
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 04 September 2020
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.02041
 Karina Tozatto-Maio1,2,3,4*
Karina Tozatto-Maio1,2,3,4* Robert Girot5
Robert Girot5 Indou Deme Ly6Ana Cristina Silva Pinto3,7Vanderson Rocha4
Indou Deme Ly6Ana Cristina Silva Pinto3,7Vanderson Rocha4 Francisco Fernandes8Ibrahima Diagne6
Francisco Fernandes8Ibrahima Diagne6 Yahia Benzerara9
Yahia Benzerara9 Carla L. Dinardo4
Carla L. Dinardo4 Julia Pavan Soler8Simone Kashima3,7
Julia Pavan Soler8Simone Kashima3,7 Itauá Leston Araujo10Chantal Kenzey1,2Guilherme H. H. Fonseca4Evandra S. Rodrigues3,7Fernanda Volt1,2
Itauá Leston Araujo10Chantal Kenzey1,2Guilherme H. H. Fonseca4Evandra S. Rodrigues3,7Fernanda Volt1,2 Luciana Jarduli7,11
Luciana Jarduli7,11 Annalisa Ruggeri1,12,13Christina Mariaselvam14Sandra F. M. Gualandro4Hanadi Rafii1,2Barbara Cappelli1,2Felipe Melo Nogueira4Graziana Maria Scigliuolo1,2
Annalisa Ruggeri1,12,13Christina Mariaselvam14Sandra F. M. Gualandro4Hanadi Rafii1,2Barbara Cappelli1,2Felipe Melo Nogueira4Graziana Maria Scigliuolo1,2 Renato Luiz Guerino-Cunha3,7
Renato Luiz Guerino-Cunha3,7 Kelen Cristina Ribeiro Malmegrim11
Kelen Cristina Ribeiro Malmegrim11 Belinda P. Simões3Eliane Gluckman1,2
Belinda P. Simões3Eliane Gluckman1,2 Ryad Tamouza14
Ryad Tamouza14Sickle cell disease (SCD), the most common monogenic disease worldwide, is marked by a phenotypic variability that is, to date, only partially understood. Because inflammation plays a major role in SCD pathophysiology, we hypothesized that single nucleotide polymorphisms (SNP) in genes encoding functionally important inflammatory proteins might modulate the occurrence of SCD complications. We assessed the association between 20 SNPs in genes encoding Toll-like receptors (TLR), NK cell receptors (NKG), histocompatibility leukocyte antigens (HLA), major histocompatibility complex class I polypeptide-related sequence A (MICA) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), and the occurrence of six SCD clinical complications (stroke, acute chest syndrome (ACS), leg ulcers, cholelithiasis, osteonecrosis, or retinopathy). This study was performed in a cohort of 500 patients. We found that the TLR2 rs4696480 TA, TLR2 rs3804099 CC, and HLA-G, rs9380142 AA genotypes were more frequent in patients who had fewer complications. Also, in logistic regression, the HLA-G rs9380142 G allele increased the risk of cholelithiasis (AG vs. AA, OR 1.57, 95%CI 1.16–2.15; GG vs. AA, OR 2.47, 95%CI 1.34–4.64; P = 0.02). For SNPs located in the NKG2D loci, in logistic regression, the A allele in three SNPs was associated with a lower frequency of retinopathy, namely, rs2246809 (AA vs. GG: OR 0.22, 95%CI 0.09–0.50; AG vs. GG: OR 0.47, 95%CI 0.31–0.71; P = 0.004, for patients of same origin), rs2617160 (AT vs. TT: OR 0.67, 95%CI 0.48–0.92; AA vs. TT: OR 0.45, 95%CI 0.23–0.84; P = 0.04), and rs2617169 (AA vs. TT: OR 0.33, 95%CI 0.13–0.82; AT vs. TT: OR 0.58, 95%CI 0.36–0.91, P = 0.049, in patients of same SCD genotype). These results, by uncovering susceptibility to, or protection against SCD complications, might contribute to a better understanding of the inflammatory pathways involved in SCD manifestations and to pave the way for the discovery of biomarkers that predict disease severity, which would improve SCD management.
Sickle cell disease (SCD) is the most common monogenic disorder (1), caused by a single nucleotide polymorphism (SNP) in the first exon of the β-globin gene (HBB). The βS allele emerged in some regions of Sub-Saharan Africa and Asia and SCD was initially restricted, mainly, to these areas. However, due to migration flows, the high morbidity and mortality and the global burden associated with the disease, SCD became a worldwide public health issue (2). Patients with SCD present several acute and chronic complications, some of them life-threatening. However, to date, with very few exceptions, such as stroke in children (3), most complications cannot be predicted.
A hallmark of SCD is the phenotype variability that occurs even within the same SCD genotype (SS, Sβ, SC), which is not fully understood (2). SCD complications occur due to vaso-occlusion, hemolysis due to red cell injury, and inflammation, resulting from various triggers, including intravascular hemolysis and ischemia-reperfusion injury (4–6). Although inflammation plays a major role in the pathophysiology of the disease, not all inflammatory pathways involved in SCD complications are known.
Some studies have addressed the influence of inflammatory markers in SCD complications, but few of them have evaluated the role of polymorphisms in genes encoding pivotal inflammatory proteins belonging to both innate and adaptive immune processes in the occurrence of complications (7–16). We hypothesize that such genes can modulate the occurrence of some complications in patients with SCD. Finding clinical and biological markers for predicting SCD complications would help to prevent them, and to decrease morbidity and mortality. Our work aimed to find associations between specific complications in patients with SCD and relevant polymorphisms in genes encoding potent inflammatory molecules.
This was a case-control retrospective study. Five hundred patients, followed at the Albert Royer Hospital, Dakar, Senegal (n = 56), Clinics Hospital of Ribeirão Preto, Ribeirão Preto, Brazil (n = 142), Clinics Hospital of São Paulo, São Paulo, Brazil (n = 88), and Tenon Hospital, Paris, France (n = 214), mainly from Sub-Saharan Africa and at a lesser extent from the French West Indies were included in the study. All patients had been previously diagnosed with SCD by hemoglobin electrophoresis or high-performance liquid chromatography (HPLC), had clinical records available for data collection and stored DNA samples.
We analyzed the potential influence of 20 SNPs on the occurrence of six clinical complications: acute chest syndrome (ACS), stroke, leg ulcers, cholelithiasis, osteonecrosis, and sickle retinopathy of any grade, occurring at any time from birth to last follow up. For each complication, a case was defined as a patient who had the specific complication, and a control was defined as a patient, from the same study cohort, who did not present the complication. ACS was defined as the presence of an acute respiratory symptom and a new pulmonary consolidation shown on chest radiography (17, 18). Stroke was defined as a new finding in neurological exam and a new image on brain computed tomography (CT). Cholelithiasis was documented by abdominal ultrasonography or CT. Osteonecrosis of any bone involving or not joint was documented by radiography, CT or magnetic resonance imaging (MRI). Retinopathy was documented by examination performed by an ophthalmologist; because the pathophysiology bases are the same, both non-proliferative and proliferative retinopathy were included.
DNA was extracted from peripheral blood samples using standard methods (19). Using TaqMan 5′-nuclease assay (Applied Biosystems, Forster City, CA, USA) pre-designed allele-specific fluorogenic oligonucleotide probes, we genotyped SNPs in genes encoding the following innate and adaptative immune molecules: Toll-like receptor (TLR)1 (rs4833095), TLR2 (rs4308099, rs4308100, rs4696480), TLR6 (rs5743810), TLR10 (rs11466653, rs11096957), all located in chromosome 4 and belonging to the TLR1 subfamily (20); natural killer (NK) group 2 member D (NKG2D) receptor (rs1982536, rs2617160, rs2617169, rs2617170, rs2617171, rs1049174, rs2246809, rs2255336), in chromosome 12; human leukocyte antigen (HLA)-G (rs9380142), HLA-E (rs2517523), major histocompatibility complex class I polypeptide-related sequence A (MICA, rs1057192), in the chromosome 6; and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) (rs5742909, rs231775), in the chromosome 2. DNA pre-amplification and real-time polymerase chain reaction (RT-PCR) were performed as previously described (15, 21). All SNPs included in this analysis were selected based on the relevance of their biological function.
For all SNPs, we compared allele distribution between the study population and the African population described on the 1,000 genomes database (22), composed by 661 healthy adult individuals, mainly from Sub-Saharan African and African-American origin. This was an exploratory comparison to determine if the frequency of risk alleles was the same in SCD and non-SCD individuals. For some SNPs, we also performed comparisons with a Brazilian non-SCD population, composed of 138 adult healthy individuals.
We performed analyses of correspondence, represented by dot charts, for indicating associations between SNPs and number of clinical complications; the significance of the association was tested using odds ratio (OR). Using item response theory (IRT) (23, 24), we also calculated a score for each patient based on the occurrence of each clinical complication; based on this score, we built characteristic curves for each clinical complication in the study population. Associations between SNPs and each complication were assessed by logistic regression; for modeling adjustment, geographical origin, SCD genotype, rate of red blood cell (RBC) transfusions (higher rate of transfusions indicating more severe disease) and gender were considered, and the significance was adjusted for multiple testing using false discovery rate (FDR). For all SNPs, two genetic models were used: the additive model, which supposes that the effect of the risk allele increases with the addition of each allele copy; and the additive-dominant model, which analyzes the effect of the heterozygous genotype vs. homozygotes. Analyses were performed using R software, version 3.4.4 (25). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, http://cran.r-project.org/doc/manuals/R-intro.pdf).
The study was approved by internal review boards, by the Eurocord scientific committee and was performed according to the declaration of Helsinki.
Five hundred patients were included in the study, originally from Brazil (n = 228), Sub-Saharan Africa (n = 200), French West Indies (n = 53), and North Africa (n = 7); 12 patients did not have their geographical origin reported. SCD genotype was available for 494 patients and was mostly SS (n = 402), followed by SC (n = 46), Sβ (n = 42), or other (n = 4). Two hundred and eighty patients were female and median age of the whole cohort was 32 (range: 0–69 years).
Ninety-seven (19%) patients did not present any of the six clinical complications analyzed in this study; among patients who presented complications, 135 (27%) had one, 134 (27%) had two, 94 (19%) had three, 34 (7%) had four, and 6 (1%) had five types of complications, respectively.
Seventy-one/500 patients (14%) presented at least one episode of stroke, 200/500 (40%) had at least one episode of ACS, 69/500 (14%) had leg ulcers, 271/500 (54%) presented cholelithiasis, 150/366 (41%) had retinopathy, and 90/500 (18%) presented osteonecrosis. For retinopathy, we have only considered patients who had retinal screening available in medical records. One hundred eighty-four patients (37%) received at least 20 RBC transfusions. Twenty-one (4%) patients underwent hematopoietic stem cell transplantation. Eleven (2%) patients died during follow-up, mostly from acute chest syndrome and hemorrhagic stroke. Table 1 shows the frequency of clinical complications according to gender, origin and SCD genotype. Figure 1A shows the score calculated for each patient and Figure 1B represents the curves of distribution of how many clinical complications were observed in this cohort. Patients who had stroke showed less probability of presenting other complications.
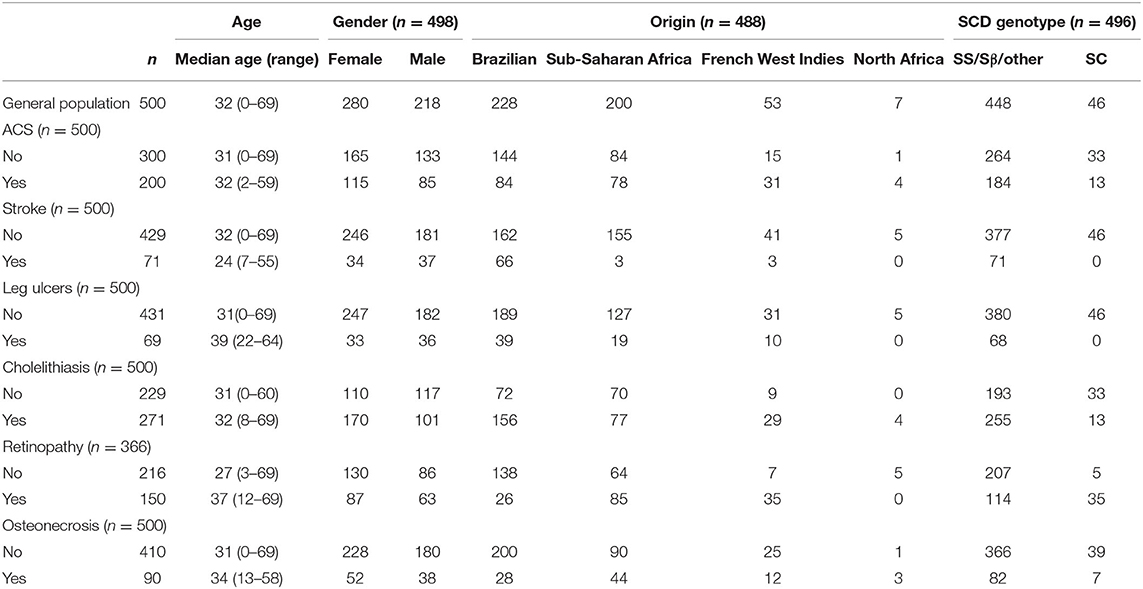
Table 1. Demography according to clinical complications.
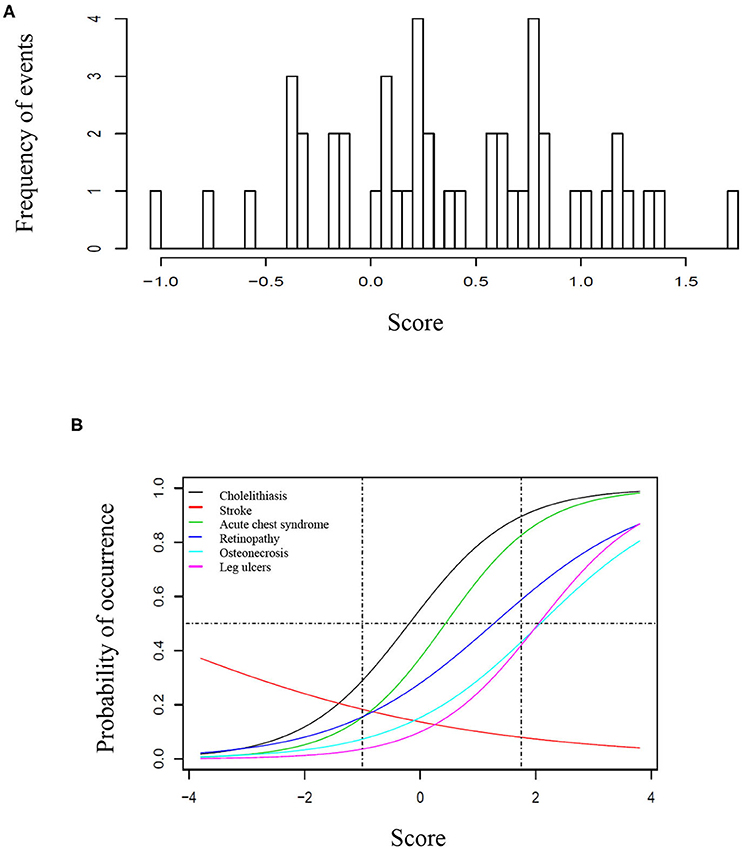
Figure 1. Score of clinical complications in the study cohort. (A) Histogram representing the score of clinical complication calculated for each patient based on the item-response theory (ITR). (B) Characteristic curves for each clinical complication, based on the IRT score. (A) Represents the score based on the distribution of complication for each patient, ranging from −1 to ~1, 8. In (B), the curves are shown from −4 to 4 to better illustrate the trend of each curve, however the vertical traced lines indicate the interval where patients are located. In this cohort, the probability of occurrence was higher for cholelithiasis, followed by acute chest syndrome, retinopathy, osteonecrosis, and leg ulcers. The horizontal line represents 50% of probability. For instance, a patient with 50% probability of having retinopathy had >50% probability of presenting cholelithiasis and acute chest syndrome, but <50% for osteonecrosis and leg ulcers. Patients who presented stroke had less probability of presenting all other clinical complications.
All SNPs had a call rate >90% and minimum allele frequency >1%. TLR2 rs4696480, TLR6 rs5743810, NKG2D rs1049174, NKG2D rs2617171, and MICA rs1051792 SNPs did not satisfy the expected Hardy-Weinberg proportions. However, because our cohort was composed only of patients with sickle cell disease, with no healthy controls, we kept these SNPs for genotype analysis. Genotype distribution of each SNP in our cohort is shown on Table 2. Linkage disequilibrium between SNPs is shown on Figure S1.
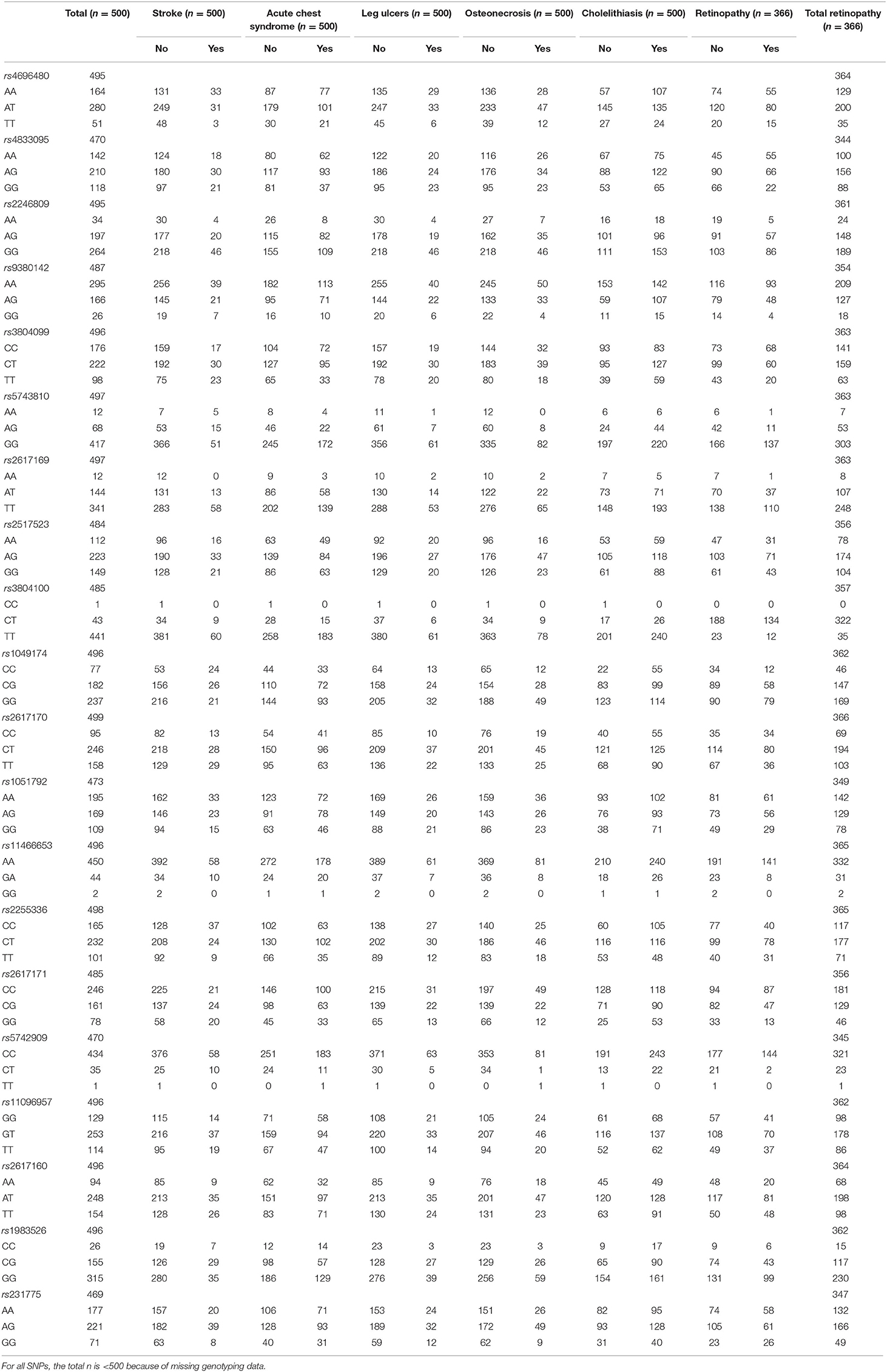
Table 2. Genotype distribution for each SNP in the study population and according to clinical complication.
Considering the indication of associations between SNPs and number of clinical complications, the TLR2 rs4696480 TA genotype was found more frequently in patients who had no complications or one complication (TA vs. AA and TT, 53 vs. 38%, OR 0.55, 95%CI 0.38–0.79, P = 0.001); The TLR2 rs3804099 CC genotype occurred more frequently in patients who had no complications (CC vs. CT or TT; 45 vs. 33%, OR 1.62, 95%CI 1.03–2.56, P = 0.03). Figure 2 shows the dot charts of the correspondence analysis. By mean of logistic regression models, we found no association between any of the SNPs in TLR genes and clinical complications. Table 3 summarizes the results of the logistic regression analyses.
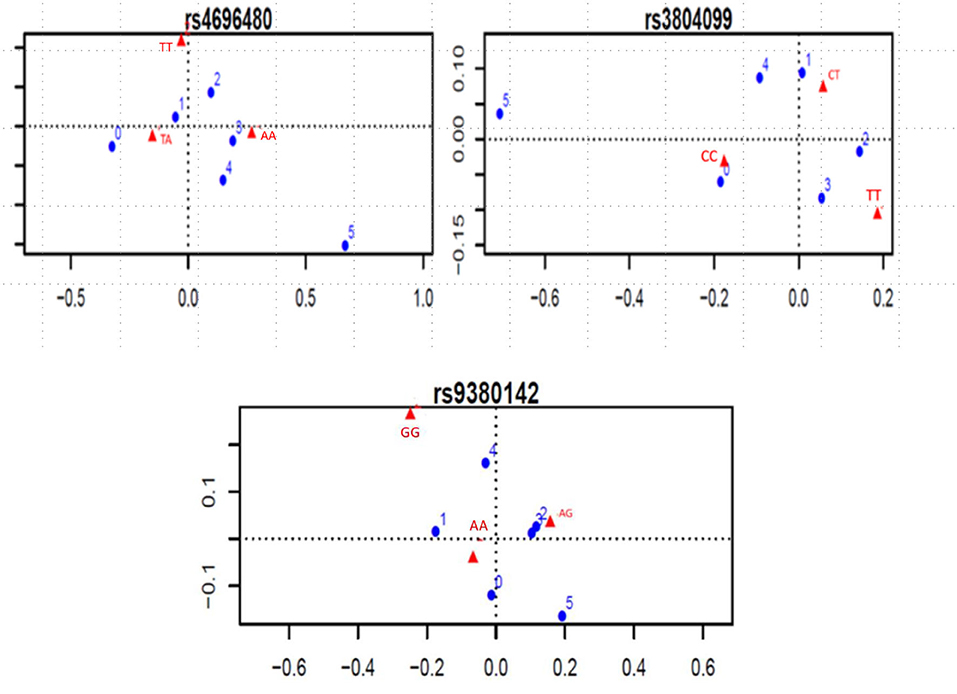
Figure 2. Significant correspondence analysis between SNP genotypes and number of complications. Blue dots represent the number of complications, ranging from 0 to 5. Red triangles represent the genotypes of each SNP.
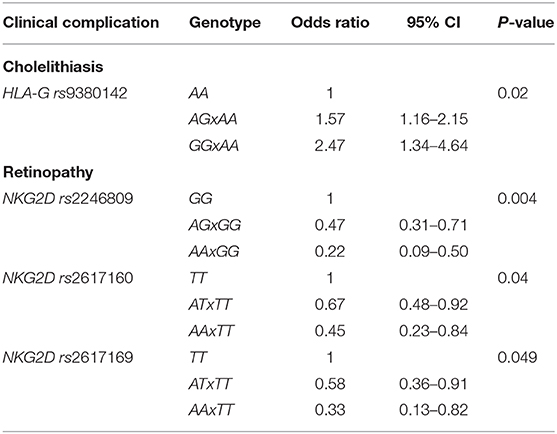
Table 3. Significant SNPs, according to the clinical variable, in logistic regression models.
Considering the associations between SNPs and each clinical complication, three NKG2D SNPs were significantly associated with the occurrence of retinopathy. The distribution of genotypes of these SNPs was: rs2246809, retinopathy group (n = 148): GG 58%, AG 39%, AA 3%; non-retinopathy group (n = 213): GG 48%, AG 43%, AA 9%; rs2617160, retinopathy group (n = 149): TT 32%, AT 55%, AA 13%; non-retinopathy group (n = 215): TT 23%, AT 55%, AA 22%; rs2617169, retinopathy group (n = 148): TT 1%, AT 25%, AA 74%; non-retinopathy group (n = 215): TT 3%, AT 33%, AA 64%. In the logistic regression additive model, the A allele in these SNPs was associated with a lower risk of retinopathy: rs2246809, AA vs. GG: OR 0.22, 95%CI 0.09–0.50; AG vs. GG: OR 0.47, 95%CI 0.31–0.71, in patients of same origin; P = 0.004; rs2617160, AT vs. TT: OR 0.67, 95%CI 0.48–0.92; AA vs. TT: OR 0.45, 95%CI 0.23–0.84; P = 0.04; rs2617169, AA vs. TT: OR 0.33, 95%CI 0.13–0.82; AT vs. TT: OR 0.58, 95%CI 0.36–0.91, P = 0.049, in patients of same SCD genotype. We did not find any association between NKG2D SNPs and other clinical complications.
The presence of the A allele for MICA rs1051792 (genotypes AA/AG) was associated with overall less complications (up to two complications), but this was not statistically significant. HLA-G rs9380142 was the only SNP significantly associated with cholelithiasis in the logistic regression additive model. The distribution of genotypes in the cholelithiasis group (n = 264) was: AA 54%, AG 40%, GG 6%, and in the non-cholelithiasis group (n = 223), AA 69%, AG 26%, and GG 5%. The G allele increased the risk of cholelithiasis (AG vs. AA, OR 1.57, 95%CI 1.16–2.15; GG vs. AA, OR 2.47, 95%CI 1.34–4.64; P = 0.02). In addition, the AA genotype of this SNP was significantly more frequent in patients who had 0–1 complication than genotypes AG and GG (65 vs. 56%, OR 1.47, 95%CI 1.02–2.12, P = 0.03). Furthermore, although MICA is a ligand for HLA-E, we did not find any significant association between the SNPs in the corresponding genes in this cohort.
No association was found between alleles or genotypes in the SNPs encoding CTLA4 and clinical manifestations of SCD.
No haplotype in this analysis was associated with complications.
Figure S2 shows a graph representing the allele distribution of SNPs in our SCD cohort and in two non-SCD populations, the one described at the 1,000 genomes database, and a previously studied Brazilian population composed by non-SCD patients (not available for all SNPs).
Patients with SCD have a steady state inflammatory background that can be boosted by ischemia-reperfusion injury, hemolysis and other situations such as infections (4). In addition, SCD has a marked phenotype variability not fully understood. It was previously demonstrated that polymorphisms in genes encoding inflammatory proteins play a role in the susceptibility to some complications in patients with SCD (7–16, 26).
NKG2D is a transmembrane receptor expressed on the surface of NK cells, iNKT cells, T CD8+ and some subsets of TCD4+ lymphocytes (27). NKG2D is encoded by a gene located in the natural killer complex (NKC) gene region on chromosome 12 and belong to the NK cell activating receptors. For instance, upon interaction with its ligand on the target cell, NK cell exert their cytotoxic functions and upregulate IFN-gamma secretion with consequent naïve cytotoxic T lymphocytes in CD8+ alpha/beta T cells activation, thus helping in the elimination of triggering event including, for example, tumor cells or pathogens (28–32). NKG2D recognizes a wide range of ligands, such as MICA and UL16-binding proteins (ULBP). However, NKG2D ligands are not spontaneously expressed but are induced upon stimulation, for example stress, viral or bacterial infections (27). Any type of cell can express NKG2D ligands when stimulated, and dysregulation of NKG2D and its ligands was observed in cancer and in autoimmune diseases such as rheumatoid arthritis and alopecia areata (30, 33, 34).
Patients with SCD have higher levels of circulating leukocytes (4, 35); higher levels of NKT cells/activated NKT cells in this population, compared with non-SCD controls, were shown in a study (36). In SCD, ischemia-reperfusion injury due to repeated episodes of vaso-occlusion triggers inflammation by activation of CD1d-restricted iNKT cells (36). NKG2D acts as a costimulatory factor for iNKT activation in response to CD1d and as an independent factor for iNKT activation (37). Haplotypes composed by SNPs scattered along the NKC region, especially NKG2D, were previously associated with high or low natural cytotoxic activity in cancer settings (38). Thus, we tested these SNPs because they have a defined role in NKG2D function. We found that three SNPs in NKG2D influence the occurrence of retinopathy. For these three SNPs, the A allele decreased the risk of retinopathy. In all cases, previously, the A allele was associated with a higher potential of cellular cytotoxicity likely in relation with enhanced inflammatory processes in the functional haplotypes studied by Hayashi et al. (38).
Retinopathy is the most prevalent vascular complication in SCD (39). It occurs in up to 46% of SS patients and 76% of SC patients (39–41). Proliferative retinopathy also occurs more in SC patients than in SS (42); it was proposed that the more severe phenotype of SS patients might lead to early occlusion of the peripheral retinal vessels, thus impairing neovascularization, whereas SC patients, because of their milder phenotype, would have retinal vessels preserved for a longer time, making neovascularization possible in this context (41). We hypothesize that patients with higher inflammatory profile might have had more severe occlusion during early years of life, leading to less neovascularization and clinically less retinopathy. Nevertheless, a recent study found that use of hydroxyurea, with HbF levels >15%, is associated with less retinopathy in SS patients (39). Hydroxyurea, besides increasing HbF expression, lowers the levels of circulating leukocytes and has anti-inflammatory effects in SCD (43, 44). Prospective studies assessing inflammatory markers and retinopathy in these settings are warranted for confirming our hypothesis.
MICA, a transmembrane glycoprotein encoded by the highly polymorphic MICA locus, located in the major histocompatibility complex (MHC) in chromosome 6. MICA, the most polymorphic non-classical HLA-class I gene, is expressed in few specific cell types upon cellular and genotoxic stress, malignant transformation, and viral infection [reviewed in Isernhagen et al. (45)]. MICA acts as a ligand for NKG2D. The SNP rs1059172 is of interest, because a missense mutation in this position (position 129 of the MICA protein, G to A) leads to an amino-acid change, from valine to methionine. The isoform with methionine has high affinity for NKG2D, resulting in enhanced NKG2D signaling, whereas valine determines low affinity (46, 47). Nevertheless, in our study, the A allele was associated with less complications, which is somewhat contra-intuitive. Isernhagen et al. (47) reviewed studies of association between rs1051792 and several diseases or clinical complications; the association of alleles and genotypes with higher or lower inflammatory responses was not consistent. Therefore, for this SNP, although the role of the amino-acid change in NKG2D activation is biologically established, its clinical repercussion in several disease settings, including SCD, needs further evaluation. One possible hypothesis, given that the main function of NK cells is the pivotal cellular cytotoxicity mediated anti-infectious responses, is that overall, the observed association could reflect a protective effect against infections acting before the occurrence of inflammatory-mediated complications.
HLA-G is involved in immunotolerance and, in physiological conditions, is expressed in some restricted human tissues and in the plasma; altered regulation of HLA-G expression has been described in several inflammatory diseases (48). Nevertheless, few studies investigated the role of HLA-G in SCD. In our study, the SNP rs9380142 AA genotype was associated with less complications, whereas the G allele was associated with more cholelithiasis. Cholelithiasis in SCD occurs due to hemolysis, which contributes to inflammatory activation in SCD settings (6). Thus, we hypothesize that the association found might reflect higher inflammatory activation because of hemolysis in patients carrying the G allele. Nevertheless, to date, there is no association between HLA-G and hemolysis in SCD. A study evaluating the association of SNPs in the HLA-G locus and the occurrence of hepatitis C virus (HCV) infection showed no effect of HLA-G SNPs in hemoglobin, lactate dehydrogenase and bilirubin levels (49). In our study, we did not have laboratory parameters of hemolysis for further comparisons.
In this cohort, most patients presented at least two SCD complications. Patients who have more complications often show a more severe phenotype, including life-threatening complications. Therefore, finding markers that predict the amount of complications is of value, because anticipating disease severity might lead to treatment optimization. Of note, the TLR2 rs4696480 TA genotype was significantly associated with occurrence of 0–1 clinical complications. Previously, our group has found that the same genotype was associated with less bacterial infections in SCD (15). Patients with SCD exhibit a marked susceptibility to infections, compared with non-SCD individuals, due to several factors, such as loss of spleen function and the chronic inflammatory status (50). This explains why comparisons between SCD and non-SCD populations, and among SCD patients, may uncover different effects of a given SNP—for instance, alleles and genotypes that modify the function of TLR and modulate the occurrence of infections in this population. Based on our findings and in previous studies on the functional role of this SNP (15, 51–56), we hypothesized that TA genotype might determine a balanced inflammatory response, whereas the TT genotype would determine a weak inflammatory response, and the AA genotype, an overwhelming inflammatory response pattern. Further studies assessing the role of this SNP and its protein expression are required in SCD settings.
Interestingly, patients who had stroke were less likely to show other complications. Life-long chronic transfusion therapy is well-established as long-term secondary prophylaxis for stroke. Thus, the lower probability of other complications in this set is probably explained by the higher rate of patients receiving chronic transfusion to lower HbS.
The retrospective nature of this study prevented us to further investigate causal relations between the SNPs and SCD complications. Furthermore, for the same reason, it was not possible to further assess the mechanisms involved in the modulation of inflammatory response promoted by the SNPs that had significant associations with SCD complications. Also, we cannot exclude that the associations found in this study might reflect the effect of SNPs in other genes inherited in LD with the SNPs tested. However, the size of the cohort improves the strength of our findings. Moreover, we have chosen SNPs that had a previously demonstrated role in inflammation. Studies including larger and/or prospective cohorts, assessing genome-wide associations and gene expression are warranted to further elucidate the influence of different inflammatory pathways in SCD pathophysiology. In addition, because the steady-state inflammatory status found in this disease, which may lower the threshold for activating some inflammatory pathways, SCD might be a good model for studying inflammatory genes.
The recent understanding about inflammation in SCD led to the development of drugs that block inflammatory pathways (57, 58). However, specific inflammatory pathways that determine the occurrence of some complications remain unknown; in addition, it is worrisome that we still cannot predict the occurrence of most complications in a disease with such a heterogeneous clinical presentation. A greater knowledge on inflammatory drivers of SCD would help not only finding biomarkers for predicting complications, but also further developing targeted therapies. Furthermore, establishing a genetic inflammatory profile of patients with severe SCD might allow the identification of patient sub-groups potentially eligible for curative hematopoietic stem cell transplantation or gene therapy.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
The studies involving human participants were reviewed and approved by Comitê de Etica em Pesquisa, Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo; Eurocord scientific committee. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
KT-M, BS, EG, RG, and RT designed the study. KT-M, IL, and CK collected clinical data. KT-M performed experiments. CM and IA advised experiments. FF, JS, and KT-M performed statistical analysis. RG, YB, ID, IL, AS, SK, ER, SG, GF, VR, FN, RC, CD, KM, and LJ provided cases and samples for the study. KT-M, FV, AR, EG, BS, RG, HR, BC, GS, and RT wrote the manuscript. All authors edited and approved the manuscript.
This work was supported by the Centre Scientifique de Monaco, by the Brazilian Coordination for the Improvement of Higher Education Personnel (CAPES, grant number 88881.133635/2016-01), and by the Center for Cell-based Therapy (CTC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank the Centre Scientifique de Monaco, CAPES, CTC, all patients, and their families.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.02041/full#supplementary-material
Figure S1. Linkage disequilibrium between SNPs. (A) Linkage disequilibrium between TLR SNPs. (B) Linkage disequilibrium between NKG SNPs. (C) Linkage disequilibrium between HLA, MICA, and CTLA4 SNPs. Linkage disequilibrium (LD) is shown on heat maps. The darker the color, the higher the LD presented between SNPs.
Figure S2. Comparison of the allele distribution of each SNP between our cohort and non-SCD populations. 1000G, 1,000 genomes database; BRZ, Brazilian non-SCD population.
1. Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. (2010) 376:2018–31. doi: 10.1016/S0140-6736(10)61029-X
2. Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. (2017) 376:1561–73. doi: 10.1056/NEJMra1510865
3. Adams R, McKie V, Nichols F, Carl E, Zhang DL, McKie K, et al. The use of transcranial ultrasonography to predict stroke in sickle cell disease. N Engl J Med. (1992) 326:605–10. doi: 10.1056/NEJM199202273260905
4. Conran N, Belcher JD. Inflammation in sickle cell disease. Clin Hemorheol Microcirc. (2018) 68:263–99. doi: 10.3233/CH-189012
5. Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, et al. Sickle cell disease. Nat Rev Dis Primers. (2018) 4:18010. doi: 10.1038/nrdp.2018.10
6. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. (2014) 123:377–90. doi: 10.1182/blood-2013-04-495887
7. Norris CF, Surrey S, Bunin GR, Schwartz E, Buchanan GR, McKenzie SE. Relationship between Fc receptor IIA polymorphism and infection in children with sickle cell disease. J Pediatr. (1996) 128:813–9. doi: 10.1016/S0022-3476(96)70334-7
8. Neonato MG, Lu CY, Guilloud-Bataille M, Lapouméroulie C, Nabeel-Jassim H, Dabit D, et al. Genetic polymorphism of the mannose-binding protein gene in children with sickle cell disease: identification of three new variant alleles and relationship to infections. Eur J Hum Genet. (1999) 7:679–86. doi: 10.1038/sj.ejhg.5200360
9. Tamouza R, Neonato MG, Busson M, Marzais F, Girot R, Labie D, et al. Infectious complications in sickle cell disease are influenced by HLA class II alleles. Hum Immunol. (2002) 63:194–9. doi: 10.1016/S0198-8859(01)00378-0
10. Hoppe C, Klitz W, Cheng S, Apple R, Steiner L, Robles L, et al. Gene interactions and stroke risk in children with sickle cell anemia. Blood. (2004) 103:2391–6. doi: 10.1182/blood-2003-09-3015
11. Fertrin KY, Costa FF. Genomic polymorphisms in sickle cell disease: implications for clinical diversity and treatment. Expert Rev Hematol. (2010) 3:443–58. doi: 10.1586/ehm.10.44
12. Tatari-Calderone Z, Gordish-Dressman H, Fasano R, Riggs M, Fortier C, Campbell AD, et al. Protective effect of HLA-DQB1 alleles against alloimmunization in patients with sickle cell disease. Hum Immunol. (2016) 77:35–40. doi: 10.1016/j.humimm.2015.10.010
13. Sippert EÂ, Visentainer JEL, Alves HV, Rodrigues C, Gilli SCO, Addas-Carvalho M, et al. Red blood cell alloimmunization in patients with sickle cell disease: correlation with HLA and cytokine gene polymorphisms. Transfusion. (2017) 57:379–89. doi: 10.1111/trf.13920
14. David S, Aguiar P, Antunes L, Dias A, Morais A, Sakuntabhai A, et al. Variants in the non-coding region of the TLR2 gene associated with infectious subphenotypes in pediatric sickle cell anemia. Immunogenetics. (2018) 70:37–51. doi: 10.1007/s00251-017-1013-7
15. Tozatto-Maio K, Girot R, Ly ID, Rocha V, Silva Pinto AC, Diagne I, et al. A Toll-like receptor 2 genetic variant modulates occurrence of bacterial infections in patients with sickle cell disease. Br J Haematol. (2019) 185:918–24. doi: 10.1111/bjh.15875
16. Oliveira VB, Dezan MR, Gomes FCA, Menosi Gualandro SF, Krieger JE, Pereira AC, et al. −318C/T polymorphism of the CTLA-4 gene is an independent risk factor for RBC alloimmunization among sickle cell disease patients. Int J Immunogenet. (2017) 44:219–24. doi: 10.1111/iji.12334
17. Vichinsky EP, Styles LA, Colangelo LH, Wright EC, Castro O, Nickerson B. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood. (1997) 89:1787–92. doi: 10.1182/blood.V89.5.1787.1787_1787_1792
18. Jain S, Bakshi N, Krishnamurti L. Acute chest syndrome in children with sickle cell disease. Pediatr Allergy Immunol Pulmonol. (2017) 30:191–201. doi: 10.1089/ped.2017.0814
19. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. (1988) 16:1215. doi: 10.1093/nar/16.3.1215
20. Matsushima N, Tanaka T, Enkhbayar P, Mikami T, Taga M, Yamada K, et al. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC Genomics. (2007) 8:124. doi: 10.1186/1471-2164-8-124
21. Oliveira J, Etain B, Lajnef M, Hamdani N, Bennabi M, Bengoufa D, et al. Combined effect of TLR2 gene polymorphism and early life stress on the age at onset of bipolar disorders. PLoS ONE. (2015) 10:e0119702. doi: 10.1371/journal.pone.0119702
22. International Genome Sample Resource. 1000 Genomes|A Deep Catalog of Human Genetic Variation. Available online at: https://www.internationalgenome.org/ (accessed April 18, 2020).
23. Baker FB. The Basics of Item Response Theory. 2nd ed. College Park, MD: ERIC Clearinghouse on Assessment and Evaluation (2001).
24. Moustaki I, Knott M. Generalized latent trait models. Psychometrika. (2000) 65:391–411. doi: 10.1007/BF02296153
25. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: http://cran.r-project.org/doc/manuals/R-intro.pdf
26. Lakkakula BVKS. Association between MTHFR 677C>T polymorphism and vascular complications in sickle cell disease: a meta-analysis. Transfus Clin Biol. (2019) 26:284–8. doi: 10.1016/j.tracli.2019.01.003
27. Lanier LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res. (2015) 3:575–82. doi: 10.1158/2326-6066.CIR-15-0098
28. Wesselkamper SC, Eppert BL, Motz GT, Lau GW, Hassett DJ, Borchers MT. NKG2D is critical for NK cell activation in host defense against Pseudomonas aeruginosa respiratory infection. J Immunol. (2008) 181:5481–9. doi: 10.4049/jimmunol.181.8.5481
29. Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJ. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat Immunol. (2003) 4:557–64. doi: 10.1038/ni929
30. Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci USA. (2003) 100:9452–7. doi: 10.1073/pnas.1632807100
31. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity. (2008) 28:571–80. doi: 10.1016/j.immuni.2008.02.016
32. Fang M, Lanier LL, Sigal LJ. A role for NKG2D in NK cell-mediated resistance to poxvirus disease. PLoS Pathog. (2008) 4:e30. doi: 10.1371/journal.ppat.0040030
33. Poggi A, Zocchi MR. NK cell autoreactivity and autoimmune diseases. Front Immunol. (2014) 5:27. doi: 10.3389/fimmu.2014.00027
34. Ito T, Ito N, Saatoff M, Hashizume H, Fukamizu H, Nickoloff BJ, et al. Maintenance of hair follicle immune privilege is linked to prevention of NK cell attack. J Invest Dermatol. (2008) 128:1196–206. doi: 10.1038/sj.jid.5701183
35. Nickel RS, Osunkwo I, Garrett A, Robertson J, Archer DR, Promislow DEL, et al. Immune parameter analysis of children with sickle cell disease on hydroxycarbamide or chronic transfusion therapy. Br J Haematol. (2015) 169:574–83. doi: 10.1111/bjh.13326
36. Wallace KL, Marshall MA, Ramos SI, Lannigan JA, Field JJ, Strieter RM, et al. NKT cells mediate pulmonary inflammation and dysfunction in murine sickle cell disease through production of IFN-gamma and CXCR3 chemokines. Blood. (2009) 114:667–76. doi: 10.1182/blood-2009-02-205492
37. Kuylenstierna C, Björkström NK, Andersson SK, Sahlström P, Bosnjak L, Paquin-Proulx D, et al. NKG2D performs two functions in invariant NKT cells: direct TCR-independent activation of NK-like cytolysis and co-stimulation of activation by CD1d. Eur J Immunol. (2011) 41:1913–23. doi: 10.1002/eji.200940278
38. Hayashi T, Imai K, Morishita Y, Hayashi I, Kusunoki Y, Nakachi K. Identification of the NKG2D haplotypes associated with natural cytotoxic activity of peripheral blood lymphocytes and cancer immunosurveillance. Cancer Res. (2006) 66:563–70. doi: 10.1158/0008-5472.CAN-05-2776
39. Mian UK, Tang J, Allende APM, Heo M, Bernstein N, Vattappally L, et al. Elevated fetal haemoglobin levels are associated with decreased incidence of retinopathy in adults with sickle cell disease. Br J Haematol. (2018) 183:807–11. doi: 10.1111/bjh.15617
40. Friberg TR, Young CM, Milner PF. Incidence of ocular abnormalities in patients with sickle hemoglobinopathies. Ann Ophthalmol. (1986) 18:150–3.
41. Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood Rev. (2003) 17:167–78. doi: 10.1016/S0268-960X(03)00003-1
42. Elagouz M, Jyothi S, Gupta B, Sivaprasad S. Sickle cell disease and the eye: old and new concepts. Surv Ophthalmol. (2010) 55:359–77. doi: 10.1016/j.survophthal.2009.11.004
43. Nevitt SJ, Jones AP, Howard J. Hydroxyurea (hydroxycarbamide) for sickle cell disease. Cochrane Database Syst Rev. (2017) 4:CD002202. doi: 10.1002/14651858.CD002202.pub2
44. Penkert RR, Hurwitz JL, Thomas P, Rosch J, Dowdy J, Sun Y, et al. Inflammatory molecule reduction with hydroxyurea therapy in children with sickle cell anemia. Haematologica. (2018) 103:50–4. doi: 10.3324/haematol.2017.177360
45. Isernhagen A, Malzahn D, Viktorova E, Elsner L, Monecke S, von Bonin F, et al. The MICA-129 dimorphism affects NKG2D signaling and outcome of hematopoietic stem cell transplantation. EMBO Mol Med. (2015) 7:1480–502. doi: 10.15252/emmm.201505246
46. Steinle A, Li P, Morris DL, Groh V, Lanier LL, Strong RK, et al. Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics. (2001) 53:279–87. doi: 10.1007/s002510100325
47. Isernhagen A, Malzahn D, Bickeböller H, Dressel R. Impact of the MICA-129Met/Val dimorphism on NKG2D-mediated biological functions and disease risks. Front Immunol. (2016) 7:588. doi: 10.3389/fimmu.2016.00588
48. Baricordi OR Stignani M Melchiorri L Rizzo R. HLA-G and inflammatory diseases. Inflamm Allergy Drug Targets. (2008) 7:67–74. doi: 10.2174/187152808785107615
49. Cordero EAA, Veit TD, da Silva MAL, Jacques SMC, Silla LMDR, Chies JAB. HLA-G polymorphism influences the susceptibility to HCV infection in sickle cell disease patients. Tissue Antigens. (2009) 74:308–13. doi: 10.1111/j.1399-0039.2009.01331.x
50. Booth C, Inusa B, Obaro SK. Infection in sickle cell disease: a review. Int J Infect Dis. (2010) 14:2–12. doi: 10.1016/j.ijid.2009.03.010
51. Eder W, Klimecki W, Yu L, von Mutius E, Riedler J, Braun-Fahrländer C, et al. Toll-like receptor 2 as a major gene for asthma in children of European farmers. J Allergy Clin Immunol. (2004) 113:482–88. doi: 10.1016/j.jaci.2003.12.374
52. Oh D-Y, Schumann RR, Hamann L, Neumann K, Worm M, Heine G. Association of the toll-like receptor 2 A-16934T promoter polymorphism with severe atopic dermatitis. Allergy. (2009) 64:1608–15. doi: 10.1111/j.1398-9995.2009.02066.x
53. Kerkhof M, Postma DS, Brunekreef B, Reijmerink NE, Wijga AH, de Jongste JC, et al. Toll-like receptor 2 and 4 genes influence susceptibility to adverse effects of traffic-related air pollution on childhood asthma. Thorax. (2010) 65:690–7. doi: 10.1136/thx.2009.119636
54. Bank S, Skytt Andersen P, Burisch J, Pedersen N, Roug S, Galsgaard J, et al. Polymorphisms in the inflammatory pathway genes TLR2, TLR4, TLR9, LY96, NFKBIA, NFKB1, TNFA, TNFRSF1A, IL6R, IL10, IL23R, PTPN22, and PPARG are associated with susceptibility of inflammatory bowel disease in a Danish cohort. PLoS ONE. (2014) 9:e98815. doi: 10.1371/journal.pone.0098815
55. Lin Y, Gao Z-X, Shen X, Chen M-J, Li Y-T, Li S-L, et al. Correlation between polymorphisms in toll-like receptor genes and the activity of hepatitis B virus among treatment-naïve patients: a case-control study in a Han Chinese population. BMC Infect Dis. (2018) 18:28. doi: 10.1186/s12879-018-2943-x
56. Loft ND, Skov L, Iversen L, Gniadecki R, Dam TN, Brandslund I, et al. Associations between functional polymorphisms and response to biological treatment in Danish patients with psoriasis. Pharmacogenomics J. (2018) 18:494–500. doi: 10.1038/tpj.2017.31
57. Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. (2017) 376:429–39. doi: 10.1056/NEJMoa1611770
58. Kutlar A. Phase-I study of ETA receptor antagonist ambrisentan in sickle cell Disease. In: ASH. Orlando, FL (2019). Available online at: https://ash.confex.com/ash/2019/webprogram/Paper130036.html (accessed December 23, 2019).
Keywords: sickle cell retinopathy, sickle cell disease, inflammation markers, NK cell receptors and ligands, toll-like receptor (TLR), non-classical HLA, CTLA 4, sickle cell complications
Citation: Tozatto-Maio K, Girot R, Ly ID, Silva Pinto AC, Rocha V, Fernandes F, Diagne I, Benzerara Y, Dinardo CL, Soler JP, Kashima S, Araujo IL, Kenzey C, Fonseca GHH, Rodrigues ES, Volt F, Jarduli L, Ruggeri A, Mariaselvam C, Gualandro SFM, Rafii H, Cappelli B, Nogueira FM, Scigliuolo GM, Guerino-Cunha RL, Malmegrim KCR, Simões BP, Gluckman E and Tamouza R (2020) Polymorphisms in Inflammatory Genes Modulate Clinical Complications in Patients With Sickle Cell Disease. Front. Immunol. 11:2041. doi: 10.3389/fimmu.2020.02041
Received: 20 April 2020; Accepted: 27 July 2020;
Published: 04 September 2020.
Edited by:
Delmiro Fernandez-Reyes, University College London, United KingdomReviewed by:
Antonio Riva, Foundation for Liver Research, United KingdomCopyright © 2020 Tozatto-Maio, Girot, Ly, Silva Pinto, Rocha, Fernandes, Diagne, Benzerara, Dinardo, Soler, Kashima, Araujo, Kenzey, Fonseca, Rodrigues, Volt, Jarduli, Ruggeri, Mariaselvam, Gualandro, Rafii, Cappelli, Nogueira, Scigliuolo, Guerino-Cunha, Malmegrim, Simões, Gluckman and Tamouza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karina Tozatto-Maio, a2FyaW5hLnRtYWlvQGhjLmZtLnVzcC5icg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.