 Igal Ifergan
Igal Ifergan Stephen D. Miller
Stephen D. Miller- 1Department of Microbiology-Immunology, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 2Interdepartmental Immunobiology Center, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
Multiple Sclerosis (MS) is characterized by immune cell infiltration to the central nervous system (CNS) as well as loss of myelin. Characterization of the cells in lesions of MS patients revealed an important accumulation of myeloid cells such as macrophages and dendritic cells (DCs). Data from the experimental autoimmune encephalomyelitis (EAE) model of MS supports the importance of peripheral myeloid cells in the disease pathology. However, the majority of MS therapies focus on lymphocytes. As we will discuss in this review, multiple strategies are now in place to target myeloid cells in clinical trials. These strategies have emerged from data in both human and mouse studies. We discuss strategies targeting myeloid cell migration, growth factors and cytokines, biological functions (with a focus on miRNAs), and immunological activities (with a focus on nanoparticles).
Introduction
Myeloid cells play critical roles in the health and diseases of the central nervous system (CNS). For example, myeloid cells constitute a significant proportion of the cells found within perivascular infiltrates in CNS lesions of Multiple Sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE) (1–3). Myeloid cells are also critically involved in the secondary damage in spinal cord injury (SCI) and traumatic brain injury (TBI) (4–6). These myeloid cells have the ability to attract other immune cells, release neurotoxic factors, phagocytose proteins and debris and promote the expansion, and polarization of antigen-specific T cells in the CNS. In addition to their capacity to induce and sustain inflammation, myeloid cells are also critically involved in communication with glial cells and neurons, as well as in promoting and maintaining peripheral tolerance (7–9).
MS is an inflammatory autoimmune disease wherein cells of the immune system initiate an attack against myelin in the CNS that supports axonal conduction. The immune response in MS is thought to be mediated by autoreactive T lymphocytes that recognize myelin peptides. Typically, demyelination is associated with an accumulation of T lymphocytes (lymphoid component of infiltrates) and monocytes/ macrophages/ dendritic cells (myeloid cells component of infiltrates) that arise from the migration of peripheral blood immune cells across the CNS microvascular endothelium (10–12).As they infiltrate the CNS, encephalitogenic T lymphocytes require the presence of these blood-derived antigen-presenting cells (APCs) to further sustain lymphocyte proliferation and cytokine polarization in the CNS compartment (13–16). The role of these peripherally-derived myeloid cells in CNS inflammation will be the focus of the present review.
Role of Myeloid Cells in the Pathogenesis of MS and Other Autoimmune Diseases
Experimental autoimmune encephalomyelitis is a commonly utilized mouse model of MS that recapitulates many aspects of the human disease such as the CNS inflammation, encephalitogenic T cell infiltration, and attack of oligodendrocytes resulting in demyelination. Although not perfect, EAE has allowed uncovering some of the molecular pathways governing the pathogenesis of MS such as elucidating the pathogenic role of TH17 lymphocytes. In addition, EAE models were critical in identifying and testing new therapeutic agents such as glatiramer acetate (GA) and Natalizumab (17).
Although myeloid APCs play a prominent role in the pathogenesis of MS, there has been little consideration given to targeting these cells as an MS therapy. Some of the current MS disease-modifying therapies may act on myeloid cells even if these cells were not the original intended targets (18). However, interfering directly with myeloid cell has proven to be efficacious in other diseases including psoriasis with multiple drugs targeting IL-23 (Guselkumab, Risankizumab, and Tildrakizumab) or IL-12 and IL-23 (Ustekinumab) (19), Crohn's disease and ulcerative colitis targeting IL-12 and IL-23 (Ustekinumab) (20), rheumatoid arthritis targeting IL-1 (Anakinra) (21), systemic juvenile idiopathic arthritis targeting IL-1β (Canakinumab) (22), and many others. There are ongoing clinical trials in rheumatoid arthritis, stroke, atherosclerosis, and cancer using agents that target myeloid cells and their products. Biber et al. have provided a recent comprehensive review of drugs in clinical trials targeting myeloid cells in CNS diseases such as Alzheimer's disease, brain tumors, and inflammatory pain, as well as for other CNS diseases (23).
As we will discuss in this review, multiple tools have been developed in the EAE models of MS demonstrating significant regulation of disease progression by various approaches blocking myeloid cell activation and effector function, but to date, these approaches have not been tested for therapeutic efficacy in MS patients.
Targeting Myeloid Cell Migration
The first strategy we will discuss is interference with peripheral myeloid cell migration to the CNS. The blood-brain barrier (BBB), composed of tightly bound endothelial cells (ECs), regulates the entry of blood-borne molecules and immune cells into the CNS. Under physiological conditions, a limited number of peripheral blood immune cells gain access to the CNS, a process called immune surveillance (24). During an inflammatory process, meningeal, and BBB-ECs amplify the migration of immune cells into the CNS parenchyma, in a multi-step process that involves selectins, chemokines and cell adhesion molecules (25). BBB-ECs express cell adhesion molecules such as intercellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM)-1, activated leucocyte cell adhesion molecule (ALCAM), and melanoma cell adhesion molecule (MCAM) which mediate at least in part, the adhesion process and the transmigration of leucocytes to the CNS through their interaction with integrins αLβ2 [leucocyte function-associated antigen (LFA)-1], α4β1 [very late antigen (VLA)-4], CD6, and MCAM respectively (26–31). Interfering with immune cell trafficking across the BBB by targeting adhesion molecules has proven to be beneficial in reducing clinical disease activity and pathological indices in MS (32). Indeed, Natalizumab, which blocks VLA-4, the ligand of VCAM-1, is reported to reduce migration of most leukocyte subtypes, including myeloid cells, into the brain.
More recently, a new adhesion molecule expressed by BBB-ECs called Nerve injury-induced protein (Ninjurin)-1 was described (33). Ninjurin-1 is a membrane protein known to interact in a homophilic manner through an extracellular residue-binding motif (34). On immune cells, Ninjurin-1 was weakly expressed by lymphocytes, but highly expressed by peripheral myeloid APCs including monocytes, macrophages and dendritic cells (DCs), in humans and mice. Interestingly, Ninjurin-1 was also found to be expressed in MS lesions. Ninjurin-1 neutralization specifically abrogated the adhesion and migration of human monocytes across a monolayer of BBB endothelial cells, without affecting lymphocyte recruitment. Moreover, Ninjurin-1 blockade during the course of EAE reduced infiltration of peripheral myeloid cells and reduced clinical disease activity and histopathological indices of EAE (33).
Another adhesion molecule involved in the migration of peripheral myeloid cells is junctional adhesion molecule (JAM)-like (JAML). JAMs are type I transmembrane proteins differentially expressed at the junctions of ECs, epithelial cells, and on various leukocytes (35). Similarly to Ninjurin-1, JAML can interact in a homophilic manner (36). It was observed that JAML is expressed by BBB-ECs, and has an increase expression in MS lesions compared to normal appearing white matter (37). In addition, human monocytes and CD8+ T cells were found to express JAML, and its level was significantly increased on RRMS patients when compared control subjects: 80 vs. 52% for monocytes, and 5.5 vs. 2.1% and for CD8+ T cells. These data reveals that JAML might be a more important adhesion molecule for monocytes than for CD8+ T cells. However, migratory capacity of both cell types was significantly compromised when JAML was blocked.
Chemotactic cytokines (chemokines) are secreted proteins that regulate the migration of leukocytes. Chemokine receptor signaling plays a central role in cell migration during inflammatory responses in autoimmune and infectious diseases as well as in cancer. There are ~50 chemokines and 20 receptors known at this time. Blockade of CCR1 and CCR2 have been the two majors targets in a half dozen MS clinical trials (38). The chemokines CCL3 (macrophage inflammatory protein-1α–MIP-1α) and CCL5 (regulated on activation, normal T cell expressed and secreted—RANTES) bind to CCR1, while CCL2 (monocyte chemoattractant protein 1—MCP-1) binds to CCR2. Both lymphoid and myeloid cells express CCR1 and CCR2, with monocytes/macrophages/DCs the cells where these chemokine receptors are most abundant (39–42). In animal models of MS, it was shown that CCR1-deficient animals developed a less severe disease (43), while CCR2-deficient mice were completely resistant to disease induction (44, 45), highlighting the importance of signaling through these chemokine receptors for disease initiation. In addition, it has been shown that CCR2+Ly-6Chi monocytes are rapidly recruited to the inflamed CNS in EAE and are crucial for the effector phase of disease. Selective depletion of this specific monocyte subpopulation through engagement of CCR2 significantly reduced disease severity (46). CCR1+ and CCR2+ macrophages were both found in active MS lesions (47, 48). The role of chemokines and their receptors are now well-characterized in MS and other inflammatory diseases. The potential for a therapy targeting this signaling pathway is well-recognized. However, none of the chemokine-directed MS clinical trials has shown robust clinical efficacy. Similar lack of clinical responses have also been reported in therapeutic trials targeting chemokines in other diseases such as rheumatoid arthritis, psoriasis, asthma, and many others (49). The issue may lie in the redundancy of chemokine/chemokine receptor action, in which case, it may be beneficial to develop strategies employing multiple antagonists simultaneously.
As innate cells, myeloid cells express pattern recognition receptors (PRRs). PRRs include Toll-like receptors (TLRs), RIG-I-like receptors, NOD-like receptors, and C-type lectin receptors (CLRs) (50). Selectins, which are part of the C-type lectins family, are known to play a crucial role in the control of leukocyte trafficking and homing to sites of inflammation (51). Selectins are particularly important for the rolling of cells on endothelial cells, an important component of migration of myeloid cells into tissue sites of inflammation (52). More recently, it was uncovered that CLEC12A, a CLR, was involved in facilitating binding and transmigration of DCs across the BBB in response to CCL2 chemotaxis (53). In EAE, CLEC12A−/− mice displayed delayed disease onset and significantly reduced disease severity. Additionally, in a chronic model of EAE, anti-CLEC12A antibody treatment initiated at disease initiation also delayed onset and lessened disease severity. Anti-CLEC12A antibody administration to mice undergoing relapsing-remitting EAE after disease onset, resulted in less severe disease relapse (53). Although the ligand of CLEC12A is currently unknown, it was suggested that the ligand is present on BBB endothelial cells (53).
Targeting Myeloid Cell Activation and Cytokines
Growth Factors
Abundance of immune cells as well as cytokines, chemokines and immunoglobulins in MS plaques and their accumulation in the cerebrospinal fluid (CSF) of MS patients, support the notion that MS is an inflammatory disorder. These observations lend support to the idea that immune cell products, especially cytokines, have an important role in both the induction and progression of MS. Targeting cytokines has been a successful strategy used in therapy of other inflammatory diseases. For example, blockade of tumor necrosis factor (TNF) has shown positive results in Rheumatoid Arthritis and Crohn's disease (54, 55). As of December 2016, TNF inhibitors were the world's leading drug class, with sales of more than US $30 billion and used in more than seven million patients (56). In MS, the first treatment approved for RRMS was interferon (IFN)-β, thus showing that cytokines manipulation is potentially a good strategy.
Current data suggests that MS, and its animal model, EAE, are driven by both TH1 lymphocytes, producing IFN-γ, interleukin (IL)-2 and TNF, and TH17 lymphocytes, producing IL-17, IL-21, IL-22, and Granulocyte-macrophage colony-stimulating factor (GM-CSF also known as CSF-2). Surprisingly, IFN-γ, IL-12, IL-17A, IL-17F, IL-21, and IL-22 have all been shown to be dispensable for the development of EAE [reviewed in (57); discussed here (58)]. However, in 2011, the CNS pathogenicity of TH17 cells was reported to be primarily associated with their production of GM-CSF (59, 60). GM-CSF production by T cells has been correlated with pathogenesis in several autoimmune diseases, including MS, rheumatoid arthritis, and myocarditis. It was reported that IL-1β- and IL-23-induced production of GM-CSF by CNS-infiltrating CD4+ T cells is essential for the induction of EAE (59, 60).
GM-CSF is a hematopoietic growth factor produced by a number of hematopoietic and non-hematopoietic cell types including activated CD4+ T cells, monocytes/macrophages, B cells, NK cells, endothelial cells and epithelial cells. GM-CSF has a wide array of functions, notably the survival and activation of myeloid cells, the ability to induce differentiation of dendritic cells (DCs), the polarization of macrophages toward a pro-inflammatory M1 phenotype, enhanced antigen presentation, the induction of complement- and antibody-mediated phagocytosis, and the mobilization of monocytes and other myeloid populations from bone marrow to blood (61–63).
The GM-CSF receptor (GM-CSF Rc) is a heterodimer comprised of a specific low-affinity α chain (CD116; GM-CSF Rα) and a common β chain (CD131; GM-CSF Rβ) that is shared by IL-3 and IL-5 (64). The GM-CSF Rc is expressed in multipotent myeloid progenitor cells and continues to be expressed throughout myeloid development on monocytes, DCs, macrophages and neutrophils (65–67). It is not expressed by T and B lymphocytes (67). Thus, most of the suspected direct effects of the GM-CSF in diseases are focused on myeloid cells (peripheral and CNS resident).
Findings related to the function of GM-CSF signaling in EAE pathology have been recently reviewed (68). In brief, in EAE, GM-CSF is necessary for disease as GM-CSF KOs were found to be resistant to disease induction (69). Disease can be rescued by the administration of recombinant GM-CSF. Adoptive transfer using cytokine-deficient mice showed that wild-type, IL-17A−/−, and IFNγ−/− T cells induced EAE with similar kinetics. By contrast, GM-CSF−/− T cells were incapable of inducing EAE and invading the CNS (59). Due to the variety of cells GM-CSF can stimulate, it became important to determine the cell population in which signaling was necessary for disease. A bone marrow chimera study determined that peripheral myeloid cells, but not microglia, are key responders (59). This corresponds with earlier observations that GM-CSF administration stimulated CD11b+ Ly6Chi inflammatory monocytes into the circulation (70). Circulating Ly6Chi monocytes traffic across the blood-brain barrier, up-regulate pro-inflammatory molecules, and differentiate into central nervous system DCs and macrophages (70). These data were confirmed recently using conditional gene targeting in which the β chain of the GM-CSF receptor (Csf2rb) was deleted in specific subpopulations throughout the myeloid lineages (71). It was found that deletion of Csf2rb in CCR2+Ly6Chi monocytes phenocopied the EAE resistance seen in complete Csf2rb-deficient mice.
In humans, GM-CSF levels in the CSF are higher in patients with active MS than in patients in remission (72). Also, untreated MS patients had significantly greater numbers of CD4+ GM-CSF+ T cells and CD8+ GM-CSF+ T cells in peripheral blood compared with healthy controls and with IFN-β-treated MS patients (73). In addition, IFN-β significantly suppressed GM-CSF production by T cells in vitro. More recently, the Canadian B cells in MS Team uncovered a subset of memory B cells producing GM-CSF (74). In vitro, GM-CSF–expressing B cells efficiently activated myeloid cells in a GM-CSF–dependent manner, and in vivo, B cell depletion therapy resulted in a GM-CSF–dependent decrease in pro-inflammatory myeloid responses of MS patients.
In light of the critical role of GM-CSF in the pathogenesis of MS and other inflammatory diseases, multiple tools have been developed targeting either the cytokine or the receptor. First tested in EAE, it has been shown that blocking antibodies against GM-CSF in chronic (C)-EAE (69) or antibodies against GM-CSF Rα in C-EAE and Relapsing-Remitting (RR)-EAE (75) were able to prevent disease if given at the time of EAE induction (day 0). Mice treated with anti-GM-CSF after disease onset completely recovered within 20 days of treatment in a model of C-EAE (69). Therapeutic treatment with anti-GM-CSF Rα ameliorated progression of C-EAE and resulted in a significant reduction of the relapse severity of RR-EAE (75). Blockade of the GM-CSF Rα led to a reduction of activated mDCs, and reduced pro-inflammatory cytokine production by CD11b+ Ly6C+ inflammatory monocytes. Additionally, anti-GM-CSF Rα altered the expression of chemokine receptors, leading to the possibility that antibody treatment may impede cell migration (75).
Logically, the next step is to test the therapeutic potential of GM-CSF targeting in humans. A review of tools developed for clinical trials can be found here (76). At this time, GM-CSF blocking antibodies have been tested in Rheumatoid Arthritis and have shown promising results. As for MS, only one drug has been tested in clinical trials: MOR-103 (also known as GSK3196165 or otilimab), a human antibody to GM-CSF. The results of a Phase Ib clinical trial employing MOR-103 in patients with relapsing-remitting or secondary-progressive MS have shown the drug to be safe and well-tolerated, although with modest efficacy (77). At this moment, there are no ongoing clinical trials targeting GM-CSF or GM-CSF receptor in MS.
Another important growth factor regulating myeloid cell function is macrophage colony-stimulating factor (M-CSF also known as CSF-1). M-CSF is ubiquitously produced in the steady state by a variety of cells, including endothelial cells, fibroblasts, osteoblasts, smooth muscle, and macrophages, and can be detected in plasma at ~10 ng/ml (78–80). The levels of circulating M-CSF are upregulated in pregnancy (81) as well as in many different pathologies including cancer, autoimmune diseases and chronic inflammation (82–86). M-CSF stimulates progenitor cells from bone marrow and plays an important regulatory role in the survival, proliferation (in mice), differentiation, phagocytosis, and chemotaxis of myeloid cells, including monocytes, macrophages, DCs, and microglia (87–89). The effects of M-CSF are mediated by signaling through the type III tyrosine kinase transmembrane receptor CSF-1R (CD115), which is encoded by the c-fms proto-oncogene (90). IL-34 is also able to bind CSF-1R with similar outcomes as to M-CSF binding (88). However, M-CSF and IL-34 present differences in their spatiotemporal expression patterns, and thus seem to play complementary roles in their biological activities on target cells (88, 91, 92). CSF-1R is expressed by myeloid cells such as monocytes, macrophages, DCs, and microglia, as well as by trophoblasts, neural progenitor cells and epithelial cells (93, 94).
There is ongoing debate about whether M-CSF is a pro-inflammatory or pro-repair cytokine. M-CSF seems to be essential for the survival and renewal of tissue-resident macrophages, but not for circulating myeloid cells. Indeed, in the osteopetrotic Csf1op/Csf1op mouse, which harbor an inactivating mutation in the coding region of the CSF-1 gene and are M-CSF deficient, the functions and numbers of several tissue macrophage populations are altered while there is no difference in monocyte populations in the blood (95). These findings were later confirmed in mice deficient for a specific enhancer for Csf-1r gene, the fms-intronic regulatory element (FIRE) (96). Csf1rΔFIRE/ΔFIRE mice present a deficit in tissue resident macrophages in the brain (microglia), skin, kidney, peritoneal, and heart without significant differences in blood monocytes. During inflammation, the presence of monocytes in inflamed tissue is critical for proper immune responses, notably due to their capacity to traffic to draining lymph nodes and their ability to present antigens to T cells (2, 97–103). While tissue resident macrophages also participate in inflammatory processes, their role in promoting tissue repair and regeneration is critical (104, 105). For example, M-CSF favors kidney and liver repair after acute injury (106–108). Moreover, M-CSF is used to drive human and in mouse macrophage differentiation in vitro into an anti-inflammatory (M2) phenotype (109–111). In EAE, it was shown that peritoneal APCs treated with M-CSF and pulsed with MOG35−55, the disease initiating peptide, were able to suppress ongoing EAE when injected at the time of disease initiation or significantly reduce the severity of the disease when injected at day 7 post-immunization (112). These M-CSF activated APCs were demonstrated to induce a Treg profile from CD4+ T cells (CD25+ FoxP3+) with increased secretion of IL-10 and decreased secretion of IL-17, IFN-γ, and TNF (112).
However, as mentioned earlier, elevated levels of M-CSF are also observed in different pathologies. There are multiple publications linking M-CSF/IL-34 and CSF-1R signaling in models of arthritis (113–116), diabetes (117), systemic lupus erythematosus (85, 118), cancer (119–121), amyotrophic lateral sclerosis (122), Parkinson's disease (123), and Alzheimer's disease (124–126). In an effort to determine the role of M-CSF/IL-34 and CSF-1R signaling in MS, different groups used potent c-fms tyrosine kinase inhibitors, which block M-CSF signaling. Ki20227 (127), imatinib (128), GW2580 (128, 129), sorafenid (128), and PLX5622 (130) are all tyrosine kinase inhibitors that have shown to effectively treat C-EAE. GW2580 has the greatest apparent specificity for CSF-1R vs. the other kinase inhibitors (131). Amelioration of EAE using Ki20227 was associated with the suppression of myeloid cell expansion in the spleen and reduction in MOG-specific T-cell proliferation (127). GW2580 and sorafenib suppressed TNF-α production by macrophages whereas imatinib and sorafenib both abrogated PDGF-induced proliferation of astrocytes (128). PLX5622 effect was associated with microglia and macrophage ablation from the white matter (130). However, in the cuprizone model of CNS demyelination, which allows study of the remyelination process with little involvement of the peripheral immune cells (132), injection of M-CSF reduced demyelination by boosting microglia activity (133). Tamoxifen-induced conditional deletion of the CSF-1R in microglia from cuprizone-fed mice caused aberrant myelin debris accumulation and reduced microglial phagocytic responses (89, 133). These data indicate that M-CSF plays an important role in ability of microglia to clear myelin debris and to support proper remyelination, and suggest M-CSF functions as a critical factor in tissue repair. These divergent results exemplify the various functions of M-CSF/IL-34 and CSF-1R signaling on cells. The possible contribution of M-CSF signaling to both inflammatory and repair processes suggest that targeting M-CSF in MS may be problematic. However, although there is an increase of myeloid cells in MS lesions, the expression of CSF-1R is lower in MS lesions when compared to normal appearing white matter (134). It is thus possible to hypothesize that a therapeutic treatment targeting M-CSF in MS would primarily target peripheral myeloid cells rather than those in the CNS.
There are now multiple tools targeting M-CSF signaling approved for human therapy, especially for cancer. Imatinib was the first tyrosine kinase inhibitor approved for the treatment of chronic myelogenous leukemia (135). Imitanib is also now in clinical trials for the treatment of different pathologies, such as rheumatoid arthritis, type I diabetes and asthma, for which positive results of a phase 2 clinical trial were recently published (136). Sorafenib is approved for the treatment of primary kidney cancer and advanced primary liver cancer (137). Although there are side effects related to these inhibitors, an important advantage of tyrosine kinase inhibitors is the fact they can be administered orally to the patients. In September 2019, a phase 3 clinical trial for RRMS was started testing the efficacy of evobrutinib, a Bruton's tyrosine kinase inhibitor. Although this is not a CSF-1R inhibitor, it shows: (1) the desire to develop oral treatments in MS, and (2) the possibility of targeting tyrosine kinases in MS. Bruton's tyrosine kinase are critical for B cell receptor signaling and is also involved in TLR signaling as well as inflammasome activation in myeloid cells (138)
Cytokines
As mentioned earlier, blockade of myeloid specific cytokines IL-1β, IL-12, and IL-23 have proven to be efficient therapies in multiple diseases such as Crohn's disease, ulcerative colitis, rheumatoid arthritis, psoriasis, and systemic juvenile idiopathic arthritis. These cytokines are all involved in CD4+ T lymphocytes differentiation. While IL-12 is critical for TH1 induction (139), IL-1β and IL-23 are both involved in TH17 differentiation and promote the encephalitogenic capacity of these cells by inducing GM-CSF expression (60, 140, 141). In EAE, mice lacking IL-1β, or the receptor, IL-1R, developed a milder disease than WT animals (140, 142–145). Moreover, specific ablation of IL-1R on CD4+ T cells resulted in significantly reduced disease severity (146), confirming the importance of IL-1β signaling on T cells to induce a full EAE. In addition, rats treated with an IL-1 receptor antagonist (IL-1Ra), which blocks the biological activity of IL-1β, developed milder signs of EAE compared to control animals (147). As IL-1β secretion is the result of inflammasome activation, mice treated with a blocking agent for the inflammasome component NLRP3 exhibited decreased EAE severity (148). In MS, it was shown that IL-1R expression is significantly higher in CD4+ T cells from RRMS patients than from healthy controls (149). IL-1β expression was also found to be significantly increased in MS lesions when compared to tissue from other neurological diseases (150). Interestingly, multiple treatments used in MS [e.g., IFN-β, glatiramer acetate, and natalizumab] have shown to increase IL-1Ra expression and/or to decrease IL-1β production (151, 152)]. Multiple tools have been developed to block IL-1β activity: the recombinant IL-1Ra Anakinra used for rheumatoid arthritis, the neutralizing IL-1β antibody Canakinumab used for systemic juvenile idiopathic arthritis as well as cryopyrin-associated periodic syndrome, and the soluble decoy IL-1 receptor (Rilonacept) also use for cryopyrin-associated periodic syndromes (153). At this time, Anakinra is the only IL-1β-targeting drug in clinical testing for MS. This Phase I/II clinical trial just started a few months ago, and at this time, it is still in the recruitment phase (NCT04025554).
IL-12 and IL-23 are heterodimeric cytokines that share a common subunit IL-12p40. The other subunit needed to form IL-12 is IL-12p35, while the other subunit to form IL-23 is IL-23p19. IL-12 signals through the IL-12 receptor (IL-12R) composed of the IL-12Rβ1 and IL-12Rβ2 subunits, while IL-23 signals through IL-23R and IL-12Rβ1 (154). Thus, IL-12Rβ1 is required for biological response to both IL-12 and IL-23. When specific gene ablation was tested for the different receptor chains of IL-12 and IL-23, it was found that IL-12Rβ1−/− mice were completely resistant to EAE (155). However, IL-12Rβ2−/− mice developed severe EAE, extensive inflammation and demyelination, and higher production of pro-inflammatory cytokines than WT animals (156). Finally, similar to IL-12Rβ1−/− mice, IL-23R−/− mice were completely resistant to EAE induction (157). As for the cytokines, mice deficient for the subunits IL-23p19 or IL-12p40 were resistant to EAE. By contrast, mice in which the subunit IL-12p35 was deleted were highly susceptible to EAE (158). In addition, treatment with anti-IL-12p40 antibodies inhibited both murine and primate models of EAE (159–161). Treatment with anti-IL-23p19 antibodies reduced the clinical severity and prevented relapsing EAE by inhibiting epitope spreading (162). These results led to the conclusion that IL-23 was a more critical factor than IL-12 in the inflammatory response observed in EAE. Nevertheless, there are multiple studies linking both cytokines to MS pathology. It was demonstrated that peripheral blood monocytes from progressive MS patients produced increased amounts of IL-12 compared to controls and that IL-12 production correlated with disease activity (163). Another study showed an augmented level of IL-12 mRNA-expressing cells in the peripheral blood and the CSF of MS patients when compared to controls (164). There was also elevated levels of IL-12p70 detected in plasma from MS patients compared to healthy individuals. A more recent report showed that both RRMS and secondary progressive MS patients had increased levels of IL-12p40 mRNA compared with controls during the development of active lesions (165). IL-12p40 and IL-23p19 have also been detected in human MS lesions (166, 167). Based on this and other data, there was hope that Ustekinumab, an IL-12p40 neutralizing antibody, would be efficacious for treatment of MS. However, disappointingly no clinical improvement in the treatment group compared to the placebo was found (168). Possible reasons for the failure of Ustekinumab are the broad range of MS patients in the trial, many having very severe symptoms and long-standing disease. Also, there may be weak bioavailability of the drug as Ustekinumab may be inefficient in crossing the BBB (169). At this time there are no ongoing trials targeting IL-12/IL-23 in MS despite the impressive results in the various animal models of the disease.
Targeting Biological Functions of Myeloid Cells
MicroRNAs (miRNAs) are small non-coding RNAs of 17–25 nucleotides that regulate gene expression by inducing mRNA degradation or by interfering with translational machinery of mRNAs (170). It is predicted that more than 60% of protein-coding genes are regulated by miRNAs (171). They are key regulators of various biological processes including immune cell lineage commitment, differentiation, maturation, and maintenance of immune homeostasis and normal function [reviewed in (60)]. Extensive evidence demonstrates that miRNAs play crucial roles in the development, differentiation, and function of different immune cells, such as B and T lymphocytes, DCs and macrophages (172–175). In the last few years, miRNAs have drawn a lot of interest due to their involvement in the pathogenesis of cancer, inflammatory and autoimmune diseases [reviewed in (176)].
In MS patients, expression studies using whole blood (177), PBMCs (178), as well as brain sections (179) identified multiple deregulated miRNAs. Of these miRNAs, three were consistently upregulated across multiple studies and directly affecting myeloid cell functions: miR-223, miR-155 and miR-146a. miR-223 is induced by the myeloid transcription factors PU.1 and CCAAT/enhancer-binding protein-β (C/EBPβ) (180). miR-223 expression is mainly confined to myeloid cells and is induced during the lineage differentiation of myeloid progenitor cells. It was shown to negatively regulate both the proliferation and activation of neutrophils (181). Moreover, miR-223−/− macrophages exhibited enhanced pro-inflammatory M1, but decreased regulatory M2 responses (182). It was later described that miR-223 is required for efficient M2-associated phenotype and function (183). Moreover, a low functional level of the miR-223 is essential for monocyte differentiation. In MS patients, miR-223 was found significantly increased in blood, PBMCs and active MS lesions compared with control subjects (177, 179). During EAE development, the expression level of miR-223 is dramatically increased in myeloid cell populations, but not in other cell types, and was maintained at comparable levels between disease onset and peak of disease (184). Surprisingly, although miR-223 expression is associated with M2 macrophages and microglia (183), it was shown that miR-223 KO mice present a milder course of EAE than WT mice (184–186). Reduced disease severity was also observed in adoptive transfer EAE induced by transfer WT T lymphocytes into miR-223 KO recipient mice compared to transfer into WT recipient mice, demonstrating the importance of miR-223 on the APCs side rather than on the T cells side (184). Our group demonstrated that while M1-like macrophages were upregulated in KO mice, DCs showed a reduced inflammatory profile characterized by increased PD-L1 expression and decreased expression of IL-1β, IL-6, and IL-23, all cytokines involved in differentiating and sustaining a TH17 profile (184). Significantly, APCs from miR-223 KO mice have a comparable ability to drive TH1 cells, but possess a reduced capacity to drive TH17 cells (184). Moreover, it was shown that monocytic-myeloid-derived suppressor cells (MO-MDSCs) isolated from miR-223−/− suppressed T cell proliferation and cytokine production in vitro and regulated EAE more efficiently than MO-MDSCs derived from WT animals (186). The enhanced suppressive function of miR-223−/− MO-MDSCs was associated with higher expression of Arg1 and Stat3, which are miR-223 target genes (186). Interestingly, Stat3 controls the expression of PD-L1 on APCs (187), consistent with the previous observation of PD-L1 upregulation on DCs in miR-223−/− animals. Although these results point to miR-223 as a potential therapeutic target in MS, it is important to note that in a model of lysolecithin-induced demyelination, the absence of miR-223 was demonstrated to lead to impaired CNS remyelination and myelin debris clearance (183). The impaired capacity of M2 polarization by macrophages and microglia is likely a significant factor contributing to the decreased remyelination capacity in miR-223 KO mice. In particular, microglia adopting an M2 profile are critical for proper remyelination (188, 189). Thus, when targeting miR-223 in MS, it is important to keep in mind the different implications of such therapy.
miR-155 has drawn a lot of attention for its possible role in MS as detailed in a recent review (190). miR-155 has been shown to be upregulated in active MS lesions (179) as well as in CD14+ monocytes isolated from the blood of RR-MS patients compared to control donors (191). While miR-223 expression is limited to myeloid cells, multiple immune cell populations express miR-155 such as B cells, T cells, macrophages and DCs (192). miR-155 is found at low levels in both myeloid and lymphoid cells, but its expression is upregulated following cellular activation via antigen, Toll-like Receptor (TLR) ligands, and inflammatory cytokines. An important target of miR-155 is Src homology 2 (SH2)-domain containing inositol-5′-phosphatase 1 (SHIP-1) (193). SHIP-1 is an enzyme that inhibits phosphoinositide 3-kinase (PI3K) activity, which governs cellular responses to multiple stimuli, cell proliferation and cell survival (194). Thus, it is believed that miR-155 dysregulation would have critical consequences. Indeed, forced expression of miR-155 in hematopoietic stem cells by a retroviral vector leads to severe splenomegaly as well as increased myeloid cell populations in the bone marrow and in circulation (195). In addition, it has been reported that in absence of miR-155, mice displayed altered immune responses to infectious agents, due to defective functions of B cells, T cells, and DCs (196). Focusing on myeloid populations, it was shown that DCs lacking miR-155 are less competent at inducing antigen-specific T cell activation (196). More recently, it was demonstrated that overexpression of miR-155 in DCs is a critical event that is alone sufficient to break self-tolerance in an animal model of diabetes, and promote a CD8-mediated autoimmune response in vivo (197). Human CD14+ monocytes and macrophages overexpressing miR-155 exhibit increased production of pro-inflammatory cytokines, including IL-1β, IL-6, and TNF, and decreased production of the anti-inflammatory cytokine IL-10 (198).
miR-155-deficient mice display a delayed course and reduced severity of clinical symptoms of EAE (199, 200). Decreased disease severity in miR-155−/− mice was associated with reduced TH1 and TH17 responses. In addition to the direct effect on T cells, it was also shown that the decreased ability of miR-155 KO mice to mount inflammatory T cell responses was linked to DCs secreting less cytokines critical for driving TH1 and TH17 responses, mainly IL-1β, IL-6, IL-12, IL-23, and TNF (199). miR-155 is induced in macrophages and DCs after exposure to a variety of inflammatory cytokines such as IFN-β, IFN-γ, and TNF-α (199, 201). It is thus possible to speculate that following the first wave of inflammation, these myeloid APCs upregulate miR-155 leading to an accentuation of the inflammatory response. In addition, more recently, it has been demonstrated that miR-155 plays an essential role in driving the inflammatory phenotype of M1 macrophages (202), which would also impact the severity of the disease. Lastly, treatment with a miR-155 inhibitor after EAE onset reduced the clinical disease severity (199). Considering the important role of miR-155 in driving inflammatory responses in general, and specifically in myeloid APCs, fine tuning the expression of this miRNA in MS would most certainly prompt beneficial results in terms of slowing the inflammatory loop. It is noteworthy that miR-155 is the most consistent miRNA found to be upregulated in MS being reported in eight independent studies (203).
A third miRNA that has been shown to regulate myeloid cell activation is miR-146a. Like miR-155, miR-146a is upregulated following cell stimulation and its induction is NF-κB dependent (204). However, contrary to miR-223 and miR-155, miR-146a represses inflammatory responses by targeting two adapter proteins, TNF receptor–associated factor 6 (TRAF6) and IL-1 receptor-associated kinase 1 (IRAK1), that are crucial for pro-inflammatory signaling (204). miR-146a KO mice develop a spontaneous autoimmune disorder, characterized by splenomegaly, lymphadenopathy, and multiorgan inflammation (205, 206). In addition, miR-146a KO mice display excessive production of myeloid cells and develop flank tumors in their secondary lymphoid organs. Consistent with the repression of inflammation, miR-146a expression promotes M2-Macrophage polarization by targeting Notch-1 (207). Multiple studies have indicated that miR-146a plays pivotal roles in the pathogenesis of several autoimmune diseases, such as systemic lupus erythematosus, rheumatoid arthritis, and Sjögren's syndrome (208). In MS, miR-146a is upregulated in active lesions (179), as well as in PBMCs of RRMS patients (209, 210). Expression of miR-146a is reported to be significantly downregulated in glatiramer acetate treated RRMS patients (210). Logically, upregulation of this miRNA would seem to be beneficial in reducing the ongoing inflammation observed in MS patients leading to the possibility that the upregulation observed in MS patients is the result of the ongoing inflammation rather than a pathological expression. However, when studied in animal models of MS, there was no consensus on the suppressive effects of miR-146a. One study using the Cuprizone-induced demyelination model found that miR-146a-deficient mice displayed reduced inflammatory responses, demyelination, axonal loss, and numbers of infiltrating macrophages compared to WT controls (211). However, a second study found that miR-146a-deficient mice developed more severe EAE characterized by exaggerated TH17 responses (212), going along the possible beneficial effect of upregulation of this miRNA in MS. More recently, it was shown that miR-146a mimic treatment of mice with RR-EAE at day 14 improved neurological function, increased the number of newly generated oligodendrocytes, which may facilitate remyelination in the CNS (213). In addition, the treatment increased the number of regulatory M2 macrophages while reducing the number of pro-inflammatory M1 macrophages (213).
Currently, targeting miRNAs is a challenge since they control a myriad of immune and non-immune related functions. However, there is a strong interest in pursuing this approach, not only in MS, but also in many different diseases. Identification of technology to target miRNAs in a cell specific manner would appear to be the desired way to safely and effectively employ this targeting strategy. In the meantime, an abundance of researchers are also exploring the use of miRNAs as biomarkers of diseases pathogenesis and therapy.
Targeting Immunological Activity of Myeloid Cells
The final strategy we will discuss is the use of nanoparticles to target myeloid cells for disease therapy which has been pioneered in our laboratory. In the recent past, many studies have focused on characterizing the ability of nanoparticles to modulate immune responses and ultimately to be used as potential therapeutics for immune-related diseases. Here we will focus on the “carboxylated” poly(lactic-co-glycolic acid) (PLGA) nanoparticles, which are particles without any protein or peptide attached to the surface or encapsulated inside. Phagocytic cells have the extraordinary ability to engulf dead cells, invading microbes and other particles, and this property of phagocyte cells led to the idea of using carriers such as apoptotic cells (214–216), liposomes (217), extracellular vesicles (218), or nanoparticles (219–223) to deliver molecules to modify the immune response. We will restrict our discussion to the use carboxylated PLGA nanoparticles for the modulation of inflammatory monocytes for treatment of CNS inflammation for multiple reasons. Firstly, they can be easily manufactured under GMP conditions. Secondly, they more specifically target inflammatory monocytes by their affinity of binding via the macrophage receptor of collagenous structure (MARCO) (224) as compared to liposomes and extracellular vesicles. Thirdly, they directly carry out immune-modulatory effects on monocytes without the need for add-on agents such as would be required with liposomes. Lastly, they have been proven to be safe and efficacious for use in celiac disease patients treated via intravenous infusion of gliadin encapsulating PLGA nanoparticles for induction of immune tolerance in a phase 1/2a clinical trial (225).
Nanoparticles have diameters between 1 and 1,500 nm. Smaller particles (<100 nm) are able to cross tissue barriers and traffic directly to the lymph nodes. Larger particles (>100 nm) require uptake by phagocytic cells (226). Nanoparticles administered subcutaneously or intradermally may be taken up by tissue resident APCs or their precursor cells and are ultimately transported to the draining lymph nodes. Systemic administration of nanoparticles favors accumulation in the organs such as the spleen and liver (227). Also, the shape of nanoparticles dictates efficiency of uptake by phagocyte cells. For example, phagocyte cells internalize spherical-shaped nanoparticles more easily than stretched-shaped structures (228). And although positively charged particles are taken up more avidly, negatively charged particles have been shown to exhibit lower toxicity (229–232). Nanoparticles can be made from different materials, metallic (e.g., silver, gold, and copper), magnetic (e.g., iron) (useful for imaging), ceramic, carbon-based, silica, lipid-based, or polymeric such as poly(amino acids), polysaccharides and poly(alpha-hydroxy acids).
Our group was one of the first to test the ability of 500 nm non-biodegradable carboxylated polystyrene (PS) particles to modulate immune responses in inflammatory settings in vivo. Intravenous infusion of PS nanoparticles led to a reduction in trafficking of Ly6Chi inflammatory monocyte into the CNS and increased survival in a mouse model of West Nile virus (WNV) encephalitis (224). It was discovered that these inflammatory monocytes were redirected to the spleen of treated animals and resulted in a dramatic reduction of mortality in WNV-infected mice by preventing the release of a pro-inflammatory “cytokine storm” in the CNS. Robust anti-inflammatory effects induced by infusion of PS nanoparticles were also observed in other inflammatory diseases such as peritoneal inflammation and inflammatory bowel disease. To enhance the clinical relevance of the nanoparticle targeting approach, we next investigated the potential of biodegradable carboxylated PLGA nanoparticles for regulation of myeloid cell-dependent inflammation.
PLGA is one of the best characterized and most used biodegradable polymers. The hydrolysis of PLGA leads to metabolite monomers, lactic acid and glycolic acid. The two monomers are endogenous and easily metabolized by the body via the Krebs cycle. There is minimal systemic toxicity associated with the use of PLGA (233, 234). Because PLGA is a safe material, it has been approved by the United States Food and Drug Administration (FDA) and European Medicine Agency (EMA) in various drug delivery systems in humans. Indeed, PLGA can be engineered to deliver, alone or in any combination with small-molecule drugs, proteins, peptides, DNA, miRNAs, and even clustered regularly interspaced short palindromic repeat (CRISPR) (227).
We have shown that administration of negatively charged 500 nm PLGA nanoparticles resulted in reduced inflammatory monocytes accumulation and overall robust beneficial effects in disease severity in multiple mouse models of inflammatory disease such as EAE (224), SCI (235), TBI (236), myocardial infarction (237), and herpes simplex virus 1 infection of the cornea (238). The exact mechanisms behind immunomodulatory effects of PLGA therapy are still under investigation. However, in all these models, it has been shown PLGA particles are selectively recognized and bound by inflammatory monocytes. These monocytes undergo sequestration and eventual apoptosis in the spleen, culminating in reduced immune pathology at sites of inflammation. Phenotypic changes were also observed on DCs and macrophages in the inflammatory sites, showing decreased expression of activation markers such as MHC II and CD86. In the SCI study, PLGA nanoparticle administration led to reduced M1 macrophage polarization.
While our group has also shown that antigen (Ag)-coupled or encapsulated PLGA nanoparticles can have important immunomodulatory effects (220, 222, 224, 239), other strategies using PLGA nanoparticles have also been shown to regulate EAE (240). For example, Cappellano et al. showed that simultaneous subcutaneous injection of PLGA nanoparticles loaded with either MOG35−55 or IL-10 ameliorated the course of EAE (241). TGF-β, another immunoregulatory molecule, coupled to the surface of PLGA nanaopartlces containing PLP139−151 peptide were shown to improve the tolerogenic effect of Ag-PLGA nanoparticles (242). Another example is Maldonaldo et al. using PLGA nanoparticles loaded PLP139−151 together with rapamycin, an inhibitor of the mTOR pathway, and demonstrating that a single dose of these particles injected at the peak of disease were able to protect from relapses (243). Also, Pei et al. aimed to develop PLGA nanoparticles which function as a direct modulator of T cells, without the involvement of APCs (244). For that purpose, TGF-β1 encapsulated nanoparticles were coupled with target antigens for CD4 and CD8 T cells (MOG40−54/H-2Db-Ig dimer and MOG35−55/I-Ab multimer), regulatory molecules (anti-Fas and PD-L1-Fc) and a “self-marker” CD47-Fc (244). These particles were injected in EAE mice on day 8, 18, 28, and 38 after immunization with MOG35−55, and induced a significant reduction in EAE symptoms that lasted for more than 100 days. Moreover, the authors observed a decrease of TH1 and TH17 MOG35−55-specific cells as well as TC1 and TC17 MOG40−55-specific cells, an increase of regulatory T cells, inhibition of T cell proliferation and augmentation of T cell apoptosis in the spleen (244). In addition to regulating the immune response, PLGA nanoparticles have also been used as a transporter to help in the remyelination process. Indeed, Rittchen et al. encapsulated leukemia inhibitory factor (LIF), which is a cytokine known to promote oligodendrocyte maturation thus favoring remyelination (245). To specifically target oligodendrocytes, the LIF-PLGA nanoparticles were coupled with anti-NG2 antibodies. The authors showed that intra-lesion delivery of LIF-PLGA nanoparticles improved CNS remyelination increasing the percentage of remyelinated axons and their thickness (245).
In conclusion, the mechanism(s) of action of PLGA nanoparticles are still incompletely understood, but studies in multiple models have shown their capacity to limit inflammatory events by targeting inflammatory monocytes. PLGA nanoparticles can also be used as delivery vectors, like liposomes and extracellular vesicles. However, a critical advantage of carboxylated PLGA nanoparticles, as compared to liposomes and extracellular vesicles, is their ability to act directly to modulate the function and trafficking of inflammatory monocytes based on their ability to engage the MARCO scavenger receptor. Because of the safety record of PLGA nanoparticles, they can be easily translated into clinical use. In fact, Cour Pharmaceuticals successfully completed a Phase IIa clinical trial for celiac disease showing the safety and efficacy of systemic infusion of PLGA nanoparticles encapsulating gliadin for inducing gluten-specific immune tolerance in celiac disease patients undergoing oral gluten challenge. Takeda Pharmaceuticals has acquired the exclusive license for future development of this therapy for celiac and other GI diseases. Cour Pharmaceuticals is currently developing antigen encapsulating PLGA nanoparticle-based tolerance clinical programs for treatment of MS and peanut allergy and clinical programs using carboxylated “naked” PLGA nanoparticles targeting inflammatory monocytes for treatment of acute respiratory distress in COVID-19 infection and treatment of TBI.
Conclusions
The importance of peripheral myeloid cells in MS pathology is profound. There is an extensive presence of these cells and their products in MS lesions as well as in the CSF of MS patients. Studies in animal models of MS have clearly demonstrated the beneficial effects in targeting peripheral myeloid cells for the different forms of the disease. Multiple tools have now been developed targeting these cells including blockade of their migration to the CNS, their activation and cytokine production, their biological functions and their immunological activity (Figure 1). However, contrary to other inflammatory disorders, no drug is currently approved targeting specifically these cells in MS. Multiple pro-inflammatory cytokines including GM-CSF, IL-1β, IL-12, IL-23, M-CSF all represent potential MS therapeutic targets. Treatments targeting these cytokines have been shown to be well-tolerated and safe in patients for different diseases. Additionally, non-specific blockade of leukocyte entry to CNS using Natalizumab is beneficial in MS, however this carries the risk of severe side-effects from infections. However, specifically impeding the migration of myeloid cells would limit such adverse effects. CLEC12A, CCR1, CCR2, JAM-L, and Ninjurin-1 represent interesting options to inhibit CNS migration of peripheral myeloid cells. Altering the biological functions of myeloid cells via through miRNA modulation is an appealing strategy for treating MS and other chronic inflammatory diseases. miR-146a, miR-155, and miR-223 are all upregulated on myeloid cells from MS patients. Lastly, nanoparticles represent one of the most exciting new tools for regulating myeloid cell functions. The biodegradable PLGA particles are particularly interesting due to their approval by the FDA and EMA for use in humans, as well as their ability to regulate many different inflammatory disorders, even those that take place in the CNS.
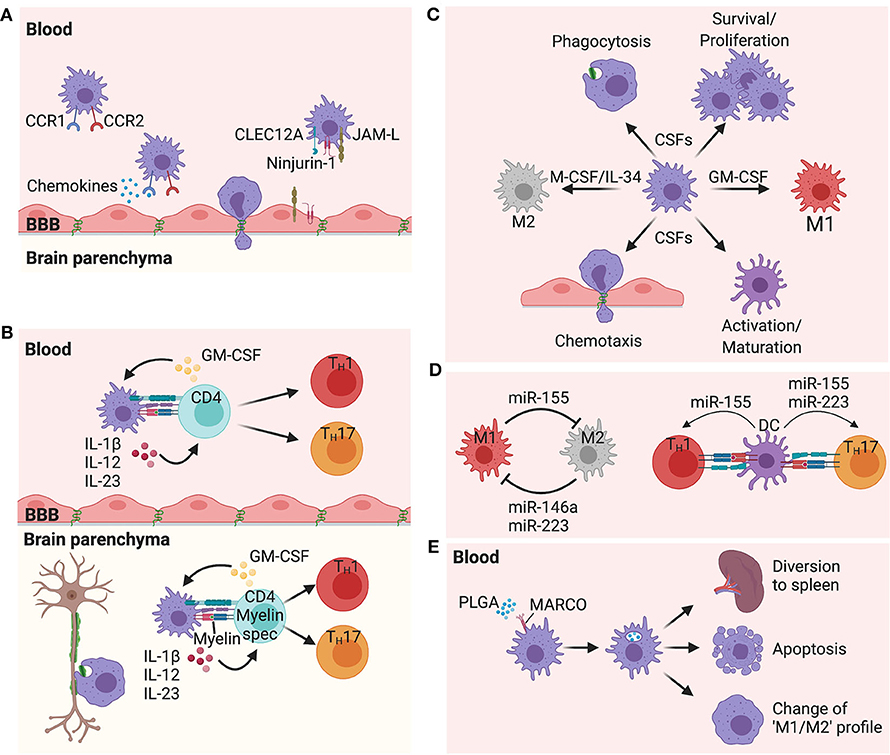
Figure 1. Proposed mechanisms of peripheral myeloid cells in multiple sclerosis pathogenesis. (A) Chemokine receptors CCR1 and CCR2, as well as the adhesion molecules Ninjurin-1 and JAM-L, and the C-lectin receptor CLEC12A have all been shown to play important roles in myeloid cell migration from the periphery to the CNS. (B) Interactions between myeloid cells and CD4 T cells are critical to shape the type of immune response. These interactions take place in the periphery as well as in the CNS. Cytokines such as IL-1β, IL-12, and IL-23 produced by myeloid cells and GM-CSF produced by activated CD4 T cells are all attractive targets for MS therapy, playing critical roles in the CNS inflammation. (C) M-CSF (CSF-1)/IL-34 and GM-CSF (CSF-2) are upregulated during inflammation and control many different functions on myeloid cells such as differentiation, phagocytosis, chemotaxis, activation, polarization, and survival. Blockade of the receptors of these growth factors are intriguing therapeutic options in MS. (D) miRNAs have the ability to regulate the function of many cells, including myeloid cells. miR-223,−155, and−146a have all shown to be upregulated in MS lesions, and are expressed by myeloid cells. While miR-146a and miR-223 promote an M2 profile, miR-155 promotes an M1 profile from macrophages/microglia. However, expression of miR-223 and miR-155 by DCs induce the ability of these cells to promote a TH17 polarization. miR-155 also has the ability to promote a TH1 polarization when expressed by DCs. (E) Intravenous injection of PLGA nanoparticles are capture by MARCO+ myeloid cells leading these cells to be sequestered in the spleen, with some cells dying by apoptosis and other cells changing their profile toward an immunoregulatory phenotype.
Author Contributions
II and SM provided intellectual contribution to the work. II wrote the manuscript. SM provided guidance, edited, and reviewed the manuscript. Both authors approved the manuscript for publication.
Funding
This work was supported in part by grants from the NIH (EB-013198 and NS-026543), Cour Pharmaceutical Development Company, the Johnnie Walker's MS Foundation, the David & Amy Fulton Foundation, the Crammer Family Foundation, and the National Multiple Sclerosis Society (RG 4952-A-5). Research support was also provided by a National Multiple Sclerosis Society Post-Doctoral Fellowship (G-1508-05965) and a National Multiple Sclerosis Society Pilot Research Grant (PP-1905-33998).
Conflict of Interest
SM is a cofounder of Cour Pharmaceutical Development Co and also a member of the scientific advisory board and consultant for Cour Pharmaceutical Development Co as well as a shareholder.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wingerchuk DM, Lucchinetti CF, Noseworthy JH. Multiple sclerosis: current pathophysiological concepts. Lab Invest. (2001) 81:263–81. doi: 10.1038/labinvest.3780235
2. Ajami B, Bennett JL, Krieger C, Mcnagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. (2011) 14:1142–9. doi: 10.1038/nn.2887
3. Moline-Velazquez V, Vila-Del Sol V, De Castro F, Clemente D. Myeloid cell distribution and activity in multiple sclerosis. Histol Histopathol. (2016) 31:357–70. doi: 10.14670/HH-11-699
4. Evans TA, Barkauskas DS, Myers JT, Hare EG, You JQ, Ransohoff RM, et al. High-resolution intravital imaging reveals that blood-derived macrophages but not resident microglia facilitate secondary axonal dieback in traumatic spinal cord injury. Exp Neurol. (2014) 254:109–20. doi: 10.1016/j.expneurol.2014.01.013
5. Bramlett HM, Dietrich WD. Long-term consequences of traumatic brain injury: current status of potential mechanisms of injury and neurological outcomes. J Neurotrauma. (2015) 32:1834–48. doi: 10.1089/neu.2014.3352
6. Gensel JC, Zhang B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. (2015) 1619:1–11. doi: 10.1016/j.brainres.2014.12.045
7. Herz J, Filiano AJ, Smith A, Yogev N, Kipnis J. Myeloid cells in the central nervous system. Immunity. (2017) 46:943–56. doi: 10.1016/j.immuni.2017.06.007
8. Prinz M, Erny D, Hagemeyer N. Ontogeny and homeostasis of CNS myeloid cells. Nat Immunol. (2017) 18:385–92. doi: 10.1038/ni.3703
9. Waisman A, Lukas D, Clausen BE, Yogev N. Dendritic cells as gatekeepers of tolerance. Semin Immunopathol. (2017) 39:153–63. doi: 10.1007/s00281-016-0583-z
10. Lucchinetti CF, Bruck W, Rodriguez M, Lassmann H. Distinct patterns of multiple sclerosis pathology indicates heterogeneity on pathogenesis. Brain Pathol. (1996) 6:259–74. doi: 10.1111/j.1750-3639.1996.tb00854.x
11. Lassmann H, Bruck W, Lucchinetti C. Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends Mol Med. (2001) 7:115–21. doi: 10.1016/S1471-4914(00)01909-2
12. Lassmann H. Multiple sclerosis pathology: evolution of pathogenetic concepts. Brain Pathol. (2005) 15:217–22. doi: 10.1111/j.1750-3639.2005.tb00523.x
13. Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. (2005) 11:328–34. doi: 10.1038/nm1197
14. Bailey SL, Schreiner B, Mcmahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides 'preferentially' polarize CD4+ T(H)-17 cells in relapsing EAE. Nat Immunol. (2007) 8:172–80. doi: 10.1038/ni1430
15. Ifergan I, Kebir H, Bernard M, Wosik K, Dodelet-Devillers A, Cayrol R, et al. The blood-brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain. (2008) 131:785–99. doi: 10.1093/brain/awm295
16. Codarri L, Greter M, Becher B. Communication between pathogenic T cells and myeloid cells in neuroinflammatory disease. Trends Immunol. (2013) 34:114–9. doi: 10.1016/j.it.2012.09.007
17. Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron. (2018) 97:742–68. doi: 10.1016/j.neuron.2018.01.021
18. Mishra MK, Yong VW. Myeloid cells—targets of medication in multiple sclerosis. Nat Rev Neurol. (2016) 12:539–51. doi: 10.1038/nrneurol.2016.110
19. Armstrong AW, Read C. Pathophysiology, clinical presentation, and treatment of psoriasis: a review. JAMA. (2020) 323:1945–60. doi: 10.1001/jama.2020.4006
20. Kashani A, Schwartz DA. The expanding role of anti-IL-12 and/or anti-IL-23 antibodies in the treatment of inflammatory bowel disease. Gastroenterol Hepatol. (2019) 15:255–65.
21. Nuki G, Bresnihan B, Bear MB, Mccabe D, European Group of Clinical I. Long-term safety and maintenance of clinical improvement following treatment with anakinra (recombinant human interleukin-1 receptor antagonist) in patients with rheumatoid arthritis: extension phase of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. (2002) 46:2838–46. doi: 10.1002/art.10578
22. Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat N, Horneff G, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. (2012) 367:2396–406. doi: 10.1056/NEJMoa1205099
23. Biber K, Moller T, Boddeke E, Prinz M. Central nervous system myeloid cells as drug targets: current status and translational challenges. Nat Rev Drug Discov. (2016) 15:110–24. doi: 10.1038/nrd.2015.14
24. Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. (2005) 23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707
25. Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. (1994) 76:301–14. doi: 10.1016/0092-8674(94)90337-9
26. Greenwood J, Wang Y, Calder VL. Lymphocyte adhesion and transendothelial migration in the central nervous system: the role of LFA-1, ICAM-1, VLA-4 and VCAM-1. Immunology. (1995) 86:408–15.
27. Wong D, Prameya R, Dorovini-Zis K. In vitro adhesion and migration of T lymphocytes across monolayers of human brain microvessel endothelial cells: regulation by ICAM-1, VCAM-1, E-selectin and PECAM-1. J Neuropathol Exp Neurol. (1999) 58:138–52. doi: 10.1097/00005072-199902000-00004
28. Biernacki K, Prat A, Blain M, Antel JP. Regulation of Th1 and Th2 lymphocyte migration by human adult brain endothelial cells. J Neuropathol Exp Neurol. (2001) 60:1127–36. doi: 10.1093/jnen/60.12.1127
29. Prat A, Biernacki K, Lavoie JF, Poirier J, Duquette P, Antel JP. Migration of multiple sclerosis lymphocytes through brain endothelium. Arch Neurol. (2002) 59:391–7. doi: 10.1001/archneur.59.3.391
30. Cayrol R, Wosik K, Berard JL, Dodelet-Devillers A, Ifergan I, Kebir H, et al. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol. (2008) 9:137–45. doi: 10.1038/ni1551
31. Larochelle C, Cayrol R, Kebir H, Alvarez JI, Lecuyer MA, Ifergan I, et al. Melanoma cell adhesion molecule identifies encephalitogenic T lymphocytes and promotes their recruitment to the central nervous system. Brain. (2012) 135:2906–24. doi: 10.1093/brain/aws212
32. Miller DH, Khan OA, Sheremata WA, Blumhardt LD, Rice GP, Libonati MA, et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. (2003) 348:15–23. doi: 10.1056/NEJMoa020696
33. Ifergan I, Kebir H, Terouz S, Alvarez JI, Lecuyer MA, Gendron S, et al. Role of Ninjurin-1 in the migration of myeloid cells to central nervous system inflammatory lesions. Ann Neurol. (2011) 70:751–63. doi: 10.1002/ana.22519
34. Araki T, Zimonjic DB, Popescu NC, Milbrandt J. Mechanism of homophilic binding mediated by ninjurin, a novel widely expressed adhesion molecule. J Biol Chem. (1997) 272:21373–80. doi: 10.1074/jbc.272.34.21373
35. Arcangeli ML, Frontera V, Aurrand-Lions M. Function of junctional adhesion molecules (JAMs) in leukocyte migration and homeostasis. Arch Immunol Ther Exp. (2013) 61:15–23. doi: 10.1007/s00005-012-0199-5
36. Moog-Lutz C, Cave-Riant F, Guibal FC, Breau MA, Di Gioia Y, Couraud PO, et al. JAML, a novel protein with characteristics of a junctional adhesion molecule, is induced during differentiation of myeloid leukemia cells. Blood. (2003) 102:3371–8. doi: 10.1182/blood-2002-11-3462
37. Alvarez JI, Kebir H, Cheslow L, Charabati M, Chabarati M, Larochelle C, et al. JAML mediates monocyte and CD8 T cell migration across the brain endothelium. Ann Clin Transl Neurol. (2015) 2:1032–7. doi: 10.1002/acn3.255
38. Sadeghian-Rizi T, Khanahmad H, Jahanian-Najafabadi A. Therapeutic targeting of chemokines and chemokine receptors in multiple sclerosis: opportunities and challenges. CNS Neurol Disord Drug Targets. (2018) 17:496–508. doi: 10.2174/1871527317666180713111100
39. Bonecchi R, Bianchi G, Bordignon PP, D'ambrosio D, Lang R, Borsatti A, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med. (1998) 187:129–34. doi: 10.1084/jem.187.1.129
40. Sorensen TL, Sellebjerg F. Distinct chemokine receptor and cytokine expression profile in secondary progressive MS. Neurology. (2001) 57:1371–6. doi: 10.1212/WNL.57.8.1371
41. Weber C, Weber KS, Klier C, Gu S, Wank R, Horuk R, et al. Specialized roles of the chemokine receptors CCR1 and CCR5 in the recruitment of monocytes and T(H)1-like/CD45RO(+) T cells. Blood. (2001) 97:1144–6. doi: 10.1182/blood.V97.4.1144
42. Sorensen TL, Ransohoff RM, Strieter RM, Sellebjerg F. Chemokine CCL2 and chemokine receptor CCR2 in early active multiple sclerosis. Eur J Neurol. (2004) 11:445–9. doi: 10.1111/j.1468-1331.2004.00796.x
43. Rottman JB, Slavin AJ, Silva R, Weiner HL, Gerard CG, Hancock WW. Leukocyte recruitment during onset of experimental allergic encephalomyelitis is CCR1 dependent. Eur J Immunol. (2000) 30:2372–7. doi: 10.1002/1521-4141(2000)30:82372::AID-IMMU23723.0.CO
44. Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med. (2000) 192:899–905. doi: 10.1084/jem.192.6.899
45. Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. (2000) 192:1075–80. doi: 10.1084/jem.192.7.1075
46. Mildner A, Mack M, Schmidt H, Bruck W, Djukic M, Zabel MD, et al. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain. (2009) 132:2487–500. doi: 10.1093/brain/awp144
47. Trebst C, Sorensen TL, Kivisakk P, Cathcart MK, Hesselgesser J, Horuk R, et al. CCR1+/CCR5+ mononuclear phagocytes accumulate in the central nervous system of patients with multiple sclerosis. Am J Pathol. (2001) 159:1701–10. doi: 10.1016/S0002-9440(10)63017-9
48. Prins M, Dutta R, Baselmans B, Breve JJ, Bol JG, Deckard SA, et al. Discrepancy in CCL2 and CCR2 expression in white versus grey matter hippocampal lesions of Multiple Sclerosis patients. Acta Neuropathol Commun. (2014) 2:98. doi: 10.1186/s40478-014-0098-6
49. Tschammer N. Chemokines: chemokines and their receptors in drug discovery. In: Topics in Medicinal Chemistry. Cham: Springer. (2015). p. 14. doi: 10.1007/978-3-319-14060-5
50. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
51. Sharon N, Lis H. History of lectins: from hemagglutinins to biological recognition molecules. Glycobiology. (2004) 14:53R−62. doi: 10.1093/glycob/cwh122
52. Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. (2003) 3:569–81. doi: 10.1038/nri1130
53. Sagar D, Singh NP, Ginwala R, Huang X, Philip R, Nagarkatti M, et al. Antibody blockade of CLEC12A delays EAE onset and attenuates disease severity by impairing myeloid cell CNS infiltration and restoring positive immunity. Sci Rep. (2017) 7:2707. doi: 10.1038/s41598-017-03027-x
54. Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, et al. Maintenance infliximab for Crohn's disease: the ACCENT I randomised trial. Lancet. (2002) 359:1541–9. doi: 10.1016/S0140-6736(02)08512-4
55. Atzeni F, Benucci M, Salli S, Bongiovanni S, Boccassini L, Sarzi-Puttini P. Different effects of biological drugs in rheumatoid arthritis. Autoimmun Rev. (2013) 12:575–9. doi: 10.1016/j.autrev.2012.10.020
57. Rodgers JM, Miller SD. Cytokine control of inflammation and repair in the pathology of multiple sclerosis. Yale J Biol Med. (2012) 85:447–68.
58. Herndler-Brandstetter D, Flavell RA. Producing GM-CSF: a unique T helper subset? Cell Res. (2014) 24:1379–80. doi: 10.1038/cr.2014.155
59. Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. (2011) 12:560–7. doi: 10.1038/ni.2027
60. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. (2011) 12:568–75. doi: 10.1038/ni.2031
61. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. (1992) 176:1693–702. doi: 10.1084/jem.176.6.1693
62. Fleetwood AJ, Cook AD, Hamilton JA. Functions of granulocyte-macrophage colony-stimulating factor. Crit Rev Immunol. (2005) 25:405–28. doi: 10.1615/CritRevImmunol.v25.i5.50
63. Zhan Y, Xu Y, Lew AM. The regulation of the development and function of dendritic cell subsets by GM-CSF: more than a hematopoietic growth factor. Mol Immunol. (2012) 52:30–7. doi: 10.1016/j.molimm.2012.04.009
64. Broughton SE, Dhagat U, Hercus TR, Nero TL, Grimbaldeston MA, Bonder CS, et al. The GM-CSF/IL-3/IL-5 cytokine receptor family: from ligand recognition to initiation of signaling. Immunol Rev. (2012) 250:277–302. doi: 10.1111/j.1600-065X.2012.01164.x
65. Kampgen E, Koch F, Heufler C, Eggert A, Gill LL, Gillis S, et al. Understanding the dendritic cell lineage through a study of cytokine receptors. J Exp Med. (1994) 179:1767–76. doi: 10.1084/jem.179.6.1767
66. Testa U, Fossati C, Samoggia P, Masciulli R, Mariani G, Hassan HJ, et al. Expression of growth factor receptors in unilineage differentiation culture of purified hematopoietic progenitors. Blood. (1996) 88:3391–406. doi: 10.1182/blood.V88.9.3391.bloodjournal8893391
67. Rosas M, Gordon S, Taylor PR. Characterisation of the expression and function of the GM-CSF receptor alpha-chain in mice. Eur J Immunol. (2007) 37:2518–28. doi: 10.1002/eji.200636892
68. Monaghan KL, Wan ECK. The role of granulocyte-macrophage colony-stimulating factor in murine models of multiple sclerosis. Cells. (2020) 9:30611. doi: 10.3390/cells9030611
69. Mcqualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med. (2001) 194:873–82. doi: 10.1084/jem.194.7.873
70. King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood. (2009) 113:3190–7. doi: 10.1182/blood-2008-07-168575
71. Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity. (2015) 43:502–14. doi: 10.1016/j.immuni.2015.08.010
72. Carrieri PB, Provitera V, De Rosa T, Tartaglia G, Gorga F, Perrella O. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacol Immunotoxicol. (1998) 20:373–82. doi: 10.3109/08923979809034820
73. Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K, et al. Expression of GM-CSF in T cells is increased in multiple sclerosis and suppressed by IFN-beta therapy. J Immunol. (2015) 194:5085–93. doi: 10.4049/jimmunol.1403243
74. Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P, et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med. (2015) 7:310ra166. doi: 10.1126/scitranslmed.aab4176
75. Ifergan I, Davidson TS, Kebir H, Xu D, Palacios-Macapagal D, Cann J, et al. Targeting the GM-CSF receptor for the treatment of CNS autoimmunity. J Autoimmun. (2017) 84:1–11. doi: 10.1016/j.jaut.2017.06.005
76. Aram J, Francis A, Tanasescu R, Constantinescu CS. Granulocyte-macrophage colony-stimulating factor as a therapeutic target in multiple sclerosis. Neurol Ther. (2019) 8:45–57. doi: 10.1007/s40120-018-0120-1
77. Constantinescu CS, Asher A, Fryze W, Kozubski W, Wagner F, Aram J, et al. Randomized phase 1b trial of MOR103, a human antibody to GM-CSF, in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e117. doi: 10.1212/NXI.0000000000000117
78. Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. (2008) 8:533–44. doi: 10.1038/nri2356
79. Ushach I, Zlotnik A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J Leukoc Biol. (2016) 100:481–9. doi: 10.1189/jlb.3RU0316-144R
80. Hume DA, Macdonald KP. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood. (2012) 119:1810–20. doi: 10.1182/blood-2011-09-379214
81. Bartocci A, Pollard JW, Stanley ER. Regulation of colony-stimulating factor 1 during pregnancy. J Exp Med. (1986) 164:956–61. doi: 10.1084/jem.164.3.956
82. Kawaji H, Yokomura K, Kikuchi K, Somoto Y, Shirai Y. Macrophage colony-stimulating factor in patients with rheumatoid arthritis. Nihon Ika Daigaku Zasshi. (1995) 62:260–70. doi: 10.1272/jnms1923.62.260
83. Scholl SM, Lidereau R, De La Rochefordiere A, Le-Nir CC, Mosseri V, Nogues C, et al. Circulating levels of the macrophage colony stimulating factor CSF-1 in primary and metastatic breast cancer patients. A pilot study. Breast Cancer Res Treat. (1996) 39:275–83. doi: 10.1007/BF01806155
84. Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol. (2006) 18:39–48. doi: 10.1016/j.coi.2005.11.006
85. Menke J, Rabacal WA, Byrne KT, Iwata Y, Schwartz MM, Stanley ER, et al. Circulating CSF-1 promotes monocyte and macrophage phenotypes that enhance lupus nephritis. J Am Soc Nephrol. (2009) 20:2581–92. doi: 10.1681/ASN.2009050499
86. Mcdermott RS, Deneux L, Mosseri V, Vedrenne J, Clough K, Fourquet A, et al. Circulating macrophage colony stimulating factor as a marker of tumour progression. Eur Cytokine Netw. (2002) 13:121–7.
87. Stanley ER, Cifone M, Heard PM, Defendi V. Factors regulating macrophage production and growth: identity of colony-stimulating factor and macrophage growth factor. J Exp Med. (1976) 143:631–47. doi: 10.1084/jem.143.3.631
88. Wei S, Nandi S, Chitu V, Yeung YG, Yu W, Huang M, et al. Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. J Leukoc Biol. (2010) 88:495–505. doi: 10.1189/jlb.1209822
89. Pons V, Rivest S. New therapeutic avenues of mCSF for brain diseases and injuries. Front Cell Neurosci. (2018) 12:499. doi: 10.3389/fncel.2018.00499
90. Sherr CJ, Rettenmier CW, Sacca R, Roussel MF, Look AT, Stanley ER. The c-fms proto-oncogene product is related to the receptor for the mononuclear phagocyte growth factor, CSF-1. Cell. (1985) 41:665–76. doi: 10.1016/S0092-8674(85)80047-7
91. Nandi S, Gokhan S, Dai XM, Wei S, Enikolopov G, Lin H, et al. The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Dev Biol. (2012) 367:100–13. doi: 10.1016/j.ydbio.2012.03.026
92. Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. (2012) 13:753–60. doi: 10.1038/ni.2360
93. Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol. (2014) 6:a021857. doi: 10.1101/cshperspect.a021857
94. Hamilton JA, Cook AD, Tak PP. Anti-colony-stimulating factor therapies for inflammatory and autoimmune diseases. Nat Rev Drug Discov. (2016) 16:53–70. doi: 10.1038/nrd.2016.231
95. Dai XM, Zong XH, Sylvestre V, Stanley ER. Incomplete restoration of colony-stimulating factor 1 (CSF-1) function in CSF-1-deficient Csf1op/Csf1op mice by transgenic expression of cell surface CSF-1. Blood. (2004) 103:1114–23. doi: 10.1182/blood-2003-08-2739
96. Rojo R, Raper A, Ozdemir DD, Lefevre L, Grabert K, Wollscheid-Lengeling E, et al. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat Commun. (2019) 10:3215. doi: 10.1038/s41467-019-11053-8
97. Hohl TM, Rivera A, Lipuma L, Gallegos A, Shi C, Mack M, et al. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe. (2009) 6:470–81. doi: 10.1016/j.chom.2009.10.007
98. Kim TS, Braciale TJ. Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PLoS ONE. (2009) 4:e4204. doi: 10.1371/journal.pone.0004204
99. Sponaas AM, Freitas Do Rosario AP, Voisine C, Mastelic B, Thompson J, Koernig S, et al. Migrating monocytes recruited to the spleen play an important role in control of blood stage malaria. Blood. (2009) 114:5522–31. doi: 10.1182/blood-2009-04-217489
100. Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell. (2010) 143:416–29. doi: 10.1016/j.cell.2010.09.039
101. Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity. (2012) 37:1076–90. doi: 10.1016/j.immuni.2012.08.026
102. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. (2013) 39:599–610. doi: 10.1016/j.immuni.2013.08.007
103. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. (2017) 17:349–62. doi: 10.1038/nri.2017.28
104. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
105. Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest. (2019) 129:2619–28. doi: 10.1172/JCI124615
106. Zhang MZ, Yao B, Yang S, Jiang L, Wang S, Fan X, et al. CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest. (2012) 122:4519–32. doi: 10.1172/JCI60363
107. Stutchfield BM, Antoine DJ, Mackinnon AC, Gow DJ, Bain CC, Hawley CA, et al. CSF1 restores innate immunity after liver injury in mice and serum levels indicate outcomes of patients with acute liver failure. Gastroenterology. (2015) 149:1896–909 e1814. doi: 10.1053/j.gastro.2015.08.053
108. Wang Y, Chang J, Yao B, Niu A, Kelly E, Breeggemann MC, et al. Proximal tubule-derived colony stimulating factor-1 mediates polarization of renal macrophages and dendritic cells, and recovery in acute kidney injury. Kidney Int. (2015) 88:1274–82. doi: 10.1038/ki.2015.295
109. Verreck FA, De Boer T, Langenberg DM, Hoeve MA, Kramer M, Vaisberg E, et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci USA. (2004) 101:4560–5. doi: 10.1073/pnas.0400983101
110. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. (2006) 177:7303–11. doi: 10.4049/jimmunol.177.10.7303
111. Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol. (2007) 178:5245–52. doi: 10.4049/jimmunol.178.8.5245
112. Guan Y, Yu S, Zhao Z, Ciric B, Zhang GX, Rostami A. Antigen presenting cells treated in vitro by macrophage colony-stimulating factor and autoantigen protect mice from autoimmunity. J Neuroimmunol. (2007) 192:68–78. doi: 10.1016/j.jneuroim.2007.09.021
113. Campbell IK, Rich MJ, Bischof RJ, Hamilton JA. The colony-stimulating factors and collagen-induced arthritis: exacerbation of disease by M-CSF and G-CSF and requirement for endogenous M-CSF. J Leukoc Biol. (2000) 68:144–50. doi: 10.1189/jlb.68.1.144
114. Paniagua RT, Chang A, Mariano MM, Stein EA, Wang Q, Lindstrom TM, et al. c-Fms-mediated differentiation and priming of monocyte lineage cells play a central role in autoimmune arthritis. Arthritis Res Ther. (2010) 12:R32. doi: 10.1186/ar2940
115. Toh ML, Bonnefoy JY, Accart N, Cochin S, Pohle S, Haegel H, et al. Bone- and cartilage-protective effects of a monoclonal antibody against colony-stimulating factor 1 receptor in experimental arthritis. Arthritis Rheumatol. (2014) 66:2989–3000. doi: 10.1002/art.38624
116. Garcia S, Hartkamp LM, Malvar-Fernandez B, Van Es IE, Lin H, Wong J, et al. Colony-stimulating factor (CSF) 1 receptor blockade reduces inflammation in human and murine models of rheumatoid arthritis. Arthritis Res Ther. (2016) 18:75. doi: 10.1186/s13075-016-0973-6
117. Carrero JA, Mccarthy DP, Ferris ST, Wan X, Hu H, Zinselmeyer BH, et al. Resident macrophages of pancreatic islets have a seminal role in the initiation of autoimmune diabetes of NOD mice. Proc Natl Acad Sci USA. (2017) 114:E10418–27. doi: 10.1073/pnas.1713543114
118. Wada Y, Gonzalez-Sanchez HM, Weinmann-Menke J, Iwata Y, Ajay AK, Meineck M, et al. IL-34-dependent intrarenal and systemic mechanisms promote lupus nephritis in MRL-Fas(lpr) mice. J Am Soc Nephrol. (2019) 30:244–59. doi: 10.1681/ASN.2018090901
119. Achkova D, Maher J. Role of the colony-stimulating factor (CSF)/CSF-1 receptor axis in cancer. Biochem Soc Trans. (2016) 44:333–41. doi: 10.1042/BST20150245
120. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. (2017) 5:53. doi: 10.1186/s40425-017-0257-y
121. Baghdadi M, Endo H, Takano A, Ishikawa K, Kameda Y, Wada H, et al. High co-expression of IL-34 and M-CSF correlates with tumor progression and poor survival in lung cancers. Sci Rep. (2018) 8:418. doi: 10.1038/s41598-017-18796-8
122. Martinez-Muriana A, Mancuso R, Francos-Quijorna I, Olmos-Alonso A, Osta R, Perry VH, et al. CSF1R blockade slows the progression of amyotrophic lateral sclerosis by reducing microgliosis and invasion of macrophages into peripheral nerves. Sci Rep. (2016) 6:25663. doi: 10.1038/srep25663
123. Neal ML, Fleming SM, Budge KM, Boyle AM, Kim C, Alam G, et al. Pharmacological inhibition of CSF1R by GW2580 reduces microglial proliferation and is protective against neuroinflammation and dopaminergic neurodegeneration. FASEB J. (2020) 34:1679–94. doi: 10.1096/fj.201900567RR
124. Dagher NN, Najafi AR, Kayala KM, Elmore MR, White TE, Medeiros R, et al. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation. (2015) 12:139. doi: 10.1186/s12974-015-0366-9
125. Olmos-Alonso A, Schetters ST, Sri S, Askew K, Mancuso R, Vargas-Caballero M, et al. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer's-like pathology. Brain. (2016) 139:891–907. doi: 10.1093/brain/awv379
126. Sosna J, Philipp S, Albay R 3rd, Reyes-Ruiz JM, Baglietto-Vargas D, Laferla FM, et al. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer's disease. Mol Neurodegener. (2018) 13:11. doi: 10.1186/s13024-018-0244-x
127. Uemura Y, Ohno H, Ohzeki Y, Takanashi H, Murooka H, Kubo K, et al. The selective M-CSF receptor tyrosine kinase inhibitor Ki20227 suppresses experimental autoimmune encephalomyelitis. J Neuroimmunol. (2008) 195:73–80. doi: 10.1016/j.jneuroim.2008.01.015
128. Crespo O, Kang SC, Daneman R, Lindstrom TM, Ho PP, Sobel RA, et al. Tyrosine kinase inhibitors ameliorate autoimmune encephalomyelitis in a mouse model of multiple sclerosis. J Clin Immunol. (2011) 31:1010–20. doi: 10.1007/s10875-011-9579-6
129. Borjini N, Fernandez M, Giardino L, Calza L. Cytokine and chemokine alterations in tissue, CSF, and plasma in early presymptomatic phase of experimental allergic encephalomyelitis (EAE), in a rat model of multiple sclerosis. J Neuroinflammation. (2016) 13:291. doi: 10.1186/s12974-016-0757-6
130. Nissen JC, Thompson KK, West BL, Tsirka SE. Csf1R inhibition attenuates experimental autoimmune encephalomyelitis and promotes recovery. Exp Neurol. (2018) 307:24–36. doi: 10.1016/j.expneurol.2018.05.021
131. Conway JG, Mcdonald B, Parham J, Keith B, Rusnak DW, Shaw E, et al. Inhibition of colony-stimulating-factor-1 signaling in vivo with the orally bioavailable cFMS kinase inhibitor GW2580. Proc Natl Acad Sci USA. (2005) 102:16078–83. doi: 10.1073/pnas.0502000102
132. Praet J, Guglielmetti C, Berneman Z, Van Der Linden A, Ponsaerts P. Cellular and molecular neuropathology of the cuprizone mouse model: clinical relevance for multiple sclerosis. Neurosci Biobehav Rev. (2014) 47:485–505. doi: 10.1016/j.neubiorev.2014.10.004
133. Laflamme N, Cisbani G, Prefontaine P, Srour Y, Bernier J, St-Pierre MK, et al. mCSF-induced microglial activation prevents myelin loss and promotes its repair in a mouse model of multiple sclerosis. Front Cell Neurosci. (2018) 12:178. doi: 10.3389/fncel.2018.00178
134. Werner K, Bitsch A, Bunkowski S, Hemmerlein B, Bruck W. The relative number of macrophages/microglia expressing macrophage colony-stimulating factor and its receptor decreases in multiple sclerosis lesions. Glia. (2002) 40:121–9. doi: 10.1002/glia.10120
135. Tridente G. Imatinib. Adverse Event Oncotarget Kinase Inhibit. (2017) 97–122. doi: 10.1016/B978-0-12-809400-6.00005-6
136. Cahill KN, Katz HR, Cui J, Lai J, Kazani S, Crosby-Thompson A, et al. KIT inhibition by imatinib in patients with severe refractory asthma. N Engl J Med. (2017) 376:1911–20. doi: 10.1056/NEJMoa1613125
137. Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. (2008) 7:3129–40. doi: 10.1158/1535-7163.MCT-08-0013
138. Weber ANR, Bittner Z, Liu X, Dang TM, Radsak MP, Brunner C. Bruton's tyrosine kinase: an emerging key player in innate immunity. Front Immunol. (2017) 8:1454. doi: 10.3389/fimmu.2017.01454
139. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. (1993) 260:547–9. doi: 10.1126/science.8097338
140. Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. (2006) 203:1685–91. doi: 10.1084/jem.20060285
141. Ghoreschi K, Laurence A, Yang XP, Tato CM, Mcgeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. (2010) 467:967–71. doi: 10.1038/nature09447
142. Schiffenbauer J, Streit WJ, Butfiloski E, Labow M, Edwards C 3rd, Moldawer LL. The induction of EAE is only partially dependent on TNF receptor signaling but requires the IL-1 type I receptor. Clin Immunol. (2000) 95:117–23. doi: 10.1006/clim.2000.4851
143. Lukens JR, Barr MJ, Chaplin DD, Chi H, Kanneganti TD. Inflammasome-derived IL-1beta regulates the production of GM-CSF by CD4(+) T cells and gammadelta T cells. J Immunol. (2012) 188:3107–15. doi: 10.4049/jimmunol.1103308
144. Levesque SA, Pare A, Mailhot B, Bellver-Landete V, Kebir H, Lecuyer MA, et al. Myeloid cell transmigration across the CNS vasculature triggers IL-1beta-driven neuroinflammation during autoimmune encephalomyelitis in mice. J Exp Med. (2016) 213:929–49. doi: 10.1084/jem.20151437
145. Ronchi F, Basso C, Preite S, Reboldi A, Baumjohann D, Perlini L, et al. Experimental priming of encephalitogenic Th1/Th17 cells requires pertussis toxin-driven IL-1beta production by myeloid cells. Nat Commun. (2016) 7:11541. doi: 10.1038/ncomms11541
146. Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. (2009) 30:576–87. doi: 10.1016/j.immuni.2009.02.007
147. Badovinac V, Mostarica-Stojkovic M, Dinarello CA, Stosic-Grujicic S. Interleukin-1 receptor antagonist suppresses experimental autoimmune encephalomyelitis (EAE) in rats by influencing the activation and proliferation of encephalitogenic cells. J Neuroimmunol. (1998) 85:87–95. doi: 10.1016/S0165-5728(98)00020-4
148. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. (2015) 21:248–55. doi: 10.1038/nm.3806
149. Sha Y, Markovic-Plese S. Activated IL-1RI signaling pathway induces Th17 cell differentiation via interferon regulatory factor 4 signaling in patients with relapsing-remitting multiple sclerosis. Front Immunol. (2016) 7:543. doi: 10.3389/fimmu.2016.00543
150. Cannella B, Raine CS. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann Neurol. (1995) 37:424–35. doi: 10.1002/ana.410370404
151. Lin CC, Edelson BT. New insights into the role of IL-1beta in experimental autoimmune encephalomyelitis and multiple sclerosis. J Immunol. (2017) 198:4553–60. doi: 10.4049/jimmunol.1700263
152. Pare A, Mailhot B, Levesque SA, Lacroix S. Involvement of the IL-1 system in experimental autoimmune encephalomyelitis and multiple sclerosis: breaking the vicious cycle between IL-1beta and GM-CSF. Brain Behav Immun. (2017) 62:1–8. doi: 10.1016/j.bbi.2016.07.146
153. Dinarello CA, Van Der Meer JW. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol. (2013) 25:469–84. doi: 10.1016/j.smim.2013.10.008
154. Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. (2000) 13:715–25. doi: 10.1016/S1074-7613(00)00070-4
155. Zhang GX, Yu S, Gran B, Li J, Siglienti I, Chen X, et al. Role of IL-12 receptor beta 1 in regulation of T cell response by APC in experimental autoimmune encephalomyelitis. J Immunol. (2003) 171:4485–92. doi: 10.4049/jimmunol.171.9.4485
156. Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, et al. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. (2003) 170:2153–60. doi: 10.4049/jimmunol.170.4.2153
157. Awasthi A, Riol-Blanco L, Jager A, Korn T, Pot C, Galileos G, et al. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol. (2009) 182:5904–8. doi: 10.4049/jimmunol.0900732
158. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. (2003) 421:744–8. doi: 10.1038/nature01355
159. Ichikawa M, Koh CS, Inoue A, Tsuyusaki J, Yamazaki M, Inaba Y, et al. Anti-IL-12 antibody prevents the development and progression of multiple sclerosis-like relapsing–remitting demyelinating disease in NOD mice induced with myelin oligodendrocyte glycoprotein peptide. J Neuroimmunol. (2000) 102:56–66. doi: 10.1016/S0165-5728(99)00153-8
160. Constantinescu CS, Hilliard B, Ventura E, Wysocka M, Showe L, Lavi E, et al. Modulation of susceptibility and resistance to an autoimmune model of multiple sclerosis in prototypically susceptible and resistant strains by neutralization of interleukin-12 and interleukin-4, respectively. Clin Immunol. (2001) 98:23–30. doi: 10.1006/clim.2000.4944
161. Brok HP, Van Meurs M, Blezer E, Schantz A, Peritt D, Treacy G, et al. Prevention of experimental autoimmune encephalomyelitis in common marmosets using an anti-IL-12p40 monoclonal antibody. J Immunol. (2002) 169:6554–63. doi: 10.4049/jimmunol.169.11.6554
162. Chen Y, Langrish CL, Mckenzie B, Joyce-Shaikh B, Stumhofer JS, Mcclanahan T, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. (2006) 116:1317–26. doi: 10.1172/JCI25308
163. Comabella M, Balashov K, Issazadeh S, Smith D, Weiner HL, Khoury SJ. Elevated interleukin-12 in progressive multiple sclerosis correlates with disease activity and is normalized by pulse cyclophosphamide therapy. J Clin Invest. (1998) 102:671–8. doi: 10.1172/JCI3125
164. Matusevicius D, Kivisakk P, Navikas V, Soderstrom M, Fredrikson S, Link H. Interleukin-12 and perforin mRNA expression is augmented in blood mononuclear cells in multiple sclerosis. Scand J Immunol. (1998) 47:582–90. doi: 10.1046/j.1365-3083.1998.00344.x
165. Van Boxel-Dezaire AH, Hoff SC, Van Oosten BW, Verweij CL, Drager AM, Ader HJ, et al. Decreased interleukin-10 and increased interleukin-12p40 mRNA are associated with disease activity and characterize different disease stages in multiple sclerosis. Ann Neurol. (1999) 45:695–703. doi: 10.1002/1531-8249(199906)45:6<695::AID-ANA3>3.0.CO
166. Windhagen A, Newcombe J, Dangond F, Strand C, Woodroofe MN, Cuzner ML, et al. Expression of costimulatory molecules B7-1 (CD80), B7-2 (CD86), and interleukin 12 cytokine in multiple sclerosis lesions. J Exp Med. (1995) 182:1985–96. doi: 10.1084/jem.182.6.1985
167. Li Y, Chu N, Hu A, Gran B, Rostami A, Zhang GX. Increased IL-23p19 expression in multiple sclerosis lesions and its induction in microglia. Brain. (2007) 130:490–501. doi: 10.1093/brain/awl273
168. Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH, et al. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. (2008) 7:796–804. doi: 10.1016/S1474-4422(08)70173-X
169. Longbrake EE, Racke MK. Why did IL-12/IL-23 antibody therapy fail in multiple sclerosis? Expert Rev Neurother. (2009) 9:319–21. doi: 10.1586/14737175.9.3.319
170. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. (2004) 116:281–97. doi: 10.1016/S0092-8674(04)00045-5
171. Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. (2009) 19:92–105. doi: 10.1101/gr.082701.108
172. Tsitsiou E, Lindsay MA. microRNAs and the immune response. Curr Opin Pharmacol. (2009) 9:514–20. doi: 10.1016/j.coph.2009.05.003
173. Davidson-Moncada J, Papavasiliou FN, Tam W. MicroRNAs of the immune system: roles in inflammation and cancer. Ann N Y Acad Sci. (2010) 1183:183–94. doi: 10.1111/j.1749-6632.2009.05121.x
174. Gracias DT, Katsikis PD. MicroRNAs: key components of immune regulation. Adv Exp Med Biol. (2011) 780:15–26. doi: 10.1007/978-1-4419-5632-3_2
175. O'connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. (2010) 10:111–22. doi: 10.1038/nri2708
176. O'connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. (2012) 30:295–312. doi: 10.1146/annurev-immunol-020711-075013
177. Keller A, Leidinger P, Lange J, Borries A, Schroers H, Scheffler M, et al. Multiple sclerosis: microRNA expression profiles accurately differentiate patients with relapsing-remitting disease from healthy controls. PLoS ONE. (2009) 4:e7440. doi: 10.1371/journal.pone.0007440
178. Martinelli-Boneschi F, Fenoglio C, Brambilla P, Sorosina M, Giacalone G, Esposito F, et al. MicroRNA and mRNA expression profile screening in multiple sclerosis patients to unravel novel pathogenic steps and identify potential biomarkers. Neurosci Lett. (2012) 508:4–8. doi: 10.1016/j.neulet.2011.11.006
179. Junker A, Krumbholz M, Eisele S, Mohan H, Augstein F, Bittner R, et al. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain. (2009) 132:3342–52. doi: 10.1093/brain/awp300
180. Fukao T, Fukuda Y, Kiga K, Sharif J, Hino K, Enomoto Y, et al. An evolutionarily conserved mechanism for microRNA-223 expression revealed by microRNA gene profiling. Cell. (2007) 129:617–31. doi: 10.1016/j.cell.2007.02.048
181. Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. (2008) 451:1125–9. doi: 10.1038/nature06607
182. Zhuang G, Meng C, Guo X, Cheruku PS, Shi L, Xu H, et al. A novel regulator of macrophage activation: miR-223 in obesity-associated adipose tissue inflammation. Circulation. (2012) 125:2892–903. doi: 10.1161/CIRCULATIONAHA.111.087817
183. Galloway DA, Blandford SN, Berry T, Williams JB, Stefanelli M, Ploughman M, et al. miR-223 promotes regenerative myeloid cell phenotype and function in the demyelinated central nervous system. Glia. (2019) 67:857–69. doi: 10.1002/glia.23576
184. Ifergan I, Chen S, Zhang B, Miller SD. Cutting edge: microRNA-223 regulates myeloid dendritic cell-driven Th17 responses in experimental autoimmune encephalomyelitis. J Immunol. (2016) 196:1455–9. doi: 10.4049/jimmunol.1501965
185. Satoorian T, Li B, Tang X, Xiao J, Xing W, Shi W, et al. MicroRNA223 promotes pathogenic T-cell development and autoimmune inflammation in central nervous system in mice. Immunology. (2016) 148:326–38. doi: 10.1111/imm.12611
186. Cantoni C, Cignarella F, Ghezzi L, Mikesell B, Bollman B, Berrien-Elliott MM, et al. Mir-223 regulates the number and function of myeloid-derived suppressor cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. (2017) 133:61–77. doi: 10.1007/s00401-016-1621-6
187. Wolfle SJ, Strebovsky J, Bartz H, Sahr A, Arnold C, Kaiser C, et al. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur J Immunol. (2011) 41:413–24. doi: 10.1002/eji.201040979
188. Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. (2013) 16:1211–8. doi: 10.1038/nn.3469
189. Lloyd AF, Miron VE. The pro-remyelination properties of microglia in the central nervous system. Nat Rev Neurol. (2019) 15:447–58. doi: 10.1038/s41582-019-0184-2
190. Mccoy CE. miR-155 dysregulation and therapeutic intervention in multiple sclerosis. Adv Exp Med Biol. (2017) 1024:111–31. doi: 10.1007/978-981-10-5987-2_5
191. Moore CS, Rao VT, Durafourt BA, Bedell BJ, Ludwin SK, Bar-Or A. miR-155 as a multiple sclerosis-relevant regulator of myeloid cell polarization. Ann Neurol. (2013) 74:709–20. doi: 10.1002/ana.23967
192. Mashima R. Physiological roles of miR-155. Immunology. (2015) 145:323–33. doi: 10.1111/imm.12468
193. O'connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci USA. (2009) 106:7113–8. doi: 10.1073/pnas.0902636106
194. Kerr WG. Inhibitor and activator: dual functions for SHIP in immunity and cancer. Ann N Y Acad Sci. (2011) 1217:1–17. doi: 10.1111/j.1749-6632.2010.05869.x
195. O'connell RM, Rao DS, Chaudhuri AA, Boldin MP, Taganov KD, Nicoll J, et al. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med. (2008) 205:585–94. doi: 10.1084/jem.20072108
196. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science. (2007) 316:608–11. doi: 10.1126/science.1139253
197. Lind EF, Millar DG, Dissanayake D, Savage JC, Grimshaw NK, Kerr WG, et al. miR-155 upregulation in dendritic cells is sufficient to break tolerance in vivo by negatively regulating SHIP1. J Immunol. (2015) 195:4632–40. doi: 10.4049/jimmunol.1302941
198. Kurowska-Stolarska M, Alivernini S, Ballantine LE, Asquith DL, Millar NL, Gilchrist DS, et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc Natl Acad Sci USA. (2011) 108:11193–8. doi: 10.1073/pnas.1019536108
199. Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J Immunol. (2011) 187:2213–21. doi: 10.4049/jimmunol.1003952
200. O'connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. (2010) 33:607–19. doi: 10.1016/j.immuni.2010.09.009
201. O'connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci USA. (2007) 104:1604–9. doi: 10.1073/pnas.0610731104
202. Jablonski KA, Gaudet AD, Amici SA, Popovich PG, Guerau-De-Arellano M. Control of the inflammatory macrophage transcriptional signature by miR-155. PLoS ONE. (2016) 11:e0159724. doi: 10.1371/journal.pone.0159724
203. Piket E, Zheleznyakova GY, Kular L, Jagodic M. Small non-coding RNAs as important players, biomarkers and therapeutic targets in multiple sclerosis: a comprehensive overview. J Autoimmun. (2019) 101:17–25. doi: 10.1016/j.jaut.2019.04.002
204. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. (2006) 103:12481–6. doi: 10.1073/pnas.0605298103
205. Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med. (2011) 208:1189–201. doi: 10.1084/jem.20101823
206. Zhao JL, Rao DS, Boldin MP, Taganov KD, O'connell RM, Baltimore D. NF-κB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc Natl Acad Sci USA. (2011) 108:9184–9. doi: 10.1073/pnas.1105398108
207. Huang C, Liu XJ, Qunzhou Xie J, Ma TT, Meng XM, Li J. MiR-146a modulates macrophage polarization by inhibiting Notch1 pathway in RAW264.7 macrophages. Int Immunopharmacol. (2016) 32:46–54. doi: 10.1016/j.intimp.2016.01.009
208. Xu WD, Lu MM, Pan HF, Ye DQ. Association of MicroRNA-146a with autoimmune diseases. Inflammation. (2012) 35:1525–9. doi: 10.1007/s10753-012-9467-0
209. Fenoglio C, Cantoni C, De Riz M, Ridolfi E, Cortini F, Serpente M, et al. Expression and genetic analysis of miRNAs involved in CD4+ cell activation in patients with multiple sclerosis. Neurosci Lett. (2011) 504:9–12. doi: 10.1016/j.neulet.2011.08.021
210. Waschbisch A, Atiya M, Linker RA, Potapov S, Schwab S, Derfuss T. Glatiramer acetate treatment normalizes deregulated microRNA expression in relapsing remitting multiple sclerosis. PLoS ONE. (2011) 6:e24604. doi: 10.1371/journal.pone.0024604
211. Martin NA, Molnar V, Szilagyi GT, Elkjaer ML, Nawrocki A, Okarmus J, et al. Experimental demyelination and axonal loss are reduced in microRNA-146a deficient mice. Front Immunol. (2018) 9:490. doi: 10.3389/fimmu.2018.00490
212. Li B, Wang X, Choi IY, Wang YC, Liu S, Pham AT, et al. miR-146a modulates autoreactive Th17 cell differentiation and regulates organ-specific autoimmunity. J Clin Invest. (2017) 127:3702–16. doi: 10.1172/JCI94012
213. Zhang J, Zhang ZG, Lu M, Zhang Y, Shang X, Chopp M. MiR-146a promotes oligodendrocyte progenitor cell differentiation and enhances remyelination in a model of experimental autoimmune encephalomyelitis. Neurobiol Dis. (2019) 125:154–62. doi: 10.1016/j.nbd.2019.01.019
214. Getts DR, Turley DM, Smith CE, Harp CT, Mccarthy D, Feeney EM, et al. Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10-producing splenic macrophages and maintained by T regulatory cells. J Immunol. (2011) 187:2405–17. doi: 10.4049/jimmunol.1004175
215. Smarr CB, Hsu CL, Byrne AJ, Miller SD, Bryce PJ. Antigen-fixed leukocytes tolerize Th2 responses in mouse models of allergy. J Immunol. (2011) 187:5090–8. doi: 10.4049/jimmunol.1100608
216. Prasad S, Kohm AP, Mcmahon JS, Luo X, Miller SD. Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9-23 epitope and involves functional epitope spreading. J Autoimmun. (2012) 39:347–53. doi: 10.1016/j.jaut.2012.04.005
217. Sercombe L, Veerati T, Moheimani F, Wu SY, Sood AK, Hua S. Advances and challenges of liposome assisted drug delivery. Front Pharmacol. (2015) 6:286. doi: 10.3389/fphar.2015.00286
218. Vader P, Mol EA, Pasterkamp G, Schiffelers RM. Extracellular vesicles for drug delivery. Adv Drug Deliv Rev. (2016) 106:148–56. doi: 10.1016/j.addr.2016.02.006
219. Getts DR, Martin AJ, Mccarthy DP, Terry RL, Hunter ZN, Yap WT, et al. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat Biotechnol. (2012) 30:1217–24. doi: 10.1038/nbt.2434
220. Hunter Z, Mccarthy DP, Yap WT, Harp CT, Getts DR, Shea LD, et al. A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease. ACS Nano. (2014) 8:2148–60. doi: 10.1021/nn405033r
221. Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. (2016) 530:434–40. doi: 10.1038/nature16962
222. Smarr CB, Yap WT, Neef TP, Pearson RM, Hunter ZN, Ifergan I, et al. Biodegradable antigen-associated PLG nanoparticles tolerize Th2-mediated allergic airway inflammation pre- and postsensitization. Proc Natl Acad Sci USA. (2016) 113:5059–64. doi: 10.1073/pnas.1505782113
223. Mccarthy DP, Yap JW, Harp CT, Song WK, Chen J, Pearson RM, et al. An antigen-encapsulating nanoparticle platform for TH1/17 immune tolerance therapy. Nanomedicine. (2017) 13:191–200. doi: 10.1016/j.nano.2016.09.007
224. Getts DR, Terry RL, Getts MT, Deffrasnes C, Muller M, Van Vreden C, et al. Therapeutic inflammatory monocyte modulation using immune-modifying microparticles. Sci Transl Med. (2014) 6:219ra217. doi: 10.1126/scitranslmed.3007563
225. Kelly C, Murray J, Leffler DA, Bledsoe A, Smithson G, Podojil J, et al. TAK-101 (TIMP-GLIA) prevents gluten challenge induced immune activation in adults with celiac disease. Gastroenterology. (2020) 158:S135. doi: 10.1016/s0016-5085(20)31017-9
226. Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol. (2008) 38:1404–13. doi: 10.1002/eji.200737984
227. Getts DR, Shea LD, Miller SD, King NJ. Harnessing nanoparticles for immune modulation. Trends Immunol. (2015) 36:419–27. doi: 10.1016/j.it.2015.05.007
228. Mathaes R, Winter G, Besheer A, Engert J. Influence of particle geometry and PEGylation on phagocytosis of particulate carriers. Int J Pharm. (2014) 465:159–64. doi: 10.1016/j.ijpharm.2014.02.037
229. Liu Y, Li W, Lao F, Liu Y, Wang L, Bai R, et al. Intracellular dynamics of cationic and anionic polystyrene nanoparticles without direct interaction with mitotic spindle and chromosomes. Biomaterials. (2011) 32:8291–303. doi: 10.1016/j.biomaterials.2011.07.037
230. Tagalakis AD, Lee DH, Bienemann AS, Zhou H, Munye MM, Saraiva L, et al. Multifunctional, self-assembling anionic peptide-lipid nanocomplexes for targeted siRNA delivery. Biomaterials. (2014) 35:8406–15. doi: 10.1016/j.biomaterials.2014.06.003
231. Ruenraroengsak P, Tetley TD. Differential bioreactivity of neutral, cationic and anionic polystyrene nanoparticles with cells from the human alveolar compartment: robust response of alveolar type 1 epithelial cells. Part Fibre Toxicol. (2015) 12:19. doi: 10.1186/s12989-015-0091-7
232. Sun Y, Guo F, Zou Z, Li C, Hong X, Zhao Y, et al. Cationic nanoparticles directly bind angiotensin-converting enzyme 2 and induce acute lung injury in mice. Part Fibre Toxicol. (2015) 12:4. doi: 10.1186/s12989-015-0080-x
233. Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Coll Surf B Biointerfaces. (2010) 75:1–18. doi: 10.1016/j.colsurfb.2009.09.001
234. Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. (2012) 161:505–22. doi: 10.1016/j.jconrel.2012.01.043
235. Jeong SJ, Cooper JG, Ifergan I, Mcguire TL, Xu D, Hunter Z, et al. Intravenous immune-modifying nanoparticles as a therapy for spinal cord injury in mice. Neurobiol Dis. (2017) 108:73–82. doi: 10.1016/j.nbd.2017.08.006
236. Sharma S, Ifergan I, Kurz JE, Linsenmeier RA, Xu D, Cooper JG, et al. Intravenous immunomodulatory nanoparticle treatment for traumatic brain injury. Ann Neurol. (2020) 87:442–55. doi: 10.1002/ana.25675
237. Deberge M, Yu S, Dehn S, Ifergan I, Yeap XY, Filipp M, et al. Monocytes prime autoreactive T cells after myocardial infarction. Am J Physiol Heart Circ Physiol. (2020) 318:H116–23. doi: 10.1152/ajpheart.00595.2019
238. Edwards RG, Kopp SJ, Ifergan I, Shui JW, Kronenberg M, Miller SD, et al. Murine corneal inflammation and nerve damage after infection with HSV-1 are promoted by HVEM and ameliorated by immune-modifying nanoparticle therapy. Invest Ophthalmol Vis Sci. (2017) 58:282–91. doi: 10.1167/iovs.16-20668
239. Prasad S, Neef T, Xu D, Podojil JR, Getts DR, Shea LD, et al. Tolerogenic Ag-PLG nanoparticles induce tregs to suppress activated diabetogenic CD4 and CD8 T cells. J Autoimmun. (2018) 89:112–24. doi: 10.1016/j.jaut.2017.12.010
240. Chountoulesi M, Demetzos C. Promising nanotechnology approaches in treatment of autoimmune diseases of central nervous system. Brain Sci. (2020) 10:60338. doi: 10.3390/brainsci10060338
241. Cappellano G, Woldetsadik AD, Orilieri E, Shivakumar Y, Rizzi M, Carniato F, et al. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine. (2014) 32:5681–9. doi: 10.1016/j.vaccine.2014.08.016
242. Casey LM, Pearson RM, Hughes KR, Liu JMH, Rose JA, North MG, et al. Conjugation of transforming growth factor beta to antigen-loaded poly(lactide- co-glycolide) nanoparticles enhances efficiency of antigen-specific tolerance. Bioconjug Chem. (2018) 29:813–23. doi: 10.1021/acs.bioconjchem.7b00624
243. Maldonado RA, Lamothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci USA. (2015) 112:E156–165. doi: 10.1073/pnas.1408686111
244. Pei W, Wan X, Shahzad KA, Zhang L, Song S, Jin X, et al. Direct modulation of myelin-autoreactive CD4(+) and CD8(+) T cells in EAE mice by a tolerogenic nanoparticle co-carrying myelin peptide-loaded major histocompatibility complexes, CD47 and multiple regulatory molecules. Int J Nanomedicine. (2018) 13:3731–50. doi: 10.2147/IJN.S164500
Keywords: myeloid cels, multiple sclerosis, chemokines, GM-CSF, miRNAs, nanoparticles, M-CSF
Citation: Ifergan I and Miller SD (2020) Potential for Targeting Myeloid Cells in Controlling CNS Inflammation. Front. Immunol. 11:571897. doi: 10.3389/fimmu.2020.571897
Received: 12 June 2020; Accepted: 03 September 2020;
Published: 06 October 2020.
Edited by:
Jorge Ivan Alvarez, University of Pennsylvania, United StatesReviewed by:
Frauke Zipp, Johannes Gutenberg University Mainz, GermanyGiacomo Casella, Thomas Jefferson University, United States
Copyright © 2020 Ifergan and Miller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen D. Miller, cy1kLW1pbGxlckBub3J0aHdlc3Rlcm4uZWR1