 Chaojun Hu1,2†
Chaojun Hu1,2† Jiuliang Zhao
Jiuliang Zhao Xiaofeng Zeng
Xiaofeng Zeng- 1Department of Rheumatology, Peking Union Medical College Hospital, Peking Union Medical College & Chinese Academy of Medical Sciences, Key Laboratory of Rheumatology & Clinical Immunology, Ministry of Education, Beijing, China
- 2National Clinical Research Center for Dermatologic and Immunologic Diseases (NCRC-DID), Beijing, China
- 3Department of Clinical Laboratory, First Affiliated Hospital of Guangxi Medical University, Nanning, China
Objective: Antiphospholipid syndrome (APS) is characterized by the presence of anti-phospholipid (aPL) antibodies. However, the relationship between the immunoglobulin (Ig) A isotype of aPL positivity and its clinical utility in APS diagnosis is controversial. Presently, we determine the clinical utility of IgA–aPL from consecutive patients in a large cohort from the Chinese population and patients with APS whose aPL profiles were obtained.
Methods: The detection of anticardiolipin (aCL) and anti-β2 glycoprotein-Ⅰ (aβ2GPⅠ) antibodies of the IgA/IgG/IgM isotype by paramagnetic particle chemiluminescent immunoassay was carried out in sera from 7293 subjects. 153 primary APS (PAPS) patients and 59 patients with secondary APS (SAPS) were included in this study.
Results: In total, 1,082 out of 7,293 (2.55%) subjects had a positive IgA–aPL test, and the prevalence of isolated IgA–aPL was 0.29% (21/7,293) in the general population. The prevalence of IgA–aPL in the PAPS patients was 12.42% (19/153); however, only one patient (0.65%) presented with isolated IgA–aPL. Fifty (25.9%) of the SAPS had IgA–aPL, none of whom lacked IgG/IgM–aPL. The combination of the IgA isotype and the IgG/IgM isotype did not increase the diagnostic performance when compared with the IgG/IgM isotype of aCL or aβ2GPⅠ, respectively. IgA–aPL was not associated with clinical manifestation in patients with APS.
Conclusion: Isolated IgA–aPL is rare in the general population as well as in patients with APS. Whether in the laboratory or in clinical practice, the presence of IgA–aPL does not provide added value for the diagnosis of APS in the Chinese population.
Introduction
Antiphospholipid syndrome (APS) is an autoimmune disease with characteristic features of recurrent thrombosis and/or obstetric complications accompanied with the persistent presence of antiphospholipid antibodies (aPL) (1). aPL are initially produced by B lymphocytes and are connected to β2 glycoprotein-I (β2GPI) in endothelial cells, which subsequently causes a series of harmful immune responses through a variety of mechanisms, including activation of inflammatory cells and endothelial cells leading to inflammation and thrombosis as well as interference with trophoblasts and decidual cells leading to pregnancy-related morbidity (2–6). According to the 2006 update of the International consensus statement on classification criteria for APS, the presence of lupus anticoagulant (LA) and/or the immunoglobulin (Ig) G and/or IgM isotypes of anticardiolipin antibodies (aCL) and/or anti-β2 glycoprotein-I (aβ2GPI) indicate APS (1).
The patients with a clinical manifestation that was highly suggestive of APS but persistently negative for routine aPL (IgG/IgM–aCL, IgG/IgM–aβ2GPI, and LA) were defined as having seronegative APS (7). Currently, researchers are trying to discover other nonstandard antibodies to improve the diagnosis of the so-called seronegative APS, such as the IgA isotype of aPL, anti-β2GPI domain I, antiphosphatidylethanolamine, and antiphosphatidylserine/prothrombin antibodies (8–10). Among them, IgA–aPL has shown much promise. On the one hand, it has been claimed that detection of IgA–aPL is significantly correlated with thrombosis and obstetric complications (10), and the presence of IgA–aβ2GPI in people with no history of thrombosis events is an independent risk factor for the development of these types of events (11–14). On the other hand, IgA–aPL (IgA–aCL and IgA–aβ2GPI) have been classified as one of the laboratory classification criteria for systemic lupus erythemathosus (SLE) (15) and can be used to distinguish seronegative APS from SLE (16, 17). Based on these findings, IgA–aPL seems to be useful for the diagnosis of APS.
However, whether IgA–aPL has the potential to become one of the diagnostic criteria is controversial. For instance, researchers have shown no association between IgA–aPL and clinical manifestations of APS (18, 19). In addition, the detection of IgA–aPL does not increase the diagnostic sensitivity of APS (20) or help diagnose patients with APS-associated SLE (21). Furthermore, the incidence rate of patients with isolated IgA–aPL who experience APS events has been reported to be as low as 3.1% per year (13). Indeed, IgA–aPL is usually accompanied with the IgG/IgM isotype, but the follow-up evidence in patients with isolated IgA–aPL is insufficient. It should be emphasized that the 14th International Congress on Antiphospholipid Antibodies Task Force pointed out that the evidence in available data from IgA aPL was level III-low quality evidence (22). Because of this controversy, researchers have not reached a consensus regarding whether or not they recommend IgA–aPL as one of the criteria for diagnosing APS; therefore, large-scale and high-quality research should be further developed to reveal the relationship between IgA–aPL and the diagnosis of APS.
The aims of this study were to evaluate the prevalence of aCL and aβ2GPI according to isotype in a large cohort of patients consecutively referred to the Rheumatology Laboratory as well as the clinical and diagnostic value of the IgA isotype of aPL in the exploration of APS. The associations between IgA–aPL and APS-related clinical manifestations were also determined.
Materials and Methods
Subjects and Patients
This retrospective cross-sectional study included 7,293 consecutive subjects who had their aPL profile determined at the Key Laboratory between May 2019 and December 2019. Duplicate patients were removed and for patients with multiple aPL profiles determined during this period, only the first test results were used in this study. This laboratory belongs to the Department of Rheumatology, Peking Union Medical College Hospital (PUMCH) and the National Clinical Research Center for Dermatologic and Immunologic Diseases (NCRC-DID). NCRC-DID is Ministry of Science and Technology of China authorized research center that have collected data on kinds of rheumatic diseases, including SLE, APS, rheumatoid arthritis (RA), ankylosing spondylitis (AS), and so on. The predecessor is Chinese Rheumatism Data Center (CRDC) (23), NCRC-DID provides real-life data to improve clinical decision-making. All the patients with APS diagnosed according to the 2006 Sydney revised classification criteria (1) who were refereed to our center were enrolled in this period. In addition, 120 healthy volunteers were recruited as healthy controls for aPL profile detection. For each subject, 4 mL of blood was collected with the help of a BD vacutainer without anticoagulants. For the next hour, the blood was allowed to clot and was later centrifuged at 4°C for 5 min at 3,000 rpm. The aPL profile was immediately determined in the separated serum. This study was approved by the Medical Ethics Committee of PUMCH, and all methods were carried out in accordance with the principles stated in the Declaration of Helsinki.
Detection of the aPL Profile
The levels of IgA/IgG/IgM–aCL and IgA/IgG/IgM–aβ2GPI were quantified by a paramagnetic particle chemiluminescent immunoassay using an iFlash 3000 Chemiluminescence Immunoassay Analyzer (YHLO Biotech Co. Ltd., Shenzhen, China). Cut-off values were determined strictly in accordance with the manufacturers. The defined cut-off levels for each isotype were as follows: IgA–aCL, 10 APL-U/mL; IgG–aCL, 10 GPL-U/mL; IgM–aCL, 10 MPL-U/mL; IgA–aβ2GPI, 20 AU/mL; IgG–aβ2GPI, 20 AU/mL; and IgM–aβ2GPI, 20 AU/mL. The manufacturer’s recommendations were followed carefully.
Statistical Analysis
The results of normally distributed data are expressed as the mean ± standard deviation. Descriptive data are presented as frequencies for categorical variables. Comparisons between two groups were performed by the Student’s t-test. Receiver operating characteristic (ROC) curves were used to calculate the area under the curve (AUC). Logistic regression analysis was employed to determine associations between aPL isotype positivity and clinical manifestation in patients with APS. SPSS 24.0 (SPSS Statistics for Windows, version 24.0; SPSS Inc., Chicago, IL, USA) was used to perform the statistical analysis of the data in this study, and two-tailed P-values of <0.05 represented statistical significance.
Results
Patient Characteristics
Consecutive serum samples from 7,293 subjects were screened for the presence of aCL and aβ2GPI antibodies with the IgA, IgG, and IgM isotypes. All patients were Chinese. The mean age was 39.5 ± 16.2 years, and 77.65% were female. The detailed demographic data of the study population are listed in Table 1. Most patients screened for aPL were from the Department of Rheumatology (52.45%). Of the 7,293 consecutive subjects, 212 patients with a confirmed diagnosis of APS according to the 2006 Sydney revised criteria of APS (1) registered in the NCRC-DID database system were identified in this study (Table 2), including 153 patients with PAPS and 59 patients with SAPS. The prevalence of thrombosis events (χ2 = 5.552, P = 0.02) was significantly higher in patients with SAPS compared to patients with PAPS. The prevalence of a smoking history (χ2 = 5.983, P = 0.018) was significantly higher in patients with PAPS compared to patients with SAPS.
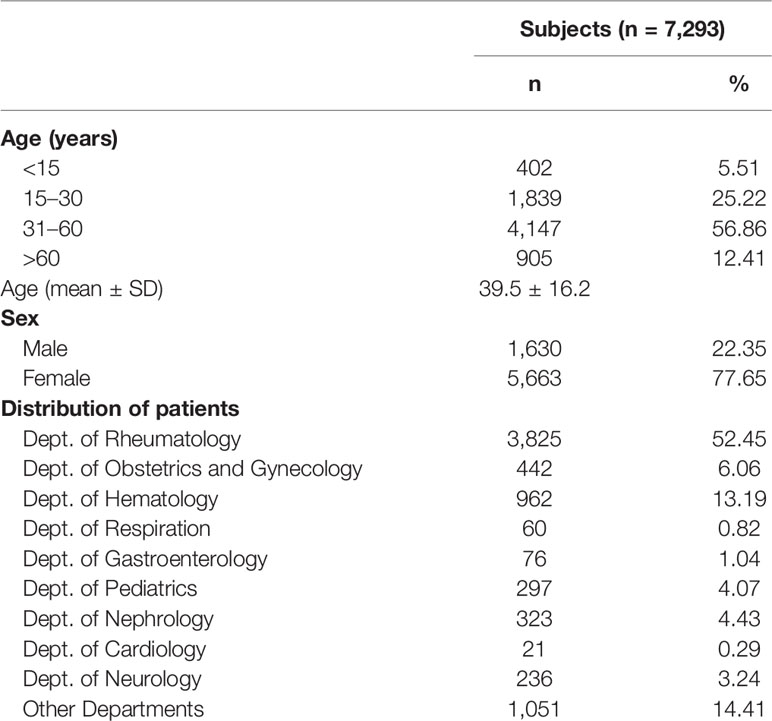
Table 1 Demographic data of 7,293 consecutive subjects.
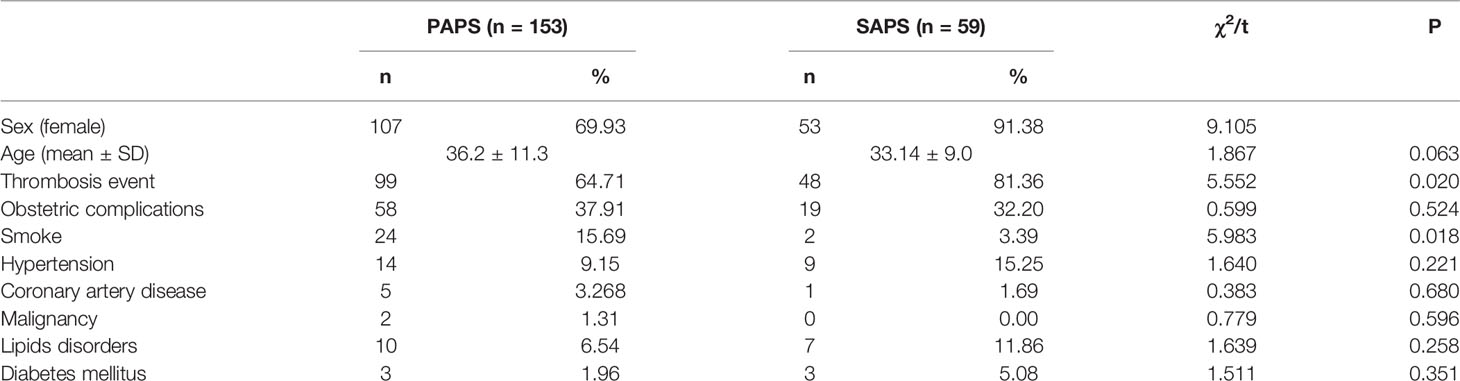
Table 2 Demographic and clinical characteristics of the 153 PAPS and 59 SAPS patients.
Prevalence of the IgA/IgG/IgM Isotypes of aCL and aβ2GPⅠ Antibodies in a Large Cohort
Based on the aPL isotype-specific antibody results, the overall prevalence of aCL was 10.97%, and the most frequent isotype was IgG–aCL (8.64%). The overall prevalence of aβ2GPI was 12.16%, and the most frequent isotype was IgM–aβ2GPI (7.72%). The overall prevalence of IgA–aCL and IgA–aβ2GPI were 2.48 and 2.13%, respectively. When the prevalence of each isolated isotype of aPL was analyzed, we found that the positive rates of IgA–aCL and IgA–aβ2GPI were very low, accounting for only 0.37 and 0.45%, respectively (Table 3).
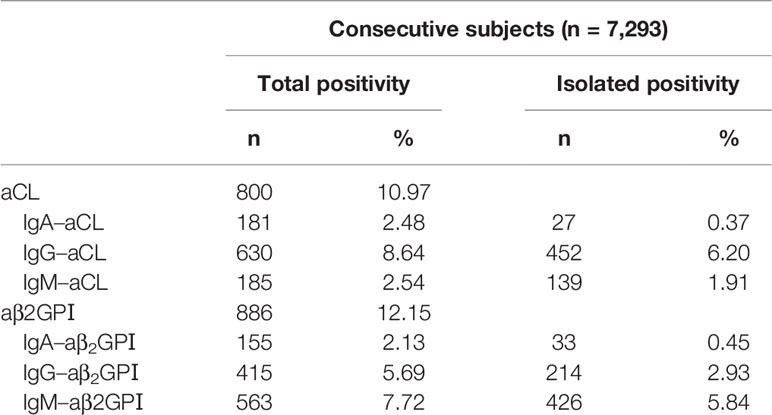
Table 3 Profile features of antiphospholipid antibodies in a large-scale cohort.
The cross-positivity for aCL and aβ2GPI according to isotype in this study is shown in Figure 1. Of the aCL-positive subjects, only 3.4% (27/800) of them presented with isolated IgA–aCL. Similarly, of the aβ2GPI-positive subjects, isolated IgA–aβ2GPI only accounted for 3.7% (33/887) of the total. Further analysis of the isotype distribution between aCL and aβ2GPI showed that the proportion of IgA–aCL (either IgA–aCL or IgA–aβ2GPI was positive) of the positive patients (any isotype of aPL was positive) was merely 1.94% (21/1082).
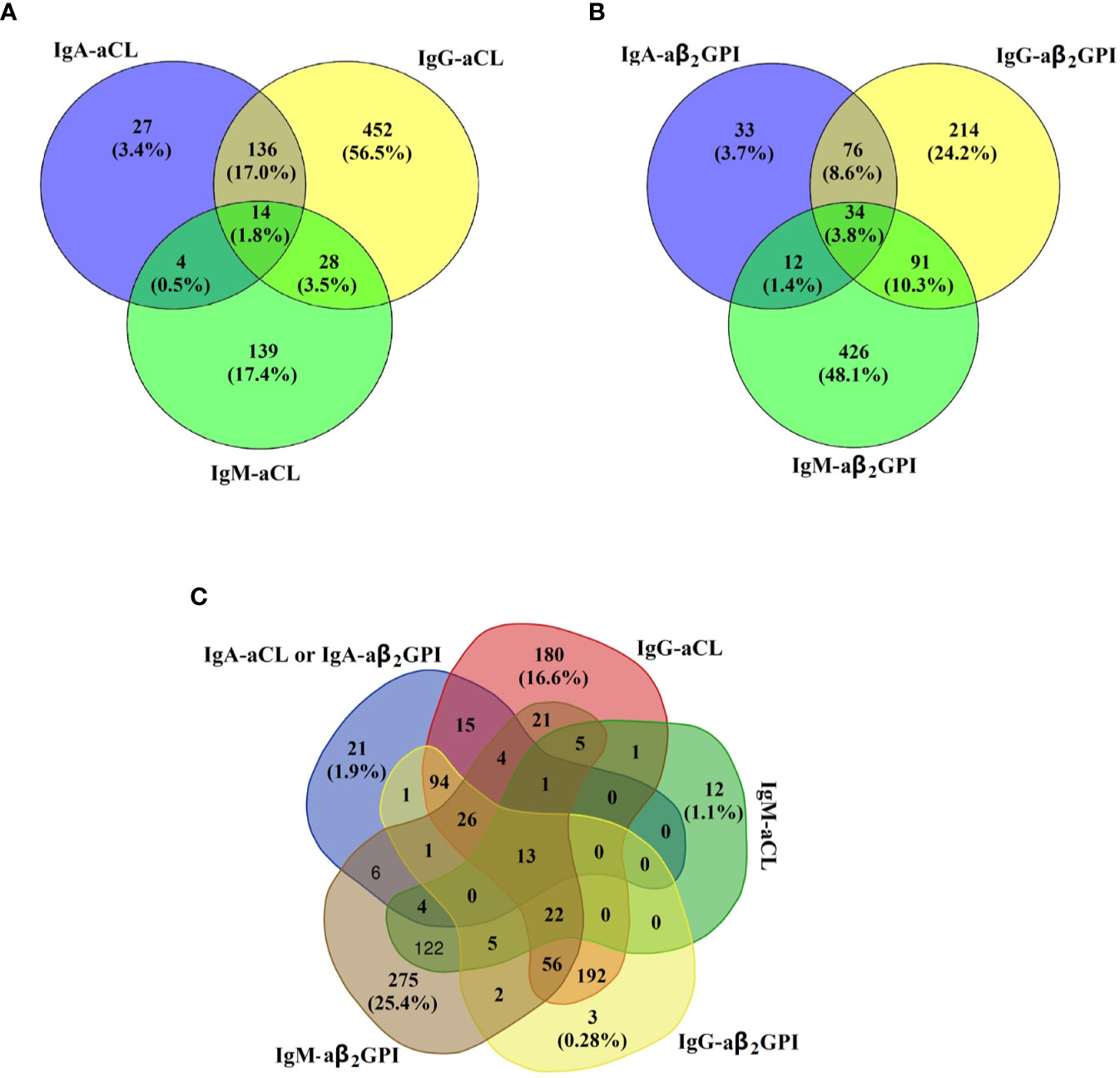
Figure 1 Cross-positivity for (A) IgA–aCL, IgG–aCL, and IgM–aCL in 800 aCL-positive subjects; (B) IgA–aβ2GPⅠ, IgG–aβ2GPⅠ, and IgM–aβ2GPI in 886 aβ2GPⅠ-positive subjects; and (C) IgA–aPL (either IgA–aCL or IgA–aβ2GPⅠ was positive), IgG–aCL, IgM–aCL, IgG–aβ2GPⅠ, and IgM–aβ2GPI in 1082 aPL-positive subjects.
Prevalence of aPL in Patients With APS and Healthy Controls
Compared with healthy controls, the total positivity of aPL isotype-specific antibodies was significantly higher in patients with APS. For isolated positivity, the positive rate of IgA–aCL and IgA–aβ2GPI were observed no differences between APS and healthy controls (Table 4). A higher frequency of positivity in patients with SAPS compared to PAPS was observed for aCL (χ2 = 16.481, P < 0.001) and aβ2GPI (χ2 = 7.868, P = 0.005). The frequency of IgA–aCL was also higher in patients with SAPS than in PAPS patients (χ2 = 6.044, P = 0.02). The presence of IgA–aβ2GPI was detected in 10.46% of PAPS patients, and it was detected in 20.34% of SAPS patients; however, there was no statistical difference in the positive rate of IgA–aβ2GPI between PAPS patients and SAPS patients (χ2 = 3.627, P = 0.07). Merely three (1.96%) patients presented with isolated IgA–aCL in the APS group, and only one (1.7%) patient presented with isolated IgA–aCL in the SAPS group. For IgA–aβ2GPI, only one (0.65%) patient in the PAPS group presented with isolated positivity, while no isolated positivity was observed in the SAPS group.
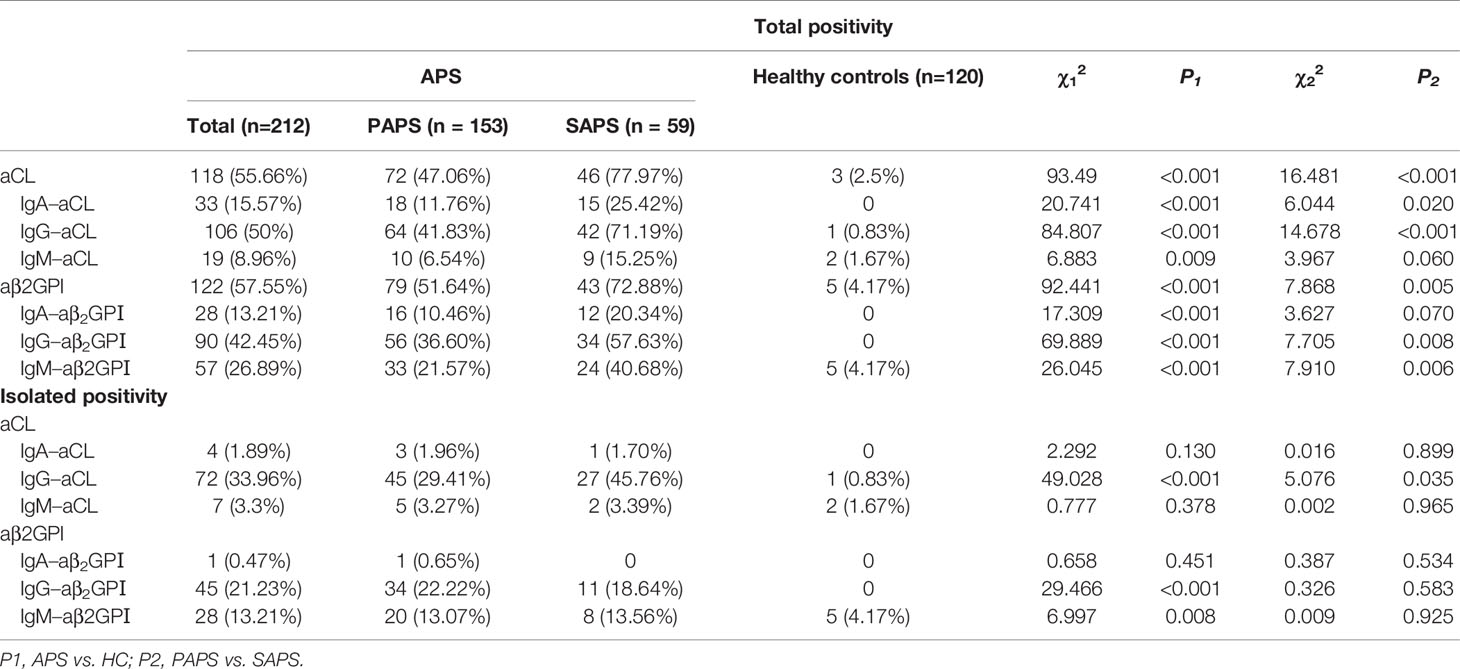
Table 4 Profile of antiphospholipid antibodies in patients with APS or healthy controls.
We also observed cross-positivity in the prevalence of the aPL isotype in the two groups (Figure 2). We found that most PAPS or SAPS patients with IgA–aPL positivity were accompanied with IgG- or IgM–aPL. In contrast, isolated IgA–aPL was rare in patients with PAPS or SAPS.
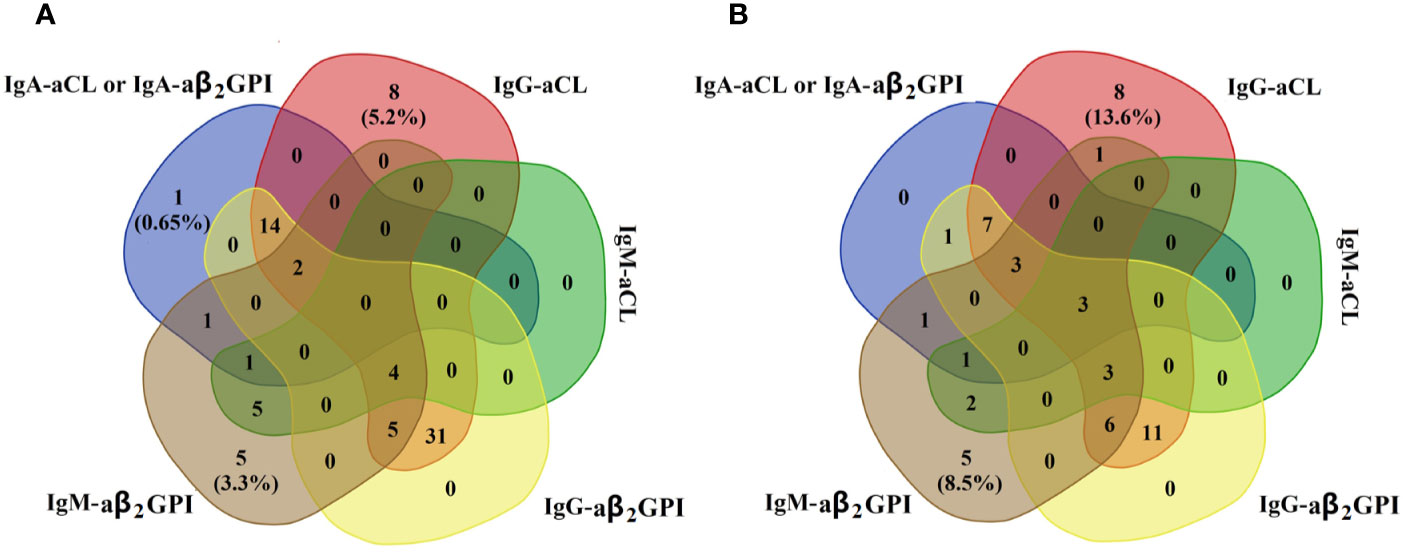
Figure 2 Distribution of IgA–aPL (either IgA–aCL or IgA–aβ2GPⅠ was positive), IgG–aCL, IgM–aCL, IgG–aβ2GPⅠ, and IgM–aβ2GPⅠ in the (A) PAPS group (n = 153) and (B) SAPS (n = 59).
Diagnostic Performance Analysis of IgA–aCL and IgA–aβ2GPⅠ Antibodies in APS Patients
We performed ROC analysis to determine the diagnostic performance of aPL isotype-specific antibodies for APS patients. The AUC of IgA–aCL was 0.670 (95% CI: 0.607–0.733, P < 0.01), which was lower than that of IgG–aCL (AUC = 0.748, 95% CI: 0.689–0.806, P < 0.01). When IgA–aCL and IgG/M-aCL were merged for combination analysis, the AUC of IgG/M/A-aCL was 0.811 (95% CI: 0.761–0.861, P < 0.01), which was similar to that of IgG/M-aCL (AUC = 0.809, 95% CI: 0.759–0.860, P < 0.01) (Figure 3A). The AUC of IgA–aβ2GPI was 0.654 (95% CI: 0.590–0.718, P < 0.01), which was lower than that of IgG–aβ2GPI (AUC = 0.826, 95% CI: 0.777–0.875, P < 0.01). When IgA–aβ2GPI and IgG/M–aβ2GPI were merged for combination analysis, the AUC of IgG/M/A–aβ2GPI was 0.861 (95% CI: 0.817–0.905, P < 0.01), which was similar to that of IgG/M–aβ2GPI (AUC = 0.861, 95% CI: 0.817–0.906, P < 0.01) (Figure 3B). Overall, IgA–aPL antibodies alone or in combination with IgG–aPL or IgG/M–aPL antibodies did not increase the diagnostic efficiency in patients with APS.
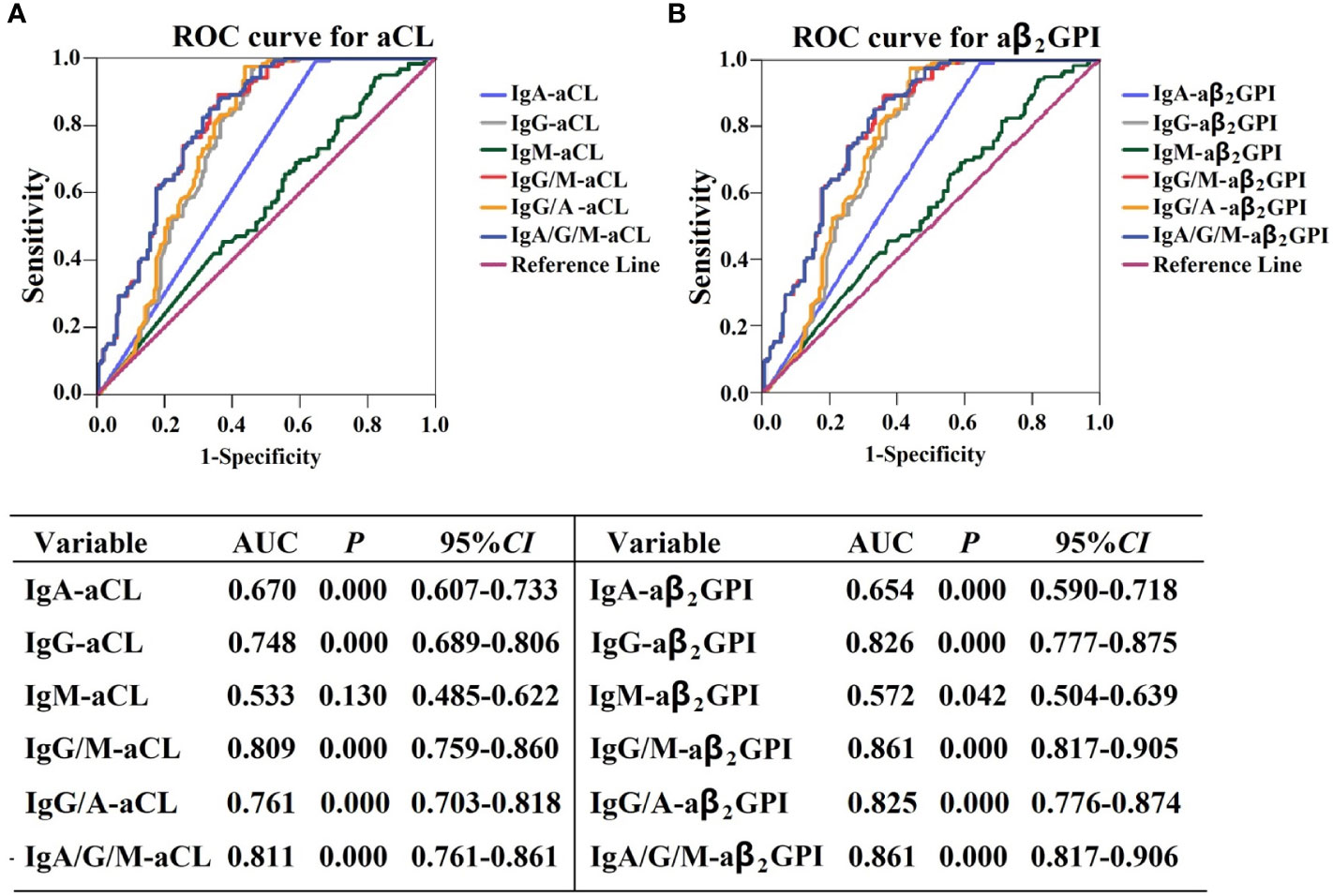
Figure 3 Receiver operating characteristic curve analysis of the aPL isotype in APS patients. (A) ROC curve for aCL and (B) ROC curve for aβ2GPI.
Associations Between APS-Related Clinical Manifestations of 212 Patients and the Presence of Each aPL Isotype
Among the various clinical manifestations presented by 212 APS, the presence of IgA–aPL wasn’t associated with any clinical manifestation. IgG–aPL was associated with cardiopulmonary involved [OR 30.512 (95% CI 1.969–472.780), P=0.015], acute coronary syndrome [OR 0.025 (0.001–0.906), P=0.025], pulmonary embolism [OR 0.058 (0.004–0.906), P=0.042] and thrombocytopenia [OR 1.992 (1.024–3.872), P=0.042]. Meanwhile, the results of IgM–aPL were different; the presence of IgM–aPL was associated with venous thrombosis [OR 3.078 (1.2–7.896), P=0.019] and thrombocytopenia [OR 4.977 (1.512–16.387), P=4.977]. The results are summarized in detail in Table 5.
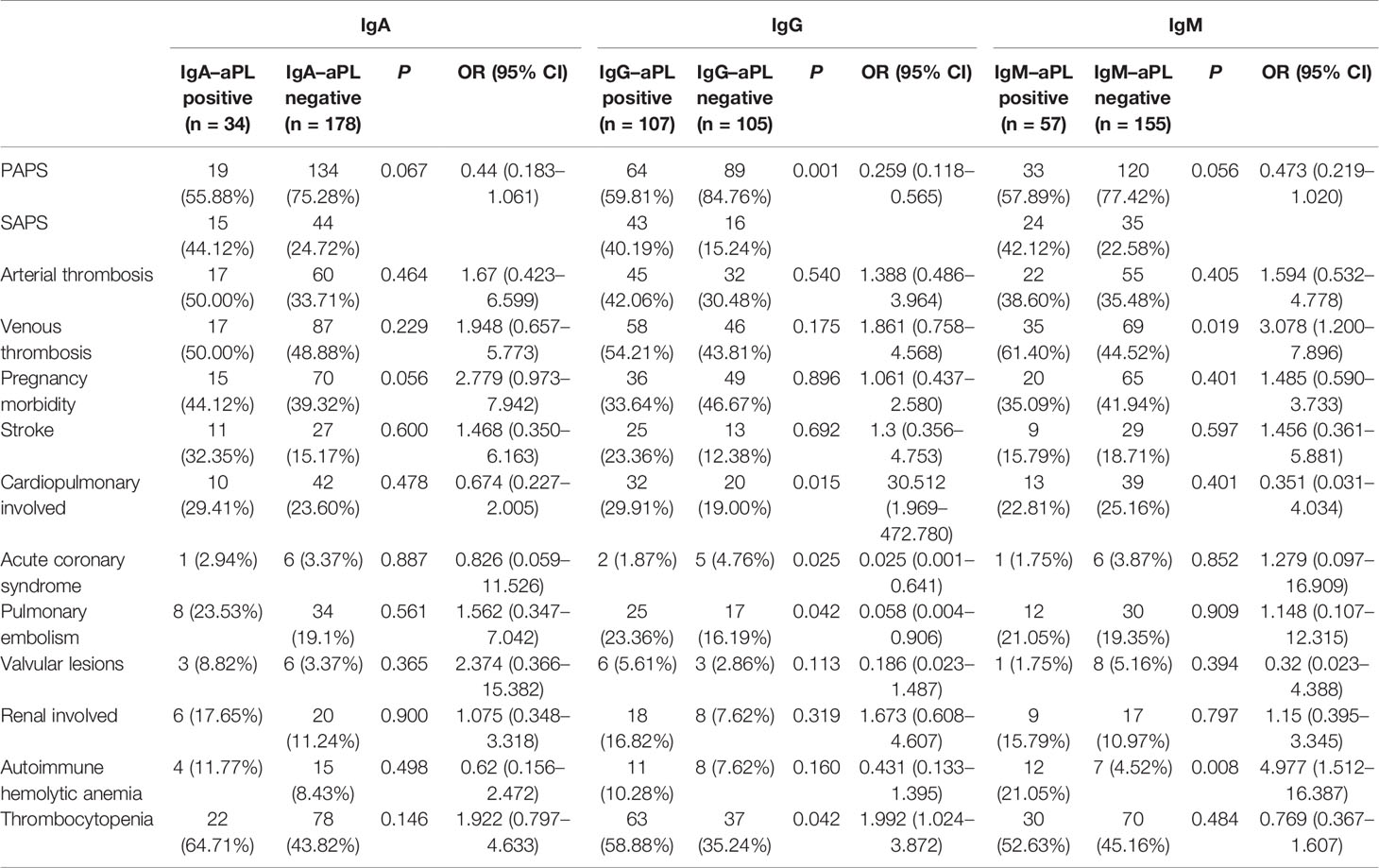
Table 5 Associations between APS-related clinical manifestations of 212 patients with aPL levels.
Discussion
APS is an autoimmune disease that affects nearly 5 in 100,000 people each year (24). aPL is a critical laboratory biomarker for the diagnosis of APS. IgA–aPL, also known as a “non-criteria” aPL, has been shown to have a controversial diagnosis performance for APS in previous studies (19, 20, 25) and a systematic review (26). To the best of our knowledge, this is the first study to analyze the aPL profile based on 7,293 consecutive subjects from a large cohort in the general Chinese population. We took advantage of this unselected large cohort and were able to analyze the relationship between the IgA isotype of aPL and the IgG/M isotype of aPL in the general population as well as in APS patients. We demonstrated that extra IgA–aPL testing did not provide any additional value for the diagnosis of APS.
In the current study, we found that the prevalences of IgA–aCL and IgA–aβ2GPI were 2.48 and 2.13%, respectively, in the general population. According to previous research, the prevalence of IgA–aCL ranges from 1.6 to 10% (27–30), and the prevalence of IgA–aβ2GPI ranges from 3 to 20.8% (29–32). These findings are consistent with our data. We also evaluated cross-positivity for the aPL profile in the general population when IgA–aCL was combined with IgA–aβ2GPI as a group; only 21 (0.29%) subjects presented isolated IgA–aPL, indicating that IgA–aPL is commonly accompanied with IgG–aPL and/or IgM–aPL and that it is rarely isolated in the general population.
We identified the patients diagnosed with APS in our cohort. Of patients with PAPS, the results showed that the proportion of those with IgA–aCL was 11.76%, and the proportion of those with IgA–aβ2GPI was 10.46%; while it was 25.42 and 20.34%, respectively, in patients with SAPS. The prevalence of IgA–aPL was consistent with that reported by the Antiphospholipid Syndrome Alliance for Clinical Trials and International Networking (33), a Swedish report (17), and the Hopkins Lupus Cohort (12).
Although the presence of IgA–aPL is not rare in PAPS or SAPS, cases with isolated IgA–aPL should receive more attention due to its clinical relevance. It stands out that only one case (0.65%) of our patients with PAPS had isolated IgA–aPL, but there were no cases of isolated IgA–aPL among the SAPS patients. These results indicate that IgA–aPL is usually accompanied by IgG/M–aPL and that isolated IgA–aPL is rare in patients with APS. Different from previous researches results, the prevalence of isolated IgA–aPL in Frodlund M (17) and Ruiz-Garcia R (25) were higher than that in this study, possible seasons for this difference should due to the patients population and detection assay used.
ROC curve analysis revealed that IgA–aCL and IgA–aβ2GPI have a similar diagnostic accuracy for APS, with AUCs of 0.670 (95%CI: 0.607–0.733, P < 0.001) and 0.654 (95%CI: 0.590–0.718, P < 0.001), respectively. It is worth noting that the triple detection of IgA/G/M-aCL or IgA/G/M–aβ2GPI does not increase the diagnostic performance of APS compared with the aPL detection criteria such as IgG isotype alone or in combination with the IgG or IgM isotype. Consistently, several previous studies have failed to show that adding the detection of IgA–aCL or IgA–aβ2GPI increases the diagnostic performance (20, 34–38). The uselessness of the aPL isotype triple detection is probably due to the relatively low prevalence of IgA–aPL and the fact that most cases of IgA–aPL were accompanied with another isotype of aPL. The diagnostic accuracy of routine detection and the correlation between IgA–aPL and APS-associated clinical manifestation could also be considered as other possible reasons.
According to the recent study, whether it is clinical manifestation criteria, mainly including arterial or venous thrombosis and pregnancy morbidity, or some other “non-criteria” clinical manifestations such as stroke and thrombocytopenia, there is no correlation between IgA–aPL positivity and clinical manifestations. In line with the previously published large cohort studies of APS patients, the results showed no association between IgA–aβ2GPI and APS manifestations in APS patients (18, 19). Of note, whether in a laboratory or clinical setting, the detection of IgA–aPL does not provide more value for the diagnosis of APS as it can be replaced by IgG–aPL detection. However, a recent study has revealed the opposite conclusion that IgA–aPL is associated with thrombosis and obstetric complications (10). Some other previous studies also have mentioned the robust association between IgA–aPL and thrombosis (13, 39–42) and that even IgA–aβ2GPI can be used as an independent risk factor for the development of APS-related events (13) and should be included as a consensus criterion for the diagnosis of APS (43). These controversial conclusions can mainly be attributed to differences in the included criteria of the study population, ethnic distribution, or statistical methods of the study. It is important to note that the proportion of isolated IgA–aPL was extremely low in our study or in controversial study (10). These findings reveal that the clinical relevance of IgA–aPL to APS is limited and uncertain. Therefore, whether a “real world” relationship between IgA–aPL and APS-related manifestations exists or whether IgA–aPL can be used as a diagnostic criterion in patients with APS requires further study.
As indicated by our results, whether it is for IgM–aCL or IgM–aβ2GPI, the AUCs were lower than those of IgG–aPL and IgA–aPL; however, the diagnostic efficacy for APS was improved when IgM–aPL was used in combination with the IgG isotype. Moreover, unlike IgA–aPL, IgM–aPL is directly related to venous thrombosis and autoimmune hemolytic anemia. A previous study has suggested an increased risk of retinal thrombosis in isolated IgM isotype-positive APS patients (44). Therefore, IgM–aPL can identify different types of APS manifestations in addition to being a diagnostic standard related to clinical manifestation criteria; it also can be used as a supplement for IgG–aPL.
There are several limitations to our study. Some of them are inherent to the overall population. Although the investigated cohort of this study was subjects with clinical suspicion of APS, both naive and follow-up patients were included in this research. Some characteristics of the patients who were followed up for several months might have changed compared to those at the first detection of aPL. Thus, our results did not reflect the true state of IgA–aPL of patients in the initial situation. Additionally, the lack of consecutive aPL profile detection on the same patients makes it impossible to estimate the clinical value of IgA–aPL for the diagnosis of APS over time. Besides, the sensitivity and diagnostic efficiency may be variable using different detection system or kits (45), which are inherent to the methodology or the selection of the antigen target (20, 46–48). These limitations will be the focus of a future study.
In conclusion, IgA–aPL is present in a small proportion of the general population. Besides, most IgA–aPL was accompanied with the IgG and/or IgM isotype of aPL, and isolated IgA–aPL rarely appeared in the general population and APS patients. Thus, extra IgA–aPL testing does not improve the diagnostic performance of aPL for APS. Moreover, the presence of IgA–aPL is not associated with clinical manifestation of APS. From a clinical point of view, these data do not support the utility of IgA–aPL as an additional biomarker for the diagnosis of APS, and IgA–aPL is not recommended as a diagnostic criterion for APS. However, additional highly sensitive aPL testing assays are needed to confirm the characteristics of different ethnic groups with IgA–aPL as well as in a large cohort of APS patients.
Data Availability Statement
All datasets presented in this study are included in the article/supplementary material.
Ethics Statement
This study was approved by the Medical Ethics Committee of PUMCH, and all methods were carried out in accordance with the principles stated in the Declaration of Helsinki. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
All authors were involved in the design of this study. CH, XL, and JZ were involved in the collection of blood samples, experimental procedures, statistical analysis, and writing of the manuscript. QW, ML, XT, XZ, and JZ were involved in the recruitment of patients and evaluation of clinical data. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Key Research and Development Program of China (2019YFC0840603, 2017YFC0907601, and 2017YFC0907602), the National Natural Science Foundation of China (81771780), and the CAMS Initiative for Innovative Medicine (2017-I2M-3-001 and 2019-I2M-2-008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost (2006) 4:295–306. doi: 10.1111/j.1538-7836.2006.01753.x
2. de Laat B, Derksen RH, Urbanus RT, de Groot PG. IgG antibodies that recognize epitope Gly40-Arg43 in domain I of beta 2-glycoprotein I cause LAC, and their presence correlates strongly with thrombosis. Blood (2005) 105:1540–5. doi: 10.1182/blood-2004-09-3387
3. Vega-Ostertag M, Casper K, Swerlick R, Ferrara D, Harris EN, Pierangeli SS. Involvement of p38 MAPK in the up-regulation of tissue factor on endothelial cells by antiphospholipid antibodies. Arthritis Rheum (2005) 52:1545–54. doi: 10.1002/art.21009
4. Lopez-Pedrera C, Buendia P, Cuadrado MJ, Siendones E, Aguirre MA, Barbarroja N, et al. Antiphospholipid antibodies from patients with the antiphospholipid syndrome induce monocyte tissue factor expression through the simultaneous activation of NF-kappaB/Rel proteins via the p38 mitogen-activated protein kinase pathway, and of the MEK-1/ERK pathway. Arthritis Rheum (2006) 54:301–11. doi: 10.1002/art.21549
5. Sorice M, Longo A, Capozzi A, Garofalo T, Misasi R, Alessandri C, et al. Anti-beta2-glycoprotein I antibodies induce monocyte release of tumor necrosis factor alpha and tissue factor by signal transduction pathways involving lipid rafts. Arthritis Rheum (2007) 56:2687–97. doi: 10.1002/art.22802
6. Perez-Sanchez C, Ruiz-Limon P, Aguirre MA, Bertolaccini ML, Khamashta MA, Rodriguez-Ariza A, et al. Mitochondrial dysfunction in antiphospholipid syndrome: implications in the pathogenesis of the disease and effects of coenzyme Q(10) treatment. Blood (2012) 119:5859–70. doi: 10.1182/blood-2011-12-400986
7. Hughes GR, Khamashta MA. Seronegative antiphospholipid syndrome. Ann Rheum Dis (2003) 62:1127. doi: 10.1136/ard.2003.006163
8. Litvinova E, Darnige L, Kirilovsky A, Burnel Y, de Luna G, Dragon-Durey MA. Prevalence and Significance of Non-conventional Antiphospholipid Antibodies in Patients With Clinical APS Criteria. Front Immunol (2018) 9:2971. doi: 10.3389/fimmu.2018.02971
9. Guo H, Zhang Y, Li A, Wang C, Yang S, Zhang Y, et al. Anti-domain 1 of beta2-glycoprotein I aids risk stratification in lupus anticoagulant-positive patients. Clin Exp Med (2019) 19:339–45. doi: 10.1007/s10238-019-00555-w
10. Zigon P, Podovsovnik A, Ambrozic A, Tomsic M, Hocevar A, Gaspersic N, et al. Added value of non-criteria antiphospholipid antibodies for antiphospholipid syndrome: lessons learned from year-long routine measurements. Clin Rheumatol (2019) 38:371–8. doi: 10.1007/s10067-018-4251-7
11. Pericleous C, Ferreira I, Borghi O, Pregnolato F, McDonnell T, Garza-Garcia A, et al. Measuring IgA Anti-beta2-Glycoprotein I and IgG/IgA Anti-Domain I Antibodies Adds Value to Current Serological Assays for the Antiphospholipid Syndrome. PLoS One (2016) 11:e0156407. doi: 10.1371/journal.pone.0156407
12. Tebo AE, Willis R, Jaskowski TD, Guerra M, Pierangeli SS, Salmon J, et al. Clinical significance and correlations between anti-beta2 glycoprotein I IgA assays in antiphospholipid syndrome and/or systemic lupus erythematosus. Clin Chim Acta (2016) 460:107–13. doi: 10.1016/j.cca.2016.06.025
13. Tortosa C, Cabrera-Marante O, Serrano M, Martinez-Flores JA, Perez D, Lora D, et al. Incidence of thromboembolic events in asymptomatic carriers of IgA anti ß2 glycoprotein-I antibodies. PLoS One (2017) 12:e0178889. doi: 10.1371/journal.pone.0178889
14. Zuo Y, Barbhaiya M, Erkan D. Primary Thrombosis Prophylaxis in Persistently Antiphospholipid Antibody-Positive Individuals: Where Do We Stand in 2018? Curr Rheumatol Rep (2018) 20:66. doi: 10.1007/s11926-018-0775-8
15. Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum (2012) 64:2677–86. doi: 10.1002/art.34473
16. Sweiss NJ, Bo R, Kapadia R, Manst D, Mahmood F, Adhikari T, et al. IgA anti-beta2-glycoprotein I autoantibodies are associated with an increased risk of thromboembolic events in patients with systemic lupus erythematosus. PLoS One (2010) 5:e12280. doi: 10.1371/journal.pone.0012280
17. Frodlund M, Vikerfors A, Grosso G, Skogh T, Wettero J, Elvin K, et al. Immunoglobulin A anti-phospholipid antibodies in Swedish cases of systemic lupus erythematosus: associations with disease phenotypes, vascular events and damage accrual. Clin Exp Immunol (2018) 194:27–38. doi: 10.1111/cei.13180
18. Danowski A, Kickler TS, Petri M. Anti-beta2-glycoprotein I: prevalence, clinical correlations, and importance of persistent positivity in patients with antiphospholipid syndrome and systemic lupus erythematosus. J Rheumatol (2006) 33:1775–9. doi: 10.1097/01.bor.0000240370.47336.ae
19. Vlagea A, Pascual-Salcedo D, Alvarez Doforno R, Lavilla P, Diez J, Padilla Merlano B, et al. IgA anti-beta2 glycoprotein I antibodies: Experience from a large center. Thromb Res (2018) 162:38–43. doi: 10.1016/j.thromres.2017.12.007
20. Tozzoli R, Villalta D. Autoantibody profiling of patients with antiphospholipid syndrome using an automated multiplexed immunoassay system. Autoimmun Rev (2014) 13:59–63. doi: 10.1016/j.autrev.2013.08.007
21. Bertolaccini ML, Atsumi T, Escudero Contreras A, Khamashta MA, Hughes GR. The value of IgA antiphospholipid testing for diagnosis of antiphospholipid (Hughes) syndrome in systemic lupus erythematosus. J Rheumatol (2001) 28:2637–43. doi: 10.1160/TH04-02-0135
22. Bertolaccini ML, Amengual O, Andreoli L, Atsumi T, Chighizola CB, Forastiero R, et al. 14th International Congress on Antiphospholipid Antibodies Task Force. Report on antiphospholipid syndrome laboratory diagnostics and trends. Autoimmun Rev (2014) 13:917–30. doi: 10.1016/j.autrev.2014.05.001
23. Li M, Tian X, Zhang W, Leng X, Zeng X. CRDC: a Chinese rheumatology research platform. Clin Rheumatol (2015) 34:1347–52. doi: 10.1007/s10067-015-3003-1
24. Houghton DE, Moll S. Antiphospholipid antibodies. Vasc Med (2017) 22:545–50. doi: 10.1177/1358863X17737374
25. Ruiz-Garcia R, Serrano M, Martinez-Flores JA, Mora S, Morillas L, Martin-Mola MA, et al. Isolated IgA anti- beta2 glycoprotein I antibodies in patients with clinical criteria for antiphospholipid syndrome. J Immunol Res (2014) 2014:704395. doi: 10.1155/2014/704395
26. Andreoli L, Fredi M, Nalli C, Piantoni S, Reggia R, Dall’Ara F, et al. Clinical significance of IgA anti-cardiolipin and IgA anti-beta2glycoprotein I antibodies. Curr Rheumatol Rep (2013) 15:343. doi: 10.1007/s11926-013-0343-1
27. Ahmed E, Stegmayr B, Trifunovic J, Weinehall L, Hallmans G, Lefvert AK. Anticardiolipin antibodies are not an independent risk factor for stroke: an incident case-referent study nested within the MONICA and Vasterbotten cohort project. Stroke (2000) 31:1289–93. doi: 10.1161/01.str.31.6.1289
28. Gonzales-Portillo F, McIntyre JA, Wagenknecht DR, Williams LS, Bruno A, Biller J. Spectrum of antiphospholipid antibodies (aPL) in patients with cerebrovascular disease. J Stroke Cerebrovasc Dis (2001) 10:222–6. doi: 10.1053/jscd.2001.29818
29. Palomo I, Pereira J, Alarcon M, Vasquez M, Pinochet C, Velez MT, et al. Prevalence and isotype distribution of antiphospholipid antibodies in unselected Chilean patients with venous and arterial thrombosis. Clin Rheumatol (2004) 23:129–33. doi: 10.1007/s10067-003-0846-7
30. Kahles T, Humpich M, Steinmetz H, Sitzer M, Lindhoff-Last E. Phosphatidylserine IgG and beta-2-glycoprotein I IgA antibodies may be a risk factor for ischaemic stroke. Rheumatol (Oxford) (2005) 44:1161–5. doi: 10.1093/rheumatology/keh698
31. Hsieh K, Knobl P, Rintelen C, Kyrle PA, Quehenberger P, Bialonczyk C, et al. Is the determination of anti-beta2 glycoprotein I antibodies useful in patients with venous thromboembolism without the antiphospholipid syndrome? Br J Haematol (2003) 123:490–5. doi: 10.1046/j.1365-2141.2003.04595.x
32. Veres K, Lakos G, Kerenyi A, Szekanecz Z, Szegedi G, Shoenfeld Y, et al. Antiphospholipid antibodies in acute coronary syndrome. Lupus (2004) 13:423–7. doi: 10.1191/0961203304lu1011oa
33. Unlu O, Erkan D, Barbhaiya M, Andrade D, Nascimento I, Rosa R, et al. The Impact of Systemic Lupus Erythematosus on the Clinical Phenotype of Antiphospholipid Antibody-Positive Patients: Results From the AntiPhospholipid Syndrome Alliance for Clinical Trials and InternatiOnal Clinical Database and Repository. Arthritis Care Res (Hoboken) (2019) 71:134–41. doi: 10.1002/acr.23584
34. Selva-O’Callaghan A, Ordi-Ros J, Monegal-Ferran F, Martinez N, Cortes-Hernandez F, Vilardell-Tarres M. IgA anticardiolipin antibodies–relation with other antiphospholipid antibodies and clinical significance. Thromb Haemost (1998) 79:282–5. doi: 10.1055/s-0037-1614978
35. Samarkos M, Davies KA, Gordon C, Loizou S. Clinical significance of IgA anticardiolipin and anti-beta2-GP1 antibodies in patients with systemic lupus erythematosus and primary antiphospholipid syndrome. Clin Rheumatol (2006) 25:199–204. doi: 10.1007/s10067-005-1156-z
36. Holc I, Hojs R, Cikes N, Ambrozic A, Cucnik S, Kveder T, et al. Antiphospholipid antibodies and atherosclerosis: insights from rheumatoid arthritis–a five-year follow-up study. Immunobiology (2011) 216:1331–7. doi: 10.1016/j.imbio.2011.05.008
37. Meijide H, Sciascia S, Sanna G, Khamashta MA, Bertolaccini ML. The clinical relevance of IgA anticardiolipin and IgA anti-beta2 glycoprotein I antiphospholipid antibodies: a systematic review. Autoimmun Rev (2013) 12:421–5. doi: 10.1016/j.autrev.2012.08.002
38. Domingues V, Magder LS, Petri M. Assessment of the independent associations of IgG, IgM and IgA isotypes of anticardiolipin with thrombosis in SLE. Lupus Sci Med (2016) 3:e000107. doi: 10.1136/lupus-2015-000107
39. Murthy V, Willis R, Romay-Penabad Z, Ruiz-Limon P, Martinez-Martinez LA, Jatwani S, et al. Value of isolated IgA anti-beta2 -glycoprotein I positivity in the diagnosis of the antiphospholipid syndrome. Arthritis Rheum (2013) 65:3186–93. doi: 10.1002/art.38131
40. Despierres L, Beziane A, Kaplanski G, Granel B, Serratrice J, Cohen W, et al. Contribution of anti-beta2glycoprotein I IgA antibodies to the diagnosis of anti-phospholipid syndrome: potential interest of target domains to discriminate thrombotic and non-thrombotic patients. Rheumatol (Oxford) (2014) 53:1215–8. doi: 10.1093/rheumatology/keu003
41. Mattia E, Ruffatti A, Tonello M, Meneghel L, Robecchi B, Pittoni M, et al. IgA anticardiolipin and IgA anti-beta2 glycoprotein I antibody positivity determined by fluorescence enzyme immunoassay in primary antiphospholipid syndrome. Clin Chem Lab Med (2014) 52:1329–33. doi: 10.1515/cclm-2014-0039
42. Serrano M, Martinez-Flores JA, Norman GL, Naranjo L, Morales JM, Serrano A. The IgA Isotype of Anti-beta2 Glycoprotein I Antibodies Recognizes Epitopes in Domains 3, 4, and 5 That Are Located in a Lateral Zone of the Molecule (L-Shaped). Front Immunol (2019) 10:1031. doi: 10.3389/fimmu.2019.01031
43. Perez D, Tincani A, Serrano M, Shoenfeld Y, Serrano A. Antiphospholipid syndrome and IgA anti-beta2-glycoprotein I antibodies: when Cinderella becomes a princess. Lupus (2018) 27:177–8. doi: 10.1177/0961203317738227
44. Del Ross T, Ruffatti A, Cuffaro S, Tonello M, Calligaro A, Favaro M, et al. The clinical relevance of the IgM isotype of antiphospholipid antibodies in the vascular antiphospholipid syndrome. Thromb Res (2015) 136:883–6. doi: 10.1016/j.thromres.2015.08.019
45. Martinez-Flores JA, Serrano M, Alfaro J, Mora S, Paz-Artal E, Morales JM, et al. Heterogeneity between diagnostic tests for IgA anti-beta2 glycoprotein I: explaining the controversy in studies of association with vascular pathology. Anal Chem (2013) 85:12093–8. doi: 10.1021/ac403194t
46. Zhang S, Wu Z, Li P, Bai Y, Zhang F, Li Y. Evaluation of the Clinical Performance of a Novel Chemiluminescent Immunoassay for Detection of Anticardiolipin and Anti-Beta2-Glycoprotein 1 Antibodies in the Diagnosis of Antiphospholipid Syndrome. Med (Baltimore) (2015) 94:e2059. doi: 10.1097/MD.0000000000002059
47. Oku K, Amengual O, Kato M, Bohgaki T, Horita T, Yasuda S, et al. Significance of fully automated tests for the diagnosis of antiphospholipid syndrome. Thromb Res (2016) 146:1–6. doi: 10.1016/j.thromres.2016.08.018
Keywords: IgA isotype of antiphospholipid antibodies, IgA isotype of anticardiolipin antibodies, antiphospholipid syndrome, diagnostic value, IgA isotype of anti-β2 glycoprotein-I
Citation: Hu C, Li X, Zhao J, Wang Q, Li M, Tian X and Zeng X (2020) Immunoglobulin A Isotype of Antiphospholipid Antibodies Does Not Provide Added Value for the Diagnosis of Antiphospholipid Syndrome in a Chinese Population. Front. Immunol. 11:568503. doi: 10.3389/fimmu.2020.568503
Received: 01 June 2020; Accepted: 14 September 2020;
Published: 05 October 2020.
Edited by:
Lazaros Ignatios Sakkas, University of Thessaly, GreeceReviewed by:
Emilio Gonzalez, University of Texas Medical Branch at Galveston, United StatesRohan Willis, University of Texas Medical Branch at Galveston, United States
Copyright © 2020 Hu, Li, Zhao, Wang, Li, Tian and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiuliang Zhao, empscHVtY0BzaW5hLmNvbQ==; Xiaofeng Zeng, emVuZ3hmcHVtY0AxNjMuY29t
†These authors have contributed equally to this work and share first authorship