 Davis M. Borucki1,2,3
Davis M. Borucki1,2,3 Amer Toutonji1,2,3
Amer Toutonji1,2,3 Christine Couch2,4
Christine Couch2,4 Khalil Mallah2
Khalil Mallah2 Baerbel Rohrer3,5,6
Baerbel Rohrer3,5,6 Stephen Tomlinson2,6*
Stephen Tomlinson2,6*- 1Medical Scientist Training Program, Medical University of South Carolina, Charleston, SC, United States
- 2Department of Microbiology and Immunology, Medical University of South Carolina, Charleston, SC, United States
- 3Department of Neurosciences, Medical University of South Carolina, Charleston, SC, United States
- 4Department of Health Sciences and Research, College of Health Professions, Medical University of South Carolina, Charleston, SC, United States
- 5Department of Ophthalmology, Medical University of South Carolina, Charleston, SC, United States
- 6Ralph H. Johnson VA Medical Center, Charleston, SC, United States
The focus of this review is the role of complement-mediated phagocytosis in retinal and neurological diseases affecting the visual system. Complement activation products opsonize synaptic material on neurons for phagocytic removal, which is a normal physiological process during development, but a pathological process in several neurodegenerative diseases and conditions. We discuss the role of complement in the refinement and elimination of synapses in the retina and lateral geniculate nucleus, both during development and in disease states. How complement and aberrant phagocytosis promotes injury to the visual system is discussed primarily in the context of multiple sclerosis, where it has been extensively studied, although the role of complement in visual dysfunction in other diseases such as stroke and traumatic brain injury is also highlighted. Retinal diseases are also covered, with a focus on glaucoma and age-related macular degeneration. Finally, we discuss the potential of complement inhibitory strategies to treat diseases affecting the visual system.
Introduction
The visual system is one of the best understood sensory/neural systems in terms of development and signal transduction and processing. The circuit itself is relatively simple (1); each part of a visual scene or light pattern is analyzed by photoreceptors that feed this information via bipolar cells to retinal ganglion cells (RGC). Interneurons (horizontal and amacrine cells) shape this response through excitation and inhibition. Signal is then passed via the optic nerve and optic tract by ganglion cell axons to the next synapse in the lateral geniculate nucleus (LGN). Neurons from the LGN terminate in the primary visual cortex for further visual processing of signal (Figure 1). As discussed later, the visual system in rodents develops and matures during the post-embryonic period for 2 weeks after birth (2). The fact that the rodent visual system develops postnatally makes it a useful experimental system for studying synaptic development and refinement.
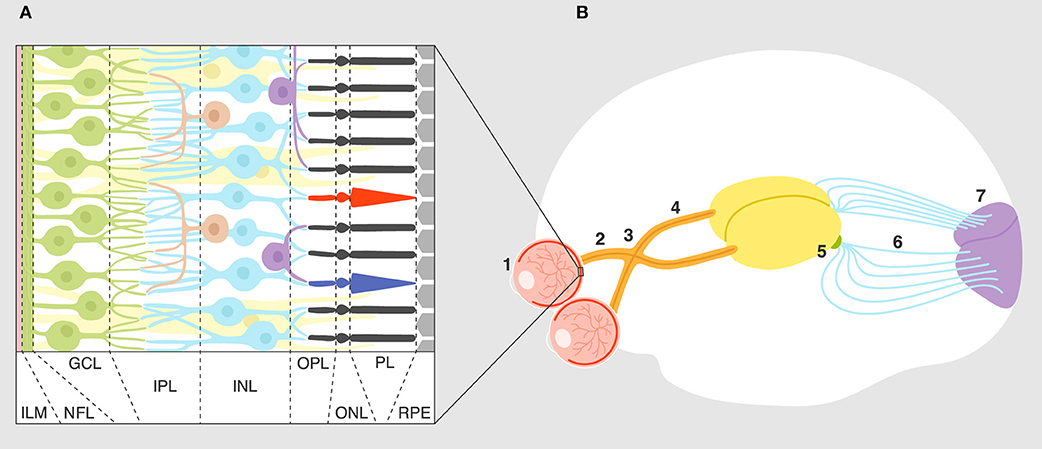
Figure 1. Overview of the visual system. (A) Layers of the retina: ILM, inner limiting membrane; NFL, nerve fiber layer; GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; PL, photoreceptor layer; RPE, retinal pigment epithelium. Connections and cell types are explained in the text. (B) Connectivity of the visual system: eye (1), optic nerve (2), optic chiasm (3), optic tract (4), lateral geniculate nucleus (5), optic radiation (6), primary visual cortex (7).
Developmental studies of the visual system show an overproduction of RGC neurons in the retina and an abundance of synaptic connections in the developing LGN late in embryonic development (3). The complement system, and specifically the classical complement pathway, is necessary for this synaptic refinement and reduction of RGC numbers and for the division of the LGN into eye-specific subregions (4, 5). Studies of neurological injury, including mechanical, ischemic and autoimmune, show that the complement system plays a role in the propagation of damage after injury (6). The role of complement in injury to the visual system in these disorders is underexplored, but recent studies in experimental autoimmune encephalomyelitis (EAE), a model of multiple sclerosis (MS) with demyelination of the retinogeniculate system, synaptic changes and visual dysfunction (7), have indicated that complement-mediated phagocytosis of neural tissue is involved in injury (8). The retina, where the cell bodies of RGCs are located and where visual information is first encoded into a spike train of action potentials, is part of the central nervous system (CNS) affected physiologically and pathogenically by complement. During development, complement is involved in the synaptic pruning of RGCs and the sculpting of neural circuits (5). However, aberrant overactivation of the complement system in retinal disease can result in damage to RGCs with degradation and loss of synaptic connections on the retinal or thalamic side (4). The role of complement-mediated inflammation in retinal disease in general and anterograde degeneration of RGCs in particular is quite well explored, and several preclinical and clinical studies show that inhibiting specific complement pathways is protective (9–12).
Complement and Phagocytosis
The complement system consists of about 50 soluble and membrane-bound proteins that include pattern recognition molecules (PRMs), enzymes, effector precursors, and regulatory proteins. Complement proteins act together to perform multiple physiological functions, such as detection and disposal of pathogens and cellular debris, while limiting off-target damage to normal tissue (13). The focus of this review is complement-dependent phagocytosis in the CNS, primarily in the visual system, but in this section, we also discuss the broader role of complement in pathologies of the CNS. Depending on the PRMs and enzymes involved, complement activation is classified into three canonical pathways: classical, lectin, and alternative. Whereas, the classical and the lectin pathway are initiated by PRMs C1q and MBL, respectively, the alternative pathway does not require a ligand-PRM interaction. Instead, the alternative pathway can spontaneously activate, but it also serves as an amplification loop for the classical and lectin pathways.
All activation pathways lead to the generation of biologic effector molecules, namely C3a, C3 opsonins, C5a, and C5b-9 (Figure 2). The C3a and C5a activation products are soluble anaphylatoxins that recruit and activate immune cells via interaction with their respective cellular receptors, C3aR and C5aR1. C5b-9, also known as the membrane attack complex (MAC), creates pores in the membrane that can lead to inflammatory signaling and/or cell lysis. The activation product C3b becomes covalently attached to activating surfaces and is the initial building block for the generation of enzymatic complexes that cleave C3 into C3a and C3b and cleave C5 into C5a and C5b. Furthermore, and of particular relevance to this review, surface-bound C3b is degraded to iC3b, C3d, and C3dg. These degradation products no longer act in the complement activation pathway, but they do function as cellular opsonins for complement receptors expressed on immune cells, including phagocytic cells. The effective clearance of synapses during circuit sculpting in development, and the removal of cellular debris and pathogens, necessary activities for normal development and for defense and recovery processes, is C3 opsonin driven.
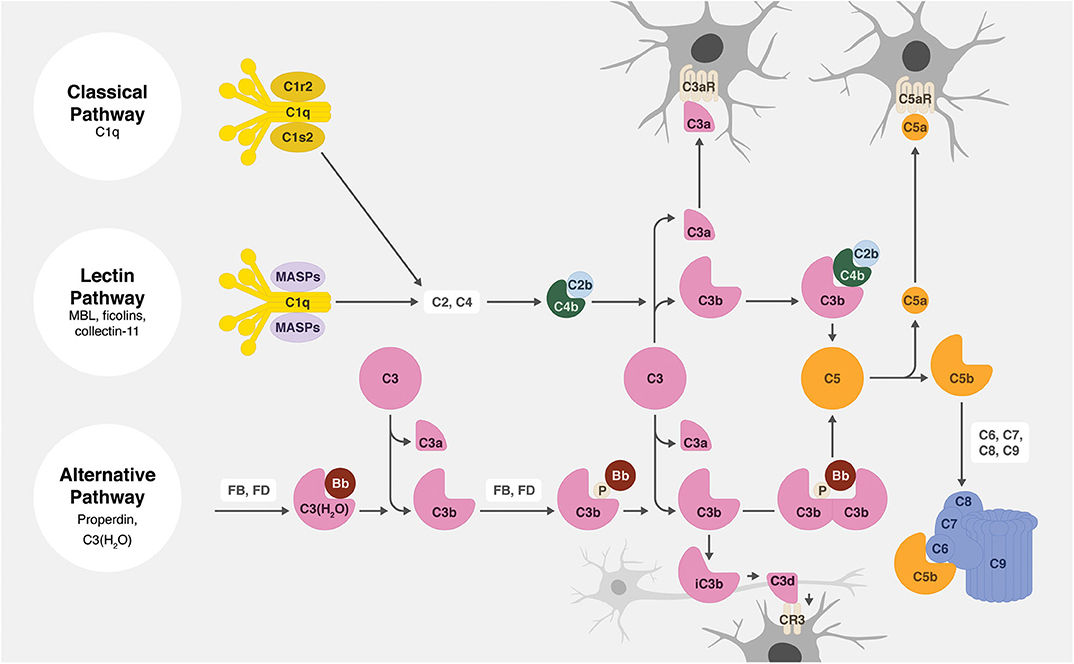
Figure 2. Overview of the complement system. There are three main complement activation pathways: the classical (top), lectin (middle), and alternative (bottom). All of these pathways lead to the cleavage of C3 and subsequently C5, leading to the opsonization of tissues (by C3b, iC3b, and C3d), the production of anaphylatoxins (C3a and C5a), and the assembly of the cytolytic membrane attack complex (C5b-9).
The main phagocytic receptor for C3 opsonins is complement receptor 3 (CR3), which consists of the two integrin chains: CD11b (itgax) and CD18 (itgb2) (14). CR3 is mainly expressed on myeloid cells and is commonly used as a lineage marker for microglia, monocytes, and macrophages. The role of CR3 in microglial phagocytosis has been studied in the context of synaptic pruning (15), the clearance of myelin (16, 17) and amyloid β (Aβ) (18, 19), as well as the removal of several pathogens of the nervous system (20–22). In Alzheimer's disease (AD), for example, CD11b immunoprecipitation from homogenates of brain samples taken postmortem from the occipital lobe shows a significant presence of CD11b/C3b/Aβ complexes as compared to age-matched controls (23). In the amyloid precursor protein (APP) transgenic mouse model of AD, eliminating C3 resulted in increased levels of Aβ and loss of neurons in the hippocampus, indicating a beneficial role for complement-mediated phagocytosis in tissue homeostasis (24). This is in contrast with findings in more recent animal studies showing that CR3 deficiency improves microglial clearance of Aβ in vivo and in vitro by increasing enzymatic degradation (25). Another study, using the double transgenic human APP/PS1 (presenilin 1) AD model, shows that despite increased levels of cerebral Aβ upon elimination of C3, mice show better cognitive performance and fewer microglia around amyloid plaques along with reduced loss of neurons and synapses (26). Together, these seemingly contradictory findings could imply that C3 is important for the clearance of Aβ plaques, the effect of which depends on the presence of a mutant presenilin 1 (PS1) protein. In addition, these studies suggest that intracellular signaling via CR3 is proinflammatory and harmful in AD and that C3-triggered phagocytosis of Aβ is beneficial when mediated by receptors other than CR3. In this context, complement receptor 4 (CR4), typically a dendritic cell marker, has also been shown to be sharply increased with Aβ plaque load throughout all stages of AD. Furthermore, 23% of activated microglia are CR4+ in AD, suggesting a potentially important role for this receptor in Aβ clearance (27). CR3-mediated phagocytosis is also implicated in synaptic phagocytosis in the APP mouse model, suggesting that the same pathway leading to clearance of Aβ is also involved in synapse loss (28).
Complement-mediated phagocytosis is not restricted to C3 opsonins and CR3 and/or CR4 engagement. In addition to being the recognition molecule of the classical pathway, C1q can also bind directly to several membrane receptors including CR1, CD91, LAIR1, SCARF1, α2β1, cC1qR, and gC1qR (29–35), interactions that have been associated with complement-independent functions of C1q. There is evidence for C1q-mediated microglial phagocytosis in the clearance of apoptotic cells (36, 37). In vivo studies show that C1q deficiency rescues neurons in a frontotemporal dementia (FTD) model, and that C1q is involved in early synaptic pruning (38, 39). One study demonstrated that treatment of cultured rat microglia with extrinsic C1q triggers an increase in intracellular calcium that is associated with a shift toward proinflammatory activation (40). This study, together with another showing microglial expression of CD93 (41), indicate that direct C1q-mediated phagocytosis remains a viable pathway (42).
The complement anaphylatoxins (C3a and C5a) can also indirectly modulate phagocytosis. A recent presentation at the 2019 Society for Neuroscience conference reported that pretreatment of transformed mouse brain microglial cells (BV-2 cells) or primary mouse microglia with a C3aR agonist increased their motility and their ability to phagocytose beads and Aβ plaques. However, RNA sequencing attributed these findings to increased expression of genes involved in cell migration and proliferation, rather than phagocytosis (43). With regard to C5aR1, it has been shown that C5aR1 antagonism in a murine AD model improves histological and cognitive outcomes, and is associated with decreased inflammatory signaling and enhanced expression of degradation/clearance pathways (41). Detrimental, proinflammatory effects of C5aR1 signaling have also been shown in spinal cord injury (44). Importantly, anaphylatoxin effects are dynamic and context-dependent and should not be generalized. For example, an in vitro study showed that whereas short-lived exposure to C3a increases Aβ phagocytosis by primary microglia, chronic exposure attenuates Aβ phagocytosis, an effect that can be reversed by C3aR antagonists (45).
Finally, it is important to note that the mere presence of complement opsonins and receptors does not necessarily indicate ongoing phagocytic activity. Efficient phagocytosis also depends on recognition of ligands, intracellular signaling by multiple receptors, successful endosomal trafficking, lysosomal digestion and product recycling, and protection of surrounding cells from bystander cytotoxic effects (46). It has been shown that intracellular PI3K signaling triggered by Galectin-3 is crucial for activating CR3-mediated microglial phagocytosis of myelin (47, 48). Microglial CR3-mediated phagocytosis has also been shown to be dependent on DAP12, PKC, DAG, cAMP, MLCK, and Rho/Rock signaling pathways, all of which could be modulated pharmacologically or by other endogenous receptors (49–52). Moreover, knockout of Trem2, a receptor implicated in the phagocytosis of Aβ in AD, decreases expression of C3 and Cd11b genes, in addition to other complement genes, indicating potential synergy between the two systems (53). Other microglial and astrocytic receptors also play a role in synaptic pruning and debris clearance independent of the complement system and have been the subject of other articles and reviews. These other receptors primarily involve TAM receptors (54), Trem2-APOE signaling (55), and the astrocytic MEGF10 and MerTK pathways (56, 57).
Complement in the Development of the Visual System
Complement, and specifically the classical activation pathway, plays a key role in the postnatal development and refinement of synaptic connections in the visual system. Here we review the normal anatomy and prenatal and postnatal development within the visual system, specifically the retina-LGN-primary visual cortex axis, with an emphasis on the role of complement in the lateral geniculate nucleus.
RGC neurons have their cell bodies in the ganglion cell layer of the retina and project to, and synapse with, neurons in the LGN in retinally mapped (retinotopic) fashion. The RGC axons extend along the innermost layer of the retina (nerve fiber layer) to the optic disc, where they form the optic nerve and exit the posterior of the eye. Axons within the eye are not myelinated and become myelinated after exiting the eye and passing through the lamina cribrosa (58). Fibers from the nasal retina cross to the contralateral optic tract in the optic chiasm, such that objects on one side of the visual field will send all related activity from both eyes to the contralateral optic tract. In humans, 50–60% of fibers cross at the optic chiasm, while the percentage is closer to 90% in mice and rats (59). In humans, the majority of RGC axons project to the LGN, although some project or send additional projections to the hypothalamus, pretectum, or superior colliculus (60). In mice, <10% of RGC axons project to the LGN, with the vast majority terminating in the superior colliculus (61). Each mature RGC synapses with one or two neurons in the LGN (62). The LGN can be subdivided into the dorsal and ventral LGN, both of which receive input from the retina and the cortex. However, only neurons with their cell bodies in the dLGN send projections through the optic radiation to the occipital lobe and synapse in defined layers of the visual cortex. Neurons from the vLGN send projections to subcortical structures involved with motor tasks (63).
During prenatal development in the C57BL/6 mouse, retinal ganglion neurons send projections from the optic cup that grow anterogradely (toward the LGN). By embryonic day 14, fibers have crossed at the optic chiasm, and by embryonic day 16 have innervated the dLGN and superior colliculus. Parturition typically occurs at embryonic day 19 (64). By postnatal day 3, temporal fibers that do not cross at the optic chiasm enter the dLGN in a small region also innervated by crossed fibers. At this time, neurons in the LGN receive inputs from as many as 9-11 RGCs coming from both eyes (3). By postnatal day 8, competition between fibers leads to a decrease in synaptic density and, eventually, the establishment of two relatively well separated regions of the dLGN innervated by each of the two eyes (65). These regions are well segregated before natural eye opening at postnatal day 12–14, and by postnatal day 19, the number of RGCs converging on a LGN neuron is reduced to 1–3 from a single eye. This synaptic pruning process is caused by spontaneous retinal activity, which causes calcium influx that reinforces synaptic connections from some neurons and leads to the degradation of others (3). In a seminal 2007 publication, Stevens et al. demonstrated a definitive role for the classical complement pathway in C3-mediated postnatal synaptic pruning in the mouse LGN (4). C1q expression is induced in retinal ganglion cell neurons in response to TGF-beta (66) released from retinal astrocytes, and is localized to the inner plexiform layer in the retina and in the LGN early in postnatal development. C1q and C3 were found to colocalize with immature synapses during early postnatal development. Congenital deletion of the C1q A chain or of C3 leads to defects in eye-specific segregation within the LGN and increased innervation of LGN neurons by RGCs, suggesting that complement is involved in refining synaptic connections in this circuit. Congenital deficiency did not lead to any observable abnormalities in this region at postnatal day 5, suggesting that complement is not involved in axonal path finding or targeting in this region. Some pruning of RGC-LGN synapses, however, did occur in C1q or C3 knockout mice, suggesting that other opsonins and phagocytic receptors may play a role in synaptic pruning. Further studies showed that synaptic pruning in the postnatal LGN is CR3/C3-dependent, and that knockout of CR3 significantly attenuates synaptic phagocytosis and leads to defects in eye-specific segregation (15). Less active synapses are phagocytosed, but the exact mechanism by which C1q labels these synapses is not yet known, although C1q localization has been linked to apoptotic-like processes at synapses (39).
The above studies demonstrate that complement is necessary in normal postnatal brain development, which in addition to its mechanistic importance, indicates the potential benefits of targeting therapeutic complement inhibitors both in location and in time. For this purpose, we and others have investigated gene therapy approaches to correct glaucoma (10) or wet age-related macular degeneration (9) by overexpressing a complement inhibitor in the retina. In the brain, the application of complement inhibitors to reduce excessive synaptic pruning in the retinogeniculate circuit due to glaucoma would ideally need to be restricted to the LGN, as complement plays a positive role in synaptic phagocytosis and memory formation in the adult hippocampus (67). Complement-pathway gene expression by microglia is also higher in gray matter relative to white matter, which may be related to complement-mediated synaptic pruning under homeostatic conditions (68). From an experimental standpoint, the fact that complement has important roles in CNS development and homeostasis also has implications for the use of complement knockout mice in these studies, since synaptic architecture might be different between knockout and wild type mice, thus potentially confounding data interpretation. Lastly, the aforementioned study on the role of complement in RGC pruning in development and glaucoma also indicated that increased expression of complement proteins seen early in development and that are subsequently suppressed upon synapse maturation could again increase under certain pathologic/disease conditions.
Complement in Demyelinating Diseases
The complement system plays a role in several demyelinating, and more broadly, neurodegenerative diseases of the CNS (69). Table 1 summarizes the diseases discussed here, and Table 2 summarizes the role of complement. Complement components are produced locally by neurons, astrocytes, and microglia, and can also access the CNS from the systemic circulation if there is blood-brain barrier (BBB) dysfunction (108). It is generally recognized that chronic inflammation is a characteristic of most neurodegenerative diseases (109). Symptoms of neurodegenerative disorders are caused in part by progressive loss of neuronal structures and functions until neuronal cell death leads to permanent disability. Here, we discuss and speculate on the role of complement in opsonizing neurons in the LGN, and myelin in the optic nerve, in demyelinating diseases of the CNS.

Table 1. Overview of the main aspects of visual system development and diseases discussed in this review.

Table 2. Overview of the influence of complement in visual system development and in disease processes discussed in this review.
Although the involvement of complement in MS has been known for decades (110), a pathological role for complement in the LGN in MS has been relatively unexplored until recently. Visual dysfunction has long been associated with MS, which commonly takes the form of optic neuritis, inflammation and demyelination of the optic nerve that can lead to permanent visual deficits and RGC loss (70). Recently, visual system degeneration in MS has been linked to polymorphisms in C3 and C1q genes, suggesting a role for complement (111). Werneburg et al. (8) recently demonstrated that in EAE models of MS, C3 colocalizes with synapses in the LGN, and that activated microglia consume synaptic material from both retinal and cortical afferents. C1q did not colocalize at synapses, leading the authors to conclude that synapse loss occurred via the alternative pathway. Notably, this process was inhibited by viral overexpression of CR2-Crry, a complement C3 inhibitor (Crry) that is targeted to sites of C3 opsonin deposition via its CR2 domain, in RGCs upon intravitreal delivery. The attenuation of vision loss in treated animals was attributed to preservation of synapses in the LGN, although a similar treatment protocol used in glaucoma suggests there would also be secretion of CR2-Crry in the retina (10), another site of complement-mediated pathology in EAE (112). It is not known if systemic administration of CR2-Crry will attenuate visual loss in EAE, but systemic administration does improve clinical scores in an EAE model (113). Jin et al. found that during the normal course of EAE there was axonal damage, but not significant neuronal/RGC death at the timepoint that Werneburg et al. observed synaptic phagocytosis, and that significant RGC death occurred later in the course of disease (112); it could thus be speculated that loss of synaptic connections in the LGN contributes to RGC death, triggering Wallerian or retrograde degeneration.
Multiple sclerosis is also accompanied by inflammation in the retina. While changes in vision in MS were previously thought to be due primarily to demyelination and inflammation in the optic nerve, recent studies suggest that complement activation on either side of the retinogeniculate pathway contributes to axonal loss. Dembla et al. (81) showed that autoantibodies directed against adhesion proteins in synapses in the retina are an early feature of EAE and possibly MS. These antibodies recruit complement to retinal synapses early in disease, and the presence of these antibodies causes visual dysfunction before noticeable demyelination or axonal loss in the optic nerve occurs, suggesting that demyelination may not be the initial cause of visual dysfunction in MS. In addition, the authors demonstrated that MAC colocalizes with synaptic markers, suggesting a role for the MAC in synapse loss. However, a role for C1q or phagocytosis was not investigated in this study, although both have been implicated in synaptic loss in other regions of the brain in MS (8, 82), as well as other retinal diseases (93).
Regional differences in microglia are also observed in MS (114). Within an active lesion, phagocytic cells are a mixture of microglia and macrophages, with the balance shifting toward the latter as the lesion progresses (115). These microglia display activated morphology and cellular markers. In an inactive lesion center, microglia express more anti-inflammatory markers. Within regions of normal-appearing white matter, there is debate as to whether nodules of microglia are activated or homeostatic (68, 115). These regional differences in microglial activation may relate to the different roles of microglia in the MS brain. The process of active demyelination in an active lesion contributes to a pro-inflammatory environment. Activated microglia in normal appearing white matter may represent early lesions or a response to Wallerian degeneration triggered by a distant lesion (116). Phagocytosis of myelin by microglia leads to a shift toward an anti-inflammatory pro-regenerative state (117), which is important for recovery and remyelination (118).
Complement and phagocytosis is involved in the clearance of myelin in MS and other demyelinating diseases. While this can occur in any region of the brain where there are active lesion, we focus here on optic neuritis. Optic neuritis is an autoimmune process characterized by transient inflammation and demyelination in the optic nerve, which causes visual dysfunction and eye pain upon movement. It is commonly associated with MS (119), although it is also a common feature of myelin oligodendrocyte glycoprotein (MOG)-antibody disease and neuromyelitis optica spectrum disorders (NMOSD) (120). MOG-antibody disease is an autoimmune demyelinating disorder characterized by an antibody against MOG, a component of the myelin sheath synthesized by oligodendrocytes. It frequently present with episodes of optic neuritis or transverse myelitis (72). NMOSD are a class of disorders that also present with optic neuritis or transverse myelitis (71), but are usually characterized by an antibody to aquaporin 4, which is expressed by astrocytes (83). In models of optic neuritis, astrocytes in the optic nerve express C3 (121), which also deposits on discarded myelin either directly or through complement-activating anti-MOG antibodies (84). In NMOSD models, although these antibodies do not directly bind myelin or oligodendrocytes, microglia/macrophages still phagocytose myelin debris (122). Unknown changes occur in the myelin of MS patients that contribute to enhanced phagocytosis (123). In these contexts, classically activated microglia are critical for myelin phagocytosis (114).
The complement system has been shown to have both deleterious and regenerative roles in the CNS. For example, although excess activation of the MAC leads to cell lysis, sub-lytic levels of the MAC have been shown to activate transcriptional programs favoring cell survival and resistance to apoptosis in oligodendrocytes (124) through increased calcium flux (125). Microglia/macrophage phagocytosis of myelin debris contributes to regeneration and remyelination by positively affecting oligodendrocyte precursor cell progression (126) and neurite outgrowth (127). Therefore, complement opsonization of myelin is essential for normal recovery from demyelinating episodes. The dual role of complement in clearance of debris on the one hand, and the clearance of functional synapses on the other, is an important consideration for any therapeutic strategy involving complement inhibition.
Complement in Neurodegenerative Diseases
The role of complement in vision loss in other neurodegenerative disease states is less well investigated. After stroke, visual deficits are common and are estimated to occur in 65% of patients (128). Stroke can directly affect parts of the visual system, such as in posterior cerebral artery stroke (129), stroke affecting the optic radiations (130), or thalamic stroke (131). However, inflammatory changes and complement activation in areas remote to the primary event could also contribute to synaptic loss, either transsynaptically or via a shared vascular bed (for example, in the LGN). In this context, Alawieh et al. (86) showed that complement activation following stroke opsonized hippocampal synapses and resulted in a loss of synaptic density in perilesional areas, with associated cognitive decline that was ongoing for at least 30 days after stroke. It is possible that a similar complement-dependent inflammatory process contributes to visual decline after stroke.
In traumatic brain injury (TBI), up to 60% of patients experience some kind of visual dysfunction after injury, which can persist for at least a year after TBI (132). TBI patients exhibit a reduction in cell size and neuronal loss in nuclei of the thalamus following severe head injury, although the LGN itself has not been analyzed histologically (133). However, functional MRI of TBI patients shows decreased connectivity between the LGN and areas of the cortex that correlates with severity of injury (134). A case study of a TBI patient showed retrograde degeneration of RGCs 2 months after injury (135). These studies show that the thalamus, and specifically visual circuitry, is affected chronically following TBI. In murine models of TBI, axonal loss affecting the LGN is observed in both retinogeniculate (136) and corticothalamic pathways (137). Inflammation, specifically gliosis and morphological alterations in microglia, is observed in the LGN 1 week after injury (138). There is also ongoing BBB dysfunction in the thalamus up to 3 months after TBI (139). Some synaptic reorganization occurs in the LGN following TBI (140), suggesting that there are competing neurodegenerative and regenerative processes occurring. While direct injury to the retina (136) or optic nerve (138) following TBI can contribute to visual dysfunction, ongoing inflammation in the LGN and axonal loss is thought to contribute to vision loss and impair recovery.
The complement system plays an important role in a secondary neuroinflammatory injury phase after the initial mechanical insult to the brain. Complement activation products, including the MAC and C3 degradation products, have been detected in postmortem brain tissue of TBI patients in close proximity to neurons (87). Elevated levels of complement proteins have been detected in the plasma of TBI patients for up to 6 months after injury, suggesting a continuous role for complement in chronic inflammation (88). In a murine model of TBI, specific inhibition of MAC reduced acute deficits, but only inhibition of complement upstream at the C3 activation step provided chronic protection (89). In addition, this study also highlighted that the alternative complement pathway plays a particularly important role as an amplification loop in chronic neuroinflammation post-TBI. However, whether there is opsonization of neurons in visual circuits that are related to delayed visual deficits after TBI has not been investigated; nevertheless, given the pathological opsonization of synapses in various disease models, it is possible that aberrant complement activation in the LGN contributes to visual dysfunction following TBI.
In summary, the role of complement in neuroinflammation is well established, but its role in visual deficits resulting from neuropathological disease states is much less well characterized. Studies modeled around analyses within the LGN have established complement-mediated phagocytosis as an important mechanism involved in CNS development, and more recently, to be a mechanism involved with visual decline in models of MS. That the alternative pathway is implicated in synaptic phagocytosis in the LGN in MS comes as somewhat of a surprise, since MS is associated with myelin-recognizing autoantibodies capable of activating complement (141), and that the classical pathway mediates hippocampal synaptic phagocytosis in MS (82). However, the classical complement pathway is involved in synaptic loss in other diseases such as AD, as well as in other regions of the brain in MS (28, 82), and both classical and lectin pathways are implicated in stroke (85, 86). There remains a paucity of data on the role of complement in opsonophagocytosis within the LGN and visual circuitry after more generalized CNS injury, even though complement-mediated synaptic phagocytosis is known to occur in other areas of the brain in injury models with accompanying visual dysfunction.
Complement in Diseases of the Retina
The role of complement in development and diseases of the retina is somewhat better characterized. During normal embryonic development, the classical complement pathway contributes to microglia-mediated RGC elimination (5), which reflect findings within the LGN (see above), and indicates that complement and microglia contribute to neuronal development and refinement in multiple regions relevant to the visual system. Of note, complement factor H-like protein 1, a truncated form of factor H, is expressed in the retinal pigment epithelium and spreads throughout the extracellular matrix in this layer of the retina (142). This protein is an alternative pathway inhibitor that functions at the C3 activation step and will thus inhibit C3 opsonization of tissues in the retina. The complement system has been implicated in several diseases of the retina, and for representation we focus here on only glaucoma and age-related macular degeneration, in which complement has a firmly established role and in which complement-based therapeutics have shown efficacy.
Glaucoma is a family of eye diseases characterized by damage to the optic nerve and vision loss (77). Complement component C1q is known to be upregulated in the retina during glaucoma (4), and upregulation of C1q and C3 expression is an early event in the development of glaucoma in mice (90, 91). C1q and C3 bind to RGCs in glaucomatous eyes in the absence of IgG: however, the target(s) for complement protein binding expressed by stressed RGCs is unknown (92). In experimental glaucoma, complement activation via the classical pathway leads to synaptic pruning in the retina by resident glial cells (93) as well as MAC-induced apoptosis of RGCs (94). Thus, the complement system appears to represent a viable therapeutic target in glaucoma. AAV-mediated expression of soluble C3d-targeted CR2-Crry by retinal ganglion cells reduces neuronal loss in a glaucoma model (10), in part by inhibiting phagocytosis of opsonized RGC synapses. Inhibition of C5 via intravitreal injection of an anti-C5 monoclonal antibody also reduced neuronal loss (11), indicating that both C3 activation products and terminal pathway products (C5a and/or MAC) may contribute to RGC loss.
Age-related macular degeneration (AMD) is another disease that can cause irreversible vision loss. It can be broadly divided into two forms: dry AMD and wet AMD. Dry AMD is characterized by progressive damage to the macula resulting from damage to the retinal pigment epithelium and choriocapillaris complex followed by photoreceptor cell loss, which results in progressive loss of central vision. Wet AMD, which sometimes follows dry AMD but can occur on its own, is characterized by the addition of VEGF-stimulated neovascularization that can grow under the retina. Drusen, an extracellular aggregate of protein and lipid and laminar deposits in Bruch's membrane, are often associated with both forms of AMD (80). Polymorphisms in multiple complement proteins including factor H (95), factor B (96) and C3 (97) are associated with increased or decreased risk of AMD, which provide strong support for the role of complement in AMD.
The alternative complement pathway and MAC have been shown to contribute to ocular injury in multiple AMD mouse models. These include the wet AMD model of laser-induced choroidal neovascularization (CNV) (98, 99), although the significance of complement is somewhat debated (143), and the dry AMD models of light damage (100), smoke exposure (101, 144) and sodium iodate-induced blood-retina barrier disruption (102). Drusen in the human retina contain complement components (103), and anaphylatoxins produced from complement activation contribute to the development of neovascularization in the mouse CNV model (104). A decline in phagocytic function of retinal pigment epithelium cells contributes to drusen and lipofuscin accumulation and AMD pathology (105). Also, sub-lytic levels of the MAC on the retinal pigment epithelium causes calcium flux that contributes to epithelial dysfunction and leakiness (106). Synaptic changes occur in the retina in AMD, specifically the retraction of photoreceptor synapses from the outer plexiform layer to the outer nuclear layer and the outgrowth of bipolar cell dendrites to this new location (145). While the mechanism of synaptic change is unclear, it has been demonstrated that C3 deposits in the outer nuclear layer in a murine model (107). It is not clear if C3-mediated phagocytosis is involved in these synaptic changes in AMD, especially since C3 does not appear to deposit in the outer plexiform layer, which is the location of the photoreceptor-to-bipolar cell synapses. In clinical trials, systemic administration of an anti-C5 monoclonal antibody did not slow the rate of progression of AMD (146), whereas intravitreal administration of C3 inhibitor pegcetacoplan did (12). However, the intravitreal administration of an alternative pathway inhibitor (anti-factor D blocking antibody) did not reach its clinical endpoint in two phase 3 trials (147). The different routes of administration notwithstanding, clinical data thus far suggest that C3 activation products, possibly C3 opsonins, may have a pathogenic role in AMD and that MAC formation contributes to macular injury.
Complement is implicated in many other diseases of the retina. To mention just two, upregulation of C3 and factor H in proliferative diabetic retinopathy suggests a role for the alternative complement pathway in this disease, and colocalization of C3 and microglia suggests the possibility of C3-mediated phagocytosis occurring in the retina (148). Classical complement pathway genes are upregulated in models of retinopathy of prematurity (149) and in the vitreous humor of human babies with this disorder (150). To our knowledge, the role of complement opsonizing retinal tissue in this condition has not been explored. Diabetic retinopathy and retinopathy of prematurity are both proliferative retinal diseases characterized by increased angiogenesis and vascular leakiness and inflammation, not unlike wet AMD.
Complement Inhibitors in Retinal and Neurologic Disease
The involvement of complement in neurodegeneration in retinal diseases, and the importance of visual function with regards to quality of life, make development of complement therapeutics an important area of study in the field of complement. A detailed understanding of complement-dependent mechanisms involved in injury to the visual circuitry, including understanding the importance of C3 opsonins and receptor recognition, will be critical for establishing an optimal complement inhibitory strategy to treat retinal and neurodegenerative disease states. A summary of the studies referenced is included in Table 3.
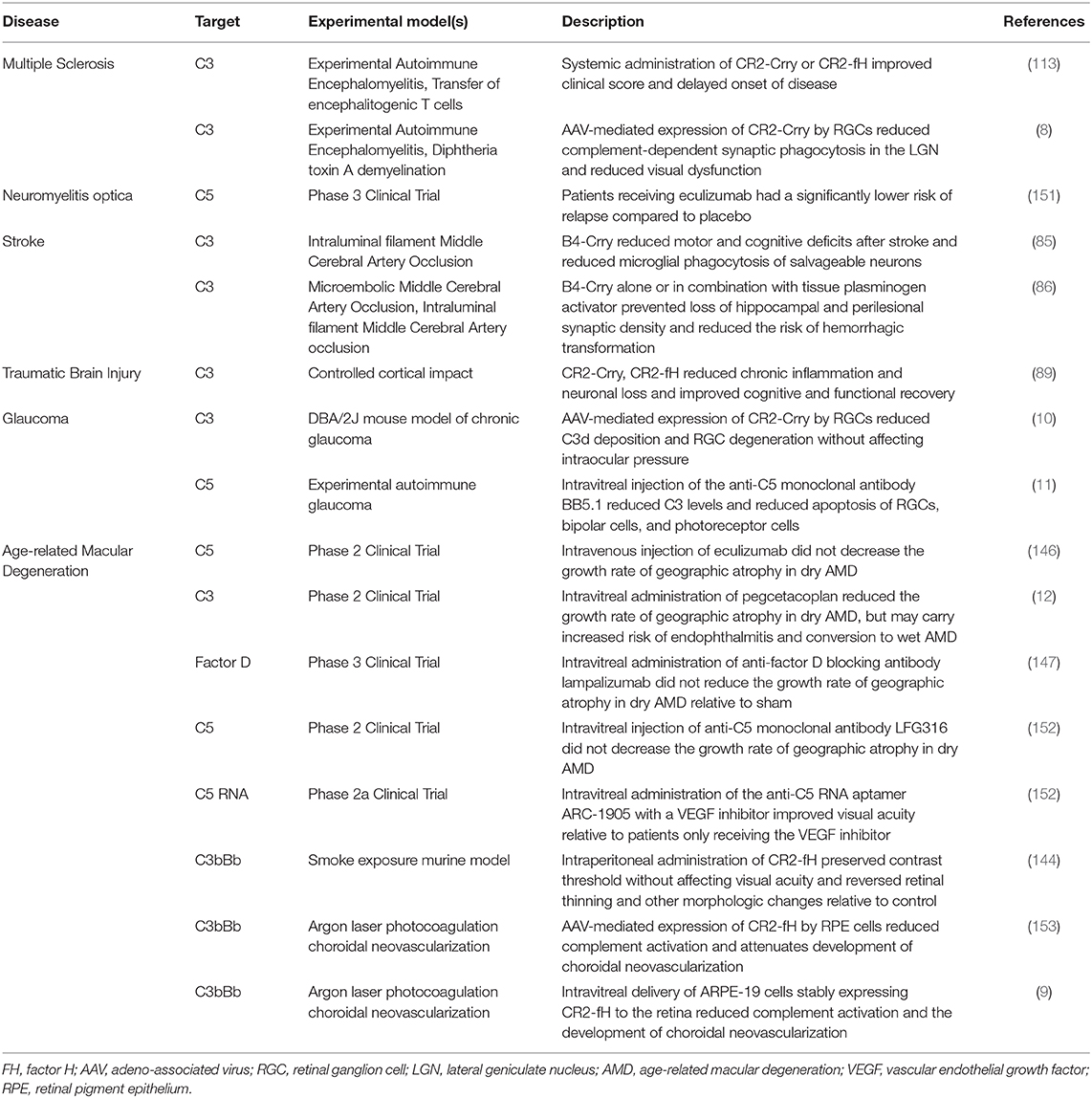
Table 3. The complement targets of the therapeutic approaches discussed in this review.
Complement-based therapeutics specific for different molecules or pathways would allow for effective treatment of disease while preserving the role of complement in other functions such as immune defense, retinal homeostasis, and other physiological functions of complement. Inhibition of complement at the C3 activation step knocks out all pathways and all enzymatically generated and assembled complement effector molecules. However, because of the multi-functionality of the complement system, a more targeted or selective approach at complement inhibition may be optimal, depending on how complement is involved in pathology. The C3 inhibitor pegcetacoplan, administered intravitreally, recently completed a phase 2 clinical trial in the treatment of geographic atrophy (GA) secondary to AMD with promising results (12). In this case, the localized administration reduced the chance of systemic complications, although endophthalmitis did occur in three participants receiving pegcetacoplan, and a significant portion of patients treated developed exudative AMD. Blockade of C5 has been a strategy for the treatment of other complement-mediated diseases in hematology (154) and nephrology (155) using an anti-C5 blocking antibody, eculizumab. Eculizumab is FDA approved for neuromyelitis optica, an autoimmune CNS disease with some clinical and pathological overlap with MS that is characterized by antibody-dependent complement activation (151). However, this and other C5-targeted strategies have failed in AMD. Anti-human C5 monoclonal antibodies [eculizumab (intravenous injection) (146) or LFG316 (intravitreal injections), summarized in (152)] did not decrease GA growth rate. Administration of an anti-C5 RNA aptamer [ARC 1905 (intravitreal injection)] with the vascular endothelial growth factor inhibitor ranibizumab preserved visual acuity better than ranibizumab alone in a phase 2a clinical trial [summarized in (152)]. Finally, lampalizumab, an anti-factor D alternative pathway inhibitor (156), failed to reduce the growth of GA in two phase 3 clinical trials for AMD when administered intravitreally (147), despite having achieved significance in a subgroup of dry AMD patients with complement factor I risk variants.
Another approach to limit systemic complement inhibition and limit potential off-target effects is to target complement inhibition specifically to the site of complement activation. Various protein inhibitors of complement fused to the C3 binding domain of complement receptor 2 (CR2) have been developed and tested in preclinical studies. The CR2 domain targets a complement inhibitor to sites of complement deposition, which prevents systemic effects of complement inhibition (157) and can function to block recognition of C3 opsonins by phagocytic cells. A CR2-factor H fusion protein, which is an alternative pathway-specific inhibitor, is effective when administered systemically in the smoke exposure dry AMD model (144). The same fusion protein, when expressed intraretinally after AAV-mediated (153) or cell-based delivery (9), reduced the growth of choroidal neovascularization in the laser-induced model of wet AMD. AAV-mediated intravitreal delivery of a CR2-Crry construct, which inhibits all complement pathways at C3 activation, provides neuroprotection to RGCs in murine models of glaucoma (10) and MS (8). Finally, intravitreal injection of a monoclonal antibody against mouse C5 also protected RGCs in a glaucoma model (11). These preclinical studies highlight the potential of targeted and/or localized delivery of complement inhibition with multiple modes of administration in reducing RGC death and visual loss in models of retinal and neurological disease.
Conclusion
The complement system and phagocytosis play essential roles in sculpting neural circuits. However, in certain disease states the complement system is aberrantly activated and contributes to opsonization of functional and/or salvageable synapses, leading to irreversible damage. In this review, we focus on these processes in the afferent visual system, specifically the retina-LGN-primary visual cortex axis, although complement plays an active role in other regions of the brain in homeostasis and disease. The studies discussed here focus on the role of the complement system in the development of the visual system and in its degeneration in various disorders, including multiple sclerosis, glaucoma, and age-related macular degeneration. Although the studies discussed here primarily focus on the negative aspects of complement activation and opsonization, complement positively contributes to disease processes by tagging myelin and other debris for phagocytic removal, which in turn contributes to removal of damaged material and an anti-inflammatory microglial phenotype. We also included a review of complement inhibitory strategies, both systemic and targeted, that have been investigated in preclinical and clinical studies in the diseases discussed herein. A deeper understanding of the mechanistic role of complement and the contribution of specific pathways and activation products in different disease processes will assist in the development of smarter and more targeted approaches for the therapeutic application of complement inhibition in the clinic.
Significance Statement
The visual system is one of the best understood neural circuits, with obvious functional importance. Over a decade ago, the centrality of the complement system in normal synaptic pruning in the afferent visual system was established. Around that time, the first links between polymorphisms in complement genes and risk of developing age-related macular degeneration were discovered. Since then, our understanding of the complement system's involvement in diseases of the brain and retina, and specifically its role in opsonizing synapses for removal by microglia, has expanded. Here, we review the current understanding of complement-mediated synaptic phagocytosis in the afferent visual system, specifically the retina and lateral geniculate nucleus, in normal development and disease. We focus on recent findings of synaptic phagocytosis in multiple sclerosis models, and review in general the involvement of complement in glaucoma and age-related macular degeneration. Lastly, we conclude with a brief look at complement inhibition in clinical trials and preclinical studies in diseases affecting the visual system.
Author Contributions
DB and ST were responsible for conceptualization of the review topic. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants by the Department of Veterans Affairs (IK6BX005235, 1BX004256, 1RX001141, and 21RX002363) to ST, grants by the National Institutes of Health (NIH) (R01EY024581), the Departments of Veterans Affairs (I01RX000444, I01BX003050, and IK6BX004858) and the South Carolina SmartState Endowment to BR, an NIH institutional training grant (2T32GM008716-21) to DB, an American Heart Association predoctoral fellowship (19PRE34450105) to AT, the NIH National Center for Advancing Translational Sciences (TL1 TR001451 and UL1 TR001450) to CC, and the NIH training grant (T32AI132164) to KM.
Conflict of Interest
ST is a cofounder and consultant for Q32Bio, a company developing complement inhibitors. ST and BR are inventors on patents that describe targeted complement inhibition.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Alison Staffin for the figure illustrations.
References
1. Demb JB, Singer JH. Functional circuitry of the retina. Ann Rev Vis Sci. (2015) 1:263–89. doi: 10.1146/annurev-vision-082114-035334
2. Zhang X, Serb JM, Greenlee MHW. Mouse retinal development: a dark horse model for systems biology research. Bioinformatics Biol Insights. (2011) 5:99–113. doi: 10.4137/BBI.S6930
3. Jaubert-Miazza L, Green E, Lo F-S, Bui KIM, Mills J, Guido W. Structural and functional composition of the developing retinogeniculate pathway in the mouse. Visual Neurosci. (2005) 22:661–76. doi: 10.1017/S0952523805225154
4. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
5. Anderson SR, Zhang J, Steele MR, Romero CO, Kautzman AG, Schafer DP, et al. Complement targets newborn retinal ganglion cells for phagocytic elimination by microglia. J Neurosci. (2019) 39:2025–40. doi: 10.1523/JNEUROSCI.1854-18.2018
6. Orsini F, De Blasio D, Zangari R, Zanier ER, De Simoni M-G. Corrigendum: versatility of the complement system in neuroinflammation, neurodegeneration, and brain homeostasis. Front Cell Neurosci. (2015) 9:263. doi: 10.3389/fncel.2015.00263
7. Quinn TA, Dutt M, Shindler KS. Optic neuritis and retinal ganglion cell loss in a chronic murine model of multiple sclerosis. Front Neurol. (2011) 2:50. doi: 10.3389/fneur.2011.00050
8. Werneburg S, Jung J, Kunjamma RB, Ha SK, Luciano NJ, Willis CM, et al. Targeted complement inhibition at synapses prevents microglial synaptic engulfment and synapse loss in demyelinating disease. Immunity. (2020) 52:167–82.e7. doi: 10.1101/841601
9. Annamalai B, Parsons N, Belhaj M, Brandon C, Potts J, Rohrer B. Encapsulated cell technology-based delivery of a complement inhibitor reduces choroidal neovascularization in a mouse model. Transl Vis Sci Technol. (2018) 7:3. doi: 10.1167/tvst.7.2.3
10. Bosco A, Anderson SR, Breen KT, Romero CO, Steele MR, Chiodo VA, et al. Complement C3-targeted gene therapy restricts onset and progression of neurodegeneration in chronic mouse glaucoma. Mol Therapy. (2018) 26:2379–96. doi: 10.1016/j.ymthe.2018.08.017
11. Reinehr S, Gomes SC, Gassel CJ, Asaad MA, Stute G, Schargus M, et al. Intravitreal therapy against the complement factor C5 prevents retinal degeneration in an experimental autoimmune glaucoma model. Front Pharmacol. (2019) 10:1381. doi: 10.3389/fphar.2019.01381
12. Liao DS, Grossi FV, El Mehdi D, Gerber MR, Brown DM, Heier JS, et al. Complement C3 inhibitor pegcetacoplan for geographic atrophy secondary to age-related macular degeneration: a randomized phase 2 trial. Ophthalmology. (2020) 127:186–95. doi: 10.1016/j.ophtha.2019.07.011
14. van den Elsen JMH, Isenman DE. A crystal structure of the complex between human complement receptor 2 and its ligand C3d. Science. (2011) 332:608. doi: 10.1126/science.1201954
15. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
16. Slobodov U, Reichert F, Mirski R, Rotshenker S. Distinct inflammatory stimuli induce different patterns of myelin phagocytosis and degradation in recruited macrophages. Exp Neurol. (2001) 167:401–9. doi: 10.1006/exnr.2000.7559
17. Reichert F, Rotshenker S. Complement-receptor-3 and scavenger-receptor-AI/II mediated myelin phagocytosis in microglia and macrophages. Neurobiol Dis. (2003) 12:65–72. doi: 10.1016/S0969-9961(02)00008-6
18. Haga S, Ikeda K, Sato M, Ishii T. Synthetic Alzheimer amyloid beta/A4 peptides enhance production of complement C3 component by cultured microglial cells. Brain Res. (1993) 601:88–94. doi: 10.1016/0006-8993(93)91698-R
19. Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. (2005) 25:8240–9. doi: 10.1523/JNEUROSCI.1808-05.2005
20. Kaur C, Too HF, Ling EA. Phagocytosis of Escherichia coli by amoeboid microglial cells in the developing brain. Acta Neuropathol. (2004) 107:204–8. doi: 10.1007/s00401-003-0798-7
21. Rambach G, Dum D, Mohsenipour I, Hagleitner M, Würzner R, Lass-Flörl C, et al. Secretion of a fungal protease represents a complement evasion mechanism in cerebral aspergillosis. Mol Immunol. (2010) 47:1438–49. doi: 10.1016/j.molimm.2010.02.010
22. Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. (2016) 534:538–43. doi: 10.1038/nature18283
23. Zabel M, Schrag M, Crofton A, Tung S, Beaufond P, Van Ornam J, et al. A shift in microglial β-amyloid binding in Alzheimer's disease is associated with cerebral amyloid angiopathy. Brain Pathol. (2013) 23:390–401. doi: 10.1111/bpa.12005
24. Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. (2008) 28:6333–41. doi: 10.1523/JNEUROSCI.0829-08.2008
25. Czirr E, Castello NA, Mosher KI, Castellano JM, Hinkson IV, Lucin KM, et al. Microglial complement receptor 3 regulates brain Aβ levels through secreted proteolytic activity. J Exp Med. (2017) 214:1081–92. doi: 10.1084/jem.20162011
26. Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, et al. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med. (2017) 9:eaaf6295. doi: 10.1126/scitranslmed.aaf6295
27. Kamphuis W, Kooijman L, Schetters S, Orre M, Hol EM. Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer's disease. Biochim Biophys Acta. (2016) 1862:1847–60. doi: 10.1016/j.bbadis.2016.07.007
28. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. (2016) 352:712–6. doi: 10.1126/science.aad8373
29. Peerschke EI, Ghebrehiwet B. Platelet C1q receptor interactions with collagen- and C1q-coated surfaces. J Immunol. (1990) 145:2984–8.
30. Klickstein LB, Barbashov SF, Liu T, Jack RM, Nicholson-Weller A. Complement receptor type 1 (CR1, CD35) is a receptor for C1q. Immunity. (1997) 7:345–55. doi: 10.1016/S1074-7613(00)80356-8
31. Stuart GR, Lynch NJ, Day AJ, Schwaeble WJ, Sim RB. The C1q and collectin binding site within C1 q receptor (cell surface calreticulin). Immunopharmacology. (1997) 38:73–80. doi: 10.1016/S0162-3109(97)00076-3
32. Gardai SJ, Xiao Y-Q, Dickinson M, Nick JA, Voelker DR, Greene KE, et al. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell. (2003) 115:13–23. doi: 10.1016/S0092-8674(03)00758-X
33. Edelson BT, Stricker TP, Li Z, Dickeson SK, Shepherd VL, Santoro SA, et al. Novel collectin/C1q receptor mediates mast cell activation and innate immunity. Blood. (2006) 107:143–50. doi: 10.1182/blood-2005-06-2218
34. Son M, Santiago-Schwarz F, Al-Abed Y, Diamond B. C1q limits dendritic cell differentiation and activation by engaging LAIR-1. Proc Natl Acad Sci. (2012) 109:E3160–7. doi: 10.1073/pnas.1212753109
35. Ramirez-Ortiz ZG, Pendergraft WF, Prasad A, Byrne MH, Iram T, Blanchette CJ, et al. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat Immunol. (2013) 14:917–26. doi: 10.1038/ni.2670
36. Trouw LA, Blom AM, Gasque P. Role of complement and complement regulators in the removal of apoptotic cells. Mol Immunol. (2008) 45:1199–207. doi: 10.1016/j.molimm.2007.09.008
37. Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem. (2010) 112:733–43. doi: 10.1111/j.1471-4159.2009.06494.x
38. Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang H-Y, et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell. (2016) 165:921–35. doi: 10.1016/j.cell.2016.04.001
39. Györffy BA, Kun J, Török G, Bulyáki É, Borhegyi Z, Gulyássy P, et al. Local apoptotic-like mechanisms underlie complement-mediated synaptic pruning. Proc Natl Acad Sci USA. (2018) 115:6303. doi: 10.1073/pnas.1722613115
40. Färber K, Cheung G, Mitchell D, Wallis R, Weihe E, Schwaeble W, et al. C1q, the recognition subcomponent of the classical pathway of complement, drives microglial activation. J Neurosci Res. (2009) 87:644–52. doi: 10.1002/jnr.21875
41. Hernandez MX, Jiang S, Cole TA, Chu S-H, Fonseca MI, Fang MJ, et al. Prevention of C5aR1 signaling delays microglial inflammatory polarization, favors clearance pathways and suppresses cognitive loss. Mol Neurodegener. (2017) 12:66. doi: 10.1186/s13024-017-0210-z
42. Webster SD, Park M, Fonseca MI, Tenner AJ. Structural and functional evidence for microglial expression of C1qR(P), the C1q receptor that enhances phagocytosis. J Leukocyte Biol. (2000) 67:109–16. doi: 10.1002/jlb.67.1.109
43. Salton SR, Audrain M. VGF-derived peptide TLQP-21 modulates microglial function through C3aR1 signaling pathways and reduces neuropathology in 5xFAD mice: Program No. 299.01. In: 2019 Neuroscience Meeting Planner. Chicago, IL: Society for Neuroscience (2019).
44. Nakamura M, Shibata R. The efficacy of C5a receptor antagonist for human iPSC-derived neural stem/progenitor cell transplantation in the injured spinal cord of mice: Program No. 302.05. 2019 In: Neuroscience Meeting Planner. Chicago, IL: Society for Neuroscience (2019).
45. Lian H, Litvinchuk A, Chiang ACA, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer's Disease. J Neurosci. (2016) 36:577–89. doi: 10.1523/JNEUROSCI.2117-15.2016
46. Rosales C, Uribe-Querol E. Phagocytosis: a fundamental process in immunity. BioMed Res Int. (2017) 2017:9042851. doi: 10.1155/2017/9042851
47. Reichert F, Rotshenker S. Galectin-3/MAC-2 in experimental allergic encephalomyelitis. Exp Neurol. (1999) 160:508–14. doi: 10.1006/exnr.1999.7229
48. Rotshenker S, Reichert F, Gitik M, Haklai R, Elad-Sfadia G, Kloog Y. Galectin-3/MAC-2, Ras and PI3K activate complement receptor-3 and scavenger receptor-AI/II mediated myelin phagocytosis in microglia. Glia. (2008) 56:1607–13. doi: 10.1002/glia.20713
49. Cohen G, Makranz C, Spira M, Kodama T, Reichert F, Rotshenker S. Non-PKC DAG/phorbol-ester receptor(s) inhibit complement receptor-3 and nPKC inhibit scavenger receptor-AI/II-mediated myelin phagocytosis but cPKC, PI3k, and PLCgamma activate myelin phagocytosis by both. Glia. (2006) 53:538–50. doi: 10.1002/glia.20304
50. Makranz C, Cohen G, Reichert F, Kodama T, Rotshenker S. cAMP cascade (PKA, Epac, adenylyl cyclase, Gi, and phosphodiesterases) regulates myelin phagocytosis mediated by complement receptor-3 and scavenger receptor-AI/II in microglia and macrophages. Glia. (2006) 53:441–8. doi: 10.1002/glia.20303
51. Gitik M, Reichert F, Rotshenker S. Cytoskeleton plays a dual role of activation and inhibition in myelin and zymosan phagocytosis by microglia. FASEB J. (2010) 24:2211–21. doi: 10.1096/fj.09-146118
52. Linnartz B, Wang Y, Neumann H. Microglial immunoreceptor tyrosine-based activation and inhibition motif signaling in neuroinflammation. Int J Alzheimer's Dis. (2010) 2010:587463. doi: 10.4061/2010/587463
53. Linnartz-Gerlach B, Bodea L-G, Klaus C, Ginolhac A, Halder R, Sinkkonen L, et al. TREM2 triggers microglial density and age-related neuronal loss. Glia. (2019) 67:539–50. doi: 10.1002/glia.23563
54. Burstyn-Cohen T, Maimon A. TAM receptors, Phosphatidylserine, inflammation, and Cancer. Cell Commun Signal. (2019) 17:156. doi: 10.1186/s12964-019-0461-0
55. Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. (2018) 18:759–72. doi: 10.1038/s41577-018-0051-1
56. Chung W-S, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. (2013) 504:394–400. doi: 10.1038/nature12776
57. Iram T, Ramirez-Ortiz Z, Byrne MH, Coleman UA, Kingery ND, Means TK, et al. Megf10 Is a receptor for C1Q that mediates clearance of apoptotic cells by astrocytes. J Neurosci. (2016) 36:5185–92. doi: 10.1523/JNEUROSCI.3850-15.2016
58. Perry VH, Lund RD. Evidence that the lamina cribrosa prevents intraretinal myelination of retinal ganglion cell axons. J Neurocytol. (1990) 19:265–72. doi: 10.1007/BF01217304
59. Kerschensteiner D, Guido W. Organization of the dorsal lateral geniculate nucleus in the mouse. Vis Neurosci. (2017) 34:E008. doi: 10.1017/S0952523817000062
60. Erskine L, Herrera E. Connecting the retina to the brain. ASN Neuro. (2014) 6:1759091414562107. doi: 10.1177/1759091414562107
61. Dhande OS, Stafford BK, Lim J-HA, Huberman AD. Contributions of retinal ganglion cells to subcortical visual processing and behaviors. Ann Rev Vis Sci. (2015) 1:291–328. doi: 10.1146/annurev-vision-082114-035502
62. Hong YK, Chen C. Wiring and rewiring of the retinogeniculate synapse. Curr Opin Neurobiol. (2011) 21:228–37. doi: 10.1016/j.conb.2011.02.007
63. LeVere TE. The primary visual system of the rat: A primer of its anatomy. Physiol Psychol. (1978) 6:142–69. doi: 10.3758/BF03326707
64. Murray SA, Morgan JL, Kane C, Sharma Y, Heffner CS, Lake J, et al. Mouse gestation length is genetically determined. PLoS ONE. (2010) 5:e12418. doi: 10.1371/journal.pone.0012418
65. Godement P, Salaün J, Imbert M. Prenatal and postnatal development of retinogeniculate and retinocollicular projections in the mouse. J Comp Neurol. (1984) 230:552–75. doi: 10.1002/cne.902300406
66. Bialas AR, Stevens B. TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci. (2013) 16:1773–82. doi: 10.1038/nn.3560
67. Wang C, Yue H, Hu Z, Shen Y, Ma J, Li J, et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science. (2020) 367:688–94. doi: 10.1126/science.aaz2288
68. van der Poel M, Ulas T, Mizee MR, Hsiao C-C, Miedema SS, Schuurman KG, et al. Transcriptional profiling of human microglia reveals grey–white matter heterogeneity and multiple sclerosis-associated changes. Nat Commun. (2019) 10:1139. doi: 10.1038/s41467-019-08976-7
69. Bonifati DM, Kishore U. Role of complement in neurodegeneration and neuroinflammation. Mol Immunol. (2007) 44:999–1010. doi: 10.1016/j.molimm.2006.03.007
70. Huang W-J, Chen W-W, Zhang X. Multiple sclerosis: pathology, diagnosis and treatments. Exp Ther Med. (2017) 13:3163–6. doi: 10.3892/etm.2017.4410
71. Jarius S, Wildemann B, Paul F. Neuromyelitis optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol. (2014) 176:149–64. doi: 10.1111/cei.12271
72. Wynford-Thomas R, Jacob A, Tomassini V. Neurological update: MOG antibody disease. J Neurol. (2019) 266:1280–6. doi: 10.1007/s00415-018-9122-2
74. Ovbiagele B, Nguyen-Huynh MN. Stroke epidemiology: advancing our understanding of disease mechanism and therapy. Neurotherapeutics. (2011) 8:319–29. doi: 10.1007/s13311-011-0053-1
75. Georges A, Booker JG. Traumatic brain injury. In: StatPearls. Treasure Island, FL: StatPearls Publishing. Copyright © 2020. StatPearls Publishing LLC (2020).
76. McKee AC, Daneshvar DH. The neuropathology of traumatic brain injury. Handbook Clin Neurol. (2015) 127:45–66. doi: 10.1016/B978-0-444-52892-6.00004-0
77. Cohen LP, Pasquale LR. Clinical characteristics and current treatment of glaucoma. Cold Spring Harbor Perspect Med. (2014) 4:a017236. doi: 10.1101/cshperspect.a017236
78. Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA. (2014) 311:1901–11. doi: 10.1001/jama.2014.3192
79. Kreft D, Doblhammer G, Guthoff RF, Frech S. Prevalence, incidence, and risk factors of primary open-angle glaucoma - a cohort study based on longitudinal data from a German public health insurance. BMC Public Health. (2019) 19:851. doi: 10.1186/s12889-019-6935-6
80. Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration–emerging pathogenetic and therapeutic concepts. Ann Med. (2006) 38:450–71. doi: 10.1080/07853890600946724
81. Dembla M, Kesharwani A, Natarajan S, Fecher-Trost C, Fairless R, Williams SK, et al. Early auto-immune targeting of photoreceptor ribbon synapses in mouse models of multiple sclerosis. EMBO Mol Med. (2018) 10:e8926. doi: 10.15252/emmm.201808926
82. Michailidou I, Willems JGP, Kooi EJ, Van Eden C, Gold SM, Geurts JJG, et al. Complement C1q-C3-associated synaptic changes in multiple sclerosis hippocampus. Ann Neurol. (2015) 77:1007–26. doi: 10.1002/ana.24398
83. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. (2005) 202:473–7. doi: 10.1084/jem.20050304
84. Van der Goes A, Kortekaas M, Hoekstra K, Dijkstra CD, Amor S. The role of anti-myelin (auto)-antibodies in the phagocytosis of myelin by macrophages. J Neuroimmunol. (1999) 101:61–7. doi: 10.1016/S0165-5728(99)00133-2
85. Alawieh A, Langley EF, Tomlinson S. Targeted complement inhibition salvages stressed neurons and inhibits neuroinflammation after stroke in mice. Sci Transl Med. (2018) 10:aao6459. doi: 10.1126/scitranslmed.aao6459
86. Alawieh A, Langley EF, Feng W, Spiotta A, Tomlinson S. Complement-dependent synaptic uptake and cognitive decline after stroke and reperfusion therapy. J Neurosci. (2020) 40:4042–58. doi: 10.1523/JNEUROSCI.2462-19.2020
87. Bellander B-M, Singhrao SK, Ohlsson M, Mattsson P, Svensson M. Complement activation in the human brain after traumatic head injury. J Neurotrauma. (2001) 18:1295–311. doi: 10.1089/08977150152725605
88. Bao W, He F, Yu L, Gao J, Meng F, Ding Y, et al. Complement cascade on severe traumatic brain injury patients at the chronic unconscious stage: implication for pathogenesis. Exp Rev Mol Diagn. (2018) 18:761–6. doi: 10.1080/14737159.2018.1471985
89. Alawieh A, Langley EF, Weber S, Adkins D, Tomlinson S. Identifying the role of complement in triggering neuroinflammation after traumatic brain injury. J Neurosci. (2018) 38:2519–32. doi: 10.1523/JNEUROSCI.2197-17.2018
90. Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, Kneeland SC, et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Investig. (2011) 121:1429–44. doi: 10.1172/JCI44646
91. Becker S, Reinehr S, Burkhard Dick H, Joachim SC. Complement activation after induction of ocular hypertension in an animal model. Ophthalmologe. (2014) 112:41–8. doi: 10.1007/s00347-014-3100-6
92. Ding QJ, Cook AC, Dumitrescu AV, Kuehn MH. Lack of immunoglobulins does not prevent C1q binding to RGC and does not alter the progression of experimental glaucoma. Investig Ophthalmol Vis Sci. (2012) 53:6370–7. doi: 10.1167/iovs.12-10442
93. Williams PA, Tribble JR, Pepper KW, Cross SD, Morgan BP, Morgan JE, et al. Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Mol Neurodegener. (2016) 11:1–13. doi: 10.1186/s13024-016-0091-6
94. Jha P, Banda H, Tytarenko R, Bora PS, Bora NS. Complement mediated apoptosis leads to the loss of retinal ganglion cells in animal model of glaucoma. Mol Immunol. (2011) 48:2151–8. doi: 10.1016/j.molimm.2011.07.012
95. Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. (2005) 102:7227–32. doi: 10.1073/pnas.0501536102
96. Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. (2006) 38:458–62. doi: 10.1038/ng1750
97. Yates JRW, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, et al. Complement C3 variant and the risk of age-related macular degeneration. New Engl J Med. (2007) 357:553–61. doi: 10.1056/NEJMoa072618
98. Bora PS, Sohn J-H, Cruz JMC, Jha P, Nishihori H, Wang Y, et al. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J Immunol. (2005) 174:491. doi: 10.4049/jimmunol.174.1.491
99. Bora NS, Kaliappan S, Jha P, Xu Q, Sohn JH, Dhaulakhandi DB, et al. Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: role of factor B and factor H. J Immunol. (2006) 177:1872–8. doi: 10.4049/jimmunol.177.3.1872
100. Rohrer BR, Guo Y, Kunchithapautham K, Gilkeson GS. Eliminating complement factor d reduces photoreceptor susceptibility to light-induced damage. Investig Ophthalmol Vis Sci. (2007) 48:5282–9. doi: 10.1167/iovs.07-0282
101. Woodell A, Coughlin B, Kunchithapautham K, Casey S, Williamson T, Ferrell WD, et al. Alternative complement pathway deficiency ameliorates chronic smoke-induced functional and morphological ocular injury. PLoS ONE. (2013) 8:e67894. doi: 10.1371/journal.pone.0067894
102. Katschke KJJr, Xi H, Cox C, Truong T, Malato Y, Lee WP, et al. Classical and alternative complement activation on photoreceptor outer segments drives monocyte-dependent retinal atrophy. Sci Rep. (2018) 8:7348. doi: 10.1038/s41598-018-30162-w
103. Mullins RF, Aptsiauri N, Hageman GS. Structure and composition of drusen associated with glomerulonephritis: implications for the role of complement activation in drusen biogenesis. Eye. (2001) 15:390–5. doi: 10.1038/eye.2001.142
104. Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci USA. (2006) 103:2328–33. doi: 10.1073/pnas.0408835103
105. Inana G, Murat C, An W, Yao X, Harris IR, Cao J. RPE phagocytic function declines in age-related macular degeneration and is rescued by human umbilical tissue derived cells. J Transl Med. (2018) 16:63. doi: 10.1186/s12967-018-1434-6
106. Lakkaraju A, Toops KA, Xu J. Should I Stay or Should I Go? Trafficking of Sub-Lytic MAC in the retinal pigment epithelium. in: Ash JD, Grimm C, Hollyfield JG, Anderson RE, LaVail MM, Bowes Rickman C, editors. Retinal Degenerative Diseases. New York, NY: Springer New York. (2014). p. 267–74.
107. Collier RJ, Wang Y, Smith SS, Martin E, Ornberg R, Rhoades K, et al. Complement deposition and microglial activation in the outer retina in light-induced retinopathy: inhibition by a 5-HT1A agonist. Investig Ophthalmol Vis Sci. (2011) 52:8108–16. doi: 10.1167/iovs.10-6418
108. Veerhuis R, Nielsen HM, Tenner AJ. Complement in the brain. Mol Immunol. (2011) 48:1592–603. doi: 10.1016/j.molimm.2011.04.003
109. Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. (2010) 129:154–69. doi: 10.1111/j.1365-2567.2009.03225.x
110. Smith ME. Phagocytic properties of microglia in vitro: implications for a role in multiple sclerosis and EAE. Microsc Res Techn. (2001) 54:81–94. doi: 10.1002/jemt.1123
111. Fitzgerald KC, Kim K, Smith MD, Aston SA, Fioravante N, Rothman AM, et al. Early complement genes are associated with visual system degeneration in multiple sclerosis. Brain. (2019) 142:2722–36. doi: 10.1093/brain/awz188
112. Jin J, Smith MD, Kersbergen CJ, Kam TI, Viswanathan M, Martin K, et al. Glial pathology and retinal neurotoxicity in the anterior visual pathway in experimental autoimmune encephalomyelitis. Acta Neuropathol Commun. (2019) 7:125. doi: 10.1186/s40478-019-0767-6
113. Hu X, Tomlinson S, Barnum SR. Targeted inhibition of complement using complement receptor 2-conjugated inhibitors attenuates EAE. Neurosci Lett. (2012) 531:35–9. doi: 10.1016/j.neulet.2012.10.012
114. Guerrero BL, Sicotte NL. Microglia in multiple sclerosis: friend or foe? Front Immunol. (2020) 11:374. doi: 10.3389/fimmu.2020.00374
115. Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, Lassmann H. Loss of ‘homeostatic' microglia and patterns of their activation in active multiple sclerosis. Brain. (2017) 140:1900–13. doi: 10.1093/brain/awx113
116. Lassmann H. Pathology of inflammatory diseases of the nervous system: human disease versus animal models. Glia. (2020) 68:830–44. doi: 10.1002/glia.23726
117. Boven LA, Van Meurs M, Van Zwam M, Wierenga-Wolf A, Hintzen RQ, Boot RG, et al. Myelin-laden macrophages are anti-inflammatory, consistent with foam cells in multiple sclerosis. Brain. (2005) 129:517–26. doi: 10.1093/brain/awh707
118. Miron VE, Boyd A, Zhao J-W, Yuen TJ, Ruckh JM, Shadrach JL, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. (2013) 16:1211–8. doi: 10.1038/nn.3469
119. Foroozan R, Buono LM, Savino PJ, Sergott RC. Acute demyelinating optic neuritis. Curr Opin Ophthalmol. (2002) 13:375–80. doi: 10.1097/00055735-200212000-00006
120. Kawachi I. Clinical characteristics of autoimmune optic neuritis. Clin Exp Neuroimmunol. (2017) 8:8–16. doi: 10.1111/cen3.12354
121. Tassoni A, Farkhondeh V, Itoh Y, Itoh N, Sofroniew MV, Voskuhl RR. The astrocyte transcriptome in EAE optic neuritis shows complement activation and reveals a sex difference in astrocytic C3 expression. Sci Rep. (2019) 9:10010. doi: 10.1038/s41598-019-46232-6
122. Bennett JL, Owens GP. Neuromyelitis optica: deciphering a complex immune-mediated astrocytopathy. J Neuro Ophthalmol. (2017) 37:291–9. doi: 10.1097/WNO.0000000000000508
123. Hendrickx DAE, Schuurman KG, van Draanen M, Hamann J, Huitinga I. Enhanced uptake of multiple sclerosis-derived myelin by THP-1 macrophages and primary human microglia. J Neuroinflamm. (2014) 11:64–64. doi: 10.1186/1742-2094-11-64
124. Rus HG, Niculescu F, Shin ML. Sublytic complement attack induces cell cycle in oligodendrocytes. J Immunol. (1996) 156:4892.
125. Wood A, Wing MG, Benham CD, Compston DA. Specific induction of intracellular calcium oscillations by complement membrane attack on oligodendroglia. J Neurosci. (1993) 13:3319–32. doi: 10.1523/JNEUROSCI.13-08-03319.1993
126. Plemel JR, Manesh SB, Sparling JS, Tetzlaff W. Myelin inhibits oligodendroglial maturation and regulates oligodendrocytic transcription factor expression. Glia. (2013) 61:1471–87. doi: 10.1002/glia.22535
127. Thallmair M, Metz GAS, Z'Graggen WJ, Raineteau O, Kartje GL, Schwab ME. Neurite growth inhibitors restrict plasticity and functional recovery following corticospinal tract lesions. Nat Neurosci. (1998) 1:124–31. doi: 10.1038/373
128. Hepworth L, Rowe F, Walker M, Rockliffe J, Noonan C, Howard C, et al. Post-stroke visual impairment: a systematic literature review of types and recovery of visual conditions. Ophthalmol Res. (2016) 5:1–43. doi: 10.9734/OR/2016/21767
129. Yamashita T, Miki A. Visual field defects and retinal changes due to cortical disorders. Neuro Ophthalmol Japan. (2016) 33:351–63. doi: 10.11476/shinkeiganka.33.351
130. Magnin E, Chamard L, Vuillier F, Tatu L, Berger E. Can chronic remote cortical hypoperfusion induced by thalamic infarction cause damage of tracts passing through those hypoperfused regions? Front Neurol. (2013) 4:156. doi: 10.3389/fneur.2013.00156
131. Kumral E, Evyapan D, Kutluhan S. Pure thalamic infarctions: clinical findings. J Stroke Cerebrovasc Dis. (2000) 9:287–97. doi: 10.1053/jscd.2000.18741
132. Armstrong RA. Visual problems associated with traumatic brain injury. Clin Exp Optom. (2018) 101:716–26. doi: 10.1111/cxo.12670
133. Maxwell WL, Pennington K, MacKinnon MA, Smith DH, McIntosh TK, Wilson JTL, et al. Differential responses in three thalamic nuclei in moderately disabled, severely disabled and vegetative patients after blunt head injury. Brain. (2004) 127:2470–8. doi: 10.1093/brain/awh294
134. Gilmore CS, Camchong J, Davenport ND, Nelson NW, Kardon RH, Lim KO, et al. Deficits in visual system functional connectivity after blast-related Mild TBI are associated with injury severity and executive dysfunction. Brain Behav. (2016) 6:1–19. doi: 10.1002/brb3.454
135. Vien L, Dalporto C, Yang D. Retrograde degeneration of retinal ganglion cells secondary to head trauma. Optometr Vis Sci. (2017) 94:125–34. doi: 10.1097/OPX.0000000000000899
136. Petras JM, Bauman RA, Elsayed NM. Visual system degeneration induced by blast overpressure. Toxicology. (1997) 121:41–9. doi: 10.1016/S0300-483X(97)03654-8
137. Ding Y, Yao B, Lai Q, McAllister JP. Impaired motor learning and diffuse axonal damage in motor and visual systems of the rat following traumatic brain injury. Neurol Res. (2001) 23:193–202. doi: 10.1179/016164101101198334
138. Evanson NK, Guilhaume-Correa F, Herman JP, Goodman MD. Optic tract injury after closed head traumatic brain injury in mice: a model of indirect traumatic optic neuropathy. PLoS ONE. (2018) 13:e0197346. doi: 10.1371/journal.pone.0197346
139. Glushakov AO, Glushakova OY, Korol TY, Acosta SA, Borlongan CV, Valadka AB, et al. Chronic upregulation of cleaved-caspase-3 associated with chronic myelin pathology and microvascular reorganization in the thalamus after traumatic brain injury in rats. Int J Mol Sci. (2018) 19:3151. doi: 10.3390/ijms19103151
140. Patel VC, Jurgens CWD, Krahe TE, Povlishock JT. Adaptive reorganization of retinogeniculate axon terminals in dorsal lateral geniculate nucleus following experimental mild traumatic brain injury. Exp Neurol. (2017) 289:85–95. doi: 10.1016/j.expneurol.2016.12.012
141. Urich E, Gutcher I, Prinz M, Becher B. Autoantibody-mediated demyelination depends on complement activation but not activatory Fc-receptors. Proc Natl Acad Sci USA. (2006) 103:18697–702. doi: 10.1073/pnas.0607283103
142. Clark SJ, Schmidt CQ, White AM, Hakobyan S, Morgan BP, Bishop PN. Identification of factor h-like protein 1 as the predominant complement regulator in Bruch's Membrane: Implications for age-related macular degeneration. J Immunol. (2014) 193:4962–70. doi: 10.4049/jimmunol.1401613
143. Poor SH, Qiu Y, Fassbender ES, Shen S, Woolfenden A, Delpero A, et al. Reliability of the mouse model of choroidal neovascularization induced by laser photocoagulation. Investig Ophthalmol Vis Sci. (2014) 55:6525–34. doi: 10.1167/iovs.14-15067
144. Woodell A, Jones BW, Williamson T, Schnabolk G, Tomlinson S, Atkinson C, et al. A targeted inhibitor of the alternative complement pathway accelerates recovery from smoke-induced ocular injury. Investig Ophthalmol Vis Sci. (2016) 57:1728–37. doi: 10.1167/iovs.15-18471
145. Sullivan RKP, WoldeMussie E, Pow DV. Dendritic and synaptic plasticity of neurons in the human age-related macular degeneration retina. Investig Ophthalmol Vis Sci. (2007) 48:2782–91. doi: 10.1167/iovs.06-1283
146. Yehoshua Z, Alexandre De Amorim Garcia Filho C, Nunes RP, Gregori G, Penha FM, Moshfeghi AA, et al. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: The COMPLETE study. Ophthalmology. (2014) 121:693–701. doi: 10.1016/j.ophtha.2013.09.044
147. Holz FG, Sadda SR, Busbee B, Chew EY, Mitchell P, Tufail A, et al. Efficacy and Safety of lampalizumab for geographic atrophy due to age-related macular degeneration: chroma and spectri phase 3 randomized clinical trials. JAMA Ophthalmol. (2018) 136:666–77. doi: 10.1001/jamaophthalmol.2018.1544
148. Shahulhameed S, Vishwakarma S, Chhablani J, Tyagi M, Pappuru RR, Jakati S, et al. A systematic investigation on complement pathway activation in diabetic retinopathy. Front Immunol. (2020) 11:154. doi: 10.3389/fimmu.2020.00154
149. Tao XY, Zheng SJ, Lei B. Activated complement classical pathway in a murine model of oxygen-induced retinopathy. Int J Ophthalmol. (2015) 8:17–22. doi: 10.3980/j.issn.2222-3959.2015.01.03
150. Rathi S, Jalali S, Patnaik S, Shahulhameed S, Musada GR, Balakrishnan D, et al. Abnormal complement activation and inflammation in the pathogenesis of retinopathy of prematurity. Front Immunol. (2017) 8:1868. doi: 10.3389/fimmu.2017.01868
151. Pittock SJ, Berthele A, Fujihara K, Kim HJ, Levy M, Palace J, et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. New Engl J Med. (2019) 381:614–25. doi: 10.1056/NEJMoa1900866
152. Nebbioso M, Lambiase A, Cerini A, Limoli PG, La Cava M, Greco A. Therapeutic approaches with intravitreal injections in geographic atrophy secondary to age-related macular degeneration: current drugs and potential molecules. Int J Mol Sci. (2019) 20:1693. doi: 10.3390/ijms20071693
153. Schnabolk G, Parsons N, Obert E, Annamalai B, Nasarre C, Tomlinson S, et al. Delivery of CR2-fH using AAV vector therapy as treatment strategy in the mouse model of choroidal neovascularization. Mol Ther Methods Clin Dev. (2018) 9:1–11. doi: 10.1016/j.omtm.2017.11.003
154. Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. (2014) 124:2804–11. doi: 10.1182/blood-2014-02-522128
155. Raina R, Krishnappa V, Blaha T, Kann T, Hein W, Burke L, et al. Atypical hemolytic-uremic syndrome: an update on pathophysiology, diagnosis, and treatment. Ther Apheresis Dialysis. (2019) 23:4–21. doi: 10.1111/1744-9987.12763
156. Yaspan BL, Williams DF, Holz FG, Regillo CD, Li Z, Dressen A, et al. Targeting factor D of the alternative complement pathway reduces geographic atrophy progression secondary to age-related macular degeneration. Sci Transl Med. (2017) 9:eaaf1443. doi: 10.1126/scitranslmed.aaf1443
Keywords: complement, synapse, phagocytosis, retina, lateral geniculate nucleus, multiple sclerosis, age-related macular degeneration, glaucoma
Citation: Borucki DM, Toutonji A, Couch C, Mallah K, Rohrer B and Tomlinson S (2020) Complement-Mediated Microglial Phagocytosis and Pathological Changes in the Development and Degeneration of the Visual System. Front. Immunol. 11:566892. doi: 10.3389/fimmu.2020.566892
Received: 28 May 2020; Accepted: 17 August 2020;
Published: 24 September 2020.
Edited by:
Marcela Pekna, University of Gothenburg, SwedenReviewed by:
Frederike Cosima Oertel, University of California, San Francisco, United StatesBenjamin Knier, Technical University of Munich, Germany
Copyright © 2020 Borucki, Toutonji, Couch, Mallah, Rohrer and Tomlinson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen Tomlinson, dG9tbGluc3MmI3gwMDA0MDttdXNjLmVkdQ==