 Maria Chitty-Lopez1
Maria Chitty-Lopez1 Emma Westermann-Clark1
Emma Westermann-Clark1 Irina Dawson1
Irina Dawson1 Boglarka Ujhazi1
Boglarka Ujhazi1 Krisztian Csomos1
Krisztian Csomos1 Kerry Dobbs2
Kerry Dobbs2 Khuong Le2
Khuong Le2 Yasuhiro Yamazaki2
Yasuhiro Yamazaki2 Amir A. Sadighi Akha3
Amir A. Sadighi Akha3 Deepak Chellapandian4Ben Oshrine4
Deepak Chellapandian4Ben Oshrine4 Luigi D. Notarangelo2
Luigi D. Notarangelo2 Gauri Sunkersett4†Jennifer W. Leiding1†
Gauri Sunkersett4†Jennifer W. Leiding1† Jolan E. Walter1,5*†
Jolan E. Walter1,5*†- 1Division of Pediatric Allergy/Immunology, University of South Florida at Johns Hopkins All Children's Hospital, St. Petersburg, FL, United States
- 2Laboratory of Clinical Immunology and Microbiology, NIAID, National Institutes of Health, Bethesda, MD, United States
- 3Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN, United States
- 4Cancer and Blood Disorders Institute, Johns Hopkins All Children's Hospital, St. Petersburg, FL, United States
- 5Division of Pediatric Allergy and Immunology, Massachusetts General Hospital for Children, Boston, MA, United States
The T-cell receptor excision circle (TREC) assay detects T-cell lymphopenia (TCL) in newborns and is especially important to identify severe combined immunodeficiency (SCID). A spectrum of SCID variants and non-SCID conditions that present with TCL are being discovered with increasing frequency by newborn screening (NBS). Recombination-activating gene (RAG) deficiency is one the most common causes of classical and atypical SCID and other conditions with immune dysregulation. We present the case of an asymptomatic male with undetectable TRECs on NBS at 1 week of age. The asymptomatic newborn was found to have severe TCL, but normal B cell quantities and lymphocyte proliferation upon mitogen stimulation. Next generation sequencing revealed compound heterozygous hypomorphic RAG variants, one of which was novel. The moderately decreased recombinase activity of the RAG variants (16 and 40%) resulted in abnormal T and B-cell receptor repertoires, decreased fraction of CD3+ TCRVα7.2+ T cells and an immune phenotype consistent with the RAG hypomorphic variants. The patient underwent successful treatment with hematopoietic stem cell transplantation (HSCT) at 5 months of age. This case illustrates how after identification of a novel RAG variant, in vitro studies are important to confirm the pathogenicity of the variant. This confirmation allows the clinician to expedite definitive treatment with HSCT in an asymptomatic phase, mitigating the risk of serious infectious and non-infectious complications.
Introduction
NBS for SCID was initially developed to identify affected infants, preferably at an asymptomatic stage, facilitating medical intervention before life-threatening complications arise (1–5). The assay is based on the absence or low count of T-cell receptor (TCR) excision circles (TREC), a by-product of TCR V (D) J rearrangement in the thymus during early T-cell development, and is universally available in the United States. Aside from SCID, low TREC detected on NBS also allows for the identification of other conditions associated with primary, secondary, and idiopathic forms of T-cell lymphopenia (TCL). Disorders associated with primary TCL include complete or partial DiGeorge syndrome, CHARGE syndrome, trisomy 21, and ataxia-telangiectasia. Examples of secondary TCL are post-surgical extravasation of fluid and lymphocytes, and gastrointestinal malformations resulting in lymphocyte loss. Finally, idiopathic TCL can occur in as many as 20% of screened cases with low TREC and these infants usually require close follow-up (6–8).
Without early intervention, such as HSCT, enzyme replacement therapy (ERT), or gene therapy (GT), a SCID patient is unlikely to survive beyond the first 2 years of life secondary to infections, failure to thrive, or immune dysregulation (4, 8). The great diversity of TCL-related conditions that can be identified via NBS for SCID in an otherwise asymptomatic child may create a degree of uncertainty regarding management strategies. This particularly applies to conditions where variants in SCID-related genes are discovered, but present with variable immune phenotypes.
Since the initiation of NBS for SCID, this condition is no longer defined by clinical symptoms in the majority of cases. Instead, variants of SCID are classified primarily by immune phenotype. The Primary Immunodeficiency Treatment Consortium (PIDTC) diagnostic algorithm subdivides SCID into three categories—typical SCID, atypical SCID, and Omenn syndrome (OS); patients are classified based on total T-cell enumeration, lymphocyte proliferation, presence of maternal T-cells, characteristic phenotypic features, and gene defects (9). SCID is further characterized by specific genotypes that can result in distinct immune phenotypes based on presence or absence of B and NK cells. X-linked interleukin 2 receptor gamma chain (IL2RG) deficiency, adenosine deaminase (ADA) deficiency, and RAG1/2 deficiencies are common genotypes in typical SCID that can be treated with HSCT. RAG1/2 deficiencies are also the most frequent genotypes to cause atypical SCID variants (4, 5).
There is an emerging category of combined immunodeficiencies (CID) with or without immune dysregulation resulting from hypomorphic variants of SCID genes. Partial RAG defects are an example of this phenomenon (10–13). In contrast with RAG1/2 null variants that typically manifest with a T-B- SCID phenotype, hypomorphic RAG deficiency has a broad spectrum of clinical presentations including OS, atypical SCID, combined immune deficiency with granuloma and/or autoimmunity (CID-G/AI), and TCL (13, 14). Characteristically, a patient with partial RAG deficiency and CID has a low naïve T-cell compartment, but relatively preserved B-cell quantities and immunoglobulin levels that may progress to B-cell lymphopenia and hypogammaglobulinemia with age (13). Patients with CID secondary to hypomorphic RAG variants often do not meet PIDTC criteria for SCID or atypical SCID. As the RAG genes are highly polymorphic and the clinical spectrum of RAG deficiencies is broad, specific functional assays and T cell receptor/B cell receptor (TCR/BCR) repertoire studies are required to establish the link between a novel RAG variant and the patient's immune phenotype.
We present an asymptomatic infant who had undetectable TREC on NBS for SCID and was found to carry compound heterozygous RAG1 variants, resulting in an abnormal immune phenotype.
Case Presentation
The patient is a 1-week-old male newborn admitted with concern for SCID due to undetectable TREC on NBS (normal range >20 copies/microliter). The patient was conceived by in vitro fertilization and born at 35 weeks of gestation. He had no family history of immunodeficiency.
Initial Evaluation
Initial laboratory evaluation was notable for a T−/B+/NK+ immune phenotype (Table 1). Due to concern for SCID, pharmacologic prophylaxis against infections was initiated per institutional protocol with acyclovir, fluconazole, and pentamidine, as well as intravenous immune globulins (IVIG). The mother was cytomegalovirus (CMV) seronegative and the patient had no detectable CMV viral load by PCR, hence breastfeeding was permitted. Further infectious evaluation confirmed undetectable serum viral loads of HIV, EBV, and adenovirus by PCR.
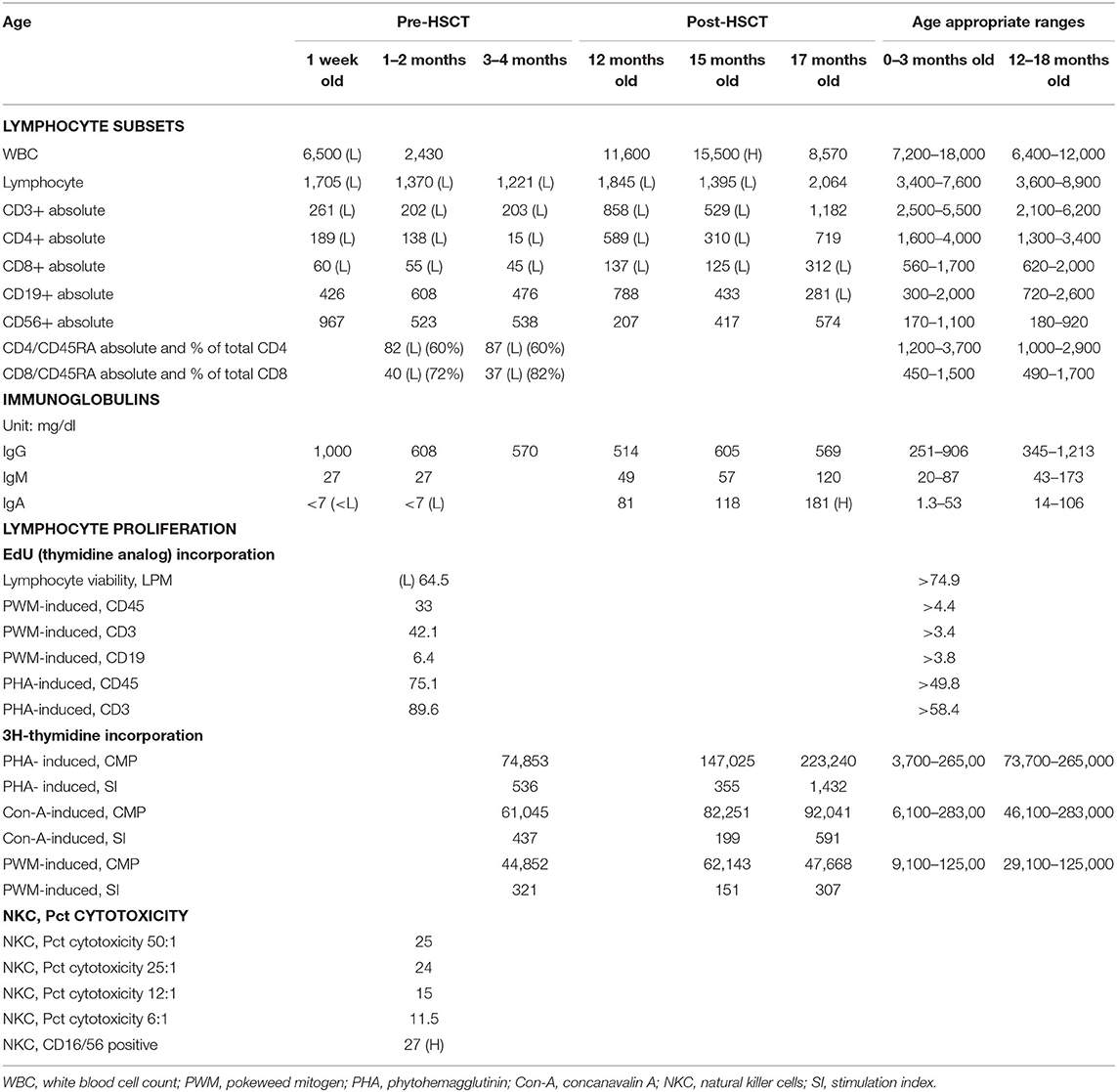
Table 1. Laboratory assessments.
Subsequent evaluation at 1 and 3 months of age revealed that profound T cell lymphopenia (TCL) persisted (<300 cells/μl) and the proportion of naïve CD45RA+CD4+ T-cells was below normal (60%, normal >90%). Maternal engraftment was excluded by short tandem repeats (STR) analysis. A microarray resulted normal. The patient continued to maintain remarkable CD4+ and CD8+ lymphopenia; however, B and NK cell quantities remained within normal range (Table 1). After the initial IVIG dose, the patient was transitioned to subcutaneous immunoglobulin replacement therapy (IgRT), which maintained IgG levels within normal range. IgA level was consistently low (<7 mg/dL) and IgM level remained normal during this time (Table 1). Lymphocyte proliferation to the mitogens phytohemagglutinin (PHA) and pokeweed mitogen (PWM) measured at 1 month of age by chemically labeled EdU (thymidine analog) incorporation method were normal. At 3 months of age, lymphocyte proliferation to PHA, concanavalin A (ConA), and PWM measured by radioactive thymidine incorporation method were normal as well (Table 1). TCR Vβ spectratyping was abnormal: of the 28 probes used for 23 TCR Vβ families and sub-families, one had no peak, 8/28 (29%) demonstrated oligoclonality (i.e., <5 peaks), and 15/28 (53%) showed a polyclonal, non-Gaussian distribution (Figure 1B).
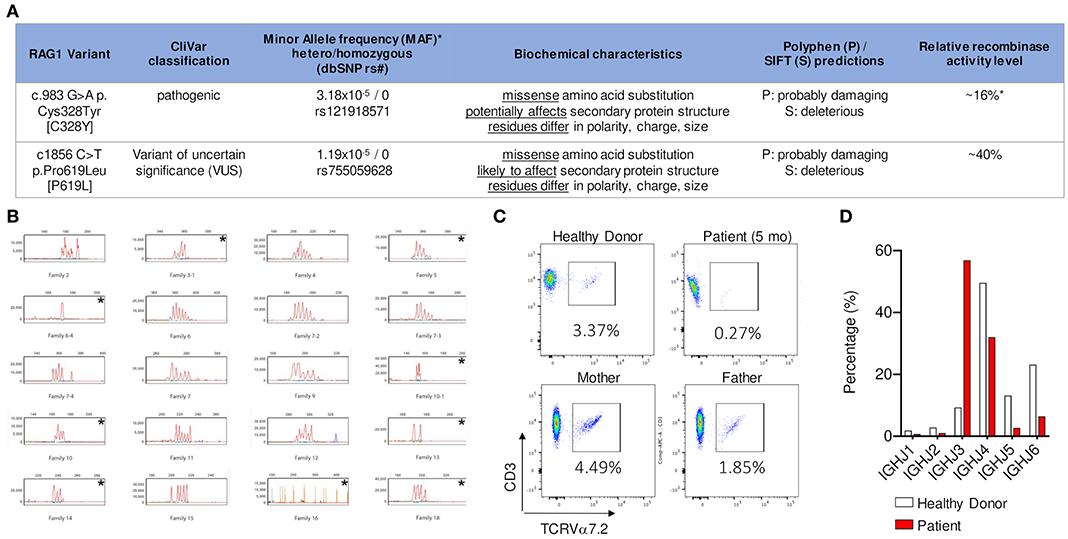
Figure 1. (A) NGS identified two heterozygous missense variants in trans in RAG1. Relative recombination activity level of the two alleles [16% (ref 16) and 40% (novel variant, measured in this study)] predict an overall relative recombinase activity of 28% that is characteristic of CID-G/AI phenotype (*based on gnomAD 12/2019). (B) CD3+ T cells analyzed by spectratyping using 28 probes to measure CDR3 length variability in 23 TCRVβ families and sub-families. Families 2 to 18 are selected for representation. The following families demonstrated an oligoclonal distribution, i.e., <5 peaks: 3–1, 5, 6–4, 10–1, 10, 13, 14, and 18; family 16 had no peak. Each is identified with an * in the upper right corner. (C) Detection of Vα7.2 segment in CD3+ T lymphocytes by flow cytometry shows that the patient had near absence of CD3+T cells expressing the TCR Vα 7.2 chain [encoded by a distal V TCR locus] compared to healthy control and family members (measured in collaboration with the NIH). (D) Frequency of individual IGHJ gene usage of memory B cell receptor repertoire shows decrease in usage of the distal IGHJ5 and IGHJ6 gene elements in patient compared to age matched healthy donor.
Genomic DNA was subjected to target enrichment using hybridization capture with a custom bait pool and sequenced using Illumina sequencing chemistry. A validated bioinformatics pipeline incorporating community standard and custom algorithms was used to simultaneously identify sequence changes and exonic deletions/duplications. Next generation sequencing identified two heterozygous missense variants in trans in RAG1 (RAG1 c.983G>A p.Cys328Tyr [C328Y] and RAG1 c.1856C>T p.Pro619Leu [P619L]). The first variant had been previously published as a homozygous trait in a patient with OS and was also present in the patient's father, who was a heterozygous carrier (15). The second variant was a novel variant of uncertain significance (VUS); the patient's mother was a heterozygous carrier for this variant. These findings confirmed the compound heterozygous status of the patient.
Evaluation of Rag Variants by Population Database Frequency, in silico Analysis, and Recombinase Activity
RAG1 C328Y has not been linked to any specific ethnicity and has a low mean allele frequency (MAF) in heterozygous form 1/31,392 (3.2e-5) (gnomAD database, June 2020). This non-conservative amino acid substitution could potentially affect secondary protein structure, as these residues differ in polarity, charge, size, and/or other properties. Polyphen and SIFT software programs predict this substitution as probably damaging and deleterious, respectively. In vitro relative recombinase activity determined by functional studies has previously shown that C328Y has reduced, but not absent, recombinase activity (16%) (16, 17). Therefore, this variant was established as a likely pathogenic hypomorphic variant.
The pathogenicity of the RAG1 P619L variant has not previously been published or fully evaluated. Population studies detected this variant in South Asian populations with a MAF of 2/30,782 (6.5e-5) and a European cohort with a MAF of 1/113,661 (8.7e-6), with an overall MAF of 3/251,358 (1.19e-5) (gnomAD database, June 2020) (18). This non-conservative amino acid substitution is likely to affect secondary protein structure, as these residues also differ in polarity, charge, size, and/or other properties. Indeed, Polyphen and SIFT predict this variant as probably damaging and deleterious, respectively. Relative recombinase activity was previously unpublished for this variant and so was evaluated using a previously described platform (17). Relative recombinase activity of the RAG1 P619L variant was reduced at 40%. In summary, based on the patient's clinical phenotype, repertoire studies, analysis of predicted genotype-phenotype implications, and relative recombinase activity measurement, it is likely that the RAG1 P619L variant should be reclassified from a VUS to a likely pathogenic hypomorphic variant. Considering one variant had 16% predicted activity and the variant on the other allele 40% predicted activity, the calculated average recombinase activity is 28% (Figure 1A).
TCR and BCR Repertoire Data
TCR/BCR repertoire studies are an in vivo readout for deficiency in V (D) J recombination activity. In particular, reduced use of distal elements can reflect decreased capacity for RAG recombinase activity (19). In comparison to healthy controls, there was also a dramatic decline in usage of the distal IGHJ5 and IGHJ6 gene elements (BCR), suggesting an abnormal V (D) J recombination consistent with partial RAG deficiency (Figure 1D). Detection of Vα7.2 segment (TCR) in CD3+ T lymphocytes and/or mucosa-associated invariant T-cells (MAIT) is a rapid method for evaluating decreased usage of distal TCRαV (TRAV) gene segments, which indicates a V (D) J recombination defect (20). Indeed, in our case we noted a decrease in CD3+ TCRVα7.2+ lymphocytes compared to controls (0.27% vs. 1.85–4.49%) (Figure 1C). The TCR/BCR repertoire studies, recombinase activity assay, and specific flow cytometric testing for distal Vα7.2 element, all support a V (D) J recombination defect with partial RAG deficiency as the underlying cause of disease.
Additional Testing
Lastly, autoantibody testing targeting IFNα, IFNω, and IL-12 was performed by ELISA, as presence of these autoantibodies have been associated with immune dysregulation in partial RAG deficiency (12, 21). These anti-cytokine antibodies were not detected in our patient. Increased natural killer (NK) cell cytotoxicity has been associated with increased rate of graft rejection after HSCT in RAG deficient patients (22). In our patient, NK cell cytotoxicity performed at 2 months of age was normal (Table 1).
Therapeutic Approach
The immune phenotype of our patient included absent TREC, TCL, normal T cell proliferation to mitogens, normal NK cell cytotoxicity, and oligoclonality with reduced relative recombinase activity. Based on his immune abnormalities, HSCT was considered as definitive treatment. The patient lacked an HLA-identical sibling, but several 10/10 HLA-matched unrelated donors (MUD) were available. The patient remained on anti-microbial prophylaxis and protective isolation, infection-free until transplant. At 5 months of age, he underwent HSCT with unmanipulated bone marrow from CMV negative 10/10 MUD (5.56 million CD34+ cells per kg of recipient body weight) with reduced intensity conditioning regimen of sub-myeloablative busulfan, fludarabine, and alemtuzumab, with a busulfan target AUC of 3,500–4,000 μM x min/L (23). Graft versus host disease (GVHD) prophylaxis included tacrolimus with a goal trough level of 10–15 ng/mL and a short-course of methotrexate given on days +1, +3, +6, and +11. The early post-HSCT period was without major complications except for Enterobacter bacteremia and Clostridium difficile colitis that resolved with antibiotics. The patient had platelet engraftment on day +22 and neutrophil engraftment on day +27 after HSCT. He demonstrated 100% donor chimerism in whole blood, myeloid, and NK lineages on day +27 after transplant and 100% donor T-cell chimerism on day +35.
Follow-Up
His post-transplant course was complicated by skin-only stage I GvHD that responded to systemic steroids and tacrolimus, and topical immunomodulating agents tacrolimus 0.03% ointment and hydrocortisone 1% cream. At 12 months of age, the patient had an absolute lymphocyte count of 1,895 cells/μL, with 858 T-cells/μL and 788 B-cell cells/μL. Over the past 2 years, he has maintained >90% donor T-cell chimerism, and 30–70% donor B-cell chimerism. The patient was weaned from IgRT 10 months after HSCT (Table 1). He has remained infection-free, off anti-microbial prophylaxis, has developed no signs of autoimmunity or immune dysregulation, and is thriving.
Discussion
Our case exemplifies the diagnostic and treatment challenges for patients with severe TCL, SCID-related gene defects (i.e., partial RAG deficiency), and atypical immune phenotype. The patient's normal B-cell count and immunoglobulin levels except for IgA, higher preserved fraction of naïve CD4+ (60%) and CD8+ (72–82%) T-cells, and normal lymphocyte proliferation with mitogen stimulation and reduced relative recombinase activity of 28% are unusual for most variants of SCID.
While multiple classifications exist for SCID and related disorders that include guidance from PIDTC, European Society for Immunodeficiencies (ESID) and International Union of Immunological Societies (IUIS) (24), these criteria are not applicable to all patients with SCID. Our patient's absolute CD3 T cell count meets the threshold for typical SCID based on PIDTC criteria. However, lymphocyte proliferation is normal, whereas it is very low in typical SCID and leaky SCID/Omenn syndrome according to the PIDTC criteria. He does not meet ESID criteria for typical SCID either, since he was asymptomatic and did not have an affected family member (25). Because of the patient's severe TCL, he was sequestered from birth until HSCT to prevent infections.
Classical SCID with RAG deficiency typically causes a T-B- phenotype as null RAG activity will impede functional TCR/BCR development and thus jeopardizes T and B cell survival. In cases of hypomorphic variants with partially preserved recombinase activity, T and B cells may survive with a restricted TCR/BCR repertoire. RAG genes are also highly polymorphic; therefore, all novel variants require confirmation of pathogenicity. This can be done through BCR or TCR repertoire studies and/or in vitro recombination assays (17, 26, 27).
Among the repertoire studies available to establish the significance of a RAG variant, detection of TCRVα7.2 segment in CD3+ T lymphocytes and/or MAIT is the most rapid and clinically feasible method for evaluating reduced usage of distal TRAV segments, indicating a V (D) J recombination defect or impaired thymocyte survival. Notably, TCRVβ spectratyping reflects skewed repertoire based on CDR3 length, commonly seen in SCID variants, but does not reflect use of distal elements, therefore would not inform of V (D) J recombination defects.
Early diagnosis of patients with RAG deficiency is of high importance. If left undiagnosed and untreated, these patients develop severe and often fatal complications of immune dysregulation and/or infections (21, 28, 29). However, there are also reports of the same compound heterozygous RAG variants resulting in variable phenotype and disease severity. The cases of two siblings, one with CID and the other with mild T-cell lymphopenia (30), and two adults with the same genetic defect, but highly divergent phenotype (antibody deficiency with mild vs. multiple autoimmune conditions) illustrate this point [patients 12 and 13 from (14) cohort]. Anti-cytokine antibodies are associated with immune deficiency, autoimmunity and/or immune dysregulation, especially in patients with partial RAG deficiency with severe herpesvirus infections (12, 21). Although tested, our patient had not yet developed anti-cytokine antibodies, presumably due to his young age and lack of infectious triggers.
Patients with partial RAG deficiency including atypical SCID and CID phenotype are increasingly recognized in young children and adults after severe infections (13, 14, 21). As with SCID, multiple classification systems for primary CID disorders exist, complicating diagnostic and management decisions. Roifman et al. distinguished CID from SCID based on a total CD3+ T-cell count of >500/μl (31). However, our patient had considerably <500 T cells/μl. In the cohort study of 103 patients by Roifman et al., only 5 had a RAG-related condition with OS or SCID phenotypes. All patients had low proliferation to mitogens and low TREC. Our patient also had low TREC but normal mitogens. The conclusions of the report implied that patients with partial RAG deficiency may be detected by NBS, further exemplified in our case. The 2019 ESID criteria for diagnosis of CID requires a symptomatic patient (infections, immune dysregulation) or history of affected family members with immune phenotype of 2 of the 4 parameters (low CD3, CD4 or CD8 T cells; low naïve CD4 and/or CD8 T cells; elevated γδT-cells; or reduced proliferation to mitogen or TCR stimulation) (25). Our patient meets the first two immunophenotypic criteria (note: γδT-cells were not measured) but not the clinical criteria as he was protected from infection by his environment and has no family history of this condition. The ESID 2019 criteria do not discuss underlying genetic defects for CID patients. The IUIS 2020 classification does list genetic defects for CID and does not include RAG deficiency (32) in the immune dysregulation group, where the CID-G/AI phenotype would best belong (24). The average recombinase activity of our patient (28%) is consistent with CID but not with typical SCID or OS (<10%) where there is typically lower activity (17, 26). Upon completing extensive immunological testing, our patient appeared to have an atypical SCID phenotype that shares several features with CID.
With the introduction of NBS for SCID, HSCT for asymptomatic SCID infants is increasingly utilized. Our patient had a unconventional immune phenotype with overlap between atypical SCID and CID that influenced the decision and design of the conditioning and transplant process. It is well-established that symptomatic patients with infectious and autoimmune complications, and CID immune phenotype secondary to RAG deficiency, do not respond well to immune modulation and ultimately require HSCT (13). However, there are no case studies nor established guidelines available on how to proceed with an asymptomatic infant with extreme TCL but normal T cell proliferation, and mostly normal humoral immunity. The decision to pursue immune reconstitution in hypomorphic RAG variants is controversial given the lack of HSCT studies in this particular population of TCL (33). Speckmann et al. published the therapeutic perspective of 51 pediatric patients (median age 9.6 years) with variants of symptomatic atypical SCID and CID characterized by infections and/or immune dysregulation and reduced, but not absent, T-cell immunity (34). The immune phenotype of these patients was not severe enough for an unambiguous early transplant decision. In this study, 29 (56%) of the 51 patients had mild disease, and six were transplanted for “prophylactic reasons” [i.e., family history or because the genetic diagnosis (DCLEREC1, LIGIV, IL2RG, NBN) was considered an HSCT indication].
Because severe T cell lymphopenia predisposes to serious infections, and hypomorphic RAG variants are linked to severe autoimmune and inflammatory complications in addition to infections, HSCT was performed electively at 5 months of age for our patient, taking into account our institutional experience and the paucity of data on timing for conditioning in this specific population. Conditioning regimens are increasingly used and streamlined for asymptomatic SCID, leaky SCID infants, and CID patients with RAG deficiency in the United States (4, 15, 33, 35, 36). Overall, using chemotherapy-based conditioning confers the advantage of facilitating T and B-cell reconstitution compared to unconditioned transplants in which B-cell dysfunction can persist with engraftment (37). The overactivated T and NK cell compartments should also be considered in the conditioning process and, in this case, inform the decision to use alemtuzumab and pre-transplant immunoablation (38).
Our case underscores the opportunity that SCID-NBS may have in capturing patients with overlapping immune phenotypes before the full spectrum of infectious and non-infectious complications develops. We also demonstrated that the pathogenicity of novel RAG variants requires high scrutiny with several methods. Awareness of the multiple presentations of immune deficiency with RAG variants is necessary for proper care of patients with this condition. HSCT can be considered for an asymptomatic infant with severe TCL but normal T cell proliferation and B cell number; however, further studies are needed to improve the knowledge of the natural course of the disease.
Ethics Statement
Written informed consent was obtained from the minor's legal guardian for the publication of any potentially identifiable images or data included in this article.
Author Contributions
MC-L, EW-C, and ID conceived the presented idea. BU, KL, KD, KC, YY, and AASA performed functional assays and assisted with data interpretation. DC and BO verified the analytical methods used and reviewed the clinical information presented. LN, GS, JL, and JW encouraged to describe this patient's entity and clinical course in the context of internationally accepted guidelines and supervised the findings of this work. All authors discussed the results and contributed and agreed to the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institutes of Health (sub-R01AI100887-05 to JW), Robert A. Good Endowment, University of South Florida (to JW), Jeffrey Modell Foundation (to JW), and by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases (to LN).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors with several of the authors JL and LN.
Acknowledgments
Maryssa Ellison for help as research coordinator, Dr. Joseph Dasso for grammar review, Dr. Attila Kumanovics for help with laboratory interpretation, Carla Duff and Jessica Trotter for coordination of care with the Florida State Newborn Screening Program.
References
1. Dvorak CC, Cowan MJ, Logan BR, Notarangelo LD, Griffith LM, Puck JM, et al. The natural history of children with severe combined immunodeficiency: baseline features of the first fifty patients of the primary immune deficiency treatment consortium prospective study 6901. J Clin Immunol. (2013) 33:1156–64. doi: 10.1007/s10875-013-9917-y
2. Pai S-Y, Logan BR, Griffith LM, Buckley RH, Parrott RE, Buckley RH, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. New Engl J Med. (2014) 371:434–46. doi: 10.1056/NEJMoa1401177
3. Heimall J, Buckley RH, Puck J, Fleisher TA, Gennery AR, Haddad E, et al. Recommendations for screening management of late effects in patients with severe combined immunodeficiency after allogenic hematopoietic cell transplantation: a consensus statement from the second pediatric blood marrow transplant consortium international conference on late effects after pediatric HCT. Biol Blood Marrow Transplant. (2017) 23:1229–40. doi: 10.1016/j.bbmt.2017.04.026
4. Heimall J, Logan BR, Cowan MJ, Notarangelo LD, Griffith LM, Puck JM, et al. Immune reconstitution and survival of 100 SCID patients post-hematopoietic cell transplant: a PIDTC natural history study. Blood. (2017) 130:2718–27. doi: 10.1182/blood-2017-05-781849
5. Haddad E, Logan BR, Griffith LM, Buckley RH, Parrott RE, Prockop SE, et al. SCID genotype and 6-month posttransplant CD4 count predict survival and immune recovery. Blood. (2018) 132:1737–49. doi: 10.1182/blood-2018-03-840702
6. Vogel BH, Bonagura V, Weinberg GA, Ballow M, Isabelle J, DiAntonio L, et al. Newborn screening for SCID in New York State: experience from the first two years. J Clin Immunol. (2014) 34:289–303. doi: 10.1007/s10875-014-0006-7
7. Kuo CY, Garcia-Lloret MI, Slev P, Bohnsack JF, Chen K. Profound T-cell lymphopenia associated with prenatal exposure to purine antagonists detected by TREC newborn screening. J Allergy Clin Immunol Pract. (2017) 5:198–200. doi: 10.1016/j.jaip.2016.09.028
8. Kobrynski LJ. Identification of nonand#x2013;severe combined immune deficiency T-cell lymphopenia at newborn screening for severe combined immune deficiency. Ann Allergy Asthma Immunol. (2019) 123:424–7. doi: 10.1016/j.anai.2019.08.006
9. Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol. (2014) 133:1092–8. doi: 10.1016/j.jaci.2013.09.044
10. Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. New Engl J Med. (2008) 358:2030–8. doi: 10.1056/NEJMoa073966
11. De Ravin SS, Cowen EW, Zarember KA, Whiting-Theobald NL, Kuhns DB, Sandler NG, et al. Hypomorphic Rag mutations can cause destructive midline granulomatous disease. Blood. (2010) 116:1263–71. doi: 10.1182/blood-2010-02-267583
12. Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest. (2015) 125:4135–48. doi: 10.1172/JCI80477
13. Farmer JR, Foldvari Z, Ujhazi B, De Ravin SS, Chen K, Bleesing JJH, et al. Outcomes and treatment strategies for autoimmunity and hyperinflammation in patients with RAG deficiency. J Allergy Clin Immunol Pract. (2019) 7:1970–85.e4. doi: 10.1016/j.jaip.2019.02.038
14. Lawless D, Geier CB, Farmer JR, Lango Allen H, Thwaites D, Atschekzei F, et al. Prevalence and clinical challenges among adults with primary immunodeficiency and recombination-activating gene deficiency. J Allergy Clin Immunol. (2018) 141:2303–6. doi: 10.1016/j.jaci.2018.02.007
15. Villa A, Sobacchi C, Notarangelo LD, Bozzi F, Abinun M, Abrahamsen TG, et al. V (D) J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood. (2001) 97:81–8. doi: 10.1182/blood.V97.1.81
16. Simkus C, Anand P, Bhattacharyya A, Jones MJ. Biochemical and folding defects in a RAG1 variant associated with Omenn syndrome. J Immunol. (2007) 179:8332–40. doi: 10.4049/jimmunol.179.12.8332
17. Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol. (2014) 133:1099–108. doi: 10.1016/j.jaci.2013.10.007
18. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. (2016) 536:285–91. doi: 10.1038/nature19057
19. IJspeert H, Driessen GJ, Moorhouse MJ, Hartwig NG, Wolska-Kusnierz B, Kalwak K, et al. Similar recombination-activating gene (RAG) mutations result in similar immunobiological effects but in different clinical phenotypes. J Allergy Clin Immunol. (2014) 133:1124–33. doi: 10.1016/j.jaci.2013.11.028
20. Berland A, Rosain J, Kaltenbach S, Allain V, Mahlaoui N, Melki I, et al. PROMIDISα: A T-cell receptor alpha signature associated with immunodeficiencies caused by V (D) J recombination defects. J Allergy Clin Immunol. (2019) 143:325–34.e2. doi: 10.1016/j.jaci.2018.05.028
21. Goda V, Malik A, Kalmar T, Maroti Z, Patel B, Ujhazi B, et al. Partial RAG deficiency in a patient with varicella infection, autoimmune cytopenia, anticytokine antibodies. J Allergy Clin Immunol Pract. (2018) 6:1769–71.e2. doi: 10.1016/j.jaip.2018.01.015
22. Gennery A. Recent advances in understanding RAG deficiencies. F1000Res. (2019) 8. doi: 10.12688/f1000research.17056.1
23. Gungor T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. (2014) 383:436–48. doi: 10.1016/S0140-6736(13)62069-3
24. Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. (2018) 38:129–43. doi: 10.1007/s10875-017-0465-8
25. Seidel MG, Kindle G, Gathmann B, Quinti I, Buckland M, van Montfrans J, et al. The European Society for Immunodeficiencies (ESID) registry working definitions for the clinical diagnosis of inborn errors of immunity. J Allergy Clin Immunol Pract. (2019) 7:1763–70. doi: 10.1016/j.jaip.2019.02.004
26. Lee YN, Frugoni F, Dobbs K, Tirosh I, Du L, Ververs FA, et al. Characterization of T and B cell repertoire diversity in patients with RAG deficiency. Sci Immunol. (2016) 1:eaah6109. doi: 10.1126/sciimmunol.aah6109
27. Bulkhi AA, Dasso JF, Schuetz C, Walter EJ. Approaches to patients with variants in RAG genes: from diagnosis to timely treatment. Expert Rev Clin Immunol. (2019) 15:1033–46. doi: 10.1080/1744666X.2020.1670060
28. Dutmer CM, Asturias EJ, Smith C, Dishop MK, Schmid DS, Bellini WJ, et al. Late onset hypomorphic RAG2 deficiency presentation with fatal vaccine-strain VZV infection. J Clin Immunol. (2015) 35:754–60. doi: 10.1007/s10875-015-0207-8
29. Geier CB, Farmer JR, Foldvari Z, Ujhazi B, Eibl MM, Wolf HM, et al. Vasculitis as a major morbidity factor in patients with hypomorphic RAG mutations. J Allergy Clin Immunol. (2019) 143:AB116. doi: 10.1016/j.jaci.2018.12.353
30. Schuetz C, Pannicke U, Jacobsen EM, Burggraf S, Albert MH, Honig M, et al. Lesson from hypomorphic recombination-activating gene (RAG) mutations: why asymptomatic siblings should also be tested. J Allergy Clin Immunol. (2014) 133:1211–5. doi: 10.1016/j.jaci.2013.10.021
31. Roifman CM, Somech R, Kavadas F, Pires L, Nahum A, Dalal I, et al. Defining combined immunodeficiency. J Allergy Clin Immunol. (2012) 130:177–83. doi: 10.1016/j.jaci.2012.04.029
32. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2020) 40:65. doi: 10.1007/s10875-020-00763-0
33. John T, Walter JE, Schuetz C, Chen K, Abraham RS, Bonfim C, et al. Unrelated hematopoietic cell transplantation in a patient with combined immunodeficiency with granulomatous disease and autoimmunity secondary to RAG deficiency. J Clin Immunol. (2016) 36:725–32. doi: 10.1007/s10875-016-0326-x
34. Speckmann C, Doerken S, Aiuti A, Albert MH, Al-Herz W, Allende LM, et al. A prospective study on the natural history of patients with profound combined immunodeficiency: an interim analysis. J Allergy Clin Immunol. (2017) 1391302–10.e4. doi: 10.1016/j.jaci.2016.07.040
35. Delmonte OM, Schuetz C, Notarangelo DL. RAG deficiency: two genes, many diseases. J Clin Immunol. (2018) 38:646–55. doi: 10.1007/s10875-018-0537-4
36. Westermann-Clark E, Grossi A, Fioredda F, Giardino S, Cappelli E, Terranova P, et al. RAG deficiency with ALPS features successfully treated with TCRαβ/CD19 cell depleted haploidentical stem cell transplant. Clin Immunol. (2018) 187:102–3. doi: 10.1016/j.clim.2017.10.012
37. Castagnoli R, Delmonte OM, Calzoni E, Notarangelo DL. Hematopoietic stem cell transplantation in primary immunodeficiency diseases: current status and future perspectives. Front Pediatr. (2019) 7:295. doi: 10.3389/fped.2019.00295
38. Dobbs K, Tabellini G, Calzoni E, Patrizi O, Martinez P, Giliani SC, et al. Natural killer cells from patients with recombinase-activating gene and non-homologous end joining gene defects comprise a higher frequency of CD56 (bright) NKG2A(+++) cells, and yet display increased degranulation and higher perforin content. Front Immunol. (2017) 8:798. doi: 10.3389/fimmu.2017.00798
Keywords: recombinase activating gene (RAG), newborn screening, SCID, asymptomatic infant, immunodeficiency, HSCT
Citation: Chitty-Lopez M, Westermann-Clark E, Dawson I, Ujhazi B, Csomos K, Dobbs K, Le K, Yamazaki Y, Sadighi Akha AA, Chellapandian D, Oshrine B, Notarangelo LD, Sunkersett G, Leiding JW and Walter JE (2020) Asymptomatic Infant With Atypical SCID and Novel Hypomorphic RAG Variant Identified by Newborn Screening: A Diagnostic and Treatment Dilemma. Front. Immunol. 11:1954. doi: 10.3389/fimmu.2020.01954
Received: 07 April 2020; Accepted: 20 July 2020;
Published: 29 September 2020.
Edited by:
Mikko Risto Juhana Seppänen, Helsinki University Central Hospital, FinlandReviewed by:
Kohsuke Imai, Tokyo Medical and Dental University, JapanDavid Buchbinder, Children's Hospital of Orange County, United States
Copyright © 2020 Chitty-Lopez, Westermann-Clark, Dawson, Ujhazi, Csomos, Dobbs, Le, Yamazaki, Sadighi Akha, Chellapandian, Oshrine, Notarangelo, Sunkersett, Leiding and Walter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jolan E. Walter, am9sYW53YWx0ZXImI3gwMDA0MDt1c2YuZWR1
†These authors share senior authorship