 Kanako Watanabe-Kusunoki
Kanako Watanabe-Kusunoki Daigo Nakazawa
Daigo Nakazawa Akihiro Ishizu
Akihiro Ishizu Tatsuya Atsumi
Tatsuya Atsumi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 16 September 2020
Sec. Molecular Innate Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.575890
This article is part of the Research TopicDAMPs across the Tree of Life, Volume 2: Regulated Cell Death and Immune ResponsesView all 10 articles
Thrombomodulin (TM), which is predominantly expressed on the endothelium, plays an important role in maintaining vascular homeostasis by regulating the coagulation system. Intravascular injury and inflammation are complicated physiological processes that are induced by injured endothelium-mediated pro-coagulant signaling, necrotic endothelial- and blood cell-derived damage-associated molecular patterns (DAMPs), and DAMP-mediated inflammation. During the hypercoagulable state after endothelial injury, TM is released into the intravascular space by proteolytic cleavage of the endothelium component. Recombinant TM (rTM) is clinically applied to patients with disseminated intravascular coagulation, resulting in protection from tissue injury. Recent studies have revealed that rTM functions as an inflammatory regulator beyond hemostasis through various molecular mechanisms. More specifically, rTM neutralizes DAMPs, including histones and high mobility group box 1 (HMGB1), suppresses excessive activation of the complement system, physiologically protects the endothelium, and influences both innate and acquired immunity. Neutrophil extracellular traps (NETs) promote immunothrombosis by orchestrating platelets to enclose infectious invaders as part of the innate immune system, but excessive immunothrombosis can cause intravascular injury. However, rTM can directly and indirectly regulate NET formation. Furthermore, rTM interacts with mediators of acquired immunity to resolve vascular inflammation. So far, rTM has shown good efficacy in suppressing inflammation in various experimental models, including thrombotic microangiopathy, sterile inflammatory disorders, autoimmune diseases, and sepsis. Thus, rTM has the potential to become a novel tool to regulate intravascular injury via pleiotropic effects.
Endothelial cells coordinate vascular homeostasis, including vessel permeability, provision of a lining surface, and coagulation system regulation. To prevent unnecessary clotting, the endothelium expresses anti-coagulant factors, such as tissue factor pathway inhibitor and thrombomodulin (TM), and regulators of platelet activation, such as nitric oxide, prostacyclin, and ADPase, at steady state. When traumatic vascular injury occurs, platelet aggregation and the activated blood coagulation system invoke a thrombus to prevent blood loss. Moreover, damaged endothelium reduces the expression of anti-coagulant and platelet molecules, and releases pro-coagulant factors via the activation of nuclear factor-kappa B (NF-κB) signaling, consequently enhancing thrombus formation. Meanwhile, during non-traumatic intravascular injury, including disseminated intravascular coagulation (DIC), atherosclerosis, and thrombotic microangiopathy (1), the endothelium collaborates with the blood coagulation system and platelets to cope with the traumatic situation, possibly forming an unwanted thrombus. In addition, cross-talk between the activated coagulation system and inflammatory signaling leads to mutual amplification (2). Accordingly, damage-associated molecular patterns (DAMPs) released from injured tissues and blood cells activate the innate immune system and elicit vascular inflammation (3, 4). DAMPs directly activate platelets and indirectly induce platelet aggregation via interaction with neutrophils, leading to an enhancement of the pre-existing pro-coagulant state. This series of events of coagulation and blood cell activation, collectively referred to as immunothrombosis, is supposed to physiologically enclose and effectively kill invading microbes as part of an innate immune response (5). The structural basis of the immunothrombotic clot is formed by fibrin, consisting of coagulant factors, platelets, and leukocytes. The immunothrombus can also be involved in the development of non-infectious diseases, including ischemia-reperfusion, drug-induced tissue damage, autoimmune diseases, and cancer as an executor of intravascular injury. In the pro-coagulant state, TM derived from altered endothelium serves to maintain vascular homeostasis by participating in the coagulation system. Furthermore, TM possesses multiple regulatory properties against inflammation beyond its anti-coagulant effect, which could possibly contribute to the termination of intravascular injury (6, 7).
TM is a transmembrane glycoprotein encoded by the THBD gene, and it is expressed on endothelium, immune cells (including neutrophils, macrophages, monocytes, and dendritic cells), vascular smooth muscle cells, keratinocytes, and lung alveolar epithelial cells (8–10). The structure of TM comprises five domains; each domain possesses a different function. Surface domains are a lectin-like domain (TMD1), a domain with six epidermal growth factor-like structures (TMD2), and a serine- and threonine-rich domain (TMD3). Certain stimuli, including tissue factor, orchestrate the coagulation cascade and produce thrombin as a coagulant executor. In response to thrombin production, thrombomodulin on the endothelium acts as a thrombin receptor to reduce the ability of thrombin that converts fibrinogen to fibrin and activates platelet. The thrombin-thrombomodulin complexes activate protein C and the activated protein C (APC) inactivates Va and VIIIa, resulting in the suppression of thrombin generation (11, 12). As such, TM naturally serves to terminate excessive intravascular coagulation.
The surface TMD1 domain has no anti-coagulant effects, but has various anti-inflammatory properties. TM directly acts as a natural regulator of inflammation via its lectin-like domain TMD1 by (1) inhibiting leukocyte-mediated intravascular injury, (2) neutralizing DAMPs, including high mobility group box 1 (HMGB1) protein and histones, (3) binding to bacteria-derived components, and (4) suppressing the complement system. (1) Transgenic mice with a genetically deleted TMD1 domain showed increased mortality in endotoxin-induced sepsis, together with the finding that adhesion molecule expression and neutrophil infiltration were increased in TMD1-deficient endothelium (13). Ex vivo studies have shown that additional TMD1 binds to endothelial antigen during inflammation, competitively inhibiting leukocyte migration and adhesion (14). Furthermore, we (15) showed that recombinant TM (rTM), containing TMD123, directly binds to neutrophils via the macrophage-1 antigen (Mac-1) receptor, and thus inhibits neutrophil activation. In addition, rTM affects lymphocytes to inhibit pro-inflammatory cytokine/chemokine production during an inflammatory response. (2) Necrotic parenchymal cells and neutrophil extracellular traps (NETs) release HMGB1 and histones into the extracellular space. The former is a nuclear chromatin-binding protein that transduces intracellular pro-inflammatory signals via toll-like receptor 4 (TLR4) and the receptor for advanced glycation endproducts (RAGE) (16). The latter exerts distinct biological effects, including direct cell toxicity, exacerbation of immune responses via TLR stimulation, and the activation of platelets, consequently exacerbating DIC, thrombosis, post-ischemic organ damage, and sepsis (17, 18). TM potentially neutralizes these DAMPs, attenuating intravascular injury and organ damage (19, 20). (3) The TMD1 domain potentially binds to the Lewis Y antigen of lipopolysaccharide (LPS) that has pro-inflammatory properties, as it can interact with CD14 and TLRs, thus inhibiting excessive inflammatory responses (21). (4) TM and its TMD1 domain regulate the complement system by eliciting complement-inhibitory signals (22). Abnormal complement activation leads to endothelial dysfunction, including thrombotic microangiopathy. TM may negatively regulate the alternative complement pathway by enhancing complement factor I-mediated inactivation of C3b. In addition, TM interferes with thrombin-mediated complement factor C5 activation, which involves the production of anaphylatoxin, and the formation of a membrane attack complex. TMD2 and TMD3 also exert indirect anti-inflammatory effects via APC production, which activates protease-activated receptor-1 on the endothelium to induce cell protection by inhibiting NF-κB signaling (23). Furthermore, TM-thrombin binding enhances the activation of thrombin activation of fibrinolysis inhibitor (TAFI) that degrades bradykinin and complement factors (24), contributing to the regulation of inflammation. Collectively, TM regulates inflammation, the complement system, and endothelial protection in addition to anti-coagulation during intravascular injury, consequently preserving intravascular homeostasis.
Various stimuli induce NETs through their own NETs-signaling mechanisms. However, regardless of the type of trigger, the NETs resulting from it could become major sources of DAMPs, and act as initiators of immunothrombosis in the face of intravascular injury (25, 26). Thus, NETs have the potential to become a therapeutic target for treatment of immunothrombosis-related diseases. Previously, rTM has been reported to downregulate several types of NET formation. Shimomura et al. showed that rTM inhibited NET formation following treatment with LPS-primed platelets by suppressing TLR4 signaling (27, 28). Studies by Shrestha et al. (29) indicated that rTM treatment ameliorated histone-induced sepsis by neutralizing extracellular histones and suppressing the formation of NETs (20). These previous reports implied indirect effects against neutrophils. Recently, we (15) could show the direct effect of rTM binding to neutrophils, which inhibited auto-antibody-mediated NET formation. In anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis, pathogenic myeloperoxidase (MPO)-ANCA binds to MPO expressed on tumor necrosis factor α-primed neutrophils, and the Fc region of ANCA crosslinks with the Fcγ receptor coupled with Mac-1 on neutrophils to activate spleen tyrosine kinase signaling and ROS production, which results in peptidylarginine deiminase 4 activation and NET formation (30–32). In this scenario, rTM binds to Mac-1 to competitively interfere with ANCA binding on neutrophils, and inhibits downstream signaling, which suppresses ANCA-induced NET formation. Thus, TM potentially has direct and indirect inhibitory effects on NET formation, which contributes to the resolution of intravascular inflammation and immunothrombosis (Figure 1A).
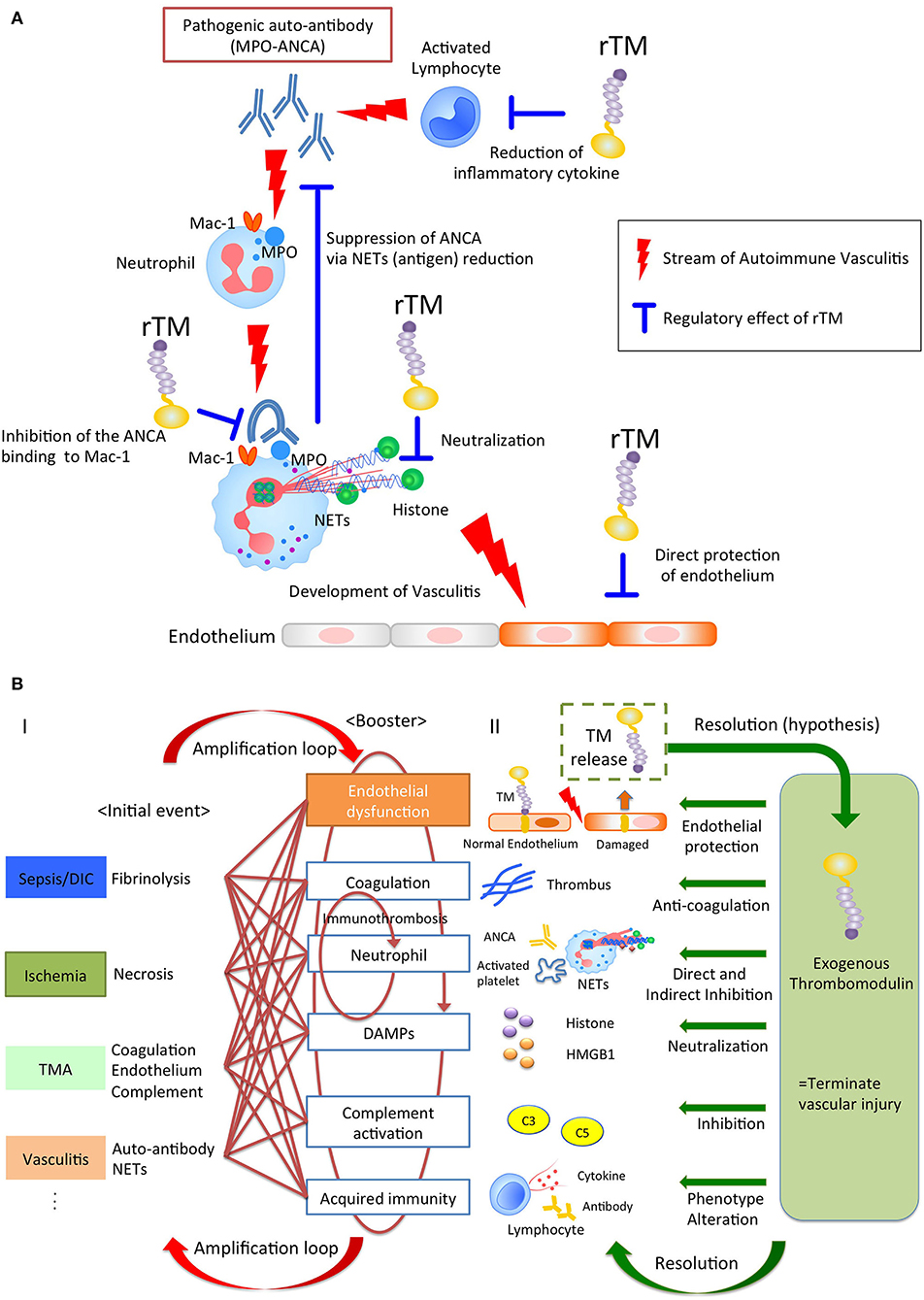
Figure 1. (A) The pleiotropic effects of rTM in autoimmune vasculitis. Pathogenic anti-neutrophil cytoplasmic antibody (ANCA) produced by lymphocytes binds to neutrophil antigen, inducing neutrophil extracellular traps (NETs). The NETs components cause vasculitis and could become auto-antigens, resulting in the further ANCA production. rTM suppresses the pro-inflammatory lymphocytes and inhibits the ANCA binding to Mac-1 on neutrophil, resulting in the suppression of NETs, which leads to the reduction of auto-antigens and ANCA production. Furthermore, rTM neutralizes cytotoxic extracellular histones in NETs and directly protects endothelium. Collectively, rTM could regulate the multiple points in pathogenesis of autoimmune vasculitis. (B) Thrombomodulin terminates auto-amplification of intravascular injury. (I) Intravascular injury in sepsis, ischemia-reperfusion injury, thrombotic microangiopathy, and vasculitis develops due to fibrinolysis, necrosis, coagulation/endothelial dysfunction, and neutrophil activation, respectively, as an initial event. In the next step, these events appear jointly with endothelial dysfunction, coagulation, neutrophil activation, damage-associated molecular patterns, complement activation, and acquired immunity to exacerbate the disease. In particular, immunity and coagulant systems collaborate to generate robust immune-thrombi, which accelerate intravascular injury, leading to an amplification loop. (II) Thrombomodulin is released into the intravascular space after endothelial injury and serves to counteract excessive coagulation and inflammation via its pleiotropic effects.
Of note, rTM containing all the extracellular domains acts not only as an anti-coagulant, but also displays anti-inflammatory properties, hence contributing to the resolution of various diseases (Figure 1B and Table 1A).
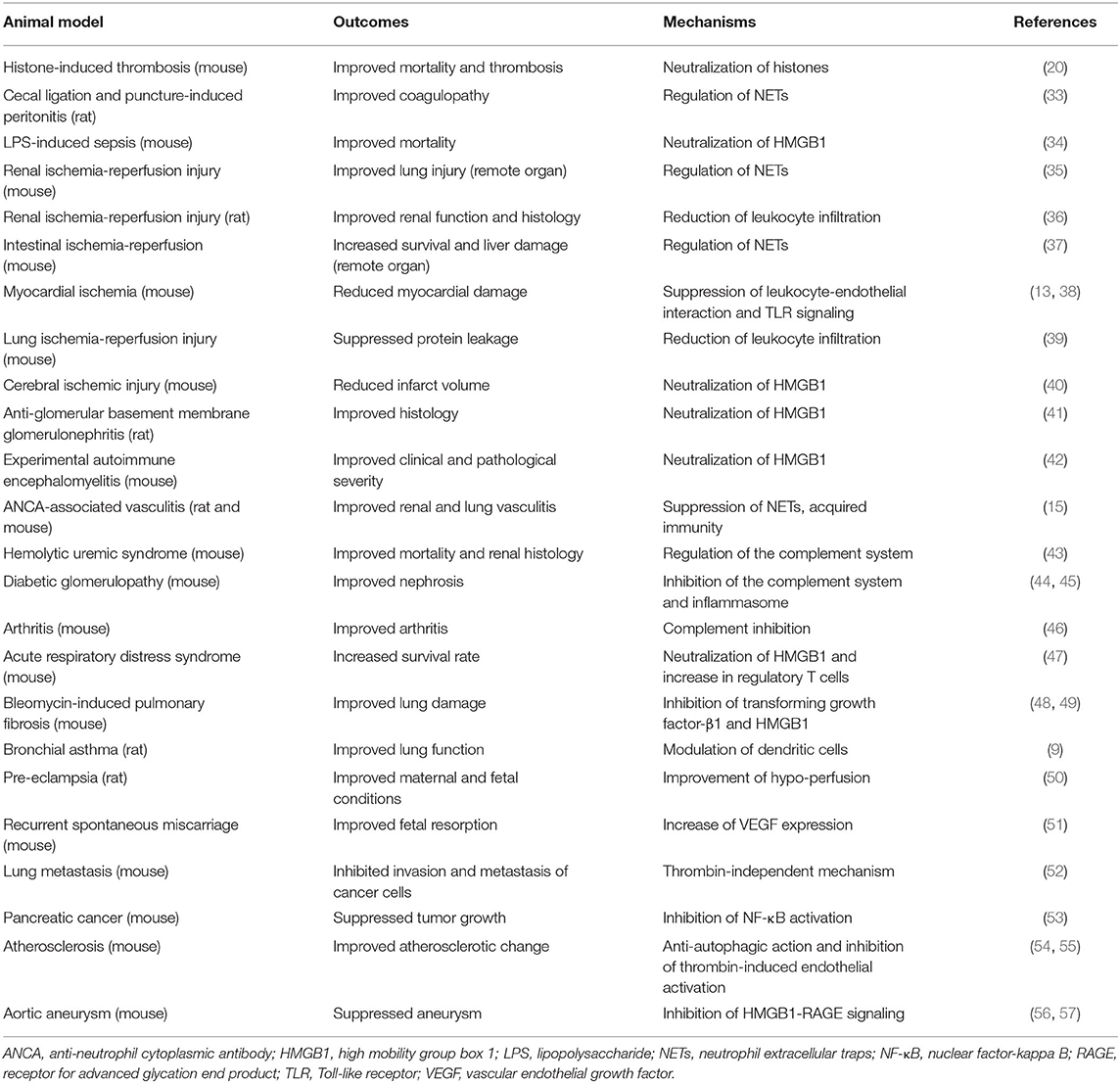
Table 1A. Experimental evidence on recombinant thrombomodulin (rTM, including TMD1, TMD23, and TMD123 domains) in animal disease models.
Sepsis involves multi-organ dysfunction with systemic inflammatory processes, immune dysregulation, coagulopathy, and other physiological responses. Among these processes, NETs and necrotic cell-derived DAMPs directly injure the endothelium and contribute to the development of immunothrombosis through the activation of platelets, coagulation systems, and recruitment of neutrophils (17, 25, 89, 90). In a mouse histone-induced septic model, pretreatment with rTM reduced mortality rates by neutralizing histones (20). In a rat sepsis/peritonitis model (33) and a murine LPS-induced septic model (34), rTM controlled sepsis-related immunothrombosis by limiting abnormal hemostasis and NET formation.
IRI occurs in response to the physiological processes that accompany tissue ischemia with inadequate oxygen supply. This is followed by reperfusion that drives regulated necrosis and subsequent inflammatory responses, leading not only to local organ damage, but also to remote organ injury in the form of necroinflammation (91, 92). In the animal brain, heart, lung, and liver, rTM (the entire ectodomain with lectin-like domain TMD1) ameliorated IRI tissue damage via anti-inflammatory effects, including neutralization of HMGB1 and histones, subsequently triggering the TLR4 signaling pathway (13, 38–40, 93). In a mouse model of renal IRI, ischemia-initiated tubular epithelial cell necrosis released extracellular histones and induced NET formation, which further contributed to remote lung injury (94). Interestingly, rTM (35) and a histone-neutralizing antibody (94) ameliorated remote organ damage, but did not have sufficient effects on local kidney injury. Conversely, inhibition of regulated necrosis, including necroptosis, mitochondrial necrosis, and ferroptosis, rescued local kidney injury at primary lesions, but had less effect on remote organ injury compared with histone neutralization (94). The discrepancy between local and remote injury was compatible with the phenomenon observed in an rTM-treated intestinal IRI mouse model, in which rTM improved remote liver injury, but not local intestinal damage (37). These findings imply that primary necrotic organ injury might develop based on the intracellular signaling cascades arising in response to IRI, but remote organ injury might mainly be caused by DAMPs and inflammatory responses, which could provide a better understanding of DAMP-related IRI pathogenicity.
TMA is characterized by thrombocytopenia, microangiopathic hemolytic anemia, and organ injury. The underlying pathogenesis of TMA is understood to be endothelial dysfunction, which is caused by bacterial toxins, deficiency or dysfunction of the complement system, deficiency or inhibition of ADAM-TS13, drug-induced reactions, and transplant complications (95). The major disorders are hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP). Escherichia coli (O157:H7) induces HUS by producing Shiga toxins, which bind to endothelial cells in the kidney and brain, triggering them to undergo cell death by inhibiting protein synthesis and inducing the secretion of Von Willebrand factor multimers, which leads to endothelial injury and microthrombi (96, 97). In mice, TM deficiency (more specifically, lectin-like domain TMD1) exacerbated Shiga toxin-producing E. coli (STEC)-HUS (98). Furthermore, in STEC-HUS-induced mice, rTM treatment protected them from kidney injury by regulating intravascular inflammation, complement dysfunction, and the coagulation system (43).
Aortic aneurysm develops in association with certain risk factors, including age, genetic predisposition, atherosclerosis, and smoking. The underlying pathogenesis is characterized by chronic vascular inflammation and degradation of collagen-producing structural matrix proteins, which weaken the aortic wall (99). In a CaCl2-induced abdominal aortic aneurysm model, rTM [entire ectodomain (56) and lectin-like domain TMD1 (57)] treatment ameliorated abdominal aortic aneurysm by suppressing inflammatory mediators, macrophage recruitment, and HMGB1-RAGE signaling. In an apolipoprotein E-deficient atherosclerosis model, rTM (TMD23) inhibited autophagy-related cell death of aortic endothelial cells, preventing the progression of atherosclerosis (54). In vitro studies have shown that rTM directly binds to fibroblast growth factor receptor 1 on the endothelium, which activates the phosphatidylinositol 3-kinase-AKT/mammalian target of rapamycin complex 1 signaling pathway, and inhibits autophagy (54, 100). These findings indicate that TM could potentially mediate large vessel homeostasis by controlling immunological responses and endothelium protection.
In previous sections, the anti-inflammatory effects of rTM against intravascular injury were mainly described. In autoimmune diseases, including Goodpasture's syndrome (41) and autoimmune encephalomyelitis (42), rTM ameliorated the disease by suppressing inflammation and neutralizing DAMPs. Interestingly, recent reports have indicated that rTM acts as an immune modulator in addition to serving as an inflammatory regulator. In our study, rTM affected acquired immunity as well as neutrophil activation to resolve autoimmune vasculitis (15). Pathogenic ANCA auto-antibodies play a pivotal role in the development of ANCA-associated vasculitis. In this regard, rTM binds to antibody-producing lymphocytes to alter their activities from pro-inflammatory to anti-inflammatory, which contributes to the reduction of ANCA production and the resolution of the disease. Furthermore, Takagi et al. (9) reported that rTM ameliorated the ovalbumin-induced asthma model by regulating pathogenic dendritic cells. In a graft-vs.-host disease (GVHD) model, rTM increased regulatory T cells via the induction of anti-apoptotic Mcl-1 expression, resulting in the improvement of GVHD (101, 102). Similarly, rTM ameliorated acute respiratory distress syndrome in mice with an increase in regulatory T cells (47). Van De Wouwer et al. (46) showed that rTM (lectin-like domain TMD1) improved mouse arthritis by suppressing excessive inflammatory responses by macrophages and complement activation. As such, rTM could potentially modulate systemic acquired immunity in response to intravascular injury separately from maintaining local vessel homeostasis.
Several studies have reported the serum TM level to examine its role in various diseases. Sepsis (58), ischemic disease (63), and autoimmune diseases (64) showed high levels of soluble TM in serum and plasma that reflected prevailing endothelial injury, indicating that soluble TM levels might be useful for disease diagnosis (Table 1B). Does endogenous soluble TM protect from intravascular injury in human disease? In coronary heart disease, the level of soluble TM is inversely correlated with disease severity (77), implying that endogenous TM might contribute to the resolution of this disease. However, because soluble TM is released from damaged endothelium to counteract the disease, soluble TM levels are often found to increase with disease severity (Table 1B) (77). Meanwhile, genetic polymorphisms of TM could influence the disease beyond the quantity of TM, which might explain the discrepancy between the titer and disease (103). It might be difficult to determine the role of endogenous TM based on soluble TM levels. However, the efficacy of additional TM has been clinically revealed with regard to several diseases during the past two decades.
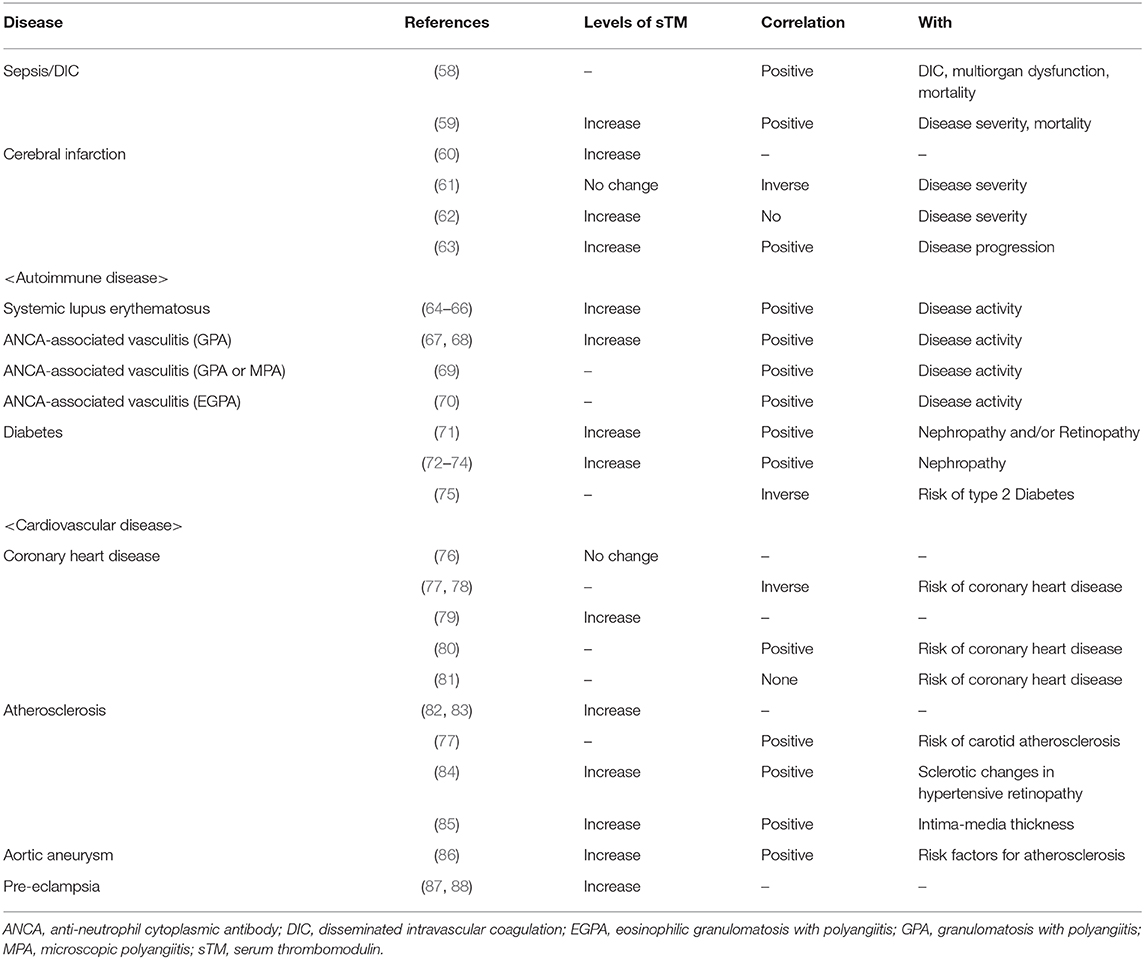
Table 1B. The levels of serum thrombomodulin (TM) in diseases with intravascular injury.
In randomized, double-blind clinical trials, in which patients with DIC associated with hematologic malignancy or infection were treated with rTM or heparin, rTM improved DIC, and alleviated hemorrhagic complications compared with heparin (104). Although rTM therapy did not reduce all-cause mortality in a large clinical trial, post-hoc subgroup analysis stratified by the persistence of abnormal coagulation showed a tendency to decrease mortality (105). Meanwhile, a one-arm prospective trial revealed the effectiveness of rTM in solid tumor-associated DIC (106). Moreover, rTM administration could potentially be useful for treatment of obstetric DIC. During pregnancy, placental abruption, bleeding, and hypoxia could drive DIC underlying obstetric disorders, which is associated with maternal and fetal morbidity and mortality (107). A retrospective comparative study revealed that rTM significantly improved clinical and laboratory findings compared with controls in patients with obstetric DIC (108).
TMA is associated with high mortality regardless of the underlying disease, including HUS, TTP, transplant complications, and drug side effects. In a case series of three patients with HUS, rTM ameliorated clinical outcomes with improvements reflected in reduced platelet counts and excessive complement activation (109). Furthermore, rTM could be beneficial for patients with transplant-associated (TA)-TMA. The latter is a severe complication after hematopoietic stem cell transplantation. The putative etiology is endothelial injury, which is caused by cytotoxic agents, infections, and GVHD (110). A case report (111) and retrospective cohort study (112, 113) showed the effectiveness of rTM with favorable clinical features and overall survival. Likewise, hepatic sinusoidal obstructive syndrome shows clinical manifestations characterized by hepatomegaly, jaundice, ascites, fluid retention, and thrombocytopenia following hematopoietic stem cell transplantation, with pathogenesis mechanisms similar to those of TA-TMA (114). Moreover, patients treated with rTM showed remission and survival rates equivalent to that of patients receiving defibrotide, which is the only recommended therapy for sinusoidal obstructive syndrome (115).
AE-IPF is a lethal condition associated with endothelial damage and abnormalities of the coagulation system (116, 117). HMGB1 is involved in the pathophysiology of pulmonary fibrosis (48). Furthermore, NETs are identified in the bronchi of patients diagnosed with AE-IPF, and are believed to contribute to disease progression (118). Kataoka et al. (119) reported that rTM therapy resulted in improved mortality rates compared with the control group (rTM vs. control: 30 vs. 65%). However, similar to the sepsis clinical trial, a large randomized phase III study in patients with AE-IPF did not show the superiority of rTM using the state of the control as primary endpoint (120). The cause is thought to be the heterogeneous pathology in the comparison group. Therefore, an appropriate study protocol with stratified risk factors is required.
Although the efficacy of rTM has not been clinically shown in autoimmune disease and inflammatory disorder, several experimental data represent the potential to overcome these diseases. In vitro and animal studies indicate that rTM possesses the direct immunomodulatory effects in innate and acquired immunity independently of anti-coagulant effect (9, 15). Based on animal studies (Table 1A), rTM is being clinically expected to contribute to resolving diseases with inflammation including diabetes mellitus, arthritis, bronchial asthma, and ischemic-reperfusion injury. In particular, autoimmune ANCA vasculitis, which is characterized by immune dysregulation and intravascular injury, might be a candidate for rTM treatment. However, the dosage of rTM in many experimental situations (15, 33, 41) is 15–50 times of therapeutic dosage in patients with DIC and the effective concentration as an anti-inflammatory and immune-regulatory property remains unclear. Thus, in the future the indications of rTM therapy and the suitable dosage with no serious complications such as bleeding tendency should be carefully addressed.
Immunothrombosis during intravascular injury leads to organ damage and further intravascular injury via cellular and molecular signaling, including excessive inflammation, coagulation, and cell activation. rTM regulates the immunothrombosis to terminate inflammation/coagulation, neutralize DAMPs, and affect immunity. The administration of rTM has the potential to become a novel therapeutic strategy for various diseases associated with immunothrombosis-mediated intravascular injury.
KW-K and DN conducted the literature research and reviewed all articles. AI and TA edited the article. All authors contributed to the article and approved the submitted version.
This study was supported by a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (grant number JP 18K15968).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Editage (www.editage.com) for English language editing.
AE-IPF, Acute exacerbation of idiopathic pulmonary fibrosis; ANCA, Anti-neutrophil cytoplasmic antibody; APC, Activated protein C; DAMPs, Damage-associated molecular patterns; DIC, Disseminated intravascular coagulation; EGPA, Eosinophilic granulomatosis with polyangiitis; GPA, Granulomatosis with polyangiitis; GVHD, Graft-vs.-host disease; HMGB1, High mobility group box 1; HUS, Hemolytic uremic syndrome; IRI, Ischemia-reperfusion injury; LPS, Lipopolysaccharide; Mac-1, Macrophage-1 antigen; MPA, Microscopic polyangiitis; MPO, Myeloperoxidase; NETs, Neutrophil extracellular traps; NF-κB, Nuclear factor-kappa B; RAGE, Receptor for advanced glycation endproducts; rTM, Recombinant thrombomodulin; STEC, Shiga toxin-producing Escherichia coli; sTM, Serum thrombomodulin; TAFI, Thrombin activatable fibrinolysis inhibitor; TA-TMA, transplant-associated thrombotic microangiopathy; TLR, Toll-like receptor; TM, Thrombomodulin; TMA, Thrombotic microangiopathy; TTP, Thrombotic thrombocytopenic purpura; VEGF, Vascular endothelial growth factor.
1. Chang JC. Acute respiratory distress syndrome as an organ phenotype of vascular microthrombotic disease: based on hemostatic theory and endothelial molecular pathogenesis. Clin Appl Thromb Hemost. (2019) 25:1076029619887437. doi: 10.1177/1076029619887437
2. Foley JH, Conway EM. Cross talk pathways between coagulation and inflammation. Circ Res. (2016) 118:1392–408. doi: 10.1161/CIRCRESAHA.116.306853
3. Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. (2016) 128:2435–49. doi: 10.1182/blood-2016-04-710632
4. Nakazawa D, Desai J, Steiger S, Muller S, Devarapu SK, Mulay SR, et al. Activated platelets induce MLKL-driven neutrophil necroptosis and release of neutrophil extracellular traps in venous thrombosis. Cell Death Discov. (2018) 5:6. doi: 10.1038/s41420-018-0073-2
5. Gaertner F, Massberg S. Blood coagulation in immunothrombosis-At the frontline of intravascular immunity. Semin Immunol. (2016) 28:561–9. doi: 10.1016/j.smim.2016.10.010
6. Loghmani H, Conway EM. Exploring traditional and nontraditional roles for thrombomodulin. Blood. (2018) 132:148–58. doi: 10.1182/blood-2017-12-768994
7. Li YH, Kuo CH, Shi GY, Wu HL. The role of thrombomodulin lectin-like domain in inflammation. J Biomed Sci. (2012) 19:34. doi: 10.1186/1423-0127-19-34
8. Maruyama I, Bell C, Majerus PW. Thrombomodulin is found on endothelium of arteries, veins, capillaries, and lymphatics, and on syncytiotrophoblast of human placenta. J Cell Biol. (1985) 101:363–71. doi: 10.1083/jcb.101.2.363
9. Takagi T, Taguchi O, Toda M, Ruiz DB, Bernabe PG, D'Alessandro-Gabazza CN, et al. Inhibition of allergic bronchial asthma by thrombomodulin is mediated by dendritic cells. Am J Respir Crit Care Med. (2011) 183:31–42. doi: 10.1164/rccm.201001-0107OC
10. Conway EM, Nowakowski B, Steiner-Mosonyi M. Human neutrophils synthesize thrombomodulin that does not promote thrombin-dependent protein C activation. Blood. (1992) 80:1254–63. doi: 10.1182/blood.V80.5.1254.1254
11. Esmon CT, Owen WG. The discovery of thrombomodulin. J Thromb Haemost. (2004) 2:209–13. doi: 10.1046/j.1538-7933.2003.00537.x
12. Fuentes-Prior P, Iwanaga Y, Huber R, Pagila R, Rumennik G, Seto M, et al. Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex. Nature. (2000) 404:518–25. doi: 10.1038/35006683
13. Conway EM, Van de Wouwer M, Pollefeyt S, Jurk K, Van Aken H, De Vriese A, et al. The lectin-like domain of thrombomodulin confers protection from neutrophil-mediated tissue damage by suppressing adhesion molecule expression via nuclear factor κB and mitogen-activated protein kinase pathways. J Exp Med. (2002) 196:565–77. doi: 10.1084/jem.20020077
14. Lin WL, Chang CF, Shi CS, Shi GY, Wu HL. Recombinant lectin-like domain of thrombomodulin suppresses vascular inflammation by reducing leukocyte recruitment via interacting with lewis Y on endothelial cells. Arterioscler Thromb Vasc Biol. (2013) 33:2366–73. doi: 10.1161/ATVBAHA.113.301221
15. Watanabe-Kusunoki K, Nakazawa D, Kusunoki Y, Kudo T, Hattanda F, Nishio S, et al. Recombinant thrombomodulin ameliorates autoimmune vasculitis via immune response regulation and tissue injury protection. J Autoimmun. (2020) 108:102390. doi: 10.1016/j.jaut.2019.102390
16. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. (2002) 418:191–5. doi: 10.1038/nature00858
17. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. (2009) 15:1318–21. doi: 10.1038/nm.2053
18. Allam R, Kumar SV, Darisipudi MN, Anders HJ. Extracellular histones in tissue injury and inflammation. J Mol Med. (2014) 92:465–72. doi: 10.1007/s00109-014-1148-z
19. Abeyama K, Stern DM, Ito Y, Kawahara K, Yoshimoto Y, Tanaka M, et al. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest. (2005) 115:1267–74. doi: 10.1172/JCI22782
20. Nakahara M, Ito T, Kawahara K, Yamamoto M, Nagasato T, Shrestha B, et al. Recombinant thrombomodulin protects mice against histone-induced lethal thromboembolism. PLoS ONE. (2013) 8:e75961. doi: 10.1371/journal.pone.0075961
21. Shi CS, Shi GY, Hsiao HM, Kao YC, Kuo KL, Ma CY, et al. Lectin-like domain of thrombomodulin binds to its specific ligand lewis Y antigen and neutralizes lipopolysaccharide-induced inflammatory response. Blood. (2008) 112:3661–70. doi: 10.1182/blood-2008-03-142760
22. Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. (2009) 361:345–57. doi: 10.1056/NEJMoa0810739
23. Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. (2007) 109:3161–72. doi: 10.1182/blood-2006-09-003004
24. Nishimura T, Myles T, Piliponsky AM, Kao PN, Berry GJ, Leung LL. Thrombin-activatable procarboxypeptidase B regulates activated complement C5a in vivo. Blood. (2007) 109:1992–7. doi: 10.1182/blood-2006-03-012567
25. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. (2013) 13:34–45. doi: 10.1038/nri3345
26. McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. (2017) 129:1357–67. doi: 10.1182/blood-2016-09-741298
27. Shimomura Y, Suga M, Kuriyama N, Nakamura T, Sakai T, Kato Y, et al. Recombinant human thrombomodulin inhibits neutrophil extracellular trap formation in vitro. J Intensive Care. (2016) 4:48. doi: 10.1186/s40560-016-0177-9
28. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
29. Shrestha B, Ito T, Kakuuchi M, Totoki T, Nagasato T, Yamamoto M, et al. Recombinant thrombomodulin suppresses histone-induced neutrophil extracellular trap formation. Front Immunol. (2019) 10:2535. doi: 10.3389/fimmu.2019.02535
30. Kallenberg CG. Pathogenesis of ANCA-associated vasculitides. Ann Rheum Dis. (2011) 70(Suppl 1):i59–63. doi: 10.1136/ard.2010.138024
31. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. (2009) 184:205–13. doi: 10.1083/jcb.200806072
32. Hewins P, Williams JM, Wakelam MJ, Savage CO. Activation of Syk in neutrophils by antineutrophil cytoplasm antibodies occurs via Fcgamma receptors and CD18. J Am Soc Nephrol. (2004) 15:796–808. doi: 10.1097/01.ASN.0000113241.98702.77
33. Helms J, Clere-Jehl R, Bianchini E, Le Borgne P, Burban M, Zobairi F, et al. Thrombomodulin favors leukocyte microvesicle fibrinolytic activity, reduces NETosis and prevents septic shock-induced coagulopathy in rats. Ann Intensive Care. (2017) 7:118. doi: 10.1186/s13613-017-0340-z
34. Takehara K, Murakami T, Kuwahara-Arai K, Iba T, Nagaoka I, Sakamoto K. Evaluation of the effect of recombinant thrombomodulin on a lipopolysaccharide-induced murine sepsis model. Exp Ther Med. (2017) 13:2969–74. doi: 10.3892/etm.2017.4308
35. Hayase N, Doi K, Hiruma T, Matsuura R, Hamasaki Y, Noiri E, et al. Recombinant thrombomodulin prevents acute lung injury induced by renal ischemia-reperfusion injury. Sci Rep. (2020) 10:289. doi: 10.1038/s41598-019-57205-0
36. Sharfuddin AA, Sandoval RM, Berg DT, McDougal GE, Campos SB, Phillips CL, et al. Soluble thrombomodulin protects ischemic kidneys. J Am Soc Nephrol. (2009) 20:524–34. doi: 10.1681/ASN.2008060593
37. Hayase N, Doi K, Hiruma T, Matsuura R, Hamasaki Y, Noiri E, et al. Recombinant thrombomodulin on neutrophil extracellular traps in murine intestinal ischemia-reperfusion. Anesthesiology. (2019) 131:866–82. doi: 10.1097/ALN.0000000000002898
38. Herzog C, Lorenz A, Gillmann HJ, Chowdhury A, Larmann J, Harendza T, et al. Thrombomodulin's lectin-like domain reduces myocardial damage by interfering with HMGB1-mediated TLR2 signalling. Cardiovasc Res. (2014) 101:400–10. doi: 10.1093/cvr/cvt275
39. Geudens N, Van de Wouwer M, Vanaudenaerde BM, Vos R, Van De Wauwer C, Verleden GM, et al. The lectin-like domain of thrombomodulin protects against ischaemia-reperfusion lung injury. Eur Respir J. (2008) 32:862–70. doi: 10.1183/09031936.00157107
40. Nakamura Y, Nakano T, Irie K, Sano K, Tanaka J, Yamashita Y, et al. Recombinant human soluble thrombomodulin ameliorates cerebral ischemic injury through a high-mobility group box 1 inhibitory mechanism without hemorrhagic complications in mice. J Neurol Sci. (2016) 362:278–82. doi: 10.1016/j.jns.2016.01.047
41. Tachibana S, Iyoda M, Matsumoto K, Wada Y, Suzuki T, Iseri K, et al. Recombinant human soluble thrombomodulin attenuates anti-glomerular basement membrane glomerulonephritis in wistar-kyoto rats through anti-inflammatory effects. Nephrol Dial Transplant. (2019) 34:774–82. doi: 10.1093/ndt/gfy201
42. Uzawa A, Mori M, Masuda H, Ohtani R, Uchida T, Kuwabara S. Recombinant thrombomodulin ameliorates experimental autoimmune encephalomyelitis by suppressing high mobility group box 1 and inflammatory cytokines. Clin Exp Immunol. (2018) 193:47–54. doi: 10.1111/cei.13123
43. Suyama K, Kawasaki Y, Miyazaki K, Kanno S, Ono A, Ohara S, et al. The efficacy of recombinant human soluble thrombomodulin for the treatment of shiga toxin-associated hemolytic uremic syndrome model mice. Nephrol Dial Transplant. (2015) 30:969–77. doi: 10.1093/ndt/gfv004
44. Wang H, Vinnikov I, Shahzad K, Bock F, Ranjan S, Wolter J, et al. The lectin-like domain of thrombomodulin ameliorates diabetic glomerulopathy via complement inhibition. Thromb Haemost. (2012) 108:1141–53. doi: 10.1160/TH12-07-0460
45. Yang SM, Ka SM, Wu HL, Yeh YC, Kuo CH, Hua KF, et al. Thrombomodulin domain 1 ameliorates diabetic nephropathy in mice via anti-NF-κB/NLRP3 inflammasome-mediated inflammation, enhancement of NRF2 antioxidant activity and inhibition of apoptosis. Diabetologia. (2014) 57:424–34. doi: 10.1007/s00125-013-3115-6
46. Van de Wouwer M, Plaisance S, De Vriese A, Waelkens E, Collen D, Persson J, et al. The lectin-like domain of thrombomodulin interferes with complement activation and protects against arthritis. J Thromb Haemost. (2006) 4:1813–24. doi: 10.1111/j.1538-7836.2006.02033.x
47. Kudo D, Toyama M, Aoyagi T, Akahori Y, Yamamoto H, Ishii K, et al. Involvement of high mobility group box 1 and the therapeutic effect of recombinant thrombomodulin in a mouse model of severe acute respiratory distress syndrome. Clin Exp Immunol. (2013) 173:276–87. doi: 10.1111/cei.12106
48. Kida T, Seno T, Nagahara H, Inoue T, Nakabayashi A, Kukida Y, et al. Roles of high-mobility group box 1 and thrombin in murine pulmonary fibrosis and the therapeutic potential of thrombomodulin. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L473–83. doi: 10.1152/ajplung.00287.2017
49. Fujiwara K, Kobayashi T, Fujimoto H, Nakahara H, D'Alessandro-Gabazza CN, Hinneh JA, et al. Inhibition of cell apoptosis and amelioration of pulmonary fibrosis by thrombomodulin. Am J Pathol. (2017) 187:2312–22. doi: 10.1016/j.ajpath.2017.06.013
50. Shin M, Hino H, Tamura M, Ishizuka B, Tanaka M, Suzuki N, et al. Thrombomodulin improves maternal and fetal conditions in an experimental pre-eclampsia rat model. J Obstet Gynaecol Res. (2014) 40:1226–34. doi: 10.1111/jog.12323
51. Sano T, Terai Y, Daimon A, Nunode M, Nagayasu Y, Okamoto A, et al. Recombinant human soluble thrombomodulin as an anticoagulation therapy improves recurrent miscarriage and fetal growth restriction due to placental insufficiency - the leading cause of preeclampsia. Placenta. (2018) 65:1–6. doi: 10.1016/j.placenta.2018.03.006
52. Hosaka Y, Higuchi T, Tsumagari M, Ishii H. Inhibition of invasion and experimental metastasis of murine melanoma cells by human soluble thrombomodulin. Cancer Lett. (2000) 161:231–40. doi: 10.1016/S0304-3835(00)00617-0
53. Shirai Y, Uwagawa T, Shiba H, Shimada Y, Horiuchi T, Saito N, et al. Recombinant thrombomodulin suppresses tumor growth of pancreatic cancer by blocking thrombin-induced PAR1 and NF-kappaB activation. Surgery. (2017) 161:1675–82. doi: 10.1016/j.surg.2016.12.008
54. Chen PS, Wang KC, Chao TH, Chung HC, Tseng SY, Luo CY, et al. Recombinant thrombomodulin exerts anti-autophagic action in endothelial cells and provides anti-atherosclerosis effect in apolipoprotein E deficient mice. Sci Rep. (2017) 7:3284. doi: 10.1038/s41598-017-03443-z
55. Wei HJ, Li YH, Shi GY, Liu SL, Chang PC, Kuo CH, et al. Thrombomodulin domains attenuate atherosclerosis by inhibiting thrombin-induced endothelial cell activation. Cardiovasc Res. (2011) 92:317–27. doi: 10.1093/cvr/cvr220
56. Lai CH, Shi GY, Lee FT, Kuo CH, Cheng TL, Chang BI, et al. Recombinant human thrombomodulin suppresses experimental abdominal aortic aneurysms induced by calcium chloride in mice. Ann Surg. (2013) 258:1103–10. doi: 10.1097/SLA.0b013e31827df7cb
57. Lai CH, Wang KC, Kuo CH, Lee FT, Cheng TL, Chang BI, et al. Recombinant adeno-associated virus vector carrying the thrombomodulin lectin-like domain for the treatment of abdominal aortic aneurysm. Atherosclerosis. (2017) 262:62–70. doi: 10.1016/j.atherosclerosis.2017.03.024
58. Lin SM, Wang YM, Lin HC, Lee KY, Huang CD, Liu CY, et al. Serum thrombomodulin level relates to the clinical course of disseminated intravascular coagulation, multiorgan dysfunction syndrome, and mortality in patients with sepsis. Crit Care Med. (2008) 36:683–9. doi: 10.1097/CCM.0B013E31816537D8
59. Lin JJ, Hsiao HJ, Chan OW, Wang Y, Hsia SH, Chiu CH. Increased serum thrombomodulin level is associated with disease severity and mortality in pediatric sepsis. PLoS ONE. (2017) 12:e0182324. doi: 10.1371/journal.pone.0182324
60. Kozuka K, Kohriyama T, Nomura E, Ikeda J, Kajikawa H, Nakamura S. Endothelial markers and adhesion molecules in acute ischemic stroke–sequential change and differences in stroke subtype. Atherosclerosis. (2002) 161:161–8. doi: 10.1016/S0021-9150(01)00635-9
61. Nomura E, Kohriyama T, Kozuka K, Kajikawa H, Nakamura S, Matsumoto M. Significance of serum soluble thrombomodulin level in acute cerebral infarction. Eur J Neurol. (2004) 11:329–34. doi: 10.1111/j.1468-1331.2004.00776.x
62. Dharmasaroja P, Dharmasaroja PA, Sobhon P. Increased plasma soluble thrombomodulin levels in cardioembolic stroke. Clin Appl Thromb Hemost. (2012) 18:289–93. doi: 10.1177/1076029611432744
63. Han Y, Wu S, Hu Q, Xiao JQ, Wei DM, Liu LL, et al. Thrombomodulin and high-sensitive C-reactive protein levels in blood correlate with the development of cerebral infarction among Asians. Mol Neurobiol. (2016) 53:2659–67. doi: 10.1007/s12035-015-9279-y
64. Boehme MW, Nawroth PP, Kling E, Lin J, Amiral J, Riedesel J, et al. Serum thrombomodulin. A novel marker of disease activity in systemic lupus erythematosus. Arthritis Rheum. (1994) 37:572–7. doi: 10.1002/art.1780370419
65. Boehme MW, Raeth U, Galle PR, Stremmel W, Scherbaum WA. Serum thrombomodulin-a reliable marker of disease activity in systemic lupus erythematosus (SLE): advantage over established serological parameters to indicate disease activity. Clin Exp Immunol. (2000) 119:189–95. doi: 10.1046/j.1365-2249.2000.01107.x
66. El-Gamal YM, Heshmat NM, El-Kerdany TH, Fawzy AF. Serum thrombomodulin in systemic lupus erythematosus and juvenile idiopathic arthritis. Pediatr Allergy Immunol. (2004) 15:270–7. doi: 10.1111/j.1399-3038.2004.00085.x
67. Boehme MW, Schmitt WH, Youinou P, Stremmel WR, Gross WL. Clinical relevance of elevated serum thrombomodulin and soluble E-selectin in patients with wegener's granulomatosis and other systemic vasculitides. Am J Med. (1996) 101:387–94. doi: 10.1016/S0002-9343(96)00230-6
68. Zycinska K, Wardyn KA, Zielonka TM, Krupa R, Lukas W. Clinical implications of serum thrombomodulin in PR3-ANCA-associated vasculitis. Eur J Med Res. (2009) 14(Suppl 4):268–70. doi: 10.1186/2047-783X-14-S4-268
69. Monach PA, Tomasson G, Specks U, Stone JH, Cuthbertson D, Krischer J, et al. Circulating markers of vascular injury and angiogenesis in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. (2011) 63:3988–97. doi: 10.1002/art.30615
70. Schmitt WH, Csernok E, Kobayashi S, Klinkenborg A, Reinhold-Keller E, Gross WL. Churg-Strauss syndrome: serum markers of lymphocyte activation and endothelial damage. Arthritis Rheum. (1998) 41:445–52. doi: 10.1002/1529-0131(199803)41:3<445::AID-ART10>3.0.CO;2-3
71. Oida K, Takai H, Maeda H, Takahashi S, Tamai T, Nakai T, et al. Plasma thrombomodulin concentration in diabetes mellitus. Diabetes Res Clin Pract. (1990) 10:193–6. doi: 10.1016/0168-8227(90)90044-T
72. Shimano H, Takahashi K, Kawakami M, Gotoda T, Harada K, Shimada M, et al. Elevated serum and urinary thrombomodulin levels in patients with non-insulin-dependent diabetes mellitus. Clin Chim Acta. (1994) 225:89–96. doi: 10.1016/0009-8981(94)90036-1
73. Inukai T, Fujiwara Y, Tayama K, Aso Y, Takemura Y. Clinical significance of measurements of urinary and serum thrombomodulins in patients with non-insulin-dependent diabetes mellitus. Diabetes Res Clin Pract. (1996) 33:99–104. doi: 10.1016/0168-8227(96)01283-1
74. Aso Y, Inukai T, Takemura Y. Mechanisms of elevation of serum and urinary concentrations of soluble thrombomodulin in diabetic patients: possible application as a marker for vascular endothelial injury. Metabolism. (1998) 47:362–5. doi: 10.1016/S0026-0495(98)90272-4
75. Thorand B, Baumert J, Herder C, Meisinger C, Koenig W. Soluble thrombomodulin as a predictor of type 2 diabetes: results from the MONICA/KORA Augsburg case-cohort study, 1984-1998. Diabetologia. (2007) 50:545–8. doi: 10.1007/s00125-006-0568-x
76. Naruse M, Kawana M, Hifumi S, Naruse K, Yoshihara I, Oka T, et al. Plasma immunoreactive endothelin, but not thrombomodulin, is increased in patients with essential hypertension and ischemic heart disease. J Cardiovasc Pharmacol. (1991) 17(Suppl 7):S471–4. doi: 10.1097/00005344-199100177-00135
77. Salomaa V, Matei C, Aleksic N, Sansores-Garcia L, Folsom AR, Juneja H, et al. Soluble thrombomodulin as a predictor of incident coronary heart disease and symptomless carotid artery atherosclerosis in the Atherosclerosis Risk in Communities (ARIC) study: a case-cohort study. Lancet. (1999) 353:1729–34. doi: 10.1016/S0140-6736(98)09057-6
78. Aleksic N, Wang YW, Ahn C, Juneja HS, Folsom AR, Wu KK. Assessment of coronary heart disease risk by combined analysis of coagulation factors. Atherosclerosis. (2008) 198:294–300. doi: 10.1016/j.atherosclerosis.2007.12.062
79. Wiśniewska E, Wodyńska T, Kulwas A, Kotschy M, Nartowicz E, Paczuski R, et al. Thrombomodulin–endothelial thrombin receptor in blood of patients with unstable angina pectoris. Med Sci Monit. (2001) 7:256–9.
80. Chan SH, Chen JH, Li YH, Lin LJ, Tsai LM. Increasing post-event plasma thrombomodulin level associates with worse outcome in survival of acute coronary syndrome. Int J Cardiol. (2006) 111:280–5. doi: 10.1016/j.ijcard.2005.09.015
81. Karakas M, Baumert J, Herder C, Rottbauer W, Meisinger C, Koenig W, et al. Soluble thrombomodulin in coronary heart disease: lack of an association in the MONICA/KORA case-cohort study. J Thromb Haemost. (2011) 9:1078–80. doi: 10.1111/j.1538-7836.2011.04229.x
82. Seigneur M, Dufourcq P, Conri C, Constans J, Mercié P, Pruvost A, et al. Levels of plasma thrombomodulin are increased in atheromatous arterial disease. Thromb Res. (1993) 71:423–31. doi: 10.1016/0049-3848(93)90116-6
83. Taylan A, Sari I, Kozaci DL, Yildiz Y, Bilge S, Coker I, et al. Evaluation of various endothelial biomarkers in ankylosing spondylitis. Clin Rheumatol. (2012) 31:23–8. doi: 10.1007/s10067-011-1760-z
84. Dohi Y, Ohashi M, Sugiyama M, Takase H, Sato K, Ueda R. Circulating thrombomodulin levels are related to latent progression of atherosclerosis in hypertensive patients. Hypertens Res. (2003) 26:479–83. doi: 10.1291/hypres.26.479
85. Pawlak K, Mysliwiec M, Pawlak D. Kynurenine pathway - a new link between endothelial dysfunction and carotid atherosclerosis in chronic kidney disease patients. Adv Med Sci. (2010) 55:196–203. doi: 10.2478/v10039-010-0015-6
86. Budzyn M, Gryszczynska B, Majewski W, Krasinski Z, Kasprzak MP, Formanowicz D, et al. The association of serum thrombomodulin with endothelial injuring factors in abdominal aortic aneurysm. Biomed Res Int. (2017) 2017:2791082. doi: 10.1155/2017/2791082
87. Hsu CD, Iriye B, Johnson TR, Witter FR, Hong SF, Chan DW. Elevated circulating thrombomodulin in severe preeclampsia. Am J Obstet Gynecol. (1993) 169:148–9. doi: 10.1016/0002-9378(93)90151-8
88. Rousseau A, Favier R, Van Dreden P. Elevated circulating soluble thrombomodulin activity, tissue factor activity and circulating procoagulant phospholipids: new and useful markers for pre-eclampsia? Eur J Obstet Gynecol Reprod Biol. (2009) 146:46–9. doi: 10.1016/j.ejogrb.2009.06.001
89. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. (2011) 118:1952–61. doi: 10.1182/blood-2011-03-343061
90. Chaput C, Zychlinsky A. Sepsis: the dark side of histones. Nat Med. (2009) 15:1245–6. doi: 10.1038/nm1109-1245
91. Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. (2011) 17:1391–401. doi: 10.1038/nm.2507
92. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. (2010) 10:826–37. doi: 10.1038/nri2873
93. Kadono K, Uchida Y, Hirao H, Miyauchi T, Watanabe T, Iida T, et al. Thrombomodulin attenuates inflammatory damage due to liver ischemia and reperfusion injury in mice in toll-like receptor 4-dependent manner. Am J Transplant. (2017) 17:69–80. doi: 10.1111/ajt.13991
94. Nakazawa D, Kumar SV, Marschner J, Desai J, Holderied A, Rath L, et al. Histones and neutrophil extracellular traps enhance tubular necrosis and remote organ injury in ischemic AKI. J Am Soc Nephrol. (2017) 28:1753–68. doi: 10.1681/ASN.2016080925
95. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. (2014) 371:654–66. doi: 10.1056/NEJMra1312353
96. Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol. (2012) 8:622–33. doi: 10.1038/nrneph.2012.195
97. Nolasco LH, Turner NA, Bernardo A, Tao Z, Cleary TG, Dong JF, et al. Hemolytic uremic syndrome-associated shiga toxins promote endothelial-cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood. (2005) 106:4199–209. doi: 10.1182/blood-2005-05-2111
98. Zoja C, Locatelli M, Pagani C, Corna D, Zanchi C, Isermann B, et al. Lack of the lectin-like domain of thrombomodulin worsens shiga toxin-associated hemolytic uremic syndrome in mice. J Immunol. (2012) 189:3661–8. doi: 10.4049/jimmunol.1102118
99. Sakalihasan N, Limet R, Defawe OD. Abdominal aortic aneurysm. Lancet. (2005) 365:1577–89. doi: 10.1016/S0140-6736(05)66459-8
100. Kuo CH, Sung MC, Chen PK, Chang BI, Lee FT, Cho CF, et al. FGFR1 mediates recombinant thrombomodulin domain-induced angiogenesis. Cardiovasc Res. (2015) 105:107–17. doi: 10.1093/cvr/cvu239
101. Ikezoe T, Yang J, Nishioka C, Honda G, Furihata M, Yokoyama A. Thrombomodulin protects endothelial cells from a calcineurin inhibitor-induced cytotoxicity by upregulation of extracellular signal-regulated kinase/myeloid leukemia cell-1 signaling. Arterioscler Thromb Vasc Biol. (2012) 32:2259–70. doi: 10.1161/ATVBAHA.112.251157
102. Ikezoe T, Yang J, Nishioka C, Yokoyama A. Thrombomodulin alleviates murine GVHD in association with an increase in the proportion of regulatory T cells in the spleen. Bone Marrow Transplant. (2015) 50:113–20. doi: 10.1038/bmt.2014.208
103. Xu J, Jin J, Tan S. Association of thrombomodulin gene polymorphisms with susceptibility to atherosclerotic diseases: a meta-analysis. Ann Hum Genet. (2016) 80:172–81. doi: 10.1111/ahg.12148
104. Saito H, Maruyama I, Shimazaki S, Yamamoto Y, Aikawa N, Ohno R, et al. Efficacy and safety of recombinant human soluble thrombomodulin (ART-123) in disseminated intravascular coagulation: results of a phase III, randomized, double-blind clinical trial. J Thromb Haemost. (2007) 5:31–41. doi: 10.1111/j.1538-7836.2006.02267.x
105. Vincent JL, Francois B, Zabolotskikh I, Daga MK, Lascarrou JB, Kirov MY, et al. Effect of a recombinant human soluble thrombomodulin on mortality in patients with sepsis-associated coagulopathy: the SCARLET randomized clinical trial. JAMA. (2019) 321:1993–2002. doi: 10.1001/jama.2019.5358
106. Tamura K, Saito H, Asakura H, Okamoto K, Tagawa J, Hayakawa T, et al. Recombinant human soluble thrombomodulin (thrombomodulin alfa) to treat disseminated intravascular coagulation in solid tumors: results of a one-arm prospective trial. Int J Clin Oncol. (2015) 20:821–8. doi: 10.1007/s10147-014-0768-1
107. Erez O, Mastrolia SA, Thachil J. Disseminated intravascular coagulation in pregnancy: insights in pathophysiology, diagnosis and management. Am J Obstet Gynecol. (2015) 213:452–63. doi: 10.1016/j.ajog.2015.03.054
108. Yoshihara M, Uno K, Tano S, Mayama M, Ukai M, Kondo S, et al. The efficacy of recombinant human soluble thrombomodulin for obstetric disseminated intravascular coagulation: a retrospective study. Crit Care. (2015) 19:369. doi: 10.1186/s13054-015-1086-3
109. Honda T, Ogata S, Mineo E, Nagamori Y, Nakamura S, Bando Y, et al. A novel strategy for hemolytic uremic syndrome: successful treatment with thrombomodulin alpha. Pediatrics. (2013) 131:e928–33. doi: 10.1542/peds.2012-1466
110. Khosla J, Yeh AC, Spitzer TR, Dey BR. Hematopoietic stem cell transplant-associated thrombotic microangiopathy: current paradigm and novel therapies. Bone Marrow Transplant. (2018) 53:129–37. doi: 10.1038/bmt.2017.207
111. Sakai M, Ikezoe T, Bandobashi K, Togitani K, Yokoyama A. Successful treatment of transplantation-associated thrombotic microangiopathy with recombinant human soluble thrombomodulin. Bone Marrow Transplant. (2010) 45:803–5. doi: 10.1038/bmt.2009.242
112. Fujiwara H, Maeda Y, Sando Y, Nakamura M, Tani K, Ishikawa T, et al. Treatment of thrombotic microangiopathy after hematopoietic stem cell transplantation with recombinant human soluble thrombomodulin. Transfusion. (2016) 56:886–92. doi: 10.1111/trf.13437
113. Nomura S, Konishi A, Tsubokura Y, Azuma Y, Hotta M, Yoshimura H, et al. Effects of recombinant thrombomodulin on long-term prognosis after allogeneic hematopoietic stem cell transplantation. Transpl Immunol. (2019) 57:101247. doi: 10.1016/j.trim.2019.101247
114. Mohty M, Malard F, Abecassis M, Aerts E, Alaskar AS, Aljurf M, et al. Sinusoidal obstruction syndrome/veno-occlusive disease: current situation and perspectives-a position statement from the European Society for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant. (2015) 50:781–9. doi: 10.1038/bmt.2015.52
115. Yakushijin K, Ikezoe T, Ohwada C, Kudo K, Okamura H, Goto H, et al. Clinical effects of recombinant thrombomodulin and defibrotide on sinusoidal obstruction syndrome after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. (2019) 54:674–80. doi: 10.1038/s41409-018-0304-4
116. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. (2000) 342:1334–49. doi: 10.1056/NEJM200005043421806
117. Collard HR, Calfee CS, Wolters PJ, Song JW, Hong SB, Brady S, et al. Plasma biomarker profiles in acute exacerbation of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. (2010) 299:L3–7. doi: 10.1152/ajplung.90637.2008
118. Ojima M, Yamamoto N, Hirose T, Hamaguchi S, Matsumoto N, Takegawa R, et al. Presence of neutrophil extracellular traps in bronchial aspirate of patients diagnosed with acute respiratory distress syndrome or acute exacerbation of idiopathic pulmonary fibrosis. Crit Care. (2015) 19(Suppl 1):P233. doi: 10.1186/cc14313
119. Kataoka K, Taniguchi H, Kondoh Y, Nishiyama O, Kimura T, Matsuda T, et al. Recombinant human thrombomodulin in acute exacerbation of idiopathic pulmonary fibrosis. Chest. (2015) 148:436–43. doi: 10.1378/chest.14-2746
Keywords: thrombomodulin, damage-associated molecular patterns, disseminated Intravascular coagulation, neutrophil extracellular traps, high mobility group box 1, immunothrombosis
Citation: Watanabe-Kusunoki K, Nakazawa D, Ishizu A and Atsumi T (2020) Thrombomodulin as a Physiological Modulator of Intravascular Injury. Front. Immunol. 11:575890. doi: 10.3389/fimmu.2020.575890
Received: 24 June 2020; Accepted: 19 August 2020;
Published: 16 September 2020.
Edited by:
S. Y. Seong, Seoul National University, South KoreaReviewed by:
Péter Gál, Hungarian Academy of Sciences (MTA), HungaryCopyright © 2020 Watanabe-Kusunoki, Nakazawa, Ishizu and Atsumi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daigo Nakazawa, ZGFpZ28tbmFAbWVkLmhva3VkYWkuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.