 Ludovico De Stefano1,2
Ludovico De Stefano1,2 Francesca Bobbio-Pallavicini1
Francesca Bobbio-Pallavicini1 Antonio Manzo1,2
Antonio Manzo1,2 Carlomaurizio Montecucco1,2
Carlomaurizio Montecucco1,2 Serena Bugatti1,2*
Serena Bugatti1,2*- 1Division of Rheumatology, IRCCS Policlinico San Matteo Foundation, Pavia, Italy
- 2Department of Internal Medicine and Therapeutics, University of Pavia, Pavia, Italy
The effects of cytokine inhibition in the different phases of the severe coronavirus disease 2019 (COVID-19) are currently at the center of intense debate, and preliminary results from observational studies and case reports offer conflicting results thus far. The identification of the correct timing of administration of anti-cytokine therapies and other immunosuppressants in COVID-19 should take into account the intricate relationship between the viral burden, the hyperactivation of the innate immune system and the adaptive immune dysfunction. The main challenge for effective administration of anti-cytokine therapy in COVID-19 will be therefore to better define a precise “window of therapeutic opportunity.” Only considering a more specific set of criteria able to integrate information on direct viral damage, the cytokine burden, and the patient’s immune vulnerability, it will be possible to decide, carefully balancing both benefits and risks, the appropriateness of using immunosuppressive drugs even in patients affected primarily by an infectious disease.
Introduction
The coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has a wide spectrum of clinical expressiveness ranging from asymptomatic or paucisymptomatic infection to life-threating multiple organ failure. In most severe forms, SARS-CoV-2 infection leads to fulminant pneumonia and acute respiratory distress syndrome (ARDS) with a mortality rate approaching 40–50% (1). Clinical deterioration generally occurs several days after the onset of symptoms, in association with declining viral titers (2), suggesting that part of pathophysiology may be driven by dysregulated immune responses rather than by direct viral damage. Uncontrolled hyperinflammation has indeed been recognized as a pivotal pathogenetic event in COVID-19 (3), with the release of inflammatory cytokines which are injurious to host cells similarly to what happens in other hyperinflammatory syndromes characterized by cytokine storms, such as the cytokine release syndrome (CRS), secondary hemophagocytic lymphohistiocytosis (sHLH) and macrophage activation syndrome (MAS) (4). The known sensitivity of these syndromes to cytokine-directed therapies has fueled many expectations also for patients with COVID-19. However, the precise nature and role of hyperinflammation in severe COVID-19 remain poorly defined, and the risk-benefit ratio of cytokine inhibition in the different phases of the disease is largely debated (5–8). As a matter of facts, preliminary clinical results on the efficacy of therapeutic blockade of interleukin (IL)-6, IL-1, tumor necrosis factor (TNF), and Janus kinase signaling are thus far mixed (9–23) (Table 1), possibly also due to the different timing of drug administration in different reports. Early immunosuppression during an infectious disease indeed incurs risks; on the other hand, however, suppression of pathogenic hyperinflammation may be more effective in the initial stages before clinical deterioration occurs. The identification of the correct timing of administration of immunosuppressive drugs remains therefore a research priority for the personalized management of COVID-19.
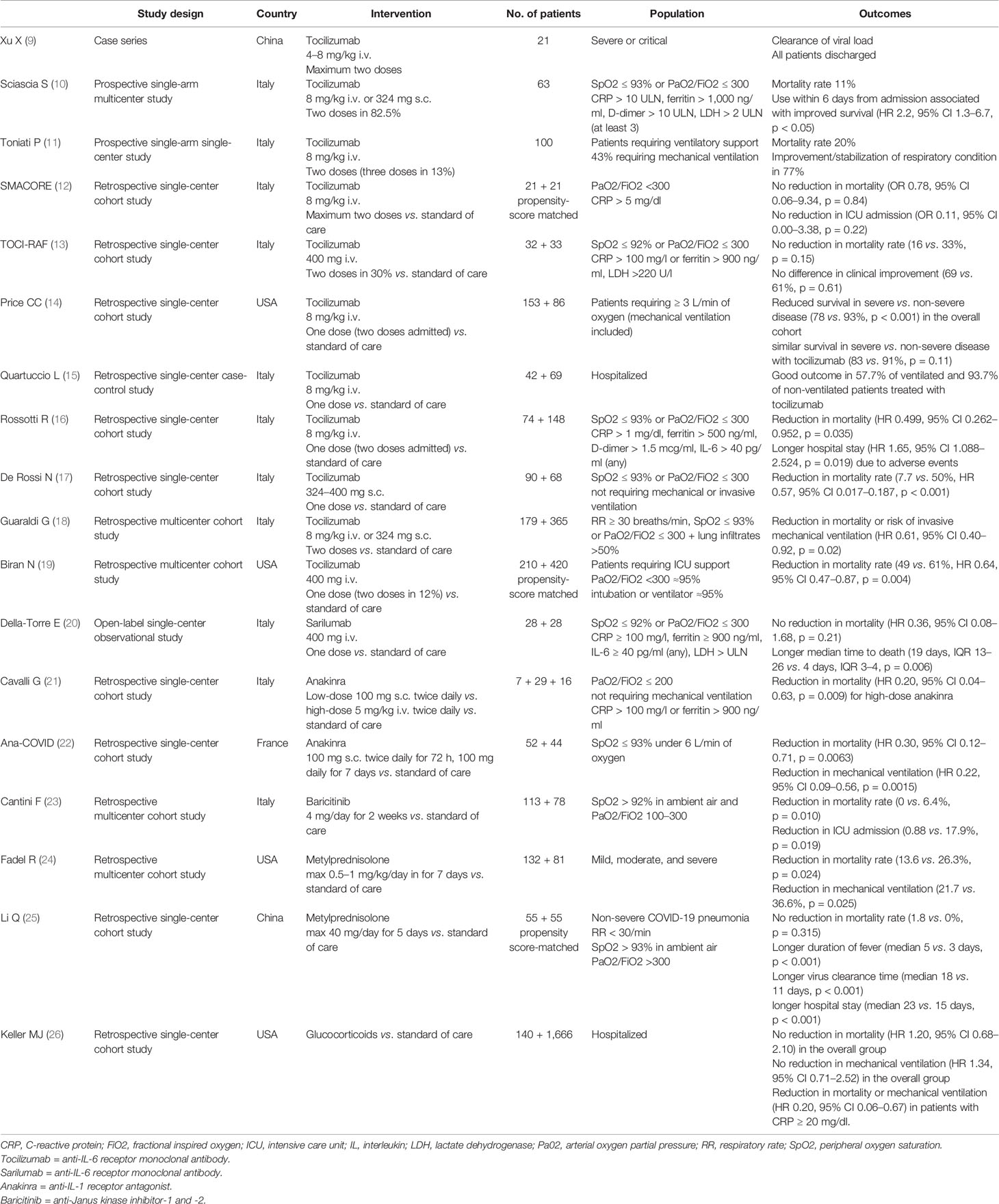
Table 1 A selection of observational cohort studies on anti-cytokine therapy and immunosuppressants in coronavirus disease 2019 (COVID-19).
The Complex Interplay Between the Viral Burden, Innate, and Adaptive Immune Responses in Coronavirus Disease 2019
The health analytic platform OpenSAFELY established in the United Kingdom has recently confirmed that COVID-19-related deaths are strongly associated with demographic factors and comorbidities such as increasing age, male gender, obesity, cardiovascular and respiratory diseases (27). Risk prediction and tailored treatment solely based on such generic parameters is however largely inaccurate, and COVID-19 outcomes likely depend on a number of other variables influencing host-pathogen interactions, including SARS-CoV-2 genetic variants as well as host genetic susceptibility. Research in this area is only in its infancy. Also building on previous experience with other Coronaviridae, several studies are investigating the link between SARS and genetic variants in innate immune response (28), including mannose‐binding lectin deficiency and polymorphisms, which are involved in the inactivation of a variety of respiratory pathogens through direct binding and complement activation (29). Similarly, adaptive immune dysfunction is appearing as a key, and recent studies have confirmed the role of CD3+ T cell cytopenia in determining COVID-19 progression and fatality (30). Discovery of virus and host genomic factors will undoubtedly support risk stratification and targeted treatment; however, as genomic studies require long times before entering clinical practice, it is urgent to integrate easily accessible information on the dynamics and pathogenicity of the immune response during the different phases of SARS-CoV-2 infection.
The relative contribution of viral damage and of innate and adaptive immune responses in course of COVID-19 greatly varies over time (31–33). In the early phases, common to all patients, pathogenic mechanisms are mainly driven by the cytotoxic role directly exerted by SARS-CoV-2, which usually affects only the upper respiratory tract. The early antiviral response mostly relying on type-I interferon (IFN) and natural killer (NK) cells, and the subsequent CD8+ T cell-mediated killing of virally infected cells as well as CD4+ T cell-dependent antibody production, allow the rapid reduction of the viral load, the paucisymptomatic character of the disease, and its recovery. However, in up to 20% of the patients, virus-induced immunosuppression occurs. This is highlighted by a marked reduction of CD4+ and CD8+ T lymphocytes in peripheral blood, defective CD8+ T cell and NK cell function, low IFN-γ production, and low specific IgG antibodies (34–37). The impairment of the initial antiviral defense mechanisms favors a sustained increase in the viral load capable of conditioning disease progression, especially in organs that have high angiotensin-converting enzyme 2 expression, such as the lungs but also the endothelium, the heart, the kidney, and the intestine (38, 39). The clinical picture can evolve rapidly, with the development of interstitial pneumonia which can progress into ARDS (40). In these early stages, lung damage is mainly driven by SARS-CoV-2 cytotoxicity, while inflammatory responses aim at eliminating the pathogen ultimately leading to tissue repair. Accordingly, longitudinal immune profiling of hospitalized COVID-19 cases with different outcomes has recently shown that, despite similar levels of inflammatory cytokines in the first 10 days from symptom onset, patients with less severe disease evolution also express mediators of wound healing and tissue repair (41). In this phase, the therapeutic strategy should thus include the use of antiviral drugs and of treatments aimed at cautiously enhance immune responses. Conversely, the use of drugs that compromise the efficiency of the immune system could be counterproductive, as patients with high viral loads and long virus-shedding periods are at higher risk of severe COVID-19 (42).
In immunocompromised patients in whom the therapeutic strategies adopted and the presence of comorbidities do not allow adequate control of the viral load, extensive tissue damage, and subsequent uncontrolled inflammatory innate responses with exaggerated myeloid-derived cytokine production can occur (43–46). The clinical picture suddenly and unexpectedly changes with fever, respiratory failure, and ARDS associated with increased levels of acute phase reactants, neutrophilia, thrombocytosis, anemia, signs of coagulopathy, and cell lysis. These acute inflammatory mechanisms may precipitate tissue damage both locally (especially at the lung level) (47) and systemically, thus playing an even greater pathogenetic role than that played by direct viral damage, and significantly affecting mortality. This hyperinflammatory syndrome shares pathophysiological similarities with cytokine storms in CRS, sHLH and MAS, conditions that sometimes complicate viral infections, systemic autoimmune and autoinflammatory diseases, hematologic diseases, and medications such as engineered T cell therapy (48–51). Experience from these conditions has encouraged the use of anti-cytokine therapy also for the management of COVID-19. However, the clinical picture of the cytokine storm in COVID-19, especially when it is not associated with multi-organ damage secondary to coagulopathy, is different from that of the classic sHLH/MAS: it is mostly anatomically compartmentalized to the lungs, not associated with organomegaly and not accompanied by pancytopenia (5). Accordingly, serum levels of IL-6 are lower in COVID-19 compared to other CRS (52). Furthermore, hypercytokinemia should be considered as a general marker of SARS-CoV-2, even in the absence of a cytokine storm, and this makes COVID-19 CRS not easily distinguishable from ARDS pathogenetically correlated only with viral cytotoxic activity, also because levels of inflammatory markers and cytokines not always correlate with outcome (53). Finally, compared to sHLH/MAS, the CRS in COVID-19 occurs in the context of immunodeficiency and immunoparalysis (5, 30, 54), thus imposing caution on further iatrogenic immunosuppression.
Challenges in the Identification of a “Window of Opportunity” for Immunosuppression in Coronavirus Disease 2019
The identification of the correct timing of administration of anti-cytokine therapies and other immunosuppressants in COVID-19 should thus take into account the intricate relationship between the viral burden, the hyperactivation of the innate immune system, and the adaptive immune dysfunction. Although the therapeutic window in CRS is narrow, and timely control of the cytokine storm is crucial to reduce short-term mortality, premature use of immunosuppressants could indeed further compromise viral shedding with the risk of increasing viral replication and tissue damage directly induced by the virus (43). Furthermore, iatrogenic immunosuppression could promote bacterial, fungus, or viral infectious complications, as shown with the targeting of IL-6 with tocilizumab in observational studies on patients not requiring mechanical ventilation (18, 55). Also at later stages, pharmacologically induced immunosuppression in already immunocompromised patients could adversely affect the course of the disease, as suggested by the finding of significantly longer periods of hospitalization and higher rates of mortality in ventilated patients treated with tocilizumab (15, 16) or high-doses corticosteroids (56, 57).
The main challenge for effective anti-cytokine therapy in COVID-19 will be therefore to better define a precise “window of therapeutic opportunity,” a phase of the disease during which the benefits of cytokine inhibition are prevalent on the inevitable consequences of immunosuppression. Although > 100 randomized clinical trials (RCTs) on different cytokine inhibitors in COVID-19 are underway, the identification of such a “window” remains elusive, and only few studies have tried to integrate further patients’ characterization beyond the clinical status among the inclusion criteria (Table 2).

Table 2 A selection of the inclusion criteria of some randomized clinical trials on anti-cytokine therapy for coronavirus disease 2019 (COVID-19).
Most of the protocols essentially require, among the inclusion criteria, the presence of pneumonia not otherwise defined if not by the extent of functional impairment, with a tendency to focus mainly on the early stages of the disease, in the absence of ARDS or systemic involvement. Some observational studies continue to suggest that immunosuppressants such as glucocorticoids and IL-6 antagonists (alone or in combination) are advantageous if administered early at hospital admission (17, 24, 58) (Table 1). However, the RECOVERY trial has clearly shown that benefits from dexamethasone are restricted to those patients with at least 7 days of symptoms and those requiring invasive or non-invasive ventilation (59), suggesting that, based on the clinical criterion alone, only a late phase of COVID-19 is dominated by pathogenic immunity. Accordingly, the use of early short-term corticosteroid therapy, even at low-doses, has been associated with worse clinical outcomes in non-severe COVID-19 pneumonia in observational studies (25) (Table 1). In line with these findings, following preliminary results, RCTs on sarilumab (another IL-6 receptor antagonist) have been amended to enroll only critical patients (https://investor.regeneron.com/news-releases/news-release-details/regeneron-and-sanofi-provide-update-us-phase-23-adaptive), and the COVACTA trial on tocilizumab recently ended recruitment because neither primary nor secondary end-points were met (https://www.roche.com/media/releases/med-cor-2020-07-29.htm).
Better and still clinically feasible characterization of those patients with an hyperinflammatory syndrome potentially susceptible to immunosuppression could derive from the integration of markers of inflammation, tissue damage, cell lysis, and coagulopathy. This has indeed been the rationale for the development of the H-score for the diagnosis of sHLH and MAS, which includes, among the others, hyperferritinemia, cytopenias, and liver damage (60). However, the many clinical and pathogenetic differences between COVID-19 hyperinflammation and sLHL/MAS limit the generalizability of the H-score to COVID-19 associated cytokine storm, and only few patients with severe COVID-19 achieve diagnostic cut-offs of >169, mainly due to lower ferritin levels, absence of pancytopenia and hypofibrinogenemia (61). Similarly, the prevalence of the hyperinflammatory phenotype in ARDS due to COVID-19 is lower compared to that observed in non-COVID-19 ARDS despite its higher mortality (53). However, laboratory indicators of hyperinflammation may still be of value in the identification of those COVID-19 patients more at risk of escalation of respiratory support and death. Using cut-off values of C-reactive protein (CRP) concentrations greater than 150 mg/L or ferritin concentrations greater than 1,500 μg/L, Manson JJ and colleagues (62) recently described an hyperinflammatory phenotype of COVID-19 characterized by poor clinical outcomes irrespective of other demographic and clinical variables. Patients’ stratification based on this approach could be of promise. Accordingly, in an observational study on hospitalized COVID-19 cases, early use of glucocorticoids was effective only in patients with high CRP levels, being instead associated with increased mortality in case of low CRP (26).
Elevation of inflammatory markers such as CRP, ferritin, and others is however non-specific for cytokine storms. Cytokine assays are ready available in the clinic and testing for serum levels of cytokines known to be involved in pathogenic inflammation could help guiding the rational use of targeted immunomodulatory therapeutic strategies. Accordingly, IL-6 and other pro-inflammatory cytokines have been shown to be predictive of patient outcomes in terms of both disease severity and survival, and integrated models taking into account demographics, comorbidities, markers of inflammation and tissue damage, and cytokines show good performance with areas under the receiver operating characteristic curve approaching 0.8 (63). Several limitations however still exist even using similar approaches. Apart from IL-6, the trend of other cytokines whose targeting is already available in the clinic does not appear uniform in COVID-19 (30, 41, 54, 63), hampering the possibility of tailored treatments and contrasting with the apparent efficacy of some agents, such as IL-1 antagonists, in preliminary observational studies (21, 22). Even in the case of IL-6, no established cut-offs exist. Furthermore, the release of inflammatory cytokines is also part of a well-conserved innate immune response necessary for efficient clearance of infectious agents. Accordingly, levels of IL-6 are similarly increased in the hyperinflammatory phenotype of non-COVID-19 ARDS irrespective of its causative mechanism, but interventions targeting single cytokines in this setting have a long history of failure (64).
Collectively, current clinical, laboratory, and even biological parameters thus do not appear optimal at distinguishing an appropriate from a dysregulated inflammatory response in the context of COVID-19. Ideally, in light of the very pulmonary-centric character of this condition, better knowledge could arise from the analysis of parameters deriving from bronchoalveolar lavage fluid and microscopic examination of transbronchial lung biopsies (Table 3). These include cytological analyses with prevalence of granulocytes, macrophages, and lymphocytes; pro-inflammatory cytokine and chemokine secretion including neutrophil recruiting mediators and other attractants of monocytes and immune cells; lung infiltration by monocytes, macrophages, and neutrophils; thrombosis (65–68). The clinical, radiological, and laboratory characteristics already identified in patients with COVID-19 would thus be put in the context of more specific histological, cytological, and immune-inflammatory criteria in order to identify those parameters more accurately signaling the presence of a predominantly immuno-inflammatory pathogenesis which might benefit from immunosuppression (Table 3). The evidence of ongoing SARS-CoV-2 replication late in disease would in any case support the associated use of antiviral therapy, even at a point when immunopathology is dominant (69).

Table 3 Possible inclusion criteria for anti-cytokine therapy in coronavirus disease 2019 (COVID-19).
In conclusion, despite the emergency situation imposes speed in the identification of the therapeutic options for patients with COVID-19, better harmonization of inclusion criteria and patient stratifications for studies on immunomodulatory therapies should remain a priority. Only considering a more specific set of clinical and pathological criteria, together with the extent of the viral load present in the alveolar bronchial lavage fluid and parameters useful to quantify the patient’s immune vulnerability (lymphocyte count, CD4+ and CD8+ T cell levels, markers of lymphocyte exhaustion, development of specific antibodies), it will be possible to decide, carefully balancing both benefits and risks, the appropriateness of using immunosuppressive drugs even in patients affected primarily by an infectious disease.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding author.
Author Contributions
All the authors contributed to the conception of the commentary. LS drafted the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This article was supported in part by fundings from the IRCCS Policlinico San Matteo Foundation, Pavia, Italy.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer LQ declared a past co-authorship with two of the authors CM and SB to the handling editor.
References
1. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med (2020) 180:1–11. doi: 10.1001/jamainternmed.2020.0994
2. Joynt GM, Wu WK. Understanding COVID-19: what does viral RNA load really mean? Lancet Infect Dis (2020) 20:635–6. doi: 10.1016/S1473-3099(20)30237-1
3. Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol (2020) 20:355–62. doi: 10.1038/s41577-020-0331-4
4. Behrens EM, Koretzky GA. Review: cytokine storm syndrome: looking toward the precision medicine era. Arthritis Rheumatol (2017) 69:1135–43. doi: 10.1002/art.40071
5. McGonagle D, Sharif K, O’Regan A, Bridgewood C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun Rev (2020) 19:102537. doi: 10.1016/j.autrev.2020.102537
6. Jamilloux Y, Henry T, Belot A, Viel S, Fauter M, El Jammal T, et al. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun Rev (2020) 19:102567. doi: 10.1016/j.autrev.2020.102567
7. Alijotas-Reig J, Esteve-Valverde E, Belizna C, Selva-O’Callaghan A, Pardos-Gea J, Quintana A, et al. Immunomodulatory Therapy for the Management of Severe COVID-19. Beyond the Anti-Viral Therapy: A Comprehensive Review. Autoimmun Rev (2020) 19:102569. doi: 10.1016/j.autrev.2020.102569
8. Ruscitti P, Berardicurti O, Iagnocco A, Giacomelli R. Cytokine storm syndrome in severe COVID-19. Autoimmun Rev (2020) 19:102562. doi: 10.1016/j.autrev.2020.102562
9. Xu X, Han M, Li T, Sun W, Wang D, Fu B, et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci U S A (2020) 117:10970–5. doi: 10.1073/pnas.2005615117
10. Sciascia S, Aprà F, Baffa A, Baldovino S, Boaro D, Boero R, et al. Pilot prospective open, single-arm multicentre study on off-label use of tocilizumab in patients with severe COVID-19. Clin Exp Rheumatol (2020) 38:529–32.
11. Toniati P, Piva S, Cattalini M, Garrafa E, Regola F, Castelli F, et al. Tocilizumab for the treatment of severe COVID-19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: A single center study of 100 patients in Brescia, Italy. Autoimmun Rev (2020) 19:102568. doi: 10.1016/j.autrev.2020.102568
12. Colaneri M, Bogliolo L, Valsecchi P, Sacchi P, Zuccaro V, Brandolino F, et al. Tocilizumab for Treatment of Severe COVID-19 Patients: Preliminary Results from SMAtteo COvid19 REgistry (SMACORE). Microorganisms (2020) 8:695. doi: 10.3390/microorganisms8050695
13. Campochiaro C, Della-Torre E, Cavalli G, De Luca G, Ripa M, Boffini N, et al. Efficacy and safety of tocilizumab in severe COVID-19 patients: a single-centre retrospective cohort study. Eur J Intern Med (2020) 76:43–9. doi: 10.1016/j.ejim.2020.05.021
14. Price CC, Altice FL, Shyr Y, Koff A, Pischel L, Goshua G, et al. Tocilizumab Treatment for Cytokine Release Syndrome in Hospitalized COVID-19 Patients: Survival and Clinical Outcomes. Chest (2020) S0012-3692(20):31670–6. doi: 10.1016/j.chest.2020.06.006
15. Quartuccio L, Sonaglia A, McGonagle D, Fabris M, Peghin M, Pecori D, et al. Profiling COVID-19 pneumonia progressing into the cytokine storm syndrome: Results from a single Italian Centre study on tocilizumab versus standard of care. J Clin Virol (2020) 129:104444. doi: 10.1016/j.jcv.2020.104444
16. Rossotti R, Travi G, Ughi N, Corradin M, Baiguera C, Fumagalli R, et al. Safety and efficacy of anti-il6-receptor tocilizumab use in severe and critical patients affected by coronavirus disease 2019: A comparative analysis. J Infect (2020) S0163-4453(20):30467–9. doi: 10.1016/j.jinf.2020.07.008
17. De Rossi N, Scarpazza C, Filippini C, Cordioli C, Rasia S, Mancinelli CR, et al. Early use of low dose tocilizumab in patients with COVID-19: A retrospective cohort study with a complete follow-up. EClinicalMedicine (2020) 17:100459. doi: 10.1016/j.eclinm.2020.100459
18. Guaraldi G, Meschiari M, Cozzi-Lepri A, Milic J, Tonelli R, Menozzi M, et al. Tocilizumab in patients with severe COVID-19: a retrospective cohort study. Lancet Rheumatol (2020) 2:e474–84. doi: 10.1016/S2665-9913(20)30173-9
19. Biran N, Ip A, Ahn J, Go RC, Wang S, Mathura S, et al. Tocilizumab among patients with COVID-19 in the intensive care unit: a multicentre observational study. Lancet Rheumatol (2020) 2(10):e603–12. doi: 10.1016/S2665-9913(20)30277-0
20. Della-Torre E, Campochiaro C, Cavalli G, De Luca G, Napolitano A, La Marca S, et al. Interleukin-6 blockade with sarilumab in severe COVID-19 pneumonia with systemic hyperinflammation: an open-label cohort study. Ann Rheum Dis (2020) 3:annrheumdis–2020-218122. doi: 10.1136/annrheumdis-2020-218122
21. Cavalli G, De Luca G, Campochiaro C, Della-Torre E, Ripa M, Canetti D, et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol (2020) 2:e325–31. doi: 10.1016/S2665-9913(20)30127-2
22. Huet T, Beaussier H, Voisin O, Jouveshomme S, Dauriat G, Lazareth I, et al. Anakinra for severe forms of COVID-19: a cohort study. Lancet Rheumatol (2020) 2:e393–400. doi: 10.1016/S2665-9913(20)30164-8
23. Cantini F, Niccoli L, Nannini C, Matarrese D, Natale MED, Lotti P, et al. Beneficial impact of Baricitinib in COVID-19 moderate pneumonia; multicentre study. J Infect (2020) S0163-4453(20):30433–3. doi: 10.1016/j.jinf.2020.06.052
24. Fadel R, Morrison AR, Vahia A, Smith ZR, Chaudhry Z, Bhargava P, et al. Early Short Course Corticosteroids in Hospitalized Patients with COVID-19. Clin Infect Dis (2020) 19:ciaa601. doi: 10.1093/cid/ciaa601
25. Li Q, Li W, Jin Y, Xu W, Huang C, Li L, et al. Efficacy Evaluation of Early, Low-Dose, Short-Term Corticosteroids in Adults Hospitalized with Non-Severe COVID-19 Pneumonia: A Retrospective Cohort Study. Infect Dis Ther (2020) 2:1–14. doi: 10.1007/s40121-020-00332-3
26. Keller MJ, Kitsis EA, Arora S, Chen JT, Agarwal S, Ross MJ, et al. Effect of Systemic Glucocorticoids on Mortality or Mechanical Ventilation in Patients With COVID-19. J Hosp Med (2020) 15:489–93. doi: 10.12788/jhm.3497
27. Williamson EJ, Walker AJ, Bhaskaran K, Bacon S, Bates C, Morton CE, et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature (2020) 584:430–6. doi: 10.1038/s41586-020-2521-4
28. Ovsyannikova IG, Haralambieva IH, Crooke SN, Poland GA, Kennedy RB. The role of host genetics in the immune response to SARS-CoV-2 and COVID-19 susceptibility and severity. Immunol Rev (2020) 296:205–19. doi: 10.1111/imr.12897
29. Ip WK, Chan KH, Law HK, Tso GHW, Kong EKP, Wong WHS, et al. Mannose-binding lectin in severe acute respiratory syndrome coronavirus infection. J Infect Dis (2005) 191:1697–704. doi: 10.1086/429631
30. Zhang X, Tan Y, Ling Y, Lu F, Yi Z, Jia X, et al. Viral and host factors related to the clinical outcome of COVID-19. Nature (2020) 583:437–00. doi: 10.1038/s41586-020-2355-0
31. Ling L, Lianfeng L, Wei C, Taisheng L. Hypothesis for potential pathogenesis of SARSCoV-2 infection -a review of immune changes in patients with viral pneumonia. Emerg Microbes Infect (2020) 9:727–32. doi: 10.1080/22221751.2020.1746199
32. Cao X. COVID-19: immunopathology and its implications for therapy. Nat Rev Immunol (2020) 9:1–2. doi: 10.1038/s41577-020-0308-3
33. Vardhana SA, Wolchok JD. The Many Faces of the anti-COVID Immune Response. J Exp Med (2020) 217:e20200678. doi: 10.1084/jem.20200678
34. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunologic features in severe and moderate forms of Coronavirus Disease 2019. J Clin Invest (2020) 130:2620–9. doi: 10.1172/JCI137244
35. Wang X, Xu W, Hu G, Xia S, Sun Z, Liu Z, et al. SARS-CoV-2 infects T lymphocytes through its spike protein-mediated membrane fusion. Cell Mol Immunol (2020) 7:1–3. doi: 10.1038/s41423-020-0424-9
36. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol (2020) 17:533–5. doi: 10.1038/s41423-020-0402-2
37. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe (2016) 19:181–93. doi: 10.1016/j.chom.2016.01.007
38. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol (2004) 203:631–7. doi: 10.1002/path.1570
39. Rivellese F, Prediletto E. ACE2 at the centre of COVID-19 from paucisymptomatic infections to severe pneumonia. Autoimmun Rev (2020) 19:102536. doi: 10.1016/j.autrev.2020.102536
40. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
41. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature (2020) 584:463–9. doi: 10.1038/s41586-020-2588-y
42. Chen W, Lan Y, Yuan X, Deng X, Li Y, Cai X, et al. Detectable 2019-nCoV viral RNA in blood is a strong indicator for the further clinical severity. Emerg Microbes Infect (2020) 9:469–73. doi: 10.1080/22221751.2020.1732837
43. Liu Y, Yan LM, Wan L, Xiang TX, Le A, Liu JM, et al. Viral dynamics in mild and severe cases of COVID-19. Lancet Infect Dis (2020) 20:656–7. doi: 10.1016/S1473-3099(20)30232-2
44. Velazquez-Salinas L, Verdugo-Rodriguez A, Rodriguez LL, Borca MV. The role of interleukin 6 during viral infections. Front Microbiol (2019) 10:1057. doi: 10.3389/fmicb.2019.01057
45. Yap JKY, Moriyama M, Iwasaki A. Inflammasomes and Pyroptosis as Therapeutic Targets for COVID-19. J Immunol (2020) 205:307–12. doi: 10.4049/jimmunol.2000513
46. McKechnie JL, Blish CA. The Innate Immune System: Fighting on the Front Lines or Fanning the Flames of COVID-19? Cell Host Microbe (2020) S1931-3128(20):30291–2. doi: 10.1016/j.chom.2020.05.009
47. Hogner K, Wolff T, Pleschka S, Plog S, Gruber AD, Kalinke U, et al. Macrophage expressed IFN-β contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PloS Pathog (2013) 9:e1003188. doi: 10.1371/journal.ppat.1003188
48. Al-Samkari H, Berliner N. Hemophagocytic Lymphohistiocytosis. Annu Rev Pathol (2018) 13:27–49. doi: 10.1146/annurev-pathol-020117-043625
49. Ravelli A, Davì S, Minoia F, Martini A, Cron RQ. Macrophage Activation Syndrome. Hematol Oncol Clin North Am (2015) 29:927–41. doi: 10.1016/j.hoc.2015.06.010
50. Huang KJ, Su IJ, Theron M, Wu YC, Lai SK, Liu CC, et al. An Interferon-Gamma-Related Cytokine Storm in SARS Patients. J Med Virol (2005) 75:185–94. doi: 10.1002/jmv.20255
51. Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J (2014) 20:119–22. doi: 10.1097/PPO.0000000000000035
52. Maude S, Barrett DM. Current status of chimeric antigen receptor therapy for haematological malignancies. Br J Haematol (2016) 172:11–22. doi: 10.1111/bjh.13792
53. Sinha P, Calfee CS, Cherian S, Brealey D, Cutler S, King C, et al. Prevalence of phenotypes of acute respiratory distress syndrome in critically ill patients with COVID-19: a prospective observational study. Lancet Respir Med (2020) 27:S2213-2600(20)30366-0. doi: 10.1016/S2213-2600(20)30366-0
54. Laing AG, Lorenc A, Del Molino Del Barrio I, Das A, Fish M, Monin L, et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat Med (2020) doi: 10.1038/s41591-020-1038-6
55. Antinori S, Bonazzetti C, Gubertini G, Capetti A, Pagani C, Morena V, et al. Tocilizumab for cytokine storm syndrome in COVID-19 pneumonia: an increased risk for candidemia? Autoimmun Rev (2020) 19:102564. doi: 10.1016/j.autrev.2020.102564
56. Russell CD, Millar JE, Baillie JK. Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet (2020) 395:473–5. doi: 10.1016/S0140-6736(20)30317-2
57. Yang Z, Liu J, Zhou Y, Zhao X, Zhao Q, Liu J. The effect of corticosteroid treatment on patients with coronavirus infection: a systematic review and meta-analysis. J Infect (2020) S0163-4453(20):30191–2. doi: 10.1016/j.jinf.2020.03.062
58. Martínez-Urbistondo D, Costa Segovia R, Suárez Del Villar Carrero R, Risco Risco C, Villares Fernández P. Early combination of Tocilizumab and Corticosteroids: An upgrade in anti-inflammatory therapy for severe COVID. Clin Infect Dis (2020) 4:ciaa910. doi: 10.1093/cid/ciaa910
59. Brightling C, Ustianowski A, Elmahi E, Prudon B, Green C, Felton T, et al. Dexamethasone in Hospitalized Patients with Covid-19 - Preliminary Report. N Engl J Med (2020) 17:NEJMoa2021436. doi: 10.1056/NEJMoa2021436
60. Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol (2014) 66:2613–20. doi: 10.1002/art.38690
61. Ruscitti P, Bruno F, Berardicurti O, Acanfora C, Pavlych V, Palumbo P, et al. Lung involvement in macrophage activation syndrome and severe COVID-19: results from a cross-sectional study to assess clinical, laboratory and artificial intelligence-radiological differences. Ann Rheum Dis (2020) 79:1152–5. doi: 10.1136/annrheumdis-2020-218048
62. Manson JJ, Crooks C, Naja M, Ledlie A, Goulden B, Liddle T, et al. COVID-19-associated hyperinflammation and escalation of patient care: a retrospective longitudinal cohort study. Lancet Rheumatol (2020) 2:e594–e602. doi: 10.1016/S2665-9913(20)30275-7
63. Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med (2020) doi: 10.1038/s41591-020-1051-9
64. Sinha P, Calfee CS. Phenotypes in acute respiratory distress syndrome: moving towards precision medicine. Curr Opin Crit Care (2019) 25:12–20. doi: 10.1097/MCC.0000000000000571
65. Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect (2020) 9:761–70. doi: 10.1080/22221751.2020.1747363
66. Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe (2020) 27:883–90. doi: 10.1016/j.chom.2020.04.017
67. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
68. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
Keywords: cytokine storm, coronavirus, tocilizumab (IL-6 inhibitor), T cell, immune activation, innate and adaptive immune response
Citation: De Stefano L, Bobbio-Pallavicini F, Manzo A, Montecucco C and Bugatti S (2020) A “Window of Therapeutic Opportunity” for Anti-Cytokine Therapy in Patients With Coronavirus Disease 2019. Front. Immunol. 11:572635. doi: 10.3389/fimmu.2020.572635
Received: 30 June 2020; Accepted: 21 September 2020;
Published: 06 October 2020.
Edited by:
Christoph T. Berger, University of Basel, SwitzerlandReviewed by:
Luca Quartuccio, University of Udine, ItalyThorsten Demberg, Marker Therapeutics, United States
Copyright © 2020 De Stefano, Bobbio-Pallavicini, Manzo, Montecucco and Bugatti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Serena Bugatti, c2VyZW5hLmJ1Z2F0dGlAdW5pcHYuaXQ=