 Torsten P. M. Scheithauer1,2*
Torsten P. M. Scheithauer1,2* Elena Rampanelli2Max Nieuwdorp1,2
Elena Rampanelli2Max Nieuwdorp1,2 Bruce A. Vallance3C. Bruce Verchere4Daniël H. van Raalte1,2
Bruce A. Vallance3C. Bruce Verchere4Daniël H. van Raalte1,2 Hilde Herrema2
Hilde Herrema2- 1Department of Internal Medicine, Amsterdam University Medical Center (UMC), Vrije Universiteit (VU) University Medical Center, Amsterdam, Netherlands
- 2Department of Experimental Vascular Medicine, Amsterdam University Medical Center (UMC), Academic Medical Center, Amsterdam, Netherlands
- 3Division of Gastroenterology, Department of Pediatrics, Child and Family Research Institute, Vancouver, BC, Canada
- 4Department of Surgery, University of British Columbia and BC Children's Hospital Research Institute, Vancouver, BC, Canada
The gut microbiota has been linked to the development of obesity and type 2 diabetes (T2D). The underlying mechanisms as to how intestinal microbiota may contribute to T2D are only partly understood. It becomes progressively clear that T2D is characterized by a chronic state of low-grade inflammation, which has been linked to the development of insulin resistance. Here, we review the current evidence that intestinal microbiota, and the metabolites they produce, could drive the development of insulin resistance in obesity and T2D, possibly by initiating an inflammatory response. First, we will summarize major findings about immunological and gut microbial changes in these metabolic diseases. Next, we will give a detailed view on how gut microbial changes have been implicated in low-grade inflammation. Lastly, we will critically discuss clinical studies that focus on the interaction between gut microbiota and the immune system in metabolic disease. Overall, there is strong evidence that the tripartite interaction between gut microbiota, host immune system and metabolism is a critical partaker in the pathophysiology of obesity and T2D.
Introduction
Type 2 diabetes (T2D) incidence, which is in large driven by the obesity pandemic, is increasing with alarming rates. In 2019, it was estimated that 463 million people were suffering from diabetes worldwide; these numbers are expected to continue to rise toward 578 million patients in 2030 (1). T2D is typically preceded by insulin resistance; a condition in which the actions of insulin on peripheral tissues including skeletal muscle, liver, and adipose, are impaired. This results in reduced insulin-stimulated glucose disposal, impaired insulin-induced suppression of hepatic glucose production and lipolysis rates, respectively (2). Insulin is produced by the beta cells of the endocrine pancreas. In early stages of insulin resistance, the pancreas can compensate for impaired peripheral insulin action by increasing insulin production. When pancreatic beta cells fail to meet the increased insulin demand, hyperglycemia develops (3). Hyperglycemia has been extensively linked to the detrimental micro- and macrovascular complications typically observed in humans with T2D (4).
In recent decades, chronic low-grade “metabolic” inflammation (local and systemic), also called metainflammation, has been identified to contribute to the development of insulin resistance and progression to T2D. As such, metainflammation has been linked to both impaired insulin action and secretion (5). While a vast body of research has provided detailed insight into regulation of glucose homeostasis by inflammatory pathways, the upstream triggers of these pathways have remained elusive for a long time. The intestinal microbiota, the collective community of microorganisms in the gastrointestinal tract, plays a critical role in human metabolism, in part by acting as an immunomodulator. Although this property is critical for human health, it can also have detrimental consequences. In accordance, the gut microbiota has been appointed as driver of metainflammation observed in obesity and T2D, which are also characterized by an altered gut microbiota composition (6–8). In this review, we extensively address the tripartite interaction between the gut microbiota, the mammalian immune system and glucometabolic pathways (Figure 1). In addition, we propose whether, based on current evidence, modulation of inflammation via the intestinal microbiota could form a target for novel therapies to reduce the current diabetes pandemic.
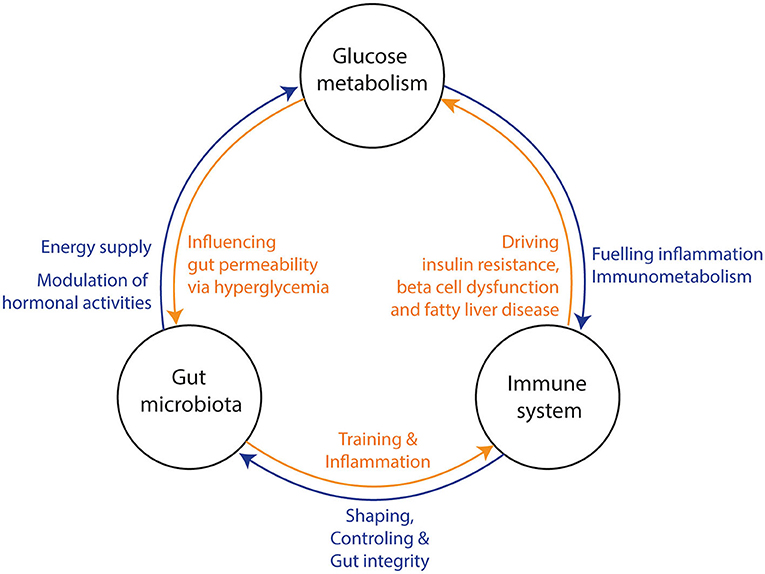
Figure 1. Three-way interaction between the gut microbiota, glucose metabolism, and the immune system. (1) The gut microbiota influences the host‘s glucose metabolism and hormone production via the production of several metabolites. Hyperglycemia increases gut permeability and thereby translocation of bacterial components into the circulation. In turn, bacterial translocation is fueling a (pro) inflammatory response of the immune system. Under normal conditions, the gut microbiota is training the immune system via several bacterial components and metabolites. (2) The immune system is shaping and controlling gut microbiota to keep a symbiotic relationship between host and microbiota. Further, it prevents bacterial translocation via promoting gut integrity. Bacterial translocation may lead to inflammation in several tissues and consequential loss of function (e.g., beta-cell dysfunction, insulin resistance and fatty liver disease). (3) The glucose metabolism can induce a pro-inflammatory response of the immune system through interplay of metabolic and inflammatory pathways (immunometabolism). Thereby, all three factors affect each other and may drive metabolic diseases.
Inflammation in Obesity and Type 2 Diabetes
Low-grade inflammation has been extensively linked to disturbances in glucometabolic pathways as observed in people with obesity and T2D. Several cytokines are increased in the circulation of people with metabolic syndrome (9–11) and have negative effects on peripheral tissue metabolism. For example, people with insulin resistance and glucose intolerance had a higher inflammatory tone and an altered response to respiratory viral infections compared to insulin sensitive individuals (12). Similarly, obese subjects had a higher inflammatory tone and more severe asthma, which was related to the gut microbiota (13). Interestingly, a 10% weight loss reduced plasma concentrations of several cytokines in obese women (10). Further, numerous pharmacological treatments aiming to reduce inflammation in metabolic diseases have positive effects on glucose tolerance in mice and human. These include the novel hybrid cytokine interleukin (IL) 233, which is produced in a genetically modified Escherichia coli by fusing murine IL-2 and murine IL-33 (14). Other treatment strategies include IL-1 receptor blockage (15), IL-1β antagonism (16), inhibition of the intracellular pro-inflammatory NF-κB pathway (17), and TNF antagonism (18) (Figure 2). These findings highlight pivotal roles of metainflammation in the development of T2D and the opportunities for (pharmacological) interventions. Below, we review a number of key immune cell types and inflammatory mediators that have been linked to impaired glucose metabolism.
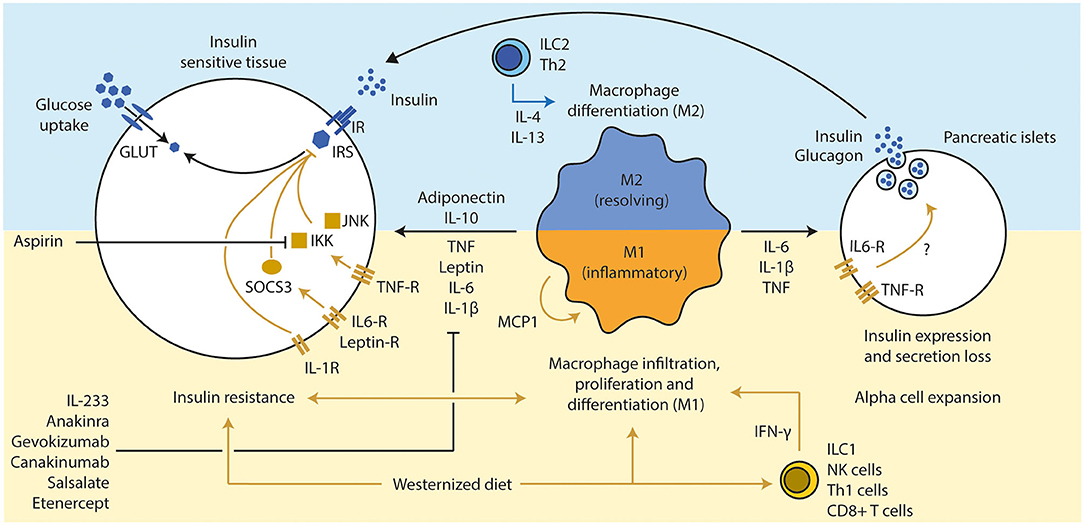
Figure 2. Inflammation influences beta cell function and insulin sensitivity. (1) A westernized diet induces insulin resistance and a pro-inflammatory immune response in metabolic active tissues. T cells (Th1 via IFN-γ and CD8+ T cells) have been discussed a secondary mediator that led to the attraction of macrophages, which are the main source of several pro-inflammatory cytokines. (2) An active pro-inflammatory response in those tissues enhances and deteriorates the extend of the insulin resistance via several inflammatory mediators (TNF, IL-6, and IL-1β), mainly secreted from M1 macrophages. Several downstream molecules (JNK, IKK, and SOCS3) interfere with the insulin signaling. Inhibition of those pro-inflammatory pathways led to improvement of insulin sensitivity and glucose tolerance (e.g., pharmacological treatments such as anakinra, gevokizumab, and aspirin). Anti-inflammatory cytokines such as IL-10 (expressed by various immune cell types, but mainly M2 macrophages) and adiponectin (from adipocytes) can resolve inflammation and improve insulin sensitivity. (3) Chronic high concentrations of pro-inflammatory cytokines lead to alpha cell expansion and beta cell dysfunction in pancreatic islets, which drives the progression toward T2D in obese subjects. Th, T helper cell; IFN, interferon; CD, cluster of differentiation; TNF, tumor necrosis factor; IL, interleukin; JNK, c-Jun N-terminal kinases; IKK, IκB kinase; SOCS, suppressor of cytokine signaling; GLUT, glucose transporter; IR, insulin receptor; IRS, insulin response substrate; MCP, monocyte chemoattractant protein 1.
Adipose tissue was one of the first tissues in which inflammation was highly correlated with insulin resistance. It contains a plethora of immune cells including T cells, eosinophils and mast cells that keep resident macrophages in an M2 polarized or alternatively activated state (19). Although these terms have fallen out of favor due to the identification of various other macrophage subtypes (20), we will stick to these terms for the sake of simplicity. Importantly, secretion of the anti-inflammatory cytokine IL-10 from M2 macrophages has been shown to protect lean mice from insulin resistance (21, 22). Obese mice and people with T2D have lower IL-10 expression and higher pro-inflammatory signals (21, 23). Obese humans with metabolic syndrome had lower levels of IL-10 compared to subjects without metabolic syndrome (24). Further, IL-10 overexpression in murine muscle tissue improved insulin sensitivity even under high fat diet conditions (22). These findings suggest a pivotal role of IL-10 in prevention of inflammation in people with metabolic abnormalities.
Other important, but less studied, anti-inflammatory cytokines include IL-4 and IL-13. IL-4 promotes glucose tolerance and inhibits adipogenesis (25). Further, it promotes an alternative macrophage activation (26, 27). Genetic variations in the IL-4 promotor have been associated with T2D susceptibility (28). However, the literature is scarce to draw conclusions. IL-13 shares a similar structure to IL-4, has anti-inflammatory properties (29) and promotes an alternative macrophage activation (30). Surprisingly, both cytokines were elevated in the blood circulation of obese humans compared to lean controls, which was associated with a lower physical activity (31). Another study supported this findings by showing increased levels in insulin resistant humans, which positively correlated with hyperglycemia (32). A study in mice points toward a disturbed IL-13 receptor activity (32). Obese animals have higher IL-13Rα2 activity, which inactivates and depletes IL-13. Therefore, the anti-inflammatory characteristics of IL-13 might be abolished despite high circulating levels. More research is necessary about anti-inflammatory cytokines in metabolic diseases.
M1 or classically activated macrophages have particularly been implicated in the metainflammation observed in metabolic diseases (33). M1 macrophages are generally responsible for secretion of pro-inflammatory cytokines and are associated with T2D development by altering local and distant tissue functions.
Certain cytokines (called chemokines) are able to attract immune cells to metabolic active tissues (34). Monocyte chemoattractant protein-1 (MCP1) is a strong chemokine for monocytes (35), and its expression was increased in obese human (36) and rodent adipose tissue (37, 38). In rodents, MCP1 overexpression in adipose tissue increased macrophage infiltration and mediated insulin resistance, whereas MCP1 knock out in combination with high fat diet feeding augmented development of insulin resistance compared to wild type mice (38). Although this observation was not observed in another study (39), higher chemokine production in obese adipose tissue has been associated with infiltration of immune cells and development of insulin resistance.
A recent study suggests that obesity-related insulin resistance precedes the infiltration of pro-inflammatory macrophages, giving insights in the cause and consequence dilemma (40). First, insulin resistance, which was genetically induced by knocking out mammalian target of rapamycin complex 2 (mTORC2), was shown to coincide with increased MCP1 expression, monocyte infiltration and differentiation into pro-inflammatory macrophages (M1). Second, insulin resistance in wild type mice preceded the accumulation of macrophages during diet-induced obesity. Thirdly, adipose tissue from obese insulin resistant patients had lower mTORC2 signaling, high MCP1 expression and high macrophage content (40). Thereby, insulin resistance might be the consequence of obesity and the cause for macrophage infiltration, which in turn amplifies the development toward diabetes.
Human pancreatic islets express and secrete MCP1 as well. MCP1 expression was increased after exposure to pro-inflammatory stimuli such as lipopolysaccharide (LPS) (41). Furthermore, overexpression of MCP1 in murine pancreatic beta cells led to robust macrophage infiltration in islets and spontaneous development of diabetes (42). These results suggest a major role of MCP1 in the pathogenesis of insulin resistance and beta cell dysfunction via immune cell infiltration.
Cytotoxic CD8 T cell infiltration into epididymal adipose tissue of obese mice precedes accumulation of macrophages. In fact, genetic depletion of CD8+ T cells lowered macrophage infiltration, adipose tissue inflammation and insulin resistance, whereas adoptive transfer of CD8+ T cells aggravated adipose inflammation (43). Further, T cells in obese adipose tissue produce more pro-inflammatory mediators compared to lean controls (44, 45). Moreover, the number of interferon (IFN) γ producing T helper (Th) 1 cells in human visceral adipose tissue positively correlates with systemic inflammatory tone, but are not associated with insulin resistance; though, the anti-inflammatory Th2 cells negatively correlated with insulin resistance (46). Further, obese mice lacking IFN-γ display lower adipose tissue inflammation and better glucose control (47, 48). Therefore, an interplay between different immune cells takes place, which has to be further eluted.
Innate lymphoid cells (ILCs) are recognized as the innate counterpart of T cells due to similar functionality (49). However, they miss the adaptive antigen receptors of T cells (50). They are divided into 5 subsets: Natural killer (NK) cells, ILC1, ILC2, ILC3, and lymphocytes tissue-inducer cells (LTi) (51). ILCs protect barrier tissues against pathogens and maintain immune homeostasis in several tissue types (52). Further, some of the ILCs have cytotoxic characteristics that are important to remove transformed cells and keep macrophages in homeostasis (51). Under steady state, cytotoxic ILCs kill adipose tissue macrophages to maintain homeostasis (53). However, this is impaired under high fat diet conditions leading to an increase of pro-inflammatory macrophages in the adipose tissue of obese mice and humans.
High fat diet increases NK cell numbers and the production of pro-inflammatory TNF in epididymal adipose tissue (54). Depletion of NK cells decreased adipose tissue macrophages, inflammation, and insulin resistance (54). Further, high fat diet drove the proliferation of ILC1 in adipose tissue and promoted a pro-inflammatory environment for macrophages via IFN-γ secretion (52). Similarly, obese subjects had higher ILC1 counts in adipose tissue and blood, which decreased after bariatric surgery (55). Therefore, NK cells and ILC1 seem to contribute to obesity phenotype by promoting a pro-inflammatory environment.
Obesity increased the activity and proliferation of NK cells, which stimulated the production of IFN-γ (56). That in turn stimulated the differentiation of macrophages and promoted insulin resistance (56). In contrast, high fat diet reduced the numbers of ILC2s in adipose tissue (57). They are important to sustain metabolic homeostasis in adipose tissue (58) and to keep macrophages in a M2 phenotype (57). IFN-γ suppressed the activity of ILC2s (58). Collectively, several recent studies indicate the involvement of ILCs in (adipose tissue) inflammation; potentially posing the unknown immunological trigger that induces a pro-inflammatory environment. However, the ILC research is relatively young to draw major conclusions yet.
Tumor necrosis factor (TNF), which is mostly expressed and secreted by adipose tissue macrophages (59), was one of the first cytokines shown to be increased in adipose tissue (60) and circulation of people with T2D and obesity (61). TNF expression in adipose tissue was inversely correlated to insulin sensitivity in obese people without T2D compared to heathy lean controls (62). Interestingly, TNF infusion in rats disturbed insulin sensitivity already on day one (63). Further, mice deficient in TNF and TNF receptor 1 & 2 were protected from diet-induced insulin resistance (64). Neutralization of TNF by infusing reactive immunoglobulins in rats improved insulin-stimulated glucose uptake (60). Lastly, weight loss in obese subjects improved insulin sensitivity and reduced TNF expression in adipose tissue (65), suggesting a pivotal role of obesity on inflammation and insulin sensitivity. TNF inhibition has been suggested for the treatment and prevention of T2D (66, 67). However, large and long-term clinical studies are warranted.
Several studies addressed the mechanism behind the insulin disturbing effects of TNF (68). TNF activates the intracellular IκB kinase (IKK), which comprises two kinases (alpha and beta), leading to the activation of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) (69) and transcription of inflammatory genes. Interestingly, inhibition of IKKβ by the anti-inflammatory agent aspirin or sodium salicylate (70) increased insulin sensitivity in mice (71) and humans (72, 73). Further, TNF is able to activate c-jun N-terminal kinase (JNK) (74), which is a direct inhibitor of the insulin signaling pathway (75). JNK activity was elevated in several tissue types of obese mice (76). Absence of JNK1 resulted in decreased adiposity and improved insulin sensitivity in mice (76–78). Both downstream molecules (IKK and JNK) of TNF interfere with insulin receptor substrate 1 (IRS1) and thereby stop insulin signaling (69, 79). Furthermore, within pancreatic islets, TNF production by macrophages drives dysfunction of insulin-producing beta cells and may directly mediate insulin resistance of pancreatic beta cells (80, 81). Therefore, TNF plays a crucial role in the development of insulin resistance in several tissue types with high glucometabolic relevance.
Adipose tissue macrophages are the main source of the highly studied pro-inflammatory cytokine IL-6, with estimated contributions of 15–35% of the total circulating IL-6 (59, 82). Mohamed-Ali et al. (82) measured the IL-6 concentration in arterial as well as venous blood from subcutaneous adipose tissue. They found two times more IL-6 in the venous blood compared to arterial blood, indicating major portions of circulating IL-6 is secreted from the adipose tissue. Surprisingly, they found no increase in TNF between both vessel types. Further, Weisberg et al. (59) confirmed that most of the IL-6 (and TNF) secreted from adipose tissue is originating from macrophages. Chronically elevated levels of IL-6 have been shown to decrease hepatic insulin sensitivity in vitro (83), to induce hyperinsulinemia in mice (84) and to mediate insulin resistance in murine muscle tissue (85). Mechanistically, IL-6 increases the activity of suppressor of cytokine signaling 3 (SOCS3), which inhibits several downstream mediators of insulin receptor signaling (86–88). Interestingly, knocking out the receptor for IL-6 in immune cells did not protect from insulin resistance, but disturbed immune homeostasis in mice (89, 90), suggesting a fine-tuned mechanism.
In the pancreas, the IL-6 receptor is mostly expressed in the endocrine portion, with higher levels in alpha cells (91). IL-6 expression was increased in pancreatic islets from obese mice (92) and associated with expansion of alpha cells, a known histological observation in the islets of people with T2D. Indeed, IL-6 knock out in obese mice inhibited the expansion of alpha cells, accompanied by reduced glucose stimulated insulin secretion (GSIS) (91). In addition, IL-6 administration in mice enhanced insulin secretion by increasing the incretin glucagon-like peptide 1 (GLP1) from intestinal L cells and pancreatic alpha cells (93, 94). Therefore, IL-6 has physiological effects on pancreatic islets, but the underlying mechanism is yet to be revealed.
The pro-inflammatory cytokine IL-1β has been implicated in the development of obesity and T2D (95–97). IL-1 receptor knockout mice are protected against high-fat diet induced glucose intolerance and adipose tissue inflammation (98). However, IL-1β has been shown to have physiological roles in glucose metabolism. Feeding increased IL-1β secretion from peritoneal macrophages in a glucose depended manner, which contributed to postprandial insulin secretion. Lack of endogenous IL-1β reduced postprandial insulin (99). Therefore, although IL-1β plays a physiological role in glucose metabolism, chronically elevated levels might lead into T2D.
In T2D, pancreatic islets are infiltrated by pro-inflammatory macrophages (92, 100), which drive the production of IL-1β (101) via the NLRP3 (NACHT, LRR, and PYD domains-containing protein 3) inflammasome (102). Initially, IL-1β at low concentrations may be beneficial by promoting β-cell proliferation (103); however, chronically elevated concentrations might lead to beta cell failure (66, 104). Administration of an IL-1 receptor antagonist improved glucose tolerance via improving beta-cell function and systemic inflammation in humans (15).
Further, chronic IL-1β administration is able to induce insulin resistance in adipose tissue in vitro (105). The mechanisms by which IL-1β mediates insulin-resistance have been attributed, at least in part, to downregulation of insulin substrate receptor-1 (IRS-1) (95) and aberrant activity of the transcription factors NF-κB and FOXO1 (Forkhead box protein O1) (106) during obesity or inflammatory conditions. Therefore, IL1β has physiological roles in glucose metabolism with deleterious consequences after chronic exposure to high concentrations.
Macrophages play a crucial role in liver inflammation. Resident hepatic macrophages undergo activation and thereby alter inflammatory pathways in obesity conditions (107). In addition, there is a pronounced increase in macrophage (and other immune cell) infiltration in the liver. This leads to production of inflammatory cytokines that generate insulin resistance in hepatocytes and drive T2D-related diseases such as non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) (108, 109). Nevertheless, a recent study in mice and humans with obesity could not confirm that obesity per se induces a pro-inflammatory phenotypic switch in liver macrophages (110), although their depletion prevents diet-induced insulin resistance (111). Interestingly, liver macrophages were shown to contribute to the development of insulin resistance independent of production of inflammatory factors. Rather, liver macrophages were shown to produce insulin-like growth factor–binding protein 7 (IGFBP7), which directly altered insulin receptor signaling in the liver (110).
Inflammation is a biological response of the immune system that can be triggered by exposure to pathogens, damaged cells and toxic compounds. The reaction can be acute or chronic, potentially leading to damage in several tissues. In particular, bacterial components such as lipopolysaccharide (LPS), a component of the cell wall of Gram-negative bacteria, have been proposed as a source for metabolic inflammation since LPS was found to be increased in the circulation of people with diabetes (112). LPS was postulated to enter the circulation via chylomicrons or via increased intestinal permeability (113) where it induces an inflammatory response in systemic sites. In mice, chronic LPS infusion perturbed glucose tolerance by inducing hepatic insulin resistance and hampering glucose-stimulated insulin secretion (114). In humans, associations between LPS concentrations and several aspects of the metabolic syndrome were also noted (112, 115).
Two phenomena have been noted: “metabolic endotoxaemia” (116, 117) and “postprandial inflammation” (118). The former describes an inflammatory response to increased systemic levels of LPS due to a “leaky gut” (114). The latter defines the increase in circulating endotoxins and other inflammatory markers after a meal, particularly meals rich in fat (119–121). Interestingly, LPS and long-chain saturated fatty acids, such as palmitate, act synergistically in activating inflammatory signaling in macrophages (122), highlighting a link between a westernized diet rich in saturated fatty acids (123) and postprandial inflammation.
From the present data, it is clear that a dysregulated immune system is strongly associated with obesity and T2D. Moreover, several inflammatory components directly alter glucose tolerance and insulin sensitivity, which provides evidence for a causal role of inflammation in these pathologies. The intertwined relation between metabolic disorders and low-grade inflammation has been coined “metainflammation.” This illustrates that, in contrast to an instrumental acute inflammatory response to pathogens and tissue damage, chronic low-grade inflammation as observed in obesity and T2D has detrimental consequences for human metabolism.
The Intestinal Microbiota in Metabolic Diseases
Within the human intestine, a complex mutualistic relation exists between trillions of microorganisms, collectively known as the gut microbiota, and the host (124). While much focus is on the bacterial component of this community, recent studies highlight the importance of other microorganisms such as bacteriophages (125, 126) and fungi (127, 128) in human health. It is beyond the scope of this review to cover the role of other members of the microbial community in human metabolism. We will here focus on the bacterial component that we will refer to as microbiota. The gut microbiota is critical for human health (129–131). It plays a key role in digestion, production of metabolites with the potential to alter human metabolism (e.g., short chain fatty acids) and development of the immune system. The immunomodulatory properties of the gut microbiota are of particular interest in the context of metainflammation as observed in obesity and T2D (132).
Germ-free mice housed in aseptic conditions, have an underdeveloped immune system. These mice typically have impaired development of gut-associated lymphoid tissues (GALT), decreased serum immunoglobulin levels, smaller spleens and mesenteric lymph nodes with low levels of tissue resident macrophages (133). In line, the mice have an insufficient immune response to pathogens (134, 135). Introducing bacteria into germ-free mice restores immune system formation and functioning which exemplifies the critical and dynamic relation between host immunity and the gut microbiota (136). These findings have not only been related to presence of bacteria but also to the microbial metabolites such as SCFA (137) or components such as commensal DNA containing CpG (unmethylated cytosine phosphate guanosine dinucleotides) motifs (138) and polysaccharide A (PSA) (139). The development of a symbiotic relationship during early years may determine the development of several diseases (140).
Importantly, germ-free mice are resistant to diet-induced obesity (141, 142). Introducing a single bacterium that has been associated with obesity (Enterobacter cloacae) into germ-free mice led to weight gain, a disturbed glucose tolerance, higher systemic lipopolysaccharide binding protein (LBP) concentrations and lower adiponectin levels (143). Similarly, germ-free mice that received the microbiota from obese donors gained more energy from the diet compared to germ-free mice that received the microbiota of lean donors (144, 145). These studies were the first to show that an obese phenotype can be transferred via the gut microbiota and hence indicate causality.
Although a definition of a “healthy gut microbiota” is generally lacking, a plethora of disease conditions are associated with a microbiota composition that is different from a healthy control group (124, 146). The most preferred term in this context is microbial “dysbiosis,” which can describe a bloom of pathobionts, loss of commensals or/and loss of diversity (147). However, the term falls out of favor due to lack of a clear definition. An altered microbiota is present in people with obesity (144, 148) and T2D (6, 149) (Figure 3). It is generally characterized by expansion of usually underrepresented microorganisms (often opportunistic pathogens) (6, 143) and lower phylogenetic alpha diversity (8, 150, 151). Diet (152) and antibiotic use (153) have been identified as drivers of these changes.
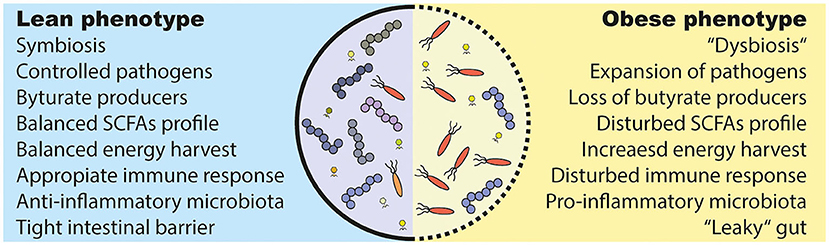
Figure 3. Alterations in the obese and diabetic gut microbiota. (1) Under healthy conditions (lean), the gut microbiota lives in symbiosis and provides the host with several beneficial functions. For example, it produces short chain fatty acids (SCFAs) that are used as an energy source and have effects on several host tissues. However, several bacteria are able to induce an inflammatory response and can even breach the intestinal barrier, which has to be prevented by a proper immune response. (2) The gut microbiota in metabolic diseases is often described as “dysbiotic,” meaning that there is an expansion of normally underrepresented bacteria (in particular opportunistic pathogens) and a lower diversity. A disturbed intestinal immune response and a westernized diet is discussed as causes. Further, a westernized diet induces a “leakiness” of the gut. Parts of (opportunistic) bacteria are able to cross the intestinal barrier and induce a pro-inflammatory response in the host. Lastly, people with obesity show an increased energy harvest by the gut microbiota and a different SCFAs profile as lean people, which might have deleterious consequences for the host health.
The obesity-associated microbiota has been shown to extract more energy from the diet compared to lean controls, proposing a role in the development of excessive energy stores (144, 145, 154). Further, an enrichment of pro-inflammatory bacteria (e.g., Escherichia coli) at the expense of anti-inflammatory bacteria (e.g., Fecalibacterium prausnitzii) in T2D has been reported several times (6, 7, 149). An increase in typical pro-inflammatory Gram negative bacteria might be a plausible source for the metainflammation seen in metabolic diseases (155, 156). However, this is only confirmed in animal studies and in associative nature in humans.
A reduced number of anti-inflammatory bacteria such as F. prausnitzii has been related to a disturbed SCFA production of the gut microbiota (6). SCFAs, which result from microbial degradation of fibers, have several beneficial effects on host metabolism (157). Surprisingly in obesity, an increased SCFA production has been observed (158, 159), which was proposed to be responsible for higher energy extraction (160) and was dependent on the diet (161). It is estimated that 5–10% of our daily energy is provided by fermentation processes by the gut microbiota (162). However, in diabetes a reduced SCFA production or uptake, in particular the anti-inflammatory acting butyrate (163), has been suggested (6). However, strong evidence for this concept is still lacking. A recent study found a causal relation between a host genetic-driven increase in butyrate production which was associated with an improved insulin response. Vice versa, abnormalities in the production or absorption of propionate were related to increased risk of developing T2D (164). Absorption of intestinal SCFAs is very efficient, leaving only 5–10% in feces (165). Large amounts of the intestinal butyrate is directly used by the colonic enterocytes as an energy source (166, 167). However, it is difficult to measure real turnover and concentrations of SCFAs due to rapid utilization by intestinal cells and microorganism. Thus, although SCFAs are involved in host metabolism, the mechanism and extent are still elusive.
In summary, changes of the gut microbial composition and a lower microbial diversity in obese subjects has been associated with higher inflammatory tone (8, 168, 169). This implicates a role for the gut microbiota in the low-grade inflammation observed in people with metabolic syndrome (170, 171).
Gut Barrier Function and Metabolic Inflammation
A proper gut barrier function is critical to prevent bacterial infiltration from the gut into the circulation and periphery. Many pathologies, including inflammatory bowel disease (172), liver diseases (173), and metabolic syndrome (114), show signs of a disturbed gut barrier function leading to bacterial translocation (174). A “leaky” gut can facilitate translocation of bacterial components form the intestine into the periphery (174). The following part summarizes evidence of bacterial leakage in metabolic diseases.
Intestinal Barrier Integrity
One of the first papers implicating a link between gut barrier function, insulin resistance and increased levels of circulating levels of LPS were derived from a study in obese mice (114) (Figure 4). Antibiotic treatment reduced intestinal and systemic LPS along with an improved glucose tolerance, underscoring the gut microbiota as the endotoxin source (155). Mechanistically, these findings were related to high-fat diet (HFD) induced reduction in expression of tight junction genes, which was linked to increased intestinal permeability (155, 175). Tight junctions are protein complexes that prevent leakage of various compounds along paracellular spaces (175). LPS directly increased intestinal permeability in vitro and in vivo in mice, suggesting a link between increased intestinal LPS and the gut tight junction expression (176).
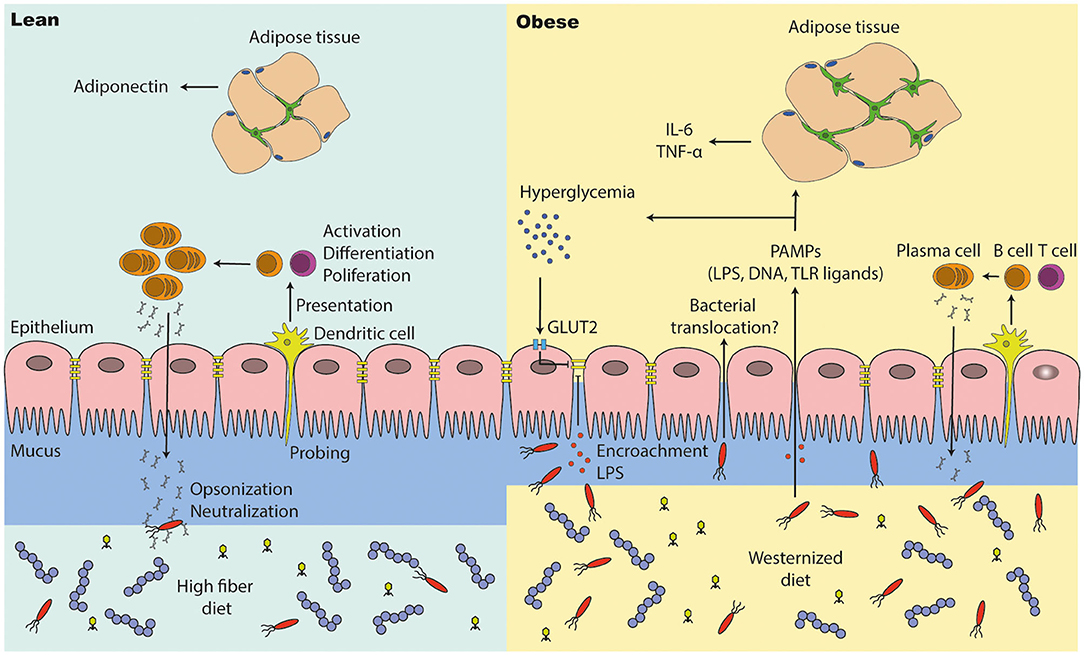
Figure 4. The intestinal barrier is disturbed in people with obesity and diabetes. (1) A high fiber diet supports intestinal barrier function by improving intestinal tight junction expression and immune cell function. Antigen presenting cells (e.g., dendritic cells) are probing the intestinal environment, present the antigens to T and B cells, which may lead into immune tolerance or an inflammatory response (cytokine and antibody expression). (2) The intestinal integrity is affected in people with metabolic syndrome. They have a thinner mucus layer, which leads to penetration of opportunistic bacteria; lower levels of IgA positive B cells and a lower IgA secretion, which may end into microbial alterations (outgrowth of opportunistic pathogens). A westernized diet decreases intestinal tight junction expression, which results into translocation of bacteria and pathogen associated molecular patterns (PAMPs). High glucose levels (hyperglycemia) reduces tight junction expression via GLUT2, promoting bacterial translocation in people with diabetes. PAMPs in the periphery induce inflammation in several other tissues such as the adipose tissue, where macrophages proliferate and accumulate. In particular, adipose tissue macrophages are responsible for low grade inflammation (high pro-inflammatory cytokine levels and less anti-inflammatory cytokines).
Interestingly, hyperglycemia directly drives intestinal barrier permeability though Glut2-dependent reprogramming of intestinal epithelial cells (177). Loss of gut integrity increased influx of bacterial products in the periphery of mice and potentially humans, proposing an interplay between inflammation and a disturbed glucose tolerance. Further, systemic toll like receptor (TLR) ligands were positively correlated with the long-term glucose marker HbA1c in humans (177). Although metabolic endotoxemia in obesity and T2D becomes more evident, human studies showing a higher intestinal permeability and disturbed tight junction expression are still lacking.
Several groups tried to identify “translocating” bacteria in peripheral organs and tissues by measuring or sequencing the 16s ribosomal RNA of bacteria. First, more bacterial DNA was found in the circulation of diabetic subjects and obese mice compared to healthy controls, with predominantly Proteobacteria identified (178). Secondly, DNA from Gram-negative bacteria was detected in adipose tissue of obese women and correlated with a decrease in adiponectin levels, suggesting a link between bacterial translocation and activation of inflammatory pathways in (adipose) tissue (179). Further, an increased adherence of pro-inflammatory bacteria to the epithelium might indicate that bacteria are passing the epithelium (178). However, the concept or bacterial translocation is still very controversial due to technical limitations to detect small amounts of bacteria in systemic tissue sides.
Immunoglobulins
Immunoglobulins control the gut microbiota and prevent bacterial invasion by binding to microorganisms directly to block the attachment to the host. In addition, immunoglobulins binding marks (so called “opsonization”) bacteria for phagocytosis and antigen presentation to dendritic cells. Moreover, immunoglobulins neutralize microbial toxins (180). A proper induction in childhood is essential to build up an immune tolerance against gut bacteria and to prevent microbial changes (181, 182). Particularly, secretory IgA are pivotal players in shaping gut microbiota composition, both in mice and humans, and defective IgA secretion has been shown to shift microbial communities (183–185). An altered intestinal immunoglobulin response has been noted for several diseases including undernutrition (186), inflammatory bowel disease (187), and obesity (188).
A recent study reported fewer IgA+ immune cells and less IgA secretion in obese mice (188). Further, IgA deficient obese mice had impaired glucose tolerance, higher macrophage content in adipose tissue and more systemic endotoxins. Antibiotic treatment improved glucose tolerance, suggesting the involvement of the gut microbiota in this phenotype. The anti-diabetic drug metformin improved IgA response in obese mice and bariatric surgery increases fecal IgA in human subjects (188). Further, a higher flagellin expression of motile bacteria and a disturbed antibody response against flagellins leading to bacterial encroachment on the intestine has been discussed in obesity and inflammatory bowel diseases (189).
Immunoglobulins are produced by B cells, which had impaired function in T2D (190). B cells accumulated in visceral adipose tissue of obese mice (191) and produced more pro-inflammatory cytokines compared to B cells in adipose tissue of lean controls (192). B cell depletion in obese mice decreased inflammation and improved glucose tolerance (192), suggesting that disturbed B cell function in obesity is linked to inflammation and glucose metabolism.
T cells, particularly follicular helper T cells, are important players in the induction of a proper immune and antibody response (193, 194). Depletion of their function led to a disturbed intestinal IgA activity and the development of metabolic syndrome with age. IgA inappropriately targeted Clostridia species and allowed for the outgrowth of Desulfovibrio. The former suppresses and the latter enhances host lipid absorption via CD36 modulation (194), which plays essential role in fatty acid uptake (195). This study gave insight in the development of microbial alterations, which are present in metabolic diseases, via IgA. It is clear that immunoglobulins play a major role in gut homeostasis (196), but only a few recent studies show the involvement of immunoglobulins in the disease progression of metabolic diseases.
Bacterial translocation, as discussed above, might be an interesting concept to explain inflammatory changes in obese and diabetic tissues that leads into insulin activity loss (beta-cell dysfunction and insulin resistance). However, bacterial translocation as a concept is still very controversial due to lack of strong evidence in humans. There currently is a trend to look into the role of bacterial metabolites as causative in the development of metabolic diseases rather than presence or absence of bacterial strains. In the following part, we will discuss evidence of metabolites involved in metabolic disease progression.
Mechanistic Insights in Microbiota-Induced Metabolic Inflammation
Low grade chronic inflammation in obesity and T2D has been studied for more than 25 years (60), with several pathways to a large extend disentangled (197). In contrast, observations on changes in the gut microbiota of obese and diabetic people were revealed no longer than 15 years ago (6, 144, 198). Particularly a westernized lifestyle is known to change not only the immunemetabolism, but also the gut microbiota (199). The connection between gut microbial changes and metainflammation in metabolic diseases was established even more recently. The following part addresses mechanistic insights from rodent studies and the few causal relationships found in humans.
Pattern Recognition Receptors
The intestinal epithelium is the first line of defense against intestinal microorganism (200). A tight intestinal wall with a properly working immune system is necessary to avoid invasion. Pattern recognition receptors (PRR) are expressed on most cells of the innate immune system. PPRs recognize microbial components and hence are crucial parts of the immune system. They include the widely-studied membrane-bound TLR superfamily. These receptors are mainly expressed on epithelial cells and cells of the innate immune system (201). TLRs detect pathogen-associated molecular patterns (PAMPS) such as LPS or flagellin and facilitate an inflammatory response (202). Acute inflammation is important to clear of infected, abnormal or damaged tissue. The process needs to be resolved to avoid unnecessary damage on healthy tissue. Usually this resolution is disturbed in conditions of chronic inflammation (203) (Figure 5).
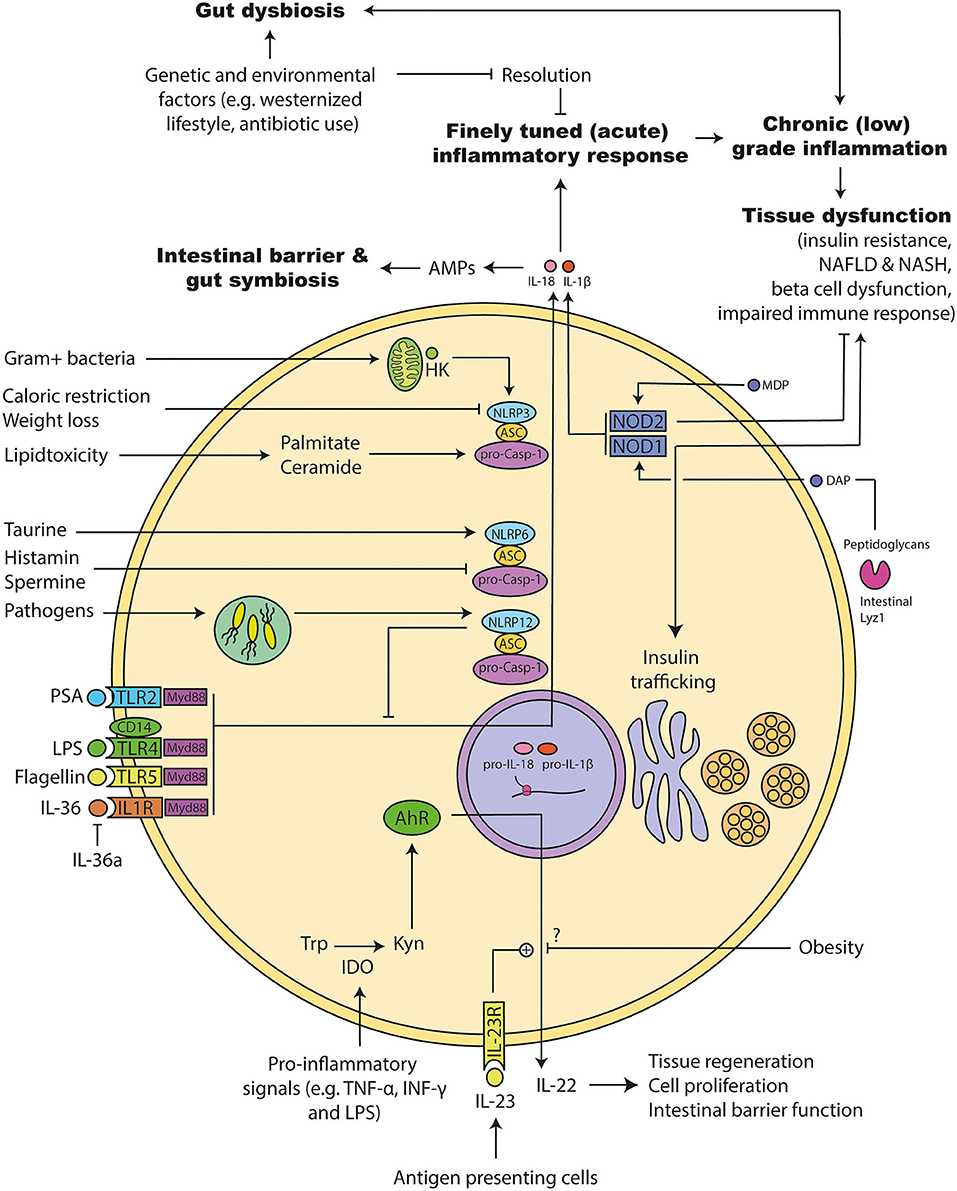
Figure 5. Molecular mechanism involved in microbiota promoted metainflammation. (1) The acute inflammation has to be resolved to avoid chronic inflammation that can induce tissue damage. Genetic and environmental factors can disturb this system leading to a chronic (low-grade) inflammation. Several of following pathways are disturbed during obesity and diabetes: (2) Tolle like receptors (TLRs) and their adapter molecules are important for recognizing bacterial components. Activation triggers different inflammasomes to initiate an inflammatory response. Similarly, interleukin (IL) 36 leads to the activation of the inflammasomes and a pro-inflammatory response that can be inhibited by the endogenous IL-36 antagonist. (3) Inflammasomes consists of different proteins: NACHT, LRR, and PYD domains-containing protein (NLRP), Apoptosis-associated speck like protein containing a caspase recruitment domain (ASC), and pro-caspase. Upon activation they can mature IL-1β and IL-18. NLRP12 has dual roles: It acts pro-inflammatory response via maturation of IL-1β and anti-inflammatory by inhibiting down-stream signals of several TLRs. NLRP6 is important for the maturation of IL-18 and antimicrobial protein expression in the intestine. Its activity can be increased by the microbial metabolites taurine and decreased by Spermidine as well as Histamine. It is important for maintaining a gut symbiosis and intestinal barrier function. NLRP3 activity can be increased during lipid accumulation. Further, hexokinases can detect intracellular particles of Gram positive bacteria and activate NLRP3, which leads to the maturation of the pro-inflammatory acting IL-1β. (4) Pro-inflammatory signals can increase the intracellular enzyme indoleamine 2,3-dioxygenase (IDO), which in turn metabolizes tryptophan to kynurenine. Kynurenine can activate the transcription factor Aryl hydrocarbon receptor (AhR), which induces the release of IL-22. IL-22 is important for the intestinal barrier function, which can be promoted via IL-23. Obesity interferes with that response, but the exact mechanism is not clear. (5) Nucleotide-binding oligomerization domain-containing protein (NOD) 1 can be activated by bacterial diaminopimelic acid (DAP). DAP can be cleaved by intestinal Lyzozyme (lyz) 1 enzymes from bacterial peptidoglycans. NOD1 has dual roles: It induces insulin resistance and insulin trafficking in beta cells. NOD2 can be activated by bacterial muramyl dipeptide (MDP). NOD2 inhibits the development of insulin resistance.
TLRs are a crucial tool of our immune system to detect invading microorganism. Several TLRs have been associated with the development of the metabolic syndrome, in particular TLR2 and TLR4 (204). TLR4 recognizes several ligands, but most predominantly LPS (205). Monocytes from people with T2D showed higher expression of TLR4, along with higher cytokine and LPS levels (206). Interestingly, insulin infusion for several hours is able to suppress the expression of TLRs on monocytes from people with T2D (207), suggesting that insulin resistance promotes higher expression of TLRs (and thereby inflammation). Further, TLR4 has been suggested to play a role in beta cell failure during diet-induced obesity in mice, as HFD-fed TLR4 knock out mice exhibit preserved insulin secretory function, lower inflammatory markers and no macrophage infiltration in pancreatic islets (208). Further, a combined knockout of TLR4 and TLR2 in mice increased proliferation of beta cells, but not glucagon-producing alpha cells, and improved glucose tolerance in obese mice (209). Interestingly, absence of CD14, a co-receptor of TLR4, protected mice from most of HFD and LPS induced metabolic deteriorations (114). Thereby, TLR4 might be an interesting treatment target and an important mediator of insulin resistance, metainflammation and beta cell failure.
TLR5 has been associated with the development of metabolic diseases (210). TLR5 is expressed on several cell types and recognizes mainly flagellins, a protein involved in the motility of bacteria (211). Knock out of TLR5 in mice led to the development of metabolic syndrome, which correlated with changes in the gut microbiota. Transfer of feces from TLR5 knockout mice to wild type mice conferred many features of the metabolic syndrome (210). Importantly, follow-up studies suggest that the observed metabolic changes were likely due to housing techniques rather than the genetic profile (212). TLR5 knock out in intestinal epithelial cells induced low grade inflammation and metabolic syndrome, which was reversed by antibiotics, implying a connection with the gut microbiota (213). TLR5 is likely involved in the development of a metainflammation.
Myeloid differentiation factor 88 (Myd88) is a downstream target of most TLR- and IL-1 receptor-mediated signaling pathways and a central player in innate immune signaling (214). Myd88 knock out in mice induced metabolic syndrome with an increase in bacterial translocation (178). Further, hepatocyte specific deletion of MyD88 predisposed mice to glucose intolerance, inflammation, hepatic insulin resistance and induced gut microbiota alterations (215). In contrast, inducible deletion of Myd88 in intestinal epithelial cells partially protected against diet-induced obesity, diabetes and inflammation. The protective phenotype was transferred by transplanting feces from intestinal Myd88 knock out mice into germ-free mice. Targeting MyD88 (inducible knock out) after onset of obesity reduced fat mass and inflammation, suggesting a link between microbiota and MyD88 in the development of obesity (216). A finely tuned MyD88 activation therefore is crucial for the homeostasis of several tissues and is involved in metabolic diseases.
Similar to TLRs, nucleotide-binding oligomerization domain-containing protein 1 (NOD1) and NOD2 recognize PAMPs inside of the cell. NOD1 and NOD2 are cytosolic receptors that respond to bacterial peptidoglycans (217) and have been associated with the development of insulin resistance (218, 219). Interestingly, NOD1 and NOD2 double knock out protected mice from inflammation and peripheral insulin intolerance. Direct NOD1 activation led to insulin resistance (218) and gut microbial derived NOD1 ligands promoted insulin trafficking in beta cells (220). The latter is most likely a compensatory mechanism after the development of an insulin resistance. On the contrary, NOD2 signaling is protective against T2D. Defective NOD2 sensing promotes diet-induced inflammation, microbial changes and insulin resistance (221). NOD2 activation via bacterial cell wall-derived muramyl dipeptide (MDP) improved insulin resistance (219). From the present data it seems that NOD2 signaling is beneficial whereas NOD1 is deleterious for insulin sensitivity and beta cell function.
Inflammasomes
Inflammasomes are a central component of the innate immune response and have been implicated in several metabolic diseases (222). Inflammasomes are intracellular multimeric complexes composed by NLRs (NOD-like receptors), ASC (Apoptosis-associated speck-like protein containing a CARD), and pro-caspase-1. They are formed in response to PAMPs and DAMPs (damage associated molecular patterns) and control the activation and secretion of IL-1β and IL-18 (223). Thereby, they act as intracellular sentinels of inflammatory and metabolic cellular disturbances. Knocking out of several NLRs in mice led to changes in the gut microbiota (224–229) and aggravation of diet-induced metabolic syndrome (229).
NLRP3 (NOD-like receptor family, pyrin domain-containing 3) is one of the most studied NLRs. It responds to a wide range of infectious and endogenous molecules, such as several bacterial cell wall components (222, 230) and saturated fatty acids (97). However, none of these molecules seem to directly interact with NLRP3. Common cellular signals might activate NLRP3 (231). That priming process leads to the activation of caspase-1 and subsequent IL-1β secretion (232). Several studies linked increased NLRP3 expression in adipose tissue and monocytes to obesity (233) and T2D (234), respectively. Interestingly, calorie restriction in obese and diabetic subjects reduced activation of NLRP3 in adipose tissue and coincided with reduced inflammation and improved insulin sensitivity (232).
Further, there is a direct connection between cell metabolism and inflammation. Hexokinases, enzymes that are mainly involved in glucose metabolism, were able to detect bacterial peptidoglycans and thereby activated NLRP3 (230). Several studies disclosed that an ablation of NLRP3 or of the inflammasome components ASC and caspase-1 in mice prevented obesity-induced inflammation in fat depots and liver, ameliorated insulin signaling, increased energy expenditure and prevented liver steatosis as well as pancreatic damage (225, 232). Mechanistically, NLRP3 inflammasome participated in metainflammation and lipotoxicity by sensing intracellular rise of the sphingosine ceramide (232), the saturated fatty acid palmitate (97) and reactive oxygen species (235). Therefore, NLRP3 is a major component in the inflammatory processes deranged in metabolic diseases.
Further, ablation of ASC, a common adapter molecule to all inflammasomes, on a db/db (leptin-deficient mice) background resulted in increased obesity and loss of glycemic control under high fat diet conditions. Moreover, this phenotype was transmissible to wild type mice by co-housing and abrogated by antibiotic treatment pointing to an essential role of gut microbiota in the phenotype. In the same paper, the authors found that deficiency in components of the inflammasome (caspase-1, ASC, NLRP3, or NLRP6) aggravated the progression from NAFLD to NASH and disease severity, and that this phenotype was transmissible to wild type mice by co-housing. These effects were attributed to a different cologenic microbiota in inflammasome deficient mice that increased the amount of TLR4 and TLR9 bacterial agonists into the portal circulation, driving hepatic TNF induction and, hence, NASH (225). However, the authors used unrelated wild-type mice instead of littermate controls. Two later studies highlight the importance of using proper controls, namely related littermates from the knock out mice, to exclude the influence of housing conditions and other genetic variations in the littermates (236, 237). Nevertheless, these findings suggest a role of inflammasomes and their components in several comorbidities of obesity.
Interestingly, Levy et al. provide evidence of a direct link between commensal-derived metabolites and NLRP6 inflammasome activity (227). The authors found that histamine and spermine inhibited NLRP6-dependent IL-18 production, while taurine enhanced it and consequentially stimulated the IL-18 driven production of antimicrobial peptides in the intestinal epithelium. However, a two later papers disputed the impact of NLRP6 and ACS-associated inflammasome in shaping the microbiota composition using littermate-controlled experimental design in two geographically separated vivarium to compare the phylogenetic composition of wild type, NLRP6−/−, ASC−/− mice microbiota and minimize non-genetic confounders (236, 237). Therefore, NLRP6 and its components are involved in metainflammation via the gut microbiota, but the exact mechanism is still elusive.
The NLRP12 inflammasome has more recently been implicated in the development of metabolic diseases. Interestingly, it was first though to induce a pro-inflammatory response, but recently a dual role became evident (238). It has been shown to dampen and resolve inflammation, in particular in the intestine, in a microbiota dependent manner (228) and by promoting the growth of beneficial bacteria (228). NLRP12 expression in adipose tissue was reduced in obese compared to lean subjects. In mice, deletion of NLRP12 increased weight gain, adipose tissue deposition, blood glucose and pro-inflammatory macrophage expansion. Interestingly, NLRP12 knock out in mice induced gut microbiota alterations with decreased number of SCFA producing bacteria. Depletion of the microbiota, germ-free condition or co-housing with wild type mice was sufficient to restrain inflammation, obesity, and insulin tolerance in knock out mice, highlighting a direct link of the gut microbiota in obesity and inflammation (229). Overall, these findings form a strong link between microbiota, inflammasomes, and metabolic diseases.
Cytokines
Several cytokines are associated with the development of the metabolic syndrome (as discussed above), but only a few studies found direct links between cytokines and the gut microbiota. IL-22 is essential for maintaining an antimicrobial response and intestinal barrier function (239). Its production is induced by intestinal type 3 innate lymphoid cells (ILC3) and influenced by bacterial tryptophan metabolism (227, 240, 241). Induction of IL-22 was impaired in obese mice, in particular during infection with Citrobacter rodentium (242), suggesting an impaired immune response against pro-inflammatory bacteria. Mice lacking the IL-22 receptor on a high fat diet are prone to develop metabolic syndrome and administration of IL-22 in obese mouse models reversed several symptoms such as hyperglycemia and insulin resistance (242, 243).
Protective effects of IL-22 on pancreatic islets may explain these findings. Administration of IL-22 reduced ER stress and inflammation with a restored glucose tolerance in mice (244). Some bacterial metabolites such as butyrate were able to induce IL-22 secretion from pancreatic innate lymphoid cells, which improved beta cell proliferation and inflammation (245). Next, inactivation of IL-22 (and IL-23) signaling in mice deteriorates intestinal barrier, microbial alterations and expansion of pathogenic bacteria causing systemic increase in LPS and Trimethylamine N-oxide (TMAO), showing direct connection of those particular cytokines, the gut microbiota and obesity related comorbidities such as atherosclerosis (246). On the other hand, T cell derived IL-22 amplified IL-1β driving inflammation in human adipose tissue (190), suggesting either a tissue specific activity of IL-22 or a disturbed action in metabolic syndrome.
Further mechanistic insights of the IL-22 signaling were given in a recent studies, where indoleamine 2,3-dioxygenase 1 (IDO), an enzyme that is present in many immune cells and activated during inflammation, was linked to the gut microbiota and obesity (247). IDO metabolizes tryptophan along the kynurenine pathway, which is a potent AhR ligand and shown to disturb an anti-tumor response (248). Interestingly, it becomes more evident that the tryptophan metabolism plays an important role in metainflammation (249) and energy homeostasis (250). Although IDO activity is important for a proper regulatory T cell function (251), a high enzymatic activation is associated with cardiovascular complications and inflammation (252). The enzyme activity was shown to be increased in obesity (247). Deletion or inhibition of the enzyme improved insulin sensitivity, improves gut barrier and chronic inflammation. Neutralization of IL-22 abrogated the protective effects of IDO deletion, highlighting the beneficial roles of IL-22 in intestinal health and glucose tolerance (247). More studies are needed to elucidate the dysregulation of the tryptophan metabolism and its metabolites in metainflammation.
ILC3s are an important source of IL-22 in the intestine, which is regulated by the adaptive immune system (CD4+ T cells) (253). The lack of CD4+ T cells led to an upregulated activity of intestinal ILCs that disturbed the host-microbe interaction and reduced intestinal lipid metabolism (253). Further, elevated levels of IL-23 and IL-22 in young mice decreased the production of pancreatic enzymes, explaining the disturbed lipid metabolism to some extend (254). However, inactivation of both cytokines led to deterioration of the intestinal barrier, dysbiosis and a pro-atherogenic environment (increase in LPS and TMAO) (246). These findings highlight a fine tuned interplay between various immune cell types and mediators. Generalizing them as pro- or anti-inflammatory as well as beneficial or deleterious is in most cases not possible.
IL-23 has mainly pro-inflammatory activities (255), which is mostly secreted from activated macrophages and dendritic cells (DCs) located in peripheral tissues (256). Further, it induces the production and secretion of IFN-γ from various cell types, upon recognition of bacterial, viral and fungal components (257). Further, IL-23 is important for the development of T helper 17 cells (Th17) (258), which are in turn critical players in the homeostasis within the gut (259). Interestingly, obese women had higher circulating levels of IL-23 compared to healthy controls (260), supporting the assumption of a pro-inflammatory environment in metabolic diseases. Surprisingly, mice lacking IL-23 on a high fat diet, gained more weight and were more glucose intolerant than the controls (261). These mice also exhibit an increased gut permeability and bacterial translocation compared to wild type mice, underlining the important of IL-23 in the gut homeostasis.
Recently, IL-36 has been discovered and associated with obesity. IL-36 belongs to the IL-1 receptor family (262) and promotes the resolution of intestinal damage (263). IL-36 expression was increased in obese patients and negatively correlated with high blood glucose levels (264). Mice lacking an endogenous inhibitor for the IL-36 receptor had better glucose tolerance and insulin sensitivity compared to controls. Lack of IL-36 inhibition increased the abundance of Akkermansia muciniphila, mucin formation and intestinal integrity (264). However, this is the sole study indicating a role of IL-36 in metabolic diseases.
Lastly, typical anti-inflammatory cytokines (as discussed above) have essential roles in maintaining the intestinal homeostasis. For example, IL-10 is important to dampen an inflammatory response against intestinal bacteria (265). Particularly, microbiota derived SCFAs can stimulate the protection of IL-10 (266). Knock out of IL-10 induces colitis in mice, which is dependent on the microbiota since germ-free mice do not develop colitis (267). However, studies in metabolic diseases focuses mostly on the circulation than the intestine of obese humans or rodents.
Although several cytokines have been associated with metainflammation in obesity and T2D, only a few show direct connection to the gut microbiota such as IL-22, IL-23, and IL-36. However, several others have been discussed in the context of intestinal health and immune function (268), which have not been associated with metabolic inflammation yet. It is plausible that more cytokines are involved in all three processes. The ongoing discovery of new bacterial metabolites will shed more light into the complex pathways.
Microbiota-Produced Metabolites and Host Inflammation
The hypothesis that bacteria physically translocate to tissues, where they may invoke an inflammatory response has generated mixed results in literature as described above. A more recent line of reasoning, which relates gut microbiota to inflammation and cardiometabolic disease, includes the production of gut-derived metabolites that enter the systemic circulation to induce several effects. These metabolites have been linked to specific intestinal microbiota. Several of these metabolites have received much attention in recent years (269). Recently, metabolites such as imidazole propionate were discovered and shown to be involved in insulin resistance (270), however only for a few bacterial metabolites a direct connection between metabolism, immune system, and gut microbiota has been made.
Fibers and Short Chain Fatty Acids
The most widely studied metabolites include the short-chain fatty acids (SCFA) acetate, butyrate and propionate, generated by gut microbiota which ferment indigestible dietary components such as complex carbohydrates (fibers) (165). In that regard, high fiber intake was associated with a protection from several metabolic diseases such as T2D (271). Further, fiber intake promoted the expansion of SCFAs producing bacteria and improved glucose tolerance (partly via GLP-1) in people with T2D (272) (Figure 6). Inulin (high fermentable fiber) supplementation increased insulin sensitivity in human subjects. Inulin propionate ester improved insulin sensitivity as well as reduced IL-8 (273), suggesting beneficial effects on inflammatory parameters. Furthermore, high fiber intake promoted gut barrier integrity in mice, which was associated with the SCFAs production (274).
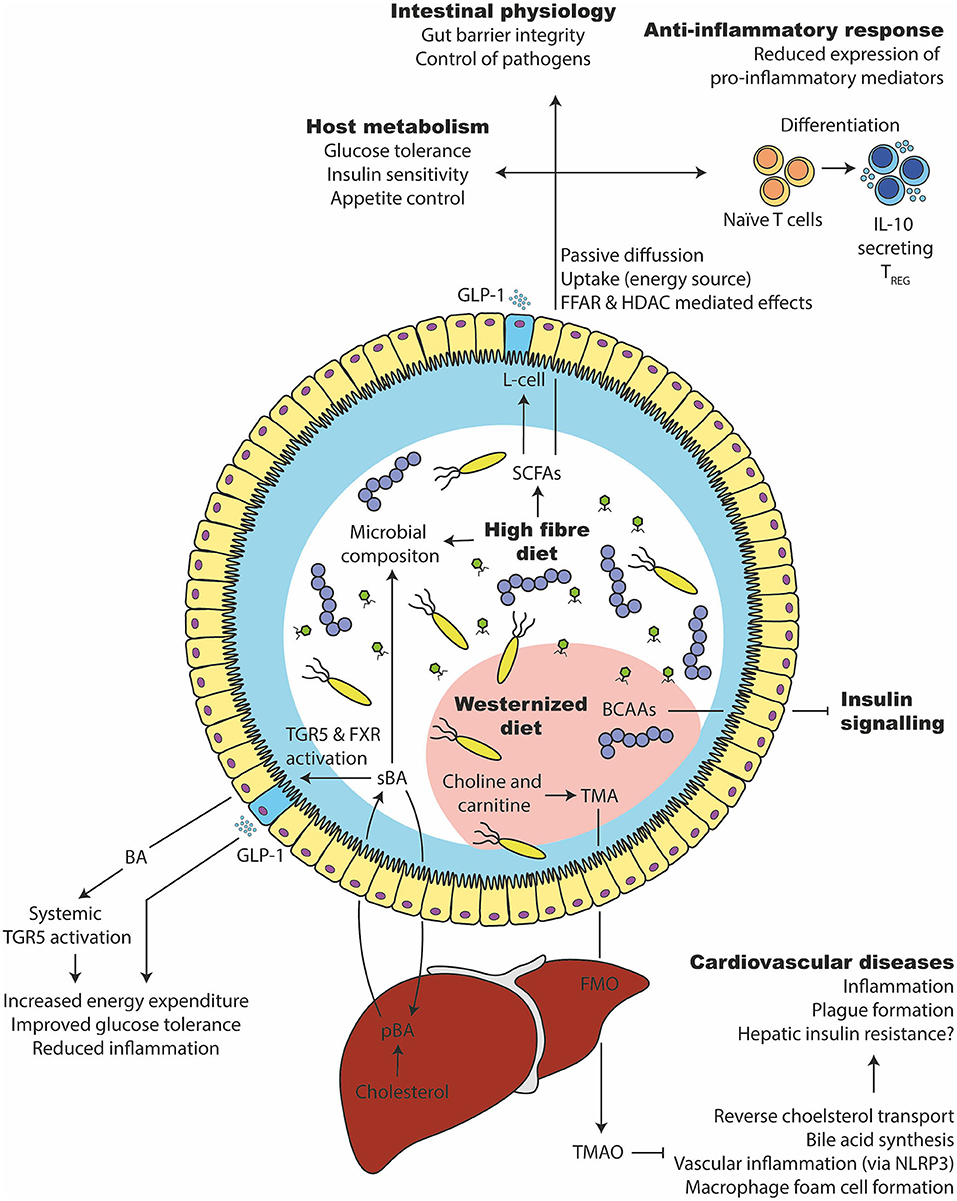
Figure 6. Microbial metabolites that affect glucose tolerance and inflammation. (1) A high fiber consumption has several beneficial effects on the gut microbiota and host health. They are degraded by the gut microbiota in short chain fatty acids such as butyrate, propionate, and acetate. SCFAs can be taken up by the enterocytes, used as an energy source or bound to free fatty acid receptors to stimulate varies responses (e.g., GLP-1 release from intestinal L-cells). Intracellularly, it can stimulate epigenetic changes via histone deacetylase (HDAC). It supports the expansion of beneficial bacteria and keeps opportunistic pathogens in control, improves glucose and appetite control, supports intestinal barrier integrity and induces an anti-inflammatory immune response in intestinal as well as systemic tissue sides. (2) Primary bile acids (pBA) are produced in the liver from cholesterol and secreted into the intestine via the gall bladder. There, they can change the gut microbiota and are transformed by bacteria to secondary bile acids (sBA). Bile acids can activate intestinal and systemic TGR5 as well as FXR, which increases the energy expenditure, lower inflammation and improves glucose tolerance. (3) A westernized diet, which is usually rich in saturated lipids, can disturb the branched chain amino acid (BCAA) catabolism of the host, which in turn inhibits the insulin signaling. (4) Further, a westernized diet is commonly rich in choline and carnitine, which the gut microbiota can metabolize to trimethylamine (TMA). After intestinal uptake, the liver transforms TMA into Trimethylamine N-oxide (TMAO) via flavin-containing monooxygenase (FMO). TMAO inhibits bile acid synthesis, reverse cholesterol transport (RCT), induce macrophage foam cell formation and inflammation via NLRP3. That in turn leads to cardiovascular complications.
SCFAs have been linked to a several metabolic processes including induction of appetite regulation (275, 276) and improving insulin resistance in muscle and adipose tissue (165, 277, 278). However, not all SCFAs seem to have beneficial effects. Acetate was shown to increase glucose-stimulated insulin secretion and weight gain in mice (279). Administration of low concentrations of propionate reduced insulin sensitivity in mice and humans by stimulating glucagon and FABP4 production (280). Instead, butyrate promotes gut epithelial integrity (281), an anti-inflammatory milieu, resistance to enteropathogens and regulatory T cell generation. Further, butyrate can inhibit the epigenetic modulator histone deacetylases (HDACs) (282) and thereby induce an anti-inflammatory response particularly in intestinal cells (283, 284). Importantly, butyrate-producing bacteria are less abundant in fecal microbiota of T2D individuals (6), which might have deleterious effects on the intestinal immune system. Fiber intake is associated with a decreased risk for metabolic diseases via increased SCFAs production, however the mechanism and extend is still elusive.
Trimethylamine-N-Oxide
Trimethylamine-N-oxide (TMAO) has attracted a lot of attention in the context of cardiometabolic diseases. TMAO is produced by the liver from trimethylamine (TMA), which on its turn is produced by intestinal microbiota from choline and carnitine-containing nutrients. TMAO has been linked to vascular inflammation (285, 286), e.g., via activation of NLRP3, and atherosclerotic plaque development, which could explain the increased risk for atherosclerotic cardiovascular disease and heart failure in people with higher levels of TMAO (287). Inhibition of the gut microbial TMA formation reduced macrophage foam cell formation and atherosclerotic lesions development in mice (288).
Further, a recent human trial found that energy-reducing diets decreased choline and L-carnitine. Those changes were associated with an improvement in fasting insulin and insulin resistance in overweight and obese adults. Further, there was a link between dietary fat intake and TMAO: increase in TMAO was related to lower improvements concerning glucose tolerance. All three metabolites (TMAO, choline and L-carnitine) were associated with changes in amino acids, in particular branched-chain (BCAAs) and aromatic amino acids (289) that have been associated with diabetes (290, 291) and changes in the gut microbiota (292).
Bile Acids
Bile acids have been proposed to be involved in glucose metabolism (293). They include different molecules that are essential for nutrient absorption in the intestine and are transformed by the microbiota (293). Bile acids are end products from cholesterol that are formed in the liver and excreted in the intestine via the gall bladder. In the intestine, the bacteria transform primary into secondary bile acids (293). Further, bile acids have an effect on the microbiota composition, for example, cholic acids changed the gut microbiota and reduced systemic adiponectin levels in rats (294). Bile acids are efficiently absorbed in the enterohepatic circulation. Small parts leave the circulation and are either excreted in feces or end in the systemic circulation (293).
In the blood circulation, bile acids can bind to the farnesoid X receptor (FXR) and G protein coupled bile acid receptor 1 (TGR5), which are involved in glucose metabolism (295) and liver regeneration (296). Using an intestinal agonist for FXR induced amongst others GLP-1 secretion, change in the gut microbiota and improved glucose tolerance in mice (295). Antibiotic treatment reversed this metabolic phenotype (295), suggesting the gut microbiota to be involved. However, effects of FXR activation are rather contradictory. It seems to improve insulin sensitivity (297, 298), but also promotes obesity (299, 300). Knocking out the FXR receptor prevented obesity and metabolic changes in mice (301). Interestingly, FXR is expressed by various immune cells and is a modulator of the intestinal innate immunity and intestinal integrity (302). More studies are needed to disentangle the role of FXR in metainflammation.
Although these metabolites have given novel insight into the interaction between nutrient intake-gut microbiota composition—host metabolism and immunity, many data are derived from rodent models and require confirmation in humans. In addition, it remains to be proven that modulation of these metabolites in fact can alter metabolic disease and inflammation in humans.
Interventions Aimed at Restoring Gut Microbiota Balance and Inflammation
Several interventions were partially able to restore gut microbiota symbiosis and associated metabolic function. In particular, dietary interventions have shown effects on microbiota function and meta-inflammatory effects. However, results from dietary interventions are complex and difficult to interpret since subjects show a low adherence and have high inter-individual variations. Therefore, other more specific approaches have been pursued to alter gut microbiota composition. Most commonly, supplementation of specific selective fermented ingredients that may alter microbiota composition (prebiotics) or supplementation of beneficial bacterial strains (probiotics) has been carried out, while in two studies, the metabolic effects of replacement of host microbiota by fecal microbiota transplantation (FMT) was investigated.
Diet
Modulation of gut microbiota has been speculated to be a novel tool to improve the metabolic abnormalities associated with obesity and T2D. As was shown in a recent study, the success of a dietary intervention could be predicted based on baseline microbiota composition (151, 303). A higher microbial gene richness was associated with a lower inflammation (151). As diet is an important factor shaping the gut microbiota (304), dietary interventions (such as increasing plant-based fibers intake and reducing food additives such as artificial sweeteners) have been put forward as an attractive target to improve functional and compositional aspects of gut microbiota (146). A dietary intervention study which included whole grain products and prebiotics to particularly target the gut microbiota decreased typical pro-inflammatory bacteria belonging to Enterobacteriaceae taxa and lowered levels of LPS binding protein (LBP), several inflammatory markers and it increased systemic adiponectin concentrations (305) (Table 1). A meta-analysis shows that fiber intake leads to a higher abundance of Bifidobacterium spp. and Lactobacillus spp., which might contribute to an increase in fecal butyrate concentration (306). Although only associative in nature, these results suggest an effect of dietary intake on the gut microbiota and inflammatory read outs (305).
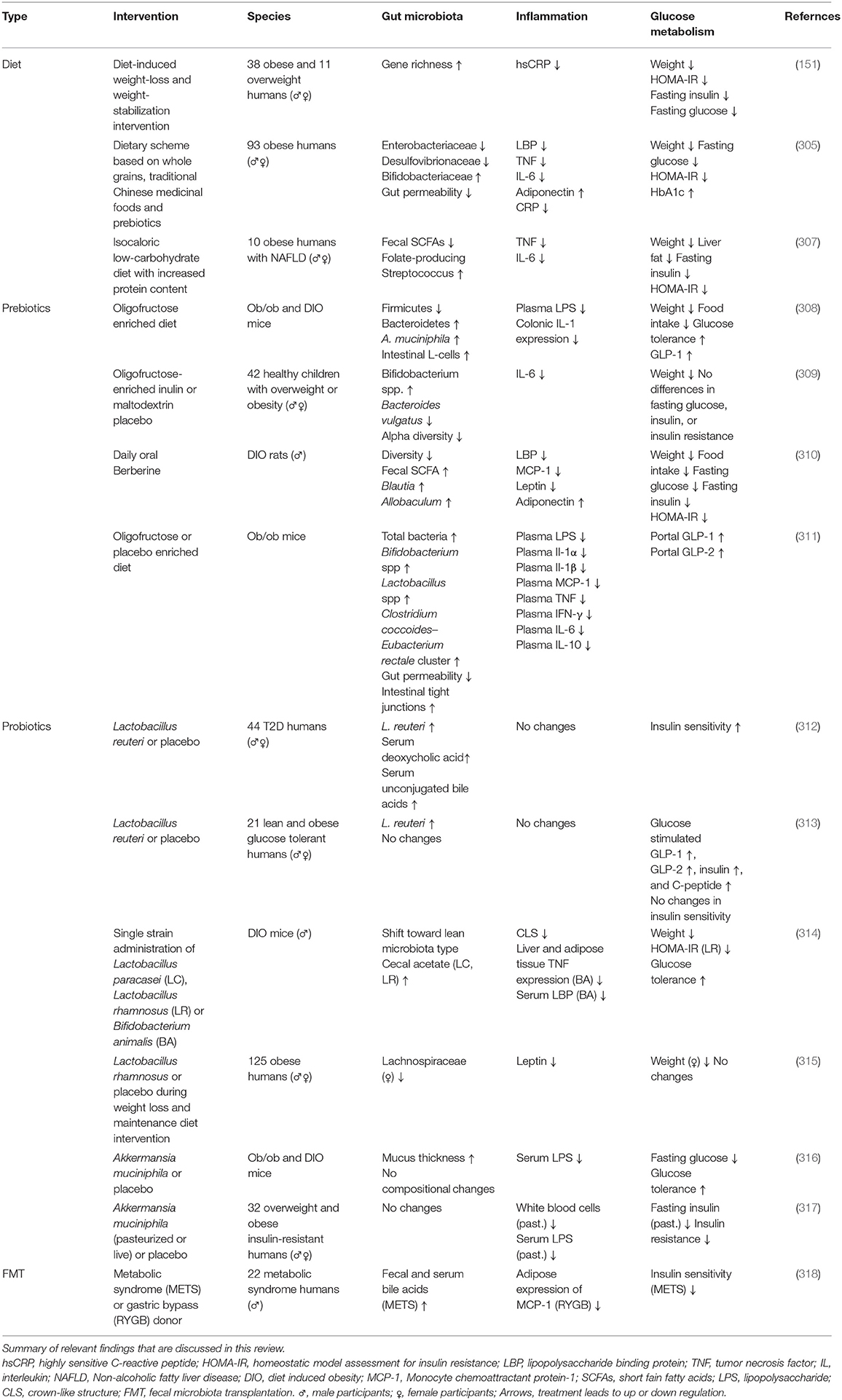
Table 1. Intervention studies that include the three way interaction (gut microbiota, inflammation, and glucose metabolism).
Changes in diet, especially the amount of indigestible food components such as fibers change the gut microbiota composition (319). A low-fiber westernized diet was associated with decrease in beneficial Firmicutes bacteria and an increase in mucosa penetrating Proteobacteria (320). Further, several different types of fibers improved gut permeability and colitis in mice (304). The diversity is only partially restorable after reintroducing fibers into the diet, which leads to increasing permanent loss of bacterial strains in mice over multiple generations (152, 319). As a result of selection in favor of particular bacteria during western diet, this loss in diversity is mostly a loss of Bacteroides, known for their capacity to break down fibers (152, 320). This permanent loss of specialized bacteria may harbor vast consequences. Indeed, a rapid reduction in SCFAs was observed after low carbohydrate intake (307). Supplementation of butyrate directly, improved insulin sensitivity in mice (277), but failed in several human studies. Therefore, fibers are positively associated with the glucose tolerance, potentially via SCFAs production; however, the mechanism is still elusive.
Prebiotics
Prebiotics have been described as selectively fermented ingredients that allow specific changes in the composition and/or activity of the gastrointestinal microflora that confers benefits upon host wellbeing and health (321). Current prebiotics are mainly complex carbohydrates, but also other compounds such as polyphenols and polyunsaturated fatty acids exert prebiotic effects (322). Next to improving stool consistency, prebiotics can be fermented to SCFAs that have various beneficial function on the host health (322).
Several prebiotics have been studied already in metabolic diseases. For example, oligofructose treatment in genetically obese mice, improved glucose homeostasis, inflammation and leptin sensitivity (308). Further, it increased L-cell content in rats with higher production of GLP-1 (323) and improved gut integrity, reduced endotoxemia as well as lowered inflammation in genetically obese mice (155). In humans, oligofructose-enriched inulin changed the gut microbiota in obese children, reduced body fat and decreased systemic IL-6 (309). Similarly, Berberine, a component of a Chinese herb, have shown beneficial effects on gut microbiota and glucose tolerance by increasing intestinal SCFA production and reducing inflammation in obese rats (310). A vast amount of animal studies suggests beneficial effect of prebiotics on the host glucose tolerance, however human studies fail to provide enough evidence (324). Further, only a few studies focused on inflammatory markers. More research is needed to assess the potential of prebiotic treatment to reduce metainflammation in humans.
Probiotics
Probiotics are defined as live microorganisms which, when administered in adequate amounts, confer a health benefit on the host. The probiotic candidate must be a taxonomically defined. Further, safety and health benefits must be supported by reproducible human studies (325). When combined with prebiotics, they may be referred to as synbiotics. Given the associations between altered gut microbiota and T2D, and more specifically, a reduction in beneficial, butyrate producing bacteria, several trials have been performed in humans to improve metabolic health supplementing these bacteria, most commonly Lactobacillus or Bifidobacterium species (326).
In mice, Lactobacillus reuteri improved insulin sensitivity, gut permeability and aryl hydrocarbon receptor ligand production (327). Administration of a mixture of different Lactobacillus and Bifidobacterium strains improved glucose homeostasis, macrophage infiltration and changed the gut microbiota (314). Similarly, a multi-strain treatment (VSL#3) improved liver function, inflammation and insulin sensitivity in genetically obese mice (328). These rodent studies point out several beneficial roles of probiotics in metabolism and inflammation; however, probiotics are only moderately effective in humans.
The randomized clinical trials that were performed up-to-date however, were mostly of short duration and included a relatively small number of participants, and as such, generated contrasting effects. In an attempt to provide more clarity, several meta-analyses have been performed focusing on different outcomes. Two meta-analyses observed small improvements in fasting glucose levels (0.3–0.5 mmol/l) and reported conflicting results on fasting insulin levels or insulin sensitivity indices, dependent on the studies included in the meta-analysis given the significant heterogeneity (329, 330). In general, effects were more pronounced on glucose metabolism when interventions were performed in T2D patients (331, 332) as compared to normoglycemic obese participants and when a probiotic cocktail using several strains was administered instead of one. Other meta analyses reported no effect of probiotics on weight (333) and small improvements in total and LDL-cholesterol (329) with most benefits again in studies using multiple bacterial strains. The effects on systemic inflammation were only investigated in a handful of studies, where reductions in C-reactive protein (CRP), but not TNF were observed (329, 334).
Akkermansia muciniphila has been extensively studied in metabolic diseases (316) and has recently been tested in humans as a probiotic. Importantly, this strain inversely correlates with body weight in humans and rodents (316). First, administration of A. muciniphila in obese mice improved insulin sensitivity, metabolic endotoxemia and adipose tissue inflammation (316). Further, a sole purified membrane protein of that particular strain was able to improve metabolism and gut barrier function in obese as well as diabetic mice (335). Supplementation in humans improved insulin sensitivity and inflammation, however, the effects were rather small, but the authors concluded from this proof of principle study that the treatment is safe and has therapeutic potential (317).
A rather new strategy is the modification of exiting bacteria to produce biological active compounds. For example, in mice L. reuteri genetically modified to produce IL-22 improved liver function, inflammation and bacterial translocation (336). These findings give completely new treatment options that might be the future for the use of probiotic.
Thus, several studies point toward a beneficial effect of pre- and probiotics in people with metabolic diseases. However, the absolute improvements in clinically relevant outcomes such as fasting glucose levels or HbA1c have been very modest, especially when compared to the efficacy of commonly used glucose lowering agents. On the other hand, definitive conclusions cannot be drawn yet due to the small studies with short duration and heterogenic treatment groups. Further, it is still not clear whether probiotics colonize the gut during consumption (334). A recent study demonstrates that the colonization of probiotic depends on the host; thus, questioning the universal usage of probiotics and highlighting the importance of personalized medicine (337).
Fecal Microbiota Transplantation
A drastic way to alter gut microbiota composition is by fecal microbiota transplantation (FMT). In this procedure, donor feces (obtained from a healthy donor following an extensive screening process) is transplanted into the recipient by upper GI (duodenal) infusion or lower GI (colonic) infusion. While anecdotally used in the past (338, 339), in more recent decades, the FMT procedure has become more common. More recently, two studies have been conducted to investigate whether transplantation of lean donor feces could improve glucose metabolism in obese metabolic syndrome patients. In a pilot study, lean donor FMT into nine obese males with metabolic syndrome induced a small but significant improvement in peripheral insulin sensitivity and a trend toward improved hepatic insulin sensitivity when compared to participants that underwent autologous gut microbiota infusion. Lean donor FMT resulted concomitantly in increased bacterial diversity and increased presence of butyrate producing bacteria (340).
The results from this pilot study were confirmed in a trial in 38 participants. Again, allogenic FMT improved insulin-stimulated glucose disposal in skeletal muscle after 6 weeks of treatment; this effect was most striking in patients with lower fecal microbiota diversity at baseline (341, 342). However, after 18 weeks, no effects could be observed on insulin sensitivity. Also, the microbiota composition had returned to the composition prior to the allogenic FMT (342). The mechanisms that underlie this beneficial effect remain to be investigated, as the FMT studies did not point to solid mechanisms responsible for these improvements. Thus, whether a reduction of bacterial translocation and/or low-grade inflammation contributes are involved remains hitherto unclear. The plasma levels of short-chain fatty acids such as butyrate were not altered.
However, a recent study compared FMTs from patients after bariatric surgery and patients with metabolic syndrome. Both donor feces were infused in people with metabolic syndrome. The latter decreased insulin sensitivity, along with an increase of several secondary bile acids (318). In contrast, the former led to a decrease in the macrophage attracting factor (MCP-1) in adipose tissue and in plasma. These results highlight the role of the gut microbiota in insulin tolerance and metabolic inflammation (318).
Future research in this area should focus on further standardization of the FMT technique (development of standard operating procedures) as well as validation in larger populations, including T2D patients. In addition, various refinements should be explored, including the transfer of specific strains (preferably in pill form) rather than feces, combination with prebiotics or antibiotics, and a better match between donors and receivers, thereby tailoring microbiota-based precision treatment. It is very likely that FMT into the upper GI tract might induce an inflammatory response since this part usually does not see fecal microbiota, however this has never been tested. Further, other microorganisms (e.g., viruses and fungi) than bacteria have been in the focus of gut microbiota related effects on host metabolism. Therefore, more studies are necessary to deceiver their role. Finally, the mechanisms that may drive the metabolic should be explored in depth.
Conclusions and Future Perspectives
It is accepted already for several decades that an increased inflammatory tone has major influences on the glucose metabolism (60, 65). For example, expansion and infiltration of pro-inflammatory immune cells is present in several metabolic active tissues during the development of T2D (21, 92). This pro-inflammatory milieu has vast consequences on the organ function as seen in the development of insulin resistance (73), beta cell dysfunction (343), and fatty liver disease (344). The trigger or origin of this inflammatory response is still elusive. Only recently, we started to understand the role of the gut microbiota in those processes.
There is a great afford to identify and describe the underlying metabolic and inflammatory pathways; however, studies are often contradictory. Particularly, murine knock out models frequently give inconclusive results (216, 237, 298) and numerous inflammatory mediators have dual roles (93, 103, 228). Further, players of the immune system serve important physiological functions (other than purely inflammation) and have tissue specific responses (9, 345). These findings highlight that the immune system is a complex organization, which is often neither pro- nor anti-inflammatory per se. Additional, a controlled (acute) inflammatory response is important for the host to fight invading pathogens and remove damaged tissue. However, this resolution is disturbed in metabolic diseases leading to metainflammation. Environmental factors such as the diet (346) and genetics (347) appear to be the main drivers for those deviations. Lastly, research in metainflammation focuses on typical pro-inflammatory cytokines such as IL-6 and TNF, with less work done on anti-inflammatory mediators. Future treatment strategies could aim to increase anti-inflammatory cytokines in obese and diabetic humans.
To sustain a symbiotic relationship with the gut microbiota, a controlled and appropriate immune response is essential to benefit from the gut microbiota's numerous functions (348). Recent studies suggest a disturbed immune intestinal immune response in obesity (188). Further, the host influences the microbiota via the diet. By ingesting a diet rich in fiber, we promote the production of SCFAs that can improve the hosts energy homeostasis (275), glucose tolerance (277) and particularly the regulation of an adequate inflammatory response (349). Not only a reduced fiber intake (271), but also a genetically dependent lower butyrate update is present in T2D (164). Both functions, the gut microbiota (6) and the intestinal immune system (188), are disturbed in metabolic diseases. However, it is not clear which disturbance comes first.
Several potential mechanisms on how the microbiota influence glucose metabolism and inflammation have been described (114, 225, 247). Though, numerous contradictory reports make it difficult to make a clear conclusion (237, 280). Understanding housing techniques and the vivarium of animals are important for the research field (350). Further, lack of technical understanding might explain several contradictory findings in microbiota research. Omics and (bio) informatical strategies are getting more complex and difficult to grasp for the majority of scientists. Standardized and transparent protocols are important to move the field forward (351).
The concept of bacterial translocation is a good example for the lack of technical understanding (178, 179, 352). Although it is a reasonable concept, low amounts of bacteria in systemic tissue sides make it difficult to trust histology or sequencing techniques due to background noise. The identification of small bacterial metabolites is more plausible (114, 270, 353). We are only at the beginning to understand the effects of microbial metabolites on the host health.
Techniques to identify and describe microorganism are getting more affordable and available for the majority of research groups (354). Thereby, several bacterial species have been discovered and were associated with metabolic abnormalities (6, 143). Identification of single relevant strains is important to decipher mechanistic interactions, however the gut microbiota is a complex ecosystem with thousands of different groups of microorganisms. For example, the role of the virome (355) or mycobiome (356) is less explored. In that respect, a recent study highlight the influence of the fecal virome on glucose tolerance during FMT (355). More effort is needed to understand the whole microbial ecosystem.
Lastly, treatments targeting the gut microbiota to expand beneficial bacteria or exchange a “dysbiotic” microbiota with a symbiotic one show promising result. FMTs display moderate effects in metabolic diseases, however with a limited duration (342). Similarly, probiotic treatment only show moderate to no success (284). Individual microbiomes and responses to bacterial treatment are major challenges that have to be further evaluated (337, 342). Some of these findings might be explained by regional and ethnical differences (357, 358). Modifying existing bacteria might be a novel way to improve the response and help to regulate inflammation (336).
The field of microbiota research is very young with several challenges, but enormous potential. The complex relationship between millions of different microorganism, thousands host cell types and molecular mediators make it difficult to grasp the mechanism, but technical advantages are moving the field forward.
Author Contributions
TS wrote the manuscript and prepared the figures. DR initiated the review. DR and HH supervised and revised the document. All authors discussed and contributed to the final manuscript.
Conflict of Interest
MN is in the Scientific Advisory Board of Caelus Pharmaceuticals, the Netherlands. DR has acted as a consultant and received honoraria from Boehringer Ingelheim, Eli Lilly, Merck, Novo Nordisk and Sanofi and has received research operating funds from the Boehringer Ingelheim–Eli Lilly Diabetes Alliance, MSD, AstraZeneca and Novo Nordisk. BV was funded by the Canadian Institutes of Health Research, Crohn's and Colitis Canada, and the National Institutes of Health (R01AI134766). CV is supported by grants from the Canadian Institutes of Health Research (PJT - 153156) and JDRF (3-SRA-2014-39-Q-R), a grant from the National Institutes of Health. HH is supported by a senior fellowship (2019.82.004) of the Dutch Diabetes Research Foundation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. (2019) 157:107843. doi: 10.1016/j.diabres.2019.107843
2. Morigny P, Houssier M, Mouisel E, Langin D. Adipocyte lipolysis and insulin resistance. Biochimie. (2016) 125:259–66. doi: 10.1016/j.biochi.2015.10.024
3. Heine RJ, Diamant M, Mbanya JC, Nathan DM. Management of hyperglycaemia in type 2 diabetes: the end of recurrent failure? BMJ. (2006) 333:1200–4. doi: 10.1136/bmj.39022.462546.80
4. Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. (2009) 58:773–95. doi: 10.2337/db09-9028
5. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. (2011) 29:415–45. doi: 10.1146/annurev-immunol-031210-101322
6. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. (2012) 490:55–60. doi: 10.1038/nature11450
7. Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. (2013) 498:99–103. doi: 10.1038/nature12198
8. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. (2013) 500:541–6. doi: 10.1038/nature12506
9. Spranger J, Kroke A, Möhlig M, Hoffmann K, Bergmann MM, Ristow M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes. Diabetes. (2003) 52:812. doi: 10.2337/diabetes.52.3.812
10. Marfella R, Esposito K, Siniscalchi M, Cacciapuoti F, Giugliano F, Labriola D, et al. Effect of weight loss on cardiac synchronization and proinflammatory cytokines in premenopausal obese women. Diabetes Care. (2004) 27:47. doi: 10.2337/diacare.27.1.47
11. Liu C, Feng X, Li Q, Wang Y, Li Q, Hua M. Adiponectin, TNF-α and inflammatory cytokines and risk of type 2 diabetes: a systematic review and meta-analysis. Cytokine. (2016) 86:100–9. doi: 10.1016/j.cyto.2016.06.028
12. Zhou W, Sailani MR, Contrepois K, Zhou Y, Ahadi S, Leopold SR, et al. Longitudinal multi-omics of host–microbe dynamics in prediabetes. Nature. (2019) 569:663–71. doi: 10.1038/s41586-019-1236-x
13. Michalovich D, Rodriguez-Perez N, Smolinska S, Pirozynski M, Mayhew D, Uddin S, et al. Obesity and disease severity magnify disturbed microbiome-immune interactions in asthma patients. Nat Commun. (2019) 10:5711. doi: 10.1038/s41467-019-13751-9
14. Sabapathy V, Stremska ME, Mohammad S, Corey RL, Sharma PR, Sharma R. Novel immunomodulatory cytokine regulates inflammation, diabetes, and obesity to protect from diabetic nephropathy. Front Pharmacol. (2019) 10:572. doi: 10.3389/fphar.2019.00572
15. Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. (2007) 356:1517–26. doi: 10.1056/NEJMoa065213
16. Cavelti-Weder C, Babians-Brunner A, Keller C, Stahel MA, Kurz-Levin M, Zayed H, et al. Effects of gevokizumab on glycemia and inflammatory markers in type 2 diabetes. Diabetes Care. (2012) 35:1654. doi: 10.2337/dc11-2219
17. Fleischman A, Shoelson SE, Bernier R, Goldfine AB. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care. (2008) 31:289–94. doi: 10.2337/dc07-1338
18. Dominguez H, Storgaard H, Rask-Madsen C, Steffen Hermann T, Ihlemann N, Baunbjerg Nielsen D, et al. Metabolic and vascular effects of tumor necrosis factor-alpha blockade with etanercept in obese patients with type 2 diabetes. J Vasc Res. (2005) 42:517–25. doi: 10.1159/000088261
19. Lu J, Zhao J, Meng H, Zhang X. Adipose tissue-resident immune cells in obesity and type 2 diabetes. Front Immunol. (2019) 10:1173. doi: 10.3389/fimmu.2019.01173
20. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime Rep. (2014) 6:13. doi: 10.12703/P6-13
21. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. (2007) 117:175–84. doi: 10.1172/JCI29881
22. Hong EG, Ko HJ, Cho YR, Kim HJ, Ma Z, Yu TY, et al. Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes. (2009) 58:2525–35. doi: 10.2337/db08-1261
23. Van Exel E, Gussekloo J, De Craen AJ, Frolich M, Bootsma-Van Der Wiel A, Westendorp RG. Low production capacity of interleukin-10 associates with the metabolic syndrome and type 2 diabetes: the Leiden 85-Plus Study. Diabetes. (2002) 51:1088–92. doi: 10.2337/diabetes.51.4.1088
24. Esposito K, Pontillo A, Giugliano F, Giugliano G, Marfella R, Nicoletti G, et al. Association of low interleukin-10 levels with the metabolic syndrome in obese women. J Clin Endocrinol Metab. (2003) 88:1055–8. doi: 10.1210/jc.2002-021437
25. Tsao C-H, Shiau M-Y, Chuang P-H, Chang Y-H, Hwang J. Interleukin-4 regulates lipid metabolism by inhibiting adipogenesis and promoting lipolysis. J Lipid Res. (2014) 55:385–97. doi: 10.1194/jlr.M041392
26. Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, et al. Adipocyte-derived Th2 cytokines and myeloid PPARδ regulate macrophage polarization and insulin sensitivity. Cell Metab. (2008) 7:485–95. doi: 10.1016/j.cmet.2008.04.002
27. Luzina IG, Keegan AD, Heller NM, Rook GAW, Shea-Donohue T, et al. Regulation of inflammation by interleukin-4: a review of “alternatives”. J Leukoc Biol. (2012) 92:753–64. doi: 10.1189/jlb.0412214
28. Ho K-T, Shiau M-Y, Chang Y-H, Chen C-M, Yang S-C, Huang C-N. Association of interleukin-4 promoter polymorphisms in Taiwanese patients with type 2 diabetes mellitus. Metabolism. (2010) 59:1717–22. doi: 10.1016/j.metabol.2010.04.010
29. Minty A, Chalon P, Derocq JM, Dumont X, Guillemot JC, Kaghad M, et al. Interleukin-13 is a new human lymphokine regulating inflammatory and immune responses. Nature. (1993) 362:248–50. doi: 10.1038/362248a0
30. Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. (2013) 31:317–43. doi: 10.1146/annurev-immunol-032712-095906
31. Schmidt FM, Weschenfelder J, Sander C, Minkwitz J, Thormann J, Chittka T, et al. Inflammatory cytokines in general and central obesity and modulating effects of physical activity. PLoS ONE. (2015) 10:e0121971. doi: 10.1371/journal.pone.0121971
32. Martínez-Reyes CP, Gómez-Arauz AY, Torres-Castro I, Manjarrez-Reyna AN, Palomera LF, Olivos-García A, et al. Serum levels of interleukin-13 increase in subjects with insulin resistance but do not correlate with markers of low-grade systemic inflammation. J Diabetes Res. (2018) 2018:7209872. doi: 10.1155/2018/7209872
33. Castoldi A, Naffah De Souza C, Câmara NOS, Moraes-Vieira PM. The macrophage switch in obesity development. Front Immunol. (2016) 6:637. doi: 10.3389/fimmu.2015.00637
34. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
35. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. (2009) 29:313–26. doi: 10.1089/jir.2008.0027
36. Kang YE, Kim JM, Joung KH, Lee JH, You BR, Choi MJ, et al. The roles of adipokines, proinflammatory cytokines, and adipose tissue macrophages in obesity-associated insulin resistance in modest obesity and early metabolic dysfunction. PLoS ONE. (2016) 11:e0154003. doi: 10.1371/journal.pone.0154003
37. Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci USA. (2003) 100:7265–70. doi: 10.1073/pnas.1133870100
38. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K-I, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. (2006) 116:1494–505. doi: 10.1172/JCI26498
39. Inouye KE, Shi H, Howard JK, Daly CH, Lord GM, Rollins BJ, et al. Absence of CC chemokine ligand 2 does not limit obesity-associated infiltration of macrophages into adipose tissue. Diabetes. (2007) 56:2242. doi: 10.2337/db07-0425
40. Shimobayashi M, Albert V, Woelnerhanssen B, Frei IC, Weissenberger D, Meyer-Gerspach AC, et al. Insulin resistance causes inflammation in adipose tissue. J Clin Invest. (2018) 128:1538–50. doi: 10.1172/JCI96139
41. Piemonti L, Leone BE, Nano R, Saccani A, Monti P, Maffi P, et al. Human pancreatic islets produce and secrete MCP-1/CCL2: relevance in human islet transplantation. Diabetes. (2002) 51:55. doi: 10.2337/diabetes.51.1.55
42. Martin AP, Rankin S, Pitchford S, Charo IF, Furtado GC, Lira SA. Increased expression of CCL2 in insulin-producing cells of transgenic mice promotes mobilization of myeloid cells from the bone marrow, marked insulitis, and diabetes. Diabetes. (2008) 57:3025–33. doi: 10.2337/db08-0625
43. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. (2009) 15:914–20. doi: 10.1038/nm.1964
44. Yang H, Youm YH, Vandanmagsar B, Ravussin A, Gimble JM, Greenway F, et al. Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity: implications for systemic inflammation and insulin resistance. J Immunol. (2010) 185:1836–45. doi: 10.4049/jimmunol.1000021
45. Sell H, Habich C, Eckel J. Adaptive immunity in obesity and insulin resistance. Nat Rev Endocrinol. (2012) 8:709–16. doi: 10.1038/nrendo.2012.114
46. McLaughlin T, Liu LF, Lamendola C, Shen L, Morton J, Rivas H, et al. T-cell profile in adipose tissue is associated with insulin resistance and systemic inflammation in humans. Arterioscler Thromb Vasc Biol. (2014) 34:2637–43. doi: 10.1161/ATVBAHA.114.304636
47. Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Vernon AH, et al. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res. (2008) 103:467–76. doi: 10.1161/CIRCRESAHA.108.177105
48. O'Rourke RW, White AE, Metcalf MD, Winters BR, Diggs BS, Zhu X, et al. Systemic inflammation and insulin sensitivity in obese IFN-gamma knockout mice. Metabolism. (2012) 61:1152–61. doi: 10.1016/j.metabol.2012.01.018
49. Panda SK, Colonna M. Innate lymphoid cells in mucosal immunity. Front Immunol. (2019) 10:861. doi: 10.3389/fimmu.2019.00861
50. Vivier E, Van De Pavert SA, Cooper MD, Belz GT. The evolution of innate lymphoid cells. Nat Immunol. (2016) 17:790–4. doi: 10.1038/ni.3459
51. Bal SM, Golebski K, Spits H. Plasticity of innate lymphoid cell subsets. Nat Rev Immunol. (2020) 20:552–65. doi: 10.1038/s41577-020-0282-9
52. O'Sullivan TE, Rapp M, Fan X, Weizman O-E, Bhardwaj P, Adams NM, et al. Adipose-resident group 1 innate lymphoid cells promote obesity-associated insulin resistance. Immunity. (2016) 45:428–41. doi: 10.1016/j.immuni.2016.06.016
53. Boulenouar S, Michelet X, Duquette D, Alvarez D, Hogan AE, Dold C, et al. Adipose type one innate lymphoid cells regulate macrophage homeostasis through targeted cytotoxicity. Immunity. (2017) 46:273–86. doi: 10.1016/j.immuni.2017.01.008
54. Lee B-C, Kim M-S, Pae M, Yamamoto Y, Eberlé D, Shimada T, et al. Adipose natural killer cells regulate adipose tissue macrophages to promote insulin resistance in obesity. Cell Metab. (2016) 23:685–98. doi: 10.1016/j.cmet.2016.03.002
55. Wang H, Shen L, Sun X, Liu F, Feng W, Jiang C, et al. Adipose group 1 innate lymphoid cells promote adipose tissue fibrosis and diabetes in obesity. Nat Commun. (2019) 10:3254. doi: 10.1038/s41467-019-11270-1
56. Wensveen FM, Jelenčić V, Valentić S, Šestan M, Wensveen TT, Theurich S, et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol. (2015) 16:376–85. doi: 10.1038/ni.3120
57. Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. (2015) 519:242–6. doi: 10.1038/nature14115
58. Molofsky AB, Van Gool F, Liang HE, Van Dyken SJ, Nussbaum JC, Lee J, et al. Interleukin-33 and Interferon-γ Counter-Regulate Group 2 innate lymphoid cell activation during immune perturbation. Immunity. (2015) 43:161–74. doi: 10.1016/j.immuni.2015.05.019
59. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. (2003) 112:1796–808. doi: 10.1172/JCI200319246
60. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. (1993) 259:87–91. doi: 10.1126/science.7678183
61. Plomgaard P, Nielsen AR, Fischer CP, Mortensen OH, Broholm C, Penkowa M, et al. Associations between insulin resistance and TNF-alpha in plasma, skeletal muscle and adipose tissue in humans with and without type 2 diabetes. Diabetologia. (2007) 50:2562–71. doi: 10.1007/s00125-007-0834-6
62. Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab. (2001) 280:E745–51. doi: 10.1152/ajpendo.2001.280.5.E745
63. Ruan H, Miles PDG, Ladd CM, Ross K, Golub TR, Olefsky JM, et al. Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor-α. Implic Insulin Resist. (2002) 51:3176–88. doi: 10.2337/diabetes.51.11.3176
64. Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. (1997) 389:610–4. doi: 10.1038/39335
65. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. (1995) 95:2409–15. doi: 10.1172/JCI117936
66. Donath MY. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov. (2014) 13:465–76. doi: 10.1038/nrd4275
67. Trinh B, Donath MY, Laubli H. Successful treatment of immune checkpoint inhibitor-induced diabetes with infliximab. Diabetes Care. (2019) 42:e153–4. doi: 10.2337/dc19-0908
68. Nieto-Vazquez I, Fernández-Veledo S, Krämer DK, Vila-Bedmar R, Garcia-Guerra L, Lorenzo M. Insulin resistance associated to obesity: the link TNF-alpha. Arch Physiol Biochem. (2008) 114:183–94. doi: 10.1080/13813450802181047
69. Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol. (2016) 12:15–28. doi: 10.1038/nrendo.2015.189
70. Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. (1998) 396:77–80. doi: 10.1038/23948
71. Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. (2001) 293:1673–7. doi: 10.1126/science.1061620
72. Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE, et al. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest. (2002) 109:1321–6. doi: 10.1172/JCI0214955
73. Shoelson SE, Lee J, Yuan M. Inflammation and the IKKβ/IκB/NF-κB axis in obesity- and diet-induced insulin resistance. Int J Obes. (2003) 27:S49–52. doi: 10.1038/sj.ijo.0802501
74. Reinhard C, Shamoon B, Shyamala V, Williams LT. Tumor necrosis factor alpha-induced activation of c-jun N-terminal kinase is mediated by TRAF2. EMBO J. (1997) 16:1080–92. doi: 10.1093/emboj/16.5.1080
75. Lee YH, Giraud J, Davis RJ, White MF. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem. (2003) 278:2896–902. doi: 10.1074/jbc.M208359200
76. Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature. (2002) 420:333–6. doi: 10.1038/nature01137
77. Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, et al. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. (2007) 6:386–97. doi: 10.1016/j.cmet.2007.09.011
78. Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science. (2008) 322:1539–43. doi: 10.1126/science.1160794
79. Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. (2005) 87:99–109. doi: 10.1016/j.biochi.2004.10.019
80. Kwon G, Xu G, Marshall CA, McDaniel ML. Tumor necrosis factor alpha-induced pancreatic beta-cell insulin resistance is mediated by nitric oxide and prevented by 15-deoxy-Delta12,14-prostaglandin J2 and aminoguanidine. A role for peroxisome proliferator-activated receptor gamma activation and inos expression. J Biol Chem. (1999) 274:18702–8. doi: 10.1074/jbc.274.26.18702
81. Watanabe Y, Nagai Y, Takatsu K. Activation and regulation of the pattern recognition receptors in obesity-induced adipose tissue inflammation and insulin resistance. Nutrients. (2013) 5:3757–78. doi: 10.3390/nu5093757
82. Mohamed-Ali V, Goodrick S, Rawesh A, Katz DR, Miles JM, Yudkin JS, et al. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J Clin Endocrinol Metab. (1997) 82:4196–200. doi: 10.1210/jc.82.12.4196
83. Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes. (2002) 51:3391. doi: 10.2337/diabetes.51.12.3391
84. Franckhauser S, Elias I, Rotter Sopasakis V, Ferre T, Nagaev I, Andersson CX, et al. Overexpression of Il6 leads to hyperinsulinaemia, liver inflammation and reduced body weight in mice. Diabetologia. (2008) 51:1306–16. doi: 10.1007/s00125-008-0998-8
85. Nieto-Vazquez I, Fernández-Veledo S, De Alvaro C, Lorenzo M. Dual role of interleukin-6 in regulating insulin sensitivity in murine skeletal muscle. Diabetes. (2008) 57:3211. doi: 10.2337/db07-1062
86. Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelle D, Hilton D, Van Obberghen E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem. (2000) 275:15985–91. doi: 10.1074/jbc.275.21.15985
87. Lagathu C, Bastard JP, Auclair M, Maachi M, Capeau J, Caron M. Chronic interleukin-6 (IL-6) treatment increased IL-6 secretion and induced insulin resistance in adipocyte: prevention by rosiglitazone. Biochem Biophys Res Commun. (2003) 311:372–9. doi: 10.1016/j.bbrc.2003.10.013
88. Jorgensen SB, O'Neill HM, Sylow L, Honeyman J, Hewitt KA, Palanivel R, et al. Deletion of skeletal muscle SOCS3 prevents insulin resistance in obesity. Diabetes. (2013) 62:56–64. doi: 10.2337/db12-0443
89. Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol. (2014) 15:423–30. doi: 10.1038/ni.2865
90. Xu E, Pereira MMA, Karakasilioti I, Theurich S, Al-Maarri M, Rappl G, et al. Temporal and tissue-specific requirements for T-lymphocyte IL-6 signalling in obesity-associated inflammation and insulin resistance. Nat Commun. (2017) 8:14803. doi: 10.1038/ncomms14803
91. Ellingsgaard H, Ehses JA, Hammar EB, Van Lommel L, Quintens R, Martens G, et al. Interleukin-6 regulates pancreatic α-cell mass expansion. Proc Natl Acad Sci USA. (2008) 105:13163–8. doi: 10.1073/pnas.0801059105
92. Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes. (2007) 56:2356–70. doi: 10.2337/db06-1650
93. Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med. (2011) 17:1481–9. doi: 10.1038/nm.2513
94. Timper K, Dalmas E, Dror E, Rutti S, Thienel C, Sauter NS, et al. Glucose-dependent insulinotropic peptide stimulates glucagon-like peptide 1 production by pancreatic islets via interleukin 6, produced by alpha cells. Gastroenterology. (2016) 151:165–79. doi: 10.1053/j.gastro.2016.03.003
95. Jager J, Gremeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology. (2007) 148:241–51. doi: 10.1210/en.2006-0692
96. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol. (2010) 11:897–904. doi: 10.1038/ni.1935
97. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. (2011) 12:408–15. doi: 10.1038/ni.2022
98. McGIllicuddy FC, Harford KA, Reynolds CM, Oliver E, Claessens M, Mills KH, et al. Lack of interleukin-1 receptor I (IL-1RI) protects mice from high-fat diet-induced adipose tissue inflammation coincident with improved glucose homeostasis. Diabetes. (2011) 60:1688–98. doi: 10.2337/db10-1278
99. Dror E, Dalmas E, Meier DT, Wueest S, Thevenet J, Thienel C, et al. Postprandial macrophage-derived IL-1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat Immunol. (2017) 18:283–92. doi: 10.1038/ni.3659
100. Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab. (2012) 15:518–33. doi: 10.1016/j.cmet.2012.01.023
101. Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. (2002) 110:851–60. doi: 10.1172/JCI200215318
102. Jourdan T, Godlewski G, Cinar R, Bertola A, Szanda G, Liu J, et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat Med. (2013) 19:1132–40. doi: 10.1038/nm.3265
103. Maedler K, Schumann DM, Sauter N, Ellingsgaard H, Bosco D, Baertschiger R, et al. Low concentration of interleukin-1beta induces FLICE-inhibitory protein-mediated beta-cell proliferation in human pancreatic islets. Diabetes. (2006) 55:2713–22. doi: 10.2337/db05-1430
104. Westwell-Roper C, Nackiewicz D, Dan M, Ehses JA. Toll-like receptors and NLRP3 as central regulators of pancreatic islet inflammation in type 2 diabetes. Immunol Cell Biol. (2014) 92:314–23. doi: 10.1038/icb.2014.4
105. Lagathu C, Yvan-Charvet L, Bastard JP, Maachi M, Quignard-Boulange A, Capeau J, et al. Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia. (2006) 49:2162–73. doi: 10.1007/s00125-006-0335-z
106. Su D, Coudriet GM, Hyun Kim D, Lu Y, Perdomo G, Qu S, et al. FoxO1 links insulin resistance to proinflammatory cytokine IL-1beta production in macrophages. Diabetes. (2009) 58:2624–33. doi: 10.2337/db09-0232
107. Lefere S, Tacke F. Macrophages in obesity and non-alcoholic fatty liver disease: crosstalk with metabolism. JHEP Rep. (2019) 1:30–43. doi: 10.1016/j.jhepr.2019.02.004
108. Lanthier N, Molendi-Coste O, Horsmans Y, Van Rooijen N, Cani PD, Leclercq IA. Kupffer cell activation is a causal factor for hepatic insulin resistance. Am J Physiol Gastrointest Liver Physiol. (2010) 298:G107–16. doi: 10.1152/ajpgi.00391.2009
109. Obstfeld AE, Sugaru E, Thearle M, Francisco A-M, Gayet C, Ginsberg HN, et al. C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes. (2010) 59:916. doi: 10.2337/db09-1403
110. Morgantini C, Jager J, Li X, Levi L, Azzimato V, Sulen A, et al. Liver macrophages regulate systemic metabolism through non-inflammatory factors. Nat Metab. (2019) 1:445–59. doi: 10.1038/s42255-019-0044-9
111. Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes. (2010) 59:347–57. doi: 10.2337/db09-0016
112. Gomes JMG, Costa JA, Alfenas RCG. Metabolic endotoxemia and diabetes mellitus: a systematic review. Metabolism. (2017) 68:133–44. doi: 10.1016/j.metabol.2016.12.009
113. Ghoshal S, Witta J, Zhong J, De Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res. (2009) 50:90–7. doi: 10.1194/jlr.M800156-JLR200
114. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. (2007) 56:1761–72. doi: 10.2337/db06-1491
115. Lassenius MI, Pietilainen KH, Kaartinen K, Pussinen PJ, Syrjanen J, Forsblom C, et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care. (2011) 34:1809–15. doi: 10.2337/dc10-2197
116. Harte AL, Varma MC, Tripathi G, McGee KC, Al-Daghri NM, Al-Attas OS, et al. High fat intake leads to acute postprandial exposure to circulating endotoxin in type 2 diabetic subjects. Diabetes Care. (2012) 35:375–82. doi: 10.2337/dc11-1593
117. Burcelin R. Gut microbiota and immune crosstalk in metabolic disease. Mol Metab. (2016) 5:771–81. doi: 10.1016/j.molmet.2016.05.016
118. Van Oostrom AJ, Sijmonsma TP, Rabelink TJ, Van Asbeck BS, Cabezas MC. Postprandial leukocyte increase in healthy subjects. Metabolism. (2003) 52:199–202. doi: 10.1053/meta.2003.50037
119. Erridge C, Attina T, Spickett CM, Webb DJ. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr. (2007) 86:1286–92. doi: 10.1093/ajcn/86.5.1286
120. Pendyala S, Walker JM, Holt PR. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology. (2012) 142:1100–01.e1102. doi: 10.1053/j.gastro.2012.01.034
121. Bakker GJ, Schnitzler JG, Bekkering S, De Clercq NC, Koopen AM, Hartstra AV, et al. Oral vancomycin treatment does not alter markers of postprandial inflammation in lean and obese subjects. Physiol Rep. (2019) 7:e14199. doi: 10.14814/phy2.14199
122. Lancaster GI, Langley KG, Berglund NA, Kammoun HL, Reibe S, Estevez E, et al. Evidence that TLR4 is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab. (2018) 27:1096–110.e1095. doi: 10.1016/j.cmet.2018.03.014
123. Cordain L, Eaton SB, Sebastian A, Mann N, Lindeberg S, Watkins BA, et al. Origins and evolution of the Western diet: health implications for the 21st century. Am J Clin Nutr. (2005) 81:341–54. doi: 10.1093/ajcn.81.2.341
124. Scheithauer TP, Dallinga-Thie GM, De Vos WM, Nieuwdorp M, Van Raalte DH. Causality of small and large intestinal microbiota in weight regulation and insulin resistance. Mol Metab. (2016) 5:759–70. doi: 10.1016/j.molmet.2016.06.002
125. Hsu BB, Gibson TE, Yeliseyev V, Liu Q, Lyon L, Bry L, et al. Dynamic modulation of the gut microbiota and metabolome by bacteriophages in a mouse model. Cell Host Microbe. (2019) 25:803–814.e805. doi: 10.1016/j.chom.2019.05.001
126. Shkoporov AN, Hill C. Bacteriophages of the human gut: the “known unknown” of the microbiome. Cell Host Microbe. (2019) 25:195–209. doi: 10.1016/j.chom.2019.01.017
127. Jiang TT, Shao TY, Ang WXG, Kinder JM, Turner LH, Pham G, et al. Commensal fungi recapitulate the protective benefits of intestinal bacteria. Cell Host Microbe. (2017) 22:809–16.e804. doi: 10.1016/j.chom.2017.10.013
128. Shao TY, Ang WXG, Jiang TT, Huang FS, Andersen H, Kinder JM, et al. Commensal Candida albicans positively calibrates systemic Th17 immunological responses. Cell Host Microbe. (2019) 25:404–17.e406. doi: 10.1016/j.chom.2019.02.004
129. Lazar V, Ditu L-M, Pircalabioru GG, Gheorghe I, Curutiu C, Holban AM, et al. Aspects of gut microbiota and immune system interactions in infectious diseases, immunopathology, and cancer. Front Immunol. (2018) 9:1830. doi: 10.3389/fimmu.2018.01830
130. Rowland I, Gibson G, Heinken A, Scott K, Swann J, Thiele I, et al. Gut microbiota functions: metabolism of nutrients and other food components. Eur J Nutr. (2018) 57:1–24. doi: 10.1007/s00394-017-1445-8
131. Valdes AM, Walter J, Segal E, Spector TD. Role of the gut microbiota in nutrition and health. BMJ. (2018) 361:k2179. doi: 10.1136/bmj.k2179
132. Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature. (2016) 535:65–74. doi: 10.1038/nature18847
133. Khosravi A, Yáñez A, Price JG, Chow A, Merad M, Goodridge HS, et al. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe. (2014) 15:374–81. doi: 10.1016/j.chom.2014.02.006
134. Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. (2005) 122:107–18. doi: 10.1016/j.cell.2005.05.007
135. Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, et al. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. (2012) 336:1325–9. doi: 10.1126/science.1222195
136. Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. (2009) 9:313–23. doi: 10.1038/nri2515
137. Erny D, Hrabe De Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. (2015) 18:965–77. doi: 10.1038/nn.4030
138. Hall JA, Bouladoux N, Sun CM, Wohlfert EA, Blank RB, Zhu Q, et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity. (2008) 29:637–49. doi: 10.1016/j.immuni.2008.08.009
139. Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, et al. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. (2011) 332:974–7. doi: 10.1126/science.1206095
140. Galazzo G, Van Best N, Bervoets L, Dapaah IO, Savelkoul PH, Hornef MW, et al. Development of the microbiota and associations with birth mode, diet, and atopic disorders in a longitudinal analysis of stool samples, collected from infancy through early childhood. Gastroenterology. (2020) 158:1584–96. doi: 10.1053/j.gastro.2020.01.024
141. Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA. (2007) 104:979. doi: 10.1073/pnas.0605374104
142. Rabot S, Membrez M, Bruneau A, Gerard P, Harach T, Moser M, et al. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. (2010) 24:4948–59. doi: 10.1096/fj.10.164921
143. Fei N, Zhao L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. (2012) 7:880. doi: 10.1038/ismej.2012.153
144. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. (2006) 444:1027–31. doi: 10.1038/nature05414
145. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. (2013) 341:1241214. doi: 10.1126/science.1241214
146. Meijnikman AS, Gerdes VE, Nieuwdorp M, Herrema H. Evaluating causality of gut microbiota in obesity and diabetes in humans. Endocr Rev. (2017) 39:133–53. doi: 10.1210/er.2017-00192
147. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol. (2017) 17:219–32. doi: 10.1038/nri.2017.7
148. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. (2006) 444:1022–3. doi: 10.1038/4441022a
149. Larsen N, Vogensen FK, Van Den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE. (2010) 5:e9085. doi: 10.1371/journal.pone.0009085
150. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. (2009) 457:480–4. doi: 10.1038/nature07540
151. Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, et al. Dietary intervention impact on gut microbial gene richness. Nature. (2013) 500:585–8. doi: 10.1038/nature12480
152. Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, Sonnenburg JL. Diet-induced extinctions in the gut microbiota compound over generations. Nature. (2016) 529:212. doi: 10.1038/nature16504
153. Azad MB, Konya T, Persaud RR, Guttman DS, Chari RS, Field CJ, et al. Impact of maternal intrapartum antibiotics, method of birth and breastfeeding on gut microbiota during the first year of life: a prospective cohort study. Bjog. (2016) 123:983–93. doi: 10.1111/1471-0528.13601
154. Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. (2004) 101:15718–23. doi: 10.1073/pnas.0407076101
155. Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. (2008) 57:1470–81. doi: 10.2337/db07-1403
156. Serino M, Luche E, Gres S, Baylac A, Berge M, Cenac C, et al. Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut. (2012) 61:543–53. doi: 10.1136/gutjnl-2011-301012
157. Chambers ES, Preston T, Frost G, Morrison DJ. Role of gut microbiota-generated short-chain fatty acids in metabolic and cardiovascular health. Curr Nutr Rep. (2018) 7:198–206. doi: 10.1007/s13668-018-0248-8
158. Rahat-Rozenbloom S, Fernandes J, Gloor GB, Wolever TM. Evidence for greater production of colonic short-chain fatty acids in overweight than lean humans. Int J Obes. (2014) 38:1525–31. doi: 10.1038/ijo.2014.46
159. De La Cuesta-Zuluaga J, Mueller NT, Álvarez-Quintero R, Velásquez-Mejía EP, Sierra JA, Corrales-Agudelo V, et al. Higher fecal short-chain fatty acid levels are associated with gut microbiome dysbiosis, obesity, hypertension and cardiometabolic disease risk factors. Nutrients. (2018) 11:51. doi: 10.3390/nu11010051
160. Schwiertz A, Taras D, Schafer K, Beijer S, Bos NA, Donus C, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity. (2010) 18:190–5. doi: 10.1038/oby.2009.167
161. Jumpertz R, Le DS, Turnbaugh PJ, Trinidad C, Bogardus C, Gordon JI, et al. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am J Clin Nutr. (2011) 94:58–65. doi: 10.3945/ajcn.110.010132
162. McNeil NI. The contribution of the large intestine to energy supplies in man. Am J Clin Nutr. (1984) 39:338–42. doi: 10.1093/ajcn/39.2.338
163. Segain J-P, De La Blétière DR, Bourreille A, Leray V, Gervois N, Rosales C, et al. Butyrate inhibits inflammatory responses through NFκB inhibition: implications for Crohn's disease. Gut. (2000) 47:397–403. doi: 10.1136/gut.47.3.397
164. Sanna S, Van Zuydam NR, Mahajan A, Kurilshikov A, Vich Vila A, Võsa U, et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nature. Genetics. (2019) 51:600–5. doi: 10.1038/s41588-019-0350-x
165. Canfora EE, Jocken JW, Blaak EE. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat Rev Endocrinol. (2015) 11:577–91. doi: 10.1038/nrendo.2015.128
166. Roediger WE. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut. (1980) 21:793–8. doi: 10.1136/gut.21.9.793
167. Ardawi MS, Newsholme EA. Fuel utilization in colonocytes of the rat. Biochem J. (1985) 231:713–9. doi: 10.1042/bj2310713
168. Furet JP, Kong LC, Tap J, Poitou C, Basdevant A, Bouillot JL, et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: links with metabolic and low-grade inflammation markers. Diabetes. (2010) 59:3049–57. doi: 10.2337/db10-0253
169. Martinez I, Lattimer JM, Hubach KL, Case JA, Yang J, Weber CG, et al. Gut microbiome composition is linked to whole grain-induced immunological improvements. ISME J. (2013) 7:269–80. doi: 10.1038/ismej.2012.104
170. Van Den Munckhof ICL, Kurilshikov A, Ter Horst R, Riksen NP, Joosten LAB, Zhernakova A, et al. Role of gut microbiota in chronic low-grade inflammation as potential driver for atherosclerotic cardiovascular disease: a systematic review of human studies. Obes Rev. (2018) 19:1719–34. doi: 10.1111/obr.12750
171. Meade KG, O'Farrelly C. β-Defensins: farming the microbiome for homeostasis and health. Front Immunol. (2019) 9:3072. doi: 10.3389/fimmu.2018.03072
172. Peyrin-Biroulet L, Gonzalez F, Dubuquoy L, Rousseaux C, Dubuquoy C, Decourcelle C, et al. Mesenteric fat as a source of C reactive protein and as a target for bacterial translocation in Crohnand#039;s disease. Gut. (2012) 61:78. doi: 10.1136/gutjnl-2011-300370
173. Fouts DE, Torralba M, Nelson KE, Brenner DA, Schnabl B. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J Hepatol. (2012) 56:1283–92. doi: 10.1016/j.jhep.2012.01.019
174. Brenchley JM, Douek DC. Microbial translocation across the GI tract. Annu Rev Immunol. (2012) 30:149–73. doi: 10.1146/annurev-immunol-020711-075001
175. Rohr MW, Narasimhulu CA, Rudeski-Rohr TA, Parthasarathy S. Negative effects of a high-fat diet on intestinal permeability: a review. Adv Nutr. (2019) 11:77–91. doi: 10.1093/advances/nmz061
176. Guo S, Al-Sadi R, Said HM, Ma TY. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am J Pathol. (2013) 182:375–87. doi: 10.1016/j.ajpath.2012.10.014
177. Thaiss CA, Levy M, Grosheva I, Zheng D, Soffer E, Blacher E, et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science. (2018) 359:1376–83. doi: 10.1126/science.aar3318
178. Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermudez-Humaran LG, et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med. (2011) 3:559–72. doi: 10.1002/emmm.201100159
179. Udayappan SD, Kovatcheva-Datchary P, Bakker GJ, Havik SR, Herrema H, Cani PD, et al. Intestinal Ralstonia pickettii augments glucose intolerance in obesity. PLoS ONE. (2017) 12:e0181693. doi: 10.1371/journal.pone.0181693
180. Kubinak JL, Round JL. Do antibodies select a healthy microbiota? Nat Rev Immunol. (2016) 16:767. doi: 10.1038/nri.2016.114
181. Hapfelmeier S, Lawson MAE, Slack E, Kirundi JK, Stoel M, Heikenwalder M, et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science. (2010) 328:1705–9. doi: 10.1126/science.1188454
182. Knoop KA, Gustafsson JK, McDonald KG, Kulkarni DH, Coughlin PE, McCrate S, et al. Microbial antigen encounter during a preweaning interval is critical for tolerance to gut bacteria. Sci Immunol. (2017) 2:eaao1314. doi: 10.1126/sciimmunol.aao1314
183. Fagarasan S, Muramatsu M, Suzuki K, Nagaoka H, Hiai H, Honjo T. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science. (2002) 298:1424–7. doi: 10.1126/science.1077336
184. Suzuki K, Meek B, Doi Y, Muramatsu M, Chiba T, Honjo T, et al. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc Natl Acad Sci USA. (2004) 101:1981–6. doi: 10.1073/pnas.0307317101
185. Catanzaro JR, Strauss JD, Bielecka A, Porto AF, Lobo FM, Urban A, et al. IgA-deficient humans exhibit gut microbiota dysbiosis despite secretion of compensatory IgM. Sci Rep. (2019) 9:13574. doi: 10.1038/s41598-019-49923-2
186. Kau AL, Planer JD, Liu J, Rao S, Yatsunenko T, Trehan I, et al. Functional characterization of IgA-targeted bacterial taxa from undernourished Malawian children that produce diet-dependent enteropathy. Sci Transl Med. (2015) 7:276ra224. doi: 10.1126/scitranslmed.aaa4877
187. Palm NW, De Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. (2014) 158:1000–10. doi: 10.1016/j.cell.2014.08.006
188. Luck H, Khan S, Kim JH, Copeland JK, Revelo XS, Tsai S, et al. Gut-associated IgA+ immune cells regulate obesity-related insulin resistance. Nat Commun. (2019) 10:3650. doi: 10.1038/s41467-019-11370-y
189. Tran HQ, Ley RE, Gewirtz AT, Chassaing B. Flagellin-elicited adaptive immunity suppresses flagellated microbiota and vaccinates against chronic inflammatory diseases. Nat Commun. (2019) 10:5650. doi: 10.1038/s41467-019-13538-y
190. Dalmas E, Venteclef N, Caer C, Poitou C, Cremer I, Aron-Wisnewsky J, et al. T cell-derived IL-22 amplifies IL-1beta-driven inflammation in human adipose tissue: relevance to obesity and type 2 diabetes. Diabetes. (2014) 63:1966–77. doi: 10.2337/db13-1511
191. Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. (2011) 17:610–7. doi: 10.1038/nm.2353
192. Defuria J, Belkina AC, Jagannathan-Bogdan M, Snyder-Cappione J, Carr JD, Nersesova YR, et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci USA. (2013) 110:5133–8. doi: 10.1073/pnas.1215840110
193. Zhao Q, Elson CO. Adaptive immune education by gut microbiota antigens. Immunology. (2018) 154:28–37. doi: 10.1111/imm.12896
194. Petersen C, Bell R, Klag KA, Lee S-H, Soto R, Ghazaryan A, et al. T cell–mediated regulation of the microbiota protects against obesity. Science. (2019) 365:eaat9351. doi: 10.1126/science.aat9351
195. Glatz JFC, Luiken JJFP. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J Lipid Res. (2018) 59:1084–93. doi: 10.1194/jlr.R082933
196. Pabst O, Slack E. IgA and the intestinal microbiota: the importance of being specific. Mucosal Immunol. (2019) 13:12–21. doi: 10.1038/s41385-019-0227-4
197. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. (2017) 542:177–85. doi: 10.1038/nature21363
198. Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. (2005) 102:11070–5. doi: 10.1073/pnas.0504978102
199. Christ A, Latz E. The Western lifestyle has lasting effects on metaflammation. Nat Rev Immunol. (2019) 19:267–8. doi: 10.1038/s41577-019-0156-1
200. Allaire JM, Crowley SM, Law HT, Chang SY, Ko HJ, Vallance BA. The intestinal epithelium: central coordinator of mucosal immunity. Trends Immunol. (2018) 39:677–96. doi: 10.1016/j.it.2018.04.002
201. Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. (2002) 168:554–61. doi: 10.4049/jimmunol.168.2.554
202. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. (2009) 22:240–73. doi: 10.1128/CMR.00046-08
203. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. (2017) 9:7204–18. doi: 10.18632/oncotarget.23208
204. Jialal I, Kaur H, Devaraj S. Toll-like receptor status in obesity and metabolic syndrome: a translational perspective. J Clin Endocrinol Metab. (2014) 99:39–48. doi: 10.1210/jc.2013-3092
205. Park BS, Lee J-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med. (2013) 45:e66. doi: 10.1038/emm.2013.97
206. Dasu MR, Devaraj S, Park S, Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care. (2010) 33:861–8. doi: 10.2337/dc09-1799
207. Ghanim H, Mohanty P, Deopurkar R, Sia CL, Korzeniewski K, Abuaysheh S, et al. Acute modulation of toll-like receptors by insulin. Diabetes Care. (2008) 31:1827–31. doi: 10.2337/dc08-0561
208. Li J, Chen L, Zhang Y, Zhang WJ, Xu W, Qin Y, et al. TLR4 is required for the obesity-induced pancreatic beta cell dysfunction. Acta Biochim Biophys Sin. (2013) 45:1030–8. doi: 10.1093/abbs/gmt092
209. Ji Y, Sun S, Shrestha N, Darragh LB, Shirakawa J, Xing Y, et al. Toll-like receptors TLR2 and TLR4 block the replication of pancreatic beta cells in diet-induced obesity. Nat Immunol. (2019) 20:677–86. doi: 10.1038/s41590-019-0396-z
210. Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. (2010) 328:228–31. doi: 10.1126/science.1179721
211. Yoon S-I, Kurnasov O, Natarajan V, Hong M, Gudkov AV, Osterman AL, et al. Structural basis of TLR5-flagellin recognition and signaling. Science. (2012) 335:859–64. doi: 10.1126/science.1215584
212. Ubeda C, Lipuma L, Gobourne A, Viale A, Leiner I, Equinda M, et al. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med. (2012) 209:1445–56. doi: 10.1084/jem.20120504
213. Chassaing B, Ley RE, Gewirtz AT. Intestinal epithelial cell toll-like receptor 5 regulates the intestinal microbiota to prevent low-grade inflammation and metabolic syndrome in mice. Gastroenterology. (2014) 147:1363–77.e1317. doi: 10.1053/j.gastro.2014.08.033
214. Deguine J, Barton GM. MyD88: a central player in innate immune signaling. F1000prime Rep. (2014) 6:97. doi: 10.12703/P6-97
215. Duparc T, Plovier H, Marrachelli VG, Van Hul M, Essaghir A, Stahlman M, et al. Hepatocyte MyD88 affects bile acids, gut microbiota and metabolome contributing to regulate glucose and lipid metabolism. Gut. (2017) 66:620–32. doi: 10.1136/gutjnl-2015-310904
216. Everard A, Geurts L, Caesar R, Van Hul M, Matamoros S, Duparc T, et al. Intestinal epithelial MyD88 is a sensor switching host metabolism towards obesity according to nutritional status. Nat Commun. (2014) 5:5648. doi: 10.1038/ncomms6648
217. Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med. (2010) 16:228–31. doi: 10.1038/nm.2087
218. Schertzer JD, Tamrakar AK, Magalhaes JG, Pereira S, Bilan PJ, Fullerton MD, et al. NOD1 activators link innate immunity to insulin resistance. Diabetes. (2011) 60:2206–15. doi: 10.2337/db11-0004
219. Cavallari JF, Fullerton MD, Duggan BM, Foley KP, Denou E, Smith BK, et al. Muramyl dipeptide-based postbiotics mitigate obesity-induced insulin resistance via IRF4. Cell Metab. (2017) 25:1063–1074.e1063. doi: 10.1016/j.cmet.2017.03.021
220. Zhang Q, Pan Y, Zeng B, Zheng X, Wang H, Shen X, et al. Intestinal lysozyme liberates Nod1 ligands from microbes to direct insulin trafficking in pancreatic beta cells. Cell Res. (2019) 29:516–32. doi: 10.1038/s41422-019-0190-3
221. Denou E, Lolmede K, Garidou L, Pomie C, Chabo C, Lau TC, et al. Defective NOD2 peptidoglycan sensing promotes diet-induced inflammation, dysbiosis, and insulin resistance. EMBO Mol Med. (2015) 7:259–74. doi: 10.15252/emmm.201404169
222. Sharma D, Kanneganti T-D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol. (2016) 213:617. doi: 10.1083/jcb.201602089
223. Man SM. Inflammasomes in the gastrointestinal tract: infection, cancer and gut microbiota homeostasis. Nat Rev Gastroenterol Hepatol. (2018) 15:721–37. doi: 10.1038/s41575-018-0054-1
224. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. (2011) 145:745–57. doi: 10.1016/j.cell.2011.04.022
225. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. (2012) 482:179–85. doi: 10.1038/nature10809
226. Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell. (2014) 156:1045–59. doi: 10.1016/j.cell.2014.01.026
227. Levy M, Thaiss CA, Zeevi D, Dohnalova L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell. (2015) 163:1428–43. doi: 10.1016/j.cell.2015.10.048
228. Chen L, Wilson JE, Koenigsknecht MJ, Chou WC, Montgomery SA, Truax AD, et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat Immunol. (2017) 18:541–51. doi: 10.1038/ni.3690
229. Truax AD, Chen L, Tam JW, Cheng N, Guo H, Koblansky AA, et al. The inhibitory innate immune sensor NLRP12 maintains a threshold against obesity by regulating gut microbiota homeostasis. Cell Host Microbe. (2018) 24:364–78.e366. doi: 10.1016/j.chom.2018.08.009
230. Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell. (2016) 166:624–36. doi: 10.1016/j.cell.2016.05.076
231. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20:3328. doi: 10.3390/ijms20133328
232. Vandanmagsar B, Youm Y-H, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature. medicine. (2011) 17:179–88. doi: 10.1038/nm.2279
233. Rheinheimer J, De Souza BM, Cardoso NS, Bauer AC, Crispim D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: a systematic review. Metabolism. (2017) 74:1–9. doi: 10.1016/j.metabol.2017.06.002
234. Lee H-M, Kim J-J, Kim HJ, Shong M, Ku BJ, Jo E-K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. (2013) 62:194. doi: 10.2337/db12-0420
235. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. (2010) 11:136–40. doi: 10.1038/ni.1831
236. Lemire P, Robertson SJ, Maughan H, Tattoli I, Streutker CJ, Platnich JM, et al. The NLR Protein NLRP6 does not impact gut microbiota composition. Cell Rep. (2017) 21:3653–61. doi: 10.1016/j.celrep.2017.12.026
237. Mamantopoulos M, Ronchi F, Van Hauwermeiren F, Vieira-Silva S, Yilmaz B, Martens L, et al. Nlrp6- and ASC-dependent inflammasomes do not shape the commensal gut microbiota composition. Immunity. (2017) 47:339–48.e334. doi: 10.1016/j.immuni.2017.07.011
238. Gharagozloo M, Gris KV, Mahvelati T, Amrani A, Lukens JR, Gris D. NLR-dependent regulation of inflammation in multiple sclerosis. Front Immunol. (2018) 8:2012. doi: 10.3389/fimmu.2017.02012
239. Ngo VL, Abo H, Maxim E, Harusato A, Geem D, Medina-Contreras O, et al. A cytokine network involving IL-36gamma, IL-23, and IL-22 promotes antimicrobial defense and recovery from intestinal barrier damage. Proc Natl Acad Sci USA. (2018) 115:E5076–85. doi: 10.1073/pnas.1718902115
240. Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. (2013) 39:372–85. doi: 10.1016/j.immuni.2013.08.003
241. Victor AR, Nalin AP, Dong W, McClory S, Wei M, Mao C, et al. IL-18 Drives ILC3 proliferation and promotes IL-22 production via NF-kappaB. J Immunol. (2017) 199:2333–42. doi: 10.4049/jimmunol.1601554
242. Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature. (2014) 514:237–41. doi: 10.1038/nature13564
243. Dudakov JA, Hanash AM, Van Den Brink MRM. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. (2015) 33:747–85. doi: 10.1146/annurev-immunol-032414-112123
244. Hasnain SZ, Borg DJ, Harcourt BE, Tong H, Sheng YH, Ng CP, et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat Med. (2014) 20:1417–26. doi: 10.1038/nm.3705
245. Miani M, Le Naour J, Waeckel-Enée E, Verma SC, Straube M, Emond P, et al. Gut microbiota-stimulated innate lymphoid cells support β-defensin 14 expression in pancreatic endocrine cells, preventing autoimmune diabetes. Cell Metab. (2018) 28:557–572.e556. doi: 10.1016/j.cmet.2018.06.012
246. Fatkhullina AR, Peshkova IO, Dzutsev A, Aghayev T, McCulloch JA, Thovarai V, et al. An interleukin-23-interleukin-22 axis regulates intestinal microbial homeostasis to protect from diet-induced atherosclerosis. Immunity. (2018) 49:943–57.e949. doi: 10.1016/j.immuni.2018.09.011
247. Laurans L, Venteclef N, Haddad Y, Chajadine M, Alzaid F, Metghalchi S, et al. Genetic deficiency of indoleamine 2,3-dioxygenase promotes gut microbiota-mediated metabolic health. Nat Med. (2018) 24:1113–20. doi: 10.1038/s41591-018-0060-4
248. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. (2011) 478:197–203. doi: 10.1038/nature10491
249. Taleb S. Tryptophan dietary impacts gut barrier and metabolic diseases. Front Immunol. (2019) 10:2113. doi: 10.3389/fimmu.2019.02113
250. Agudelo LZ, Ferreira DMS, Cervenka I, Bryzgalova G, Dadvar S, Jannig PR, et al. Kynurenic acid and Gpr35 regulate adipose tissue energy homeostasis and inflammation. Cell Metab. (2018) 27:378–92.e375. doi: 10.1016/j.cmet.2018.01.004
251. Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, et al. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol. (2009) 183:2475–83. doi: 10.4049/jimmunol.0900986
252. Metghalchi S, Ponnuswamy P, Simon T, Haddad Y, Laurans L, Clement M, et al. Indoleamine 2,3-dioxygenase fine-tunes immune homeostasis in atherosclerosis and colitis through repression of interleukin-10 production. Cell Metab. (2015) 22:460–71. doi: 10.1016/j.cmet.2015.07.004
253. Mao K, Baptista AP, Tamoutounour S, Zhuang L, Bouladoux N, Martins AJ, et al. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature. (2018) 554:255–9. doi: 10.1038/nature25437
254. Chen L, Strohmeier V, He Z, Deshpande M, Catalan-Dibene J, Durum SK, et al. Interleukin 22 disrupts pancreatic function in newborn mice expressing IL-23. Nat Commun. (2019) 10:4517. doi: 10.1038/s41467-019-12540-8
255. Kleinschek MA, Muller U, Brodie SJ, Stenzel W, Kohler G, Blumenschein WM, et al. IL-23 enhances the inflammatory cell response in Cryptococcus neoformans infection and induces a cytokine pattern distinct from IL-12. J Immunol. (2006) 176:1098–106. doi: 10.4049/jimmunol.176.2.1098
256. Tang C, Chen S, Qian H, Huang W. Interleukin-23: as a drug target for autoimmune inflammatory diseases. Immunology. (2012) 135:112–24. doi: 10.1111/j.1365-2567.2011.03522.x
257. Ziblat A, Nuñez SY, Raffo Iraolagoitia XL, Spallanzani RG, Torres NI, Sierra JM, et al. Interleukin (IL)-23 stimulates IFN-γ secretion by CD56bright natural killer cells and enhances IL-18-driven dendritic cells activation. Front Immunol. (2018) 8:1959. doi: 10.3389/fimmu.2017.01959
258. Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol. (2008) 181:5948–55. doi: 10.4049/jimmunol.181.9.5948
259. Ivanov II, Frutos RDL, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. (2008) 4:337–49. doi: 10.1016/j.chom.2008.09.009
260. Sumarac-Dumanovic M, Stevanovic D, Ljubic A, Jorga J, Simic M, Stamenkovic-Pejkovic D, et al. Increased activity of interleukin-23/interleukin-17 proinflammatory axis in obese women. Int J Obes. (2009) 33:151–6. doi: 10.1038/ijo.2008.216
261. Martins LMS, Perez MM, Pereira CA, Costa FRC, Dias MS, Tostes RC, et al. Interleukin-23 promotes intestinal T helper type17 immunity and ameliorates obesity-associated metabolic syndrome in a murine high-fat diet model. Immunology. (2018) 154:624–36. doi: 10.1111/imm.12946
262. Walsh PT, Fallon PG. The emergence of the IL-36 cytokine family as novel targets for inflammatory diseases. Ann N Y Acad Sci. (2018) 1417:23–34. doi: 10.1111/nyas.13280
263. Medina-Contreras O, Harusato A, Nishio H, Flannigan KL, Ngo V, Leoni G, et al. Cutting Edge: IL-36 receptor promotes resolution of intestinal damage. J Immunol. (2016) 196:34–8. doi: 10.4049/jimmunol.1501312
264. Giannoudaki E, Hernandez-Santana YE, Mulfaul K, Doyle SL, Hams E, Fallon PG, et al. Interleukin-36 cytokines alter the intestinal microbiome and can protect against obesity and metabolic dysfunction. Nat Commun. (2019) 10:4003. doi: 10.1038/s41467-019-11944-w
265. Mishima Y, Oka A, Liu B, Herzog JW, Eun CS, Fan T-J, et al. Microbiota maintain colonic homeostasis by activating TLR2/MyD88/PI3K signaling in IL-10-producing regulatory B cells. The J Clin Invest. (2019) 129:3702–16. doi: 10.1172/JCI93820
266. Sun M, Wu W, Chen L, Yang W, Huang X, Ma C, et al. Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat Commun. (2018) 9:3555. doi: 10.1038/s41467-018-05901-2
267. Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. (1998) 66:5224–31. doi: 10.1128/IAI.66.11.5224-5231.1998
268. Andrews C, McLean MH, Durum SK. Cytokine tuning of intestinal epithelial function. Front Immunol. (2018) 9:1270. doi: 10.3389/fimmu.2018.01270
269. Cani PD. Microbiota and metabolites in metabolic diseases. Nat Rev Endocrinol. (2019) 15:69–70. doi: 10.1038/s41574-018-0143-9
270. Koh A, Molinaro A, Stahlman M, Khan MT, Schmidt C, Manneras-Holm L, et al. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell. (2018) 175:947–961.e917. doi: 10.1016/j.cell.2018.09.055
271. Reynolds A, Mann J, Cummings J, Winter N, Mete E, Te Morenga L. Carbohydrate quality and human health: a series of systematic reviews and meta-analyses. Lancet. (2019) 393:434–45. doi: 10.1016/S0140-6736(18)31809-9
272. Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science. (2018) 359:1151–6. doi: 10.1126/science.aao5774
273. Chambers ES, Byrne CS, Morrison DJ, Murphy KG, Preston T, Tedford C, et al. Dietary supplementation with inulin-propionate ester or inulin improves insulin sensitivity in adults with overweight and obesity with distinct effects on the gut microbiota, plasma metabolome and systemic inflammatory responses: a randomised cross-over trial. Gut. (2019) 68:1430–8. doi: 10.1136/gutjnl-2019-318424
274. Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun. (2015) 6:6734. doi: 10.1038/ncomms7734
275. Byrne CS, Chambers ES, Morrison DJ, Frost G. The role of short chain fatty acids in appetite regulation and energy homeostasis. In J Obes. (2015) 39:1331–8. doi: 10.1038/ijo.2015.84
276. Chambers ES, Viardot A, Psichas A, Morrison DJ, Murphy KG, Zac-Varghese SE, et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut. (2015) 64:1744–54. doi: 10.1136/gutjnl-2014-307913
277. Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. (2009) 58:1509–17. doi: 10.2337/db08-1637
278. Den Besten G, Bleeker A, Gerding A, Van Eunen K, Havinga R, Van Dijk TH, et al. Short-chain fatty acids protect against high-fat diet-induced obesity via a PPARgamma-dependent switch from lipogenesis to fat oxidation. Diabetes. (2015) 64:2398–408. doi: 10.2337/db14-1213
279. Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL, et al. Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature. (2016) 534:213–7. doi: 10.1038/nature18309
280. Tirosh A, Calay ES, Tuncman G, Claiborn KC, Inouye KE, Eguchi K, et al. The short-chain fatty acid propionate increases glucagon and FABP4 production, impairing insulin action in mice and humans. Sci Transl Med. (2019) 11:eaav0120. doi: 10.1126/scitranslmed.aav0120
281. Peng L, Li Z-R, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr. (2009) 139:1619–25. doi: 10.3945/jn.109.104638
282. Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem. (2008) 19:587–93. doi: 10.1016/j.jnutbio.2007.08.002
283. Lin MY, De Zoete MR, Van Putten JP, Strijbis K. Redirection of epithelial immune responses by short-chain fatty acids through inhibition of histone deacetylases. Front Immunol. (2015) 6:554. doi: 10.3389/fimmu.2015.00554
284. Freedman SB, Williamson-Urquhart S, Farion KJ, Gouin S, Willan AR, Poonai N, et al. Multicenter trial of a combination probiotic for children with gastroenteritis. N Engl J Med. (2018) 379:2015–26. doi: 10.1056/NEJMoa1802597
285. Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, et al. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-kappaB. J Am Heart Assoc. (2016) 5:e002767. doi: 10.1161/JAHA.115.002767
286. Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J Am Heart Assoc. (2017) 6:e006347. doi: 10.1161/JAHA.117.006347
287. Tang WHW, Backhed F, Landmesser U, Hazen SL. Intestinal microbiota in cardiovascular health and disease: JACC state-of-the-art review. J Am Coll Cardiol. (2019) 73:2089–105. doi: 10.1016/j.jacc.2019.03.024
288. Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, et al. Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell. (2015) 163:1585–95. doi: 10.1016/j.cell.2015.11.055
289. Heianza Y, Sun D, Li X, Didonato JA, Bray GA, Sacks FM, et al. Gut microbiota metabolites, amino acid metabolites and improvements in insulin sensitivity and glucose metabolism: the POUNDS Lost trial. Gut. (2019) 68:263. doi: 10.1136/gutjnl-2018-316155
290. Bloomgarden Z. Diabetes and branched-chain amino acids: What is the link? J Diabetes. (2018) 10:350–2. doi: 10.1111/1753-0407.12645
291. Flores-Guerrero JL, Osté MCJ, Kieneker LM, Gruppen EG, Wolak-Dinsmore J, Otvos JD, et al. Plasma branched-chain amino acids and risk of incident type 2 diabetes: results from the PREVEND prospective cohort study. J Clin Med. (2018) 7:513. doi: 10.3390/jcm7120513
292. Neis EPJG, Dejong CHC, Rensen SS. The role of microbial amino acid metabolism in host metabolism. Nutrients. (2015) 7:2930–46. doi: 10.3390/nu7042930
293. Shapiro H, Kolodziejczyk AA, Halstuch D, Elinav E. Bile acids in glucose metabolism in health and disease. J Exp Med. (2018) 215:383. doi: 10.1084/jem.20171965
294. Islam KB, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology. (2011) 141:1773–81. doi: 10.1053/j.gastro.2011.07.046
295. Pathak P, Xie C, Nichols RG, Ferrell JM, Boehme S, Krausz KW, et al. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology. (2018) 68:1574–88. doi: 10.1002/hep.29857
296. Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, Liu J, et al. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science. (2006) 312:233. doi: 10.1126/science.1121435
297. Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci USA. (2006) 103:1006–11. doi: 10.1073/pnas.0506982103
298. Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature. (2014) 509:183–8. doi: 10.1038/nature13135
299. Prawitt J, Abdelkarim M, Stroeve JH, Popescu I, Duez H, Velagapudi VR, et al. Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes. (2011) 60:1861–71. doi: 10.2337/db11-0030
300. Watanabe M, Horai Y, Houten SM, Morimoto K, Sugizaki T, Arita E, et al. Lowering bile acid pool size with a synthetic farnesoid X receptor (FXR) agonist induces obesity and diabetes through reduced energy expenditure. J Biol Chem. (2011) 286:26913–20. doi: 10.1074/jbc.M111.248203
301. Parséus A, Sommer N, Sommer F, Caesar R, Molinaro A, Ståhlman M, et al. Microbiota-induced obesity requires farnesoid X receptor. Gut. (2017) 66:429. doi: 10.1136/gutjnl-2015-310283
302. Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol. (2009) 183:6251–61. doi: 10.4049/jimmunol.0803978
303. Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, et al. Personalized nutrition by prediction of glycemic responses. Cell. (2015) 163:1079–94. doi: 10.1016/j.cell.2015.11.001
304. Llewellyn SR, Britton GJ, Contijoch EJ, Vennaro OH, Mortha A, Colombel JF, et al. Interactions between diet and the intestinal microbiota alter intestinal permeability and colitis severity in mice. Gastroenterology. (2018) 154:1037–46.e1032. doi: 10.1053/j.gastro.2017.11.030
305. Xiao S, Fei N, Pang X, Shen J, Wang L, Zhang B, et al. A gut microbiota-targeted dietary intervention for amelioration of chronic inflammation underlying metabolic syndrome. FEMS Microbiol Ecol. (2014) 87:357–67. doi: 10.1111/1574-6941.12228
306. So D, Whelan K, Rossi M, Morrison M, Holtmann G, Kelly JT, et al. Dietary fiber intervention on gut microbiota composition in healthy adults: a systematic review and meta-analysis. Am J Clin Nutr. (2018) 107:965–83. doi: 10.1093/ajcn/nqy041
307. Mardinoglu A, Wu H, Bjornson E, Zhang C, Hakkarainen A, Rasanen SM, et al. An integrated understanding of the rapid metabolic benefits of a carbohydrate-restricted diet on hepatic steatosis in humans. Cell Metab. (2018) 27:559–71.e555. doi: 10.1016/j.cmet.2018.01.005
308. Everard A, Lazarevic V, Derrien M, Girard M, Muccioli GG, Neyrinck AM, et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes. (2011) 60:2775–86. doi: 10.2337/db11-0227
309. Nicolucci AC, Hume MP, Martinez I, Mayengbam S, Walter J, Reimer RA. Prebiotics reduce body fat and alter intestinal microbiota in children who are overweight or with obesity. Gastroenterology. (2017) 153:711–22. doi: 10.1053/j.gastro.2017.05.055
310. Zhang X, Zhao Y, Zhang M, Pang X, Xu J, Kang C, et al. Structural changes of gut microbiota during berberine-mediated prevention of obesity and insulin resistance in high-fat diet-fed rats. PLoS ONE. (2012) 7:e42529. doi: 10.1371/journal.pone.0042529
311. Cani PD, Possemiers S, Van De Wiele T, Guiot Y, Everard A, Rottier O, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. (2009) 58:1091. doi: 10.1136/gut.2008.165886
312. Mobini R, Tremaroli V, Stahlman M, Karlsson F, Levin M, Ljungberg M, et al. Metabolic effects of Lactobacillus reuteri DSM 17938 in people with type 2 diabetes: a randomized controlled trial. Diabetes Obes Metab. (2017) 19:579–89. doi: 10.1111/dom.12861
313. Simon MC, Strassburger K, Nowotny B, Kolb H, Nowotny P, Burkart V, et al. Intake of Lactobacillus reuteri improves incretin and insulin secretion in glucose-tolerant humans: a proof of concept. Diabetes Care. (2015) 38:1827–34. doi: 10.2337/dc14-2690
314. Wang J, Tang H, Zhang C, Zhao Y, Derrien M, Rocher E, et al. Modulation of gut microbiota during probiotic-mediated attenuation of metabolic syndrome in high fat diet-fed mice. ISME J. (2015) 9:1–15. doi: 10.1038/ismej.2014.99
315. Sanchez M, Darimont C, Drapeau V, Emady-Azar S, Lepage M, Rezzonico E, et al. Effect of Lactobacillus rhamnosus CGMCC1.3724 supplementation on weight loss and maintenance in obese men and women. Br J Nutr. (2014) 111:1507–19. doi: 10.1017/S0007114513003875
316. Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA. (2013) 110:9066–71. doi: 10.1073/pnas.1219451110
317. Depommier C, Everard A, Druart C, Plovier H, Van Hul M, Vieira-Silva S, et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat Med. (2019) 25:1096–103. doi: 10.1038/s41591-019-0495-2
318. De Groot P, Scheithauer T., Bakker G. J., Prodan A., Levin E., Khan M. T., et al. (2019). Donor metabolic characteristics drive effects of faecal microbiota transplantation on recipient insulin sensitivity, energy expenditure and intestinal transit time. Gut. 69:502–12. doi: 10.1136/gutjnl-2019-318320
319. Makki K, Deehan EC, Walter J, Backhed F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe. (2018) 23:705–15. doi: 10.1016/j.chom.2018.05.012
320. Simpson HL, Campbell BJ. Review article: dietary fibre-microbiota interactions. Aliment Pharmacol Ther. (2015) 42:158–79. doi: 10.1111/apt.13248
321. Roberfroid M, Gibson GR, Hoyles L, McCartney AL, Rastall R, Rowland I, et al. Prebiotic effects: metabolic and health benefits. Br J Nutr. (2010) 104(Suppl. 2):S1–63. doi: 10.1017/S0007114510003363
322. Sanders ME, Merenstein DJ, Reid G, Gibson GR, Rastall RA. Probiotics and prebiotics in intestinal health and disease: from biology to the clinic. Nat Rev Gastroenterol Hepatol. (2019) 16:605–16. doi: 10.1038/s41575-019-0173-3
323. Cani PD, Hoste S, Guiot Y, Delzenne NM. Dietary non-digestible carbohydrates promote L-cell differentiation in the proximal colon of rats. Br J Nutr. (2007) 98:32–7. doi: 10.1017/S0007114507691648
324. Beserra BT, Fernandes R, Do Rosario VA, Mocellin MC, Kuntz MG, Trindade EB. A systematic review and meta-analysis of the prebiotics and synbiotics effects on glycaemia, insulin concentrations and lipid parameters in adult patients with overweight or obesity. Clin Nutr. (2015) 34:845–58. doi: 10.1016/j.clnu.2014.10.004
325. Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol. (2014) 11:506–14. doi: 10.1038/nrgastro.2014.66
326. Suez J, Zmora N, Segal E, Elinav E. The pros, cons, and many unknowns of probiotics. Nat Med. (2019) 25:716–29. doi: 10.1038/s41591-019-0439-x
327. Natividad JM, Agus A, Planchais J, Lamas B, Jarry AC, Martin R, et al. Impaired aryl hydrocarbon receptor ligand production by the gut microbiota is a key factor in metabolic syndrome. Cell Metab. (2018) 28:737–749.e734. doi: 10.1016/j.cmet.2018.07.001
328. Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. (2003) 37:343–50. doi: 10.1053/jhep.2003.50048
329. Ruan Y, Sun J, He J, Chen F, Chen R, Chen H. Effect of probiotics on glycemic control: a systematic review and meta-analysis of randomized, controlled trials. PLoS ONE. (2015) 10:e0132121. doi: 10.1371/journal.pone.0132121
330. Sun J, Buys NJ. Glucose- and glycaemic factor-lowering effects of probiotics on diabetes: a meta-analysis of randomised placebo-controlled trials. Br J Nutr. (2016) 115:1167–77. doi: 10.1017/S0007114516000076
331. Akbari V, Hendijani F. Effects of probiotic supplementation in patients with type 2 diabetes: systematic review and meta-analysis. Nutr Rev. (2016) 74:774–84. doi: 10.1093/nutrit/nuw039
332. Samah S, Ramasamy K, Lim SM, Neoh CF. Probiotics for the management of type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetes Res Clin Pract. (2016) 118:172–82. doi: 10.1016/j.diabres.2016.06.014
333. Park S, Bae JH. Probiotics for weight loss: a systematic review and meta-analysis. Nutr Res. (2015) 35:566–75. doi: 10.1016/j.nutres.2015.05.008
334. Yao K, Zeng L, He Q, Wang W, Lei J, Zou X. Effect of probiotics on glucose and lipid metabolism in type 2 diabetes mellitus: a meta-analysis of 12 randomized controlled trials. Med Sci Monit. (2017) 23:3044–53. doi: 10.12659/MSM.902600
335. Plovier H, Everard A, Druart C, Depommier C, Van Hul M, Geurts L, et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med. (2017) 23:107–13. doi: 10.1038/nm.4236
336. Hendrikx T, Duan Y, Wang Y, Oh JH, Alexander LM, Huang W, et al. Bacteria engineered to produce IL-22 in intestine induce expression of REG3G to reduce ethanol-induced liver disease in mice. Gut. (2019) 68:1504–15. doi: 10.1136/gutjnl-2018-317232
337. Zmora N, Zilberman-Schapira G, Suez J, Mor U, Dori-Bachash M, Bashiardes S, et al. Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell. (2018) 174:1388–405.e1321. doi: 10.1016/j.cell.2018.08.041
338. Eiseman B, Silen W, Bascom GS, Kauvar AJ. Fecal enema as an adjunct in the treatment of pseudomembranous enterocolitis. Surgery. (1958) 44:854–9.
339. Zhang F, Luo W, Shi Y, Fan Z, Ji G. Should we standardize the 1,700-year-old fecal microbiota transplantation? Am J Gastroenterol. (2012) 107, 1755; author reply 1755–6. doi: 10.1038/ajg.2012.251
340. Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. (2012) 143:913–6 e917. doi: 10.1053/j.gastro.2012.06.031
341. Li SS, Zhu A, Benes V, Costea PI, Hercog R, Hildebrand F, et al. Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science. (2016) 352:586–9. doi: 10.1126/science.aad8852
342. Kootte RS, Levin E, Salojarvi J, Smits LP, Hartstra AV, Udayappan SD, et al. Improvement of insulin sensitivity after lean donor feces in metabolic syndrome is driven by baseline intestinal microbiota composition. Cell Metab. 26, 611–9 e616. doi: 10.1016/j.cmet.2017.09.008
343. Donath MY, Böni-Schnetzler M, Ellingsgaard H, Ehses JA. Islet inflammation impairs the pancreatic β-Cell in type 2 diabetes. Physiology. (2009) 24:325–31. doi: 10.1152/physiol.00032.2009
344. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology. (2010) 52:1836–46. doi: 10.1002/hep.24001
345. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta. (2011) 1813:878–88. doi: 10.1016/j.bbamcr.2011.01.034
346. Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. (2018) 555:210–5. doi: 10.1038/nature25973
347. Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human genetics shape the gut microbiome. Cell. (2014) 159:789–99. doi: 10.1016/j.cell.2014.09.053
348. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. (2014) 157:121–41. doi: 10.1016/j.cell.2014.03.011
349. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. (2013) 341:569–73. doi: 10.1126/science.1241165
350. Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K, et al. Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell. (2017) 171:1015–1028.e1013. doi: 10.1016/j.cell.2017.09.016
351. Allaband C, McDonald D, Vázquez-Baeza Y, Minich JJ, Tripathi A, Brenner DA, et al. Microbiome 101: studying, analyzing, and interpreting gut microbiome data for clinicians. Clin Gastroenterol Hepatol. (2019) 17:218–30. doi: 10.1016/j.cgh.2018.09.017
352. Amar J, Serino M, Lange C, Chabo C, Iacovoni J, Mondot S, et al. Involvement of tissue bacteria in the onset of diabetes in humans: evidence for a concept. Diabetologia. (2011) 54:3055–61. doi: 10.1007/s00125-011-2329-8
353. Tang WHW, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. (2013) 368:1575–84. doi: 10.1056/NEJMoa1109400
354. Malla MA, Dubey A, Kumar A, Yadav S, Hashem A, Abd_Allah EF. Exploring the human microbiome: the potential future role of next-generation sequencing in disease diagnosis and treatment. Front Immunol. (2019) 9:2868. doi: 10.3389/fimmu.2018.02868
355. Rasmussen TS, Mentzel CMJ, Kot W, Castro-Mejia JL, Zuffa S, Swann JR, et al. Faecal virome transplantation decreases symptoms of type 2 diabetes and obesity in a murine model. Gut. (2020) 1–9. doi: 10.1101/792556
356. Huseyin CE, O'Toole PW, Cotter PD, Scanlan PD. Forgotten fungi—the gut mycobiome in human health and disease. FEMS Microbiol Rev. (2017) 41:479–511. doi: 10.1093/femsre/fuw047
357. Deschasaux M, Bouter KE, Prodan A, Levin E, Groen AK, Herrema H, et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat Med. (2018) 24:1526–31. doi: 10.1038/s41591-018-0160-1
Keywords: microbiota, obesity, metainflammation, metabolism, diabetes
Citation: Scheithauer TPM, Rampanelli E, Nieuwdorp M, Vallance BA, Verchere CB, van Raalte DH and Herrema H (2020) Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 11:571731. doi: 10.3389/fimmu.2020.571731
Received: 11 June 2020; Accepted: 11 September 2020;
Published: 16 October 2020.
Edited by:
Yang Mao-Draayer, University of Michigan, United StatesReviewed by:
Andy Wullaert, Ghent University, BelgiumZhengxiang He, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2020 Scheithauer, Rampanelli, Nieuwdorp, Vallance, Verchere, van Raalte and Herrema. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Torsten P. M. Scheithauer, t.p.scheithauer@amsterdamumc.nl