université Laval, Quebec City, QC, Canada
- 2Center for Immunity and Immunotherapies, Seattle Children’s Research Institute, Seattle, WA, United States
- 3Department of Immunology, Benaroya Research Institute, University of Washington School of Medicine, Seattle, WA, United States
The main function of the lung is to perform gas exchange while maintaining lung homeostasis despite environmental pathogenic and non-pathogenic elements contained in inhaled air. Resident cells must keep lung homeostasis and eliminate pathogens by inducing protective immune response and silently remove innocuous particles. Which lung cell type is crucial for this function is still subject to debate, with reports favoring either alveolar macrophages (AMs) or lung epithelial cells (ECs) including airway and alveolar ECs. AMs are the main immune cells in the lung in steady-state and their function is mainly to dampen inflammatory responses. In addition, they phagocytose inhaled particles and apoptotic cells and can initiate and resolve inflammatory responses to pathogens. Although AMs release a plethora of mediators that modulate immune responses, ECs also play an essential role as they are more than just a physical barrier. They produce anti-microbial peptides and can secrete a variety of mediators that can modulate immune responses and AM functions. Furthermore, ECs can maintain AMs in a quiescent state by expressing anti-inflammatory membrane proteins such as CD200. Thus, AMs and ECs are both very important to maintain lung homeostasis and have to coordinate their action to protect the organism against infection. Thus, AMs and lung ECs communicate with each other using different mechanisms including mediators, membrane glycoproteins and their receptors, gap junction channels, and extracellular vesicles. This review will revisit characteristics and functions of AMs and lung ECs as well as different communication mechanisms these cells utilize to maintain lung immune balance and response to pathogens. A better understanding of the cross-talk between AMs and lung ECs may help develop new therapeutic strategies for lung pathogenesis.
Introduction
The main function of the lung is to perform gas exchange while maintaining lung homeostasis despite environmental pathogenic and non-pathogenic elements contained in inhaled air. Considering that the volume of air inhaled every day is 5 to 8 L a minute, the prevalence of inflammation and pulmonary diseases is surprisingly low. Resident lung cells must discriminate between innocuous and harmful particles without creating unnecessary inflammation against inoffensive particles, while initiating an immune response against pathogens when necessary. Inappropriate or imbalanced immune response may underpin respiratory diseases. To maintain a proper balance, the lung needs specialized cells that can efficiently initiate and resolve inflammatory responses. Alveolar macrophages (AMs) and lung epithelial cells (ECs) are described in the literature as being the most important cells in the maintenance of lung homeostasis. AMs are the main immune cell type in the lung that determine the orientation and the magnitude of the immune response (1). In addition, they eliminate pathogens, apoptotic cells, and debris. On the other hand, a great number of publications claim that lung ECs are the main cell type keeping lung homeostasis with their antimicrobial activities acting as a barrier and a sensor of lung environment content (2). In reality, both AMs and ECs are the gatekeepers of the lung as they together are the first line of host defense and innate immunity. Furthermore, they communicate with each other to coordinate their actions to preserve lung homeostasis and gas exchange (3). Thus, in this review, we will briefly delineate the role of AMs and ECs and then focus on the everlasting cross-talk between AMs and lung ECs to maintain lung immune balance.
AMs
Origin and Heterogeneity
There are two types of macrophages found in the lung and named according to their location. AMs are found in the alveoli and airways and are easy to collect by bronchoalveolar lavage making them a highly studied cell type (4). There are also macrophages located in the alveolar septa and in the vascular adventitia that can be isolated from digested lung (5). These two macrophage populations can be distinguished using autofluorescence and surface markers (6). For the purpose of this review, we will focus on AMs that are found throughout the respiratory tract and are intimately associated with epithelial surfaces of both terminal airspaces and conducting airways (7).
For many years it was believed that AMs came from the differentiation of monocytes in the lung. Although this is true for some AMs (8), it is now well established that AMs mainly derive from embryo yolk sac and fetal liver cells (9–11). AMs are long-lived cells and a subpopulation of them can proliferate in situ to replenish themselves with a turnover rate of around 40% in one year (8, 12–14). This in situ proliferation required granulocyte-macrophage colony-stimulating factor (GM-CSF) and is controlled by mechanistic target of rapamycin complex 1 (mTORC1), a regulator of cell growth and proliferation (6, 15). Interestingly, GM-CSF also increases the expression of anti-apoptotic genes leading to long-lived cells (6). This maintenance of AM number is observed during homeostasis and under stress condition, but during acute inflammatory responses more macrophages are needed. Thus, there is rapid recruitment of monocyte-derived macrophages to the lung to eliminate pathogens (8, 16). These recruited macrophages promote lung inflammation whereas resident AMs dampen it (14, 17). During the resolution of inflammation, the majority of monocyte-derived macrophages undergo programmed cell death, while resident AMs survive and persist after the resolution phase (8). However, some recruited macrophages become phenotypically and functionally similar to resident AMs two months after infection showing that a part of AM population can also be replenished by monocyte-derived macrophages (18).
AMs are characterized by high expression of GM-CSFR, CD200R, and SIRP1α (Figure 1). Like other tissue macrophages, AMs show functional heterogeneity and plasticity depending on the microenvironment (1, 19). Tissue macrophages were first divided similarly to T helper lymphocytes; M1 being classically activated macrophages and M2 alternatively activated macrophages (20). M1 macrophages express high levels of pro-inflammatory mediators, whereas M2 express anti-inflammatory and wound healing mediators (19, 21) (Figure 1). The characterization of these populations of macrophages mostly results from in vitro stimulation. Macrophages stimulated with lipopolysaccharide (LPS) or inflammatory cytokines, such as interferon-γ (IFN-γ), display M1 phenotype with high expression of inducible nitric oxide synthase (iNOS), interleukin-1β (IL-1β), and tumor necrosis factor α (TNFα) (Figure 1) (22–24). These cells also express high level of MHCII and CD200 (25–27) and have cytotoxic and antitumoral properties (28, 29). On the other side, M2 macrophages arise in response to Th2 cytokines, IL-4, and/or IL-13 (30, 31). These macrophages express high level of arginase and anti-inflammatory cytokines such as IL-10 and transforming growth factor β (TGFβ) (Figure 1). They are involved in wound healing, tissue repair, and fibrosis, but have poor anti-microbicidal activity (32, 33), including low nitric oxide production (NO), as high arginase expression is known to inhibit NO production (34). Further studies demonstrated the existence of different subpopulations of M2 macrophages (35). Benoit et al. suggested dividing M2 macrophage subpopulations according to exposure agents; M2a referring to macrophages exposed to IL-4/IL-13, M2b to immune complexes and toll like receptor (TLR) agonists, M2c to IL-10 and glucocorticoid hormones, and M2d to TLR agonists through adenosine receptor (33, 35, 36). Thus, the dichotomous classification of M1/M2 does not align perfectly with Th1/Th2 immune response as previously suggested (37). Although M1 and M2a,b,c,d macrophages are phenotypically and functionally distinct macrophages (38, 39), they lack specific surface markers [reviewed by Wang et al. (40)]. Indeed, macrophage subpopulation identification relies on the relative intensity of marker expression and not on induction of expression. Thus, best practice characterization of macrophage subpopulations should include several markers of each phenotype (19).
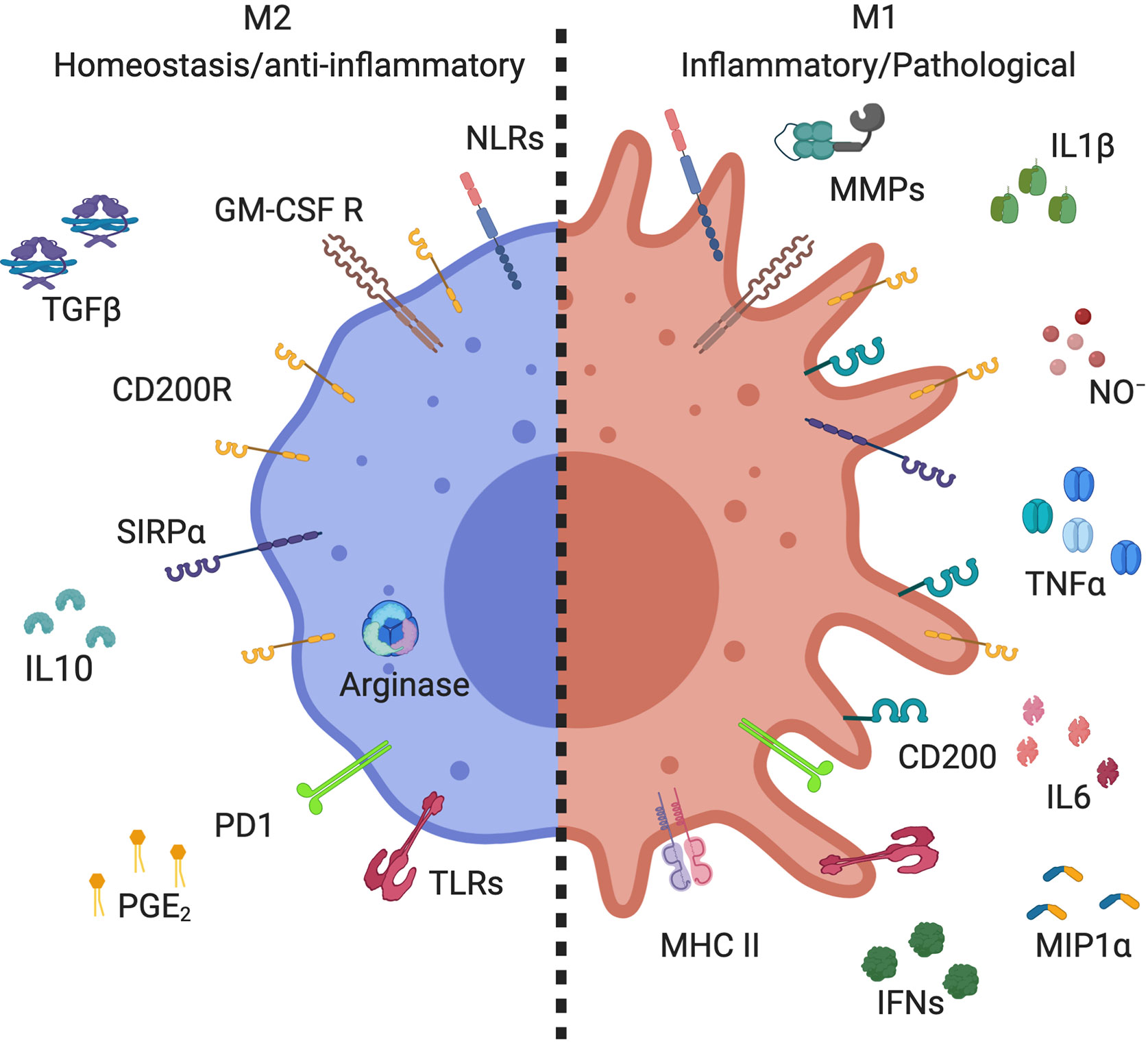
Figure 1 Markers expressed by alveolar macrophage subsets. Alveolar macrophages (AMs) expressed high levels of CD200R, SIRPα, GM-GSF R, and PPRs, including TLRs and NLRs. M2 AMs (homeostasis/anti-inflammatory) have high expression of arginase, and secrete anti-inflammatory mediators, such as IL-10, PGE2, and TGFβ. M1 AMs (inflammatory/pathological) increase their expression of CD200 and MHC II, as well as producing inflammatory mediators, such as metallopeptidase (MMPs), NO-, IL-1β, IFNs, IL-6, TNFα, and MIP-1α. Created with BioRender.com.
M1/M2 classification of macrophages in vitro is easily done by adding specific stimuli to differentiate them, but identification of macrophage subpopulations is more complicated in vivo where numerous cytokines are present, particularly in inflammatory diseases. In reality, there is a continuum of macrophage populations with various functions and phenotypes that can be shifted from one to the other phenotype depending on the microenvironment and according to macrophage plasticity (41). In steady-state lung, AMs are in an immunosuppressed state and their phenotype is tightly control by the lung microenvironment. They express high level of CD200 receptor (CD200R) which is associated with M2 phenotype (42, 43) and are involved in the downregulation of immune inflammation (44, 45). This may be important for tolerating innocuous inhaled agent. However, in humans, there is no consensus on AM phenotypes. Studies showed data ranging from 8% to 50% of AMs are M2 in human steady-state lung (46, 47). These discrepancies may be caused by the infection/inflammation history long-term impact on AM functions and phenotypes. Alternatively, it could suggest the presence of multiple AM phenotypes in healthy lung responding to their specific environment to perform a variety of functions (14).
AM Functions and Immune Response
AMs are well known for their role in maintaining lung homeostasis by phagocytosing microbes, dead cells, and other airborne particles to prevent unnecessary inflammation (48–51). Although AMs are considered poor antigen presenting cells (52–54), they can transport antigens to the draining lymph nodes (55). However, in the lung, antigen presentation is mainly mediated by dendritic cells. Interestingly, AMs suppress dendritic cell function and migration in and out of the airways to avoid immune responses against innocuous particles (53, 56, 57). Furthermore, AMs are known to downregulate T cell-dependent immune response in the lung by inducing FoxP3 expression in T cells (58, 59). A defect in this function is observed in asthma patients demonstrating the importance of AMs in the initiation of tolerance (60, 61). Depletion of AMs potentiates allergic asthma development and the severity of influenza infection, showing the significant role of these cells to dampen immune responses (48, 50, 51).
In steady-state, AMs execute anti-inflammatory functions to avoid immunopathology and the development of specific immune responses to harmless antigens. They are also the source of anti-inflammatory mediators such as IL-10, TGFβ, and prostaglandin E2 (PGE2) (1, 62, 63), facilitating the resolution of inflammation (64, 65). However, AMs maintain their capacity to be activated by pathogens and other danger signals via immune recognition of pathogen-associated molecular patterns by pattern recognition receptors (PRRs) including TLR, NOD-like receptors (NLRs) and C-type lectin receptors to initiate innate and adaptive immune responses (66–68). AMs are a primary source of cytokines and chemokines initiating immune responses, including TNFα, NO, IL-1β, IL-6, IFNs, and macrophage inflammatory protein (MIP)-1α (69, 70) (Figure 1). However, over production of these mediators contributes to the pathogenesis of inflammatory lung diseases such as acute lung injury, asthma, and chronic obstructive pulmonary diseases (COPD) (71–73). Thus, a rigorous regulation of AM secretory mediators is required to maintain lung homeostasis.
In addition to their role in modulating the immune response, AMs maintain lung homeostasis, in part, by internalizing and catabolizing lung surfactant which is critical for lung biomechanic and immunity (74, 75). A deficiency in GM-CSF signaling can lead to the dysregulation of AM surfactant clearance and causes accumulation of proteins and phospholipids in airspaces leading to pulmonary alveolar proteinosis (76). Interestingly, transplantation of functional AMs reduces alveolar proteinosis supporting the assumption that AMs are essential for surfactant metabolism (77). However, AMs are not the only cells that control surfactant levels. Alveolar epithelial type 2 cells are also involved in production and active removal of surfactant demonstrating the collaboration between these two cell types to maintain lung homeostasis (78).
Alveolar and Airway ECS
Origin and Heterogeneity, and Characteristics
Airway epithelium represents a tight barrier separating the organism from the external environment. In the tracheobronchial airway, the epithelium is pseudostratified, ciliated, and contains secretory cells. In the small airway, the epithelium becomes more cuboidal with increased club cells (79) formerly named Clara cells, which should be avoided given the origin of the experimentation (80). The alveoli are composed of two distinct EC types, alveolar epithelial type I cells that are thin and cover around 95% of the internal surface of the lung, and alveolar epithelial type II cells that are cuboidal secreting cells located between type I cells (81). Alveolar type I cells are specialized in gas exchange and alveolar fluid regulation (82, 83), whereas type II cells have secretory functions and constitute the progenitor cells of the epithelium (84).
The predominant cell types constituting the bronchial airway epithelium include basal progenitor cells, ciliated cells, secretory club cells, and goblet cells (79, 85). However, rarer and more specialized airway EC types have recently been better characterized such as neuroendocrine cells, tuft-like cells, and ionocytes (85–88). Over the past several years newer methodologies including single cell RNA sequencing (scRNA-Seq) and lineage tracing using pulse-Seq have allowed for better characterization, identification of cell type markers, and improved understanding of the evolution of these cell types in the airway epithelium in in vivo, ex vivo, and in vitro model systems (86, 88). Basal cells (identified by expression of P63 and KRT5) are the airway progenitors or stem cells that have the ability to differentiate and replenish all subtypes of cells of the airway epithelium (89). Goblet cells (expressing MUC5B and/or MUC5AC) secrete mucins (90, 91), and ciliated cells (identified by expression of FOXJ1 and AcTub) (90, 92) serve the critical physiologic role of facilitating mucociliary transport by propelling the airway mucus gel layer that overlies airway surface liquid proximally in the airways. Secretory club cells (expressing CCSP, SCGB1A1, and SCGB3A2) serve a protective role by both metabolizing inhaled toxins using cytochrome P450 in their smooth endoplasmic reticulum and through secretion of glycosaminoglycans, uteroglobin, and a surfactant-like substance (90, 93). Tuft cells, originally described in the intestine as chemosensory cells that facilitate Th2 inflammation through their production of IL-25 and thymic stromal lymphopoietin (TSLP), have recently been identified as a rare airway EC type (identified by expression of POU2F3) (94). The role of pulmonary neuroendocrine cells (PNECs), present in bronchial airway epithelium (identified by expression of SYP, CHGA, PGP9.5, ROBO2, and ENO2), and their secretion of bioactive amines and peptides, remains poorly understood (95). The recently identified airway EC type ionocyte (co-expressing FOXI1 and CFTR), although rare appears to be a major source of CFTR expression and function in the airway (86, 88). Recent elegant studies employing scRNA-Seq together with lineage tracing with pulse-Seq to track differentiation of airway ECs in vivo in mice, have described how airway basal cells can directly differentiate into club cells, tuft cells, PNECs, and ionocytes, whereas ciliated cells and goblet cells are derived secondarily from club cells (86). In the alveolar lung compartment where gas exchange occurs, the epithelium is squamous and consists of alveolar type I cells, identified through their expression of HOPX, PDPN, AQP5, and alveolar type II cells identified by their expression of SPB, SPC, and HT2-280 (96, 97).
The effectiveness of the lung epithelial barrier arises from its capacity to elaborate apical tight junctions with underlying adherent junctions (98). These intercellular junctions establish cell polarity and provide a selective permeability barrier regulating the movement of ions and macromolecules between the apical and basolateral face of the epithelium (99). Several membrane proteins are involved in these tight junctions such as claudin family, occludin, zonula occludens, and junction adhesion molecules (100, 101). A disruption in the epithelial barrier or a dysfunction/dysregulation of junction proteins contributes to lung pathologies such as asthma, cystic fibrosis, COPD, and acute respiratory distress syndrome (102–108).
Another type of intercellular contact that enable intercellular communication and metabolites and signaling molecules exchange between ECs is the gap junctions (109). These junctions, formed by channel proteins called connexins, play a major role in cellular coordination of cell functions and ensure the integration of metabolic activity of attached cells. Lung cellular interactions also involve pannexin glycoproteins and connexin proteins unopposed to gap junctions that form hemichannels allowing paracrine cell-cell communication (110, 111). These various junctions between airway ECs are important for epithelium barrier and functions.
Lung EC Model Systems
The current “gold-standard” for studying the airway epithelium in vitro or ex vivo using primary airway ECs is the air-liquid interface (ALI) model system. In this approach, airway ECs are seeded into collagen-coated permeable transwells, and when they become confluent apical media is removed and cultures are maintained in an ALI environment and epithelial differentiation medium (e.g. PneumaCult ALI™; Stemcell™) for at least 21 days, generating an organotypic differentiated airway epithelial culture that closely resembles the in vivo airway (97, 112, 113). This model can be used to study EC responses to environmental insults (114, 115) and viral infection (116, 117), and can be modified to support co-cultures of airway ECs with stromal and/or immune cells (118–121). Model systems that closely approximate the in vivo characteristics of alveolar ECs are less well developed. When primary cultures of pediatric alveolar lung ECs are cultured in vitro they expand as KRT5 expressing basal-like cells, however, when cultured at an ALI they demonstrate increased expression of markers for airway, but not alveolar ECs (122). Recently progress has been made developing distal lung directed differentiation protocols that utilize lung progenitor cells cultured in a 3D organoid phase, with or without mesenchymal cells (97, 123, 124). Although some of these models do generate alveolar type II-like cells (123, 124), these cultures are grown in submerged culture conditions and therefore are suboptimal in that they do not yet model the in vivo ALI environment (97).
Alveolar and Airway EC Functions
In addition to its role as a physical barrier, the epithelium is crucial for lung biomechanics (78, 125). Secretion of surfactant by alveolar epithelial type II cells is critical to stabilize the structure of alveoli by reducing surface tension at the air-liquid interface to avoid alveolar collapse (74). The absence or deficiency/inactivation of surfactant causes severe respiratory disorders such as neonatal respiratory distress syndrome and acute respiratory distress syndrome (126). On the other hand, impaired surfactant catabolism by AMs and/or alveolar epithelial type II cells leads to accumulation of surfactant in alveoli and is associated with lung proteinosis causing respiratory failure (127). Pulmonary surfactant is composed of around 90% lipids, mainly phospholipids (80%–85%) and some neutral lipids (5%–10%), and 8%–10% proteins, including two hydrophilic proteins, surfactant protein (SP)-A and SP-D, and two hydrophobic proteins, SP-B and SP-C. The proportion of all the constituents of pulmonary surfactant is important for the biomechanical functions of the film at the air-liquid interface reducing surface tension (125) and also for protecting against pathogens.
Protection against microbes entering the lung is crucial to maintain gas exchange. One line of defense of airway ECs is the production of a plethora of antimicrobial proteins and peptides such as β defensins, LL-37, lysozyme, lactoferrin, NO, and secretory leukocyte proteinase inhibitor (128, 129). The surfactant proteins, SP-A and SP-D, produce by type II alveolar ECs are also involved in pathogen clearance via several mechanisms including binding, agglutination, anti-microbial and fungal effects, and enhancement of neutrophil and AM phagocytosis and killing (130, 131). The production of antimicrobial peptides is stimulated by the activation of PRRs on lung ECs such as TLR. There are at least 10 TLRs expressed by lung ECs that recognise distinct pathogen-associated molecular patterns derived from viruses, bacteria, mycobacteria, fungi, and parasites (129, 132). Lung ECs express other PRRs such as the RIG-I-like receptor viral sensors RIG-1, MDA5, and LPG2, in addition to NLRs, C-type lectin, and inflammasome components (133–137) that participate to the immune responses.
Modulation of Immune Response
Lung ECs contribute to local immune response through the production of a multitude of modulatory mediators. The stimulation of PRRs on lung ECs by viruses induces the production of type I and III IFNs which in turn induce expression of hundreds of interferon-stimulated genes (ISGs) (117), the protein products of some of which have local direct antiviral effects while others promote cellular immunity. Stimulation of lung EC PRRs also induce a number of pro-inflammatory cytokines and chemokines including IL-1β, IL-6, IL-8, TNF, GM-CSF, MIP-1α, RANTES, and monocyte chemoattractant protein (MCP)-1 (138–140). Together these ISGs and mediators promote the recruitment and activation of inflammatory cells to eliminate pathogens.
In addition to the production of inflammatory mediators, lung ECs contribute to the regulation of inflammation and airway remodeling through the production of anti-inflammatory products including IL-10, TGFβ, lipoxins, resolvins, protectins, and PGE2 (139, 141–143). Overall, the secretion of cytokines, chemokines, and lipid mediators by lung ECs help to shape a balanced immune response. However, EC aberrant secretion of pro-inflammatory cytokines lead to lung pathologies, as observed in pulmonary allergic inflammation where lung EC production of IL-25, IL-33, and TSLP are linked to the initiation and progression of the disease (116, 144, 145). Lung ECs also contribute to the pathogenesis of COPD through the production of a plethora of inflammatory mediators (146).
Lung ECs can also modulate immune cells through physical interaction via immunomodulatory surface molecules such as CD200, program death-ligand (PDL)-1, and CD47 (147–149). The binding of CD200 to its receptor, CD200R which is highly expressed on AMs, downregulate the secretion of inflammatory cytokines upon LPS stimulation (42), suggesting an immunomodulatory function of lung ECs. This regulation affects other immune cells such as T cells and dendritic cells that also express CD200R (150). Similarly, the binding of PDL-1 to its receptor PD-1 dampens AM secretion of inflammatory cytokines and negatively regulates T cell effector functions (151, 152), whereas binding of CD47 to SIRP1α expressing macrophages downregulate phagocytosis (153, 154). Thus, there is a breadth of evidence demonstrating the importance of lung ECs in regulating local immune responses, by interacting/communicating with immune cells, including AMs.
Cross-Talk Between AMS AND LUNG ECS
Given that lung ECs and AMs are the first cell types being in contact with pathogens, they must coordinate their actions to eliminate pathogens without causing too much damage to the lung, either via direct cell-cell contact or secretion of molecules (Figure 2). In steady-state, ECs maintain AMs in a quiescent state. AMs adhere to the epithelium via extracellular membrane proteins such as CD200R, PD-1, and SIRP1α and their ligands on ECs, respectively CD200, PDL-1, and CD47 (42, 147, 149, 151, 155, 156). In addition to maintaining cells in close proximity to increase paracrine communication, these protein interactions downregulate AM activation. The contiguity of these two cell types may also allow the regulation of AMs by anti-inflammatory cytokines secreted by lung ECs such as IL-10 and TGFβ (139, 141). The loss of these regulatory interactions, either due to epithelial damage or sensing of pathogens in the airways, lead to the activation of AMs and initiation of inflammatory response. Whether the epithelium is the first to stimulate AM pro-inflammatory reaction is still a matter of debate, and may depend on the nature of the pathogen, but both of them are needed for adequate immune response.
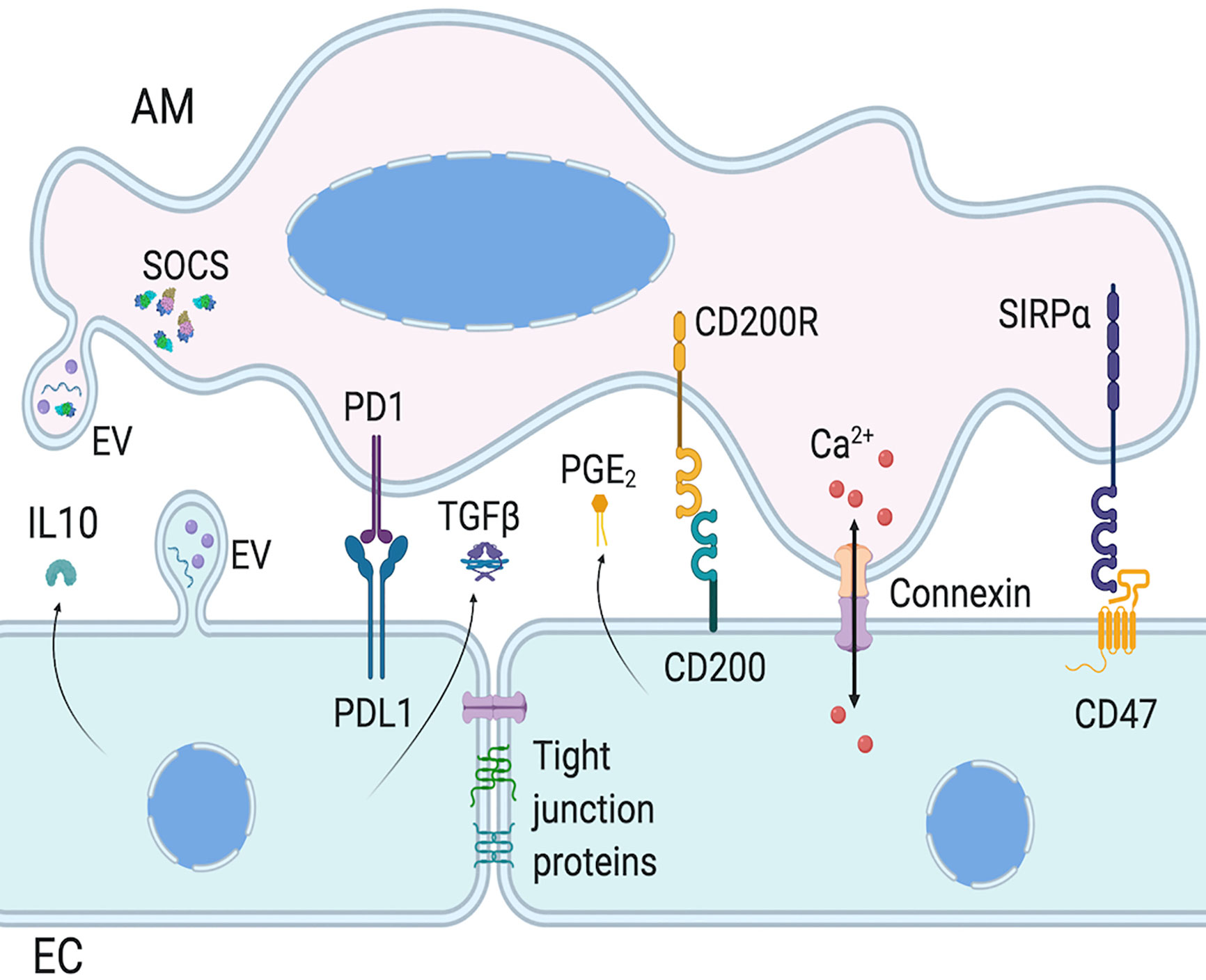
Figure 2 Intercellular communication between AMs and ECs. Epithelial cells (ECs) and alveolar macrophages (AMs) communication involves surface protein interaction, as well as mediator secretion and extracellular vesicles (EVs). AM activation is regulated by EC expression of PDL1, CD200, and CD47, which activate AM cognate receptor, respectively PD1, CD200R, and SIRPα. EC also regulate AM functions via paracrine secretion of mediators, such as IL-10 and PGE2. AM and EC functions can also be regulated with the release of EVs which can have inflammatory or anti-inflammatory functions, via EV surface proteins, cytokines or miRNAs. Of note, AMs constitutively secrete EVs containing SOCS. Finally, AMs and ECs can form gap junctions to allow bi-directional intercellular metabolic synchronicity, including Ca2+ waves. Created with BioRender.com.
Alveolar Gap Junction Channel
Gap junctions are usually an intercellular contact between ECs, but they are also found between AMs and alveolar ECs. In mouse, a subset of AMs express connexin 43 enabling the formation of gap junctions with ECs (157). These gap junctions allow waves of Ca2+ signal to travel from AMs to ECs and vice-versa, as observed with LPS stimulation causing cyclic and synchronized calcium spikes in both cell types. Mice with macrophages deficient in connexin 43 had higher levels of pro-inflammatory cytokines in bronchoalveolar lavage that originated from both AMs and airway ECs (157). This supports the bi-directional anti-inflammatory role of AM-epithelium gap junction channel, and is essential to reduce LPS induced lung inflammation and injury. The presence of connexin 43 gap junctions between macrophages and ECs was also shown using human cells (158, 159), suggesting its involvement in human lung cell communication. However, the exact role of this communication is still undetermined.
Extracellular Vesicles
Recently, extracellular vesicles (EVs) have emerged as a novel communication mechanism to exchange proteins, lipids, and genetic material between cells. These vesicles are divided into three categories, apoptotic bodies, exosomes, and microvesicles. The release of apoptotic bodies during apoptosis is well known and will not be discussed here [reviewed by Battistelli M et al. (160)]. Exosomes and microvesicles are released in steady-state conditions. The main difference between these two EVs resides in their size and origin. Exosomes are 40-120 nm in diameter and are released from endosomes fusing with the membrane, whereas microvesicles (also called microparticles or ectosomes) are 50–1,000 nm and are formed by the outward budding and fission of the plasma membrane (161, 162). Given the overlapping range of size and composition, and the difficulty of isolating exosomes and microvesicles separately to discriminate their specific functions (163), we will refer to EVs as an umbrella term for both exosomes and microvesicles in this review.
EVs are found in most human body fluids, including bronchoalveolar lavages (164). These EVs express membrane surface proteins, cytoskeletal and cytoplasmic proteins, cytokines, mRNAs, and miRNAs (Table 1). EVs may explain in part how cytokines/chemokines reach physiologic concentrations to affect target cells. In steady-state, EVs in bronchoalveolar lavages come largely from AMs and lung ECs (Figure 2), but during inflammatory response, infiltrating cell types also produce them (166–168, 171, 172, 174–177), as they are involved in immune responses, inflammation, bacterial and viral sequestration.
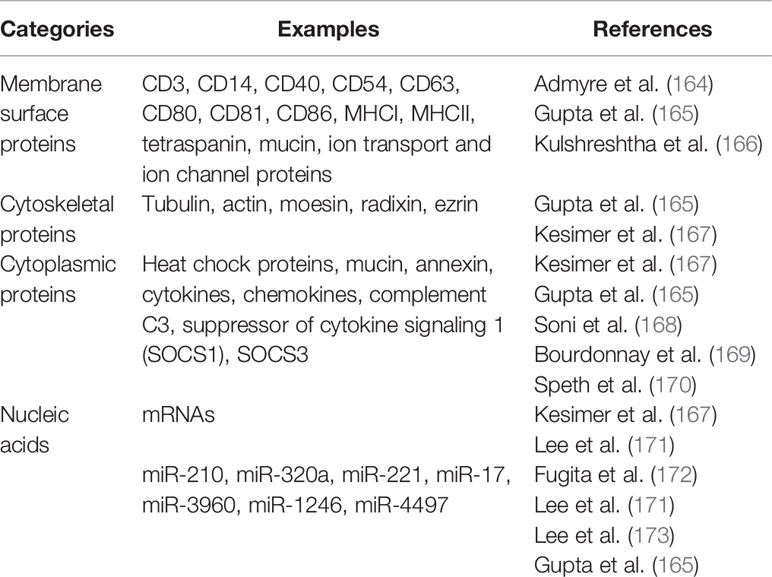
Table 1 Extracellular vesicle content.
EVs released by AMs and lung ECs can modulate each other in a pro- and anti-inflammatory manner. AMs constitutively secrete EVs containing suppressor of cytokine signaling (SOCS) proteins, which inhibit the inflammatory STAT pathway (Figure 2) (169). Lung ECs barely express SOCS proteins, however they secrete mediators, such as PGE2 and IL-10, that increase SOCS protein secretion by AMs. EVs containing SOCS proteins are taken up by lung ECs and downregulate cytokine-induced STAT activation, maintaining ECs in a quiescent state and limiting tumor transformation of normal ECs (170). Furthermore, vesicular SOCS can dampen allergic airway inflammation by inhibiting EC production of type 2 cytokines (178). Thus, EVs are crucial to maintain lung homeostasis through cell-cell communication, but they can also be involved in lung pathogenesis.
Indeed, content and concentration of EVs in bronchoalveolar lavage change during lung inflammation. However, there is still controversy on which cell types initiate immune responses. Under hyperoxia-induced oxidative stress, lung ECs secrete EVs to activate pro-inflammatory functions of AMs, facilitating the recruitment of immunomodulatory cells involved in lung injury (171, 176). In contrast, after pulmonary LPS exposure, AMs are the first producers of EVs containing inflammatory mediators which activate lung EC inflammatory response (168). In allergic inflamed lung, inhibition of EV secretion alleviates asthma features, although the cellular origin of EVs is unknown (166). Interestingly, intranasal transfer of EVs from bronchoalveolar lavage of tolerized mice prevent allergic sensitization, including production of IgE, Th2 cytokines, and lung inflammation (179). Yet, the functions of EVs produced by AMs and lung ECs are underexplored, and may have beneficial or detrimental effects depending on the context. More research is needed to better understand the communication between these two cell types in lung homeostasis and diseases.
Conclusion
AMs close proximity to lung ECs allow them to communicate using gap junctions, surface membrane molecules, soluble mediators, and EVs. In steady-state, AMs and ECs downregulate each other to avoid unnecessary inflammation. However, each cell type can activate the other one to initiate an immune response when required.
There is a plethora of publications on AMs and lung ECs; yet, very few of them investigate their interactions under lung homeostatic conditions and how this interaction is altered in pathological conditions. Furthermore, numerous studies use macrophages derived from bone marrow or monocytes as surrogate for AMs, even though AMs are functionally and phenotypically different from other macrophages. Thus, to extend our knowledge on the interaction between AMs and ECs in lung steady-state and diseases, it is essential to perform in vivo experiments or to harvest AMs from bronchoalveolar lavage for in vitro co-culture with human primary lung ECs which show normal differentiation patterns (unlike epithelial cell lines) (180). In addition, in vitro co-culture systems of AMs and ECs should be performed at air-liquid surface, which mimics more closely the in vivo situation, including the formation of functional tight junctions (181). This more representative model may help improving our knowledge on AMs and lung ECs communication and their collaboration in the maintenance of lung homeostasis and response to pathogens and injuries. A better understanding of AMs and lung ECs cross-talk may help develop new therapeutic strategies for lung pathogenesis.
Author Contributions
EB wrote the core of the manuscript, J-FL-J and JD made substantial intellectual contribution, J-FL-J drew the figures, and SZ reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
EB is funded by Canadian Institutes of Health Research (grant PJT-152876) and Quebec Respiratory Research Health Network. J-FL-J is funded by Quebec Respiratory Research Health Network. JD is funded by the National Institutes of Health (grants U19AI125378, K24AI150991, and R01HL128361), and SZ is funded by the National Institutes of Health (grant U19AI125378).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol (2014) 14(2):81–93. doi: 10.1038/nri3600
2. Leiva-Juarez MM, Kolls JK, Evans SE. Lung epithelial cells: therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol (2018) 11(1):21–34. doi: 10.1038/mi.2017.71
3. Tao F, Kobzik L. Lung macrophage-epithelial cell interactions amplify particle-mediated cytokine release. Am J Respiratory Cell Mol Biol (2002) 26(4):499–505. doi: 10.1165/ajrcmb.26.4.4749
4. Joshi N, Walter JM, Misharin AV. Alveolar Macrophages. Cell Immunol (2018) 330:86–90. doi: 10.1016/j.cellimm.2018.01.005
5. Hume PS, Gibbings SL, Jakubzick CV, Tuder RM, Curran-Everett D, Henson PM, et al. Localization of Macrophages in the Human Lung via Design-based Stereology. Am J Respiratory Crit Care Med (2020) 201(10):1209–17. doi: 10.1164/rccm.201911-2105OC
6. Draijer C, Penke LRK, Peters-Golden M. Distinctive Effects of GM-CSF and M-CSF on Proliferation and Polarization of Two Major Pulmonary Macrophage Populations. J Immunol (2019) 202(9):2700–9. doi: 10.4049/jimmunol.1801387
7. Parra SC, Burnette R, Price HP, Takaro T. Zonal distribution of alveolar macrophages, type II pneumonocytes, and alveolar septal connective tissue gaps in adult human lungs. Am Rev Respiratory Dis (1986) 133(5):908–12.
8. Janssen WJ, Barthel L, Muldrow A, Oberley-Deegan RE, Kearns MT, Jakubzick C, et al. Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am J Respiratory Crit Care Med (2011) 184(5):547–60. doi: 10.1164/rccm.201011-1891OC
9. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity (2013) 38(1):79–91. doi: 10.1016/j.immuni.2012.12.001
10. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Sci (N Y NY) (2012) 336(6077):86–90. doi: 10.1126/science.1219179
11. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity (2014) 41(1):21–35. doi: 10.1016/j.immuni.2014.06.013
12. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity (2013) 38(4):792–804. doi: 10.1016/j.immuni.2013.04.004
13. Maus UA, Janzen S, Wall G, Srivastava M, Blackwell TS, Christman JW, et al. Resident alveolar macrophages are replaced by recruited monocytes in response to endotoxin-induced lung inflammation. Am J Respiratory Cell Mol Biol (2006) 35(2):227–35. doi: 10.1165/rcmb.2005-0241OC
14. Mould KJ, Jackson ND, Henson PM, Seibold M, Janssen WJ. Single cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI Insight (2019) 4(5):1–14. doi: 10.1172/jci.insight.126556
15. Deng W, Yang J, Lin X, Shin J, Gao J, Zhong XP. Essential Role of mTORC1 in Self-Renewal of Murine Alveolar Macrophages. J Immunol (2017) 198(1):492–504. doi: 10.4049/jimmunol.1501845
16. Goritzka M, Makris S, Kausar F, Durant LR, Pereira C, Kumagai Y, et al. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J Exp Med (2015) 212(5):699–714. doi: 10.1084/jem.20140825
17. Zaslona Z, Przybranowski S, Wilke C, van Rooijen N, Teitz-Tennenbaum S, Osterholzer JJ, et al. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol (2014) 193(8):4245–53. doi: 10.4049/jimmunol.1400580
18. Aegerter H, Kulikauskaite J, Crotta S, Patel H, Kelly G, Hessel EM, et al. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat Immunol (2020) 21(2):145–57. doi: 10.1038/s41590-019-0568-x
19. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
20. Mills CD, Kincaid K, Alt JM, Heilman MJ. Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol (2000) 164(12):6166–73. doi: 10.4049/jimmunol.164.12.6166
21. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol (2011) 11(11):723–37. doi: 10.1038/nri3073
22. Hibbs JB Jr., Taintor RR, Vavrin Z, Rachlin EM. Nitric oxide: a cytotoxic activated macrophage effector molecule. Biochem Biophys Res Commun (1988) 157(1):87–94. doi: 10.1016/s0006-291x(88)80015-9
23. Nau GJ, Richmond JF, Schlesinger A, Jennings EG, Lander ES, Young RA. Human macrophage activation programs induced by bacterial pathogens. Proc Natl Acad Sci U States America (2002) 99(3):1503–8. doi: 10.1073/pnas.022649799
24. Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med (1983) 158(3):670–89. doi: 10.1084/jem.158.3.670
25. Lauzon-Joset JF, Marsolais D, Tardif-Pellerin E, Patoine D, Bissonnette EY. CD200 in asthma. Int J Biochem Cell Biol (2019) 112:141–4. doi: 10.1016/j.biocel.2019.05.003
26. Mukhopadhyay S, Pluddemann A, Hoe JC, Williams KJ, Varin A, Makepeace K, et al. Immune inhibitory ligand CD200 induction by TLRs and NLRs limits macrophage activation to protect the host from meningococcal septicemia. Cell Host Microbe (2010) 8(3):236–47. doi: 10.1016/j.chom.2010.08.005
27. Xie X, Luo X, Liu N, Li X, Lou F, Zheng Y, et al. Monocytes, microglia, and CD200-CD200R1 signaling are essential in the transmission of inflammation from the periphery to the central nervous system. J Neurochem (2017) 141(2):222–35. doi: 10.1111/jnc.13972
28. Celada A, Gray PW, Rinderknecht E, Schreiber RD. Evidence for a gamma-interferon receptor that regulates macrophage tumoricidal activity. J Exp Med (1984) 160(1):55–74. doi: 10.1084/jem.160.1.55
29. Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U States America (2000) 97(16):8841–8. doi: 10.1073/pnas.97.16.8841
30. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med (1992) 176(1):287–92. doi: 10.1084/jem.176.1.287
31. Doherty TM, Kastelein R, Menon S, Andrade S, Coffman RL. Modulation of murine macrophage function by IL-13. J Immunol (1993) 151(12):7151–60.
32. Rutschman R, Lang R, Hesse M, Ihle JN, Wynn TA, Murray PJ. Cutting edge: Stat6-dependent substrate depletion regulates nitric oxide production. J Immunol (2001) 166(4):2173–7. doi: 10.4049/jimmunol.166.4.2173
33. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233(9):6425–40. doi: 10.1002/jcp.26429
34. Durante W, Johnson FK, Johnson RA. Arginase: a critical regulator of nitric oxide synthesis and vascular function. Clin Exp Pharmacol Physiol (2007) 34(9):906–11. doi: 10.1111/j.1440-1681.2007.04638.x
35. Benoit M, Desnues B, Mege JL. Macrophage polarization in bacterial infections. J Immunol (2008) 181(6):3733–9. doi: 10.4049/jimmunol.181.6.3733
36. Pinhal-Enfield G, Ramanathan M, Hasko G, Vogel SN, Salzman AL, Boons GJ, et al. An angiogenic switch in macrophages involving synergy between Toll-like receptors 2, 4, 7, and 9 and adenosine A(2A) receptors. Am J Pathol (2003) 163(2):711–21. doi: 10.1016/S0002-9440(10)63698-X
37. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi: 10.12703/P6-13
38. Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol (2006) 80(6):1298–307. doi: 10.1189/jlb.0406249
39. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25(12):677–86. doi: 10.1016/j.it.2004.09.015
40. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol (2019) 106(2):345–58. doi: 10.1002/JLB.3RU1018-378RR
41. Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol (2005) 175(1):342–9. doi: 10.4049/jimmunol.175.1.342
42. Snelgrove RJ, Goulding J, Didierlaurent AM, Lyonga D, Vekaria S, Edwards L, et al. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat Immunol (2008) 9(9):1074–83. doi: 10.1038/ni.1637
43. Koning N, van Eijk M, Pouwels W, Brouwer MS, Voehringer D, Huitinga I, et al. Expression of the inhibitory CD200 receptor is associated with alternative macrophage activation. J Innate Immun (2010) 2(2):195–200. doi: 10.1159/000252803
44. Lauzon-Joset J, Langlois A, Lai LJ, Santerre K, Lee-Gosselin A, Bosse Y, et al. Lung CD200R activation abrogates airway hyperresponsiveness in experimental asthma. Am J Respiratory Cell Mol Biol (2015) 53(2):276–84. doi: 10.1165/rcmb.2014-0229OC
45. Zhu B, Yu Y, Liu X, Han Q, Kang Y, Shi L. CD200 Modulates S. aureus-Induced Innate Immune Responses Through Suppressing p38 Signaling. Int J Mol Sci (2019) 20(3):1–15. doi: 10.3390/ijms20030659
46. Melgert BN, ten Hacken NH, Rutgers B, Timens W, Postma DS, Hylkema MN. More alternative activation of macrophages in lungs of asthmatic patients. J Allergy Clin Immunol (2011) 127(3):831–3. doi: 10.1016/j.jaci.2010.10.045
47. Pechkovsky DV, Prasse A, Kollert F, Engel KM, Dentler J, Luttmann W, et al. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin Immunol (2010) 137(1):89–101. doi: 10.1016/j.clim.2010.06.017
48. Purnama C, Ng SL, Tetlak P, Setiagani YA, Kandasamy M, Baalasubramanian S, et al. Transient ablation of alveolar macrophages leads to massive pathology of influenza infection without affecting cellular adaptive immunity. Eur J Immunol (2014) 44(7):2003–12. doi: 10.1002/eji.201344359
49. Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, et al. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PloS Pathog (2014) 10(4):e1004053. doi: 10.1371/journal.ppat.1004053
50. Careau E, Bissonnette EY. Adoptive transfer of alveolar macrophages abrogates bronchial hyperresponsiveness. Am J Respiratory Cell Mol Biol (2004) 31(1):22–7. doi: 10.1165/rcmb.2003-0229OC
51. Thepen T, McMenamin C, Oliver J, Kraal G, Holt PG. Regulation of immune response to inhaled antigen by alveolar macrophages: differential effects of in vivo alveolar macrophage elimination on the induction of tolerance vs. immunity. Eur J Immunol (1991) 21(11):2845–50. doi: 10.1002/eji.1830211128
52. MacLean JA, Xia W, Pinto CE, Zhao L, Liu HW, Kradin RL. Sequestration of inhaled particulate antigens by lung phagocytes. A mechanism for the effective inhibition of pulmonary cell-mediated immunity. Am J Pathol (1996) 148(2):657–66.
53. Jakubzick C, Tacke F, Llodra J, van Rooijen N, Randolph GJ. Modulation of dendritic cell trafficking to and from the airways. J Immunol (2006) 176(6):3578–84. doi: 10.4049/jimmunol.176.6.3578
54. Chelen CJ, Fang Y, Freeman GJ, Secrist H, Marshall JD, Hwang PT, et al. Human alveolar macrophages present antigen ineffectively due to defective expression of B7 costimulatory cell surface molecules. J Clin Invest (1995) 95(3):1415–21. doi: 10.1172/JCI117796
55. Kirby AC, Coles MC, Kaye PM. Alveolar macrophages transport pathogens to lung draining lymph nodes. J Immunol (2009) 183(3):1983–9. doi: 10.4049/jimmunol.0901089
56. Holt PG, Oliver J, Bilyk N, McMenamin C, McMenamin PG, Kraal G, et al. Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. J Exp Med (1993) 177(2):397–407. doi: 10.1084/jem.177.2.397
57. Lauzon-Joset JF, Marsolais D, Langlois A, Bissonnette EY. Dysregulation of alveolar macrophages unleashes dendritic cell-mediated mechanisms of allergic airway inflammation. Mucosal Immunol (2014) 7(1):155–64. doi: 10.1038/mi.2013.34
58. Strickland DH, Thepen T, Kees UR, Kraal G, Holt PG. Regulation of T-cell function in lung tissue by pulmonary alveolar macrophages. Immunology (1993) 80(2):266–72.
59. Strickland D, Kees UR, Holt PG. Regulation of T-cell activation in the lung: isolated lung T cells exhibit surface phenotypic characteristics of recent activation including down-modulated T-cell receptors, but are locked into the G0/G1 phase of the cell cycle. Immunology (1996) 87(2):242–9. doi: 10.1046/j.1365-2567.1996.460541.x
60. Coleman MM, Ruane D, Moran B, Dunne PJ, Keane J, Mills KH. Alveolar macrophages contribute to respiratory tolerance by inducing FoxP3 expression in naive T cells. Am J Respiratory Cell Mol Biol (2013) 48(6):773–80. doi: 10.1165/rcmb.2012-0263OC
61. Tang C, Ward C, Reid D, Bish R, O’Byrne PM, Walters EH. Normally suppressing CD40 coregulatory signals delivered by airway macrophages to TH2 lymphocytes are defective in patients with atopic asthma. J Allergy Clin Immunol (2001) 107(5):863–70. doi: 10.1067/mai.2001.114987
62. Zissel G, Homolka J, Schlaak J, Schlaak M, Muller-Quernheim J. Anti-inflammatory cytokine release by alveolar macrophages in pulmonary sarcoidosis. Am J Respiratory Crit Care Med (1996) 154(3 Pt 1):713–9. doi: 10.1164/ajrccm.154.3.8810610
63. Menard G, Turmel V, Bissonnette EY. Serotonin modulates the cytokine network in the lung: involvement of prostaglandin E2. Clin Exp Immunol (2007) 150(2):340–8. doi: 10.1111/j.1365-2249.2007.03492.x
64. Aggarwal NR, Tsushima K, Eto Y, Tripathi A, Mandke P, Mock JR, et al. Immunological priming requires regulatory T cells and IL-10-producing macrophages to accelerate resolution from severe lung inflammation. J Immunol (2014) 192(9):4453–64. doi: 10.4049/jimmunol.1400146
65. Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest (2019) 129(7):2619–28. doi: 10.1172/JCI124615
66. Gordon SB, Read RC. Macrophage defences against respiratory tract infections. Br Med Bull (2002) 61:45–61. doi: 10.1093/bmb/61.1.45
67. Martinez EC, Garg R. van Drunen Littel-van den Hurk S. Innate immune protection from pneumonia virus of mice induced by a novel immunomodulator is prolonged by dual treatment and mediated by macrophages. Antiviral Res (2019) 171:104594. doi: 10.1016/j.antiviral.2019.104594
68. Liu CH, Liu H, Ge B. Innate immunity in tuberculosis: host defense vs pathogen evasion. Cell Mol Immunol (2017) 14(12):963–75. doi: 10.1038/cmi.2017.88
69. Kolli D, Gupta MR, Sbrana E, Velayutham TS, Chao H, Casola A, et al. Alveolar macrophages contribute to the pathogenesis of human metapneumovirus infection while protecting against respiratory syncytial virus infection. Am J Respiratory Cell Mol Biol (2014) 51(4):502–15. doi: 10.1165/rcmb.2013-0414OC
70. Menard G, Bissonnette EY. Priming of alveolar macrophages by leukotriene D(4): potentiation of inflammation. Am J Respiratory Cell Mol Biol (2000) 23(4):572–7. doi: 10.1165/ajrcmb.23.4.4152
71. Lomas-Neira J, Chung CS, Perl M, Gregory S, Biffl W, Ayala A. Role of alveolar macrophage and migrating neutrophils in hemorrhage-induced priming for ALI subsequent to septic challenge. Am J Physiol (2006) 290(1):L51–8. doi: 10.1152/ajplung.00028.2005
72. Fricker M, Gibson PG. Macrophage dysfunction in the pathogenesis and treatment of asthma. Eur Respir J (2017) 50(3):1–16. doi: 10.1183/13993003.00196-2017
73. Russell RE, Culpitt SV, DeMatos C, Donnelly L, Smith M, Wiggins J, et al. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respiratory Cell Mol Biol (2002) 26(5):602–9. doi: 10.1165/ajrcmb.26.5.4685
74. Lopez-Rodriguez E, Gay-Jordi G, Mucci A, Lachmann N, Serrano-Mollar A. Lung surfactant metabolism: early in life, early in disease and target in cell therapy. Cell Tissue Res (2017) 367(3):721–35. doi: 10.1007/s00441-016-2520-9
75. Stern N, Riklis S, Kalina M, Tietz A. The catabolism of lung surfactant by alveolar macrophages. Biochim Biophys Acta (1986) 877(3):323–33. doi: 10.1016/0005-2760(86)90196-7
76. Carey B, Trapnell BC. The molecular basis of pulmonary alveolar proteinosis. Clin Immunol (2010) 135(2):223–35. doi: 10.1016/j.clim.2010.02.017
77. Happle C, Lachmann N, Ackermann M, Mirenska A, Gohring G, Thomay K, et al. Pulmonary Transplantation of Human Induced Pluripotent Stem Cell-derived Macrophages Ameliorates Pulmonary Alveolar Proteinosis. Am J Respiratory Crit Care Med (2018) 198(3):350–60. doi: 10.1164/rccm.201708-1562OC
78. Knudsen L, Ochs M. The micromechanics of lung alveoli: structure and function of surfactant and tissue components. Histochem Cell Biol (2018) 150(6):661–76. doi: 10.1007/s00418-018-1747-9
79. Crystal RG, Randell SH, Engelhardt JF, Voynow J, Sunday ME. Airway epithelial cells: current concepts and challenges. Proc Am Thorac Soc (2008) 5(7):772–7. doi: 10.1513/pats.200805-041HR
80. Winkelmann A, Noack T. The Clara cell: a “Third Reich eponym”? Eur Respir J (2010) 36(4):722–7. doi: 10.1183/09031936.00146609
81. Wong MH, Johnson MD. Differential response of primary alveolar type I and type II cells to LPS stimulation. PloS One (2013) 8(1):e55545. doi: 10.1371/journal.pone.0055545
82. Maina JN, West JB. Thin and strong! The bioengineering dilemma in the structural and functional design of the blood-gas barrier. Physiol Rev (2005) 85(3):811–44. doi: 10.1152/physrev.00022.2004
83. Johnson MD, Widdicombe JH, Allen L, Barbry P, Dobbs LG. Alveolar epithelial type I cells contain transport proteins and transport sodium, supporting an active role for type I cells in regulation of lung liquid homeostasis. Proc Natl Acad Sci U States America (2002) 99(4):1966–71. doi: 10.1073/pnas.042689399
84. Fehrenbach H. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir Res (2001) 2(1):33–46. doi: 10.1186/rr36
85. Zaragosi LE, Deprez M, Barbry P. Using single-cell RNA sequencing to unravel cell lineage relationships in the respiratory tract. Biochem Soc Trans (2020) 48(1):327–36. doi: 10.1042/BST20191010
86. Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature (2018) 560(7718):319–24. doi: 10.1038/s41586-018-0393-7
87. Deprez M, Zaragosi LE, Truchi M, Becavin C, Ruiz Garcia S, Arguel MJ, et al. A Single-cell Atlas of the Human Healthy Airways. Am J Respiratory Crit Care Med (2020). doi: 10.1164/rccm.201911-2199OC
88. Plasschaert LW, Zilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature (2018) 560(7718):377–81. doi: 10.1038/s41586-018-0394-6
89. Parekh KR, Nawroth J, Pai A, Busch SM, Senger CN, Ryan AL. Stem Cells and Lung Regeneration. Am J Physiol Cell Physiol (2020). doi: 10.1152/ajpcell.00036.2020
90. Yang J, Zuo WL, Fukui T, Chao I, Gomi K, Lee B, et al. Smoking-Dependent Distal-to-Proximal Repatterning of the Adult Human Small Airway Epithelium. Am J Respiratory Crit Care Med (2017) 196(3):340–52. doi: 10.1164/rccm.201608-1672OC
91. Rogers DF. The airway goblet cell. Int J Biochem Cell Biol (2003) 35(1):1–6. doi: 10.1016/s1357-2725(02)00083-3
92. Schamberger AC, Staab-Weijnitz CA, Mise-Racek N, Eickelberg O. Cigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interface. Sci Rep (2015) 5:8163. doi: 10.1038/srep08163
93. Boers JE, Ambergen AW, Thunnissen FB. Number and proliferation of clara cells in normal human airway epithelium. Am J Respiratory Crit Care Med (1999) 159(5 Pt 1):1585–91. doi: 10.1164/ajrccm.159.5.9806044
94. Goldfarbmuren KC, Jackson ND, Sajuthi SP, Dyjack N, Li KS, Rios CL, et al. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat Commun (2020) 11(1):2485. doi: 10.1038/s41467-020-16239-z
95. Hor P, Punj V, Calvert BA, Castaldi A, Miller AJ, Carraro G, et al. Efficient Generation and Transcriptomic Profiling of Human iPSC-Derived Pulmonary Neuroendocrine Cells. iScience (2020) 23(5):101083. doi: 10.1016/j.isci.2020.101083
96. Crapo JD, Young SL, Fram EK, Pinkerton KE, Barry BE, Crapo RO. Morphometric characteristics of cells in the alveolar region of mammalian lungs. Am Rev Respiratory Dis (1983) 128(2 Pt 2):S42–6. doi: 10.1164/arrd.1983.128.2P2.S42
97. Varma R, Soleas JP, Waddell TK, Karoubi G, McGuigan AP. Current strategies and opportunities to manufacture cells for modeling human lungs. Adv Drug Delivery Rev (2020). doi: 10.1016/j.addr.2020.08.005
98. Georas SN, Rezaee F. Epithelial barrier function: at the front line of asthma immunology and allergic airway inflammation. J Allergy Clin Immunol (2014) 134(3):509–20. doi: 10.1016/j.jaci.2014.05.049
99. Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol (2016) 17(9):564–80. doi: 10.1038/nrm.2016.80
100. Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol (2004) 286(6):C1213–28. doi: 10.1152/ajpcell.00558.2003286/6/C1213[pii
101. Davies DE. Epithelial barrier function and immunity in asthma. Ann Am Thoracic Soc (2014) 11(Suppl 5):S244–51. doi: 10.1513/AnnalsATS.201407-304AW
102. Gon Y, Hashimoto S. Role of airway epithelial barrier dysfunction in pathogenesis of asthma. Allergol Int (2018) 67(1):12–7. doi: 10.1016/j.alit.2017.08.011
103. Ruan YC, Wang Y, Da Silva N, Kim B, Diao RY, Hill E, et al. CFTR interacts with ZO-1 to regulate tight junction assembly and epithelial differentiation through the ZONAB pathway. J Cell Sci (2014) 127(Pt 20):4396–408. doi: 10.1242/jcs.148098
104. Molina SA, Stauffer B, Moriarty HK, Kim AH, McCarty NA, Koval M. Junctional abnormalities in human airway epithelial cells expressing F508del CFTR. Am J Physiol (2015) 309(5):L475–87. doi: 10.1152/ajplung.00060.2015
105. De Rose V, Molloy K, Gohy S, Pilette C, Greene CM. Airway Epithelium Dysfunction in Cystic Fibrosis and COPD. Mediators Inflammation (2018) 2018:1309746. doi: 10.1155/2018/1309746
106. Gohy ST, Hupin C, Pilette C, Ladjemi MZ. Chronic inflammatory airway diseases: the central role of the epithelium revisited. Clin Exp Allergy (2016) 46(4):529–42. doi: 10.1111/cea.12712
107. Rezaee F, Georas SN. Breaking barriers. New insights into airway epithelial barrier function in health and disease. Am J Respiratory Cell Mol Biol (2014) 50(5):857–69. doi: 10.1165/rcmb.2013-0541RT
108. Sapoznikov A, Gal Y, Falach R, Sagi I, Ehrlich S, Lerer E, et al. Early disruption of the alveolar-capillary barrier in a ricin-induced ARDS mouse model: neutrophil-dependent and -independent impairment of junction proteins. Am J Physiol (2019) 316(1):L255–L68. doi: 10.1152/ajplung.00300.2018
109. Evans WH, Martin PE. Gap junctions: structure and function (Review). Mol Membr Biol (2002) 19(2):121–36. doi: 10.1080/09687680210139839
110. Wang N, De Bock M, Decrock E, Bol M, Gadicherla A, Vinken M, et al. Paracrine signaling through plasma membrane hemichannels. Biochim Biophys Acta (2013) 1828(1):35–50. doi: 10.1016/j.bbamem.2012.07.002
111. Sosinsky GE, Boassa D, Dermietzel R, Duffy HS, Laird DW, MacVicar B, et al. Pannexin channels are not gap junction hemichannels. Channels (Austin) (2011) 5(3):193–7. doi: 10.4161/chan.5.3.15765
112. Gruenert DC, Finkbeiner WE, Widdicombe JH. Culture and transformation of human airway epithelial cells. Am J Physiol (1995) 268(3 Pt 1):L347–60. doi: 10.1152/ajplung.1995.268.3.L347
113. Lopez-Guisa JM, Powers C, File D, Cochrane E, Jimenez N, Debley JS. Airway epithelial cells from asthmatic children differentially express proremodeling factors. J Allergy Clin Immunol (2012) 129(4):990–7 e6. doi: 10.1016/j.jaci.2011.11.035
114. Iwanaga K, Elliott MS, Vedal S, Debley JS. Urban particulate matter induces pro-remodeling factors by airway epithelial cells from healthy and asthmatic children. Inhal Toxicol (2013) 25(12):653–60. doi: 10.3109/08958378.2013.827283
115. Warner SM, Hackett TL, Shaheen F, Hallstrand TS, Kicic A, Stick SM, et al. Transcription factor p63 regulates key genes and wound repair in human airway epithelial basal cells. Am J Respiratory Cell Mol Biol (2013) 49(6):978–88. doi: 10.1165/rcmb.2012-0447OC
116. Lee HC, Headley MB, Loo YM, Berlin A, Gale M Jr., Debley JS, et al. Thymic stromal lymphopoietin is induced by respiratory syncytial virus-infected airway epithelial cells and promotes a type 2 response to infection. J Allergy Clin Immunol (2012) 130(5):1187–96 e5. doi: 10.1016/j.jaci.2012.07.031
117. Altman MC, Reeves SR, Parker AR, Whalen E, Misura KM, Barrow KA, et al. Interferon response to respiratory syncytial virus by bronchial epithelium from children with asthma is inversely correlated with pulmonary function. J Allergy Clin Immunol (2018) 142(2):451–9. doi: 10.1016/j.jaci.2017.10.004
118. Reeves SR, Kolstad T, Lien TY, Elliott M, Ziegler SF, Wight TN, et al. Asthmatic airway epithelial cells differentially regulate fibroblast expression of extracellular matrix components. J Allergy Clin Immunol (2014) 134(3):663–70 e1. doi: 10.1016/j.jaci.2014.04.007
119. Reeves SR, Kang I, Chan CK, Barrow KA, Kolstad TK, White MP, et al. Asthmatic bronchial epithelial cells promote the establishment of a Hyaluronan-enriched, leukocyte-adhesive extracellular matrix by lung fibroblasts. Respir Res (2018) 19(1):146. doi: 10.1186/s12931-018-0849-1
120. Reeves SR, Barrow KA, Kolstad TK, White MP, Rich LM, Wight TN, et al. Fibroblast gene expression following asthmatic bronchial epithelial cell conditioning correlates with epithelial donor lung function and exacerbation history. Sci Rep (2018) 8(1):15768. doi: 10.1038/s41598-018-34021-6
121. Altman MC, Lai Y, Nolin JD, Long S, Chen CC, Piliponsky AM, et al. Airway epithelium-shifted mast cell infiltration regulates asthmatic inflammation via IL-33 signaling. J Clin Invest (2019) 129(11):4979–91. doi: 10.1172/JCI126402
122. Wang Q, Bhattacharya S, Mereness JA, Anderson C, Lillis JA, Misra RS, et al. A novel in vitro model of primary human pediatric lung epithelial cells. Pediatr Res (2020) 87(3):511–7. doi: 10.1038/s41390-019-0340-9
123. Yamamoto Y, Gotoh S, Korogi Y, Seki M, Konishi S, Ikeo S, et al. Long-term expansion of alveolar stem cells derived from human iPS cells in organoids. Nat Methods (2017) 14(11):1097–106. doi: 10.1038/nmeth.4448
124. Jacob A, Morley M, Hawkins F, McCauley KB, Jean JC, Heins H, et al. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell (2017) 21(4):472–88 e10. doi: 10.1016/j.stem.2017.08.014
125. Autilio C, Perez-Gil J. Understanding the principle biophysics concepts of pulmonary surfactant in health and disease. Arch Dis Child Fetal Neonatal Ed (2019) 104(4):F443–F51. doi: 10.1136/archdischild-2018-315413
126. Echaide M, Autilio C, Arroyo R, Perez-Gil J. Restoring pulmonary surfactant membranes and films at the respiratory surface. Biochim Biophys Acta Biomembr (2017) 1859(9 Pt B):1725–39. doi: 10.1016/j.bbamem.2017.03.015
127. Suzuki T, Trapnell BC. Pulmonary Alveolar Proteinosis Syndrome. Clin Chest Med (2016) 37(3):431–40. doi: 10.1016/j.ccm.2016.04.006
128. Hiemstra PS. Epithelial antimicrobial peptides and proteins: their role in host defence and inflammation. Paediatr Respir Rev (2001) 2(4):306–10. doi: 10.1053/prrv.2001.0165
129. Bals R, Hiemstra PS. Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur Respir J (2004) 23(2):327–33. doi: 10.1183/09031936.03.00098803
130. Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, et al. Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol Immunol (2006) 43(9):1293–315. doi: 10.1016/j.molimm.2005.08.004
131. Casals C, Campanero-Rhodes MA, Garcia-Fojeda B, Solis D. The Role of Collectins and Galectins in Lung Innate Immune Defense. Front Immunol (2018) 9:1998:1998. doi: 10.3389/fimmu.2018.01998
132. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34(5):637–50. doi: 10.1016/j.immuni.2011.05.006
133. Matikainen S, Siren J, Tissari J, Veckman V, Pirhonen J, Severa M, et al. Tumor necrosis factor alpha enhances influenza A virus-induced expression of antiviral cytokines by activating RIG-I gene expression. J Virol (2006) 80(7):3515–22. doi: 10.1128/JVI.80.7.3515-3522.2006
134. Sorrentino R, de Souza PM, Sriskandan S, Duffin C, Paul-Clark MJ, Mitchell JA. Pattern recognition receptors and interleukin-8 mediate effects of Gram-positive and Gram-negative bacteria on lung epithelial cell function. Br J Pharmacol (2008) 154(4):864–71. doi: 10.1038/bjp.2008.139
135. Unger BL, Ganesan S, Comstock AT, Faris AN, Hershenson MB, Sajjan US. Nod-like receptor X-1 is required for rhinovirus-induced barrier dysfunction in airway epithelial cells. J Virol (2014) 88(7):3705–18. doi: 10.1128/JVI.03039-13
136. Yeh CC, Horng HC, Chou H, Tai HY, Shen HD, Hsieh SL, et al. Dectin-1-Mediated Pathway Contributes to Fusarium proliferatum-Induced CXCL-8 Release from Human Respiratory Epithelial Cells. Int J Mol Sci (2017) 18(3):1–10. doi: 10.3390/ijms18030624
137. Hirota JA, Hirota SA, Warner SM, Stefanowicz D, Shaheen F, Beck PL, et al. The airway epithelium nucleotide-binding domain and leucine-rich repeat protein 3 inflammasome is activated by urban particulate matter. J Allergy Clin Immunol (2012) 129(4):1116–25 e6. doi: 10.1016/j.jaci.2011.11.033
138. Chan MC, Cheung CY, Chui WH, Tsao SW, Nicholls JM, Chan YO, et al. Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir Res (2005) 6:135. doi: 10.1186/1465-9921-6-135
139. Matsukura S, Kokubu F, Kurokawa M, Kawaguchi M, Ieki K, Kuga H, et al. Synthetic double-stranded RNA induces multiple genes related to inflammation through Toll-like receptor 3 depending on NF-kappaB and/or IRF-3 in airway epithelial cells. Clin Exp Allergy (2006) 36(8):1049–62. doi: 10.1111/j.1365-2222.2006.02530.x
140. Stegemann-Koniszewski S, Jeron A, Gereke M, Geffers R, Kroger A, Gunzer M, et al. Alveolar Type II Epithelial Cells Contribute to the Anti-Influenza A Virus Response in the Lung by Integrating Pathogen- and Microenvironment-Derived Signals. mBio (2016) 7(3):1–11. doi: 10.1128/mBio.00276-16
141. Mayer AK, Bartz H, Fey F, Schmidt LM, Dalpke AH. Airway epithelial cells modify immune responses by inducing an anti-inflammatory microenvironment. Eur J Immunol (2008) 38(6):1689–99. doi: 10.1002/eji.200737936
142. Holtzman MJ, Hansbrough JR, Rosen GD, Turk J. Uptake, release and novel species-dependent oxygenation of arachidonic acid in human and animal airway epithelial cells. Biochim Biophys Acta (1988) 963(3):401–13. doi: 10.1016/0005-2760(88)90308-6
143. Bonnans C, Gras D, Chavis C, Mainprice B, Vachier I, Godard P, et al. Synthesis and anti-inflammatory effect of lipoxins in human airway epithelial cells. BioMed Pharmacother (2007) 61(5):261–7. doi: 10.1016/j.biopha.2007.02.016
144. Lloyd CM, Saglani S. Epithelial cytokines and pulmonary allergic inflammation. Curr Opin Immunol (2015) 34:52–8. doi: 10.1016/j.coi.2015.02.001
145. Lai JF, Thompson LJ, Ziegler SF. TSLP drives acute TH2-cell differentiation in lungs. J Allergy Clin Immunol (2020). doi: 10.1016/j.jaci.2020.03.032
146. Gao W, Li L, Wang Y, Zhang S, Adcock IM, Barnes PJ, et al. Bronchial epithelial cells: The key effector cells in the pathogenesis of chronic obstructive pulmonary disease? Respirology (2015) 20(5):722–9. doi: 10.1111/resp.12542
147. Jiang-Shieh YF, Chien HF, Chang CY, Wei TS, Chiu MM, Chen HM, et al. Distribution and expression of CD200 in the rat respiratory system under normal and endotoxin-induced pathological conditions. J Anat (2010) 216(3):407–16. doi: 10.1111/j.1469-7580.2009.01190.x
148. Stanciu LA, Bellettato CM, Laza-Stanca V, Coyle AJ, Papi A, Johnston SL. Expression of programmed death-1 ligand (PD-L) 1, PD-L2, B7-H3, and inducible costimulator ligand on human respiratory tract epithelial cells and regulation by respiratory syncytial virus and type 1 and 2 cytokines. J Infect Dis (2006) 193(3):404–12. doi: 10.1086/499275
149. Shinohara M, Ohyama N, Murata Y, Okazawa H, Ohnishi H, Ishikawa O, et al. CD47 regulation of epithelial cell spreading and migration, and its signal transduction. Cancer Sci (2006) 97(9):889–95. doi: 10.1111/j.1349-7006.2006.00245.x
150. El-Mokhtar MA, Bauer A, Madela J, Voigt S. Cellular distribution of CD200 receptor in rats and its interaction with cytomegalovirus e127 protein. Med Microbiol Immunol (2018) 207(5-6):307–18. doi: 10.1007/s00430-018-0552-3
151. Hu JF, Zhang W, Zuo W, Tan HQ, Bai W. Inhibition of the PD-1/PD-L1 signaling pathway enhances innate immune response of alveolar macrophages to mycobacterium tuberculosis in mice. Pulmonary Pharmacol Ther (2020) 60:101842. doi: 10.1016/j.pupt.2019.101842
152. Li J, Jin C, Wu C, Huang J. PD-1 modulating Mycobacterium tuberculosis-specific polarized effector memory T cells response in tuberculosis pleurisy. J Leukoc Biol (2019) 106(3):733–47. doi: 10.1002/JLB.MA1118-450RR
153. Okazawa H, Motegi S, Ohyama N, Ohnishi H, Tomizawa T, Kaneko Y, et al. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J Immunol (2005) 174(4):2004–11. doi: 10.4049/jimmunol.174.4.2004
154. Janssen WJ, McPhillips KA, Dickinson MG, Linderman DJ, Morimoto K, Xiao YQ, et al. Surfactant proteins A and D suppress alveolar macrophage phagocytosis via interaction with SIRP alpha. Am J Respiratory Crit Care Med (2008) 178(2):158–67. doi: 10.1164/rccm.200711-1661OC
155. Oumouna M, Weitnauer M, Mijosek V, Schmidt LM, Eigenbrod T, Dalpke AH. Cell-contact dependent inhibition of monocytes by airway epithelial cells and reversion by infection with Respiratory Syncytial Virus. Immunobiology (2015) 220(11):1240–5. doi: 10.1016/j.imbio.2015.06.017
156. Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol (2014) 32:25–50. doi: 10.1146/annurev-immunol-032713-120142
157. Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature (2014) 506(7489):503–6. doi: 10.1038/nature12902
158. Al-Ghadban S, Kaissi S, Homaidan FR, Naim HY, El-Sabban ME. Cross-talk between intestinal epithelial cells and immune cells in inflammatory bowel disease. Sci Rep (2016) 6:29783. doi: 10.1038/srep29783
159. Beckmann A, Grissmer A, Meier C, Tschernig T. Intercellular communication between alveolar epithelial cells and macrophages. Ann Anat (2020) 227:151417. doi: 10.1016/j.aanat.2019.151417
160. Battistelli M, Falcieri E. Apoptotic Bodies: Particular Extracellular Vesicles Involved in Intercellular Communication. Biol (Basel) (2020) 9(1):1–10. doi: 10.3390/biology9010021
161. ELA S, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discovery (2013) 12(5):347–57. doi: 10.1038/nrd3978
162. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol (2013) 200(4):373–83. doi: 10.1083/jcb.201211138
163. van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol (2018) 19(4):213–28. doi: 10.1038/nrm.2017.125
164. Admyre C, Grunewald J, Thyberg J, Gripenback S, Tornling G, Eklund A, et al. Exosomes with major histocompatibility complex class II and co-stimulatory molecules are present in human BAL fluid. Eur Respir J (2003) 22(4):578–83. doi: 10.1183/09031936.03.00041703
165. Gupta R, Radicioni G, Abdelwahab S, Dang H, Carpenter J, Chua M, et al. Intercellular Communication between Airway Epithelial Cells Is Mediated by Exosome-Like Vesicles. Am J Respiratory Cell Mol Biol (2019) 60(2):209–20. doi: 10.1165/rcmb.2018-0156OC
166. Kulshreshtha A, Ahmad T, Agrawal A, Ghosh B. Proinflammatory role of epithelial cell-derived exosomes in allergic airway inflammation. J Allergy Clin Immunol (2013) 131(4):1194–203, 203 e1-14. doi: 10.1016/j.jaci.2012.12.1565
167. Kesimer M, Scull M, Brighton B, DeMaria G, Burns K, O’Neal W, et al. Characterization of exosome-like vesicles released from human tracheobronchial ciliated epithelium: a possible role in innate defense. FASEB J (2009) 23(6):1858–68. doi: 10.1096/fj.08-119131
168. Soni S, Wilson MR, O’Dea KP, Yoshida M, Katbeh U, Woods SJ, et al. Alveolar macrophage-derived microvesicles mediate acute lung injury. Thorax (2016) 71(11):1020–9. doi: 10.1136/thoraxjnl-2015-208032
169. Bourdonnay E, Zaslona Z, Penke LR, Speth JM, Schneider DJ, Przybranowski S, et al. Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. J Exp Med (2015) 212(5):729–42. doi: 10.1084/jem.20141675
170. Speth JM, Penke LR, Bazzill JD, Park KS, de Rubio RG, Schneider DJ, et al. Alveolar macrophage secretion of vesicular SOCS3 represents a platform for lung cancer therapeutics. JCI Insight (2019) 4(20):1–16. doi: 10.1172/jci.insight.131340
171. Lee H, Zhang D, Zhu Z, Dela Cruz CS, Jin Y. Epithelial cell-derived microvesicles activate macrophages and promote inflammation via microvesicle-containing microRNAs. Sci Rep (2016) 6:35250. doi: 10.1038/srep35250
172. Fujita Y, Araya J, Ito S, Kobayashi K, Kosaka N, Yoshioka Y, et al. Suppression of autophagy by extracellular vesicles promotes myofibroblast differentiation in COPD pathogenesis. J Extracell Vesicles (2015) 4:28388. doi: 10.3402/jev.v4.28388
173. Lee H, Zhang D, Wu J, Otterbein LE, Jin Y. Lung Epithelial Cell-Derived Microvesicles Regulate Macrophage Migration via MicroRNA-17/221-Induced Integrin beta1 Recycling. J Immunol (2017) 199(4):1453–64. doi: 10.4049/jimmunol.1700165
174. Guervilly C, Lacroix R, Forel JM, Roch A, Camoin-Jau L, Papazian L, et al. High levels of circulating leukocyte microparticles are associated with better outcome in acute respiratory distress syndrome. Crit Care (2011) 15(1):R31. doi: 10.1186/cc9978
175. Bastarache JA, Fremont RD, Kropski JA, Bossert FR, Ware LB. Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome. Am J Physiol (2009) 297(6):L1035–41. doi: 10.1152/ajplung.00214.2009
176. Moon HG, Cao Y, Yang J, Lee JH, Choi HS, Jin Y. Lung epithelial cell-derived extracellular vesicles activate macrophage-mediated inflammatory responses via ROCK1 pathway. Cell Death Dis (2015) 6:e2016. doi: 10.1038/cddis.2015.282
177. Kadota T, Fujita Y, Yoshioka Y, Araya J, Kuwano K, Ochiya T. Extracellular Vesicles in Chronic Obstructive Pulmonary Disease. Int J Mol Sci (2016) 17(11):1–16. doi: 10.3390/ijms17111801
178. Draijer C, Speth JM, Penke LRK, Zaslona Z, Bazzill JD, Lugogo N, et al. Resident alveolar macrophage-derived vesicular SOCS3 dampens allergic airway inflammation. FASEB J (2020) 34(3):4718–31. doi: 10.1096/fj.201903089R
179. Prado N, Marazuela EG, Segura E, Fernandez-Garcia H, Villalba M, Thery C, et al. Exosomes from bronchoalveolar fluid of tolerized mice prevent allergic reaction. J Immunol (2008) 181(2):1519–25. doi: 10.4049/jimmunol.181.2.1519
180. Hiemstra PS, Tetley TD, Janes SM. Airway and alveolar epithelial cells in culture. Eur Respir J (2019) 54(5):1–5. doi: 10.1183/13993003.00742-2019
Keywords: alveolar macrophages, airway epithelial cells, cytokines, extracellular vesicles, inflammation, macrophage heterogeneity
Citation: Bissonnette EY, Lauzon-Joset J-F, Debley JS and Ziegler SF (2020) Cross-Talk Between Alveolar Macrophages and Lung Epithelial Cells is Essential to Maintain Lung Homeostasis. Front. Immunol. 11:583042. doi: 10.3389/fimmu.2020.583042
Received: 29 July 2020; Accepted: 30 September 2020;
Published: 15 October 2020.
Edited by:
Guochang Hu, University of Illinois at Chicago, United StatesReviewed by:
Beth Garvy, University of Kentucky, United StatesYang Jin, Boston University, United States
Jennifer Speth, University of Michigan, United States
Copyright © 2020 Bissonnette, Lauzon-Joset, Debley and Ziegler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elyse Y. Bissonnette, ZWx5c2UuYmlzc29ubmV0dGVAZm1lZC51bGF2YWwuY2E=