
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 25 September 2020
Sec. Microbial Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.582044
This article is part of the Research TopicFighting an Elusive Enemy: Staphylococcus aureus and its Antibiotic Resistance, Immune-Evasion and Toxic MechanismsView all 20 articles
 Andrea R. Teufelberger1†
Andrea R. Teufelberger1† Sharon Van Nevel1
Sharon Van Nevel1 Paco Hulpiau2,3Maria Nordengrün4
Paco Hulpiau2,3Maria Nordengrün4 Savvas N. Savvides5,6Sarah De Graeve1
Savvas N. Savvides5,6Sarah De Graeve1 Srinivas Akula7Gabriele Holtappels1Natalie De Ruyck1Wim Declercq2,8Peter Vandenabeele2,8
Srinivas Akula7Gabriele Holtappels1Natalie De Ruyck1Wim Declercq2,8Peter Vandenabeele2,8 Lars Hellman7
Lars Hellman7 Barbara M. Bröker4
Barbara M. Bröker4 Dmitri V. Krysko9,10,11Claus Bachert1,12,13*‡
Dmitri V. Krysko9,10,11Claus Bachert1,12,13*‡ Olga Krysko1,11*‡
Olga Krysko1,11*‡Staphylococcus aureus (S. aureus) can secrete a broad range of virulence factors, among which staphylococcal serine protease-like proteins (Spls) have been identified as bacterial allergens. The S. aureus allergen serine protease-like protein D (SplD) induces allergic asthma in C57BL/6J mice through the IL-33/ST2 signaling axis. Analysis of C57BL/6J, C57BL/6N, CBA, DBA/2, and BALB/c mice treated with intratracheal applications of SplD allowed us to identify a frameshift mutation in the serine (or cysteine) peptidase inhibitor, clade A, and member 3I (Serpina3i) causing a truncated form of SERPINA3I in BALB/c, CBA, and DBA/2 mice. IL-33 is a key mediator of SplD-induced immunity and can be processed by proteases leading to its activation or degradation. Full-length SERPINA3I inhibits IL-33 degradation in vivo in the lungs of SplD-treated BALB/c mice and in vitro by direct inhibition of mMCP-4. Collectively, our results establish SERPINA3I as a regulator of IL-33 in the lungs following exposure to the bacterial allergen SplD, and that the asthma phenotypes of mouse strains may be strongly influenced by the observed frameshift mutation in Serpina3i. The analysis of this protease-serpin interaction network might help to identify predictive biomarkers for type-2 biased airway disease in individuals colonized by S. aureus.
Several type 2 airway diseases, such as allergic rhinitis, chronic rhinosinusitis with nasal polyps, and allergic asthma are associated with nasal colonization by Staphylococcus aureus (S. aureus) (1, 2). S. aureus can secrete a wide range of virulence factors, among which staphylococcal serine protease-like proteins (Spls) have been identified as bacterial allergens and inducers of allergic asthma in C57BL/6J mice by activating the IL-33/ST2 signaling axis (3, 4). Spl-specific IgE levels are increased in asthmatic patients compared to healthy controls, and detectable amounts of Spls have been found in nasal polyp tissue (3, 4). Strikingly, 25–30% of the general population are persistently colonized with S. aureus and up to 69% are intermittent or occasional S. aureus carriers (5). Even though there is strong evidence for S. aureus being an inducer, enhancer, and driver of chronic inflammatory airway diseases (3, 6–9), it is not understood why not all persistent S. aureus carriers develop a chronic inflammatory response toward it. Since S. aureus exposure on its own cannot explain the pathogenesis, a genetic predisposition might be underlying the response toward S. aureus.
IL-33 is an innate cytokine of the IL-1 family, which is expressed in epithelial, endothelial (10), and smooth muscle cells (11), as well as in fibroblasts, activated mast cells (12), and dendritic cells (13), and released upon allergen exposure or during necrosis (14, 15). IL-33 mediates Th2 cytokine production in innate lymphoid cells type 2 (ILC2s), Th2 cells, and invariant natural killer T cells by binding to its membrane bound receptor ST2 (13, 16–19). Like other IL-1 cytokine family members, the cytokine activity of IL-33 can be regulated by proteolytic cleavage (20). When it is cleaved by endogenous proteases or proteolytic allergens in the cleavage/activation domain, the cytokine activity of IL-33 is increased (21–25). Inactivation of IL-33 occurs by cleavage in the IL-1-like cytokine domain through endogenous proteases such as the human neutrophil proteinase 3, the human mast cell chymase and the murine chymase mouse mast cell protease 4 (mMCP-4), or by the caspases 3 and 7 in humans and mice (26–29).
Endogenous protease activity is, in turn, under tight regulation of serine protease inhibitors, such as serpins. Serpins inhibit proteases with their C-terminal reactive center loop (RCL). Upon cleavage in the RCL, they form SDS-stable complexes with their target proteases by covalent binding (30). The murine Serpina3i is expressed at low levels in the lungs, thymus, and spleen during homeostasis (31).
In the present study we have identified SERPINA3I as a novel regulator of IL-33 processing in a murine asthma model. Interestingly, BALB/c mice carry a frameshift mutation in the Serpina3i gene, leading to faster cleavage of IL-33 upon serine protease-like protein D (SplD) exposure. This observation may explain the lack of a type 2 inflammatory response upon SplD challenge of the lungs. Our results suggest that SERPINA3I is important for controlling inflammatory reactions in mice.
The aim of this study was to find the underlying genetic difference between BALB/c and C57BL/6J mice leading to the different response toward SplD.
Recombinant SplD was produced in the Bacillus subtilis strain 6051HGW LS8P-D, which lacks the proprietary proteases WprA, Epr, Bpr, NprB, NprE, Vpr, Mpr, and AprE, as previously described in detail (3, 4). SplD was purified from the cell-free supernatants by means of ion-exchange chromatography on an SP Sepharose Fast Flow column (GE Healthcare, Fairfield, CT, United States), followed by a 2-step purification with centrifugal filter units (Amicon Ultra 30K/10K, Merck Millipore, Billerica, MA, United States). The buffer was exchanged to PBS and the quality of the native SplD preparation was verified by using SDS-PAGE. Recombinant full-length, C-terminal HIS tagged SERPINA3I (isoform X3; NCBI reference sequence: XP_017170642.1) was produced in HEK293 cells by ProSpec-Tany Technogene Ltd. (Rehovot, Israel). The purified protein was endotoxin low (<0.01 EU/μg) and provided in PBS. Proteolytically active mMCP-4 was produced and activated essentially as previously described in Andersson et al. (32).
Female C57BL/6JRj (C57BL/6J), C57BL/6NRj (C57BL/6N), BALB/cJRj (BALB/c), CBA/JRj (CBA), and DBA/2 JRj (DBA/2) wild-type mice (Janvier Labs, Saint-Berthevin Cedex, France) were 6–8 weeks old during the treatment protocols. Animals were kept under standard conditions, in the individually ventilated (IVC) cages, 12 h light/dark cycle, access to food and water ad libitum. All animal experiments were approved by the local ethics committees of Ghent University.
For the short-term treatment, mice were treated with an intratracheal (i.t.) application of either 50 μL PBS or 50 μg SplD in 50 μL PBS once and euthanized 6 or 12 h after the single treatment. For the long-term sensitization protocol, C57BL/6J, C57BL/6N, CBA, DBA/2, and BALB/c mice were treated with six i.t. applications of either 50 μL PBS (Gibco, Erembodegem, Belgium) or 45 μg SplD in 50 μL PBS with one additional application of 50 μL PBS every 2 days. Additional groups of BALB/c mice were treated with either 50 μL PBS and 200 ng murine IL-33 (Peprotech, Rocky Hill, NJ, United States) in 50 μL PBS or with 45 μg SplD in combination with 200 ng of IL-33 in 50 μL PBS (SplD + IL-33) in the same manner as the other treatment groups. All mice underwent light anesthesia by inhalation of isoflurane/air (Ecuphar, Oostkamp, Belgium) during every application. 48 h after the last i.t. application, mice were euthanized with an intraperitoneal injection of 100 μL nembutal (Ceva Santé Animale, Libourne, France). For the reconstitution of SERPINA3I BALB/c, mice received 50 μg SplD in combination with 5 μg SERPINA3I (SplD + SERPINA3I), or sole 5 μg SERPINA3I, and were euthanized after 12 h. Serum, bronchoalveolar lavage fluid (BALF) and lungs were collected and processed as previously described (4). All experimental protocols are summarized in Supplementary Table 2.
Blood was collected in EDTA Microvettes 200Z (Sarstedt, Nümbrecht, Germany) and centrifuged for 10 min at 3000 rpm. Serum was collected and stored at -20°C before further analysis. BALF was collected by rinsing the airways three times with 0.3 mL of 1% bovine serum albumin (BSA; Sigma Aldrich, Diegem, Belgium) in PBS with complete protease inhibitor cocktail (Roche Diagnostics, Anderlecht, Belgium) and twice with 1 mL of 0.2% EDTA (Sigma-Aldrich) in PBS and kept at 4°C. Red blood cells were lysed for 2 min with VersaLyse buffer (Beckman Coulter, Krefeld, Germany), and BALF cells were used for flow cytometry analysis. The left lobe of the perfused lungs was fixed with 10% formalin (Sigma-Aldrich) for paraffin embedding, while the right lobes were either snap-frozen or minced and digested with 1 mg/mL collagenase type II (Worthington Biochemical, Lakewood, NJ, United States) for flow cytometry analysis. Snap-frozen lungs were either used for protein or RNA extraction. For protein extraction, they were homogenized by using the Tissue Lyser LT (Qiagen, Antwerp, Belgium) for 2 min at 50 oscillations/s with 10 times more w/v T-Per Tissue Protein Extraction Reagent (Thermo Fisher Scientific) with HALT protease inhibitor cocktail kit (Thermo Fisher Scientific). After 10 min of centrifugation at 3000 rpm, supernatants were collected and repeatedly centrifuged at 15,000 rpm for three in each. The total protein concentration of the lung homogenates was determined with the Bio-Rad Protein assay (Bio-Rad Laboratories, Hercules, CA, United States) in the collected supernatants.
Thirty μg protein of lung homogenates were denatured for 10 min at 95°C with sample buffer under reducing conditions (sample buffer: Laemmli buffer with 5% β-mercapto-ethanol and 0.25% Bromophenol blue) and loaded on 4–15% mini-PROTEAN TGX Stain-Free Protein gels (Bio-Rad), separated by means of SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad). For Western Blotting, mouse IL-33 antigen affinity-purified polyclonal goat IgG (dilution 1:300; R&D Systems) antibody was used with polyclonal peroxidase labeled anti-goat IgG (H + L; 1:1000; Vector Laboratories Inc., Burlingame, CA, United States); and mouse anti β-actin primary antibody, clone AC-74 (1:5000; Sigma-Aldrich), was used with polyclonal peroxidase labeled anti-mouse IgG (1:2000; Invitrogen). Bands were visualized with the Immobilon Western Chemiluminescence HRP substrate (Merck Millipore). Semiquantitative analysis of band intensities was performed by measuring the area under the peak of plotted lanes with the ImageJ software (National Institutes of Health, Bethesda, MD, United States). For all Western blot quantifications, n = 4.
Concentrations of murine IL-4, IL-5, and IL-13, and IL-33 in lung homogenates were analyzed by using Luminex Performance assays (R&D Systems, Abingdon, United Kingdom).
Deparaffinized and rehydrated 5-μm lung sections were stained for mucus producing goblet cells by using the periodic acid–Schiff kit (Sigma-Aldrich). Positively stained cells in airway with a perimeter between 800 and 2000 μm were counted using the cell counting tool in ImageJ.
Total serum IgE was measured with the Mouse IgE Ready-Set-Go! ELISA Kit (affymetrix eBioscience, Vienna, Austria). The SplD-specific IgE ELISA was performed as described previously in detail by coating the plates with 5 μg/mL SplD, and specific IgE was measured in serum.
The following antibodies were used for identification of immune cells: anti-mouse CD16/CD32 clone 93 (eBioscience), Gr1-FITC (Ly-66) clone RB6-8C5 (eBioscience), SiglecF-PE clone ES22-10D8 (Miltenyi Biotec, Bergisch Gladbach, Germany), CD11b–peridinin-chlorophyll-protein complex (PerCP)–Cy5.5 (eBioscience), F4/80-APC (BD Biosciences), CD8a-PerCp-Cy5.5 clone 53-6.7 (eBioscience), CD4-FITC clone RM4-4 (eBioscience), MHCII clone M5/114.15.2 FITC (eBioscience), CD11c PE-Cy7 clone HL3 (eBioscience), and LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Thermo Fisher Scientific). Cells were stained for 30 min at room temperature and washed with PBS; LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Thermo Fisher Scientific) was used for 10 min to exclude dead cells. Cells were washed and analyzed by using flow cytometry with the FACS Canto II (BD Biosciences).
For RNA extraction, cells were lysed with RLT buffer (Qiagen), or frozen lung tissues were ground in liquid nitrogen, prior to lysis in RLT (Qiagen). Lysed lung tissues were loaded on the QIAshredder Mini Spin Columns (Qiagen). The RNA was extracted from the cell lysate or QIAshredder Mini Spin Columns eluate following the manufacturer’s instructions of the RNeasy Mini Kit (Qiagen). DNA was digested using the RNase-Free DNase Set (Qiagen). RNA concentrations were measured with Nanodrop, and cDNA was synthesized with the iScriptTM Advanced cDNA Synthesis Kit for RT-qPCR (Bio-Rad). For RT-qPCR, mIL-33, and serpina3i were measured and normalized to HRPT1 and RTL32. The following primers were used (5′-3′ direction):
HPRT1-Fwd: CCTAAGATGAGCGCAAGTTGAA; HPRT1- Rev: CCACAGGACTAGAACACCTGCTAA; RTL32-Fwd: GG CACCAGTCAGACCGATATG; RTL32-Rev: CCTTCTCCG CACCCTGTTG; IL-33-Fwd: GCTGCGTCTGTTGACACATT; and IL-33-Rev: CACCTGGTCTTGCTCTTGGT. Serpina3i primers were purchased from Bio-Rad, intron-spanning (qMmuCID0022819), and Il1rl1 (encoding ST2) Taqman (QuantiTect Probe PCR Kit; Mm00516117_m1) was purchased from Applied Biosystems. Amplification reactions were performed on the Roche Light Cycler 480. PCR reactions containing 5 ng/well sample cDNA were used with 1 × SsoAdvancedTM Universal SYBR® Green Supermix (Bio-Rad) and 250 nM primer pairs in a final volume of 5 μL. The PCR protocol consisted of 1 cycle at 95°C, 2 min, followed by 44 cycles at 95°C, 5 s; 30 s at 60°C; and 1 s at 72°C. Ct and Tm were calculated by the Roche LightCycler 480 software (Roche Molecular Systems Inc., Pleasanton, CA, United States), and the relative expression of the genes of interest was normalized to the expression of the reference genes HPRT1 and RTL32 in qbase+ (Biogazelle, Zwijnaarde, Belgium).
One and a half μg of recombinant activated mMCP-4 (0.1 mg/mL in PBS) were incubated alone or with 1.5 μg (1:1 ratio), 7.5 μg (1:5 ratio), or 15 μg (1:10 ratio) of recombinant SERPINA3I (0.1 mg/mL in PBS) for 1 h at 37°C; 12.5 ng of recombinant murine IL-33 (Peprotech, 210-33, Rocky Hill, NJ, United States) in PBS were added to each mixture and incubated again for 1 h at 37°C. Thereafter, samples were denatured (10 min, 95°C with sample buffer) under reducing conditions and loaded on a precast gel (4–15% Mini-PROTEAN TGX Stain-Free Precast Gels, Bio-Rad) to perform SDS-PAGE. IL-33 Western Blot was performed as mentioned above.
Sequence variation data (release v5) from the Mouse Genome Project (33) was used for comparative analyses of the C57BL/6N, CBA, DBA/2, and BALB/c strains relative to the C57BL/6J reference strain. In total, a list of 352 indels affecting the protein coding sequence in at least one of the mouse strains was retrieved using a custom Perl script.
Analysis of data obtained in animal studies was performed using the GraphPad prism version 7 software (GraphPad Software, La Jolla, CA, United States) and one-way ANOVA or Kruskal–Wallis test followed by Tukey multiple comparison or Dunn’s multiple comparison tests, respectively.
C57BL/6J mice respond to repeated intratracheal applications of SplD with key features of allergic asthma (Figure 1A). However, BALB/c mice merely increased total BALF cell numbers (Figure 1A). Eosinophils and neutrophils were nearly absent in SplD-treated BALB/c mice, while C57BL/6J mice showed a strong eosinophilic response toward SplD in the BALF (Figures 1B,C). Goblet cell hyperplasia was not observed in SplD-treated BALB/c mice, in contrast to C57BL/6J mice (Figure 1D). Measurable titers of SplD-specific serum IgE could only be detected in SplD-treated C57BL/6J mice (Figure 1E), and total IgE titers were not different between SplD-sensitized and PBS control BALB/c mice, while a significant up-regulation occurred in the SplD-sensitized C57BL/6J mice (Figure 1F).
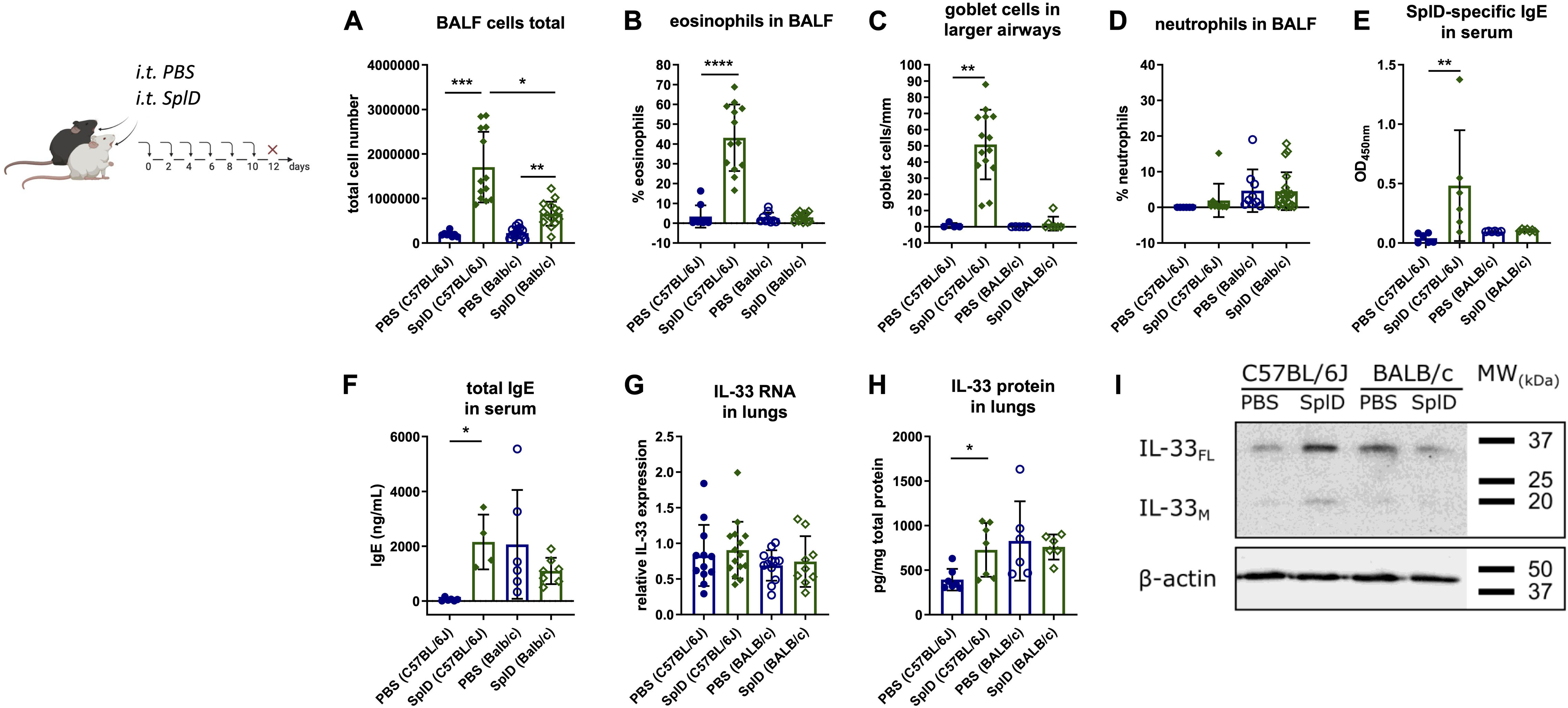
Figure 1. Lack of type 2 airway inflammation and increased IL-33 cleavage in SplD-exposed BALB/c mice. C57BL/6J and BALB/c mice received six i.t. applications of PBS or SplD and 48 h after the last application the samples were collected. (A) total cell count, (B) percentage of eosinophils, (C) Goblet cell count in airways with 800–2000 μm perimeter of periodic acid–Schiff-stained lung sections, and (D) neutrophils of bronchoalveolar lavage fluid (BALF). (E) SplD-specific and (F) total IgE measured by ELISA in sera of mice. (G) relative mRNA levels and (H) protein levels of IL-33 in lungs determined by RT-qPCR and Luminex, respectively. (I) Western blotting of mouse lungs stained for IL-33 and β-actin. One representative blot is shown. Data are presented as mean ± SD. n = 4–15. *P < 0.05; **P < 0.01; ***P < 0.001.
Basal and SplD-induced IL-33 mRNA levels, as determined by quantitative RT-qPCR, were similar in the lungs of BALB/c and C57BL/6J mice after 2 weeks of treatment with PBS or SplD (Figure 1G). In contrast, IL-33 protein levels were significantly elevated in SplD-treated C57BL/6J mice but did not change significantly in BALB/c mice compared to the PBS-treated controls (Figure 1H). Less prominent bands of full length (30 kDa; IL-33FL) and mature IL-33 (18 kDa; IL-33M) was observed by Western blotting in SplD-treated BALB/c mice compared to C57BL/6J mice after 2 weeks of treatment (Figure 1I).
IL-33 expression was measured in the lungs of C57BL/6J and BALB/c mice 6 and 12 h after a single i.t. application of SplD. 6 h after SplD exposure, the IL-33 mRNA levels were not significantly increased and were comparable between C57BL/6J and BALB/c mice. However, 12 h after SplD exposure, significantly higher IL-33 mRNA levels were observed in BALB/c mice, while in C57BL/6J mice a statistically insignificant trend to increased IL-33 mRNA levels was observed (Figure 2A). In contrast to mRNA expression, IL-33 protein levels were significantly up-regulated in both mouse strains 6 h after a single i.t. application of SplD. The levels were significantly decreased 12 h after SplD application in both mouse strains compared to 6 h after SplD exposure (Figure 2B). When comparing full length IL-33FL of BALB/c and C57BL/6J mice after 6 and 12 h of a single application of SplD using western blotting analysis, there was a strong increase of IL-33FL in both strains, but in BALB/c mice, the proteolytic processing of IL-33FL and IL-33M was accelerated (Figure 2C). In BALB/c mice, the bands representing IL-33 FL and IL-33M were most pronounced after 6 h and declined 12 h after SplD treatment. On the contrary, IL-33FL levels were comparable after 6 and 12 h of SplD treatment in C57BL/6J mice, and IL-33M was increased only after 12 h (Figure 2C).
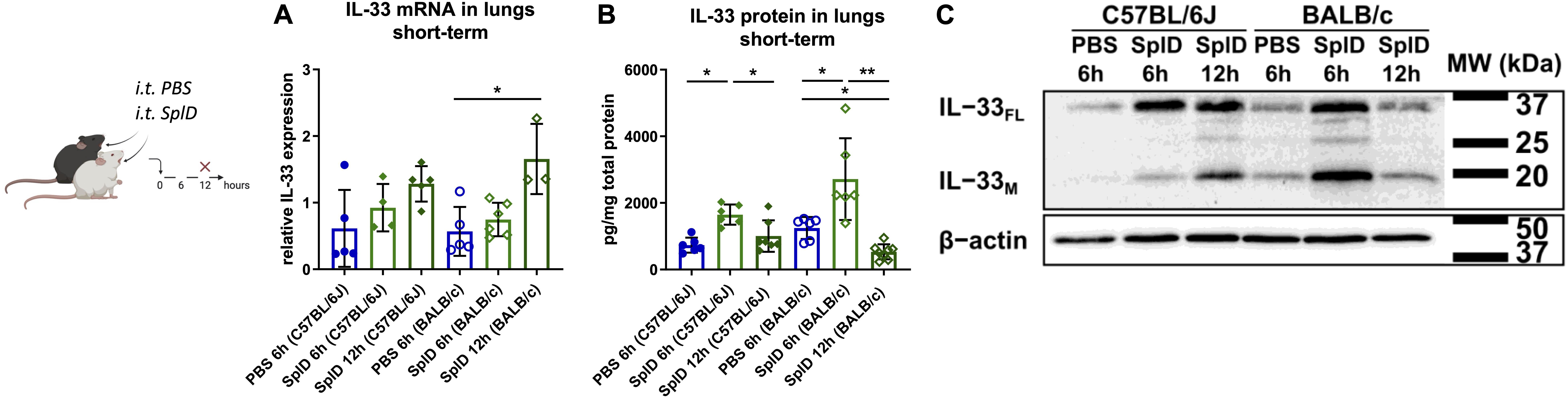
Figure 2. Early degradation of IL-33 in SplD-exposed BALB/c mice. (A) mRNA levels and (B) protein levels of IL-33 6 and 12 h after PBS or SplD challenge in lungs, determined by RT-qPCR and Luminex, respectively, and (C) western blotting of mouse lungs collected 6 and 12 h after a single application of SplD in C57BL/6J or BALB/c mice stained for IL-33 and β-actin. One representative blot is shown. Data are presented as mean ± SD. n = 3–8. *P < 0.05; **P < 0.01.
Taken together, the lower IL-33 protein amounts in BALB/c mice at 12 h after a single application and 48 h after repeated applications could be indicative of an increased activity protease cleaving IL-33 in the lungs of BALB/c.
We aimed to compensate the lack of IL-33 in BALB/c mice by intratracheal applications of exogenous IL-33 directly after each SplD application. After the addition of IL-33 to SplD, the percentage of eosinophils in the BALF was significantly elevated, while the percentage of neutrophils was decreased without major effect on the total cell number in BALF (Figures 3A–C). The combined treatment with SplD + IL-33 induced an up-regulation of CD4+ T cells in the BALF, compared to the SplD or IL-33 only treatment groups (Figure 3D). The numbers of CD8+ cells in the BALF did not significantly differ between the treatment groups (Figure 3E). In the lungs, the percentage of eosinophils was significantly elevated in mice that received SplD + IL-33 compared to all control groups (Figure 3F). BALB/c mice treated with IL-33 alone or in combination with SplD showed elevated percentages of MHCII+CD11c+ dendritic cells in the lungs (Figure 3G). The levels of IL-5 were below detection levels in the lungs of BALB/c mice treated with SplD, and no up-regulation could be seen after treatment with IL-33 or a combination of IL-33 and SplD (Supplementary Figure 2B). The levels of IL-4 and IL-13 also remained very low in all experimental groups without significant difference compared between the groups (Supplementary Figures 2A,C). SplD-specific serum IgE (Figure 3H) and total serum IgE (Figure 3I) were significantly up-regulated in the SplD + IL-33 group. Thus, the addition of exogenous IL-33 could sensitize BALB/c mice to SplD, while SplD given alone did not stimulate IgE production in these mice. Goblet cell hyperplasia was observed in the IL-33 only and the SplD + IL-33 groups, indicating that goblet cell formation and mucus production are strongly dependent on IL-33 (Figures 3J,L). IL-33 supplementation restored the up-regulation of Il1rl1 mRNA in BALB/c mice to a comparable degree as in C57BL/6J mice, suggesting that the rapid cleavage of IL-33 explains the absence of ST2 induction in BALB/c mice that were treated with SplD alone (Figure 3K).
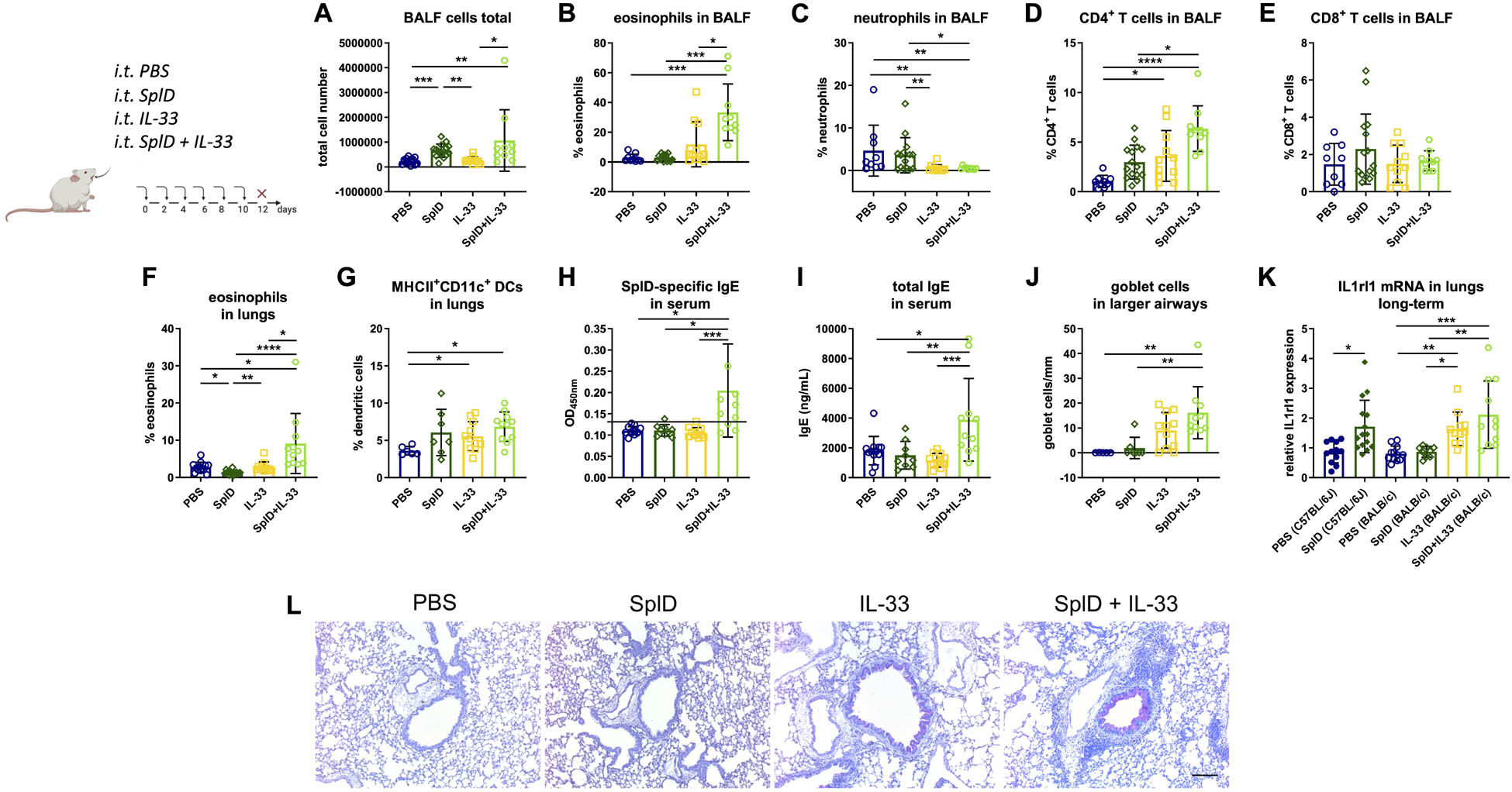
Figure 3. Supplementation with IL-33 restores the asthmatic response and has a synergistic effect on SplD-induced allergic sensitization in BALB/c mice. BALB/c wild-type mice were treated with six i.t. applications of PBS, SplD, IL-33 or SplD + IL-33 every second day. 48 h after the last application, mice were euthanized, and samples were collected. (A) Total cell count and percentage of (B) eosinophils, (C) neutrophils, (D) CD4 + T cells, and (E) CD8 + T cells in the BALF were determined by flow cytometry. Percentage of (F) eosinophils and (G) MHCII + CD11c + dendritic cells (DCs) in the lungs were determined by flow cytometry. (H) SplD-specific IgE and (I) total IgE in sera of mice, measured by ELISA; the black line indicates the background optical density. (J) Goblet cell count in airways of 800–2000-μm perimeter in periodic acid–Schiff-stained lung sections. (K) Relative Il1rl1 mRNA expression in lungs. (L) representative images of periodic acid–Schiff-stained lung sections. Data are presented as mean ± SD. n = 4–15. *P < 0.05; **P < 0.01; ***P < 0.001.
C57BL/6N, CBA, and DBA/2 mice were tested in addition to C57BL/6J and BALB/c mice to define SplD responders and non-responders in order to facilitate the search for the genetic cause underlying the different inflammatory responses toward SplD. We compared the relative magnitude of inflammatory response parameters in different mouse strains and presented the data in radar graphs where the highest mean value of each parameter amongst all five tested strains was taken as one hundred percent (Figure 4A). SplD-treated C57BL/6J mice showed the strongest response in all measured parameters, which indicate a Th2-biased inflammation, but not in those reflecting their neutrophil and CD8+ T cell responses (Figure 4A). C57BL/6N mice show a comparable reaction pattern as C57BL/6J mice upon SplD treatment, even though the magnitude of response was significantly reduced and the total cell numbers in BALF cells were not increased (Figure 4B). BALB/c mice did not develop a Th2 inflammation in response to SplD. There was, however, an increase in CD8+ T cells, but as shown in Figure 3E, the numbers of CD8+ T cells in the BALF were low (Figure 4C). DBA/2 mice presented with a very weak response toward SplD, which was, however, neutrophilic rather than eosinophilic (Figure 4D). CBA mice responded in a manner comparable to DBA/2 mice, but neutrophil and T cell counts were lower than in the PBS control group of this strain (Figure 4E). Goblet cell hyperplasia was only found in SplD-treated C57BL/6J and C57BL/6N mice (Figure 4). The data of each measured parameter of C57BL/6N, DBA/2, and CBA mice with the statistical analysis can be found in Supplementary Figure 1. We classified BALB/c, CBA, and DBA/2 mice as SplD non-responder strains due to the weak or absent type 2 inflammatory response in comparison to C57BL/6J and C57BL/6N mice, which were defined as SplD-responder strains.
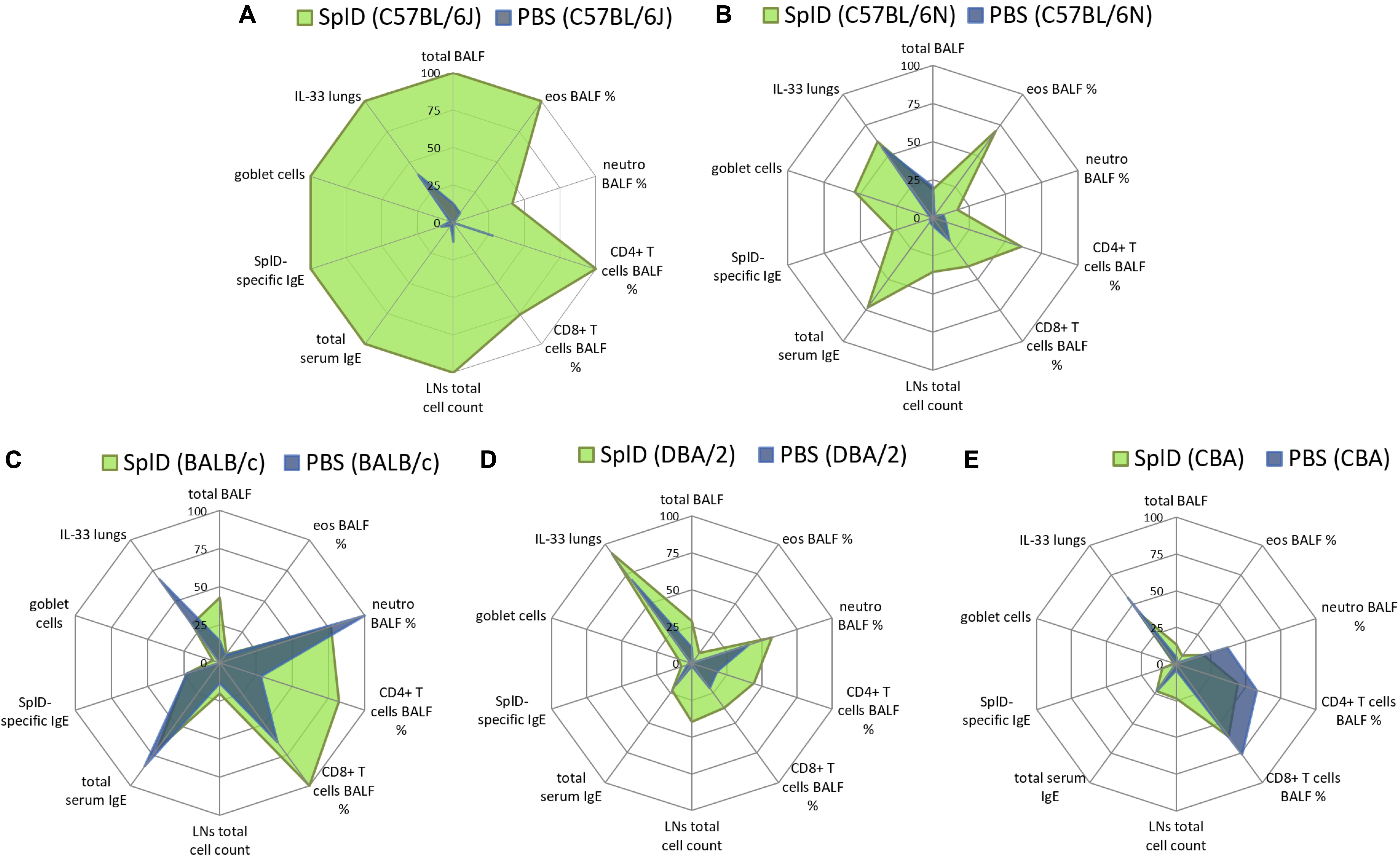
Figure 4. C57BL/6J and C57BL/6N mice respond to SplD with key features of allergic asthma, while BALB/c, CBA, and DBA/2 mice lack a clear type 2 response. Radar graphs, presenting relative values of measured parameters from (A) C57BL/6J, (B) C57BL/6N, (C) BALB/c, (D) DBA/2, and (E) CBA mice treated repeatedly with PBS or SplD. Per parameter, the highest mean value between the groups was set at one hundred percent.
Sequence variation data (release v5) from the Mouse Genome Pro (33) was used for comparative analyses of the C57BL/6N, CBA, DBA/2, and BALB/c strains relative to the C57BL/6J reference strain. In total, a list of 352 indels affecting the protein coding sequence in at least one of the mouse strains was retrieved using a custom Perl script. Out of this list, 116 frameshift mutations and nine stop-codon-inducing frameshift mutations were selected for further analysis as these mutations are expected to exert the most severe effects on the function of the encoded proteins. These variant consequences, predicted by the Ensembl Variant Effect Predictor (VEP), were further investigated using CLC Main Workbench to predict if the main transcript is affected and if the frameshift possibly affects the function of the mutated gene product. In 20 genes, frameshift or stop frameshift mutations were identified upstream or in their active domain. These 20 genes were studied by literature research to find pro- and contra-arguments for them being crucial for the asthmatic response toward SplD (Supplementary Table 1). Our workflow (Figure 5) allowed us to identify Serpina3i and CXCL-11 as the most promising candidates. CXCL-11 mRNA was not detectable by RT-qPCR in the lungs of SplD-treated mice in any of the five mouse strains (data not shown) and was therefore excluded.
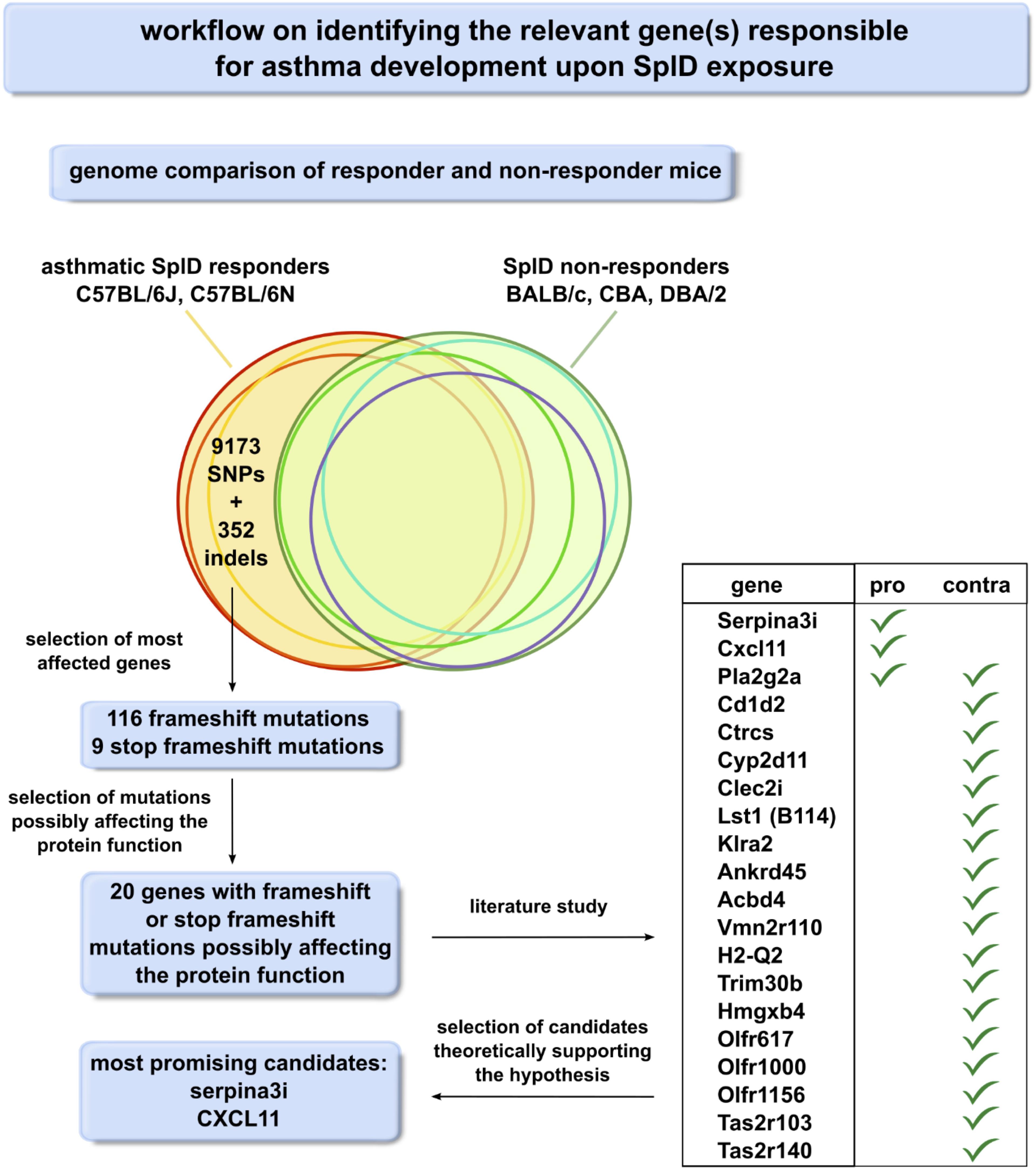
Figure 5. Workflow scheme for the identification of the relevant gene(s) responsible for asthma development upon SplD exposure. Genomes of SplD responders (C57BL/6J and C57BL/6N mouse strains) were compared to the genomes of SplD non-responders (BALB/c, CBA and DBA/2 mouse strains). The indels specific for SplD responders were further analyzed for their consequence on the gene product, and the frameshift mutations were selected as the mutations affecting protein function the most.
Mice harbor an unusually large group of Serpina3 genes as a result of extensive gene duplication and diversification, which in turn encode for 13 closely related SERPINA3 protein products, albeit with differing target specificities and biodistribution. The high level of sequence similarity shared by murine SERPINA3 proteins essentially guarantees a highly conserved core structure. To this end, murine SERPINA3I displays a 74% sequence identity to SERPINA3N (Figure 6A) for which the high resolution crystal structure is available. We harnessed the high sequence homology of the two proteins and the available structural information to construct a homology model for SERPINA3I (Figure 6B). Thus, SERPINA3I is expected to adopt a fold displaying a main central beta-sandwich consisting of two beta-sheets flanked by a cluster of 7–9 alfa helices. This sets the structural stage for a third beta-sheet at the top of the fold to help project the flexible RCL. The RCL contains an enzyme cleavage site (P1-P1’), which is located at the C-terminus of the protein. RCL is expected to be cleaved by a target protease at P1 and P1’, leading to its covalent attachment via the main carbonyl carbon of the P1 residue of the SERPINA3I. This would be expected to establish a stable complex between SERPINA3I and a protease, such as mMCP-4 leading to the inhibition of mMCP-4. The frameshift mutation in serpina3i was stated in the sanger mouse genome sequencing database (rs242560633). The sequence lacks a cytosine at bp747, which results in a frameshift at residue Cys249 and a stop codon after residue 251. As the RCL starts at G357 (31), the mutated Serpina3i is predicted to result in an unfunctional protein. In C57BL/6J and C57BL/6N mice, SERPINA3I is predicted to be full length, comprising 408 amino acids (NP_001186869.1).

Figure 6. Protein sequence alignment of murine SERPINA3N and SERPINA3I from C57BL/6J and BALB/c mice and structural mapping of SERPINA3I deficiency in BALB/c mice. (A) Alignment of the sequences of murine SERPINA3N (Uniprot sequence Q91WP6) and the sequences for SERPINA3I from mouse strains C57BL/6J and BALB/c. The sequence segment absent in BALB/c SERPINA3I as a result of the frameshift mutation at Cys249 and the introduction of a stop codon after residues 251 is colored in red. The reactive center loop (RCL) locates within the deleted sequence and is colored in orange. (B) Cartoon representation of a predicted model for murine SERPINA3I based on the crystal structure of murine SERPINA3N (Horvath et al. 2005) as computed by the program RaptorX (Källberg et al. 2012). The figure was produced with PyMOL v. 2.3.0.
To obtain insights into the structural and functional consequences of the identified frameshift mutation, we compared the sequences of full-length and truncated SERPINA3I to the closely related SERPINA3N (Figure 6A) and constructed a homology model of SERPINA3I based on the crystal structure of the mouse SERPINA3N (34) (Figure 6B). Our analyses illustrate that the predicted protein product of Serpina3i in mouse strain BALB/c would display a debilitating sequence truncation from Cys259 onward that includes the RCL and would prevent the protein from folding to a functional form (Figures 6A,B). Thus, the observed deletion would ultimately cause deficiency in SERPINA3I in BALB/c mice and other mouse strains bearing the same mutation. A public online tool1 is available to analyze different mouse strains having the same Serpina3i frameshift mutation (35). Gene expression of Serpina3i in mouse lungs collected 6 h after a single i.t. application of PBS or SplD showed no significant up-regulation in SplD-treated mice. Furthermore, Serpina3i mRNA was up-regulated in response to IL-33 + SplD treatment in BALB/c mice suggesting a possible involvement of SERPINA3I in IL-33 signaling or regulation (Figure 7A). In other mouse strains, no difference in gene expression of Serpina3i after a single i.t. application SplD was observed. Importantly, the observed difference in Serpina3i gene expression difference does not fully reflect the observed phenotypic difference since a truncation of the gene in the non-responder mouse strains BALB/c, CBA, and DBA/2 would result in an unfunctional protein.
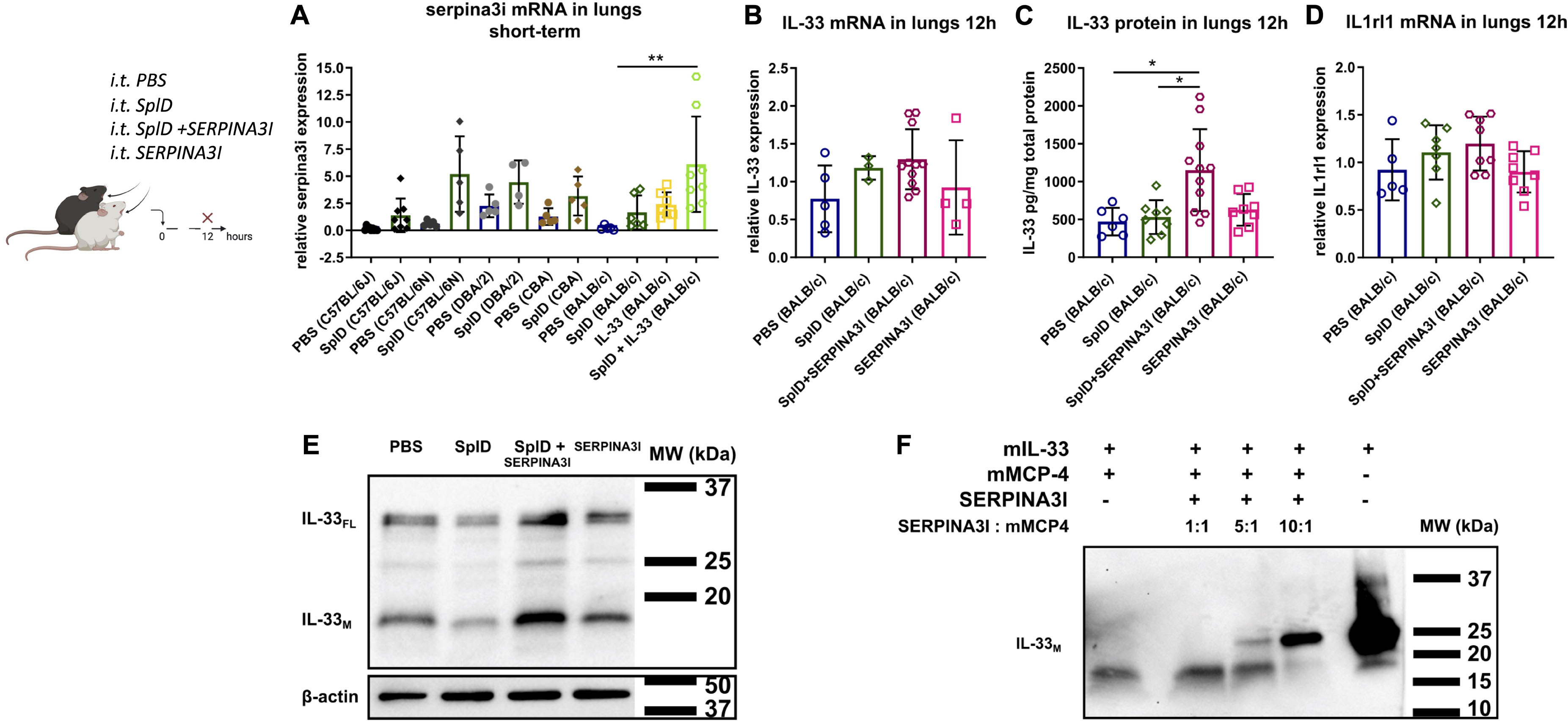
Figure 7. Serpina3i mRNA expression is up-regulated in mouse lungs upon SplD and IL-33 exposure and decelerates IL-33 degradation in vivo and in vitro. (A) relative mRNA expression of Serpina3i in lungs 6 h after a single i.t. application of PBS or SplD in C57BL/6J, C57BL/6N, DBA/2, CBA, and BALB/c mice as well as BALB/c mice 6 h after IL-33 or SplD + IL-33 i.t. treatment determined by RT-qPCR. Relative mRNA levels determined by RT-qPCR of IL-33 (B) and Il1rl1 (D) determined by RT-PCR. IL-33 protein determined by (C) Luminex and (E) Western blot in mouse lungs of BALB/c mice 12 h after PBS, SplD, SERPINA3I, or SplD + SERPINA3I treatment. Data are presented as mean ± SD. n = 5–9. (F) IL-33 Western blot of recombinant mature murine IL-33 (20 kDa) co-incubated with mMCP-4 for 1 h with and without 1 h preincubation with increasing concentrations of SERPINA3I. One representative blot of three independent experiments is shown. *P < 0.05; **P < 0.01.
To test the functional activity in vivo, recombinant full-length SERPINA3I was administered intratracheally directly after SplD to BALB/c mice. SERPINA3I treatment did not cause any up-regulation of IL-4, IL-5, or IL-13 in the lungs of BALB/c mice (Supplementary Figures 2D–F), while, IL-33FL and IL-33M protein levels were significantly higher in the lungs of mice receiving SplD + SERPINA3I than when SplD was administered alone (Figure 7C). As expected the IL-33 and Il1rl1 mRNA expression levels were not significantly changed (Figures 7B,D). The analysis of the cleavage bands of IL-33 by Western blot revealed that both IL-33FL and IL-33M were increased in SplD + SERPINA3I-treated mice (Figure 7E). These results indicate that SERPINA3I can decelerate the degradation of IL-33FL that was induced by in vivo application of SplD. Moreover, preincubation of mMCP-4 with SERPINA3I prevents mMCP-4-induced enzymatic degradation of recombinant murine IL-33M in a dose-dependent manner (Figure 7F).
In this study, we have demonstrated that the allergic response toward the S. aureus protease SplD is mouse strain dependent. Our data suggest that rather than differences in S. aureus strains, the genetic variability of the host could be an important determinant of the immune response to Spls in human S. aureus carriers. In humans that are naturally exposed to S. aureus, the quality of the Spl-specific antibody and T-cell response is strongly biased toward a Th2 profile on average with extensive variation between individuals (3). This could partially be explained by differences in exposure to Spls, which are encoded in around 80% of clinical S. aureus isolates. Our data, however, suggest that genetic differences affecting the posttranslational IL-33 regulation could also play a role.
While C57BL/6J and C57BL/6N mice show typical features of allergic asthma, this response is lacking or significantly diminished in BALB/c, CBA, and DBA/2 mouse strains. C57BL/6 and BALB/c mice have often been referred to as Th1-prone or Th2-prone strains, respectively (36). In striking contrast, C57BL/6 mice presented a higher eosinophilic Th2-biased response than BALB/c mice in our SplD sensitization model. A stronger eosinophilic response in C57BL/6 mice compared to BALB/c mice has been described previously in other murine asthma models using OVA/alum, house dust mite extract or the house dust mite allergen Der p 1 (37–40). In the mentioned models, however, BALB/c mice still developed a weak eosinophilic response, which might be due to the activation of other pathways next to the IL-33/ST2 signaling axis. We have shown previously that the type 2 inflammatory response toward SplD and lung infiltration with IL-13+ type innate lymphoid cells and IL13+ CD4+ T cells in C57BL/6J mice is mainly mediated by IL-33 (4). In this study, we found that C57BL/6J and BALB/c mice also differ in their post-translational regulation of IL-33. A single intratracheal application of SplD caused an up-regulation of IL-33 in both mouse strains after 6 h; however, IL-33 was degraded more rapidly in BALB/c than in C57BL/6J mice. In line with these results, C57BL/6 mice responded to long-term exposure to SplD with significantly increased Il1rl1 expression, while this was not the case in BALB/c mice. We excluded an impairment in ST2 expression downstream of IL-33 in BALB/c mice, because the administration of IL-33 restored the up-regulation of Il1rl1 and, in combination with SplD, led to a strong eosinophilic response in BALB/c mice. Our observed mouse strain dependence of the phenotype in the SplD asthma model illustrates the importance of the genetic background when testing a potential allergen in mice (Figure 8). The observed loss of function mutation in Serpina3i could also explain other mouse strain-dependent differences in allergic asthma, which have previously been reported. Genetic differences in the regulation of IL-33 could be determinants of allergic sensitization or severity of airway diseases. In humans there are several single-nucleotide polymorphisms (SNPs) upstream of the IL-33 gene, which are associated with asthma prevalence (41); however, the functional consequences of these SNPs is largely unknown. The human SERPINA3 gene has expanded by gene duplication into a cluster of 14 equivalent mouse SERPINA3 genes, Serpina3a-n. Based on protein expression patterns, especially in the brain, Serpina3n is considered as the murine orthologue of the human SERPINA3 (31, 34). The murine SERPINA3N is known to inhibit leukocyte elastase and cathepsin G (34), but the mast cell chymase inhibitory function of α-1 anti-chymotrypsin, however, could potentially be conserved in SERPINA3I and could explain our observed mechanism. Mutations in SERPINA3 have been described to have an impact in lung diseases. An association between childhood onset asthma and a 1.4 kb gain mutation downstream of SERPINA3 has been reported (42). Mutations in SERPINA3 leading to decreased α-1 anti-chymotrypsin serum levels were associated with the inflammatory lung diseases COPD and emphysema (43). Our study revealed a novel IL-33 regulation pathway of SERPINA3I inhibiting the IL-33 degrading mast cell chymase mMCP-4, which could be, with the equivalent human proteins, of general importance in allergic airway diseases (44). The human SERPINA3 is a known mast cell chymase inhibitor and could thus be relevant in IL-33 regulation (45).
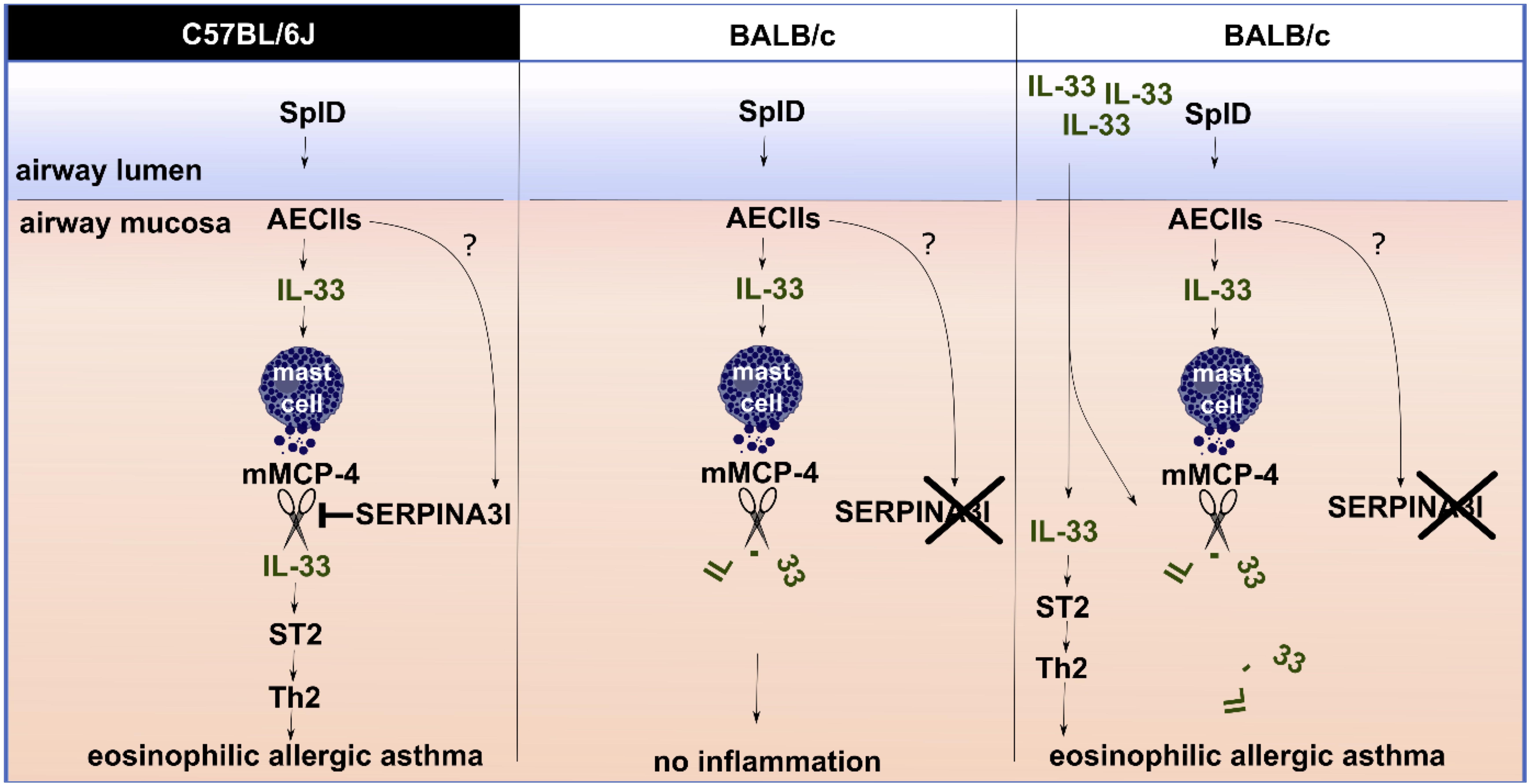
Figure 8. The role of SERPINA3I in the IL-33 regulation mechanisms in C57BL/6J and BALB/c mice upon SplD exposure. SplD administration to the murine airway mucosa causes IL-33 release from alveolar epithelial cells type 2 (AECIIs), and Serpina3i up-regulation in the lungs. In the lungs, IL-33 triggers mast cells to release murine mast cell protease 4 (mMCP-4), which can degrade IL-33. In C57BL/6J mice, functional SERPINA3I is present; it inhibits mMCP-4 and thereby prevents IL-33 degradation (shown in vitro). As a result, IL-33 triggers a type 2 inflammatory response via the IL-33/ST2 axis. BALB/c mice lack a functional SERPINA3I protein due to a mutation. Consequently, SplD-triggered IL-33 is degraded, and no type 2 inflammation is triggered. In BALB/c mice the administration of an active form of recombinant IL-33 in excess additionally to SplD is leading to an inflammatory response comparable to C57BL/6J mice since the degradation of IL-33 can be circumvented to the degree that the IL-33/ST2 signaling can take place.
The study of individual differences in this protease-serpin interaction network may help to identify predictive biomarkers for Th2-biased airway disease development, especially in individuals colonized by S. aureus.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Ethics committee of Faculty of Medicine and Health Sciences, Ghent University.
AT designed and performed experiments, analyzed data, and wrote the original draft. SV performed experiments. PH performed genome-wide comparison analysis and predicted protein function affecting frameshift mutations. SS performed protein sequence and structural analyses. MN provided SplD. GH and SA performed experiments. ND performed experiments. SD performed experiments. LH provided recombinant Mcpt-4. WD and PV provided reagents. DK provided analysis tools and supervised experiments. BB provided reagents. CB supervised experiments and edited the manuscript. OK designed, performed, supervised, and analyzed experiments, and edited the manuscript. All authors discussed the results and commented on the manuscript.
AT is a doctoral fellow paid by the Fund for Scientific Research Flanders. The study has been supported by an FWO grant 3G065319N to CB, and OK, CB, WD, and DK are supported by BOF 14-GOA-019; SS, by BOF17-GOA-028; PV, by EOS 30826052 MODEL-IDI, G.0C31.14N, G.0C31.14N, G.0C37.14N, FWO G0E04.16N, G.0C76.18N, G.0B71.18N, BOF16/MET_V/007, FAF-F/2016/865, and VIB. BB is supported by the German Research Council (DFG: GRK 1870, TRR34). MN is a doctoral fellow supported by the German Research Council (GRK 1870) and the Exzellenzförder program Mecklenburg-Vorpommern, Germany (Card-ii-Omics). Research in the DK group is supported by FWO-Vlaanderen 1506218N, 1507118N, RF No. 13.1902.21.0026, G043219, and BOF 01/O3618.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.582044/full#supplementary-material
Supplementary Figure 1 | Inflammatory profile of C57BL/6N, DBA/2 and CBA mice upon repeated intratracheal applications of PBS or SplD for 2 weeks. (A) Total cell count, (B) percentage of eosinophils, (C) neutrophils, (D) CD4+ T cells, and (E) CD8 + T cells in the BALF analyzed by flow cytometry. (F) Total cell count of local draining lymph nodes, (G) total, and (H) SplD-specific IgE in serum measured by ELISA. (I) Goblet cells in airways with a perimeter of 800–2000 μm, analyzed in periodic acid–Schiff-stained lungs. (J) IL-33 protein levels in lungs measured by Luminex. (K) Relative Il1rl1 mRNA expression in lungs. Data are presented as mean ± SD. n = 4–6.
Supplementary Figure 2 | Analysis of type-2 cytokines in the lung homogenates of mice by Luminex. The levels of IL-4 (A), IL-5 (B), and IL-13 (C) cytokines in the lungs of C57BL/6J, and BALB/c mice treated with six i.t. applications of PBS, SplD, IL-33, or SplD + IL-33 every second day for 2 weeks. The lung homogenates of BALB/c mice after 12 h after a single i.t. application of PBS, SplD, SERPINA3I, or SplD + SERPINA3I treatment were analyzed for IL-4 (D), IL-5 (E), and IL-13 (F). Data are presented as mean ± SD. n = 6. The limit of detection of each cytokine is indicated by a dotted line.
Supplementary Table 1 | A list of mutated genes between SplD-responder and non-responder strains. Mutation, predicted consequence for the protein function, pro- and contra arguments for being a candidate gene responsible for asthma development according to literature research.
Supplementary Table 2 | A summary of experimental procedures per figure.
1. Pastacaldi C, Lewis P, Howarth P. Staphylococci and staphylococcal superantigens in asthma and rhinitis: a systematic review and meta-analysis. Allergy. (2011) 66:549–55. doi: 10.1111/j.1398-9995.2010.02502.x
2. Van Zele T, Gevaert P, Watelet JB, Claeys G, Holtappels G, Claeys C, et al. Staphylococcus aureus colonization and IgE antibody formation to enterotoxins is increased in nasal polyposis. J Allergy Clin Immunol. (2004) 114:981–3. doi: 10.1016/j.jaci.2004.07.013
3. Stentzel S, Teufelberger A, Nordengrün M, Kolata J, Schmidt F, van Crombruggen K, et al. Staphylococcal serine protease-like proteins are pacemakers of allergic airway reactions to Staphylococcus aureus. J Allergy Clin Immunol. (2017) 139:492–500.e8. doi: 10.1016/j.jaci.2016.03.045
4. Teufelberger AR, Nordengrün M, Braun H, Maes T, De Grove K, Holtappels G, et al. The IL-33/ST2 axis is crucial in type 2 airway responses induced by Staphylococcus aureus-derived serine protease-like protein D. J Allergy Clin Immunol. (2018) 141:549–59.e7. doi: 10.1016/j.jaci.2017.05.004
5. Mehraj J, Witte W, Akmatov MK, Layer F, Werner G, Krause G. Epidemiology of Staphylococcus aureus nasal carriage patterns in the community. Curr Top Microbiol Immunol. (2016) 398:55–87. doi: 10.1007/82_2016_497
6. Teufelberger AR, Bröker BM, Krysko DV, Bachert C, Krysko O. Staphylococcus aureus orchestrates type 2 airway diseases. Trends Mol Med. (2019) 25:696–707. doi: 10.1016/j.molmed.2019.05.003
7. Bachert C, Zhang N. Chronic rhinosinusitis and asthma: novel understanding of the role of IgE ‘above atopy’. J Intern Med. (2012) 272:133–43. doi: 10.1111/j.1365-2796.2012.02559.x
8. Tomassen P, Vandeplas G, Zele T. Van, Cardell LO, Arebro J, Olze H, et al. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol. (2016) 137:1449–56.e4. doi: 10.1016/j.jaci.2015.12.1324
9. Lan F, Zhang N, Holtappels G, De Ruyck N, Krysko O, Van Crombruggen K, et al. Staphylococcus aureus induces a mucosal type 2 immune response via epithelial cell-derived cytokines. Am J Respir Crit Care Med. (2018) 198:452–63. doi: 10.1164/rccm.201710-2112OC
10. Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS One. (2008) 3:e3331. doi: 10.1371/journal.pone.0003331
11. Préfontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, et al. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol. (2009) 183:5094–103. doi: 10.4049/jimmunol.0802387
12. Hsu CL, Neilsen CV, Bryce PJ. IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS One. (2010) 5:e11944. doi: 10.1371/journal.pone.0011944
13. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
14. Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol. (2011) 186:4375–87. doi: 10.4049/jimmunol.1003020
15. Lamkanfi M, Dixit VM. IL-33 raises alarm. Immunity. (2009) 31:5–7. doi: 10.1016/j.immuni.2009.06.011
16. Chackerian AA, Oldham ER, Murphy EE, Schmitz J, Pflanz S, Kastelein RA. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J Immunol. (2007) 179:2551–5. doi: 10.4049/jimmunol.179.4.2551
17. Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. (2011) 12:631–8. doi: 10.1038/ni.2045
18. Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, et al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol. (2012) 129:216–27.e1-6. doi: 10.1016/j.jaci.2011.10.036
19. Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol. (2008) 20:1019–30. doi: 10.1093/intimm/dxn060
20. Talabot-Ayer D, Martin P, Seemayer CA, Vigne S, Lamacchia C, Finckh A, et al. Immune-mediated experimental arthritis in IL-33 deficient mice. Cytokine. (2014) 69:68–74. doi: 10.1016/j.cyto.2014.05.007
21. Cayrol C, Duval A, Schmitt P, Roga S, Camus M, Stella A, et al. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat Immunol. (2018) 19:375–85. doi: 10.1038/s41590-018-0067-5
22. Hayakawa M, Hayakawa H, Matsuyama Y, Tamemoto H, Okazaki H, Tominaga S. Mature interleukin-33 is produced by calpain-mediated cleavage in vivo. Biochem Biophys Res Commun. (2009) 387:218–22. doi: 10.1016/j.bbrc.2009.07.018
23. Lefrançais E, Duval A, Mirey E, Roga S, Espinosa E, Cayrol C, et al. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci USA. (2014) 111:15502–7. doi: 10.1073/pnas.1410700111
24. Scott IC, Majithiya JB, Sanden C, Thornton P, Sanders PN, Moore T, et al. Interleukin-33 is activated by allergen- and necrosis-associated proteolytic activities to regulate its alarmin activity during epithelial damage. Sci Rep. (2018) 8:3363. doi: 10.1038/s41598-018-21589-2
25. Lefrançais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard JP, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA. (2012) 109:1673–8. doi: 10.1073/pnas.1115884109
26. Clancy DM, Sullivan GP, Moran HBT, Henry CM, Reeves EP, McElvaney NG, et al. Extracellular Neutrophil proteases are efficient regulators of IL-1, IL-33, and IL-36 cytokine activity but poor effectors of microbial killing. Cell Rep. (2018) 22:2937–50. doi: 10.1016/j.celrep.2018.02.062
27. Fu Z, Thorpe M, Alemayehu R, Roy A, Kervinen J, de Garavilla L, et al. Highly selective cleavage of cytokines and chemokines by the human mast cell chymase and neutrophil cathepsin G. J Immunol. (2017) 198:1474–83. doi: 10.4049/jimmunol.1601223
28. Lüthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. (2009) 31:84–98. doi: 10.1016/j.immuni.2009.05.007
29. Roy A, Ganesh G, Sippola H, Bolin S, Sawesi O, Dagälv A, et al. Mast cell chymase degrades the alarmins heat shock protein 70, biglycan, HMGB1, and interleukin-33 (IL-33) and limits danger-induced inflammation. J Biol Chem. (2014) 289:237–50. doi: 10.1074/jbc.M112.435156
30. Heit C, Jackson BC, McAndrews M, Wright MW, Thompson DC, Silverman GA, et al. Update of the human and mouse SERPIN gene superfamily. Hum Genomics. (2013) 7:22. doi: 10.1186/1479-7364-7-22
31. Horvath AJ, Forsyth SL, Coughlin PB. Expression patterns of murine antichymotrypsin-like genes reflect evolutionary divergence at the Serpina3 locus. J Mol Evol. (2004) 59:488–97. doi: 10.1007/s00239-004-2640-9
32. Andersson MK, Karlson U, Hellman L. The extended cleavage specificity of the rodent beta-chymases rMCP-1 and mMCP-4 reveal major functional similarities to the human mast cell chymase. Mol Immunol. (2008) 45:766–75. doi: 10.1016/j.molimm.2007.06.360
33. Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. (2011) 477:289–94. doi: 10.1038/nature10413
34. Horvath AJ, Irving JA, Rossjohn J, Law RH, Bottomley SP, Quinsey NS, et al. The murine orthologue of human antichymotrypsin: a structural paradigm for clade A3 serpins. J Biol Chem. (2005) 280:43168–78. doi: 10.1074/jbc.M505598200
35. Timmermans S, Van Montagu M, Libert C. Complete overview of protein-inactivating sequence variations in 36 sequenced mouse inbred strains. Proc Natl Acad Sci USA. (2017) 114:9158–63. doi: 10.1073/pnas.1706168114
36. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
37. Gueders MM, Paulissen G, Crahay C, Quesada-Calvo F, Hacha J, Van Hove C, et al. Mouse models of asthma: a comparison between C57BL/6 and BALB/c strains regarding bronchial responsiveness, inflammation, and cytokine production. Inflamm Res. (2009) 58:845–54. doi: 10.1007/s00011-009-0054-2
38. Van Hove CL, Maes T, Cataldo DD, Guéders MM, Palmans E, Joos GF, et al. Comparison of acute inflammatory and chronic structural asthma-like responses between C57BL/6 and BALB/c mice. Int Arch Allergy Immunol. (2009) 149:195–207. doi: 10.1159/000199715
39. Kelada SN, Wilson MS, Tavarez U, Kubalanza K, Borate B, Whitehead GS, et al. Strain-dependent genomic factors affect allergen-induced airway hyperresponsiveness in mice. Am J Respir Cell Mol Biol. (2011) 45:817–24. doi: 10.1165/rcmb.2010-0315OC
40. Serra-Pages M, Torres R, Plaza J, Herrerias A, Costa-Farré C, Marco A, et al. Activation of the Prostaglandin E2 receptor EP2 prevents house dust mite-induced airway hyperresponsiveness and inflammation by restraining mast cells’ activity. Clin Exp Allergy. (2015) 45:1590–600. doi: 10.1111/cea.12542
41. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. (2010) 363:1211–21. doi: 10.1056/NEJMoa0906312
42. Rogers AJ, Chu JH, Darvishi K, Ionita-Laza I, Lehmann H, Mills R, et al. Copy number variation prevalence in known asthma genes and their impact on asthma susceptibility. Clin Exp Allergy. (2013) 43:455–62. doi: 10.1111/cea.12060
43. Ishii T, Matsuse T, Teramoto S, Matsui H, Hosoi T, Fukuchi Y, et al. Association between alpha-1-antichymotrypsin polymorphism and susceptibility to chronic obstructive pulmonary disease. Eur J Clin Invest. (2000) 30:543–8. doi: 10.1046/j.1365-2362.2000.00655.x
44. Krysko O, Teufelberger A, Van Nevel S, Krysko DV, Bachert C. Protease/antiprotease network in allergy: the role of Staphylococcus aureus protease-like proteins. Allergy. (2019) 74:2077–86. doi: 10.1111/all.13783
Keywords: allergy, asthma, IL-33, S. aureus, SplD, type 2 immunity
Citation: Teufelberger AR, Van Nevel S, Hulpiau P, Nordengrün M, Savvides SN, De Graeve S, Akula S, Holtappels G, De Ruyck N, Declercq W, Vandenabeele P, Hellman L, Bröker BM, Krysko DV, Bachert C and Krysko O (2020) Mouse Strain-Dependent Difference Toward the Staphylococcus aureus Allergen Serine Protease-Like Protein D Reveals a Novel Regulator of IL-33. Front. Immunol. 11:582044. doi: 10.3389/fimmu.2020.582044
Received: 10 July 2020; Accepted: 24 August 2020;
Published: 25 September 2020.
Edited by:
Rachel McLoughlin, Trinity College Dublin, IrelandReviewed by:
Gaurav Gupta, NIIT University, IndiaCopyright © 2020 Teufelberger, Van Nevel, Hulpiau, Nordengrün, Savvides, De Graeve, Akula, Holtappels, De Ruyck, Declercq, Vandenabeele, Hellman, Bröker, Krysko, Bachert and Krysko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olga Krysko, b2xnYS5rcnlza29AdWdlbnQuYmU=; Claus Bachert, Y2xhdXMuYmFjaGVydEB1Z2VudC5iZQ==
†Present address: Andrea R. Teufelberger, Department of Medicine Solna, Immunology and Allergy Division, Karolinska Institutet, Solna, Sweden
‡These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.