 Elizabeth L. Siegler1,2
Elizabeth L. Siegler1,2 Saad S. Kenderian1,2,3,4*
Saad S. Kenderian1,2,3,4*- 1T Cell Engineering, Mayo Clinic, Rochester, MN, United States
- 2Division of Hematology, Mayo Clinic, Rochester, MN, United States
- 3Department of Immunology, Mayo Clinic, Rochester, MN, United States
- 4Department of Molecular Medicine, Mayo Clinic, Rochester, MN, United States
Chimeric antigen receptor T (CART) cell immunotherapy has been remarkably successful in treating certain relapsed/refractory hematological cancers. However, CART cell therapy is also associated with toxicities which present an obstacle to its wider adoption as a mainstay for cancer treatment. The primary toxicities following CART cell administration are cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS). New insights into the mechanisms of these toxicities have spurred novel treatment options. In this review, we summarize the available literature on the clinical manifestations, mechanisms, and treatments of CART-associated CRS and ICANS.
Introduction
Three mainstays of cancer therapy – surgery, chemotherapy, and radiation – are often insufficient to induce long-term remissions. Within the last few decades, cancer immunotherapy has made great strides in harnessing and enhancing the patient’s own immune system to fight cancer. One such type of cancer immunotherapy is chimeric antigen receptor T (CART) cell therapy, in which T cells are engineered to express a synthetic receptor which confers specificity against a particular antigen overexpressed on the cancer cell surface. Since the late 1980s, CART cells have gone from a little-known academic project (1) to an FDA approved therapy which has treated over 1000 patients with cancer in the United States alone.
Initially, CARs consisted of an antibody-derived extracellular antigen recognition domain linked to an intracellular CD3ζ signaling domain. These first-generation CART cells showed little therapeutic effects in early clinical trials (2–5). A major breakthrough in the field occurred when researchers developed second-generation CART cells, which included an additional costimulatory domain such as 4-1BB or CD28 to improve persistence and potency.
In the last decade, second-generation CART cells have led to great clinical success in treating B-cell malignancies. The first promising CART clinical trial showed durable remission in a patient with chronic lymphocytic leukemia treated with CD19-targeted CART (CART19) cells (6). Other early clinical trials showed similar potential in treating B-cell cancers (7, 8). These early successes paved the way for pivotal trials, such as the ELIANA trial for pediatric patients with acute lymphoblastic leukemia (ALL) (9) and the ZUMA-1, JULIET, and TRANSCEND trials for patients with B-cell lymphomas (10–12). CART pivotal trials demonstrated astounding overall response rates (ORR) and complete responses (CR) and led to the FDA approval of two CART products, tisagenlecleucel and axicabtagene ciloleucel, in 2017.
Despite the astonishing initial success in treating many B-cell malignancies, CART cell therapy often fails to yield durable responses. Although ORR and CR are generally high in CART cell clinical trials against blood cancers, 30–60% of patients will relapse (13) due to limited CART persistence (14) or antigen escape (15, 16). These relapse mechanisms have been observed in both blood cancers and solid tumors following CART cell treatment. Additionally, solid tumors are surrounded by an immunosuppressive tumor microenvironment composed of immune cells, vasculature, and stromal cells, which create physical and immunological barriers to CART cells. As such, clinical trials of CART cell therapy in solid tumors have been largely disappointing.
In addition to a lack of antitumor efficacy or durable remission, severe toxicities following CART cell administration pose significant threats to patients. Despite remarkable clinical success, incidences of CART-associated toxicities remain high. These toxicities are often severe and occasionally fatal. One analysis of over 1000 patients treated with tisagenlecleucel or axicabtagene ciloleucel reported the sobering statistic that 7% of patients died due to non-relapse mortality within 30 days of initial CART cell administration (17). Life-threatening adverse events present a challenge to more widespread adoption of CART cell therapy in the clinic. In this review, we will discuss types and mechanisms of the two most common CART-associated toxicities, cytokine release syndrome (CRS) and neurotoxicity, as well as existing and emerging management strategies.
Cytokine Release Syndrome
Clinical Manifestations
The most common toxicity after CART cell infusion is CRS, which has been reported to be as high as 100% in some CART19 clinical trials (9, 10, 18–20). Various grading scales have been used in clinical trials by different groups to determine the severity of CRS; consequently, the reported rates of CRS vary among the different scales. The American Society for Transplantation and Cellular Therapy (ASTCT) created a grading scale in order to standardize the assessment and treatment of CART-associated toxicities, including CRS. CRS of any severity can include constitutional symptoms such as headache, nausea, dyspnea, myalgias, and malaise. According to ASTCT guidelines, Grade 1 CRS includes fever (≥38.0°C) without hypotension or hypoxia. Grade 2 CRS is described as fever plus hypotension without requiring vasopressors and/or hypoxia requiring oxygen via low-flow nasal cannula. Grade 3 CRS presents with fever, hypotension requiring one vasopressor, and hypoxia requiring high-flow nasal cannula. Finally, Grade 4 CRS escalates to fever plus hypotension requiring multiple vasopressors and/or hypoxia requiring positive pressure (21).
Acquired hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS) also stems from immune hyperactivation. Patients who develop HLH/MAS present with fever as well as elevated serum levels of ferritin, triglycerides, and cytokines including interferon (IFN)γ, interleukin (IL)-6, IL-10, macrophage inflammatory protein (MIP)1β, and monocyte chemoattractant protein (MCP)-1 (22, 23). Most patients with severe CRS display laboratory markers that meet the criteria for HLH/MAS, and in most cases, HLH/MAS symptoms fade with CRS resolution (24), indicating that these two hyperimmune disorders share common pathways and features and are not easily untangled from each other. As such, HLH/MAS is not treated as a separate CART-associated adverse event according to ASTCT guidelines.
Cytokine release syndrome onset typically occurs during the first week after CART cell treatment. However, patients treated with 4-1BB-costimulated CART cells often experience later onset of CRS than patients treated with CD28-costimulated CART cells (25). CRS onset usually coincides near the peak of CART cell expansion and cytokine production. In general, CD28-costimulated CART cells expand more rapidly than slower growing but more persistent 4-1BB-costimulated CART cells (26–28), contributing to the difference in CRS onset between these two constructs.
C-reactive protein (CRP) and ferritin are diagnostic markers of CRS and are monitored daily after CART cell infusion (29). Elevated D-dimer and low fibrinogen are indicative of coagulopathy following CART cell therapy and are also checked daily (30). Elevated triglycerides are a symptom of HLH/MAS and are monitored post-CART cell infusion as well (29). In addition to these clinical markers, several predictive biomarkers of CRS have been proposed. These predictive markers may lead to preemptive treatment in high-risk patients; preliminary data of early intervention in pediatric patients with mild CRS demonstrated trends toward reducing the incidence of subsequent severe (Grade 3+) CRS (31). Scientists reported a predictive algorithm of CRS in pediatric patients based on serum levels of IFNγ, IL-13, and MIP1α within 72 h after CART cell infusion (24). Researchers found that cytokines including IFNγ, IL-6, IL-8, IL-10, and MCP-1 were significantly higher within 36 h of CART cell administration in patients who went on to experience severe (Grade 4+) CRS compared to patients who developed mild or moderate CRS. They found that, in particular, elevated MCP-1 levels above a certain threshold were the most accurate in predicting severe CRS development. These researchers also identified several independent predictors of CRS: high tumor burden, prior lymphodepletion with fludarabine and cyclophosphamide, high CART dose, and high peak CART blood counts all correlated with CRS development (32). Some of these risks can be mitigated, for example, by debulking the tumor prior to CART cell infusion (22, 33, 34). However, many of the factors that contribute to a higher risk of CRS also help optimize CART cell efficacy. Lymphodepleting regimens such as fludarabine/cyclophosphamide are associated with greater CRS occurrence, yet are important in creating a favorable immune environment for CART cell activity. Fludarabine/cyclophosphamide conditioning has been shown to influence the cytokine milieu, possibly by eliminating cells that serve as cytokine sinks for IL-7 and IL-15 and subsequently increasing the availability of these pro-survival cytokines to therapeutic lymphocytes, in addition to eliminating immunosuppressive cells such as regulatory T cells (29, 35–37). Combination of CART cell therapy with checkpoint blockade has displayed evidence of boosting CART cell efficacy but also has the potential to increase the risk of CRS (38, 39), although some clinical trials have not reported excess toxicity (40, 41). Furthermore, although it is an undesired and occasionally severe side effect, the occurrence of CRS is also linked to improved clinical response to CART cell therapy.
It is important to note that while most clinical experience involves CART19 cell therapy, CART cell therapies targeting other hematological antigens such as BCMA have resulted in CRS incidence at comparable levels to CART19 cells (42). CRS-like symptoms, including fever, lymphopenia, myalgia, and headache, were detailed in a case study of a patient who received intracranial IL-13Rα2-targeted CART cell infusions for glioblastoma (43); however, these symptoms were mild, and the localized routes of administration did not appear to increase unwanted toxicity. In a phase I trial of EGFRvIII-targeted CART cells given intravenously to patients with glioblastoma, symptoms including fever, hypotension, and elevated CRP and IL-6 were observed but attributed to localized intracranial CRS rather than systemic CRS (44). Other than these isolated observations during the treatment of glioblastoma, CRS has not been reported in CART cell therapy for solid tumors to date. CRS results from massive CART cell activation and ensuing systemic inflammation; CART cells are more localized and less stimulated in solid tumors than in hematological cancers and as such, do not trigger CRS. Rather, the main CART-associated toxicity in solid tumors is on-target off-tumor effects, in which CART cells attack not only tumor cells but also healthy cells expressing the target antigen. On-target off-tumor toxicity has been noted in clinical trials of CART cells targeting CAIX (45), Her2 (46, 47), CEA (48), IL13Rα2 (49), EGFR (50), EGFRvIII (51), CD171 (52), and TAG-72 (53).
Mechanisms
Greater insight into the mechanisms behind CART-associated toxicities has been made through correlative science and retrospective analysis of clinical trials and through improvements in preclinical models. Key cells and cytokines involved in CART-related toxicities are shown in Figure 1, and symptoms, markers, and mechanisms are summarized in Table 1. Mechanisms behind CRS are complicated and poorly understood. After the patient is infused, CART cells come into contact with target cells and release inflammatory cytokines such as tumor necrosis factor (TNF)α and IFNγ, which in turn activate monocytes and macrophages to secrete cytokines including IL-1, IL-6, and inducible nitric oxide synthase (iNOS).
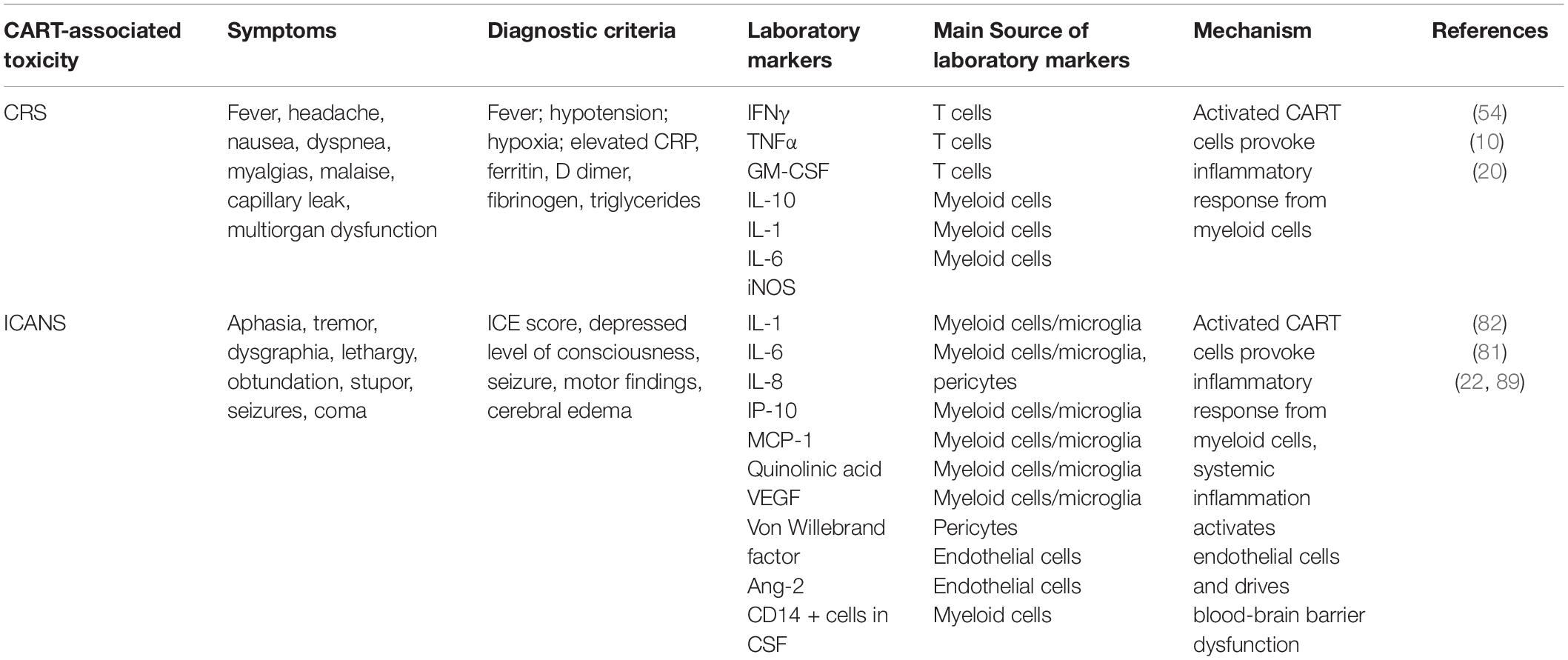
Table 1. Summary of CART cell toxicities.
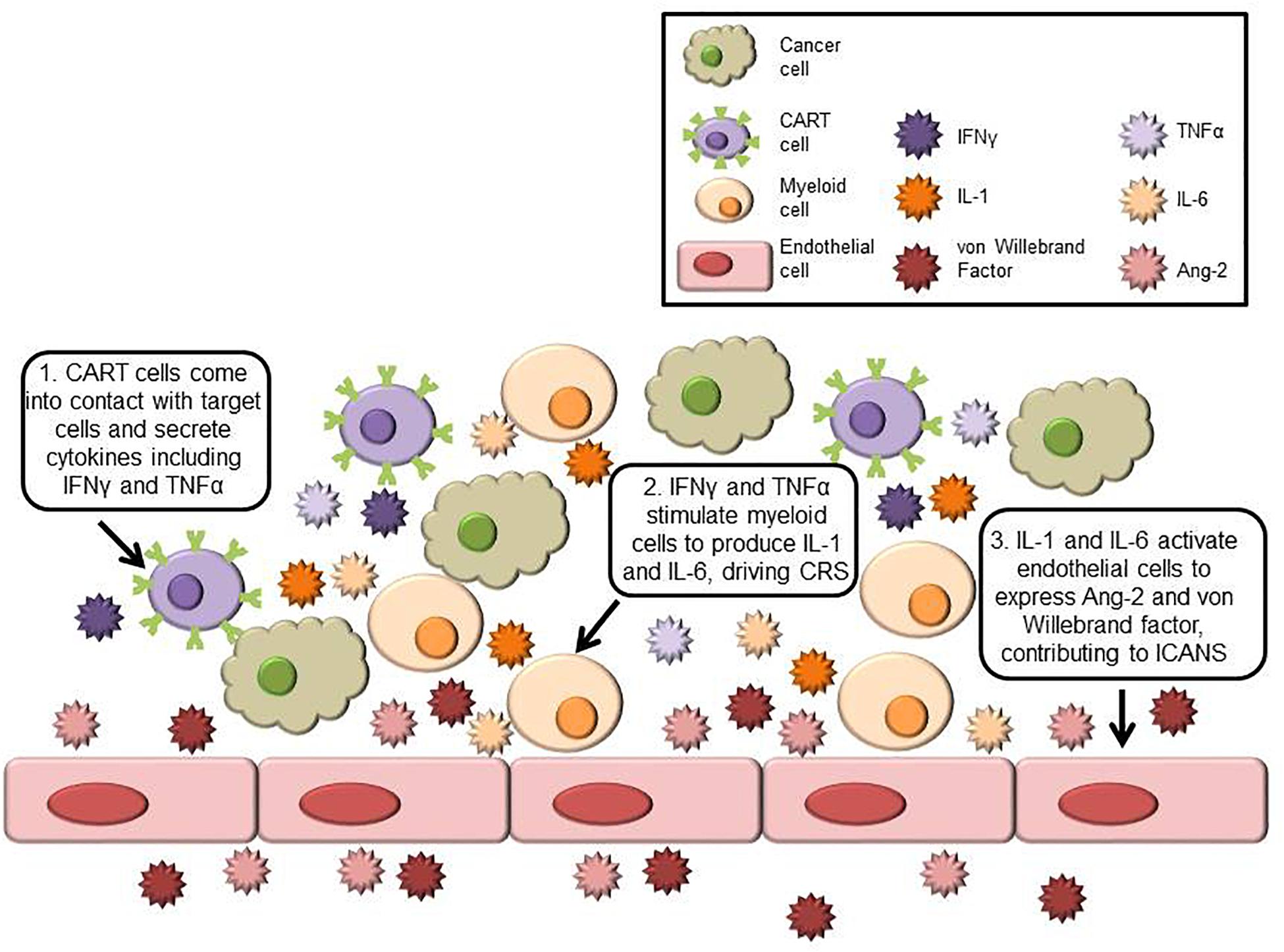
Figure 1. Schematic of key cells and cytokines involved in CRS and ICANS. Cancer cells expressing the target antigen stimulate CART cells to secrete inflammatory cytokines including IFNγ and TNFα. These cytokines activate myeloid cells, which produce CRS-linked cytokines such as IL-1 and IL-6. In ICANS, activated endothelial cells produce von Willebrand factor and Ang-2 and contribute to blood-brain barrier dysfunction.
Recent studies are shedding more light on the cytokines secreted by myeloid cells in relation to CRS development. The inflammatory cytokine IL-6 is highly elevated in patients experiencing CRS (54), indicating an important role in mediating CRS onset. Preclinical models of CRS have shown that engrafted human CART cells produce IFNγ and granulocyte-macrophage colony-stimulating factor (GM-CSF), which in turn activate myeloid cells. Host myeloid cells release large amounts of IL-6 and iNOS when colocalized with tumor and CART cells. Furthermore, engrafting mice with CART cells expressing murine CD40L, which interacts with host myeloid cells, exacerbates CRS symptoms (55). These data suggest that direct interactions between CART and host myeloid cells – primarily macrophages – lead to increased IL-6 and iNOS production and subsequent CRS; however, earlier in vitro studies have concluded that direct contact between CART and myeloid cells is not required to produce IL-6 (56). Animal models have also shown a correlation between monocyte count and CRS mortality after CART cell administration. Monocytes were found to be the primary source of IL-1, which preceded monocyte production of IL-6. Depleting monocytes prior to CART cell treatment decreased CRS incidence but also dampened CART cell therapeutic effects (57). Many HLH/MAS symptoms and markers overlap with those of severe CRS; the association of HLH/MAS with CRS also points to the importance of macrophage hyperactivation as a trigger of CRS.
Treatments
Elucidating the mechanisms of CART-related toxicities has facilitated the development of more effective treatment protocols and of novel treatment approaches. Table 2 summarizes the current and investigational approaches for the treatment of CART-associated toxicities. Treatment plans can be guided by the ASTCT grading system for CART toxicities (21). Current guidelines for CRS management after CART cell therapy vary between clinics but typically involve supportive care and treatment with the anti-IL-6R antibody, tocilizumab. Originally used to treat rheumatoid and juvenile arthritis, tocilizumab was FDA approved alongside tisagenlecleucel in 2017 to treat CRS after CART cell therapy (9, 10). Tocilizumab does not appear to affect CART cell efficacy in mice (57) or therapeutic outcomes in patients (10, 19, 58, 59). Tocilizumab often resolves symptoms of CRS within hours and has become the standard of care. Siltuxumab is a clinically available antibody against IL-6 and has also been used to treat CRS, although less frequently than tocilizumab (60). Corticosteroids have been used to treat severe CRS if unresponsive to tocilizumab (7, 10, 33, 61).
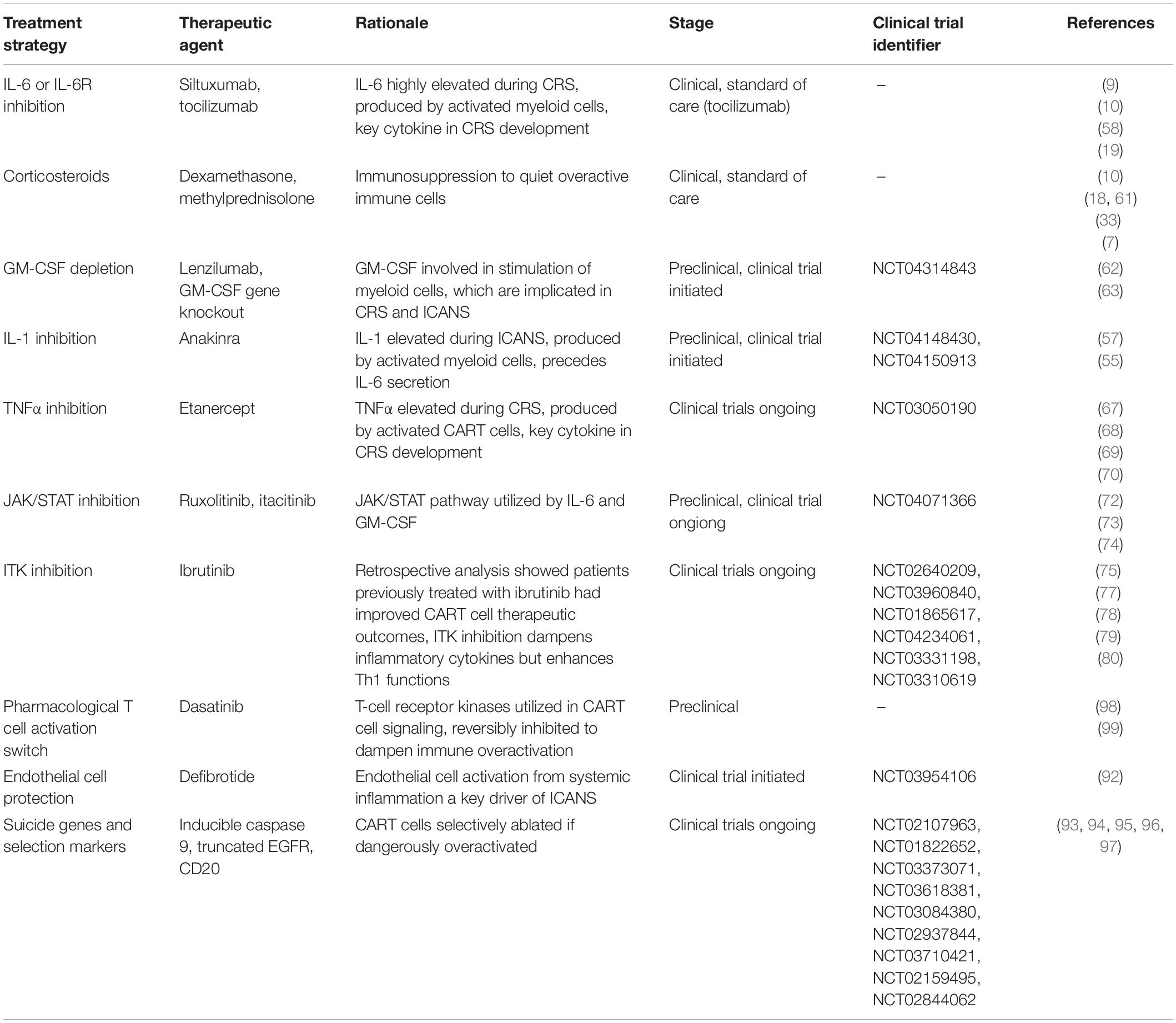
Table 2. Summary of current and investigational approaches to CART-associated toxicities.
Patients refractory to tocilizumab and corticosteroids remain difficult to treat, and new strategies for the management or prevention of CRS are ongoing. Given the success of tocilizumab in treating CRS, there is a strong rationale for the selective inhibition of additional key cytokines involved in CART-related toxicities. GM-CSF has been implicated in the stimulation of myeloid cells, which greatly contribute to CRS development. Preclinical studies have shown that treatment with the anti-GM-CSF antibody, lenzilumab, had no negative effects on CART cell function in vitro or in vivo and even improved leukemic disease control in mice. Furthermore, GM-CSF neutralization reduced CRS symptoms in a patient-derived xenograft model. GM-CSF was also genetically nullified by using a CRISPR-Cas9 platform; GM-CSF knockout CART cells led to improved overall survival in mice, indicating additional potential for next-generation gene-edited CART cells (62). Another study demonstrated that GM-CSF neutralized by antibodies or knocked out with TALEN technology ablated macrophage-associated cytokines linked to CRS development, including MCP-1, IL-6, and IL-8 (63). A clinical trial has been designed using lenzilumab to prevent toxicities in patients receiving axicabtagene ciloleucel.
IL-1 is an inflammatory cytokine produced by myeloid cells and has been linked to CRS. Anakinra, another drug used to treat rheumatoid arthritis, is an IL-1R antagonist and has been explored to treat CART-associated toxicities. Researchers found that monocytes produced IL-1 earlier than IL-6 when cocultured with CART cells. When mice were treated with anakinra, CRS was eliminated while CART cell anticancer efficacy was preserved (57). In another preclinical study, anakinra downregulated iNOS expression by macrophages and reduced mortality due to CRS in CART-treated mice (55). Anakinra has been shown to be effective in treating patients with HLH (64–66), and clinical trials have been initiated to investigate this promising strategy for CART-related CRS.
Treatment with the soluble TNFα receptor, etanercept, helped rapidly resolve CRS symptoms in one pediatric patient (67) but had no clear clinical benefit in an adult patient (68), both of whom experienced severe CRS after CART19 cell infusion. However, etanercept is more widely used to treat CART-associated CRS in clinical trials in China: several patients were treated with etanercept alone or in combination with tocilizumab during phase I/II trials (69, 70).
Another approach to managing CRS is to modulate the T-cells with small molecule inhibitors. IL-6 and GM-CSF utilize the JAK/STAT signaling pathway, and inhibiting this pathway has shown to be effective at dampening CRS after CART cell treatment. Ruxolitinib is an FDA approved JAK/STAT pathway inhibitor which has been shown to reduce inflammatory cytokines in preclinical studies and clinical trials for myeloproliferative neoplasms (71). Ruxolitinib diminished inflammatory cytokines such as IFNγ and TNFα, alleviated symptoms of CRS, and prolonged overall survival in a mouse model of CART-induced CRS (72). However, non-specific targeting of the JAK/STAT pathway might be detrimental to T-cell functions. JAK-1 inhibitors have been investigated as well: itacitinib exerted greater control over inflammatory cytokines than tocilizumab in vitro and reduced serum levels of CRS-linked cytokines without impacting CART cell function in vivo (73). An ongoing phase II clinical trial is investigating itacitinib for the prevention of CRS in patients treated with CART cells (74).
Bruton’s tyrosine kinase (BTK) is a critical component in B-cell receptor signaling, and the BTK inhibitor, ibrutinib, is FDA approved to treat B-cell chronic lymphocytic leukemia. Researchers found that patients treated with ibrutinib for at least 1 year prior to CART cell infusion had decreased T-cell exhaustion markers, better CART cell expansion, and improved clinical outcomes (75). These observations spurred investigation into the effects of ibrutinib on CART cell therapy. Ibrutinib also inhibits IL-2 inducible T-cell kinase (ITK), a tyrosine kinase in the same family as BTK, and appears to enhance Th1 functions by preferentially inhibiting Th2 CD4 T-cells (76). Animal models also demonstrated that combination therapy of ibrutinib with CART cells was more effective than either therapy alone, and CART cells administered with ibrutinib expressed lower levels of exhaustion marker PD1. Peripheral T-cell and CART cell counts were also increased in ibrutinib-treated mice, possibly due to inhibition of the CXCR4 pathway and subsequent peripheral blood lymphocytosis (77). Ibrutinib was further found to drastically reduce IL-6, IFNγ, TNFα, and GM-CSF, preventing CRS and prolonging survival in CART-treated mice. ITK inhibition by ibrutinib is likely the mechanism behind the observed decrease in T-cell inflammatory cytokines, and ibrutinib has the potential to both enhance efficacy and improve safety of CART cell therapy (78). Clinical trials of combination ibrutinib and CART cell therapy are ongoing, and initial results are promising (79, 80).
Neurotoxicity
Clinical Manifestations
Neurotoxicity, also referred to as immune effector cell-associated neurotoxicity syndrome (ICANS), is another common and unique toxicity following CART cell therapy, occurring in up to 67% of patients with leukemia and 62% of patients with lymphoma (81). ICANS usually appears within one to 3 weeks after CART cell infusion, although there have been reports of delayed ICANS development. ICANS often accompanies and correlates with CRS, but it has also been occasionally reported to occur independently from CRS. Early manifestations of ICANS include expressive aphasia, tremor, dysgraphia, and lethargy; these symptoms can progress to global aphasia, seizures, obtundation, stupor, and coma. ASTCT published guidelines for ICANS consensus grading based on immune effector cell-associated encephalopathy (ICE) score, depressed level of consciousness, seizure, motor findings, and cerebral edema (21). Typically, these symptoms will resolve within a week with treatment, but severe ICANS can lead to fatal intracerebral hemorrhage and malignant cerebral edema.
Scientists developed a predictive algorithm based on early fever onset and elevated IL-6 and MCP-1 serum concentrations within 36 h of CART cell infusion to identify patients at risk of developing severe ICANS. These scientists also discovered several baseline characteristics predictive of subsequent ICANS development: younger patient age, B-cell ALL, high marrow disease burden, high CART cell dose, and any pre-existing neurologic comorbidity (82). Researchers also found that elevated IL-2 and IL-5 at day 3 post-CART cell infusion were unique predictors of severe ICANS development (83). These observations may form a basis for preemptive ICANS treatment or adjustment of CART dose in high-risk patients. Most treatment centers use the anti-seizure medication levetiracetam prophylactically on the first day of infusion with CART products linked with higher ICANS incidence (84), and recently discovered predictive biomarkers have the potential to further expand prophylactic treatment protocols to reduce ICANS incidence and severity.
CART cells utilizing CD28 costimulatory domains appear to pose a greater risk for development of ICANS, although the reasons are unclear and the correlations are inconclusive (81). Efforts to develop one CD28-costimulated CART19 product were abandoned due to the ICANS-related deaths of five adult patients with ALL during the ROCKET clinical trial (85). ICANS most commonly occurs with CART19 cell therapy, and ICANS has not been observed in CART cell therapy for solid tumors to date.
Mechanisms
Initially, there had been some concerns that CD19 expression in parts of the central nervous system contributes to the development of ICANS. ICANS can occur in patients treated with CD19-targeting bispecific antibodies as well as with CART19 cells. However, cases of ICANS have been documented with CART cell therapies targeting CD22 (86) and BCMA (87) as well as CD19, making it unlikely that ICANS occurrence is due to the nature of the target antigen and suggests an alternate mechanism. Additionally, a non-human primate study found that ICANS development was not CD19 antigen-specific, as CD20-targeted CART cells also led to ICANS in rhesus macaques (88). This study also found a marked accumulation of both CART and endogenous T cells within the cerebrospinal fluid and brain during ICANS. However, based on clinical trial data, CART cell infiltration in the central nervous system has not been found to correlate with ICANS (81, 89).
There is increasing evidence that myeloid cells have a role in the development of ICANS after CART cell administration. Based on data from mouse models, monocyte-derived IL-1 appeared to mediate ICANS as well as CRS (57), and GM-CSF-mediated stimulation of monocytes after CART cell treatment was linked to neuroinflammation in mice (62). Non-human primate models of ICANS have also shown high levels of IL-6 and GM-CSF in the cerebrospinal fluid after CART cell administration (88). In the clinic, myeloid cell-derived cytokines, most notably IL-1 and IL-6, have been shown to drive systemic inflammation which correlates with severe ICANS development (82). GM-CSF was the cytokine most significantly associated with the development of ICANS after CART cell therapy in the ZUMA-1 clinical trial (10). Analysis of the cerebrospinal fluid of patients who developed severe ICANS showed a 17-fold increase in the infiltration of CD14+ myeloid cells compared to patients with low-grade neurotoxicity (90). Additionally, patients experiencing severe ICANS often display elevated levels of the NMDA receptor agonist quinolinic acid, which is produced by stimulated macrophages. High concentrations of MCP-1, IP-10, IL-6, and IL-8 found during severe ICANS are indicative of activated macrophages and microglia (81). Collectively, these data indicate that myeloid cell hyperactivation contributes significantly to the development of ICANS.
Myeloid cell-driven inflammation may lead to endothelial activation, systemic capillary leak, and subsequent dysfunction of the blood-brain barrier frequently observed in patients with ICANS. Aberrant macrophages were found clustered around brain vasculature in a case study of fatal CART-associated ICANS (91). In another analysis, patients with severe ICANS had elevated serum concentrations of von Willebrand factor and Ang-2, which are released when endothelial cells are activated by inflammatory cytokines. Pericytes exposed to CART-generated inflammatory cytokines such as IFNγ can also produce IL-6 and VEGF, further contributing to blood-brain barrier disruption and allowing the cerebrospinal fluid to be exposed to systemic cytokines (82). It appears that endothelial activation occurs shortly after CART cell administration and precedes ICANS.
Treatments
The standard of care for ICANS includes supportive care and the administration of corticosteroids. Paradoxically, treatment with tocilizumab may worsen ICANS symptoms. Cohort 3 of the Zuma-1 trial treated patients with tocilizumab prophylactically in combination with CART19. While this resulted in a reduction in severe CRS, there was a trend toward increased ICANS rates and severity (90). Tocilizumab is a monoclonal antibody that cannot cross the blood-brain barrier, and it blocks IL-6R in peripheral tissues that act as a systemic sink for IL-6 (20). Instead, corticosteroids are administered to treat ICANS symptoms, and shorter, rapidly tapered doses used in more recent studies have not been shown to suppress CART cell therapeutic response (10). Dexamethasone or methylprednisolone are recommended to treat moderate to severe ICANS (Grade 2+). However, there is no clinical consensus on recommendations of further treatments for patients who do not respond to high-dose steroids.
As with CRS, targeting key cytokines has shown promise in preventing or reducing ICANS after CART cell therapy. GM-CSF secretion contributes to pro-inflammatory myeloid cells, which are associated with ICANS occurrence. Not only did GM-CSF neutralization dampen CRS, but it also resulted in decreased myeloid and T-cell infiltration in the central nervous system and reduction of neuroinflammation in a patient-derived xenograft model. IL-1 secreted by activated myeloid cells has been associated with ICANS. In addition to preventing CRS, the IL-1 antagonist anakinra abolished ICANS symptoms in mice treated with CART cells (57) and is being evaluated in the clinic.
There is strong evidence of the role of endothelial activation and subsequent blood-brain barrier disruption during the development of ICANS. As such, some researchers have considered protecting endothelial cells with defibrotide, an FDA-approved drug for the treatment of hepatic veno-occlusive disease (92). While there is currently no preclinical data available on the effects of defibrotide on CART-related toxicities, a clinical trial has been initiated to investigate the prevention of ICANS with defibrotide in patients receiving axicabtagene ciloleucel.
Additional Methods to Enhance CART Safety
Significant progress has also been made in creating safer CART cells through remote control of CART cell activation or apoptosis. Suicide genes, such as inducible caspase 9, trigger apoptotic pathways upon pharmacological activation and induce rapid and precise apoptosis in CART cells expressing the transgene (93, 94). Similarly, selection markers expressed on the CART cell surface allow CART cell elimination after administration of a neutralizing antibody, such as cetixumab for truncated EGFR-expressing CART cells (95), or rituximab for CD20-expressing CART cells (96). These methods have been explored to ablate CART cells in the event of severe toxicity, including CRS or ICANS, and clinical trials are ongoing. However, these are largely permanent, one-time approaches that will likely nullify the therapeutic effects of CART cells, although more recent studies have demonstrated dose-dependent rather than binary triggering of suicide genes (97).
It is desirable to have tunable and reversible pharmacological inhibition of CART cells in the event of severe toxicities. Researchers have shown that dasatinib, an FDA-approved tyrosine kinase inhibitor, can serve as a switch for the reversible inhibition of CART cell activity in the event of toxicities. Dasatinib temporarily suppresses T-cell activation by inhibiting T-cell receptor signaling kinases, which are also used for signal transduction in CART cells. In vitro, CART cells displayed a dose-dependent decrease in activation and degranulation markers, target cell killing, and cytokine secretion when dasatinib was added to cell cocultures, but regained antitumor functions within hours of dasatinib removal. Similarly, mice treated with dasatinib and CART cells displayed a reduction in CART cell function and tumor control which was restored when dasatinib administration ceased. Furthermore, these mice had significantly lower serum levels of CRS-linked inflammatory cytokines. These studies showed that dasatinib inhibition of CART cell function is dose-dependent and reversible, presenting possibilities for more precise control over CART cell activation over the course of treatment (98). Dasatinib was further tested in vitro and reversibly halted CART cell activity shortly after CART cell stimulation, even after sequential interactions with target cells. In a mouse model of CRS, dasatinib lowered levels of inflammatory cytokines produced by both CART cells and host myeloid cells, and mice receiving dasatinib had significantly prolonged survival (99). Additionally, dasatinib crosses the blood-brain barrier and has potential to mitigate ICANS as well as CRS. A rapid, reversible, titratable method to control CART cell activation is an appealing prospect both in the management of CART cell toxicities and in preventing CART cell exhaustion, and clinical trials are needed to further validate these findings in patients.
Summary and Conclusion
CART cell therapy has yielded impressive outcomes in treating relapsed/refractory B-cell malignancies and has exploded with the field of immunotherapy in the last decade. However, severe and occasionally lethal toxicities, especially CRS and ICANS, remain serious issues in the clinic. As the mechanisms behind these toxicities are unraveled, improved treatment strategies can be formulated. Mounting evidence points to an important role of myeloid cells in CRS and ICANS. Many of the main inflammatory cytokines contributing to these toxicities, such as IL-1 and IL-6, appear to be primarily released by monocytes and macrophages. As such, many newer treatment strategies for CRS and ICANS are aimed at reducing myeloid cell activation. In preclinical models, GM-CSF neutralization reduced myeloid cell-derived inflammatory cytokines and ameliorated CRS and ICANS, and IL-1 inhibition reduced macrophage activity and abolished CRS and ICANS symptoms. Toxicity management strategies targeting T-cells directly have also gained momentum. JAK/STAT inhibitors decrease CRS-associated cytokines and symptoms in mice, and ITK inhibitors lengthen survival in mice and improve clinical outcomes in patients, with reduced incidence of CRS. Additionally, preventing ICANS by protecting endothelial cells with defibrotide during CART cell therapy is currently being tested in the clinic. More precise pharmacological control over CART cell activation has also gained traction in preclinical studies.
Adverse events associated with CART cell therapy, particularly CRS and ICANS, are complex and have overlapping features. Further distillation of the mechanisms behind these toxicities and identification of predictive biomarkers have provided more insight and inspiration for novel management and prevention methods. Ideally, these strategies will become preventative rather than reactive, leading to simplified clinical management, widespread implementation of CART cell therapy outside of specialized treatment centers, reduction of medical and financial burden, and improved patient outcomes.
Author Contributions
ES wrote the initial manuscript. ES and SK edited and approved the final manuscript. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported through K12CA090628 (SK), the National Comprehensive Cancer Network (SK), the Mayo Clinic Center for Individualized Medicine (SK), the Predolin Foundation (SK), and the Exact Foundation (SK).
Conflict of Interest
SK is an inventor on patents in the field of CAR immunotherapy that are licensed to Novartis (through an agreement between Mayo Clinic, University of Pennsylvania, and Novartis), and patents in the field of CAR immunotherapy that are licensed to Humanigen and Mettaforge (through Mayo Clinic). SK receives research funding from Kite, Gilead, Juno, Celgene, Novartis, Humanigen, MorphoSys, Tolero, Sunesis, and Lentigen.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA. (1989) 86:10024–8. doi: 10.1073/pnas.86.24.10024
2. Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. (2000) 96:785–93. doi: 10.1182/blood.V96.3.785
3. Walker RE, Bechtel CM, Natarajan V, Baseler M, Hege KM, Metcalf JA, et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood. (2000) 96:467–74. doi: 10.1182/blood.V96.2.467
4. Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Ther. (2002) 5:788–97. doi: 10.1006/mthe.2002.0611
5. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. (2006) 12(20 Pt 1):6106–15. doi: 10.1158/1078-0432.CCR-06-1183
6. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
7. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. (2011) 3:95ra73. doi: 10.1126/scitranslmed.3002842
8. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. (2012) 119:2709–20. doi: 10.1182/blood-2011-10-384388
9. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
10. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
11. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang ML, Arnason JE, et al. Pivotal safety and efficacy results from transcend NHL 001, a multicenter phase 1 study of lisocabtagene maraleucel (liso-cel) in relapsed/refractory (R/R) large B cell lymphomas. Blood. (2019) 134:241. doi: 10.1182/blood-2019-127508
12. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
13. Xu X, Sun Q, Liang X, Chen Z, Zhang X, Zhou X, et al. Mechanisms of relapse after CD19 CAR T-cell therapy for acute lymphoblastic leukemia and its prevention and treatment strategies. Front Immunol. (2019) 10:2664. doi: 10.3389/fimmu.2019.02664
14. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. (2019) 16:372–85. doi: 10.1038/s41571-019-0184-6
15. Jacoby E, Nguyen SM, Welp KM, Qin H, Yang Y, Chien CD, et al. Lineage switch as a relapse mechanism of Pre-B acute lymphoblastic leukemia following CD19 CAR. Blood. (2015) 126:2524–2524. doi: 10.1182/blood.V126.23.2524.2524
16. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. (2015) 5:1282–95. doi: 10.1158/2159-8290.CD-15-1020
17. Burns E, Anand K, Westin JR, Pingali SRK, Ensor J, Sano D, et al. Comparative review of 30 day non-relapse mortality (NRM) in B-cell lymphomas associated with Anti-CD19 chimeric antigen receptor T-cells (CAR-T) from FDA database, clinical studies, and MD Anderson. Blood. (2019) 134:1931. doi: 10.1182/blood-2019-131077
18. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. (2015) 125:4017–23. doi: 10.1182/blood-2014-12-580068
19. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. (2017) 377:2545–54. doi: 10.1056/NEJMoa1708566
20. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. (2018) 15:47–62. doi: 10.1038/nrclinonc.2017.148
21. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. (2019) 25:625–38. doi: 10.1016/j.bbmt.2018.12.758
22. Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. (2014) 20:119–22. doi: 10.1097/PPO.0000000000000035
23. Fitzgerald JC, Weiss SL, Maude SL, Barrett DM, Lacey SF, Melenhorst JJ, et al. Cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Crit Care Med. (2017) 45:e124–31. doi: 10.1097/CCM.0000000000002053
24. Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. (2016) 6:664–79. doi: 10.1158/2159-8290.CD-16-0040
25. Hirayama AV, Turtle CJ. Toxicities of CD19 CAR-T cell immunotherapy. Am J Hematol. (2019) 94:S42–9. doi: 10.1002/ajh.25445
26. Zhao Z, Condomines M, van der Stegen SJ, Perna F, Kloss CC, Gunset G, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T Cells. Cancer Cell. (2015) 28:415–28. doi: 10.1016/j.ccell.2015.09.004
27. Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
28. Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunology. (2019) 8:e1049. doi: 10.1002/cti2.1049
29. Mahadeo KM, Khazal SJ, Abdel-Azim H, Fitzgerald JC, Taraseviciute A, Bollard CM, et al. Management guidelines for paediatric patients receiving chimeric antigen receptor T cell therapy. Nat Rev Clin Oncol. (2019) 16:45–63. doi: 10.1038/s41571-018-0075-2
30. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. (2016) 127:3321–30. doi: 10.1182/blood-2016-04-703751
31. Gardner RA, Ceppi F, Rivers J, Annesley C, Summers C, Taraseviciute A, et al. Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation of antileukemic efficacy. Blood. (2019) 134:2149–58. doi: 10.1182/blood.2019001463
32. Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. (2017) 130:2295–306. doi: 10.1182/blood-2017-06-793141
33. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. (2014) 6:224ra225. doi: 10.1126/scitranslmed.3008226
34. Yanez L, Sanchez-Escamilla M, Perales MA. CAR T cell toxicity: current management and future directions. Hemasphere. (2019) 3:e186. doi: 10.1097/HS9.0000000000000186
35. Dummer W, Niethammer AG, Baccala R, Lawson BR, Wagner N, Reisfeld RA, et al. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest. (2002) 110:185–92. doi: 10.1172/JCI15175
36. Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol/. (2005) 23:2346–57. doi: 10.1200/JCO.2005.00.240
37. Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. (2005) 26:111–7. doi: 10.1016/j.it.2004.12.003
38. Yoon DH, Osborn MJ, Tolar J, Kim CJ. Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-Ts): combination or built-In CAR-T. Int J Mol Sci. (2018) 19:340. doi: 10.3390/ijms19020340
39. Ceschi A, Noseda R, Palin K, Verhamme K. Immune checkpoint inhibitor-related cytokine release syndrome: analysis of WHO global pharmacovigilance database. Front Pharmacol. (2020) 11:557. doi: 10.3389/fphar.2020.00557
40. Chong EA, Svoboda J, Nasta S, Landsburg D, Winchell N, Napier E, et al. Sequential Anti-CD19 directed chimeric antigen receptor modified T-cell therapy (CART19) and PD-1 blockade with pembrolizumab in patients with relapsed or refractory B-cell non-hodgkin lymphomas. Blood. (2018) 132:4198. doi: 10.1182/blood-2018-99-119502
41. Li A, Hucks G, Dinofia A, Sief A, Teachey D, Baniewicz D, et al. Checkpoint inhibitors augment CD19-directed chimeric antigen receptor (CAR) T cell therapy in relapsed B-cell acute lymphoblastic leukemia. Blood. (2018) 132:556. doi: 10.1182/blood-2018-99-112572
42. Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. (2018) 36:2267–80. doi: 10.1200/JCO.2018.77.8084
43. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
44. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
45. Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. (2006) 24:e20–2. doi: 10.1200/JCO.2006.05.9964
46. Morgan R, Yang J, Kitano M, Dudley M, Laurencot C, Rosenberg S. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
47. Feng K, Liu Y, Guo Y, Qiu J, Wu Z, Dai H, et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell. (2018) 9:838–47. doi: 10.1007/s13238-017-0440-4
48. Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. (2017) 66:1425–36. doi: 10.1007/s00262-017-2034-7
49. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. (2015) 21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428
50. Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci. (2016) 59:468–79. doi: 10.1007/s11427-016-5023-8
51. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. (2019) 42:126–35. doi: 10.1097/CJI.0000000000000260
52. Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. (2007) 15:825–33. doi: 10.1038/sj.mt.6300104
53. Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. (2017) 5:22. doi: 10.1186/s40425-017-0222-9
54. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. (2013) 5:177ra138. doi: 10.1126/scitranslmed.30059305/177/177ra38
55. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. (2018) 24:731–8. doi: 10.1038/s41591-018-0041-7
56. Singh N, Hofmann TJ, Gershenson Z, Levine BL, Grupp SA, Teachey DT, et al. Monocyte lineage-derived IL-6 does not affect chimeric antigen receptor T-cell function. Cytotherapy. (2017) 19:867–80. doi: 10.1016/j.jcyt.2017.04.001
57. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. (2018) 24:739–48. doi: 10.1038/s41591-018-0036-4
58. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
59. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
60. Mahmoudjafari Z, Hawks KG, Hsieh AA, Plesca D, Gatwood KS, Culos KA. American society for blood and marrow transplantation pharmacy special interest group survey on chimeric antigen receptor T cell therapy administrative, logistic, and toxicity management practices in the United States. Biol Blood Marrow Transplant. (2019) 25:26–33. doi: 10.1016/j.bbmt.2018.09.024
61. Maude SL, Barrett DM, Ambrose DE, Rheingold SR, Aplenc R, Teachey DT, et al. Efficacy and safety of humanized chimeric antigen receptor (CAR)-modified T cells targeting CD19 in children with relapsed/refractory all. Blood. (2015) 126:683–683. doi: 10.1182/blood.V126.23.683.683
62. Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. (2019) 133:697–709. doi: 10.1182/blood-2018-10-881722
63. Sachdeva M, Duchateau P, Depil S, Poirot L, Valton J. Granulocyte-macrophage colony-stimulating factor inactivation in CAR T-cells prevents monocyte-dependent release of key cytokine release syndrome mediators. J Biol Chem. (2019) 294:5430–7. doi: 10.1074/jbc.AC119.007558
64. Rajasekaran S, Kruse K, Kovey K, Davis AT, Hassan NE, Ndika AN, et al. Therapeutic role of anakinra, an interleukin-1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children∗. Pediatr Crit Care Med. (2014) 15:401–8. doi: 10.1097/PCC.0000000000000078
65. Kumar B, Aleem S, Saleh H, Petts J, Ballas ZK. A personalized diagnostic and treatment approach for macrophage activation syndrome and secondary hemophagocytic lymphohistiocytosis in adults. J Clin Immunol. (2017) 37:638–43. doi: 10.1007/s10875-017-0439-x
66. Wohlfarth P, Agis H, Gualdoni GA, Weber J, Staudinger T, Schellongowski P, et al. Interleukin 1 receptor antagonist anakinra, intravenous immunoglobulin, and corticosteroids in the management of critically Ill adult patients with hemophagocytic lymphohistiocytosis. J Intensive Care Med. (2019) 34:723–31. doi: 10.1177/0885066617711386
67. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter D, Rheingold S, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
68. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
69. Dong L, Chang L, Gao Z, Lu D, Zhang J, Wang J, et al. Chimeric antigen receptor 4SCAR19-modified T cells in acute lymphoid leukemia: a phase II multi-center clinical trial in China. Blood. (2015) 126:3774–3774. doi: 10.1182/blood.V126.23.3774.3774
70. Zhang JP, Zhang R, Tsao ST, Liu YC, Chen X, Lu DP, et al. Sequential allogeneic and autologous CAR-T-cell therapy to treat an immune-compromised leukemic patient. Blood Adv. (2018) 2:1691–5. doi: 10.1182/bloodadvances.2018017004
71. Elli EM, Barate C, Mendicino F, Palandri F, Palumbo GA. Mechanisms underlying the anti-inflammatory and immunosuppressive activity of ruxolitinib. Front Oncol. (2019) 9:1186. doi: 10.3389/fonc.2019.01186
72. Kenderian SS, Ruella M, Shestova O, Kim MY, Klichinsky M, Chen F, et al. Ruxolitinib prevents cytokine release syndrome after CART cell therapy without impairing the anti-tumor effect in a Xenograft model. Blood. (2016) 128:652. doi: 10.1182/blood.V128.22.652.652
73. Huarte E, O’Connor RS, Parker MM, Huang T, Milone M, Smith P. Prophylactic itacitinib (INCB039110) for the prevention of cytokine release syndrome induced by chimeric antigen receptor T-cells (CAR-T-cells) therapy. Blood. (2019) 134:1934. doi: 10.1182/blood-2019-128288
74. Park JH, Frigault MJ, Maziarz RT, Naim A, Burke L, Tian C, et al. Trial in progress: a phase 2, single-arm, open-label study of itacitinib (ITA) for the prevention of chimeric antigen receptor (CAR) T-cell–induced cytokine release syndrome (CRS). Biol Blood Marrow Transpl. (2020) 26:S269. doi: 10.1016/j.bbmt.2019.12.436
75. Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. (2016) 127:1117–27. doi: 10.1182/blood-2015-11-679134
76. Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. (2013) 122:2539–49. doi: 10.1182/blood-2013-06-507947
77. Ruella M, Kenderian SS, Shestova O, Fraietta JA, Qayyum S, Zhang Q, et al. The addition of the BTK inhibitor ibrutinib to Anti-CD19 chimeric antigen receptor T Cells (CART19) improves responses against mantle cell lymphoma. Clin Cancer Res. (2016) 22:2684–96. doi: 10.1158/1078-0432.CCR-15-1527
78. Ruella M, Kenderian SS, Shestova O, Klichinsky M, Melenhorst JJ, Wasik MA, et al. Kinase inhibitor ibrutinib to prevent cytokine-release syndrome after anti-CD19 chimeric antigen receptor T cells for B-cell neoplasms. Leukemia. (2017) 31:246–8. doi: 10.1038/leu.2016.262
79. Gill S, Vides V, Frey N, Metzger ML, O’Brien M, Hexner EO, et al. Prospective clinical trial of Anti-CD19 CAR T cells in combination with ibrutinib for the treatment of chronic lymphocytic leukemia shows a high response rate. Blood. (2018) 132:298. doi: 10.1182/blood-2018-99-115418
80. Gauthier J, Hirayama AV, Purushe J, Hay KA, Lymp J, Li D, et al. Feasibility and efficacy of CD19-targeted CAR-T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood. (2020) 135:1650–60. doi: 10.1182/blood.2019002936
81. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. (2018) 8:958–71. doi: 10.1158/2159-8290.CD-17-1319
82. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. (2017) 7:1404–19. doi: 10.1158/2159-8290.CD-17-0698
83. Park J, Santomasso B, Riviere I, Senechal B, Wang X, Purdon T, et al. Baseline and early post-treatment clinical and laboratory factors associated with severe neurotoxicity following 19-28z CAR T cells in adult patients with relapsed B-ALL. J Clin Oncol. (2017) 35(15 Suppl.):7024. doi: 10.1200/JCO.2017.35.15_suppl.7024
84. Neelapu SS. Managing the toxicities of CAR T-cell therapy. Hematol Oncol. (2019) 37(Suppl. 1):48–52. doi: 10.1002/hon.2595
85. Gilbert MJ. Severe neurotoxicity in the phase 2 trial of JCAR015 in adult B-ALL (ROCKET Study): analyses of patient, protocol and product attributes. Proceedings of the 32nd Annual Meeting and Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2017). National Harbor, MD. (2017).
86. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. (2018) 24:20–8. doi: 10.1038/nm.4441
87. Alfred L, Garfall E, Edward A, Lacey SF, Dengel K, Ambrose D, et al. Posterior reversible encephalopathy syndrome (PRES) after infusion of anti-Bcma CAR T Cells (CART-BCMA) for multiple myeloma: successful treatment with cyclophosphamide. Blood. (2016) 128:5702. doi: 10.1182/blood.V128.22.5702.5702
88. Taraseviciute A, Tkachev V, Ponce R, Turtle CJ, Snyder JM, Liggitt HD, et al. Chimeric antigen receptor T cell-mediated neurotoxicity in nonhuman primates. Cancer Discov. (2018) 8:750–63. doi: 10.1158/2159-8290.CD-17-1368
89. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
90. Locke FL, Neelapu SS, Bartlett NL, Lekaris LJ, Jacobson CA, Braunschweig I, et al. Preliminary results of prophylactic tocilizumab after axicabtageneciloleucel (axi-cel; KTE-C19) treatment for patients with refractory, aggressive non-hodgkin lymphoma (NHL). Blood. (2017) 130:1547. doi: 10.1182/blood.V130.Suppl_1.1547.1547
91. Torre M, Solomon IH, Sutherland CL, Nikiforow S, DeAngelo DJ, Stone RM, et al. Neuropathology of a case with fatal CAR T-cell-associated cerebral edema. J Neuropathol Exp Neurol. (2018) 77:877–82. doi: 10.1093/jnen/nly064
92. Richardson PG, Riches ML, Kernan NA, Brochstein JA, Mineishi S, Termuhlen AM, et al. Phase 3 trial of defibrotide for the treatment of severe veno-occlusive disease and multi-organ failure. Blood. (2016) 127:1656–65. doi: 10.1182/blood-2015-10-676924
93. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. (2010) 24:1160–70. doi: 10.1038/leu.2010.75
94. Budde LE, Berger C, Lin Y, Wang J, Lin X, Frayo SE, et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One. (2013) 8:e82742. doi: 10.1371/journal.pone.0082742
95. Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. (2011) 118:1255–63. doi: 10.1182/blood-2011-02-337360
96. Tasian SK, Kenderian SS, Shen F, Ruella M, Shestova O, Kozlowski M, et al. Optimized depletion of chimeric antigen receptor T-cells in murine xenograft models of human acute myeloid leukemia. Blood. (2017) 129:2395–407. doi: 10.1182/blood-2016-08-736041
97. Diaconu I, Ballard B, Zhang M, Chen Y, West J, Dotti G, et al. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol Ther. (2017) 25:580–92. doi: 10.1016/j.ymthe.2017.01.011
98. Weber EW, Lynn RC, Sotillo E, Lattin J, Peng X, Mackall C. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. (2019) 3:711–7. doi: 10.1182/bloodadvances.2018028720
Keywords: CART, chimeric antigen receptor, immunotherapy, CRS, neurotoxicity
Citation: Siegler EL and Kenderian SS (2020) Neurotoxicity and Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy: Insights Into Mechanisms and Novel Therapies. Front. Immunol. 11:1973. doi: 10.3389/fimmu.2020.01973
Received: 09 April 2020; Accepted: 21 July 2020;
Published: 28 August 2020.
Edited by:
Ester Lozano, Institut de Recerca Biomèdica August Pi i Sunyer (IDIBAPS), SpainReviewed by:
Shoba Alaska Navai, Baylor College of Medicine, United StatesSujith K. Joseph, Baylor College of Medicine, United States
Copyright © 2020 Siegler and Kenderian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saad S. Kenderian, S2VuZGVyaWFuLnNhYWRAbWF5by5lZHU=