
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 15 October 2020
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.582106
This article is part of the Research TopicThe State-of-Art in Immuno-oncology, what to do with Glioblastoma?View all 27 articles
 Joshua R. D. Pearson1,2
Joshua R. D. Pearson1,2 Stefania Cuzzubbo3,4
Stefania Cuzzubbo3,4 Simon McArthur5
Simon McArthur5 Lindy G. Durrant6Jason Adhikaree7
Lindy G. Durrant6Jason Adhikaree7 Chris J. Tinsley1,2
Chris J. Tinsley1,2 A. Graham Pockley1,2
A. Graham Pockley1,2 Stephanie E. B. McArdle1,2*
Stephanie E. B. McArdle1,2*Glioblastoma multiforme (GBM) is the most frequently occurring primary brain tumor and has a very poor prognosis, with only around 5% of patients surviving for a period of 5 years or more after diagnosis. Despite aggressive multimodal therapy, consisting mostly of a combination of surgery, radiotherapy, and temozolomide chemotherapy, tumors nearly always recur close to the site of resection. For the past 15 years, very little progress has been made with regards to improving patient survival. Although immunotherapy represents an attractive therapy modality due to the promising pre-clinical results observed, many of these potential immunotherapeutic approaches fail during clinical trials, and to date no immunotherapeutic treatments for GBM have been approved. As for many other difficult to treat cancers, GBM combines a lack of immunogenicity with few mutations and a highly immunosuppressive tumor microenvironment (TME). Unfortunately, both tumor and immune cells have been shown to contribute towards this immunosuppressive phenotype. In addition, current therapeutics also exacerbate this immunosuppression which might explain the failure of immunotherapy-based clinical trials in the GBM setting. Understanding how these mechanisms interact with one another, as well as how one can increase the anti-tumor immune response by addressing local immunosuppression will lead to better clinical results for immune-based therapeutics. Improving therapeutic delivery across the blood brain barrier also presents a challenge for immunotherapy and future therapies will need to consider this. This review highlights the immunosuppressive mechanisms employed by GBM cancers and examines potential immunotherapeutic treatments that can overcome these significant immunosuppressive hurdles.
Glioblastoma multiforme (GBM, WHO grade 4) is the most frequently occurring primary brain tumor. Although primarily a disease associated with old age, it can also occur in children. The prognosis for GBM patients is poor and the disease is almost uniformly fatal with only around 5% of patients surviving for a period of 5 years after diagnosis (1). The current course of therapy for GBM patients is surgical resection of the tumor (where possible) followed by concomitant radiotherapy and temozolomide chemotherapy, followed by adjuvant temozolomide. Despite aggressive multimodal therapy, GBM tumors nearly always recur, the majority close to the site of resection (2–4). This recurrence is most likely, and most often, due to the infiltrative nature of GBM making complete resection of tumor cells incredibly difficult. Although progress to improve the surgical removal of tumor cells has been made, such as the use of 5-aminolevulinic acid (5-ALA) which is approved for intraoperative imaging of GBM cells increasing their removal, it is not possible to visualize all individual cancer cells that have migrated further into healthy areas of the brain (5).
GBM tumors are histopathologically characterized by an abundance of poorly differentiated and pleomorphic astrocytes with nuclear atypia and high mitotic activity. GBM tumors are highly vascular and necrosis is often evident within these tumors (6). Metastasis is rarely seen in GBM tumors; however, they are highly invasive, and these tumors employ a plethora of mechanisms to avoid immune detection.
In order to understand the complexity of the brain’s interaction with the immune system, the presence of the blood-brain barrier (BBB) needs to be considered and understood. The endothelial cells of the brain vasculature are connected by tight junctions that control the permeability of the endothelium. Although these tight junctions under normal physiological conditions are highly regulated, under inflammatory conditions (such as those in GBM) these junctions are not as tightly connected making the endothelium ‘leaky’ (7). The BBB, a multi-component structure found in the wall of cerebral blood vessels, selectively restricts passage of cells and molecules into the brain from the circulation. The major, but not sole, players in this defense are the endothelial cells of the cerebral vessels, which differ from their peripheral counterparts by the presence of intercellular tight junctions that essentially prevent paracellular transfer of all, but the smallest gases and ions, and the absence of fenestrations and pinocytic mechanisms that restricts bulk transcytosis (8, 9). These features are then reinforced by the presence of numerous efflux transporters that remove xenobiotics and metabolic waste from the brain into the circulation. Beyond the endothelium, the BBB is further composed of a bi-layered basement membrane within which reside pericytes and perivascular macrophages that regulate endothelial function and pose a further barrier to cellular entry, ultimately surrounded by a tight glia limitans formed of astrocyte end-feet that appose and encircle the blood vessel (8, 9).
GBM tumors contain areas of highly metabolic cells that drive local hypoxia, triggering production of vascular endothelial growth factor and angiogenesis (10). This process involves disruption of inter-endothelial tight junctions to permit vascular growth, hence the core of the tumor is associated with a weakened blood-tumor barrier (BTB) with an increased permeability (11, 12). Nevertheless, areas of GBM tumors distal from the hypoxic core, which in diffuse tumors can be a significant proportion, remain behind a BTB that is highly reminiscent of the true BBB, and are thus protected from the entry of chemotherapeutic agents, including most therapeutic antibodies (13). However, these difficulties do not mean that the delivery of effective therapeutics for GBM is futile, and a wide variety of approaches to achieve this are under active exploration.
The brain has traditionally been considered as being an immunoprivileged organ due to a variety of factors, however it is now accepted that there is an active interaction between the brain and the immune system (14, 15). Despite this active immune interaction, the brain is immunologically unique in that immune cells do not freely access the brain parenchyma. Although activated immune cells can cross the BBB, only those specific for antigens within the brain remain there. T cells cross the BBB in a capture, crawl, cross manner with integrins and selectin ligands on T cells binding to selectins and integrin ligands on endothelial cells ‘capturing’ them (16). Leukocytes are then activated by chemokine secretion resulting in their slowing and eventual transmigration. Once T cells have transmigrated, they downregulate their integrin expression and upregulate expression of matrix metalloproteinases (MMPs) enabling them to break down matrix components allowing cell penetration of the brain parenchyma (7). Inflammation within the brain has been shown to lead to an upregulation of adhesion molecules on the BBB endothelial cells (16, 17). The endothelial cells of the brain vasculature do not just control the immune response by physically excluding immune cells, these cells have also been shown to contribute to immunosuppression in GBM. FasL expression has been seen on GBM associated vascular endothelial cells, and the FasL expressed on these cells is linked to a reduced T cell infiltrate, most likely due to the FasL induced death of T cells (18). There are very few immune cells normally present within the brain, however the microglia can act as antigen presenting cells (APCs). The brain traditionally has low major histocompatibility complex (MHC) class I and class II expression meaning that antigen expression is reduced when compared to other tissues (19). It is important to note, however, that GBM cells themselves have been shown to express MHC class I and II molecules meaning that these cells present antigens to antigen specific CD8+ and CD4+ T cells (20).
Not only does the unique physiology of the brain create an unusual immune environment but it is important to note that the tumors themselves create their own microenvironment. Tumor cells can co-opt stromal cells in order to support their growth and survival (21). The brain extracellular matrix is comprised of proteoglycans, glycoproteins and glycosaminoglycans. In the GBM setting, significant increases in heparan sulphate proteoglycans (HSPG) have been seen in the tumor microenvironment (TME) (22). The increase of HSPGs in the TME leads to greater retention of growth factors such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF), thereby supporting tumor nutrition and growth. The increased local concentration of VEGF within the GBM TME results in upregulation of periostin and tenascin C within blood vessels trapping T cells and preventing tumor penetrance (23).
In the case of GBM, as with many cancers T cells are frequently exhausted and dysfunctional and therefore are inadequate at exerting an anti-tumor immune response. Persistent stimulation of T cells by tumor cells results in T cell senescence as indicated by the presence of CD57 on the surface of T cells (24). CD57 positive T cells can secrete cytokines when stimulated by their cognate peptides however they do not proliferate when stimulated (25). Tumor resident senescent T cells have also been shown to down regulate the co-stimulatory molecules CD27 and CD28 contributing to immune dysfunction, causing changes in APC phenotype such as a down regulation of CD80 and CD86 reducing their ability to stimulate T cells further exacerbating the local immune dysfunction (26). When compared to healthy donors GBM patients have a lower number of circulating CD3+ T cells in their peripheral blood mononuclear cells (PBMCs) further indicating a disease related immune dysfunction (27). Glioblastoma multiforme is more frequent in the older population with most cases occurring between the ages of 55 and 60 (28). Increased age is linked to T cell dysfunction; with elderly patients having a higher number of senescent T cells and thymic shrinkage being apparent (24, 29). The chronic stimulation of T cells by tumor cells also leads to the exhaustion of these cells, rendering them ineffective at tumor control. This exhaustion leads to an upregulation of immune checkpoint markers such as PD-1, LAG-3, TIGIT, and CD39 on GBM infiltrating CD8+ T cells (30). TILs isolated from murine GBM tumors show impaired cytokine production compared to peripheral T cells, with reduced levels of interferon gamma, tumor necrosis factor alpha and interleukin 2 being detected via flow cytometry when cells are stimulated in vitro (30). Transformed tumor cells also compete with other cells within the TME for glucose, GBM cells have an increased rate of glucose uptake when compared to non-transformed cells. T cells within the TME require glucose in order to perform effector functions and therefore the depletion of glucose by tumor cells results in impaired T cell function and exhaustion (31).
The current standard of care for GBM is maximal surgical resection (where possible) followed by concomitant radiotherapy and temozolomide chemotherapy (32). Patients are also given anti-inflammatory steroids such as dexamethasone to help control peritumoral edema (33). The US Food and Drug Administration (FDA) has also approved the use of tumor treating fields (TTFs) to treat GBMs. This involves using alternating electric fields administered via scalp electrodes to disrupt GBM tumor cell division (34).
Dexamethasone has been shown to lead to the upregulation of the immunosuppressive checkpoint cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) on the surface of T cells, thereby reducing their anti-tumor activity. Dexamethasone has also been shown to lead to a reduction of T cell proliferation (35). Dexamethasone has also been shown to dampen patients’ immune responses to immune checkpoint blockade (36).
As previously mentioned, the standard of care involves the use of the chemotherapeutic drug temozolomide (TMZ), which is known to influence the immune system. High dose temozolomide induces lymphopenia, an issue that is exacerbated when TMZ is combined with radiotherapy (37). TMZ has also been shown to result in T and B cell dysfunction in a murine model of GBM (38).
In the GBM setting, radiotherapy can be administered in a variety of ways such as whole brain radiotherapy, stereotactic radiosurgery, image guided radiotherapy and hypofractinated radiotherapy (39). Radiotherapy is known to have a number immune modulating effects (40–42), importantly brain tumor exposure to radiotherapy has been shown to upregulate MHC class I expression by brain tumors, and this improves the antigen presentation capability of these cells. Radiotherapy also increases the repertoire of peptides presented by tumor cells and the phenomenon of antigen spreading can occur – i.e. tumor cells die, and their antigens are taken up by nearby immune cells (43). Research has shown that radiotherapy is less efficient in mice lacking T cells, thereby highlighting the additive effect that radiotherapy has in immune cell-mediated control of cancer (44). Radiotherapy is often thought of as an in-situ vaccination that makes tumors susceptible to immune attack (44–46). Although a large amount of evidence points towards radiotherapy stimulating an anti-tumor immune response, radiotherapy can also unfortunately result in the secretion of immunosuppressive cytokines such as IL-6 and IL-10 from treated tumor cells (47, 48).
Combined TMZ, radiotherapy and dexamethasone therapy in GBM patients has been shown to induce a persistent lowering of CD4+ cell counts which is associated with increased rates of infection and poorer survival (49).
GBM cells secrete many immunosuppressive proteins and express many cell surface and cytoplasmic immune inhibitory proteins (as summarized in Figure 1). Intracellular adhesion molecule 1 (ICAM-1), a key regulator of cell-cell interactions, is commonly upregulated within GBM tumors, when compared to immunohistochemically stained normal brain (50). ICAM-1 interacts with lymphocyte function-associated antigen 1 (LFA-1) expressed on myeloid cells to promote migration of these cells into tumors, thereby enhancing intratumoral immune suppression (51). Myeloid derived suppressor cell (MDSC) accumulation in GBM tumors further contributes to local immune suppression (52). The presence of MDSCs circulating in the blood of GBM patients is also elevated when compared to non-diseased individuals (53). These MDSCs express many immunosuppressive molecules that suppress anti-tumor T cells such as TGF-β and arginase (52). GBM cells have been shown to overexpress galectin-1 (Gal-1), another protein important for the maintenance of cell-cell interactions. Expression of Gal-1 by GBM cells promotes the proliferation and migration of tumor cells (54, 55). Gal-1 expressing GBM cells have also been shown to induce T cell death when the two types of cells are co-cultured (55). Gal-1 interacts with CD45 and CD43 on T cells resulting in their clustering. Gal-1 also binds to CD7 on the T cells and these interactions result in T cell death (56–58).
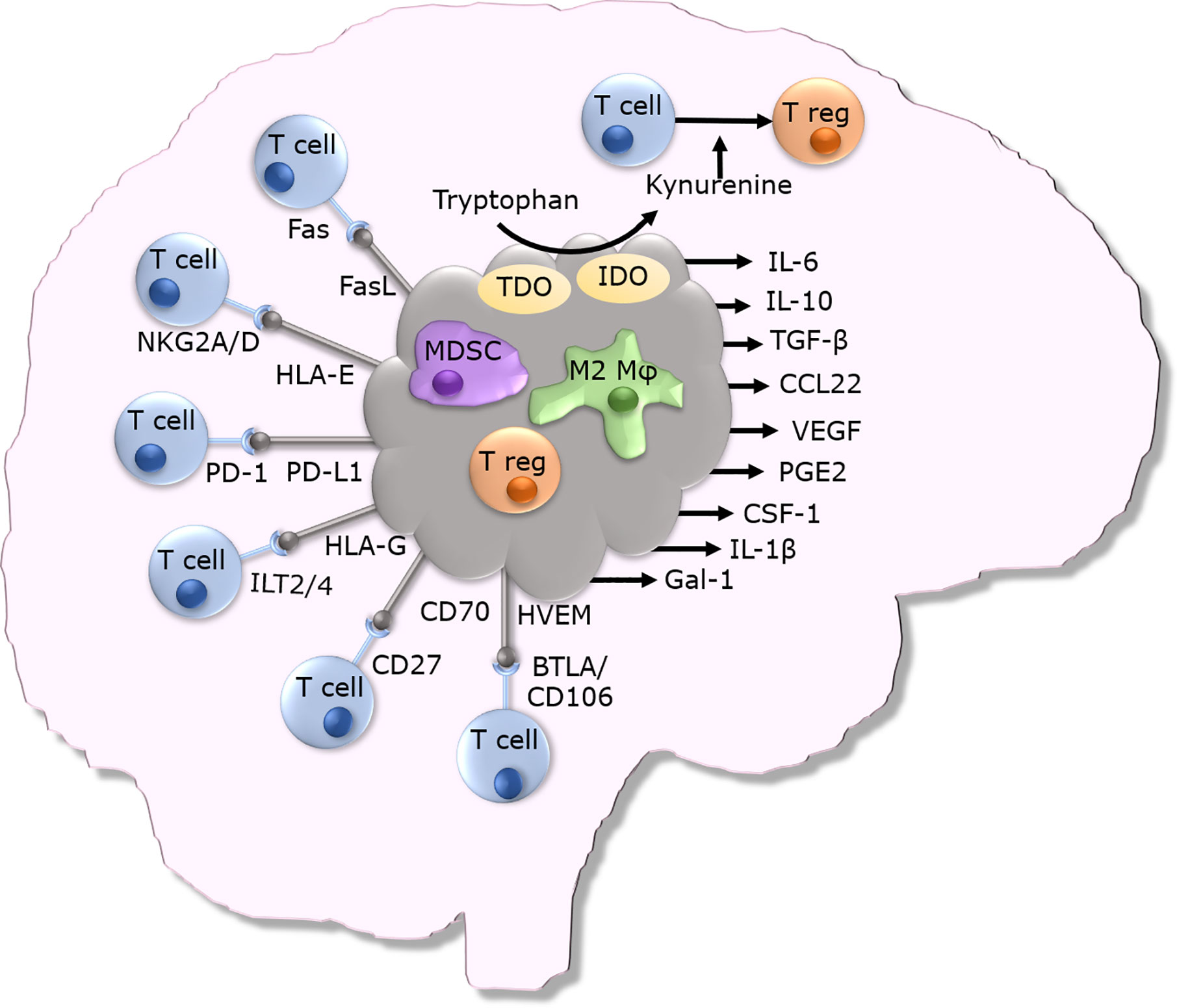
Figure 1 Overview of immunosuppressive mechanisms utilized by GBM tumors.
GBM cells have also been shown to express non-classical MHC class I molecules on their surface which enables them to evade immune cell mediated killing. HLA-G is one such non-classical MHC class I molecule that is involved in immunogenic tolerance of trophoblasts and prevents immune response to the developing semi-allogeneic fetus. In the adult, HLA-G is expressed in thymic epithelial cells, nail matrix and cornea (59). Although HLA-G expression is tightly controlled in the human body, it appears that GBM cells can express HLA-G (59). HLA-G is not just expressed on the cell surface - a soluble isoform that is secreted has been detected in plasma, cerebrospinal fluid and seminal plasma. GBM tumors are frequently infected with cytomegalovirus (hCMV), and hCMV infection has been associated with high levels of HLA-G expression (60). Cytomegalovirus infection is prevalent in the population and infection is lifelong. The immunosuppression linked with GBM results in reduced control of hCMV and this results in reactivation of the virus (60). HLA-G can bind to several receptors, namely the inhibitory receptors Ig-Like Transcript 2 (ILT2) and Ig-Like Transcript 4 (ILT4) (61). HLA-G can also bind the non-inhibitory receptors CD8, CD160, and KIR2DL4. Binding of soluble HLA-G to CD8 on T cells induces a signaling cascade that results in Fas-FasL mediated apoptosis of CD8+ T cells (61). HLA-G binding to ILT2 on natural killer (NK) cells inhibits the polarization of lytic granules and the microtubule-organizing center at the contact zone, ultimately preventing NK cell-mediated lysis (61). HLA-E is another non-classical MHC class I molecule; it is a ligand for both NKG2A and NKG2C expressed on NK cells, CD8+ αβ and γδ T cells. Binding of HLA-E to NKG2C can lead to immune cell activation, and its binding to NKG2A leads to immune cell inhibition. HLA-E, much like HLA-G, is believed to play a role in maternal tolerance of the fetus (62). HLA-E has been shown to be expressed on GBM cells and this HLA-E expression has been shown to prevent NK cell mediated lysis of these tumor cells. Blockade of the NKG2A – HLA-E interaction has been shown to improve NK cell mediated killing of GBM tumor cells (62).
GBM tumors have also been shown to express Fas ligand (CD95L) on their surface, the binding of which to Fas (CD95/APO-1) on T cells leads to apoptosis of the T cells, thereby enabling GBM cells to evade lysis by Fas-expressing T cells (63). GBMs can also induce T cell death via their expression of CD70. CD70 on GBM cells binds to CD27 on T cells inducing death of activated T cells, and blockade of this interaction has been shown to partially protect T cells from GBM cell induced death (64). GBMs have also been shown to express the immune dampening checkpoint ligand programmed death ligand 1 (PD-L1). PD-L1 binds to its cognate receptor programmed death 1 (PD-1) expressed on activated T cells, and this leads to inhibition of T cell responses to PD-L1 expressing GBM cells. It has been reported that as many as 88% of patient GBM samples express PD-L1 (65). This high level of PD-L1 expression has been shown to be linked with poorer patient survival (66).
Herpes virus entry mediator (HVEM) is an example of another immune checkpoint molecule that has been proven to be expressed in the GBM microenvironment (67). HVEM is usually expressed on T cells, it can have both co-stimulatory and inhibitory effects, depending upon its binding partner (67). HVEM exerts an immune inhibitory effect when bound to B and T lymphocyte attenuator (BTLA) or CD160 expressed by other immune cells (67). High expression of HVEM in GBM tumors has been linked to regulatory T cell differentiation, negatively associated with the regulation of T cell mediated cytotoxicity and with a decreased survival time (67).
Indoleamine 2,3-dioxygenase (IDO) is another protein involved in immunoregulation and prevention of fetal rejection. IDO catabolizes tryptophan into immune-regulatory kynurenines. IDO expression can be induced by a variety of receptors such as the toll like receptors (TLRs), tumor necrosis factor receptor superfamily members (TNFRs), interferon gamma receptors (IFNGRs), transforming growth factor beta receptors (TGFBRs) and aryl hydrocarbon receptors (AhRs) (68). The depletion of tryptophan by IDO activity inhibits immune cell function and prevents dendritic cell (DC) maturation (68). IDO expression is upregulated in recurrent GBMs, with 100% of patients being studied expressing IDO at the time of the second surgery (69). The expression of IDO within GBM tumors is associated with an increased infiltration of CD4+ regulatory T cells, immune escape and a poorer prognosis (70). Increased kynurenine production driven by IDO activity induces the differentiation of naïve CD4+ T cells into immunosuppressive regulatory CD4+ T cells triggered by the binding of kynurenine to the aryl hydrocarbon receptor (AHR) on naïve CD4+ T cells (71). Tryptophan 2,3-dioxygenase (TDO), another enzyme involved in the degradation of tryptophan into kynurenine, can also contribute to an immunosuppressive microenvironment high in kynurenine. TDO is expressed in brain tumors and represents a druggable target for reversing the immunosuppressive microenvironment (72).
GBM tumors also secrete numerous other immunosuppressive factors that shape the immune TME and enable immune evasion. GBM tumors secrete IL-6 (73, 74) and their expression of the IL-6 receptor is upregulated (75). IL-6 mediates signaling via the transcription factor STAT3. Upon activation, STAT3 is phosphorylated and persistent phosphorylation is linked with brain tumor grade; with GBM showing the highest levels of STAT3 phosphorylation. Knockdown of STAT3 in GBM cell lines slows in vitro and in vivo tumor cell growth (76). Human GBM cells isolated from tumors were shown to secrete the chemokine CCL22 (77) which attracts regulatory CD4+ CD25+ FoxP3+ T cells to the TME (78). GBM tumor cells also secrete the immunosuppressive cytokine TGF-β which reduces ICAM-1 and VCAM-1 expression on GBM endothelial cells and thereby T cell infiltration (79, 80). The active form of TGF-β secreted by GBM cells increases the activity of MMP2 and MMP9 on the surface of GBM cells which in turn increases cell motility and promotes the invasion of GBM cells into the surrounding brain (81). GBM tumor cells also secrete the anti-inflammatory cytokine IL-10 which, in the normal setting prevents excessive inflammation and reduces tissue damage by suppressing the activity of Th1 and CD8+ T cells. Immune cells such as regulatory T cells secrete IL-10 to quell the immune response (82). IL-10 mRNA is highly expressed in GBM tissues (83). More concerning is that IL-10 not only suppresses the immune system, but also increases the proliferation and migration of GBM cells. Intratumoral microglia/macrophages are major contributors to the IL-10 production within GBM tumors (84).
Human cytomegalovirus (hCMV) is a herpes virus that has been shown to persistently infect 50% to 90% of the adult population. Analysis of GBM tumors has revealed that a large proportion of tumors express hCMV proteins indicating the presence of hCMV within these tumors (85, 86). Human cytomegalovirus secretes a homolog of IL-10, known as cmvIL-10 which has the same immunoinhibitory properties as human IL-10 (87). The attenuation of the immune response by cmvIL-10 prevents eradication of the tumor as well as the virus itself. The secretion of cmvIL-10 leads to the differentiation of CD14+ monocytes to macrophages, thereby further supporting hCMV infection (88). In vitro studies have revealed that cmvIL-10 affects the maturation and life span of DCs, in that although monocytic DCs exposed to cmvIL-10 reach maturation, their cytokine production is impaired in a non-reversible manner (88). The presence of IL-10 and TGF-β in GBM tumors is believed to downregulate the expression of MHC class I in the TME (89). GBM cells express macrophage migration inhibitory factor (MIF) which renders GBM cells resistant to NK cell mediated killing (90). VEGF secretion by GBM cells stimulates the growth of new blood vessels supplying oxygen and nutrients to rapidly dividing and often hypoxic tumor cells (91, 92). As well as increasing tumor vasculature, VEGF also upregulates expression of the macromolecules tenascin C (TNC) and periostin. TNC blocks the migration of T cells across the blood tumor barrier thereby preventing them from penetrating the tumor parenchyma (23). Periostin also recruits circulating immunoinhibitory M2 macrophages into the tumor parenchyma (93). GBM stem cells secrete the macrophage attracting cytokine periostin. These macrophages support tumor growth and result in a poorer prognosis (93). GBM cells exposed to radiotherapy and chemotherapy have been shown to display increased immunosuppression. This phenomenon has been shown to be due to increased prostaglandin E2 secretion by cells. Blockade of this secreted PGE2 reverses the immunosuppressive capacity of treated cells (47). Colony stimulating factor 1 (CSF-1) is a growth factor that has been shown to be expressed in GBM tumors and by GBM cell lines (94). CSF-1 can either be secreted by cells or expressed as a transmembrane variant on the cell surface. CSF-1 is secreted by astrocytes within the brain during acute inflammatory responses. CSF-1 can bind to its receptor (CSF-1R) on the surface of macrophages and microglia within the brain promoting their switch to the immunosuppressive M2 phenotype (94, 95). GBM cells also secrete interleukin-1α and -1β (IL-1 α and β) (96). The down regulation of HLA class II expression on the U-105 MG GBM cell line by IL-1β suggests that this could be another mechanism which reduces immune recognition by CD4+ T cells (97).
Whilst GBM tumor cells contribute to immunosuppression, the immune cells recruited to the tumor can also exacerbate the immune evasive properties of these tumors. Although immune cells can contribute to tumor control, immunosuppressive populations can also contribute to the immune escape of GBM tumors. Indeed, many of the anti-tumor immune cells recruited to the TME adopt an immunosuppressive phenotype due to the cytokines secreted by the GBM tumors and the unique microenvironment which these tumors create.
Myeloid-derived suppressor cells (MDSCs) can be found within GBM tumors, and these cells contribute to the immunosuppressive phenotype of GBMs (98). MDSCs can be divided into two main types, monocytic and granulocytic. Granulocytic MDSCs are rarely found in GBM tumors, whereas the monocytic subtype are more prevalent (99). Monocytic MDSCs support tumor growth by increasing the recruitment of CD4+ regulatory T cells via chemokine release in the TME (100). CD4+ regulatory T cells are well known immunosuppressive immune cells that dampen the immune response. When compared to healthy controls, the prevalence of regulatory T cells in the peripheral blood is higher in GBM patients. Of even more relevance is that the prevalence of regulatory T cells in lymphocyte populations infiltrating GBM tumors is significantly greater than that in lymphocyte populations from ‘normal’ brain tissue obtained from seizure patients (101, 102). Although immune cell infiltration is often viewed as a positive prognostic marker, it can also contribute to the pathology of GBM. Lymphocytes entering the tumor have been shown to downregulate costimulatory molecules such as CD28 and CD62L (103). The presence of immunosuppressive regulatory T cells within GBM tumors has been correlated with shorter recurrence-free survival. GBM associated microglia/macrophages, which constitute up to 30% of the GBM tumor bulk are of the immunosuppressive M2 phenotype (103, 104). The expression of PD-L1 by these immunosuppressive M2 cells further contributes to local immunosuppression, as does their secretion of CCL22 which recruits regulatory T cells and MDSCs into the TME (103, 104).
Vaccination presents an attractive method for immunotherapeutically targeting GBMs (ongoing trials are detailed in Tables 1–3). One issue that can arise with peptide vaccinations is that immune escape variants can develop, and tumors can overcome the immune pressure applied to them. This phenomenon has been seen in the case of Rindopepimut, an EGFRvIII-keyhole limpet hemocyanin peptide conjugate. When Rindopepimut was used to treat GBM patients with EGFRvIII positive tumors, their median overall survival was 26 months compared to the 15 months of matched controls. Although vaccination prolonged the overall survival of patients, tumors recurred in a large proportion of these patients. When the recurrent tumors were analyzed immunohistochemically for EGFRvIII expression, 82% of the tumors examined had lost expression of EGFRvIII and the other 18% only displayed EGFRvIII expression in less than 1% of their tumor cells (114). These data suggest that the targeting of a single antigen can lead to the generation of immune escape variants, as a consequence of which multiple antigens need to be employed in the formulation of such vaccines.
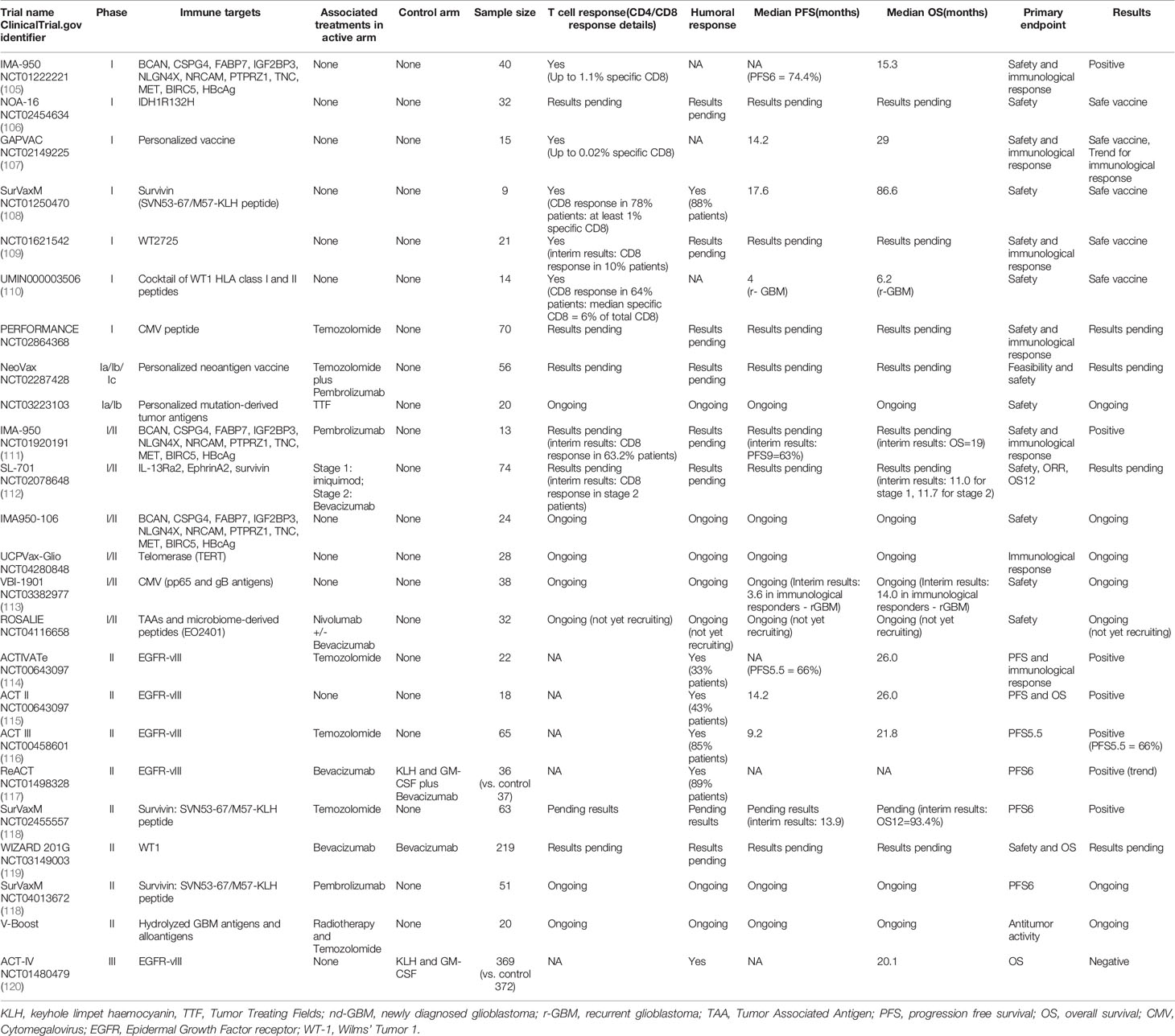
Table 1 Peptide vaccine trials for glioblastoma.
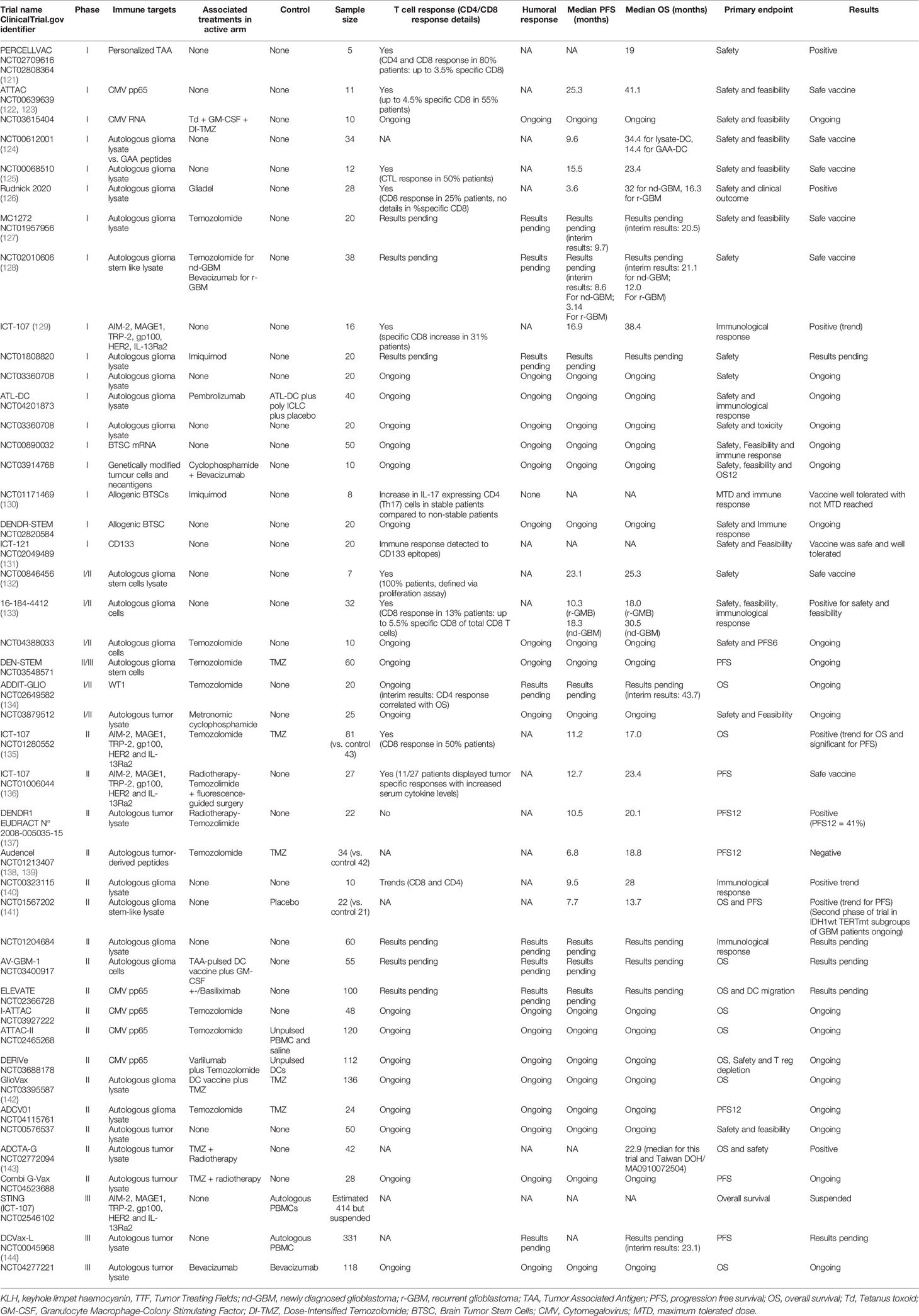
Table 2 Dendritic cell vaccine trials for glioblastoma.

Table 3 Other types of vaccine trials for glioblastoma.
IMA950 is one such multi-peptide vaccine that is being investigated in GBM. IMA950 is made up of 9 CD8 specific peptides derived from BCAN, CSPG4, FABP7, IGF2BP3, NRCAM, NLGN4X, PTPRZ1, and TNC as well as two CD4 specific peptides derived from survivin and c-met (150). This multi-peptide vaccine was given in conjunction with the immune boosting adjuvant poly-ICLC to GBM patients in a phase I/II clinical trial (111). This vaccination was well tolerated by patients and induced antigen specific CD8+ and CD4+ T cell responses (111). The level of response seen in patients varied, and analysis of five tumor samples revealed that no vaccine-specific T cells were present in the TIL population, meaning that there may be issues with the homing of vaccine-induced T cells (111). When samples from the recurrent tumors were tested for expression of the target antigens, no change in the levels of these antigens compared to the pre-vaccination tumor samples was observed, further suggesting that the issues are with T cells not trafficking to the tumor site (111). The ability of tumor cells to present immunogenic epitopes at their surface may also explain the failure of peptide vaccine treatments. Whilst tumors may express the target antigen, they may not present the target epitope on their surface meaning that vaccine generated T cells will not target these tumors. Although vaccination with these peptides generates antigen-specific T cells, there appears to be an issue with the immune TME of these tumors. As a result, the combination of IMA950 vaccination with other modalities, such as immune activating anti-CD27 and anti-PD-1 are being explored in the clinic (111).
A ‘personalized’ peptide vaccination approach has also been explored in the GBM setting. In a phase I clinical trial, GBM patients were treated with a cocktail of manufactured peptides derived from known GBM antigens followed by a vaccination that targeted neoepitopes derived from analysis of the patients’ tumor immunopeptidome and transcriptome (107). Each patient received a vaccine that was tailored to their tumor antigen expression profile. Vaccines were administered with the adjuvants Poly-ICLC and GM-CSF. The cocktail of ‘off the shelf’ peptides known as APVAC1 generated CD8+ T cell responses in twelve out of the thirteen patients studied and CD4+ T cell responses were found in nine of the thirteen patients studied (107). The neoepitope vaccine known as APVAC2 generated a predominantly CD4+ T cell response in eight out of the ten patients evaluated. The overall median overall survival of patients receiving this vaccination regime was 29 months (107). Although these findings are promising, these peptide vaccinations are far from curative. Whilst CD4+ and CD8+ responses were detected, these were at a relatively low level, with the frequency of antigen specific T cells being below 4 percent for CD4+ T cells and 1 percent for CD8+ T cells (107). The low frequency of target specific T cells may explain the failure of this therapy to act in a curative manner. Targeting of multiple antigens helps prevent the development of antigen escape variants, however combinatorial methods that enable vaccine-induced T cells to penetrate tumors and overcome the immunosuppressive microenvironment need to be explored.
The contribution of macrophages/microglia to the immunosuppressive TME of GBM and their prevalence within the tumor bulk suggest them to be attractive therapeutic targets for the immunotherapeutic targeting of GBM. As mentioned previously, microglia and macrophages in the TME adopt an immunosuppressive M2 phenotype (103, 104). As also previously mentioned, microglia/macrophages express the CSF-1R and GBM cells secrete CSF-1 resulting in the switching of GBM macrophages/microglia to the immune inhibitory M2 phenotype. The blockade of this CSF-1/CSF-1R interaction presents an attractive approach for preventing the switching of tumor resident macrophages/microglia to the immunoinhibitory M2 phenotype. In this regard, blockade of the CSF-1R with the chemical BLZ945 has been shown to improve survival and reduce tumor development in GBM bearing mice without any visible deleterious side-effects. BLZ945 treatment did not alter macrophage numbers within the implanted tumors but reduced the polarization of these macrophages to the M2 phenotype (95). As a result, combining BLZ945 with active immunotherapy represents an exciting therapeutic option for GBM.
As previously discussed GBM cells are known to overexpress MIF, making them resistant to NK cell mediated killing (90). Not only does MIF protect GBM cells from NK cell mediated killing it also exerts effects on macrophages/microglia within the tumors. MIF has been shown to interact with CD74 on microglia resulting in the adoption of the immunosuppressive M2 phenotype. Disruption of the CD74/MIF pathway prevents this M2 phenotype switch and prolongs the survival of GBM tumor bearing mice (151). Immunotoxins have also been used to target tumor associated macrophages (TAMs); Nagai et al. (2009) utilized this methodology to selectively target TAMs. Activated TAMs were shown to express folate receptor beta (FRβ), thereby providing a macrophage-specific target. The heavy and light chains of an anti-FRβ antibody were conjugated to the toxin Pseudomonas exotoxin (152). The abundance of macrophages within the tumor allows delivery of the toxin to the tumor resulting in the death of tumor cells and the potentially immunosuppressive macrophages. Administration of this immunotoxin intratumorally to a subcutaneous rat C6 glioma tumor reduced tumor growth and the number of TAMs in these tumors (152). It is important to note that this treatment was injected directly into subcutaneous tumors which reduces the potential for any deleterious off-target effects. Although FRβ was not detected in the normal brain, it was detected on macrophages resident in the heart and liver (152). This presents a potential hurdle to the systemic delivery of this immunoconjugate. The ability of this drug to cross the blood brain barrier is also unknown since this study utilized a subcutaneous model. In patients, this immunoconjugate could be administered intratumorally during surgery, or intraventricularly utilizing an Ommaya reservoir (an intracranial catheter device that allows direct delivery of drugs to the ventricles), thereby bypassing the blood brain barrier. However, this method is highly invasive and not without risks (153, 154).
Propentofylline (PPF) is a synthetic methylxanthine drug that is known to reduce the proliferation (155) and expression of inflammatory cytokines (155) by microglia in response to lipopolysaccharide. PPF could therefore be a novel therapeutic for targeting microglia within GBM tumors. In a rat model of GBM utilizing the CNS-1 cell line, a cell line which recapitulates the features of human GBM with minimal immunogenicity, systemic PPF administration reduced the volume of intracranial CNS-1 tumors (156). In vitro analysis revealed that PPF did not exert its effects on the CSF-1 cell line, rather its anti-tumor effects were attributed to its effect on microglial migration and the contribution of microglia to tumor cell migration (156). Rather than trying to remove microglia/macrophages from the TME, switching immunosuppressive M2 cells to the immune activating M1 phenotype also represents an attractive therapeutic option.
IL-12 represents an excellent immunotherapeutic candidate due to its ability to activate T-cells and NK cells and provoke antigen-specific immunity (157). As systemic administration of recombinant IL-12 was associated with adverse effects (such as damage to vital organs), gene transfer of IL-12 was achieved by the intracranial administration of an adeno-associated virus (AAV) encoding IL-12 to rats, after which they were challenged by intracranial injection of rat RG2 GBM cells. Treatment improved the survival of tumor challenged mice when compared to PBS injected control mice. Analysis of treated tumors revealed an increase in the microglial activation markers ED1 and TNF-related apoptosis-inducing ligand (TRAIL), and this was accompanied by a downregulation of the proliferation marker Ki67 and an increase in TUNEL staining - an indicator of apoptosis (157).
The blockade of TGF-β presents an attractive adjunct for active immunotherapy, due to its immunoregulating and tumor promoting effects. Trabedersen is an anti-sense RNA for human TGF-β2 mRNA that has been administered via convection-enhanced delivery to patients with recurrent GBM. Although Trabedersen improved the median survival compared to chemotherapy alone, this difference was not of statistical significance (158). In a pre-clinical murine model of metastatic pancreatic cancer, active vaccination was combined with antibody blockade of TGF-β. Soares et al. (2015) treated a murine model of pancreatic cancer using a vaccine comprised of GM-CSF secreting irradiated pancreatic cancer cells known as GVAX. This vaccine was used to treat two models of pancreatic cancer, the Panc02 model and KPC model. When GVAX vaccination was combined with TGF-β blockade, the cure rate of tumor bearing mice was improved in both models when compared to mice given GVAX with an IgG isotype antibody. The anti-tumor effects of GVAX were even further improved when the vaccine was combined with both an anti-TGF-β and anti-PD-1 antibody. This blockade of TGF-β in combination with GVAX reduced the regulatory T cell infiltrate into these tumors, a trend not seen when either therapy was used alone (159).
Due to the expression of numerous immunosuppressive checkpoints within the GBM TME, many checkpoint blockade antibodies have been tested in the GBM setting. Immune checkpoint blockade also represents a method for rescuing exhausted T cells. As monotherapies, immune checkpoints have provided lackluster results (160–162). One interesting method for altering the responsiveness to immune checkpoint blockade is to administer these immune checkpoint blocking antibodies in a neoadjuvant setting, as opposed to an adjuvant setting. Neoadjuvant administration of checkpoint blockade involves the dosing of the patient prior to tumor resection and standard therapy as opposed to after surgery and alongside standard therapy. In the GBM setting, neoadjuvant PD-1 blockade has been explored patients with recurrent disease - these patients received neoadjuvant PD-1 and therapy was then continued in the adjuvant setting post-surgery. Neoadjuvant treatment prolonged the overall survival when compared to adjuvant PD-1 blockade, and increased CD8+ T cell infiltration into tumors. An upregulation in the expression of interferon gamma related genes was also seen in the tumors of these patients (163).
Combining checkpoint blockade modalities or combining active immunotherapy with checkpoint blockade are also attractive methods for enhancing protective anti-GBM immunity. In a pre-clinical murine model of GBM, PD-1 blockade was combined with DC vaccination to great effect. Mice bearing intracranial GL261 tumors were vaccinated with DCs loaded with murine GL261 tumor cell lysate. Although this approach increased the infiltration of tumor cells into these intracranial tumors, this did not lead to improved survival in mice with an elevated tumor burden. It was hypothesized that local immune suppression within the TME was preventing tumor-specific lymphocytes from inducing tumor cell death. TILs were shown to have up-regulated their expression of PD-1, as a result of which it was decided to combine anti-PD-1 antibody therapy with DC vaccination. This combination increased the percentage of activated CD8+ T cells within the intracranial tumors and improved the survival of mice when compared to mice given vaccination alone (164).
As CSF-1R inhibition has been shown to reduce polarization of macrophages to the immunosuppressive M2 phenotype (95), combining CSF-1R inhibition with active vaccination and PD-1 blockade has been explored in the GBM setting. Myeloid derived cells recruited to the tumor were shown to express PD-L1 and contribute to the immunosuppressive environment seen in murine GL261 tumors. The presence of vaccine-induced TILs increased the recruitment of these immunosuppressive PD-L1 expressing myeloid cells. As a result, Antonios et al. (2017) combined PD-1 antibody and a CSF-1R inhibitor with active DC vaccination. CSF-1R inhibition increased the presence of TILs within tumors, whereas PD-1 blockade improved the activation of TILs. This triple therapy significantly increased the survival of GL261 tumor bearing mice when compared to non-treated, DC vaccinated and DC vaccinated mice with either CSF-1R or PD-1 blockade alone (165).
As detailed earlier, IDO and TDO expression within GBM tumors contributes to the immunosuppressive nature of these tumors. Targeting IDO alone or as part of a combinatorial strategy therefore also represents an attractive treatment avenue. The anti-viral drug acyclovir has been shown to inhibit both IDO and TDO and preventing the recruitment of regulatory T cells to the TME (166). In a pre-clinical murine model of GBM, the combined blockade of IDO, CTLA-4 and PD-1 reduced regulatory T cell infiltration into tumors and led to 100% long-term survival in mice harboring intracranial GL261 tumors (167).
Chimeric antigen receptor (CAR) T cells provide an avenue for generating tumor targeted T cells that can function in the defective tumor microenvironment. CAR T cells are generated by transfecting autologous T cells taken from patients with a construct combining a single chain variable fragment specific to a tumor cell target with costimulatory domains that enable T cell activation without the need for a secondary co-stimulatory signal (168). Numerous antigens have been targeted utilizing CAR T cells and the design of CAR T cells has been fine-tuned in order to optimize their anti-tumor activity. Traditionally CAR T cells’ intracellular signaling domain was derived from the CD3ζ chain of the T cell receptor (first generation), as progress has been made further costimulatory domains have been added to the intracellular region in order to improve the functionality of CAR T cells (second and third generation). These costimulatory domains are often derived from costimulatory CD28, OX-40, ICOS, and 4-1BB (169, 170). Whilst the design of the targeting domain of the CAR T cells has evolved so has the general design of these cells, with the knock in of other genes that enhance anti-tumor function being explored (see Table 4). As mentioned previously, GBM tumors frequently upregulate their expression of FasL (63, 175). CAR T cells generated from patient derived T cells often express Fas, which makes these T cells susceptible to FasL mediated cytotoxicity when entering the TME (176). The development of CAR T cells expressing Fas dominant negative receptors by Yamamoto and colleagues resulted in the persistence of cells without any deleterious side-effects such as autoimmunity or lymphoproliferative disease (177). CAR T cells expressing a dominant negative receptor for TGF-β have also been developed for the treatment of prostate cancer. These CAR T cells target a prostate antigen known as prostate-specific membrane antigen (PSMA) and they also express the dominant negative TGF-βRII that blocks TGF-β signaling. These CAR T cells displayed improved anti-tumor function when compared to CAR T cells that did not have the dominant negative TGF-βRII transfected into them. These CAR T cells appeared to exhibit long-term persistence and resistance to exhaustion (178). CAR T cells have also been engineered to secrete a PD-1 blocking antibody single chain variable fragment (scFv) that binds to PD-1 on the surface of activated T cells (both CAR and bystander T cells), thereby preventing PD-L1 on tumor cells from dampening T cell anti-tumor responses (179). These CAR T cells enhance the survival of PD-L1 expressing tumor bearing mice when compared to CAR T cells that do not secrete the PD-1 scFv combined with an anti-PD-1 antibody. This is believed to be due to the increased amount of PD-1 blockade within the TME when compared to systemic checkpoint blockade. These CAR T cells displayed efficacy against both hematologic and solid tumors (179). CAR T cells have also been modified to express the immune-stimulatory molecule CD40L to improve the anti-tumor function of these cells (180). The interaction of CD40L on these T-cells with CD40 on DCs results in the secretion of the immunostimulatory cytokine IL-12 (180). CD19 directed CAR T cells armed with the CD40 ligand have been shown to lyse CD19 negative cells and prevent their expansion and the development of antigen negative variants that escape an immune response (180). In order to prevent the development of antigen escape variants, CAR T cells have also been developed to produce bi-specific T cell engagers (BiTEs) in the GBM setting. EGFRvIII targeting CAR T cells have been developed to secrete BiTEs that target the wild type epidermal growth factor receptor (EGFR). These BiTEs contain an anti-EGFR domain along with an anti-CD3 domain, homing T cells onto EGFR expressing tumor cells. The secretion of these BiTEs recruits bystander cells that target tumor cells, these CAR T cells can also eradicate tumors that do not express the EGFRvIII antigen, thereby highlighting the importance of the BiTEs produced by these CAR T cells (181). One study looked at utilizing CD123 (IL-3 Receptor α chain) directed CAR T cells to target Hodgkin lymphoma cells. The investigators also hypothesized that as CD123 is expressed on myeloid cells, these CAR T cells could also target these cells and overcome the local immune suppression induced by MDSCs and M2 macrophages. These CAR T cells targeted lymphoma cells in vitro and in vivo. What was even more interesting was that these CAR T cells were resistant to inhibition by M2 macrophages when compared to classical CD19 targeting CAR T cells (182).
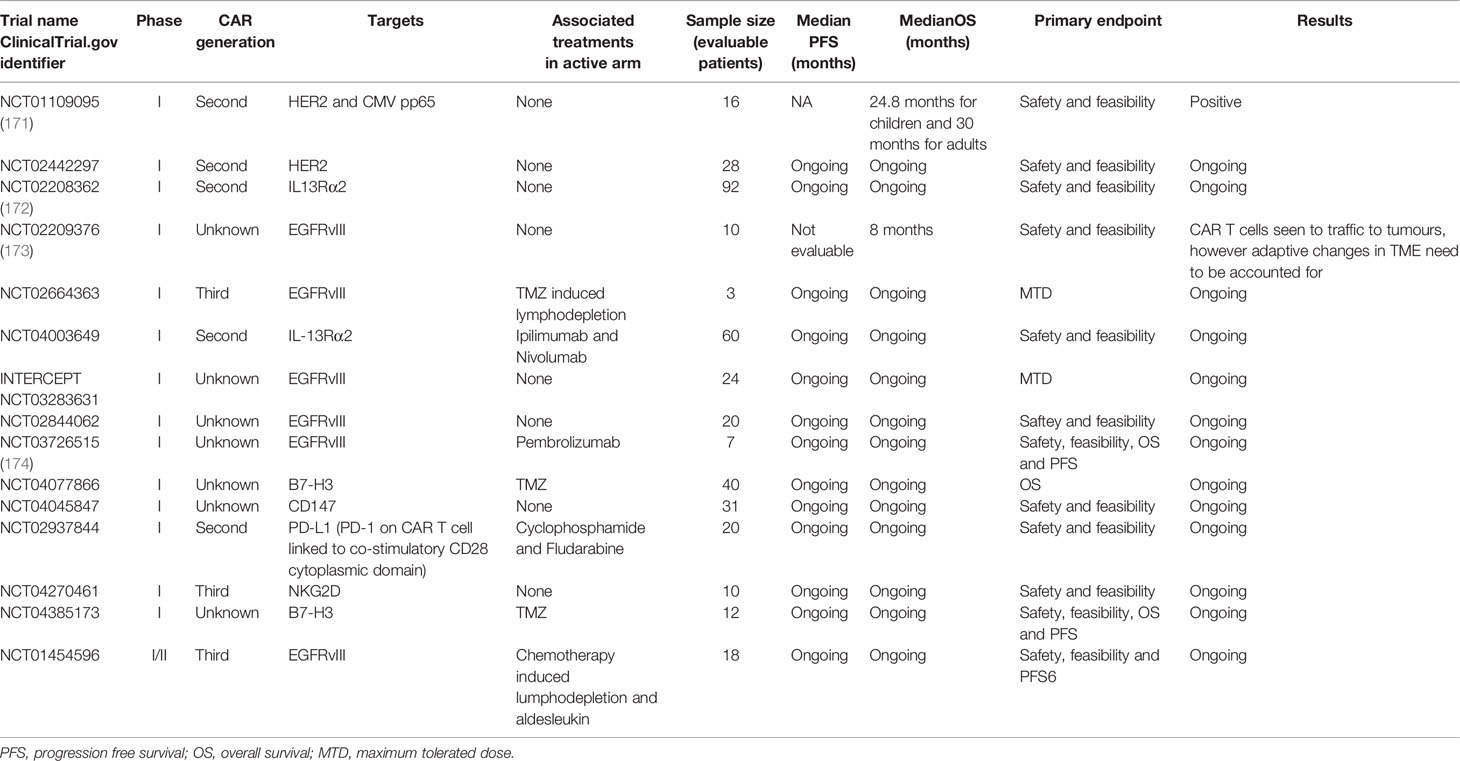
Table 4 CAR T cell trials for glioblastoma.
The design and delivery of immunotherapies must consider the pronounced immunosuppressive environment of the TME in GBM. The use of oncolytic viruses, which can selectively infect and kill tumor cells, is beginning to generate increased interest due to its tumor specificity and the ability of these viruses to turn an immunosuppressive microenvironment into an immune supporting environment (183). Oncolytic viruses are genetically altered to not infect non-transformed cells, and, in some cases, other genes may be knocked down or knocked in to enhance the immune stimulatory properties of these viruses. For example, the oncolytic herpes simplex virus T-VEC has been transfected with the human granulocyte-macrophage colony-stimulating factor (GM-CSF) gene. GM-CSF secreted by the virus increases the recruitment of DCs into the TME and thereby enhances antigen presentation and T cell activation (184). Tumor cell lysis by oncolytic viruses also triggers inflammatory immune responses involving the release of antigens, danger associated molecular patterns (DAMPs) and pathogen associated molecular patterns (PAMPs) within the TME (184). Several different types of viruses have been used in the oncolytic virotherapy of GBM, viruses such as the herpes simplex virus (HSV), Newcastle disease virus (NDV), poliovirus, reovirus, adenovirus, measles virus and H1 parvovirus (185) (see Table 5). Not only have viruses been used to directly induce the death of tumor cells they have also been used to transfer genes to tumor cells that enable these cells to be targeted. One such example of one of these viruses is Toca 511, a retroviral vector that delivers cytosine deaminase to rapidly dividing malignant cells (193). The transferred cytosine deaminase enzyme then converts the pro-drug 5-fluorocytosine to the active antineoplastic compound 5-fluorouracil resulting in the death of tumor cells (193). The use of this virus pro-drug combination has also been shown to result in an increase of immune cell activity within murine brain tumors, with a decrease in immunosuppressive cells and an increase in interferon gamma positive CD8 T cells within the tumor microenvironment (194). Whilst in treating preclinical models of GBM Toca 511 showed great promise recent however results from a phase II/III clinical trial revealed that Toca 511 in combination with 5-fluorocytosine did not improve overall survival when compared to standard therapy (195). Although oncolytic viral therapy represents an exciting avenue for GBM therapy, it is not without obstacles and as a result combinatorial therapy utilizing oncolytic viruses needs to be considered. Very little virus crosses the blood brain barrier when oncolytic viruses are delivered systemically, yet these therapies are still efficacious in brain tumor models. Oncolytic herpes simplex viruses (HSVs) can be used in combination with various other therapeutics for the treatment of GBM. The virus can also be altered with immunomodulating transgenes to improve anti-tumor efficacy and enable modulation of the TME (196). ‘Arming’ an oncolytic HSV with the murine IL-4 gene has been shown to increase the survival of mice bearing intracranial GL-261 cells. Conversely, no survival benefit compared to sham treated animals was observed when immunosuppressive IL-10 was transfected into this oncolytic virus (197). Clinical testing of oncolytic viruses remains in its relative infancy, with several viral therapies undergoing phase I/II clinical trials (198). The prospect of genetically modifying these viruses provides great hope for the future treatment of GBM. Not only can viruses be genetically manipulated but they can also be combined with other immunotherapeutic modalities to help overcome the immunosuppressive TME. One such example is combination of oncolytic measles virus therapy with anti-PD-1 checkpoint blockade (199). This combination was shown to increase survival in C57BL/6 mice bearing intracranial GL261 tumors when compared to either monotherapy, as well as increasing survival this combinatorial therapy increased T cell infiltrate into these tumors (199). Checkpoint blockade has also been combined with the IL-12 expressing oncolytic HSV in a pre-clinical model of GBM to great effect (200). This IL-12 secreting HSV was combined with both anti-CTLA-4 and anti-PD-1 checkpoint blockade for the treatment of two murine GBM models, this triple therapy reduced the number of regulatory T cells present within tumors and increased the influx of immune supporting M1 macrophages resulting in the complete cure of these mice (200). As previously mentioned GBMs frequently overexpress PGE2 which promotes an immunosuppressive environment and provides an attractive target for therapy. An oncolytic vaccina virus has been developed that expresses 15-(NAD)-hydroxy-prostaglandin-inactivating enzyme (HPGD); an enzyme that inactivates PGE2 (201). This modified vaccinia virus was tested in a variety of mouse solid tumor models and it was found to reduce the number of MDSCs and regulatory T cells within these tumors increasing the response of these tumors to viral therapy and adoptive T cell transfer (201). Whilst viral therapy is in its relative infancy with regards to clinical approval these early findings provide great hope for the future of this treatment modality.
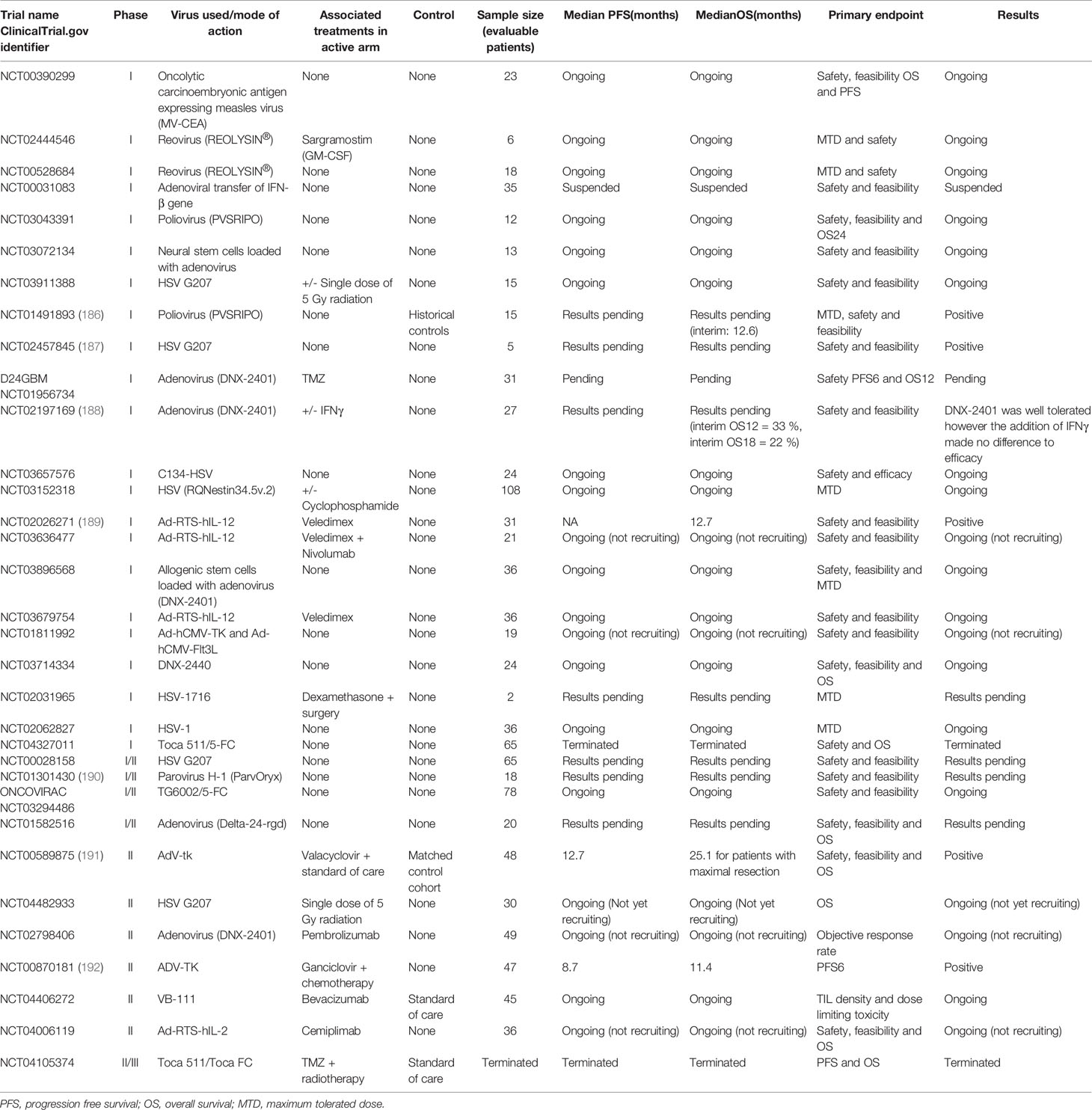
Table 5 Viral therapy trials for glioblastoma.
Adapting current therapies also needs to be considered in the context of immunotherapy for GBM, especially given the likelihood that all new approaches will need to be delivered in the context of current ‘standard’ therapy. Both TMZ and radiotherapy have immune augmenting effects that can be capitalized upon when considering the immunotherapeutic treatment of GBM. As mentioned previously, TMZ can induce lymphodepletion in patients. This lymphodepletion can be capitalized on to potentially enhance the efficacy of CAR T cell therapy. In a murine model of GBM, EGFRvIII CAR T cells failed to confer a survival advantage for mice bearing intracranial EGFRvIII expressing tumors, despite that fact that these cells were shown to have anti-tumor cell activity in vitro. Lymphodepletion with radiotherapy administered prior to CAR T cell therapy was shown to improve the efficacy of CAR T cell therapy by resulting in long-term survival of mice (202). Similarly, TMZ was used to lymphodeplete prior to CAR T cell administration. TMZ was either used in a standard or high dose, with the higher dose inducing more marked lymphodepletion. The lymphodepletion caused by high dose TMZ increased the survival of mice bearing established intracranial tumors when given CAR T cell therapy. This lymphodepletion to led to persistence of the injected CAR T cells within the blood of treated mice and this correlated with lower tumor burden (202). As well as using high dose TMZ to lymphodeplete, the dosing can also be given as low frequent doses, known as metronomic dosing. Ouyang and colleagues (2016) designed immune activating CpG carbon nanotube conjugates (SWCNT/CpG-2) that prolonged the survival of mice bearing intracranial GL261 tumors. This SWCNT/CpG-2 was used to treat a more invasive GBM model using the KR158B cell line, a model that more faithfully represents the characteristics of human GBM within a murine model. Although this intracranial SWCT/CpG-2 therapy was not curative for this KR158B model, as it was in the case of the GL261 model, when this SWCT/CpG-2 was combined with low dose daily TMZ, it significantly improved survival when compared to SWCT/CpG-2 monotherapy (203). Splenocytes taken from mice that had received metronomic TMZ in combination with SWCT/CpG-2 were more efficient at inducing in vitro KR158B tumor cell death than splenocytes from mice given either SWCT/CpG-2 or TMZ alone. This dual therapy did not reduce the number of regulatory T cells in the tumors. However, both SWCT/CpG-2 therapy and dual therapy induced an increased macrophage infiltrate into the tumors. The researchers hypothesized that the metronomic TMZ dosing increased the relative proportions of immune activating M1 macrophages to immune inhibitory M2 macrophages within the tumors (203). Radiotherapy can also be used as an adjunct to immunotherapy in order to boost the anti-tumor immune response. Weiss et al. (2018) generated an NKG2D expressing CAR T cell therapy that when systemically administered penetrated brain tumors in a murine GL261 GBM model. These NKG2D CAR T cells were shown to cure 22% of GL261 bearing mice treated. Radiotherapy upregulated the expression of NKG2D ligands on the surface of GBM cells and, as a result, it was decided to combine radiotherapy with NKG2D CAR T cells. Mice were given a single 4 gray (Gy) dose of radiotherapy on day 7 after tumor implant and CAR T cells were given on days 5, 7 and 10. The single radiotherapy dose alone did not alter the survival of tumor bearing mice compared to control mice, however it increased the survival of mice harboring intracranial GL261 cells when combined with the NKG2D CAR T cell therapy. This effect was also shown in mice bearing intracranial SMA-560 tumor cells (204). Another alternative that has been considered is the intratumoral administration of TMZ, as opposed to systemically administered TMZ. This local delivery of TMZ could theoretically prevent the profound lymphodepletion seen in systemic administration due to the therapy being mainly confined to the tumor. This method of administering TMZ was shown to improve the survival of mice bearing GL261 cell-derived tumors when compared to mice given intraperitoneal TMZ. CD4 and CD8 blocking antibodies revealed that T cells are responsible for this improved survival, with mice receiving intracranial TMZ failing to show improved survival if T cell blocking antibodies were used. Survival was improved even further when intracranial TMZ was combined with active immunotherapy using irradiated GL261 cells transfected to express GM-CSF (205). This combined intracranial TMZ and immunotherapy increased CD8+ T cell infiltrate and decreased MDSCs (205).
The BBB can act as a significant barrier for systemically administered therapeutics, including immune checkpoint blocking antibodies. Several approaches can be used to address this issue. These include direct modification and masking of therapeutic agents, encapsulation of therapeutics within vesicle-based delivery systems, and targeted opening of the BBB/BTB by physical or biochemical disruption. Conceptually, the simplest route to bypass the BTB is direct administration to the brain parenchyma or the cerebrospinal fluid (CSF). Although this is a commonly used approach in pre-clinical, experimental work, it is clinically problematic. Direct intra-parenchymal injection is rarely performed outside of intensive care medicine due to the difficulties associated with infection risk and needle damage. Moreover, although GBM are rarely metastatic (206), direct administration to the tumor site is contra-indicated due to the slow rate of diffusion of therapeutic molecules through compact brain tissue, injected substances rarely travelling more than a few millimeters beyond the injection site (207–209). This route is therefore unlikely to be sufficient for treating GBM, given both the likely tumor size on diagnosis and accessibility issues. Intracerebroventricular or intrathecal injection, delivery to the CSF, has similarly poor distribution issues (210). Passage of drugs from the CSF to the parenchymal tissue is primarily diffusive, which, coupled with rapid removal from the ependymal surface via bulk flow and the glymphatic system, results in minimal transfer of therapeutic agents into the tissue (211, 212). These restrictions are even more relevant to the delivery of large molecules such as therapeutic antibodies (213).
Working on the principle that the simplest way to overcome the BBB/BTB is to remove it, a number of methods of disrupting barrier function have been investigated for their potential use in the treatment of GBM and other neurological disorders. Such techniques were first begun over 50 years ago, with studies employing hypertonic solutions of osmolytes such as mannitol to induce osmotic endothelial shrinkage and tight junction opening (214). Such untargeted disruption, whilst effective in permitting increased therapeutic access to the brain, is also indiscriminate and enables the entry of pro-inflammatory and potentially toxic serum proteins such as albumin and complement factors (215), thereby rendering this non-specific approach unsuitable for clinical use. More targeted methods of inducing increased BBB/BTB permeability have used endogenous bioactive agents such as bradykinin or its synthetic analogues (216). Although such approaches have increased permeability of chemotherapeutic agents to the brain in preclinical models, they have not translated into clinical practice, possibly due to having too brief a duration of action (217).
Rather than chemical or osmotic-mediated disruption, another technique used to circumvent the BBB/BTB is the use of high-power focused ultrasound (218) to generate foci of increased tissue permeability. Although this approach is effective in opening the barrier to therapeutic antibodies (219), it suffers from producing bystander tissue distortion and damage in experimental animals (220). In an attempt to overcome this issue, the technique has been refined to improve specificity and reduce energy transfer through the use of injected microbubbles (221, 222). In this case, lower frequency ultrasound is used to stimulate microbubble oscillation and cavitation, disrupting the endothelial wall through local shock wave production and permitting access of therapeutic agents to the brain. Although promising, it is not yet clear to what degree brain penetration can be enhanced as efflux transport systems remain active (223), and the long-term consequences of disruption have not yet been studied.
As an alternative to BBB/BTB disruption, numerous attempts have been made to modify the therapeutic agents themselves or their delivery systems to permit greater transfer across an intact BBB/BTB. Building on the rationale that more lipophilic agents are better able to cross the BBB/BTB, initial approaches aimed to improve therapeutic agent lipid solubility. Although such modifications do indeed improve CNS access, this was achieved at the cost of increased non-specific membrane permeability and a consequent rise in off-target effects (224, 225).
To overcome these difficulties, ongoing attempts at achieving effective drug delivery across the BBB/BTB have employed a wide range of different nanocarriers, also termed nanoparticles. These are diverse molecular structures, including lipid micelles, liposome composites of phospholipid and other molecules, and polymer-based particles, with the common property that they form a vesicle that can be loaded with therapeutic agents and which can then cross the BBB to enter the parenchyma (226). Once within the brain, variation in environmental pH at the tumor site, amongst other conditions destabilize the nanocarrier structure and trigger release of the cargo within the tissue (227). Although effective, these nanocarriers are indiscriminate and passively deliver their cargo widely across the brain, a drawback that has spurred the development of more effectively targeted nanocarrier delivery systems.
Targeting can be substantially enhanced by including molecular tags within the vesicle wall using proteins, peptides, nucleic acids or small molecules that specifically recognize tumor-associated receptors, thereby minimizing off-target actions. A wide range of different molecular tags have been exploited for this purpose in vivo, including for example, the interaction of nanocarrier borne transferrin with the transferrin receptor TfR-1 on GBM cells (228), the EGFP-EGF1 fusion protein on nanoparticles with tissue factor in tumor cells (229), and cholera toxin with tumor-expressed chloride channels and matrix metalloproteinase-2 (230). Such strategies hold significant promise as they allow for both the concentration of therapeutic agents at the tumor site and, by virtue of the encapsulation, protect therapeutic agents from hepatic metabolism (230). Although a number of nanocarrier-encapsulated small molecule approaches are currently undergoing clinical trial in GBM, as yet none have been approved for use (231). Questions about the efficiency of large molecule, i.e. therapeutic antibody, encapsulation efficiency remain.
As direct lipophilic modification of therapeutic agents and encapsulation strategies have relatively broad specificity, even with improved targeting strategies, interest has grown in the use of direct molecular tagging of the therapies themselves to permit recognition by specific endothelial transporters, e.g. the transferrin receptor, insulin receptor or low density lipoprotein receptor (232, 233), a process sometimes termed receptor mediated transcytosis.
This approach has proven to hold significant promise for the experimental delivery of protein agents, including therapeutic antibodies. Exposure of ‘normal’ CNS to circulating biologic agents is restricted to less than 0.5% of the concentrations that are present in serum (234, 235), a level at which target engagement is unlikely to occur (236). However, molecular engineering of therapeutic antibodies has enabled significant enhancements in uptake across the BBB. These approaches include the development of bispecific antibodies in which one F(ab) binds the target of interest and the other binds and is transported by an endothelial transporter (237), therapeutic antibodies in which a transporter recognition domain is linked to the immunoglobulin heavy or light chain (238, 239), or, more recently, molecules in which the Fc domain itself is directly recognized by an endothelial transporter (240, 241). Such an approach has not yet been tested directly for the clinical delivery of immunotherapies targeting GBM but does hold significant promise. BBB penetrating Nano immunoconjugates have been developed with the aim of crossing the BBB and penetrating intracranial tumors. Galstyan and colleagues (2019) developed anti-CTLA-4 and anti-PD-1 IgG antibodies conjugated to poly (β-L-malic acid), a natural biopolymer scaffold. These Nano immunoconjugates cross the BBB more efficiently than the anti-PD-1 and CTLA-4 IgG antibodies without polymer conjugation, increased the CD4+ and CD8+ T cell infiltrate into tumors and improved the survival of mice bearing intracranial GL261 tumors compared to those treated with the non-conjugated antibodies (242). Antibody delivery to intracranial tumors can also be improved by disrupting the BBB using focused ultrasound and microbubbles to physically disrupt the tight junctions enabling penetrance of the brain parenchyma. The combination of focused ultrasound (FUS) with microbubbles improves entry of the anti-HER2 antibody Herceptin into brains (243). Similar results have also shown with FUS in combination with microbubbles increases the penetrance of anti-amyloid beta antibodies into the brains of mice in two separate models of Alzheimer’s disease (244). FUS is an attractive option since the opening of BBB is transient (245), thereby minimizing the potential for damage to the brain. More interestingly, FUS itself can be used therapeutically to target intracranial tumors due to its immunomodulation action. Ultrasound waves can expand and contract air bubbles present within cells to generate heat and physically damage cells, inducing cell death, leading to the release of antigenic material and an up-regulation of immune activating molecules such as heat shock proteins (246).
Glioblastoma multiforme (GBM) is the most frequently occurring primary brain tumor. It is uniformly fatal due to its highly invasive nature and resistance to standard therapies. GBM tumors employ several mechanisms to avoid being detected and killed by immune cells. These include the downregulation of important immune activating molecules such as MHC molecules, as well as upregulating expression of molecules that induce the death of immune cells such as Fas ligand, non-classical MHC molecules such as HLA-E and -G, and PD-L1. GBM cells also secrete numerous immunoinhibitory cytokines such as IL-10, TGF-β, Gal-1, IL-6 and PGE2, to name a few. These cytokines result in the inactivation/death of immune cells as well as the recruitment of inhibitory cells such as regulatory T cells and MDSCs to the TME. These cytokines also lead to a conversion of tumor resident macrophages from the immune activating M1 phenotype to the immunosuppressive M2 phenotype further dampening the anti-tumor immune response.
The plethora of immunosuppressive mechanisms that GBM tumors utilize, as well as their physiological location, make treating them with immunotherapy a daunting task. Although these tumors are immunosuppressive, this immunosuppression can be leveraged to try and boost the anti-tumor immune response. The concept of combining immune checkpoint blockade with active vaccination is one such method that can be used, or the use of genetically modified oncolytic viruses and CAR T cells that actively attack tumors whilst overcoming the local immunosuppression, either via the secretion of immune activating cytokines or immune blocking scFvs. Combinatorial immunotherapy along with improvement of BBB penetration represents an encouraging avenue for GBM therapy in the future. The only caveat to these combined therapies is the possibility of an overactive immune response and potential autoimmunity, this will have to be monitored when moving combinatorial immunotherapy forward.
JP wrote the initial draft with SC and SM making significant contributions. All authors contributed to the article and approved the submitted version.
This work was supported by the Headcase Cancer Trust (UK), the John and Lucille van Geest Foundation, Nottingham Trent University QR allocation and the John van Geest Cancer Research Centre (Nottingham Trent University).
Author LD is a director and shareholder of Scancell Ltd and is a named inventor on the ImmunoBody patents.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, et al. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol (2017) 19(suppl_5):v1–v88. doi: 10.1093/neuonc/nox158
2. Petrecca K, Guiot MC, Panet-Raymond V, Souhami L. Failure pattern following complete resection plus radiotherapy and temozolomide is at the resection margin in patients with glioblastoma. J Neurooncol (2013) 111(1):19–23. doi: 10.1007/s11060-012-0983-4
3. Brandes AA, Tosoni A, Franceschi E, Sotti G, Frezza G, Amista P, et al. Recurrence pattern after temozolomide concomitant with and adjuvant to radiotherapy in newly diagnosed patients with glioblastoma: correlation With MGMT promoter methylation status. J Clin Oncol (2009) 27(8):1275–9. doi: 10.1200/JCO.2008.19.4969
4. Sherriff J, Tamangani J, Senthil L, Cruickshank G, Spooner D, Jones B, et al. Patterns of relapse in glioblastoma multiforme following concomitant chemoradiotherapy with temozolomide. Br J Radiol (2013) 86(1022):20120414. doi: 10.1259/bjr.20120414
5. Hadjipanayis CG, Stummer W. 5-ALA and FDA approval for glioma surgery. J Neurooncol (2019) 141(3):479–86. doi: 10.1007/s11060-019-03098-y
6. Young RM, Jamshidi A, Davis G, Sherman JH. Current trends in the surgical management and treatment of adult glioblastoma. Ann Transl Med (2015) 3(9):121–5839. doi: 10.3978/j.issn.2305-5839.2015.05.10
7. Pachter JS, de Vries HE, Fabry Z. The blood-brain barrier and its role in immune privilege in the central nervous system. J Neuropathol Exp Neurol (2003) 62(6):593–604. doi: 10.1093/jnen/62.6.593
8. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis (2010) 37(1):13–25. doi: 10.1016/j.nbd.2009.07.030
9. McArthur S, Loiola RA, Maggioli E, Errede M, Virgintino D, Solito E. The restorative role of annexin A1 at the blood-brain barrier. Fluids Barriers CNS (2016) 13(1):17–016-0043-0. doi: 10.1186/s12987-016-0043-0
10. Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci (2007) 8(8):610–22. doi: 10.1038/nrn2175
11. Anderson JC, McFarland BC, Gladson CL. New molecular targets in angiogenic vessels of glioblastoma tumours. Expert Rev Mol Med (2008) 10:e23. doi: 10.1017/S1462399408000768
12. Dhermain FG, Hau P, Lanfermann H, Jacobs AH, van den Bent MJ. Advanced MRI and PET imaging for assessment of treatment response in patients with gliomas. Lancet Neurol (2010) 9(9):906–20. doi: 10.1016/S1474-4422(10)70181-2
13. Henderson JT, Piquette-Miller M. Blood-brain barrier: an impediment to neuropharmaceuticals. Clin Pharmacol Ther (2015) 97(4):308–13. doi: 10.1002/cpt.77
14. Dantzer R. Neuroimmune Interactions: From the Brain to the Immune System and Vice Versa. Physiol Rev (2018) 98(1):477–504. doi: 10.1152/physrev.00039.2016
15. Heimberger AB, Sampson JH. Immunotherapy coming of age: what will it take to make it standard of care for glioblastoma? Neuro Oncol (2011) 13(1):3–13. doi: 10.1093/neuonc/noq169
16. Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol (2012) 33(12):579–89. doi: 10.1016/j.it.2012.07.004
17. Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest (2010) 120(5):1368–79. doi: 10.1172/JCI41911
18. Ichinose M, Masuoka J, Shiraishi T, Mineta T, Tabuchi K. Fas ligand expression and depletion of T-cell infiltration in astrocytic tumors. Brain Tumor Pathol (2001) 18(1):37–42. doi: 10.1007/BF02478923
19. Vega EA, Graner MW, Sampson JH. Combating immunosuppression in glioma. Future Oncol (2008) 4(3):433–42. doi: 10.2217/14796694.4.3.433
20. Mullins C, Walter A, Schmitt M, Classen C, Linnebacher M. Tumor antigen and MHC expression in glioma cells for immunotherapeutic interventions. World J Immunol (2013) 11/27:3. doi: 10.5411/wji.v3.i3.62
21. Moserle L, Casanovas O. Anti-angiogenesis and metastasis: a tumour and stromal cell alliance. J Intern Med (2013) 273(2):128–37. doi: 10.1111/joim.12018
22. Steck PA, Moser RP, Bruner JM, Liang L, Freidman AN, Hwang TL, et al. Altered expression and distribution of heparan sulfate proteoglycans in human gliomas. Cancer Res (1989) 49(8):2096–103.
23. Quail DF, Joyce JA. The Microenvironmental Landscape of Brain Tumors. Cancer Cell (2017) 31(3):326–41. doi: 10.1016/j.ccell.2017.02.009
24. Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin Cancer Res (2018) 24(16):3792–802. doi: 10.1158/1078-0432.CCR-18-0047
25. Brenchley JM, Karandikar NJ, Betts MR, Ambrozak DR, Hill BJ, Crotty LE, et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood (2003) 101(7):2711–20. doi: 10.1182/blood-2002-07-2103
26. Huff WX, Kwon JH, Henriquez M, Fetcko K, Dey M. The Evolving Role of CD8(+)CD28(-) Immunosenescent T Cells in Cancer Immunology. Int J Mol Sci (2019) 20(11):2810. doi: 10.3390/ijms20112810
27. Chen PY, Wu CY, Fang JH, Chen HC, Feng LY, Huang CY, et al. Functional Change of Effector Tumor-Infiltrating CCR5(+)CD38(+)HLA-DR(+)CD8(+) T Cells in Glioma Microenvironment. Front Immunol (2019) Oct 910:2395. doi: 10.3389/fimmu.2019.02395
28. Hanif F, Muzaffar K, Perveen K, Malhi SM, Simjee S. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac J Cancer Prev (2017) 18(1):3–9. doi: 10.22034/APJCP.2017.18.1.3
29. Pangrazzi L, Weinberger B. T cells, aging and senescence. Exp Gerontol (2020) 134:110887. doi: 10.1016/j.exger.2020.110887
30. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin Cancer Res (2018) 24(17):4175–86. doi: 10.1158/1078-0432.CCR-17-1846
31. Mirzaei R, Sarkar S, Yong VW. T Cell Exhaustion in Glioblastoma: Intricacies of Immune Checkpoints. Trends Immunol (2017) 38(2):104–15. doi: 10.1016/j.it.2016.11.005
32. Ramirez YP, Weatherbee JL, Wheelhouse RT, Ross AH. Glioblastoma multiforme therapy and mechanisms of resistance. Pharmaceuticals (Basel) (2013) 6(12):1475–506. doi: 10.3390/ph6121475
33. Lee EQ, Wen PY. Corticosteroids for peritumoral edema: time to overcome our addiction? Neuro Oncol (2016) 18(9):1191–2. doi: 10.1093/neuonc/now167
34. Branter J, Basu S, Smith S. Tumour treating fields in a combinational therapeutic approach. Oncotarget (2018) 9(93):36631–44. doi: 10.18632/oncotarget.26344
35. Giles AJ, Hutchinson MND, Sonnemann HM, Jung J, Fecci PE, Ratnam NM, et al. Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy. J Immunother Cancer (2018) 6(1):51–018-0371-5. doi: 10.1186/s40425-018-0371-5
36. McGranahan T, Therkelsen KE, Ahmad S, Nagpal S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr Treat Options Oncol (2019) 20(3):24–019-0619-4. doi: 10.1007/s11864-019-0619-4
37. Sengupta S, Marrinan J, Frishman C, Sampath P. Impact of temozolomide on immune response during malignant glioma chemotherapy. Clin Dev Immunol (2012) 2012:831090. doi: 10.1155/2012/831090
38. Karachi A, Dastmalchi F, Mitchell DA, Rahman M. Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro Oncol (2018) 20(12):1566–72. doi: 10.1093/neuonc/noy072
39. Gzell C, Back M, Wheeler H, Bailey D, Foote M. Radiotherapy in Glioblastoma: the Past, the Present and the Future. Clin Oncol (R Coll Radiol) (2017) 29(1):15–25. doi: 10.1016/j.clon.2016.09.015
40. Foulds GA, Radons J, Kreuzer M, Multhoff G, Pockley AG. Influence of tumors on protective anti-tumor immunity and the effects of irradiation. Front Oncol (2013) 3:14. doi: 10.3389/fonc.2013.00014
41. Multhoff G, Rödel F, Pockley AG, Gaipl US. Frontiers research topic: radiation-induced effects and the immune system. Front Oncol (2013) 3:55. doi: 10.3389/fonc.2013.00055
42. Gaipl US, Multhoff G, Pockley AG, Rödel F. Editorial: Radioimmunotherapy-Translational Opportunities and Challenges. Front Oncol (2020) 10:190. doi: 10.3389/fonc.2020.00190
43. Chow KK, Hara W, Lim M, Li G. Combining immunotherapy with radiation for the treatment of glioblastoma. J Neurooncol (2015) 123(3):459–64. doi: 10.1007/s11060-015-1762-9
44. Demaria S, Formenti SC. Role of T lymphocytes in tumor response to radiotherapy. Front Oncol (2012) 2:95. doi: 10.3389/fonc.2012.00095
45. Formenti SC, Demaria S. Radiation therapy to convert the tumor into an in situ vaccine. Int J Radiat Oncol Biol Phys (2012) 84(4):879–80. doi: 10.1016/j.ijrobp.2012.06.020
46. Vanpouille-Box C, Pilones KA, Wennerberg E, Formenti SC, Demaria S. In situ vaccination by radiotherapy to improve responses to anti-CTLA-4 treatment. Vaccine (2015) 33(51):7415–22. doi: 10.1016/j.vaccine.2015.05.105
47. Authier A, Farrand KJ, Broadley KW, Ancelet LR, Hunn MK, Stone S, et al. Enhanced immunosuppression by therapy-exposed glioblastoma multiforme tumor cells. Int J Cancer (2015) 136(11):2566–78. doi: 10.1002/ijc.29309
48. Tabatabaei P, Visse E, Bergstrom P, Brannstrom T, Siesjo P, Bergenheim AT. Radiotherapy induces an immediate inflammatory reaction in malignant glioma: a clinical microdialysis study. J Neurooncol (2017) 131(1):83–92. doi: 10.1007/s11060-016-2271-1
49. Grossman SA, Ye X, Lesser G, Sloan A, Carraway H, Desideri S, et al. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin Cancer Res (2011) 17(16):5473–80. doi: 10.1158/1078-0432.CCR-11-0774
50. Gingras MC, Roussel E, Bruner JM, Branch CD, Moser RP. Comparison of cell adhesion molecule expression between glioblastoma multiforme and autologous normal brain tissue. J Neuroimmunol (1995) 57(1-2):143–53. doi: 10.1016/0165-5728(94)00178-Q
51. Piao Y, Henry V, Tiao N, Park SY, Martinez-Ledesma J, Dong JW, et al. Targeting intercellular adhesion molecule-1 prolongs survival in mice bearing bevacizumab-resistant glioblastoma. Oncotarget (2017) 8(57):96970–83. doi: 10.18632/oncotarget.18859
52. Kamran N, Kadiyala P, Saxena M, Candolfi M, Li Y, Moreno-Ayala MA, et al. Immunosuppressive Myeloid Cells’ Blockade in the Glioma Microenvironment Enhances the Efficacy of Immune-Stimulatory Gene Therapy. Mol Ther (2017) 25(1):232–48. doi: 10.1016/j.ymthe.2016.10.003
53. Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC, et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol (2011) 13(6):591–9. doi: 10.1093/neuonc/nor042
54. Jung TY, Jung S, Ryu HH, Jeong YI, Jin YH, Jin SG, et al. Role of galectin-1 in migration and invasion of human glioblastoma multiforme cell lines. J Neurosurg (2008) 109(2):273–84. doi: 10.3171/JNS/2008/109/8/0273
55. Kovacs-Solyom F, Blasko A, Fajka-Boja R, Katona RL, Vegh L, Novak J, et al. Mechanism of tumor cell-induced T-cell apoptosis mediated by galectin-1. Immunol Lett (2010) 127(2):108–18. doi: 10.1016/j.imlet.2009.10.003
56. Stillman BN, Hsu DK, Pang M, Brewer CF, Johnson P, Liu FT, et al. Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J Immunol (2006) 176(2):778–89. doi: 10.4049/jimmunol.176.2.778
57. Pace KE, Hahn HP, Pang M, Nguyen JT, Baum LG. CD7 delivers a pro-apoptotic signal during galectin-1-induced T cell death. J Immunol (2000) 165(5):2331–4. doi: 10.4049/jimmunol.165.5.2331
58. Hernandez JD, Nguyen JT, He J, Wang W, Ardman B, Green JM, et al. Galectin-1 binds different CD43 glycoforms to cluster CD43 and regulate T cell death. J Immunol (2006) 177(8):5328–36. doi: 10.4049/jimmunol.177.8.5328
59. Wastowski IJ, Simoes RT, Yaghi L, Donadi EA, Pancoto JT, Poras I, et al. Human leukocyte antigen-G is frequently expressed in glioblastoma and may be induced in vitro by combined 5-aza-2’-deoxycytidine and interferon-gamma treatments: results from a multicentric study. Am J Pathol (2013) 182(2):540–52. doi: 10.1016/j.ajpath.2012.10.021
60. Rahman M, Dastmalchi F, Karachi A, Mitchell D. The role of CMV in glioblastoma and implications for immunotherapeutic strategies. Oncoimmunology (2018) 8(1):e1514921. doi: 10.1080/2162402X.2018.1514921
61. Carosella ED, Rouas-Freiss N, Tronik-Le Roux D, Moreau P, LeMaoult J. HLA-G: An Immune Checkpoint Molecule. Adv Immunol (2015) 127:33–144. doi: 10.1016/bs.ai.2015.04.001
62. Wischhusen J, Friese MA, Mittelbronn M, Meyermann R, Weller M. HLA-E protects glioma cells from NKG2D-mediated immune responses in vitro: implications for immune escape in vivo. J Neuropathol Exp Neurol (2005) 64(6):523–8. doi: 10.1093/jnen/64.6.523
63. Didenko VV, Ngo HN, Minchew C, Baskin DS. Apoptosis of T lymphocytes invading glioblastomas multiforme: a possible tumor defense mechanism. J Neurosurg (2002) 96(3):580–4. doi: 10.3171/jns.2002.96.3.0580
64. Chahlavi A, Rayman P, Richmond AL, Biswas K, Zhang R, Vogelbaum M, et al. Glioblastomas induce T-lymphocyte death by two distinct pathways involving gangliosides and CD70. Cancer Res (2005) 65(12):5428–38. doi: 10.1158/0008-5472.CAN-04-4395
65. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol (2015) 17(8):1064–75. doi: 10.1093/neuonc/nou307
66. Xue S, Song G, Yu J. The prognostic significance of PD-L1 expression in patients with glioma: A meta-analysis. Sci Rep (2017) 7(1):4231–017-04023-x. doi: 10.1038/s41598-017-04023-x
67. Han MZ, Wang S, Zhao WB, Ni SL, Yang N, Kong Y, et al. Immune checkpoint molecule herpes virus entry mediator is overexpressed and associated with poor prognosis in human glioblastoma. EBioMedicine (2019) 43:159–70. doi: 10.1016/j.ebiom.2019.04.002
68. Mbongue JC, Nicholas DA, Torrez TW, Kim NS, Firek AF, Langridge WH. The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity. Vaccines (Basel) (2015) 3(3):703–29. doi: 10.3390/vaccines3030703
69. Mitsuka K, Kawataki T, Satoh E, Asahara T, Horikoshi T, Kinouchi H. Expression of indoleamine 2,3-dioxygenase and correlation with pathological malignancy in gliomas. Neurosurgery (2013) 72(6):1031–8; discussion 1038-9. doi: 10.1227/NEU.0b013e31828cf945
70. Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res (2012) 18(22):6110–21. doi: 10.1158/1078-0432.CCR-12-2130
71. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol (2010) 185(6):3190–8. doi: 10.4049/jimmunol.0903670
72. Pilotte L, Larrieu P, Stroobant V, Colau D, Dolusic E, Frederick R, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci USA (2012) 109(7):2497–502. doi: 10.1073/pnas.1113873109
73. Van Meir E, Sawamura Y, Diserens AC, Hamou MF, de Tribolet N. Human glioblastoma cells release interleukin 6 in vivo and in vitro. Cancer Res (1990) 50(20):6683–8.
74. Tchirkov A, Rolhion C, Bertrand S, Doré JF, Dubost JJ, Verrelle P. IL-6 gene amplification and expression in human glioblastomas. Br J Cancer (2001) 85(4):518–22. doi: 10.1054/bjoc.2001.1942
75. Kudo M, Jono H, Shinriki S, Yano S, Nakamura H, Makino K, et al. Antitumor effect of humanized anti-interleukin-6 receptor antibody (tocilizumab) on glioma cell proliferation. Lab Investigation J Neurosurg (2009) 111(2):219–25. doi: 10.3171/2008.12.JNS081284
76. Luwor RB, Stylli SS, Kaye AH. The role of Stat3 in glioblastoma multiforme. J Clin Neurosci (2013) 20(7):907–11. doi: 10.1016/j.jocn.2013.03.006
77. Jacobs JF, Idema AJ, Bol KF, Grotenhuis JA, de Vries IJ, Wesseling P, et al. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J Neuroimmunol (2010) 225(1-2):195–9. doi: 10.1016/j.jneuroim.2010.05.020
78. Crane CA, Ahn BJ, Han SJ, Parsa AT. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: implications for immunotherapy. Neuro Oncol (2012) 14(5):584–95. doi: 10.1093/neuonc/nos014
79. Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res (2011) 17(13):4296–308. doi: 10.1158/1078-0432.CCR-10-2557
80. Bodmer S, Strommer K, Frei K, Siepl C, de Tribolet N, Heid I, et al. Immunosuppression and transforming growth factor-beta in glioblastoma. Preferential production of transforming growth factor-beta 2. J Immunol (1989) 143(10):3222–9.
81. Platten M, Wick W, Weller M. Malignant glioma biology: role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microsc Res Tech (2001) 52(4):401–10. doi: 10.1002/1097-0029(20010215)52:4<401::AID-JEMT1025>3.0.CO;2-C
82. Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol (2008) 180(9):5771–7. doi: 10.4049/jimmunol.180.9.5771
83. Huettner C, Paulus W, Roggendorf W. Messenger RNA expression of the immunosuppressive cytokine IL-10 in human gliomas. Am J Pathol (1995) 146(2):317–22.
84. Wagner S, Czub S, Greif M, Vince GH, Suss N, Kerkau S, et al. Microglial/macrophage expression of interleukin 10 in human glioblastomas. Int J Cancer (1999) 82(1):12–6. doi: 10.1002/(SICI)1097-0215(19990702)82:1<12::AID-IJC3>3.0.CO;2-O
85. Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res (2002) 62(12):3347–50.
86. Lucas KG, Bao L, Bruggeman R, Dunham K, Specht C. The detection of CMV pp65 and IE1 in glioblastoma multiforme. J Neurooncol (2011) 103(2):231–8. doi: 10.1007/s11060-010-0383-6
87. Kotenko SV, Saccani S, Izotova LS, Mirochnitchenko OV, Pestka S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc Natl Acad Sci USA (2000) 97(4):1695–700. doi: 10.1073/pnas.97.4.1695
88. Chang WL, Barry PA. Attenuation of innate immunity by cytomegalovirus IL-10 establishes a long-term deficit of adaptive antiviral immunity. Proc Natl Acad Sci USA (2010) 107(52):22647–52. doi: 10.1073/pnas.1013794108
89. Brown NF, Carter TJ, Ottaviani D, Mulholland P. Harnessing the immune system in glioblastoma. Br J Cancer (2018) 119(10):1171–81. doi: 10.1038/s41416-018-0258-8
90. Mittelbronn M, Platten M, Zeiner P, Dombrowski Y, Frank B, Zachskorn C, et al. Macrophage migration inhibitory factor (MIF) expression in human malignant gliomas contributes to immune escape and tumour progression. Acta Neuropathol (2011) 122(3):353–65. doi: 10.1007/s00401-011-0858-3
91. Neurath KM, Keough MP, Mikkelsen T, Claffey KP. AMP-dependent protein kinase alpha 2 isoform promotes hypoxia-induced VEGF expression in human glioblastoma. Glia (2006) 53(7):733–43. doi: 10.1002/glia.20326
92. Oka N, Soeda A, Inagaki A, Onodera M, Maruyama H, Hara A, et al. VEGF promotes tumorigenesis and angiogenesis of human glioblastoma stem cells. Biochem Biophys Res Commun (2007) 360(3):553–9. doi: 10.1016/j.bbrc.2007.06.094
93. Wu T, Luo Q, Ouyang G. Periostin: a potent chemotactic factor for recruiting tumor-associated macrophage. Protein Cell (2015) 6(4):235–7. doi: 10.1007/s13238-015-0141-9
94. Alterman RL, Stanley ER. Colony stimulating factor-1 expression in human glioma. Mol Chem Neuropathol (1994) 21(2-3):177–88. doi: 10.1007/BF02815350
95. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med (2013) 19(10):1264–72. doi: 10.1038/nm.3337
96. Lu T, Tian L, Han Y, Vogelbaum M, Stark GR. Dose-dependent cross-talk between the transforming growth factor-beta and interleukin-1 signaling pathways. Proc Natl Acad Sci USA (2007) 104(11):4365–70. doi: 10.1073/pnas.0700118104
97. Cowan EP, Pierce ML, Dhib-Jalbut S. Interleukin-1 beta decreases HLA class II expression on a glioblastoma multiforme cell line. J Neuroimmunol (1991) 33(1):17–28. doi: 10.1016/0165-5728(91)90030-B
98. Kohanbash G, Okada H. Myeloid-derived suppressor cells (MDSCs) in gliomas and glioma-development. Immunol Invest (2012) 41(6-7):658–79. doi: 10.3109/08820139.2012.689591
99. Chen Z, Hambardzumyan D. Immune Microenvironment in Glioblastoma Subtypes. Front Immunol (2018) 9:1004. doi: 10.3389/fimmu.2018.01004
100. Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol (2012) 189(12):5602–11. doi: 10.4049/jimmunol.1201018
101. El Andaloussi A, Lesniak MS. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro Oncol (2006) 8(3):234–43. doi: 10.1215/15228517-2006-006
102. Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W, et al. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin Cancer Res (2008) 14(16):5166–72. doi: 10.1158/1078-0432.CCR-08-0320
103. Caponegro MD, Tetsuo Miyauchi J, E. Tsirka S. Contributions of immune cell populations in the maintenance, progression, and therapeutic modalities of glioma. AIMS Allergy Immunol (2018) 2(1):24–44. doi: 10.3934/Allergy.2018.1.24
104. Kennedy BC, Showers CR, Anderson DE, Anderson L, Canoll P, Bruce JN, et al. Tumor-associated macrophages in glioma: friend or foe? J Oncol (2013) 2013:486912. doi: 10.1155/2013/486912
105. Rampling R, Peoples S, Mulholland PJ, James A, Al-Salihi O, Twelves CJ, et al. A Cancer Research UK First Time in Human Phase I Trial of IMA950 (Novel Multipeptide Therapeutic Vaccine) in Patients with Newly Diagnosed Glioblastoma. Clin Cancer Res (2016) 22(19):4776–85. doi: 10.1158/1078-0432.CCR-16-0506
106. Platten M, Schilling D, Bunse L, Wick A, Bunse T, Riehl D, et al. ATIM-33. NOA-16: A FIRST-IN-MAN MULTICENTER PHASE I CLINICAL TRIAL OF THE GERMAN NEUROONCOLOGY WORKING GROUP EVALUATING A MUTATION-SPECIFIC PEPTIDE VACCINE TARGETING IDH1R132H IN PATIENTS WITH NEWLY DIAGNOSED MALIGNANT ASTROCYTOMAS. Neuro-oncology (2018) 20(suppl_6):8–9. doi: 10.1093/neuonc/noy148.028
107. Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature (2019) 565(7738):240–5. doi: 10.1038/s41586-018-0810-y
108. Fenstermaker RA, Ciesielski MJ, Qiu J, Yang N, Frank CL, Lee KP, et al. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol Immunother (2016) 65(11):1339–52. doi: 10.1007/s00262-016-1890-x
109. Fu S, Piccioni DE, Liu H, Lukas RV, Aregawi D, Yamaguchi K, et al. Initial phase 1 study of WT2725 dosing emulsion in patients with advanced malignancies. JCO (2017) 35(15):2066–6. doi: 10.1200/JCO.2017.35.15_suppl.2066
110. Tsuboi A, Hashimoto N, Fujiki F, Morimoto S, Kagawa N, Nakajima H, et al. A phase I clinical study of a cocktail vaccine of Wilms’ tumor 1 (WT1) HLA class I and II peptides for recurrent malignant glioma. Cancer Immunol Immunother (2019) 68(2):331–40. doi: 10.1007/s00262-018-2274-1
111. Migliorini D, Dutoit V, Allard M, Grandjean Hallez N, Marinari E, Widmer V, et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro Oncol (2019) 21(7):923–33. doi: 10.1093/neuonc/noz040
112. Peereboom D, Nabors LB, Kumthekar P, Badruddoja M, Fink K, Lieberman F, et al. Results of phase II trial of SL-701, a novel immunotherapy targeting IL-13Ra2, EphA2, and survivin, in adults with second-line recurrent glioblastoma (GBM). Ann Oncol Off J Eur Soc Med Oncol (2018) 29:viii122–viii123. doi: 10.1093/annonc/mdy273.361
113. Lassman AB, Reardon DA, Lee EQ, Iwamoto FM, Diaz-Mitoma F, Anderson DE, et al. Interim results of a phase I/IIa trial of a therapeutic CMV vaccine against recurrent glioblastoma (GBM). JCO (2019) 37(15):2048–8. doi: 10.1200/JCO.2019.37.15_suppl.2048
114. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol (2010) 28(31):4722–9. doi: 10.1200/JCO.2010.28.6963
115. Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol (2011) 13(3):324–33. doi: 10.1093/neuonc/noq157
116. Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol (2015) 17(6):854–61. doi: 10.1093/neuonc/nou348
117. Reardon DA, Desjardins A, Vredenburgh JJ, O’Rourke DM, Tran DD, Fink KL, et al. Rindopepimut with Bevacizumab for Patients with Relapsed EGFRvIII-Expressing Glioblastoma (ReACT): Results of a Double-Blind Randomized Phase II Trial. Clin Cancer Res (2020) 26(7):1586–94. doi: 10.1158/1078-0432.CCR-18-1140
118. Ahluwalia MS, Peereboom DM, Ciolfi M, Schilero C, Hobbs B, Ciesielski MJ, et al. Phase II study of pembrolizumab plus SurVaxM for glioblastoma at first recurrence. JCO (2020) 38(15):TPS2581–TPS2581. doi: 10.1200/JCO.2020.38.15_suppl.TPS2581
119. De Groot JF, Cloughesy TF, Pitz MW, Narita Y, Nonomura T. A randomized, multicenter phase 2 study of DSP-7888 dosing emulsion in combination with bevacizumab (Bev) versus Bev alone in patients with recurrent or progressive glioblastoma. JCO (2018) 36(15):TPS2071–TPS2071. doi: 10.1200/JCO.2018.36.15_suppl.TPS2071
120. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol (2017) 18(10):1373–85. doi: 10.1016/S1470-2045(17)30517-X
121. Wang QT, Nie Y, Sun SN, Lin T, Han RJ, Jiang J, et al. Tumor-associated antigen-based personalized dendritic cell vaccine in solid tumor patients. Cancer Immunol Immunother (2020) 69(7):1375–87. doi: 10.1007/s00262-020-02496-w
122. Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature (2015) 519(7543):366–9. doi: 10.1038/nature14320
123. Batich KA, Reap EA, Archer GE, Sanchez-Perez L, Nair SK, Schmittling RJ, et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin Cancer Res (2017) 23(8):1898–909. doi: 10.1158/1078-0432.CCR-16-2057
124. Prins RM, Wang X, Soto H, Young E, Lisiero DN, Fong B, et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J Immunother (2013) 36(2):152–7. doi: 10.1097/CJI.0b013e3182811ae4
125. Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, Giovannone AJ, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res (2005) 11(15):5515–25. doi: 10.1158/1078-0432.CCR-05-0464
126. Rudnick JD, Sarmiento JM, Uy B, Nuno M, Wheeler CJ, Mazer MJ, et al. A phase I trial of surgical resection with Gliadel Wafer placement followed by vaccination with dendritic cells pulsed with tumor lysate for patients with malignant glioma. J Clin Neurosci (2020) 74:187–93. doi: 10.1016/j.jocn.2020.03.006
127. Parney IF, Gustafson MP, Peterson T, Steinmetz SM, Dietz AB. ATIM-13. ALLOGENEIC TUMOR LYSATE/AUTOLOGOUS DENDRITIC CELL VACCINES IN NEWLY DIAGNOSED GLIOBLASTOMA: RESULTS OF CLINICAL TRIAL MC1272. Neuro-oncology (2017) 19(Suppl 6):28–9. doi: 10.1093/neuonc/nox168.109
128. Hu J, Rudnick J, Wheeler C, Mazer M, Wang H, Black K, et al. ATIM-18. A PHASE I TRIAL OF VACCINATION WITH AUTOLOGOUS DENDRITIC CELLS PULSED WITH LYSATE DERIVED FROM AN ALLOGENEIC GLIOBLASTOMA STEMLIKE CELL LINE FOR PATIENTS WITH NEWLY DIAGNOSED OR RECURRENT GLIOBLASTOMA. Neuro-oncology (2018) 20(suppl_6):4–4. doi: 10.1093/neuonc/noy148.013
129. Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, Nuño MA, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother (2013) 62(1):125–35. doi: 10.1007/s00262-012-1319-0
130. Olin MR, Low W, McKenna DH, Haines SJ, Dahlheimer T, Nascene D, et al. Vaccination with dendritic cells loaded with allogeneic brain tumor cells for recurrent malignant brain tumors induces a CD4(+)IL17(+) response. J Immunother Cancer (2014) 2:4–1426-2-4. doi: 10.1186/2051-1426-2-4
131. Rudnick JD, Fink KL, Landolfi JC, Markert J, Piccioni DE, Glantz MJ, et al. Immunological targeting of CD133 in recurrent glioblastoma: A multi-center phase I translational and clinical study of autologous CD133 dendritic cell immunotherapy. JCO (2017) 35(15):2059–2059. doi: 10.1200/JCO.2017.35.15_suppl.2059
132. Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC, Due-Tønnesen P, Suso EM, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother (2013) 62(9):1499–509. doi: 10.1007/s00262-013-1453-3
133. Akasaki Y, Kikuchi T, Homma S, Koido S, Ohkusa T, Tasaki T, et al. Phase I/II trial of combination of temozolomide chemotherapy and immunotherapy with fusions of dendritic and glioma cells in patients with glioblastoma. Cancer Immunol Immunother (2016) 65(12):1499–509. doi: 10.1007/s00262-016-1905-7
134. Berneman ZN, Anguille S, Willemen Y, de Velde AV, Germonpre P, Huizing M, et al. Vaccination of cancer patients with dendritic cells electroporated with mRNA encoding the wilms’ tumor 1 protein (WT1): correlation of clinical effect and overall survival with T-cell response. Cytotherapy (2019) 21(5, Supplement):S10. doi: 10.1016/j.jcyt.2019.03.565
135. Wen PY, Reardon DA, Armstrong TS, Phuphanich S, Aiken RD, Landolfi JC, et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients with Glioblastoma. Clin Cancer Res (2019) 25(19):5799–807. doi: 10.1158/1078-0432.CCR-19-0261
136. Inogés S, Tejada S, de Cerio AL, Gállego Pérez-Larraya J, Espinós J, Idoate MA, et al. A phase II trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J Transl Med (2017) 15(1):104–017-1202-z. doi: 10.1186/s12967-017-1202-z
137. Pellegatta S, Eoli M, Cuccarini V, Anghileri E, Pollo B, Pessina S, et al. Survival gain in glioblastoma patients treated with dendritic cell immunotherapy is associated with increased NK but not CD8(+) T cell activation in the presence of adjuvant temozolomide. Oncoimmunology (2018) 7(4):e1412901. doi: 10.1080/2162402X.2017.1412901
138. Buchroithner J, Erhart F, Pichler J, Widhalm G, Preusser M, Stockhammer G, et al. Audencel Immunotherapy Based on Dendritic Cells Has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase II Randomized Trial. Cancers (Basel) (2018) 10(10):372. doi: 10.3390/cancers10100372
139. Erhart F, Buchroithner J, Reitermaier R, Fischhuber K, Klingenbrunner S, Sloma I, et al. Immunological analysis of phase II glioblastoma dendritic cell vaccine (Audencel) trial: immune system characteristics influence outcome and Audencel up-regulates Th1-related immunovariables. Acta Neuropathol Commun (2018) 6(1):135–018-0621-2. doi: 10.1186/s40478-018-0621-2
140. Fadul CE, Fisher JL, Hampton TH, Lallana EC, Li Z, Gui J, et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J Immunother (2011) 34(4):382–9. doi: 10.1097/CJI.0b013e318215e300
141. Yao Y, Luo F, Tang C, Chen D, Qin Z, Hua W, et al. Molecular subgroups and B7-H4 expression levels predict responses to dendritic cell vaccines in glioblastoma: an exploratory randomized phase II clinical trial. Cancer Immunol Immunother (2018) 67(11):1777–88. doi: 10.1007/s00262-018-2232-y
142. Rapp M, Grauer OM, Kamp M, Sevens N, Zotz N, Sabel M, et al. A randomized controlled phase II trial of vaccination with lysate-loaded, mature dendritic cells integrated into standard radiochemotherapy of newly diagnosed glioblastoma (GlioVax): study protocol for a randomized controlled trial. Trials (2018) 19(1):293–018-2659-7. doi: 10.1186/s13063-018-2659-7
143. Huang YC, Chang CN, Wei KC, Lee HC, Chen CC, Cho DY, et al. Atim-42. Safety and Efficacy of Autologous Dendritic Cells/tumor Cell Antigen Adjuvant Therapy of Glioblastoma Multiforme: Results of 59 Cases. Neuro Oncol (2018) 20(Suppl 6):vi11. doi: 10.1093/neuonc/noy148.037
144. Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med (2018) 16(1):142–018-1507-6. doi: 10.1186/s12967-018-1507-6
145. Bota DA, Chung J, Dandekar M, Carrillo JA, Kong XT, Fu BD, et al. Phase II study of ERC1671 plus bevacizumab versus bevacizumab plus placebo in recurrent glioblastoma: interim results and correlations with CD4(+) T-lymphocyte counts. CNS Oncol (2018) 7(3):CNS22–2018-0009. doi: 10.2217/cns-2018-0009
146. Crane CA, Han SJ, Ahn B, Oehlke J, Kivett V, Fedoroff A, et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res (2013) 19(1):205–14. doi: 10.1158/1078-0432.CCR-11-3358
147. Bloch O, Raizer JJ, Lim M, Sughrue M, Komotar R, Abrahams J, et al. Newly diagnosed glioblastoma patients treated with an autologous heat shock protein peptide vaccine: PD-L1 expression and response to therapy. J Clin Oncol (2015) 33(15\_suppl):2011–1. doi: 10.1200/jco.2015.33.15_suppl.2011
148. Bloch O, Crane CA, Fuks Y, Kaur R, Aghi MK, Berger MS, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol (2014) 16(2):274–9. doi: 10.1093/neuonc/not203
149. Bloch O, Lim M, Sughrue ME, Komotar RJ, Abrahams JM, O’Rourke DM, et al. Autologous Heat Shock Protein Peptide Vaccination for Newly Diagnosed Glioblastoma: Impact of Peripheral PD-L1 Expression on Response to Therapy. Clin Cancer Res (2017) 23(14):3575–84. doi: 10.1158/1078-0432.CCR-16-1369
150. Dutoit V, Herold-Mende C, Hilf N, Schoor O, Beckhove P, Bucher J, et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain (2012) 135(Pt 4):1042–54. doi: 10.1093/brain/aws042
151. Ghoochani A, Schwarz MA, Yakubov E, Engelhorn T, Doerfler A, Buchfelder M, et al. MIF-CD74 signaling impedes microglial M1 polarization and facilitates brain tumorigenesis. Oncogene (2016) 35(48):6246–61. doi: 10.1038/onc.2016.160
152. Nagai T, Tanaka M, Tsuneyoshi Y, Xu B, Michie SA, Hasui K, et al. Targeting tumor-associated macrophages in an experimental glioma model with a recombinant immunotoxin to folate receptor beta. Cancer Immunol Immunother (2009) 58(10):1577–86. doi: 10.1007/s00262-009-0667-x
153. Lishner M, Perrin RG, Feld R, Messner HA, Tuffnell PG, Elhakim T, et al. Complications associated with Ommaya reservoirs in patients with cancer. The Princess Margaret Hospital experience and a review of the literature. Arch Intern Med (1990) 150(1):173–6. doi: 10.1001/archinte.1990.00390130145023
154. Kennedy BC, Brown LT, Komotar RJ, McKhann G. Stereotactic catheter placement for Ommaya reservoirs. J Clin Neurosci (2016) 27:44–7. doi: 10.1016/j.jocn.2015.11.005
155. Si QS, Nakamura Y, Schubert P, Rudolphi K, Kataoka K. Adenosine and propentofylline inhibit the proliferation of cultured microglial cells. Exp Neurol (1996) 137(2):345–9. doi: 10.1006/exnr.1996.0035
156. Jacobs VL, Landry RP, Liu Y, Romero-Sandoval EA, De Leo JA. Propentofylline decreases tumor growth in a rodent model of glioblastoma multiforme by a direct mechanism on microglia. Neuro Oncol (2012) 14(2):119–31. doi: 10.1093/neuonc/nor194
157. Chiu TL, Wang MJ, Su CC. The treatment of glioblastoma multiforme through activation of microglia and TRAIL induced by rAAV2-mediated IL-12 in a syngeneic rat model. J BioMed Sci (2012) 19:45–0127-19-45. doi: 10.1186/1423-0127-19-45
158. Bogdahn U, Hau P, Stockhammer G, Venkataramana NK, Mahapatra AK, Suri A, et al. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol (2011) 13(1):132–42. doi: 10.1093/neuonc/noq142
159. Soares KC, Rucki AA, Kim V, Foley K, Solt S, Wolfgang CL, et al. TGF-beta blockade depletes T regulatory cells from metastatic pancreatic tumors in a vaccine dependent manner. Oncotarget (2015) 6(40):43005–15. doi: 10.18632/oncotarget.5656
160. Blumenthal DT, Yalon M, Vainer GW, Lossos A, Yust S, Tzach L, et al. Pembrolizumab: first experience with recurrent primary central nervous system (CNS) tumors. J Neurooncol (2016) 129(3):453–60. doi: 10.1007/s11060-016-2190-1
161. Chamberlain MC, Kim BT. Nivolumab for patients with recurrent glioblastoma progressing on bevacizumab: a retrospective case series. J Neurooncol (2017) 133(3):561–9. doi: 10.1007/s11060-017-2466-0
162. Filley AC, Henriquez M, Dey M. Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget (2017) 8(53):91779–94. doi: 10.18632/oncotarget.21586
163. Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med (2019) 25(3):477–86. doi: 10.1038/s41591-018-0337-7
164. Antonios JP, Soto H, Everson RG, Orpilla J, Moughon D, Shin N, et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight (2016) 1(10):e87059. doi: 10.1172/jci.insight.87059
165. Antonios JP, Soto H, Everson RG, Moughon D, Orpilla JR, Shin NP, et al. Immunosuppressive tumor-infiltrating myeloid cells mediate adaptive immune resistance via a PD-1/PD-L1 mechanism in glioblastoma. Neuro Oncol (2017) 19(6):796–807. doi: 10.1093/neuonc/now287
166. Soderlund J, Erhardt S, Kast RE. Acyclovir inhibition of IDO to decrease Tregs as a glioblastoma treatment adjunct. J Neuroinflammation (2010) 7:44–2094-7-44. doi: 10.1186/1742-2094-7-44
167. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res (2014) 20(20):5290–301. doi: 10.1158/1078-0432.CCR-14-0514
168. Migliorini D, Dietrich PY, Stupp R, Linette GP, Posey AD, June CH. CAR T-Cell Therapies in Glioblastoma: A First Look. Clin Cancer Res (2018) 24(3):535–40. doi: 10.1158/1078-0432.CCR-17-2871
169. Filley AC, Henriquez M, Dey M. CART Immunotherapy: Development, Success, and Translation to Malignant Gliomas and Other Solid Tumors. Front Oncol (2018) 8:453. doi: 10.3389/fonc.2018.00453
170. Hyrenius-Wittsten A, Roybal KT. Paving New Roads for CARs. Trends Cancer (2019) 5(10):583–92. doi: 10.1016/j.trecan.2019.09.005
171. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Wakefield A, Ghazi A, et al. Autologous HER2 CMV bispecific CAR T cells are safe and demonstrate clinical benefit for glioblastoma in a Phase I trial. J Immunother Cancer (2015) 3(Suppl 2):O11–1426-3-S2-O11. doi: 10.1186/2051-1426-3-S2-O11
172. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med (2016) 375(26):2561–9. doi: 10.1056/NEJMoa1610497
173. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984
174. Bagley S, Desai A, Binder Z, Nasrallah M, Hwang W, Maloney-Wilensky E, et al. RBTT-12. A PHASE I STUDY OF EGFRVIII-DIRECTED CAR T CELLS COMBINED WITH PD-1 INHIBITION IN PATIENTS WITH NEWLY, DIAGNOSED, MGMT-UNMETHYLATED GLIOBLASTOMA: TRIAL IN PROGRESS. Neuro-oncology (2019) 21:vi221–1. doi: 10.1093/neuonc/noz175.923
175. Jansen T, Tyler B, Mankowski JL, Recinos VR, Pradilla G, Legnani F, et al. FasL gene knock-down therapy enhances the antiglioma immune response. Neuro Oncol (2010) 12(5):482–9. doi: 10.1093/neuonc/nop052
176. Tschumi BO, Dumauthioz N, Marti B, Zhang L, Lanitis E, Irving M, et al. CART cells are prone to Fas- and DR5-mediated cell death. J Immunother Cancer (2018) 6(1):71–018-0385-z. doi: 10.1186/s40425-018-0410-2
177. Yamamoto TN, Lee PH, Vodnala SK, Gurusamy D, Kishton RJ, Yu Z, et al. T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J Clin Invest (2019) 129(4):1551–65. doi: 10.1172/JCI121491
178. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-Negative TGF-beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol Ther (2018) 26(7):1855–66. doi: 10.1016/j.ymthe.2018.05.003
179. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol (2018) 36(9):847–56. doi: 10.1038/nbt.4195
180. Kuhn NF, Purdon TJ, van Leeuwen DG, Lopez AV, Curran KJ, Daniyan AF, et al. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell (2019) 35(3):473–488.e6. doi: 10.1016/j.ccell.2019.02.006
181. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol (2019) 37(9):1049–58. doi: 10.1038/s41587-019-0192-1
182. Ruella M, Klichinsky M, Kenderian SS, Shestova O, Ziober A, Kraft DO, et al. Overcoming the Immunosuppressive Tumor Microenvironment of Hodgkin Lymphoma Using Chimeric Antigen Receptor T Cells. Cancer Discovery (2017) 7(10):1154–67. doi: 10.1158/2159-8290.CD-16-0850
183. Pearl TM, Markert JM, Cassady KA, Ghonime MG. Oncolytic Virus-Based Cytokine Expression to Improve Immune Activity in Brain and Solid Tumors. Mol Ther Oncolytics (2019) 13:14–21. doi: 10.1016/j.omto.2019.03.001
184. Marchini A, Daeffler L, Pozdeev VI, Angelova A, Rommelaere J. Immune Conversion of Tumor Microenvironment by Oncolytic Viruses: The Protoparvovirus H-1PV Case Study. Front Immunol (2019) 10:1848. doi: 10.3389/fimmu.2019.01848
185. Wollmann G, Ozduman K, van den Pol AN. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer J (2012) 18(1):69–81. doi: 10.1097/PPO.0b013e31824671c9
186. Desjardins A, Sampson JH, Peters KB, Vlahovic G, Randazzo D, Threatt S, et al. Patient survival on the dose escalation phase of the Oncolytic Polio/Rhinovirus Recombinant (PVSRIPO) against WHO grade IV malignant glioma (MG) clinical trial compared to historical controls. JCO (2016) 34(15):2061–1. doi: 10.1200/JCO.2016.34.15_suppl.2061
187. Friedman G, Bag A, Madan-Swain A, Li R, Kachurak K, Osorio D, et al. IMMU-08. PHASE I TRIAL (NCT02457845) SAFETY, TOLERABILITY AND PRELIMINARY EFFICACY OF IMMUNOVIROTHERAPY WITH HSV G207 IN CHILDREN WITH PROGRESSIVE MALIGNANT SUPRATENTORIAL BRAIN TUMORS. Neuro Oncol (2018) 20:i100–0. doi: 10.1093/neuonc/noy059.324
188. Lang FF, Tran ND, Puduvalli VK, Elder JB, Fink KL, Conrad CA, et al. Phase 1b open-label randomized study of the oncolytic adenovirus DNX-2401 administered with or without interferon gamma for recurrent glioblastoma. JCO (2017) 35(15):2002–2. doi: 10.1200/JCO.2017.35.15_suppl.2002
189. Chiocca EA, Yu JS, Lukas RV, Solomon IH, Ligon KL, Nakashima H, et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci Transl Med (2019) 11(505):eaaw5680. doi: 10.1126/scitranslmed.aaw5680
190. Geletneky K, Huesing J, Rommelaere J, Schlehofer JR, Leuchs B, Dahm M, et al. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of Parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer (2012) 12:99–2407-12-99. doi: 10.1186/1471-2407-12-99
191. Wheeler LA, Manzanera AG, Bell SD, Cavaliere R, McGregor JM, Grecula JC, et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro Oncol (2016) 18(8):1137–45. doi: 10.1093/neuonc/now002
192. Ji N, Weng D, Liu C, Gu Z, Chen S, Guo Y, et al. Adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of recurrent high-grade glioma. Oncotarget (2016) 7(4):4369–78. doi: 10.18632/oncotarget.6737
193. Ostertag D, Amundson KK, Lopez Espinoza F, Martin B, Buckley T, Galvão da Silva AP, et al. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro Oncol (2012) 14(2):145–59. doi: 10.1093/neuonc/nor199
194. Mitchell LA, Lopez Espinoza F, Mendoza D, Kato Y, Inagaki A, Hiraoka K, et al. Toca 511 gene transfer and treatment with the prodrug, 5-fluorocytosine, promotes durable antitumor immunity in a mouse glioma model. Neuro Oncol (2017) 19(7):930–9. doi: 10.1093/neuonc/nox037
195. Cloughesy T, Petrecca K, Walbert T, Butowski N, Salacz M, Perry J, et al. LTBK-08. TOCA 511 & TOCA FC VERSUS STANDARD OF CARE IN PATIENTS WITH RECURRENT HIGH GRADE GLIOMA. Neuro Oncol (2019) 21:vi284–4. doi: 10.1093/neuonc/noz219.1199
196. Kanai R, Rabkin SD. Combinatorial strategies for oncolytic herpes simplex virus therapy of brain tumors. CNS Oncol (2013) 2(2):129–42. doi: 10.2217/cns.12.42
197. Andreansky S, He B, van Cott J, McGhee J, Markert JM, Gillespie GY, et al. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Ther (1998) 5(1):121–30. doi: 10.1038/sj.gt.3300550
198. Martikainen M, Essand M. Virus-Based Immunotherapy of Glioblastoma. Cancers (Basel) (2019) 11(2):186. doi: 10.3390/cancers11020186
199. Hardcastle J, Mills L, Malo CS, Jin F, Kurokawa C, Geekiyanage H, et al. Immunovirotherapy with measles virus strains in combination with anti-PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro Oncol (2017) 19(4):493–502. doi: 10.1093/neuonc/now179
200. Saha D, Martuza RL, Rabkin SD. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell (2017) 32(2):253–267.e5. doi: 10.1016/j.ccell.2017.07.006
201. Hou W, Sampath P, Rojas JJ, Thorne SH. Oncolytic Virus-Mediated Targeting of PGE2 in the Tumor Alters the Immune Status and Sensitizes Established and Resistant Tumors to Immunotherapy. Cancer Cell (2016) 30(1):108–19. doi: 10.1016/j.ccell.2016.05.012
202. Suryadevara CM, Desai R, Abel ML, Riccione KA, Batich KA, Shen SH, et al. Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. Oncoimmunology (2018) 7(6):e1434464. doi: 10.1080/2162402X.2018.1434464
203. Ouyang M, White EE, Ren H, Guo Q, Zhang I, Gao H, et al. Metronomic Doses of Temozolomide Enhance the Efficacy of Carbon Nanotube CpG Immunotherapy in an Invasive Glioma Model. PloS One (2016) 11(2):e0148139. doi: 10.1371/journal.pone.0148139
204. Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-Based CAR T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res (2018) 78(4):1031–43. doi: 10.1158/0008-5472.CAN-17-1788
205. Fritzell S, Sanden E, Eberstal S, Visse E, Darabi A, Siesjo P. Intratumoral temozolomide synergizes with immunotherapy in a T cell-dependent fashion. Cancer Immunol Immunother (2013) 62(9):1463–74. doi: 10.1007/s00262-013-1449-z
206. Carvalho JADV, Barbosa CCL, Feher O, Maldaun MVC, Camargo VP, Moraes FY, et al. Systemic dissemination of glioblastoma: literature review. Rev Assoc Med Bras (1992) (2019) 65(3):460–8. doi: 10.1590/1806-9282.65.3.460
207. Dykstra KH, Arya A, Arriola DM, Bungay PM, Morrison PF, Dedrick RL. Microdialysis study of zidovudine (AZT) transport in rat brain. J Pharmacol Exp Ther (1993) 267(3):1227–36.
208. Morrison PF, Laske DW, Bobo H, Oldfield EH, Dedrick RL. High-flow microinfusion: tissue penetration and pharmacodynamics. Am J Physiol (1994) 266(1 Pt 2):R292–305. doi: 10.1152/ajpregu.1994.266.1.R292
209. Krewson CE, Klarman ML, Saltzman WM. Distribution of nerve growth factor following direct delivery to brain interstitium. Brain Res (1995) 680(1-2):196–206. doi: 10.1016/0006-8993(95)00261-N
210. Pardridge WM. CSF, blood-brain barrier, and brain drug delivery. Expert Opin Drug Delivery (2016) 13(7):963–75. doi: 10.1517/17425247.2016.1171315
211. Rizk ML, Zou L, Savic RM, Dooley KE. Importance of Drug Pharmacokinetics at the Site of Action. Clin Transl Sci (2017) 10(3):133–42. doi: 10.1111/cts.12448
212. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523(7560):337–41. doi: 10.1038/nature14432
213. Larson SM, Carrasquillo JA, Cheung NK, Press OW. Radioimmunotherapy of human tumours. Nat Rev Cancer (2015) 15(6):347–60. doi: 10.1038/nrc3925
214. Rapoport SI. Effect of concentrated solutions on blood-brain barrier. Am J Physiol (1970) 219(1):270–4. doi: 10.1152/ajplegacy.1970.219.1.270
215. Siegal T, Rubinstein R, Bokstein F, Schwartz A, Lossos A, Shalom E, et al. In vivo assessment of the window of barrier opening after osmotic blood-brain barrier disruption in humans. J Neurosurg (2000) 92(4):599–605. doi: 10.3171/jns.2000.92.4.0599
216. Raymond JJ, Robertson DM, Dinsdale HB. Pharmacological modification of bradykinin induced breakdown of the blood-brain barrier. Can J Neurol Sci (1986) 13(3):214–20. doi: 10.1017/S0317167100036301
217. Warren K, Jakacki R, Widemann B, Aikin A, Libucha M, Packer R, et al. Phase II trial of intravenous lobradimil and carboplatin in childhood brain tumors: a report from the Children’s Oncology Group. Cancer Chemother Pharmacol (2006) 58(3):343–7. doi: 10.1007/s00280-005-0172-7
218. Hynynen K, McDannold N, Sheikov NA, Jolesz FA, Vykhodtseva N. Local and reversible blood-brain barrier disruption by noninvasive focused ultrasound at frequencies suitable for trans-skull sonications. Neuroimage (2005) 24(1):12–20. doi: 10.1016/j.neuroimage.2004.06.046
219. Janowicz PW, Leinenga G, Götz J, Nisbet RM. Ultrasound-mediated blood-brain barrier opening enhances delivery of therapeutically relevant formats of a tau-specific antibody. Sci Rep (2019) 9(1):9255–019-45577-2. doi: 10.1038/s41598-019-45577-2
220. Vykhodtseva NI, Hynynen K, Damianou C. Histologic effects of high intensity pulsed ultrasound exposure with subharmonic emission in rabbit brain in vivo. Ultrasound Med Biol (1995) 21(7):969–79. doi: 10.1016/0301-5629(95)00038-S
221. Hynynen K, McDannold N, Vykhodtseva N, Jolesz FA. Noninvasive MR imaging-guided focal opening of the blood-brain barrier in rabbits. Radiology (2001) 220(3):640–6. doi: 10.1148/radiol.2202001804
222. Kung Y, Huang HY, Liao WH, Huang AP, Hsiao MY, Wu CH, et al. A Single High-Intensity Shock Wave Pulse With Microbubbles Opens the Blood-Brain Barrier in Rats. Front Bioeng Biotechnol (2020) 8:402. doi: 10.3389/fbioe.2020.00402
223. Goutal S, Gerstenmayer M, Auvity S, Caillé F, Mériaux S, Buvat I, et al. Physical blood-brain barrier disruption induced by focused ultrasound does not overcome the transporter-mediated efflux of erlotinib. J Control Release (2018) 292:210–20. doi: 10.1016/j.jconrel.2018.11.009
224. Manallack DT. The pK(a) Distribution of Drugs: Application to Drug Discovery. Perspect Medicin Chem (2007) 1:25–38. doi: 10.1177/1177391X0700100003
225. Nagpal K, Singh SK, Mishra DN. Drug targeting to brain: a systematic approach to study the factors, parameters and approaches for prediction of permeability of drugs across BBB. Expert Opin Drug Delivery (2013) 10(7):927–55. doi: 10.1517/17425247.2013.762354
226. Meng J, Agrahari V, Youm I. Advances in Targeted Drug Delivery Approaches for the Central Nervous System Tumors: The Inspiration of Nanobiotechnology. J Neuroimmune Pharmacol (2017) 12(1):84–98. doi: 10.1007/s11481-016-9698-1
227. White BD, Duan C, Townley HE. Nanoparticle Activation Methods in Cancer Treatment. Biomolecules (2019) 9(5):202. doi: 10.3390/biom9050202
228. Luo M, Lewik G, Ratcliffe JC, Choi CHJ, Mäkilä E, Tong WY, et al. Systematic Evaluation of Transferrin-Modified Porous Silicon Nanoparticles for Targeted Delivery of Doxorubicin to Glioblastoma. ACS Appl Mater Interfaces (2019) 11(37):33637–49. doi: 10.1021/acsami.9b10787
229. Zhang B, Wang H, Liao Z, Wang Y, Hu Y, Yang J, et al. EGFP-EGF1-conjugated nanoparticles for targeting both neovascular and glioma cells in therapy of brain glioma. Biomaterials (2014) 35(13):4133–45. doi: 10.1016/j.biomaterials.2014.01.071
230. Fang C, Wang K, Stephen ZR, Mu Q, Kievit FM, Chiu DT, et al. Temozolomide nanoparticles for targeted glioblastoma therapy. ACS Appl Mater Interfaces (2015) 7(12):6674–82. doi: 10.1021/am5092165
231. Jena L, McErlean E, McCarthy H. Delivery across the blood-brain barrier: nanomedicine for glioblastoma multiforme. Drug Delivery Transl Res (2020) 10(2):304–18. doi: 10.1007/s13346-019-00679-2
232. Guo L, Ren J, Jiang X. Perspectives on brain-targeting drug delivery systems. Curr Pharm Biotechnol (2012) 13(12):2310–8. doi: 10.2174/138920112803341770
233. Pardridge WM. Delivery of Biologics Across the Blood-Brain Barrier with Molecular Trojan Horse Technology. BioDrugs (2017) 31(6):503–19. doi: 10.1007/s40259-017-0248-z
234. Poduslo JF, Curran GL, Berg CT. Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc Natl Acad Sci USA (1994) 91(12):5705–9. doi: 10.1073/pnas.91.12.5705
235. St-Amour I, Paré I, Alata W, Coulombe K, Ringuette-Goulet C, Drouin-Ouellet J, et al. Brain bioavailability of human intravenous immunoglobulin and its transport through the murine blood-brain barrier. J Cereb Blood Flow Metab (2013) 33(12):1983–92. doi: 10.1038/jcbfm.2013.160
236. Yu YJ, Watts RJ. Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics (2013) 10(3):459–72. doi: 10.1007/s13311-013-0187-4
237. Yu YJ, Atwal JK, Zhang Y, Tong RK, Wildsmith KR, Tan C, et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med (2014) 6(261):261ra154. doi: 10.1126/scitranslmed.3009835
238. Boado RJ, Zhang Y, Zhang Y, Pardridge WM. Humanization of anti-human insulin receptor antibody for drug targeting across the human blood-brain barrier. Biotechnol Bioeng (2007) 96(2):381–91. doi: 10.1002/bit.21120
239. Niewoehner J, Bohrmann B, Collin L, Urich E, Sade H, Maier P, et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron (2014) 81(1):49–60. doi: 10.1016/j.neuron.2013.10.061
240. Ullman JC, Arguello A, Getz JA, Bhalla A, Mahon CS, Wang J, et al. Brain delivery and activity of a lysosomal enzyme using a blood-brain barrier transport vehicle in mice. Sci Transl Med (2020) 12(545):eaay1163. doi: 10.1126/scitranslmed.aay1163
241. Kariolis MS, Wells RC, Getz JA, Kwan W, Mahon CS, Tong R, et al. Brain delivery of therapeutic proteins using an Fc fragment blood-brain barrier transport vehicle in mice and monkeys. Sci Transl Med (2020) 12(545):eaay1359. doi: 10.1126/scitranslmed.aay1359
242. Galstyan A, Markman JL, Shatalova ES, Chiechi A, Korman AJ, Patil R, et al. Blood-brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat Commun (2019) 10(1):3850–019-11719-3. doi: 10.1038/s41467-019-11719-3
243. Kinoshita M, McDannold N, Jolesz FA, Hynynen K. Noninvasive localized delivery of Herceptin to the mouse brain by MRI-guided focused ultrasound-induced blood-brain barrier disruption. Proc Natl Acad Sci USA (2006) 103(31):11719–23. doi: 10.1073/pnas.0604318103
244. Raymond SB, Treat LH, Dewey JD, McDannold NJ, Hynynen K, Bacskai BJ. Ultrasound enhanced delivery of molecular imaging and therapeutic agents in Alzheimer’s disease mouse models. PloS One (2008) 3(5):e2175. doi: 10.1371/journal.pone.0002175
245. Burgess A, Shah K, Hough O, Hynynen K. Focused ultrasound-mediated drug delivery through the blood-brain barrier. Expert Rev Neurother (2015) 15(5):477–91. doi: 10.1586/14737175.2015.1028369
Keywords: GBM - Glioblastoma multiforme, immune escape, immunotherapy, combinatorial therapy, treatment, overview
Citation: Pearson JRD, Cuzzubbo S, McArthur S, Durrant LG, Adhikaree J, Tinsley CJ, Pockley AG and McArdle SEB (2020) Immune Escape in Glioblastoma Multiforme and the Adaptation of Immunotherapies for Treatment. Front. Immunol. 11:582106. doi: 10.3389/fimmu.2020.582106
Received: 10 July 2020; Accepted: 28 September 2020;
Published: 15 October 2020.
Edited by:
Jian Wang, University of Bergen, NorwayReviewed by:
Nahum Puebla-Osorio, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2020 Pearson, Cuzzubbo, McArthur, Durrant, Adhikaree, Tinsley, Pockley and McArdle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephanie E. B. McArdle, U3RlcGhhbmllLm1jYXJkbGVAbnR1LmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.