 Laura Passerini
Laura Passerini Silvia Gregori
Silvia Gregori- Mechanisms of Peripheral Tolerance Unit, San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), IRCCS San Raffaele Scientific Institute, Milan, Italy
The development of novel approaches to control unwanted immune responses represents an ambitious goal in the management of a number of clinical conditions, including autoimmunity, autoinflammatory diseases, allergies and replacement therapies, in which the T cell response to self or non-harmful antigens threatens the physiological function of tissues and organs. Current treatments for these conditions rely on the use of non-specific immunosuppressive agents and supportive therapies, which may efficiently dampen inflammation and compensate for organ dysfunction, but they require lifelong treatments not devoid of side effects. These limitations induced researchers to undertake the development of definitive and specific solutions to these disorders: the underlying principle of the novel approaches relies on the idea that empowering the tolerogenic arm of the immune system would restore the immune homeostasis and control the disease. Researchers effort resulted in the development of cell-free strategies, including gene vaccination, protein-based approaches and nanoparticles, and an increasing number of clinical trials tested the ability of adoptive transfer of regulatory cells, including T and myeloid cells. Here we will provide an overview of the most promising approaches currently under development, and we will discuss their potential advantages and limitations. The field is teaching us that the success of these strategies depends primarily on our ability to dampen antigen-specific responses without impairing protective immunity, and to manipulate directly or indirectly the immunomodulatory properties of antigen presenting cells, the ultimate in vivo mediators of tolerance.
Introduction
The identification of novel approaches designed to selectively control antigen(Ag)-specific effector T (Teff) cell responses and promote or restore tolerance in T cell mediated diseases is an unsolved issue in the management of autoimmune diseases in humans. On this line, a new version of vaccination, also called “inverse vaccination,” aims at inducing or restoring an immunological state of unresponsiveness, either toward foreign Ags (e.g., protein therapeutics, allergens, or transgenes) or autoAgs (1). The overall goal of inverse vaccination strategies is to dampen the adverse response, through deletion, inhibition or deviation of Ag-specific Teff cells, and to support the induction and/or expansion of Ag-specific T regulatory cells (Tregs). Tregs are recognized as a cell population responsible for induction and maintenance of immune tolerance. The best characterized subsets are the Forkhead box P3 expressing Tregs (FOXP3+ Tregs) (2) and the IL-10-producing type 1 regulatory (Tr1) cells (3).
A number of different strategies have been proposed as inverse vaccination: (i) cell-free based approaches, including gene vaccination and protein or peptide delivery; (ii) vehicle approaches, to deliver Ags by means of apoptotic cells, liposomes, or nanoparticles; (iii) cell-based approaches, aimed at providing specialized cells to reinforce the regulatory arm of the immune system. This Review aims to provide an overview of the most promising approaches currently under development and clinical testing (Figure 1 and Table 1) and their potential advantages and limitations.
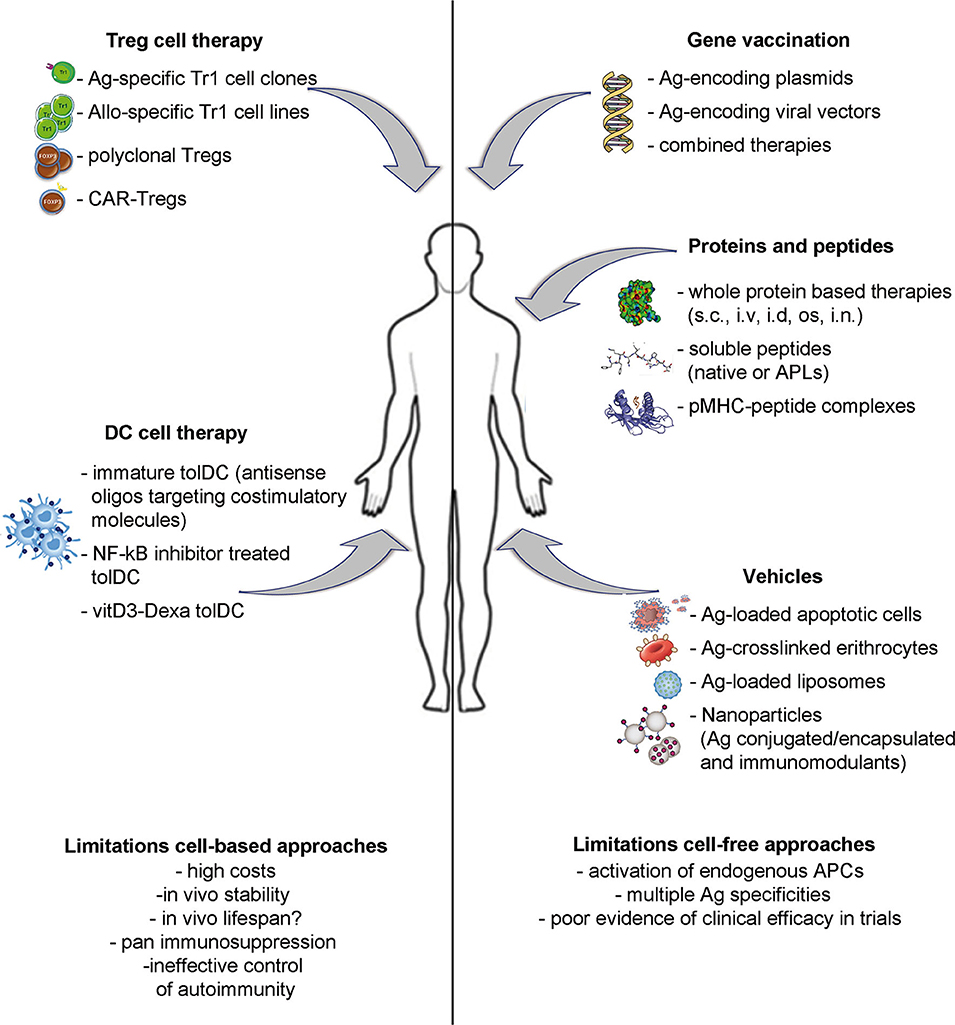
Figure 1. Strategies to induce Ag-specific tolerance in T cell mediated diseases. Approaches under development include: inverse vaccination with autoantigen-encoding DNA or viral vectors; in vivo administration of whole Ags, unmodified peptides or altered peptide ligads (APLs); autoantigen-loaded vehicles; transfer of polyclonal or Ag-specific Tregs or of tolerogenic DC loaded with disease-relevant Ags.
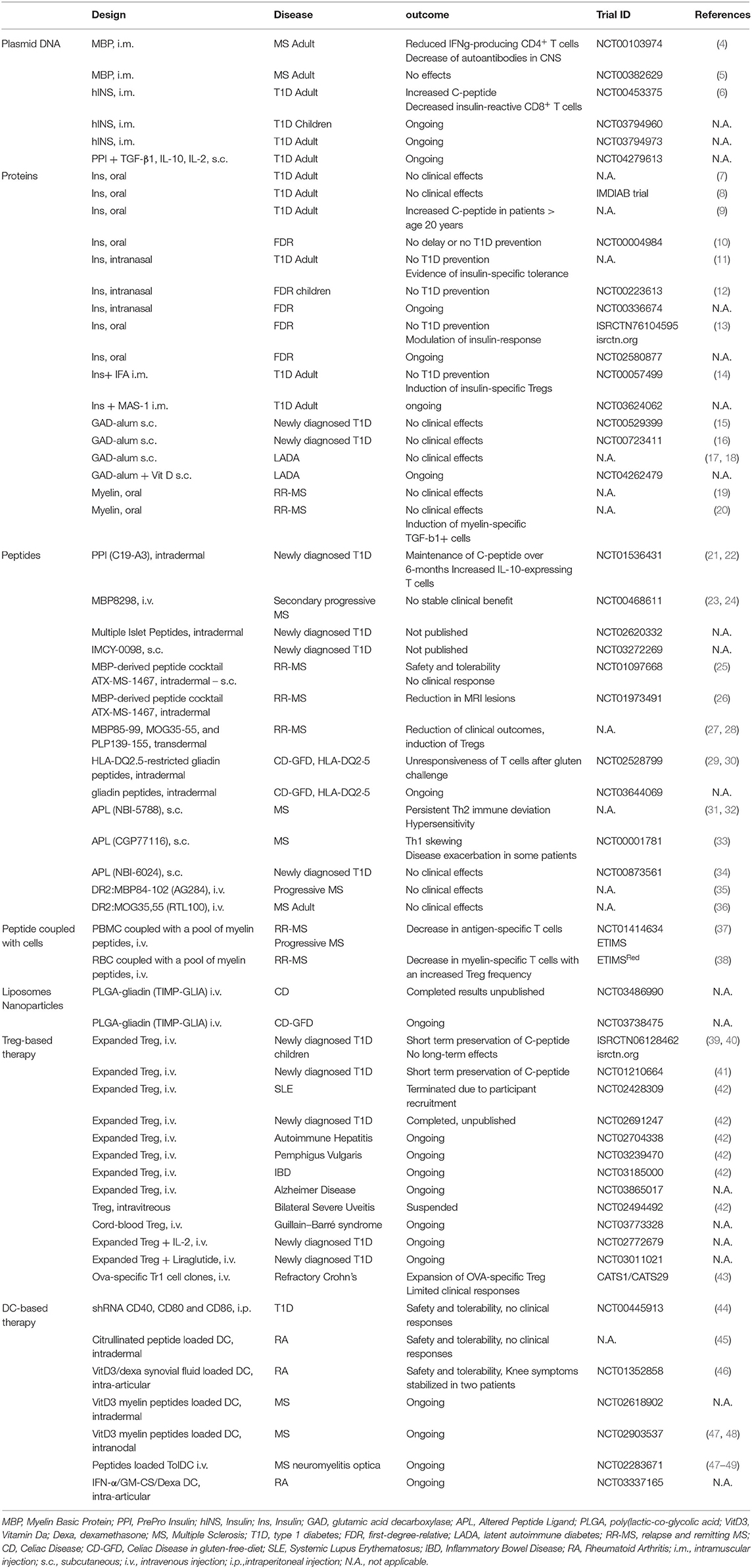
Table 1. Clinical trials using antigen-specific approaches.
Cell Free Strategies
Inverse gene vaccination strategies aim at the induction of tolerance to a relevant Ag by means of transient expression of whole proteins or epitopes from DNA or RNA vectors in the absence of pro-inflammatory stimuli. Once injected, the coding sequence needs to enter the cytoplasm of the target cells, and, in case of DNA vectors, translocate to the nucleus for transcription, followed by translation in the cytoplasm and presentation of the Ag in the context of HLA class I molecules. The balance between an inflammatory immune response and the induction of tolerance can be controlled by several factors, including the route of administration, the target tissue, and the vector design. For example, direct transfection or transduction of professional antigen presenting cells (APCs) may result in efficient presentation to Ag-specific CD8+ T cells (50) or, as a consequence of cell death or tissue damage, the Ag may be taken up by professional APCs, processed as exogenous Ags, and presented to Ag-specific CD4+ T cells in the context of HLA class II (51, 52). On the same line, the vector backbone itself may contain immunostimulatory sequences, which could impact on gene expression, intracellular localization of the product and APCs activation via TLRs (53). Hence, the activation status of APCs is pivotal for the final outcome of the response: protection vs. tolerance. Two strategies for the delivery of the Ag-coding sequences have been used in preclinical studies, plasmids and viral vectors [reviewed in (42)].
Plasmid DNA
Intramuscular plasmid DNA vaccination has been the most studied, likely due to the short persistence in the host, the low immunogenicity, and the low costs of plasmid production. This strategy was first tested in experimental autoimmune encephalitis (EAE), the murine model of multiple sclerosis (MS): immunization with plasmid encoding for an EAE epitope of myelin basic protein (MPB) prevented disease development, via T helper (Th)2 cell skewing of the Ag-specific T cell response (54). The initial preclinical studies led to clinical testing of this strategy not only in MS (4, 5), but also in Type 1 Diabetes (T1D) (6) (Table 1). A DNA vaccine (BHT-3009, Bayhill Therapeutics) containing full-length sequence of the human MBP was tested in two trials in MS patients (4, 5). In the first trial no severe adverse events were reported. Results indicated a trend of lower lesion activity, reduced IFNγ-producing CD4+ T cells up to 50 weeks after initiation, and a decrease of autoantibodies in the cerebrospinal fluid (4). Nonetheless, in the second trial the intervention did not result in any differences in the time to first relapse, rate of relapses per year, disability progression, and the treatment showed a deleterious effect at high vaccine dose, likely due to a greater percentage of immunostimulatory CpG motifs in the DNA plasmid (5). A similar approach was tested in T1D with a bacterial plasmid encoding for pro-insulin [BHT-3021, Bayhill Therapeutics; (6)]. No serious adverse events were observed, and the treatment resulted in improvement of endogenous insulin production, measured as 28% increase in C-peptide, and decreased frequency of proinsulin-reactive CD8+ T cells (6). Despite encouraging results, insulin requirements did not change substantially, and demonstration of efficacy is still pending. The same product (under the name TOL-3021, Tolerion Inc.) is going to be tested in two distinct phase II trials in T1D children and adults (NCT03794960 and NCT03794973). On the same line, DNA vaccines based on oral administration of recombinant live attenuated bacteria expressing diabetes autoAgs in combination with inhibitory cytokines, such as transforming growth factor (TGF-β1) and IL-10 or with anti-CD3 mAb have also been tested to prevent or revert the onset of diabetes in non obese diabetic (NOD) mice, showing induction of Tregs (both FOXP3-expressing and Tr1 cells) and suppression of autoimmunity (55, 56). A phase I trial will test the safety of subcutaneous injection of a plasmid co-encoding for T1D Ag and adjuvant cytokines (NNC0361-0041: plasmid encoding pre-proinsulin, TGF-β1, IL-10, and IL-2, Novo Nordisk A/S, NCT04279613).
Overall, thus far the plasmid DNA delivery approach showed the ability to skew the immune response, with no evidence of stable tolerance induction. The combination with immunomodulatory cytokines, which should sustain Ag-specific Treg induction, is expected to boost the induction of active tolerance. Results of ongoing clinical trials will shed light on the valuability of this approach.
Viral Vectors
As alternative to plasmids, the use of viral vectors allows to restrict expression of the autoAg to specific tissues and avoid unwanted expression in activated APCs. In this context, the liver is an ideal target, due to its intrinsic tolerogenic properties [reviewed in (57)]. Two types of viral vectors have been used to target gene expression specifically to hepatocytes: the recombinant adeno associated vectors (AAV) and the lentiviral vectors (LVs). Although widely used as vector systems for liver directed in vivo gene therapy, few groups explored the use of AAV to induce tolerance to autoAgs in autoimmune diseases. Liver gene therapy with an AAV vector encoding for the full sequence of myelin oligodendrocyte glycoprotein (MOG) prevented development of and reversed preexisting EAE via the induction/expansion of Ag-specific FoxP3+ Tregs (58). Earlier studies of intramuscular injection in NOD mice of AAV encoding for glutamic acid decarboxylase (GAD) peptides prevented the development of overt diabetes in NOD mice via skewing of Teff cells to Th2 responses, but those studies were not further developed and active tolerance was not demonstrated (59).
The use of LVs to induce Ag-specific tolerance upon liver targeting was also investigated in NOD mice. Intraveneous injection of LV encoding the insulin B chain (InsB) 9–23 epitope led to specific expression of the autoAg in hepatocytes, thanks to the use of tissue-specific promoter and concomitant de-targeting of Ag expression in professional APCs by miR142 target sequences. This treatment prevented diabetes development by induction of Ag-specific FoxP3+ Tregs. Although highly efficient in prevention, the control of overt disease required a combination therapy with anti-CD3 mAb, to block Teff cells from destroying the target organ (60).
Gene vaccination strategies present several advantages in terms of cost-efficient production and long shelf life for plasmid-based vaccines and available (although expensive) large scale and clinical grade protocols for LV production. However, administration of the therapeutic products invariably leads to deleterious activation of professional APCs and the innate immune system (61) and may not be sufficient to counteract the burden of expanded Teff cells with multiple Ag-specificity. The future of these approaches points at combined therapies to overcome these hurdles.
Protein Delivery Approaches
The direct administration of autoAgs in non-inflammatory conditions to induce tolerance in T cell mediated diseases has been widely investigated, especially in EAE and NOD pre-clinical models (62, 63). The underlying idea is that repetitive administration or exposure to large amounts of protein Ag, as whole protein, native or altered peptide alone or combined to carrier complexes, in the absence of pro-inflammatory adjuvants, will favor the deletion or clonal anergy of autoreactive Teff cells and the induction of Ag-specific Tregs, via uptake and presentation of the Ag by endogenous tolerogenic APCs (62, 63). In this context the route of administration is a key issue: the positive results obtained in allergic diseases by oral, intranasal and subcutaneous administration of allergens [reviewed in (64)] led to parallel attempts in autoimmune diseases.
Due to the early recognition of insulin epitopes as antigenic targets in NOD mice (65), insulin was the first Ag investigated for the development of protein-based immunotherapy of T1D. Initial promising results in murine models (66–68) led to the clinical testing of oral (7–10) and intranasal insulin [(11, 12), and INITII, NCT00336674], as tolerizing protocols in subjects at risk to develop the disease [(10, 13), NCT00336674 and TN20, NCT02580877] or in recent onset T1D patients (7–9, 11) (Table 1). Although results of few trials are still unpublished (NCT00336674; NCT02580877), thus far, none of them resulted in preserved insulin secretion in T1D patients. Inverse vaccination with InsB has also been tested as intramuscolar injection with Incomplete Freund's Adjuvant [IBS-VS01, (14)]: despite induction of InsB-specific Tregs, C-peptide levels were unaltered by the treatment. A new formulation of the vaccine in combination with MAS-1, an emulsion-based adjuvant, known to promote Th2 responses (69), is currently being tested in a Phase I study (MER3101, NCT03624062). Several trials betted on GAD65 as key Ag and on different routes: Dyamid, a GAD-Alum vaccine, was administered subcutaneous in recent onset T1D (15, 16) and in adults with latent autoimmune diabetes (LADA) (17) without achievement of clinically desirable results (18). Combination of Dyamid with vitamin D in LADA is currently being tested in a Phase II trial (NCT04262479). Similarly, attempts of oral tolerization with myelin Ags in MS, which date back to the early 90's, showed modulation of Ag-specific immune response, but no evidence of efficacy (19, 20).
Peptide Delivery Approaches
In parallel to whole protein-based approaches, administration of peptides derived from disease-causing Ags was also tested both in T1D and MS (Table 1). Intradermal administration of a HLA-DR4-restricted native peptide derived from proinsulin (C19-A3) allowed maintenance of C-peptide levels in new-onset T1D over a 6-month treatment and resulted in increased frequencies of IL-10-expressing T cells [MonoPepT1De, (21, 22)]. The HLA-DR2-restricted immunodominant synthetic peptide MBP8298, containing the MBP immune-dominant epitope 85–96, was extensively tested in patients with MS, without stable clinical benefit (23, 24).
Overall these initial peptide-based approaches resulted in modulation of Ag-specific immune responses, but poor clinically relevant results, likely because autoimmune diseases are not caused by single T cell clones, as a result of epitope spreading (70). Given this phenomenon, recent studies have pointed at mixture of peptides from multiple autoAgs for the modulation of autoimmune diseases: in the context of T1D the MultiPepT1De (NCT02620332) and the IMCY-0098 trial (NCT03272269) have been completed, although results are still unpublished. The same approach was tested in MS: following promising results in humanized mice (25), the MBP-derived peptide cocktail ATX-MS-1467 (Aptiope (https://apitope.com/multiple-sclerosis/) was tested for safety and efficacy in relapsing MS patients. Results showed association of treatment with reduction in Magnetic Resonance Imaging (MRI) lesions (26). Moreover, transdermal application of a mixture of 3 myelin peptides showed significant effect in reducing the MRI and clinical outcomes (27) via the induction of Tregs (28). Similarly, NexVax2, composed of three HLA-DQ2.5-restricted immunodominant gliadin peptides, NPL001, NPL002, and NPL003, has been tested in a phase I clinical trial in Celiac Disease (CD) patients. Despite some gluten-related gastrointestinal side effects, the treatment was safe and well tolerated (29, 30). In treated patients functional unresponsiveness of T cells after gluten challenge was observed, indicating induction of tolerance. Currently, a phase II quadruple blind clinical trial (NCT03644069) is underway (71).
Modification of native peptides alters the way peptides interact with TCR and, therefore, influences subsequent T cell activation and T cell fate. Increasing knowledge of both MHC binding registers and TCR interacting residues of peptides allowed the development of altered peptide ligands (APL), with the aim of favoring the expansion and/or induction of Tregs upon peptide recognition [reviewed in (72, 73)]. Following studies in murine disease models, showing that specific APLs were capable of eliciting cytokine release and affecting T cell polarization (74, 75), APLs were tested in vivo in autoimmune diseases. Indeed, two altered peptides of MBP83-99 have already been tested in MS. NBI-5788 (Neurocrine Biosciences Inc), in which L-amino acids were changed to D-amino acids at positions 83, 84, 89, 91, was known to stimulate Th2-type responses in MS patients' PBMC (76). Clinical testing confirmed Th2 immune deviation in treated patients (31), but several patients developed hypersensitivity and antibodies that cross-reacted with native MBP83–99 peptide (32). The second MBP-derived APL tested is CGP77116 (Ala D-amino acids at positions 83, 84, 89, 91). It caused a Th1 skewing of CD4+ T cells cross-reacting with the native peptides, thus raising issues on the APL design (33). On the same line, the use of an insulin β-chain-derived APL (NBI-6024) did not improve or maintain beta cell function in recent onset T1D patients (34). Despite the promising results obtained in murine models and the improvements in the development of algorithms for peptide-HLA-binding prediction, thus far clinical trials using APLs were unsuccessful. The design of APLs currently represents a major caveat: the ability to predict the consequence of peptide binding to HLA molecules on APCs or Ag-receptors on T and B cells is still limited and it needs to be empirically determined for each peptide.
Regardless of the type, origin, number of Ags or the route of administration used, the outcome of the administration of whole proteins or peptides is strictly dependent on the activation status of the host's APCs the Ag is binding to. APCs in vivo exist in several different flavors (77), expressing different ranges of activatory or inhibitory cell surface molecules and soluble mediators, which play a critical role on the outcome of the cognate T-APC interaction. Peptide-based therapy showed immunological effects, including increased frequency of Treg cells and of IL-10, suggesting modulation of pathogenic responses. The beneficial effects, although observed only short-term after treatment, are compatible with immune tolerance, thus suggesting that endogenous APCs function was modulated, likely indirectly by a bystander suppression mechanism. We believe that to better sustain long term tolerance protein or peptide based approaches could benefit from strategies designed to keep APCs in check.
One of the strategies tested to address this limitation is the injection of soluble peptide-MHC (pMHC) complexes to target directly T cells. Vaccination with pMHC complexes is predicted to induce tolerance either by deletion of naive and memory Teff cells that recognize the self-peptide, or by induction of Tregs (78). This strategy was applied in preclinical models of Myastenia Gravis (79), in EAE (80–82) and in NOD mice (83) resulting in reduction of T cell responsiveness. Phase I trials were performed in MS patients (35, 36), but further testing is necessary to assess clinical efficacy.
The experience with administration of soluble Ags was further developed using different types of vehicles designed to deliver the Ag specifically to steady-state or tolerogenic APCs, as outlined below.
Vehicle Approaches to Deliver Antigens
A number of different approaches to deliver Ag specifically to APCs in vivo have been investigated in pre-clinical studies and some of them have been translated into clinical application.
Peptide Coupled to Cells
The first approach tested was the intravenous administration of antigenic peptides cross-linked to peripheral blood or splenic leukocytes using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (ECDI), which promotes Ag coupling and induces cell apoptosis (Ag-SP) (84). Once injected in vivo, apoptotic Ag-SP are taken up by APCs and trigger the production and secretion of IL-10 and TGF-β and the up-regulation of PD-L1, leading to T cell anergy and apoptosis of pathogenic T cells and Treg induction (84). The efficacy of Ag-SP has been demonstrated in pre-clinical models, including EAE and NOD mice [reviewed in (84)]. The translation to the clinic of this approach was the administration of autologous peripheral blood cells coupled with seven MS-related peptides to MS patients in a Phase I-II clinical trial (ETIMS) (Table 1). Results demonstrated the feasibility, safety and tolerability of the treatment, and a decrease in Ag-specific T cell responses (37).
An alternative approach to deliver Ags to APCs in a tolerogenic manner is the administration of Ag-loaded erythrocytes, thus exploiting the natural tolerization mechanisms of dying red blood cells (85). To facilitate the binding of peptide Ags to erythrocytes, peptides were designed to contain the 12aa sequence ERY1 that binds to glycophorin A or sortase A on erythrocytes. Testing in EAE, in NOD and in transgenic mice demonstrated the deletion of Ag-specific T cells in vivo (86–88). Using this approach a phase Ib trial (ETIMSRed) has been completed; mechanistic studies demonstrated a reduction in myelin-specific T cell responses with an increased frequency of Tr1 and nTreg cells, thus pointing toward active induction of immune tolerance (38). Possible clinical translation of the erythrocyte binding technology is currently pursued by Anokion (www.anokion.com).
Liposomes and Nanoparticle
As an alternative, to mimic the features of apoptotic cells, liposomes containing phosphatidylserine have been developed and loaded with antigenic peptides. Injection of liposomes loaded with MS-related peptides reduced symptoms in the EAE model (89). Instead, phosphatidycholine liposomes loaded with Ag and NF-kB inhibitors reduced disease severity in a mouse model of arthritis (90). Poly-(lactic-co-glycolic acid) (PLGA) microspheres carrying anti-sense oligonucleotides for the costimulatory molecules CD40, CD80, and CD86 delivered to NOD mice prevented T1D development (91, 92). Notably, the Authors showed that the Ag was not required to elicit Ag-specific Tregs, since, upon microsphere administration, DC migrate from the site of injection to the pancreatic lymph nodes, where auto-Ags are captured and presented to T cells, thus leading to Ag-specific Treg induction (91). This approach is under development for the treatment of T1D (DiaVac. Inc, https://www.angelmd.co/en/startups/diavacsinc).
The discovery that polymeric biodegradable nanoparticles (NPs) could efficiently deliver molecules in vivo, prompted investigators to develop NPs suitable for tolerance induction. PLGA-NPs can encapsulate immune-modulatory agents, such as rapamycin, alone or in combination with peptide Ags. Once injected in vivo these NPs target DC, thus allowing Ag-presentation in a tolerogenic manner (93). Pre-clinical studies showed that in vivo delivery of PLGA-NPs containing MS-related peptide Ags prevents and treats EAE by up-regulating PD-L1 on APCs and inhibiting the production of pro-inflammatory cytokines by Ag-specific pathogenic T cells (93). PLGA particles encapsulating gliadin (TIMP-GLIA) were developed for application as a therapy for CD and tested in a Phase I clinical trial (NCT03486990). The results of this trial are yet to be published, and the Phase II trial is currently underway (NCT03738475).
The tolerogenic effects of NPs depend on size, which dictates their trafficking and biodistribution: (i) particles smaller than 6 nm drain to the blood; (ii) particles larger than 9 nm preferentially drain to lymphatics; (iii) particles in the range of 20–100 nm accumulate in liver sinusoidal endothelial cells (LSECs) or macrophages; (iv) particles from 100 to 200 nm can traffic to the spleen and liver; (v) particles from 200 nm to 5 μm accumulate in the spleen. Moreover, NPs biodistribution is also affected by the route of administration: intravenous injection targets APCs in the spleen and liver, whereas upon subcutaneous injection NPs are taken up by DC that accumulate in draining lymph nodes (94). The ability of LSECs to promote induction of FoxP3+ Tregs, prompted the development of NPs to deliver Ags to LSECs for autoimmune disease treatment (95). NP-based autoAg delivery to LSECs prevented the onset of clinical EAE and, in therapeutic settings, mice with already established EAE improved rapidly (95).
More recently, a further evolution on the NP approach to deliver Ag and promote tolerance was described by Santamaria et al. (96, 97). This approach consists on coating NPs with MHC class I or MHC class II molecules coupled with antigenic peptides (pMHC-coated NPs) (98). In pre-clinical models, the administration of pMHC-coated NPs promoted the differentiation of Ag-specific Tr1 cells and the conversion of Ag-specific Th1 cells into Tr1 cells, following massive expansion. Expanded Tr1 cells were activated by autologous APCs presenting the cognate Ag and induced bystander IL-10-mediated suppression (98). These pMHC-coated NPs (Navacims™) have been validated in different pre-clinical models of autoimmunity (96, 97) and are currently under clinical development.
The application of nanotechnology to advance treatment of autoimmunity is likely to undergo major development in coming years. Nanotechnology will create new materials for NP-related products. However, NPs are highly reactive, leading to their potentially harmful interaction with biological systems and the environment, thereby increasing the risk of toxicity. Detection of adverse effects is complex, since they depend on the route of administration, doses and size of NPs. NPs accumulate in the reticuloendothelial system and their long-term effects are not yet fully elucidated. Moreover, the small size of nanomaterial allows their penetrance into deeper areas of biological systems that are usually inaccessible to larger particles. Thus, due to the different properties of NPs, their application for therapeutic purposes, especially the long-term effect on the immune system, requires further attention and research (99, 100).
Cell-Based Approaches
Cell-based therapies are clinically attractive for promoting or restoring tolerance in T cell mediated diseases as they can theroretically control several inflammatory cells, including T and B lymphocytes, NK cells and APCs, leading to the control of unwanted immune responses. Therapies based on adoptive transfer of regulatory cells (T, macrophages, and DC) entered the clinical trial arena in the last years with the goal to investigate the safety and feasibility of the approach, and several studies are still ongoing.
Treg-Based Therapies
The increasing knowledge on the biology of Tregs, on their mode of action and their ability to control autoimmune responses when adoptively transferred in vivo in pre-clinical models of autoimmunity allowed the growth of a number of clinical trials to investigate the safety and feasibility of the approach (42, 101). The literature on Treg cell therapy is extensive and will not be reviewed here in depth. Tregs were first used in clinical trials to treat patients with graft vs. host disease (GvHD) after hematopoietic stem cell transplantation (HSCT). Results demonstrated that Tregs are safe, with some concern about the occurrence of mild to moderate infections (101). Treg therapy is currently applied to reduce dependency on immunosuppressive drugs in patients after organ transplantation (101, 102). In the context of autoimmune diseases both FOXP3+ Tregs and Tr1 cells have been tested in clinical trials (Table 1). The infusion of ex-vivo expanded polyclonal FOXP3+ Tregs in patients with recently diagnosed T1D showed improved beta-cell function and reduced exogenous insulin requirement only short-term (39–41). The limited efficacy of Treg-based immunotherapy in T1D may depend on the limited number of residual functional beta-cells at time of treatment, the inadequate availability of IL-2 in vivo (40), or, more importantly, on the lack of antigen-specificity of the infused Tregs. A number of clinical trials with expanded autologous Tregs are ongoing, have been closed, or have been completed but results have not been published yet [NCT02428309; NCT02494492; NCT02691247; NCT02704338; NCT03239470; NCT03185000; NCT03773328; NCT03865017; (42)].
Pre-clinical studies indeed showed that Ag-specificity may offer an advantage for Treg function compared to polyclonal Tregs (103). The first experience with Ag-specific Tregs was in Crohn's disease: ovalbumin-specific Tr1 cells (Ovasave®) expanded in vitro were infused in patients, who ingested ovalbumin to allow Treg activation and inhibitory function in the gut, with no side effects, but limited clinical effects (43). Beside the use of T cell clones, several other approaches have been investigated and applied to generate Ag-specific Tregs. The most advanced strategies were applied to the transplantation area: alloAg-specific Tregs can be generated using tolerogenic DC (104–106) or by engineering Tregs with a chimeric antigen receptor (CAR) recognizing HLA-A2 (107, 108). These approaches are currently under clinical investigation (NCT03198234; TX200, www.sangamo.com). Translation of the latter strategy to autoimmune settings is more challenging because (i) the Ags inducing the disease are often unknown; (ii) Tregs and pathogenic T cells are driven by different epitopes; and (iii) while disease progresses, epitope spreading occurs.
Results of the pioneer trials of adoptive Treg cell therapies in transplantation and T1D taught the field that transfer of Tregs alone may not be sufficient to control immune responses in the long-term, thus combined therapies with growth factors or repetitive Treg injections are currently under investigation. Based on the evidence that low doses of IL-2 can increase the endogenous pool of Tregs (109), the combination of a single infusion of autologous ex-vivo expanded polyclonal Tregs with IL-2 or with Liraglutide in patients with T1D is currently under clinical testing (NCT02772679 and NCT03011021).
Overall, Treg-based clinical trials demonstrated the safety and feasibility of the approach with some clinical benefit. However, several open issues remain to be solved specifically in the application of polyclonal ex-vivo expanded Tregs: (i) their potential to mediate pan immunosuppression in vivo, due to the phenomenon of bystander immune suppression; (ii) their intrinsic instability when exposed to strong inflammatory conditions in vivo, thereby the risk of pathogenic conversion and exacerbation of the disease; (iii) the overall impact of long-lasting Tregs on infections and malignancies (110).
DC-Based Approaches
It is now widely accepted that DC, either naturally arising or experimentally induced, play a critical role in the maintenance of tissue homeostasis and in promoting tolerance [reviewed in (111–113)], thus acting as regulatory cells. DC can acquire regulatory capacity upon treatment with immunosuppressive mediators, genetic manipulation or signals from other immune cells (114). DC with regulatory properties are generally indicated as tolerogenic DC (tolDC): they present Ags and prime Ag-specific T cells, while down-regulating the expression of costimulatory molecules and pro-inflammatory cytokines, and up-regulating the expression of inhibitory and/or modulatory receptors and anti-inflammatory cytokines. As a result, priming or activation of T cells by tolDC leads to induction of Ag-specific Tregs (114). On the other hand, DC sense environmental signals, which can impact their maturation and activation status and can modulate their microenvironment by release of soluble factors, thus indirectly impacting the outcome of Ag recognition by T cells.
A better understanding of the biology of tolDC and the development of protocols for the generation of tolDC in vitro, opened the possibility to translate their use as immunotherapy in clinical trials for immune-mediated diseases (115, 116). These therapies are not simple alternatives to Treg-based therapies, but they are complementary. Ex-vivo generated tolDC have the potential to induce, enhance, or restore Ag-specific tolerance in vivo since, once loaded with Ags, they act in an Ag-specific manner. TolDC can regulate pathogenic T cell responses via several mechanisms, including T cell deletion or inhibition, induction of T cell anergy, de novo Treg generation or expansion of pre-existing Tregs, and modulation of APCs (Figure 2). TolDC can delete Teff cells by inducing T cell apoptosis via Fas/FasL pathway. Furthermore, tolDC can inhibit Teff cell function either directly, via production of the enzyme indoleamine 2,3-dioxygenase (IDO), which degrades the amino acid tryptophan (L-Trp) causing starvation of pathogenic T cells (117), or indirectly, by activating pre-existing Tregs via interaction between CD80/CD86 and CTLA-4 to exert their suppressive function. TolDC can also promote the induction of T cell anergy into Teff cells via the secretion of anti-inflammatory cytokines, such as IL-10, or signals via inhibitory molecules, such as HLA-G and ILT3/4 (104, 118). Moreover, tolDC promote the expansion of pre-existing Tregs and de novo induction of both Tr1 cells and FOXP3+ Tregs, via the secretion of IL-10, TGF-β and active kynurenines, products of IDO-mediated L-Trp degradation (119). Finally, tolDC, via the expression and secretion of regulatory molecules, can also modulate APCs, rendering them pro-tolerogenic (e.g., modulation of resident macrophages into an M2 phenotype, or dampening the maturation of resident DC), a process that generates a self-sustaining tolerogenic microenvironment, which can promote long-term tolerance. Beside exerting their effect on immune cells, tolDC secrete several factors (e.g. pro-angiogenic cytokines) which promote tissue repairing and regeneration (Figure 2). Altogether, these properties rendered tolDC the cells of choice to restore tolerance in autoimmune diseases.
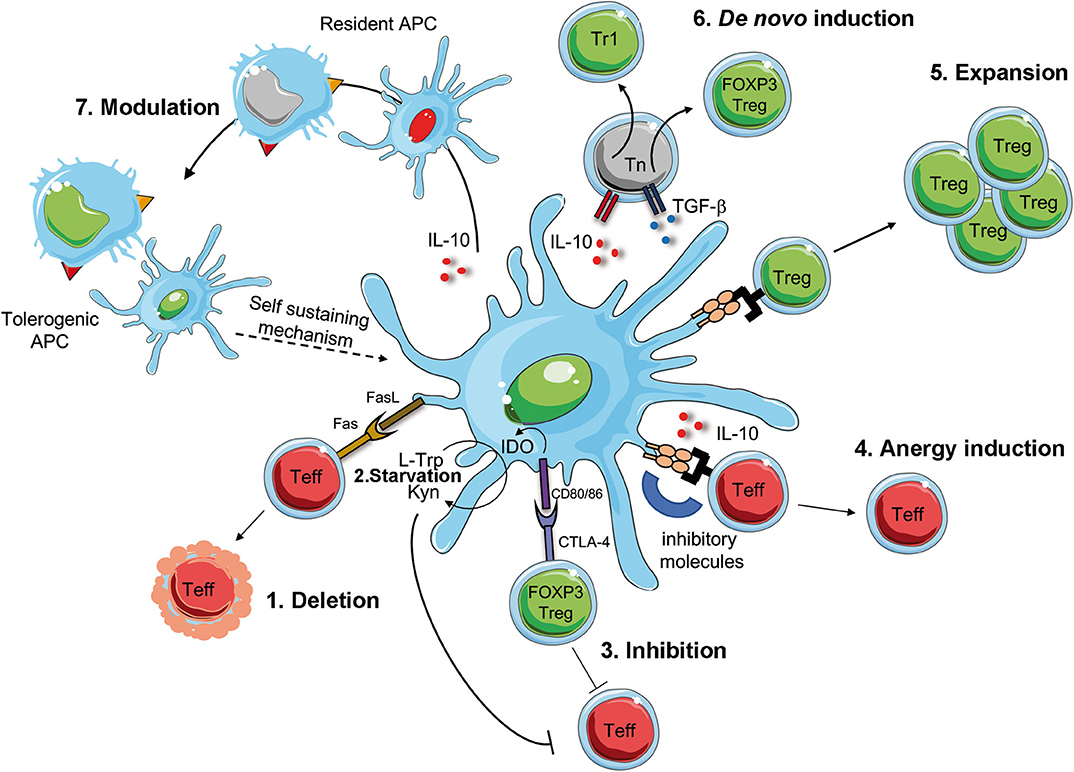
Figure 2. Mechanism of DC-mediated tolerance. Tolerogenic (Tol)DC promote deletion or modulate Teff cells via Fas/FasL interaction [1], starvation of Teff cells via IDO production that degrades tryptophan (L-Trp) into kynurenine (Kyn) [2]. IDO is induced by the interaction between CD80/CD86 on tolDC and CTLA-4 on regulatory T cell (FOXP3 Treg), which concur to the suppression of Teff cells [3]. The interaction of inhibitory molecules on tolDC and Teff cells in the presence of IL-10 secretion promotes T cell anergy [4]. TolDC favor the activation and expansion of pre-existing Tregs [5] of de novo induction of FOXP3 Treg of Tr1 cells [6]. Finally, surface expression of inhibitory molecules and secretion of regulatory mediators promote the conversion of resident APCs into tolerogenic APCs, which sustain tolerance [7]. Teff, effector T cells; CTLA-4, cytotoxic T-lymphocyte antigen-4; IDO, indoleamine 2,3-dioxygenase; L-Trp, L-tryptophan, Kyn, kynurenins; Fas, first apoptosis signal; FasL, Fas ligand.
Pioneer clinical trials with adoptive transfer of tolDC demonstrated the safety, feasibility and efficacy of the treatment and some clinical benefits [reviewed in (115)] (Table 1). Several tolerogenic approaches have been used in the past. In the first-in-man study, autologous tolDC treated with antisense oligonucleotides targeting CD40, CD80 and CD86 to maintain their immature state were infused in T1D patients [NCT00445913, (44)]. The group of Thomas treated DC with a nuclear factor-kB (NF-kB) inhibitor and pulsed them with citrullinated peptide Ags before injection into RA patients (45). More recently DC differentiated with vitamin D3 and dexamethasone alone or in combination have been or are currently used to treat RA, Crohn's disease, and MS patients [NCT01352858-AutoDECRA (46); NCT02618902-MS-tolDC; NCT02283671-TolDecEM/NMO (47–49); NCT02903537-Tolervit-MS (48, 49); NCT03337165-TolDCfoRA; (120)].
Despite these encouraging results, phase II/III clinical trials are needed to address several open issues and to allow comparison to current available treatments. Indeed, a number of open questions remain before tolDC-based therapies can be routinely used to treat or cure autoimmune diseases (101, 116). A variety of routes for tolDC administration have been tested in the past, including intradermal, intraperitoneal, intravenous and intra-articular (121). These administration routes are indeed required to allow tolDC to reach the relevant draining lymph nodes or the disease-specific site of inflammation. However, if direct administration to the relevant tissue is challenging, such as in the case of T1D, intraperitoneal administration has been preferred.
As for any Ag-specific approach for tolerance induction, an additional major hurdle in developing an effective tolDC-based therapy is the selection of the Ag critical for a given disease. As in the case of peptide-based approaches, the use of broad spectrum disease-related peptides has been postulated to overcome this limitation [reviewed in (101)]. Interestingly, in the context of T1D the identification of neoepitopes opened new perspectives in the field. The peptides characterized by improved MHC binding register, such as the insulin peptide InsB9−23 with combined substitutions in positions 14, 21, and 22 (122), those generated by fusion of peptides, such as the Hybrid Insulin Peptides (HIPs) (123), or by aberrant translation, such as INS-DriP peptide (124), have been shown to trigger strong specific T cell responses. These highly immunogenic peptides presented by tolDC are promising tools for the reprogramming of pathogenic T cells and induction of tolerance in T1D.
Besides the critical issues discussed above some additional considerations should be taken into account when designing tolDC-based therapies: (i) the necessity of multiple cell infusions to allow the induction of the self-sustained mechanisms described above will invariably lead to high manufacturing costs; (ii) the generation of autologous tolDC implies the use of patient-derived monocytes, which may not be as functional as those isolated from healthy subjects (105); (iii) the stability of the cell product to be infused must be evaluated for limiting in vivo side effects or disease exacerbation.
Conclusive Remarks
In recent years the development of in vivo and ex-vivo Ag-specific approaches to modulate detrimental immune responses has made striking progress. Results obtained in Phase I/II demonstrated the safety and tolerability of the approaches with, thus far, limited clinical responses. Phase II/III clinical trials will help in defining whether the strategies outlined here will reach the goal of completely reversing the course of T cell mediated diseases.
Overall, results obtained thus far highlighted common requirements for achieving the desired effectiveness of the Ag-specific based therapy, either peptide or protein delivery, or the vehicle strategies to delivery Ags or the regulatory cell-based approaches: the repetitive administrations and the use of multiple Ags to effectively activate the tolerogenic branch of the immune response and to tackle the epitope spreading, respectively. Moreover, the selection of the most suitable epitope/s to be used might be challenging, because different patients may display preferential response to specific Ags. This issue opens the need for the identification of peptide Ags that can be used across different HLA-type patients [e.g., (21, 22)] or for deeper characterization of patients' reactivity before enrollment in trials.
The field is rapidly evolving, and the upcoming clinical trials will confirm the safety and feasibility and will shed light on the efficacy of Ag-specific approaches. Several issues remain to be clarified for each of the approaches in the pipeline. Regardless of the tolerogenic approach used, one of the open questions in the field of tolerance induction is the definition of common parameters to monitor the response to treatment, and to allow comparison of different approaches. In the context of cell-based tolerance-inducing therapies an initiative of the European scientific community brought together the leader scientists in the field of cell-based therapies and autoimmune diseases under the umbrella of the European Cooperation in Science and Technology (COST). The main objective of A-FACTT Action was to coordinate efforts to minimize overlap and maximize comparison of the diverse cell-based approaches through establishment of consensus monitoring parameters (https://www.cost.eu/actions/BM1305/#tabs|Name:overview). More of such initiatives could help the field to address this relevant point. On the same line, the definition of “tolerogenic treatment” should be unambiguouosly referred to therapies inducing long-term active tolerance. Indeed, several treatments have been shown to modulate immune responses in the short term, but fail in controlling disease signs long-term. Tolerogenic therapies should promote long-lasting effects, and this can be achieved by different mode of action, including the conversion of pathogenic Teff cells into Tregs, or the de novo induction of Tregs. As discussed above, we believe that modulated DC, or APCs, represent the population of cells able to prevent activation of pathogenic Teff cells, to promote de novo induction of Tregs, and to re-educate Teff cells to become Tregs, thus maintaining tolerance long-term. To achieve these long lasting effects possible repetitive injection of the tolerogenic treatment might be required. Based on the central role of APCs in determining the outcome of Ag-specific T cell activation, inverse vaccination strategies are unlikely to be successful, unless the underlying mechanism allows boosting of the immunomodulatory properties of DC or, more generally, of APCs.
Author Contributions
LP and SG wrote the manuscritpt. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Telethon Foundation (Tele20-G1 to SG) and the Italian Ministry of Health (RF-2016- 02361372 to SG).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Giada Amodio for critical reading of the manuscript.
References
1. Steinman L. Inverse vaccination, the opposite of Jenner's concept, for therapy of autoimmunity. J Intern Med. (2010) 267:441–51. doi: 10.1111/j.1365-2796.2010.02224.x
2. Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, present, and future of regulatory T cell therapy in transplantation and autoimmunity. Front Immunol. (2019) 10:43. doi: 10.3389/fimmu.2019.00043
3. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M, Gagliani N. The Biology of T Regulatory Type 1 Cells and Their Therapeutic Application in Immune-Mediated Diseases. Immunity. (2018) 49:1004–19. doi: 10.1016/j.immuni.2018.12.001
4. Bar-Or A, Vollmer T, Antel J, Arnold DL, Bodner CA, Campagnolo D. Induction of antigen-specific tolerance in multiple sclerosis after immunization with DNA encoding myelin basic protein in a randomized, placebo-controlled phase 1/2 trial. Arch Neurol. (2007) 64:1407–15. doi: 10.1001/archneur.64.10.nct70002
5. Garren H, Robinson WH, Krasulova E, Havrdova E, Nadj C, Selmaj K, et al. Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann Neurol. (2008) 63:611–20. doi: 10.1002/ana.21370
6. Roep BO, Solvason N, Gottlieb PA, Abreu JRF, Harrison LC, Eisenbarth GS, et al. Plasmid-encoded proinsulin preserves C-peptide while specifically reducing proinsulin-specific CD8(+) T cells in type 1 diabetes. Sci Transl Med. (2013) 5:191ra82. doi: 10.1126/scitranslmed.3006103
7. Chaillous L, Lefevre H, Thivolet C, Boitard C, Lahlou N, Atlan-Gepner C, et al. Oral insulin administration and residual beta-cell function in recent-onset type 1 diabetes: a multicentre randomised controlled trial. Diabete Insuline Orale group. Lancet. (2000) 356:545–9. doi: 10.1016/S0140-6736(00)02579-4
8. Pozzilli P, Pitocco D, Visalli N, Cavallo MG, Buzzetti R, Crino A, et al. No effect of oral insulin on residual beta-cell function in recent-onset type I diabetes (the IMDIAB VII). IMDIAB Group. Diabetologia. (2000) 43:1000–4. doi: 10.1007/s001250051482
9. Ergun-Longmire B, Marker J, Zeidler A, Rapaport R, Raskin P, Bode B, et al. Oral insulin therapy to prevent progression of immune-mediated (type 1) diabetes. Ann N Y Acad Sci. (2004) 1029:260–77. doi: 10.1196/annals.1309.057
10. Skyler JS, Krischer JP, Wolfsdorf J, Cowie C, Palmer JP, Greenbaum C, et al. Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial–Type 1. Diabetes Care. (2005) 28:1068–76. doi: 10.2337/diacare.28.5.1068
11. Fourlanos S, Perry C, Gellert SA, Martinuzzi E, Mallone R, Butler J, et al. Evidence that nasal insulin induces immune tolerance to insulin in adults with autoimmune diabetes. Diabetes. (2011) 60:1237–45. doi: 10.2337/db10-1360
12. Nanto-Salonen K, Kupila A, Simell S, Siljander H, Salonsaari T, Hekkala A, et al. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial. Lancet. (2008) 372:1746–55. doi: 10.1016/S0140-6736(08)61309-4
13. Bonifacio E, Ziegler AG, Klingensmith G, Schober E, Bingley PJ, Rottenkolber M, et al. Effects of high-dose oral insulin on immune responses in children at high risk for type 1 diabetes: the Pre-POINT randomized clinical trial. JAMA. (2015) 313:1541–9. doi: 10.1001/jama.2015.2928
14. Orban T, Farkas K, Jalahej H, Kis J, Treszl A, Falk B, et al. Autoantigen-specific regulatory T cells induced in patients with type 1 diabetes mellitus by insulin B-chain immunotherapy. J Autoimmun. (2010) 34:408–15. doi: 10.1016/j.jaut.2009.10.005
15. Wherrett DK, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: a randomised double-blind trial. Lancet. (2011) 378:319–27. doi: 10.1016/S0140-6736(11)60895-7
16. Ludvigsson J, Krisky D, Casas R, Battelino T, Castano L, Greening J, et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N Engl J Med. (2012) 366:433–42. doi: 10.1056/NEJMoa1107096
17. Krause S, Landherr U, Agardh CD, Hausmann S, Link K, Hansen JM, et al. GAD autoantibody affinity in adult patients with latent autoimmune diabetes, the study participants of a GAD65 vaccination trial. Diabetes Care. (2014) 37:1675–80. doi: 10.2337/dc13-1719
18. Beam CA, MacCallum C, Herold KC, Wherrett DK, Palmer J, Ludvigsson J, et al. GAD vaccine reduces insulin loss in recently diagnosed type 1 diabetes: findings from a Bayesian meta-analysis. Diabetologia. (2017) 60:43–9. doi: 10.1007/s00125-016-4122-1
19. Weiner HL, Mackin GA, Matsui M, Orav EJ, Khoury SJ, Dawson DM, et al. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science. (1993) 259:1321–4. doi: 10.1126/science.7680493
20. Hafler DA, Kent SC, Pietrusewicz MJ, Khoury SJ, Weiner HL, Fukaura H. Oral administration of myelin induces antigen-specific TGF-beta 1 secreting T cells in patients with multiple sclerosis. Ann N Y Acad Sci. (1997) 835:120–31. doi: 10.1111/j.1749-6632.1997.tb48623.x
21. Thrower SL, James L, Hall W, Green KM, Arif S, Allen JS, et al. Proinsulin peptide immunotherapy in type 1 diabetes: report of a first-in-man Phase I safety study. Clin Exp Immunol. (2009) 155:156–65. doi: 10.1111/j.1365-2249.2008.03814.x
22. Alhadj Ali M, Liu YF, Arif S, Tatovic D, Shariff H, Gibson VB, et al. Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci Transl Med. (2017) 9:eaaf7779. doi: 10.1126/scitranslmed.aaf7779
23. Warren KG, Catz I, Ferenczi LZ, Krantz MJ. Intravenous synthetic peptide MBP8298 delayed disease progression in an HLA Class II-defined cohort of patients with progressive multiple sclerosis: results of a 24-month double-blind placebo-controlled clinical trial and 5 years of follow-up treatment. Eur J Neurol. (2006) 13:887–95. doi: 10.1111/j.1468-1331.2006.01533.x
24. Freedman MS, Bar-Or A, Oger J, Traboulsee A, Patry D, Young C. A phase III study evaluating the efficacy and safety of MBP8298 in secondary progressive MS. Neurology. (2011) 77:1551–60. doi: 10.1212/WNL.0b013e318233b240
25. Streeter HB, Rigden R, Martin KF, Scolding NJ, Wraith DC. Preclinical development and first-in-human study of ATX-MS-1467 for immunotherapy of MS. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e93. doi: 10.1212/NXI.0000000000000093
26. Chataway J, Martin K, Barrell K, Sharrack B, Stolt P, Wraith DC, et al. Effects of ATX-MS-1467 immunotherapy over 16 weeks in relapsing multiple sclerosis. Neurology. (2018) 90:e955–62. doi: 10.1212/WNL.0000000000005118
27. Walczak A, Siger M, Ciach A, Szczepanik M, Selmaj K. Transdermal application of myelin peptides in multiple sclerosis treatment. JAMA Neurol. (2013) 70:1105–9. doi: 10.1001/jamaneurol.2013.3022
28. Jurynczyk M, Walczak A, Jurewicz A, Jesionek-Kupnicka D, Szczepanik M, Selmaj K. Immune regulation of multiple sclerosis by transdermally applied myelin peptides. Ann Neurol. (2010) 68:593–601. doi: 10.1002/ana.22219
29. Tye-Din JA, Stewart JA, Dromey JA, Beissbarth T, van Heel DA, Tatham A, et al. Comprehensive, quantitative mapping of T cell epitopes in gluten in celiac disease. Sci Transl Med. (2010) 2:41ra51. doi: 10.1126/scitranslmed.3001012
30. Daveson AJM, Ee HC, Andrews JM, King T, Goldstein KE, Dzuris JL, et al. Epitope-specific immunotherapy targeting CD4-positive T cells in celiac disease: safety, pharmacokinetics, and effects on intestinal histology and plasma cytokines with escalating dose regimens of Nexvax2 in a randomized, double-blind, placebo-controlled phase 1 study. EBioMedicine. (2017) 26:78–90. doi: 10.1016/j.ebiom.2017.11.018
31. Kim HJ, Antel JP, Duquette P, Alleva DG, Conlon PJ, Bar-Or A. Persistence of immune responses to altered and native myelin antigens in patients with multiple sclerosis treated with altered peptide ligand. Clin Immunol. (2002) 104:105–14. doi: 10.1006/clim.2002.5258
32. Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group. Nat Med. (2000) 6:1176–82. doi: 10.1038/80525
33. Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. (2000) 6:1167–75. doi: 10.1038/80516
34. Walter M, Philotheou A, Bonnici F, Ziegler AG, Jimenez R, Group NBIS. No effect of the altered peptide ligand NBI-6024 on beta-cell residual function and insulin needs in new-onset type 1 diabetes. Diabetes Care. (2009) 32:2036–40. doi: 10.2337/dc09-0449
35. Goodkin DE, Shulman M, Winkelhake J, Waubant E, Andersson P, Stewart T, et al. A phase I trial of solubilized DR2:MBP84-102 (AG284) in multiple sclerosis. Neurology. (2000) 54:1414–20. doi: 10.1212/WNL.54.7.1414
36. Yadav V, Bourdette DN, Bowen JD, Lynch SG, Mattson D, Preiningerova J, et al. Recombinant T-cell receptor ligand (RTL) for treatment of multiple sclerosis: a double-blind, placebo-controlled, phase 1, dose-escalation study. Autoimmune Dis. (2012) 2012:954739. doi: 10.1155/2012/954739
37. Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, et al. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med. (2013) 5:188ra75. doi: 10.1126/scitranslmed.3006168
38. Lutterotti A, Ludersdorfer T.H., Docampo M.J., Hohmann M., Selles Moreno C., Hayward-Koennecke H, et al. Establish Tolerance in MS With Myelin-Peptide Coupled Red Blood Cells - The Phase Ib ETIMSredtrial. Stockholm: ECTRIMS Online Library (2019). p. 279584.
39. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, et al. Administration of CD4+CD25highCD127- regulatory T cells preserves beta-cell function in type 1 diabetes in children. Diabetes Care. (2012) 35:1817–20. doi: 10.2337/dc12-0038
40. Marek-Trzonkowska N, Mysliwiec M, Iwaszkiewicz-Grzes D, Gliwinski M, Derkowska I, Zalinska M, et al. Factors affecting long-term efficacy of T regulatory cell-based therapy in type 1 diabetes. J Transl Med. (2016) 14:332. doi: 10.1186/s12967-016-1090-7
41. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
42. Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next-generation regulatory T cell therapy. Nat Rev Drug Discov. (2019) 18:749–69. doi: 10.1038/s41573-019-0041-4
43. Desreumaux P, Foussat A, Allez M, Beaugerie L, Hebuterne X, Bouhnik Y, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn's disease. Gastroenterology. (2012) 143:1207–17 e1–2. doi: 10.1053/j.gastro.2012.07.116
44. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care. (2011) 34:2026–32. doi: 10.2337/dc11-0472
45. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Transl Med. (2015) 7:290ra87. doi: 10.1126/scitranslmed.aaa9301
46. Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA, et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheum Dis. (2017) 76:227–34. doi: 10.1136/annrheumdis-2015-208456
47. Zubizarreta I, Florez-Grau G, Vila G, Cabezon R, Espana C, Andorra M, et al. Immune tolerance in multiple sclerosis and neuromyelitis optica with peptide-loaded tolerogenic dendritic cells in a phase 1b trial. Proc Natl Acad Sci USA. (2019) 116:8463–70. doi: 10.1073/pnas.1820039116
48. Willekens B, Presas-Rodriguez S, Mansilla MJ, Derdelinckx J, Lee WP, Nijs G, et al. Tolerogenic dendritic cell-based treatment for multiple sclerosis (MS): a harmonised study protocol for two phase I clinical trials comparing intradermal and intranodal cell administration. BMJ Open. (2019) 9:e030309. doi: 10.1136/bmjopen-2019-030309
49. Willekens B, Presas-Rodríguez S, Mansilla M, Derdelinckx J, Lee WP, Nijs G, et al. Safety and Feasibility of a Tolerogenic Dendritic Cell-Based Treatment of Multiple Sclerosis (MS): A Collaborative Initiative Comparing Intranodal and Intradermal Cell Administration in Two Phase I Clinical Trials. Stockholm: ECTRIMS Online Library (2019).
50. Porgador A, Irvine KR, Iwasaki A, Barber BH, Restifo NP, Germain RN. Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization. J Exp Med. (1998) 188:1075–82. doi: 10.1084/jem.188.6.1075
51. Sudowe S, Dominitzki S, Montermann E, Bros M, Grabbe S, Reske-Kunz AB. Uptake and presentation of exogenous antigen and presentation of endogenously produced antigen by skin dendritic cells represent equivalent pathways for the priming of cellular immune responses following biolistic DNA immunization. Immunology. (2009) 128(1 Suppl.):e193–205. doi: 10.1111/j.1365-2567.2008.02947.x
52. Lazzaro S, Giovani C, Mangiavacchi S, Magini D, Maione D, Baudner B, et al. CD8 T-cell priming upon mRNA vaccination is restricted to bone-marrow-derived antigen-presenting cells and may involve antigen transfer from myocytes. Immunology. (2015) 146:312–26. doi: 10.1111/imm.12505
53. Solvason N, Lou YP, Peters W, Evans E, Martinez J, Ramirez U, et al. Improved efficacy of a tolerizing DNA vaccine for reversal of hyperglycemia through enhancement of gene expression and localization to intracellular sites. J Immunol. (2008) 181:8298–307. doi: 10.4049/jimmunol.181.12.8298
54. Waisman A, Ruiz PJ, Hirschberg DL, Gelman A, Oksenberg JR, Brocke S, et al. Suppressive vaccination with DNA encoding a variable region gene of the T-cell receptor prevents autoimmune encephalomyelitis and activates Th2 immunity. Nat Med. (1996) 2:899–905. doi: 10.1038/nm0896-899
55. Mbongue JC, Rawson J, Garcia PA, Gonzalez N, Cobb J, Kandeel F, et al. Reversal of new onset type 1 diabetes by oral salmonella-based combination therapy and mediated by regulatory T-cells in NOD mice. Front Immunol. (2019) 10:320. doi: 10.3389/fimmu.2019.00320
56. Husseiny MI, Du W, Mbongue J, Lenz A, Rawson J, Kandeel F, et al. Factors affecting Salmonella-based combination immunotherapy for prevention of type 1 diabetes in non-obese diabetic mice. Vaccine. (2018) 36:8008–18. doi: 10.1016/j.vaccine.2018.10.101
57. Dou L, Ono Y, Chen YF, Thomson AW, Chen XP. Hepatic dendritic cells, the tolerogenic liver environment, and liver disease. Semin Liver Dis. (2018) 38:170–80. doi: 10.1055/s-0038-1646949
58. Keeler GD, Kumar S, Palaschak B, Silverberg EL, Markusic DM, Jones NT, et al. Gene therapy-induced antigen-specific tregs inhibit neuro-inflammation and reverse disease in a mouse model of multiple sclerosis. Mol Ther. (2018) 26:173–83. doi: 10.1016/j.ymthe.2017.09.001
59. Han G, Wang R, Chen G, Wang J, Xu R, Feng J, et al. Gene delivery GAD 500 autoantigen by AAV serotype 1 prevented diabetes in NOD mice: transduction efficiency do not play important roles. Immunol Lett. (2008) 115:110–6. doi: 10.1016/j.imlet.2007.10.004
60. Akbarpour M, Goudy KS, Cantore A, Russo F, Sanvito F, Naldini L, et al. Insulin B chain 9-23 gene transfer to hepatocytes protects from type 1 diabetes by inducing Ag-specific FoxP3+ Tregs. Sci Transl Med. (2015) 7:289ra81. doi: 10.1126/scitranslmed.aaa3032
61. Annoni A, Goudy K, Akbarpour M, Naldini L, Roncarolo MG. Immune responses in liver-directed lentiviral gene therapy. Transl Res. (2013) 161:230–40. doi: 10.1016/j.trsl.2012.12.018
62. Pozsgay J, Szekanecz Z, Sarmay G. Antigen-specific immunotherapies in rheumatic diseases. Nat Rev Rheumatol. (2017) 13:525–37. doi: 10.1038/nrrheum.2017.107
63. Zhang N, Nandakumar KS. Recent advances in the development of vaccines for chronic inflammatory autoimmune diseases. Vaccine. (2018) 36:3208–20. doi: 10.1016/j.vaccine.2018.04.062
64. Bachmann MF, Mohsen MO, Kramer MF, Heath MD. Vaccination against allergy: a paradigm shift? Trends Mol Med. (2020) 26:357–68. doi: 10.1016/j.molmed.2020.01.007
65. Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. (2005) 435:220–3. doi: 10.1038/nature03523
66. Zhang ZJ, Davidson L, Eisenbarth G, Weiner HL. Suppression of diabetes in nonobese diabetic mice by oral administration of porcine insulin. Proc Natl Acad Sci USA. (1991) 88:10252–6. doi: 10.1073/pnas.88.22.10252
67. Sai P, Rivereau AS. Prevention of diabetes in the nonobese diabetic mouse by oral immunological treatments. Comparative efficiency of human insulin and two bacterial antigens, lipopolysacharide from Escherichia coli and glycoprotein extract from Klebsiella pneumoniae. Diabetes Metab. (1996) 22:341–8.
68. Bergerot I, Arreaza GA, Cameron MJ, Burdick MD, Strieter RM, Chensue SW, et al. Insulin B-chain reactive CD4+ regulatory T-cells induced by oral insulin treatment protect from type 1 diabetes by blocking the cytokine secretion and pancreatic infiltration of diabetogenic effector T-cells. Diabetes. (1999) 48:1720–9. doi: 10.2337/diabetes.48.9.1720
69. Zhang L, Londono P, Yu L, Grimes S, Blackburn P, Gottlieb P, et al. MAS-1 adjuvant immunotherapy generates robust Th2 type and regulatory immune responses providing long-term protection from diabetes in late-stage pre-diabetic NOD mice. Autoimmunity. (2014) 47:341–50. doi: 10.3109/08916934.2014.910768
70. Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. (2002) 2:85–95. doi: 10.1038/nri724
71. Yoosuf S, Makharia GK. Evolving therapy for celiac disease. Front Pediatr. (2019) 7:193. doi: 10.3389/fped.2019.00193
72. Candia M, Kratzer B, Pickl WF. On peptides and altered peptide ligands: from origin, mode of action and design to clinical application (immunotherapy). Int Arch Allergy Immunol. (2016) 170:211–33. doi: 10.1159/000448756
73. Antunes DA, Abella JR, Devaurs D, Rigo MM, Kavraki LE. Structure-based methods for binding mode and binding affinity prediction for peptide-MHC complexes. Curr Top Med Chem. (2018) 18:2239–55. doi: 10.2174/1568026619666181224101744
74. Sloan-Lancaster J, Evavold BD, Allen PM. Induction of T-cell anergy by altered T-cell-receptor ligand on live antigen-presenting cells. Nature. (1993) 363:156–9. doi: 10.1038/363156a0
75. Pfeiffer C, Stein J, Southwood S, Ketelaar H, Sette A, Bottomly K. Altered peptide ligands can control CD4 T lymphocyte differentiation in vivo. J Exp Med. (1995) 181:1569–74. doi: 10.1084/jem.181.4.1569
76. Crowe PD, Qin Y, Conlon PJ, Antel JP. NBI-5788, an altered MBP83-99 peptide, induces a T-helper 2-like immune response in multiple sclerosis patients. Ann Neurol. (2000) 48:758–65.
77. Banchereau J, Klechevsky E, Schmitt N, Morita R, Palucka K, Ueno H. Harnessing human dendritic cell subsets to design novel vaccines. Ann N Y Acad Sci. (2009) 1174:24–32. doi: 10.1111/j.1749-6632.2009.04999.x
78. Gojanovich GS, Murray SL, Buntzman AS, Young EF, Vincent BG, Hess PR. The use of peptide-major-histocompatibility-complex multimers in type 1 diabetes mellitus. J Diabetes Sci Technol. (2012) 6:515–24. doi: 10.1177/193229681200600305
79. Spack EG, McCutcheon M, Corbelletta N, Nag B, Passmore D, Sharma SD. Induction of tolerance in experimental autoimmune myasthenia gravis with solubilized MHC class II:acetylcholine receptor peptide complexes. J Autoimmun. (1995) 8:787–807. doi: 10.1016/S0896-8411(95)80018-2
80. Vandenbark AA, Rich C, Mooney J, Zamora A, Wang C, Huan J, et al. Recombinant TCR ligand induces tolerance to myelin oligodendrocyte glycoprotein 35-55 peptide and reverses clinical and histological signs of chronic experimental autoimmune encephalomyelitis in HLA-DR2 transgenic mice. J Immunol. (2003) 171:127–33. doi: 10.4049/jimmunol.171.1.127
81. Sinha S, Subramanian S, Proctor TM, Kaler LJ, Grafe M, Dahan R, et al. A promising therapeutic approach for multiple sclerosis: recombinant T-cell receptor ligands modulate experimental autoimmune encephalomyelitis by reducing interleukin-17 production and inhibiting migration of encephalitogenic cells into the CNS. J Neurosci. (2007) 27:12531–9. doi: 10.1523/JNEUROSCI.3599-07.2007
82. Huan J, Subramanian S, Jones R, Rich C, Link J, Mooney J, et al. Monomeric recombinant TCR ligand reduces relapse rate and severity of experimental autoimmune encephalomyelitis in SJL/J mice through cytokine switch. J Immunol. (2004) 172:4556–66. doi: 10.4049/jimmunol.172.7.4556
83. Lin M, Stoica-Nazarov C, Surls J, Kehl M, Bona C, Olsen C, et al. Reversal of type 1 diabetes by a new MHC II-peptide chimera: “Single-epitope-mediated suppression” to stabilize a polyclonal autoimmune T-cell process. Eur J Immunol. (2010) 40:2277–88. doi: 10.1002/eji.200940094
84. Getts DR, McCarthy DP, Miller SD. Exploiting apoptosis for therapeutic tolerance induction. J Immunol. (2013) 191:5341–6. doi: 10.4049/jimmunol.1302070
85. Cremel M, Guerin N, Horand F, Banz A, Godfrin Y. Red blood cells as innovative antigen carrier to induce specific immune tolerance. Int J Pharm. (2013) 443:39–49. doi: 10.1016/j.ijpharm.2012.12.044
86. Kontos S, Kourtis IC, Dane KY, Hubbell JA. Engineering antigens for in situ erythrocyte binding induces T-cell deletion. Proc Natl Acad Sci USA. (2013) 110:E60–8. doi: 10.1073/pnas.1216353110
87. Grimm AJ, Kontos S, Diaceri G, Quaglia-Thermes X, Hubbell JA. Memory of tolerance and induction of regulatory T cells by erythrocyte-targeted antigens. Sci Rep. (2015) 5:15907. doi: 10.1038/srep15907
88. Pishesha N, Bilate AM, Wibowo MC, Huang NJ, Li Z, Deshycka R, et al. Engineered erythrocytes covalently linked to antigenic peptides can protect against autoimmune disease. Proc Natl Acad Sci USA. (2017) 114:3157–62. doi: 10.1073/pnas.1701746114
89. Pujol-Autonell I, Mansilla MJ, Rodriguez-Fernandez S, Cano-Sarabia M, Navarro-Barriuso J, Ampudia RM, et al. Liposome-based immunotherapy against autoimmune diseases: therapeutic effect on multiple sclerosis. Nanomedicine. (2017) 12:1231–42. doi: 10.2217/nnm-2016-0410
90. Capini C, Jaturanpinyo M, Chang HI, Mutalik S, McNally A, Street S, et al. Antigen-specific suppression of inflammatory arthritis using liposomes. J Immunol. (2009) 182:3556–65. doi: 10.4049/jimmunol.0802972
91. Engman C, Wen Y, Meng WS, Bottino R, Trucco M, Giannoukakis N. Generation of antigen-specific Foxp3+ regulatory T-cells in vivo following administration of diabetes-reversing tolerogenic microspheres does not require provision of antigen in the formulation. Clin Immunol. (2015) 160:103–23. doi: 10.1016/j.clim.2015.03.004
92. Phillips B, Nylander K, Harnaha J, Machen J, Lakomy R, Styche A, et al. A microsphere-based vaccine prevents and reverses new-onset autoimmune diabetes. Diabetes. (2008) 57:1544–55. doi: 10.2337/db07-0507
93. Pearson RM, Podojil JR, Shea LD, King NJC, Miller SD, Getts DR. Overcoming challenges in treating autoimmuntity: Development of tolerogenic immune-modifying nanoparticles. Nanomedicine. (2019) 18:282–91. doi: 10.1016/j.nano.2018.10.001
94. Kishimoto TK, Maldonado RA. Nanoparticles for the induction of antigen-specific immunological tolerance. Front Immunol. (2018) 9:230. doi: 10.3389/fimmu.2018.00230
95. Carambia A, Freund B, Schwinge D, Bruns OT, Salmen SC, Ittrich H, et al. Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice. J Hepatol. (2015) 62:1349–56. doi: 10.1016/j.jhep.2015.01.006
96. Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. (2016) 530:434–40. doi: 10.1038/nature16962
97. Umeshappa CS, Singha S, Blanco J, Shao K, Nanjundappa RH, Yamanouchi J, et al. Suppression of a broad spectrum of liver autoimmune pathologies by single peptide-MHC-based nanomedicines. Nat Commun. (2019) 10:2150. doi: 10.1038/s41467-019-09893-5
98. Serra P, Santamaria P. Antigen-specific therapeutic approaches for autoimmunity. Nat Biotechnol. (2019) 37:238–51. doi: 10.1038/s41587-019-0015-4
99. Gómez López A. Nanotechnology and autoimmunity. In: Rosario UE, editor. Autoimmunity: From Bench to Bedside. Bogota: El Rosario University Press. (2013). p. 727–38.
100. Rezaei R, Safaei M, Mozaffari HR, Moradpoor H, Karami S, Golshah A, et al. The role of nanomaterials in the treatment of diseases and their effects on the immune system. Open Access Maced J Med Sci. (2019) 7:1884–90. doi: 10.3889/oamjms.2019.486
101. Ten Brinke A, Martinez-Llordella M, Cools N, Hilkens CMU, van Ham SM, Sawitzki B, et al. Ways forward for tolerance-inducing cellular therapies- an AFACTT perspective. Front Immunol. (2019) 10:181. doi: 10.3389/fimmu.2019.00181
102. Geissler EK, Hutchinson JA. Cell therapy as a strategy to minimize maintenance immunosuppression in solid organ transplant recipients. Curr Opin Organ Transplant. (2013) 18:408–15. doi: 10.1097/MOT.0b013e328363319d
103. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. (2004) 199:1455–65. doi: 10.1084/jem.20040139
104. Gregori S, Tomasoni D, Pacciani V, Scirpoli M, Battaglia M, Magnani CF, et al. Differentiation of type 1 T regulatory cells (Tr1) by tolerogenic DC-10 requires the IL-10-dependent ILT4/HLA-G pathway. Blood. (2010) 116:935–44. doi: 10.1182/blood-2009-07-234872
105. Mfarrej B, Tresoldi E, Stabilini A, Paganelli A, Caldara R, Secchi A, et al. Generation of donor-specific Tr1 cells to be used after kidney transplantation and definition of the timing of their in vivo infusion in the presence of immunosuppression. J Transl Med. (2017) 15:40. doi: 10.1186/s12967-017-1133-8
106. Putnam AL, Safinia N, Medvec A, Laszkowska M, Wray M, Mintz MA, et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am J Transplant. (2013) 13:3010–20. doi: 10.1111/ajt.12433
107. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. (2016) 126:1413–24. doi: 10.1172/JCI82771
108. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA Class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. (2017) 17:931–43. doi: 10.1111/ajt.14185
109. Dwyer CJ, Ward NC, Pugliese A, Malek TR. Promoting immune regulation in type 1 diabetes using low-dose interleukin-2. Curr Diab Rep. (2016) 16:46. doi: 10.1007/s11892-016-0739-1
110. Tang Q, Vincenti F. Transplant trials with Tregs: perils and promises. J Clin Invest. (2017) 127:2505–12. doi: 10.1172/JCI90598
111. Gregori S. Dendritic cells in networks of immunological tolerance. Tissue Antigens. (2011) 77:89–99. doi: 10.1111/j.1399-0039.2010.01615.x
112. Osorio F, Fuentes C, Lopez MN, Salazar-Onfray F, Gonzalez FE. Role of dendritic cells in the induction of lymphocyte tolerance. Front Immunol. (2015) 6:535. doi: 10.3389/fimmu.2015.00535
113. Raker VK, Domogalla MP, Steinbrink K. Tolerogenic dendritic cells for regulatory T cell induction in man. Front Immunol. (2015) 6:569. doi: 10.3389/fimmu.2015.00569
114. Liu J, Cao X. Regulatory dendritic cells in autoimmunity: a comprehensive review. J Autoimmun. (2015) 63:1–12. doi: 10.1016/j.jaut.2015.07.011
115. Ten Brinke A, Hilkens CM, Cools N, Geissler EK, Hutchinson JA, Lombardi G, et al. Clinical use of tolerogenic dendritic cells-harmonization approach in European Collaborative Effort. Mediators Inflamm. (2015) 2015:471719. doi: 10.1155/2015/471719
116. Amodio G, Cichy J, Conde P, Matteoli G, Moreau A, Ochando J, et al. Role of myeloid regulatory cells (MRCs) in maintaining tissue homeostasis and promoting tolerance in autoimmunity, inflammatory disease and transplantation. Cancer Immunol Immunother. (2019) 68:661–72. doi: 10.1007/s00262-018-2264-3
117. Obregon C, Kumar R, Pascual MA, Vassalli G, Golshayan D. Update on dendritic cell-induced immunological and clinical tolerance. Front Immunol. (2017) 8:1514. doi: 10.3389/fimmu.2017.01514
118. Amodio G, Comi M, Tomasoni D, Gianolini ME, Rizzo R, Lemaoult J, et al. Hla-g expression levels influence the tolerogenic activity of human DC-10. Haematologica. (2015) 100:548–57. doi: 10.3324/haematol.2014.113803
119. Mellor AL, Lemos H, Huang L. Indoleamine 2,3-Dioxygenase and tolerance: where are we now? Front Immunol. (2017) 8:1360. doi: 10.3389/fimmu.2017.01360
120. Spiering R, Jansen MAA, Wood MJ, Fath AA, Eltherington O, Anderson AE, et al. Targeting of tolerogenic dendritic cells to heat-shock proteins in inflammatory arthritis. J Transl Med. (2019) 17:375. doi: 10.1186/s12967-019-2128-4
121. Domogalla MP, Rostan PV, Raker VK, Steinbrink K. Tolerance through education: how tolerogenic dendritic cells shape immunity. Front Immunol. (2017) 8:1764. doi: 10.3389/fimmu.2017.01764
122. Wang Y, Sosinowski T, Novikov A, Crawford F, Neau DB, Yang J, et al. C-terminal modification of the insulin B:11-23 peptide creates superagonists in mouse and human type 1 diabetes. Proc Natl Acad Sci USA. (2018) 115:162–7. doi: 10.1073/pnas.1716527115
123. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science. (2016) 351:711–4. doi: 10.1126/science.aad2791
Keywords: tolerance, dendritic cells, autoimmunity, cell therapy, immunomodulation, antigen-specific
Citation: Passerini L and Gregori S (2020) Induction of Antigen-Specific Tolerance in T Cell Mediated Diseases. Front. Immunol. 11:2194. doi: 10.3389/fimmu.2020.02194
Received: 11 June 2020; Accepted: 11 August 2020;
Published: 29 September 2020.
Edited by:
Fang Zhou, CAS Lamvac Biotech Co., Ltd., ChinaReviewed by:
Jan Nilsson, Lund University, SwedenBarbara M. P. Willekens, Antwerp University Hospital, Belgium
Carolin Daniel, Helmholtz Zentrum München, Germany
David Markusic, Indiana University, Purdue University Indianapolis, United States
Copyright © 2020 Passerini and Gregori. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Gregori, gregori.silvia@hsr.it