 Abdelouaheb Benani
Abdelouaheb Benani Sanae Ben Mkaddem
Sanae Ben Mkaddem- 1Unité de Biologie Moléculaire, Institut Pasteur du Maroc, Casablanca, Morocco
- 2U978 Institut National de la Santé et de la Recherche Médicale, Bobigny, France
- 3UFR SMBH, Université Sorbonne Paris Nord, Bobigny, France
Coronavirus disease (COVID-19) is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is a betacoronavirus, and is associated with cytokine storm inflammation and lung injury, leading to respiratory distress. The transmission of the virus is mediated by human contact. To control and prevent the spread of this virus, the majority of people worldwide are facing quarantine; patients are being subjected to non-specific treatments under isolation. To prevent and stop the COVID-19 pandemic, several clinical trials are in the pipeline. The current clinical trials either target the intracellular replication and spread of the virus or the cytokine storm inflammation seen in COVID-19 cases during the later stages of the disease. Since both targeting strategies are different, the window drug administration plays a crucial role in the efficacy of the treatment. Here, we review the mechanism underlying SARS-CoV-2 cell infection and potential future therapeutic approaches.
Introduction
The members of the Coronaviridae family cause mild respiratory disease, and infection with these viruses can be transmitted between humans (1). Severe acute respiratory syndrome coronavirus (SARS-CoV) is transmitted from animals to humans, leading to severe respiratory diseases in individuals (2). SARS was discovered in Guangdong Province, China, in 2002 (3). Chinese bats serve as the natural reservoir hosts of SARS-CoV-2 (4). The human transmission of SARS-CoV requires intermediate hosts, such as animal food sources, including pangolin and cats (5). No specific antivirals or effective vaccines are available to treat or prevent SARS. In 2002 and 2003, the SARS pandemic was controlled by travel restrictions and patient isolation.
Recently, a new virus strain from the same virus family was discovered in Wuhan, Hubei Province, China, that causes coronavirus disease 19 (COVID-19) (6). It has been suggested that the human transmission of this strain was linked to the Hunan seafood market. The infection is very contagious and results in the development of the disease and fatalities (7). SARS-CoV-2 is closely related to SARS-CoV, and COVID-19 has been described as a new lung disease (8). Infections have also been detected in several countries globally and are linked to international travel. Elucidating the mechanisms through which the virus gains entry into target cells and how this process can be inhibited would allow the development of new therapeutics or vaccines to rapidly curb the ongoing pandemic. A significant number of clinical trials have been started to explore potential therapeutic strategies for COVID-19 to identify as quickly as possible high-quality efficient treatments to stop the ongoing pandemic. Here, we present a brief overview of the SARS-CoV infection mechanism and potential strategies to prevent virus entry along with the effects of infection, such as inflammatory cytokine storms, on lung injury. We discuss some published data and the mechanism of the ongoing clinical trials.
Mechanism of SARS-CoV Cell Infection
Basically, the entry of coronavirus is mediated by the interaction of cellular receptor proteins and the S1 unit of the viral spike (S) protein, which, in turn, promotes viral attachment to the target cell surface. Furthermore, viral attachment requires cellular proteases to prime the S protein, which entails its cleavage at the S1/S2 and S2' sites, resulting in the fusion of the viral and cellular membranes. It has been shown that the S protein from SARS interacts with angiotensin-converting enzyme 2 (ACE2) as its receptor and uses the cellular serine protease TMPRSS2 to prime the S protein (9, 10). Additionally, it has been demonstrated that the SARS-S/ACE2 interaction favors the spread of the virus, leading to severe acute respiratory syndrome (11). ACE and ACE2 have high homology with metalloproteases that play a role in the renin-angiotensin system (RAS) to maintain blood pressure homeostasis. The renin protease cleaves angiotensinogen to generate angiotensin I (Ang I). The two C-terminal amino acids of Ang I are cleaved by ACE to generate angiotensin II (Ang II), whereas ACE2 cleaves Ang II. Ang II acts specifically through Ang II receptor type 1 (AT1R) and Ang II receptor type 2 (AT2R) (12, 13). ACE also degrades additional substrates such as bradykinin or apelin (14). ACE2 has been identified as the key determinant of SARS-CoV transmissibility (15). The SARS-S and SARS-2-S proteins have 76% amino acid homology. However, it is not yet clear whether SARS-2-S and SARS-S use ACE2 and TMPRSS2 for host cell binding. A recent study demonstrated that SARS-CoV-2 uses the same ACE2 receptor as SARS-CoV to enter the target cell and also uses the same cellular protease, the serine protease TMPRSS2, to prime the S protein. The study also suggested a treatment strategy based on the inhibition of S protein priming by targeting TMPRSS2 to block entry. Moreover, the study showed that sera from convalescent SARS patients cross-neutralized the S protein to block SARS-2 entry (3).
COVID-19 and Cytokine Storm Syndrome
The antiviral response is mediated by both innate and acquired immunity, which recognize pathogen-associated molecular patterns (PAMPs) and the antigen-specific adaptive immune response. The viral response is based on the release of inflammatory mediators (cytokines, chemokines, leukotrienes, proteases, and reactive oxygen species) and on the clearance of virus through internalization and killing of the virus. Cell responses are in many ways controlled by the balance between antagonistic signals, which may affect the immune response to pathogens. The resulting balance is of great importance to prevent damage to tissues through immunopathology and to ensure the return of activated cells to a resting state. However, exaggerated and excessive synthesis of cytokines can lead to an acute, severe systemic inflammatory response known as a “cytokine storm” and cause severe damage to multiple organs (16). The cytokine profile of COVID-19 patients with differences in disease severity has been investigated, and a subset of patients with severe COVID-19 develop profound inflammation and multiorgan dysfunction that is consistent with a “cytokine storm.” Recently, a large panel of cytokines (IFN-γ, TNF-α, IL-2, IL-4, IL-6, and IL-10) and C-reactive protein (CRP) have been analyzed and compared with serum samples from a control group and from COVID-19 patients. The values for cytokines and CRP were significantly higher in patients with COVID-19 than those in healthy controls. However, using univariate logistic regression analysis, only two cytokines, IL-6 and IL-10, were found to be predictive of disease severity, suggesting that a higher level of cytokine storm is associated with severe disease development. Improving the understanding of hypercytokinemia (i.e., IL-6 levels from 100 to 5,000 pg/mL) and immune dysregulation associated with COVID-19 is urgent. Investigations of different potential therapeutic strategies for COVID-19 cytokine storm syndrome are ongoing that use corticosteroids, IL-6 blockade and IL-1 inhibition (17). A series of clinical trials of IL-6 inhibitors such as tocilizumab, sarilumab and siltuximab are also underway (see Table 1).
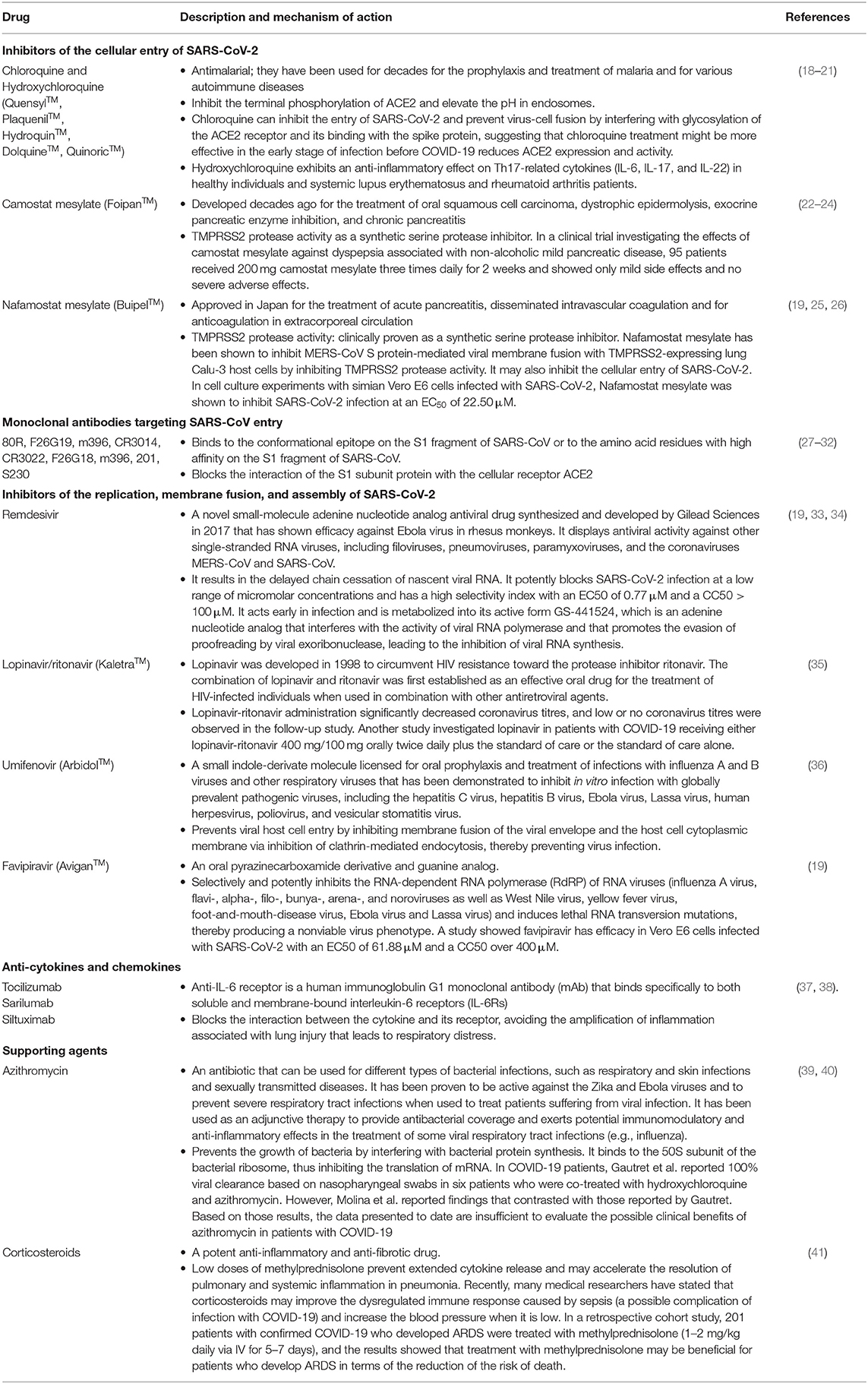
Table 1. Current therapeutic drugs used to treat COVID-19.
Coagulation and Fibrinolysis in COVID-19
The host defense against viral infection activates the coagulation cascade to limit the spread of pathogens. During the first phase of infection, an adaptive haemostatic response occurs that is associated with the activation of a systemic inflammatory response, which is characterized by an increase in inflammatory activity and thrombin and fibrinogen generation. The increase in cytokine production during virus infection induces additional procoagulant effects, such as the expression of tissue factors that are major initiators of coagulation activation. Moreover, other factors, such as DAMPs and neutrophil extracellular traps, may also contribute to the procoagulant profile in COVID-19. During pulmonary infection, the measurement of coagulation and fibrinolysis factors in bronchoalveolar lavage fluid has demonstrated an increase in thrombin generation, an insufficient balance in physiologic anticoagulation, and the suppression of fibrinolysis, mediating the pathogenesis of respiratory distress. Endothelial injury of the pulmonary capillary is also caused by vascular endothelial damage. SARS-CoV-2 infects endothelial cells through the ACE2 receptor, and viral spread and rapid viral replication leads to massive endothelial cell apoptosis and inhibits the anticoagulant function of the vascular lumen. Moreover, endothelial dysfunction contributes to procoagulant changes in COVID-19 (42).
Platelets play a dual role; they contribute to haemostasis but also to inflammation and the host defense response, especially during lung infection. Recently, many cases of thrombocytopenia have been observed in COVID-19 patients, and the baseline platelet levels and changes were associated with subsequent mortality. However, the mechanism of SARS-CoV-2 involved in thrombocytopenia is not yet clear (43). One of the possibilities is that lung tissue injury could cause platelet activation and aggregation, and thrombi formation at the site of the injury may lead to the consumption of platelets and megakaryocytes. In addition, SARS-CoV-2 induces increases in D-dimer and fibrinogen and further increases the consumption of platelets in damaged lungs.
Potential Therapeutic Approaches
Monoclonal Antibodies Targeting SARS-CoV Entry
The spike proteins of SARS-CoV-2 play a major role in the interaction between the virus and the ACE2 receptor expressed by the host cell. The binding of the spike protein to ACE2 leads to membrane fusion and the initiation of the viral life cycle. To inhibit SARS-CoV-2 binding to ACE2, several neutralizing monoclonal antibodies (MAbs) targeting the spike protein of SARS-CoV-2 have been developed. Among them, the 80R MAb binds to the S1 fragment of SARS-CoV at the conformational epitope (amino acid residues 426–492) and blocks the binding of the viral S1 subunit to the ACE2 receptor, thereby preventing the entry and spread of the virus (44). These findings have been demonstrated by in vitro and in vivo studies (27, 45). Other MAbs targeting different epitopes of the S1 subunit have also been developed and tested by in vitro and in vivo studies, such as CR3022, F26G18, F26G19, m396, 1A9, and CR3014 (27–32).
A recent study suggested the involvement of similar mechanisms of host entry in infection with SARS-CoV-2, and consequently, different studies are currently investigating single MAbs or combinations of different MAbs. Such antibodies recognize different epitopes on the SARS-CoV-2 surface, which should be assessed first by in vitro and in vivo (mouse) approaches prior to different clinical trials. However, several neutralizing MAbs also bind to IgG Fc receptors (FcγR). The antibody/FcγR interaction might lead to virus entry that could infect other cells expressing this receptor independently of the ACE2-specific virus receptor. Recently, it has been demonstrated that FcγRIIA plays a major role in viral entry via antibody-dependent enhancement (ADE) using in vitro strategies (46). However, the signaling pathway associated with the MAbs/virus/receptor interaction is not yet clear. ADE viral entry in the presence of neutralizing MAbs has been demonstrated for many viruses, especially for those expressing the coronavirus spike protein. Understanding the effect of this interaction on the activation of human cells expressing the Fc receptor and viral proliferation may help to establish new vaccination strategies in the future.
Treatment of Inflammatory Cytokine Storm
MAbs Against the IL-6 Receptor
To explore the pathophysiological mechanisms and development of novel therapeutic approaches for sepsis, a recent study using caecal ligation and puncture (CLP) was performed in a septic mouse model. The mouse models demonstrated classical inflammatory symptoms associated with an increase in soluble triggering receptors expressed on immune cells, including interleukin (IL)-6, IL-10, TNF-α, macrophage inflammatory protein (MIP)-1α, MIP-1β, and MIP-2. These results were similar to those found in human patients with sepsis (47). IL-6 plays an important role in host defense during infections. However, exacerbation of IL-6 production favors acute severe systemic inflammation, which is named 'cytokine storm' (48). During the COVID-19 pandemic, a recent study explored the levels of cytokines, including IL-6, and the T cell frequency in three groups of individuals: healthy individuals and patients with moderate and severe COVID-19 cases. The moderate cases presented an increase in IL-6 and a decrease in the total T lymphocyte frequency. However, the severe COVID-19 cases showed an increase in IL-6, IL-2R, IL-10, and TNFα secretion associated with a severe decrease in T cells, particularly CD4+ T cells (49). These results suggest that IL-6 plays a key role in the amplification of inflammation associated with lung injury, leading to respiratory distress (37, 38). Moreover, this antibody has been used in the treatment of rheumatoid arthritis and was approved by the FDA 10 years ago, and the side effects have been extensively studied (50). Taken together, these findings suggest that IL-6 or its receptor present a potent target of interest for the treatment of COVID-19-associated acute respiratory distress syndrome (ARDS). In this context, treatment of one case of COVID-19 associated with respiratory failure with an anti-interleukin-6 receptor inhibitor named tocilizumab resulted in favorable recovery (51). To explore whether tocilizumab can be used as a treatment for COVID-19, clinical trials with a large number of patients with the correct groups should be conducted robustly to prevent mortality. However, the optimal disease stage for the administration of tocilizumab must be defined carefully. Since it has been shown that IL-6 can either suppress or facilitate viral replication (52), one crucial issue to address will be the optimal timing of anti-IL6 administration. If it occurs too early, the drugs may affect viral clearance. If it occurs too late, the drugs may not be effective. The optimal timing of the administration of anti-IL-6 must be assessed in trials. Several randomized controlled trials of tocilizumab, sarilumab and siltuximab, either alone or in combination, are now being proposed in patients with severe COVID-19 and are underway mainly in China, Western Europe, USA, Russia, Malaysia, and Australia (53). Moreover, different clinical trials are under way to evaluate the safety and efficacy of IL-6 inhibitors with various protocols and comparators. The identifiers of the clinical trials are NCT04332913, NCT04335071, NCT04317092, NCT04324073, NCT04320615, NCT04306705, NCT04315298, NCT04315480, NCT04321993, NCT04348500, NCT04329650, NCT04330638, NCT04345289, NCT04327388, NCT04341870, and NCT04322773 (ClinicalTrials.gov).
MAbs Against Chemokine Receptors
Several clinical trials are also ongoing to examine the effect of blocking other proinflammatory cytokines, such as TNF (54) and granulocyte–macrophage colony-stimulating factor (GM–CSF), with the clinical trial identifier NCT04341116. The aim of this study is to interfere with cytokine signaling, leading to decreased hyperinflammation in patients with severe COVID-19. Indeed, the most highly pathological macrophages are derived from the circulating monocytes infiltrating the lung. Moreover, CCR2 plays a central role in the recruitment and accumulation of monocytes in inflamed tissues (55). Altogether, these results suggest that CCR2 blockade could potentially help to reduce the accumulation of pathological monocytes in inflamed tissues. A new clinical trial (NCT04343651) targeting CCR5, another chemokine receptor that regulates monocyte and T cell recruitment, is ongoing in patients with COVID-19 with mild-to-moderate symptoms of respiratory illness.
Chloroquine
Chloroquine (CQ) or hydroxychloroquine (HCQ) (a more soluble and less toxic metabolite of CQ) are antimalarial products that have been tested in humans (56). CQ and HCQ are also used in the treatment of several autoimmune diseases, such as systemic lupus erythematosus and rheumatoid arthritis. Additionally, CQ inhibits autophagy, favoring the apoptosis of cancer cells (57).
Promising studies have demonstrated that the CQ/HCQ compounds have the ability to inhibit certain coronaviruses, such as SARS-CoV-1 (58). Additional in vitro studies have shown that CQ and HCQ have antiviral activity against SARS-CoV-2, with more side effects being observed for CQ than for HCQ (58). In contrast, others have demonstrated that HCQ has greater antiviral activity than CQ during SARS-CoV-2 infection (18). Basically, CQ or HCQ exert their effects on eukaryotic cells by increasing the vacuolar pH of organelles such as endosomes and lysosomes. The increase in pH neutralizes the acidic lysosomal pH, decreasing autophagosome-lysosome fusion and autophagic degradation (59, 60). Autophagosome-lysosome fusion is essential for virus/cell fusion and immune-modulating activity (61). CQ and HCQ can also modify the glycosylation of ACE2, which binds to the spike protein S of SARS-CoV. This may interfere with the virus-receptor interaction (19). Additionally, an in vitro approach demonstrated that CQ inhibits COVID-19 virus infection (62). Some studies have indicated that HCQ also reduces the levels of some proinflammatory cytokines, such as IL-6, IL-18, and TNF-α (63). Indeed, CQ and HCQ inhibit endosomal TLRs and have anti-inflammatory effects by inhibiting prostaglandin synthesis or lipid peroxidation (64).
Hence, it was suggested that CQ and HCQ represent a potential new drug treatment for COVID-19. However, there are some limitations in performing clinical trials in patients owing to the restrictions on research studies using cell culture or animals and side effects, such as cardiotoxicity and liver cytotoxicity, due to the half-life of these compounds of ~3.1 days (65). However, the risk of toxicity in patients treated for 10 years with HCQ for systemic lupus erythematosus was shown to be approximately 7.5% and to be higher in patients treated for longer periods (66). In COVID-19-associated acute infection, CQ and HCQ are used for a very short time (~5 days). Nevertheless, acute adverse events, such as hypersensitivity and gastrointestinal intolerance, require attention, especially in critically ill patients who may develop similar clinical manifestations due to COVID-19. Additionally, CQ and HCQ can be safely used during pregnancy (67). Recently, a clinical trial with a small sample size showed that HCQ treatment is associated with a decrease in viral load in COVID-19 patients, and the effect is reinforced by azithromycin (39). Because of the low number of patients and the lack of some group controls during this recent study, new national and international clinical trials are being conducted to confirm the authenticity of these findings. A current clinical trial of CQ and HCQ therapy in the treatment of COVID-19 in Europe may reveal new possibilities for antiviral therapy for COVID-19 to stop the pandemic. Although the antiviral activity of hydroxychloroquine remains uncertain, there have been several controversies regarding the clinical benefits of this drug in patients with COVID-19. Recently, a new publication showed the beneficial effects of hydroxychloroquine or chloroquine when used alone or with a macrolide on in-hospital outcomes for COVID-19. Each of the drug regimens was associated with a decrease in in-hospital survival and an increased frequency of ventricular arrhythmias when used for the treatment of COVID-19. However, this study was retracted from the Lancet journal (https://doi.org/10.1016/S0140-6736(20)31174-0). In contrast, an approved study by the Ethics Committee of Shanghai Public Health Clinical Center under the number NCT04261517 demonstrated that the prognosis of COVID-19 patients with moderate cases is good. However, a large sample size study is needed to investigate the effects of HCQ in the treatment of COVID-19 (68). A new study is ongoing and can be found on ClinicalTrials.gov under the identifier NCT04303507 with the official title “Chloroquine/Hydroxychloroquine Prevention of Coronavirus Disease (COVID-19).” This study is a double-blinded, randomized, and placebo-controlled trial that will be conducted in a healthcare setting. A total of 40,000 participants will be recruited, and the investigators predict an average of 400–800 participants per site at 50–100 sites. However, the estimated completion date is April 2021.
Anticoagulant Treatments
Since recent findings revealed that most COVID-19 patients with severe cases admitted to the intensive care unit for respiratory failure present predominantly with hypercoagulation, anticoagulant drugs could a potentially prevent a state that could lead to arterial and venous thromboembolic complications (69). Antithrombin and activated protein C for the treatment of classical acute respiratory distress syndrome can be used as anticoagulants for inflammatory thrombus prevention. Platelets may be involved in systemic and local thrombotic responses. Antiplatelet therapies may present a new therapeutic approach. This is a known phenomenon in acute coronary syndromes, where anticoagulant therapy along with antiplatelet therapy decreases arterial thrombosis, but it is associated with an increase in bleeding risk (42).
Therapies Targeting Viral Replication
Remdesivir is an antiviral molecule with a chemical formula of C27H35N6O8P. Remdesivir prevents viral replication by inhibiting viral DNA polymerase. Its antiviral activity has been demonstrated against Ebola virus in multiple human cell types, including primary macrophages and human endothelial cells, with low half-maximal effective concentration (EC50) values of 0.06–0.14 μM (33). It has also been shown that remdesivir inhibits SARS-CoV in primary human airway epithelial cell cultures, which are a biologically relevant in vitro model of pulmonary infection (70). Moreover, remdesivir has exhibited antiviral activity against the Marburg virus (33). SARS-CoV and SARS-CoV-2 present 82% RNA sequence homology, and their RNA-dependent RNA polymerase (RdRp) sequences share 96% sequence similarity. Therefore, drugs targeting the viral RdRp proteins of SARS-CoV are also suspected to be effective against SARS-CoV-2. According to the in vitro antiviral activity of remdesivir, the in vivo tests showed the suppression of Ebola virus replication and the protection of all infected animals against lethal infection (33). In addition, remdesivir decreased the viral load in the lungs and preserved the pulmonary function of mice during SARS-CoV infection (70). These findings suggest that remdesivir can be used as a potential new therapeutic approach for human infections caused by coronaviruses, including SARS-CoV-2. In fact, the first case of COVID-19 in Washington, USA, was treated with intravenous remdesivir. During the treatment, no obvious adverse effects were observed (71). However, we cannot comment yet on the efficiency of the treatment effect of remdesivir during the COVID-19 outbreak.
There are four clinical trials currently enrolling patients in the United States. Moreover, two clinical trials in China have been registered on ClinicalTrials.gov: NCT04257656 for severe disease and NCT04252664 for mild-to-moderate disease (72). Recently, Yeming et al. published the results of the NCT04257656 clinical trial, which showed no clear outcome because of the death or discharge of patients (73). Moreover, in another clinical trial, the benefit in terms of the time to clinical improvement was not statistically significant (21 vs. 23 days), even though the study was underpowered (74). There are limited safety data for remdesivir, which should be obtained in further studies.
Therapies Targeting Viral Transcription
Ribavirin is a broad-spectrum nucleoside antiviral drug that is phosphorylated in virus-infected cells. Basically, the entry of the product into virus-infected cells leads to its phosphorylation. This product acts as a competitive inhibitor of the viral synthetase, interfering with early viral transcription events and thereby hindering the synthesis of ribonucleoproteins and subsequent viral spread. Several controversial in vitro studies investigating ribavirin have been conducted. While a few of them have demonstrated that ribavirin has an antiviral effect on SARS, others have revealed no evidence of its antiviral role (75, 76). Additionally, a clinical trial reported no significant antiviral effects on SARS-infected patients (77). In fact, the same study reported side effects, such as haemolytic anemia, resulting from the clinical administration of ribavirin (77). During the COVID-19 pandemic, ribavirin combined with interferon was used based on the Chinese treatment guidelines.
BCG Vaccine
The Bacillus Calmette-Guerin (BCG) vaccine against tuberculosis has been demonstrated to reduce mortality during other infections. The protective mechanism involved in tuberculosis infection has been explored in vivo. It was demonstrated that BCG vaccination increased IFN-γ production by CD4+ cells (78). T cells play a crucial role in viral infections; CD4 T cells provide B cell help for antibody production and control the response of other immune cell subsets, whereas CD8 T cells kill infected cells to reduce the viral burden. To better understand the role of T cell responses in SARS-CoV-2 infection, some studies are beginning to be conducted. During SARS-CoV-1 infection, the occurrence of lymphopenia with drastically reduced numbers of both CD4 and CD8 T cells in moderate and severe COVID-19 cases has been described in several current reports (79). Th1 and Th17 cells play a crucial role in the induction of CD4+ and CD8+ memory cells that are involved in the control of the immune system response during non-mycobacterial secondary infections. Interestingly, BCG vaccination continued to increase Th1 and Th17 responses at least 1 year after vaccination in healthy subjects (80). COVID-19 infection severity is associated with a sharp decrease in the frequency of CD4+ and CD8+ cells and the expression of INF-γ on the surface of CD4+ cells (78). The nonspecific effects of the BCG vaccine present a potential therapeutic method to increase memory responses and enhance the immune system during viral infections that might aid in combating the COVID-19 pandemic.
Discussion
Improved understanding of the viral entry mechanisms and the inflammatory response generated during infection would allow the development of appropriate therapeutic strategies to manage patients with COVID-19. The different therapeutic strategies (Table 1) discussed in this review are encouraging and have been proposed to treat or prevent the spread of COVID-19. In addition, most of the described compounds are readily available, and they are known to result in a minor risk of adverse events. Several clinical trials are in process to validate the results. However, these strategies are not without risks, and special attention to factors such as age, sex, and associations with other chronic diseases must be considered during patient selection. Non-specific proinflammatory cytokine targeting during COVID-19 treatment using corticoids, e.g., may favor viral spread. However, targeting specific individual cytokines does not increase viral infection and prevents cytokine storm inflammation-mediated tissue injury, notably in the lung. Since observations have indicated that there are two stages of disease, the first of which is characterized by virus spread and the second by the hyperproinflammatory response responsible for respiratory distress, the timing of the initiation of therapy needs to be carefully defined.
In this review, we also mentioned that virus-neutralizing MAbs represent a therapeutic method with a high potential to prevent viral spread. However, the use of immunoglobulin class G (IgG) MAbs may contribute to an ADE mechanism favoring the spread of the virus during treatment. In parallel, these antibodies can also induce anaphylactic shock that is mediated by the FcγR receptor; FcγRIIA is expressed by neutrophils and platelets, in particular (81). These side effects remain poorly studied. The development of IgG4 or F(ab)′2 antibodies to neutralize the virus or to target proinflammatory antibodies that cannot interact with FcγR may prevent this risk.
Finally, the treatment duration should be well-defined in terms of the half-life of molecules to prevent liver toxicity and the immunosuppressive effect.
Thus, the use of monotherapy or combinatorial therapeutic strategies during different stages of COVID-19 infection represent a potential therapeutic strategy to stop the ongoing pandemic.
Author Contributions
SB and AB wrote this review. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from INSERM, ANR JC (ANR-17-CE17-0002-01), and DIM1 HEALTH action financée par la Région Ile-de France.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Corman VM, Lienau J, Witzenrath M. [Coronaviruses as the cause of respiratory infections]. Internist (Berl). (2019) 60:1136–45. doi: 10.1007/s00108-019-00671-5
2. Fehr AR, Channappanavar R, Perlman S. Middle east respiratory syndrome: emergence of a pathogenic human coronavirus. Annu Rev Med. (2017) 68:387–99. doi: 10.1146/annurev-med-051215-031152
3. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80.e8. doi: 10.1016/j.cell.2020.02.052
4. Lau JT, Yang X, Pang E, Tsui HY, Wong E, Wing YK. SARS-related perceptions in Hong Kong. Emerg Infect Dis. (2005) 11:417–24. doi: 10.3201/eid1103.040675
5. Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. (2003) 302:276–8. doi: 10.1126/science.1087139
6. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
7. Chan JF, Yuan S, Kok KH, To KK, Chu H, Yang J, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. (2020) 395:514–23. doi: 10.1016/S0140-6736(20)30154-9
8. Zhu F, Cao Y, Xu S, Zhou M. Reply to Comments on 'Co-infection of SARS-CoV-2 and HIV in a patient in Wuhan city, China'. J Med Virol. (2020) doi: 10.1002/jmv.25838. [Epub ahead of print].
9. Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol. (2010) 84:12658–64. doi: 10.1128/JVI.01542-10
10. Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol. (2011) 85:873–82. doi: 10.1128/JVI.02062-10
11. Imai Y, Kuba K, Penninger JM. Angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Cell Mol Life Sci. (2007) 64:2006–12. doi: 10.1007/s00018-007-6228-6
12. Skeggs LT, Dorer FE, Levine M, Lentz KE, Kahn JR. The biochemistry of the renin-angiotensin system. Adv Exp Med Biol. (1980) 130:1–27. doi: 10.1007/978-1-4615-9173-3_1
13. Corvol P, Williams TA, Soubrier F. Peptidyl dipeptidase a: angiotensin I-converting enzyme. Methods Enzymol. (1995) 248:283–305. doi: 10.1016/0076-6879(95)48020-X
14. Tschope C, Schultheiss HP, Walther T. Multiple interactions between the renin-angiotensin and the kallikrein-kinin systems: role of ACE inhibition and AT1 receptor blockade. J Cardiovasc Pharmacol. (2002) 39:478–87. doi: 10.1097/00005344-200204000-00003
15. Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. (2005) 309:1864–8. doi: 10.1126/science.1116480
16. Gu Y, Hsu AC, Pang Z, Pan H, Zuo X, Wang G, et al. Role of the innate cytokine storm induced by the influenza A virus. Viral Immunol. (2019) 32:244–51. doi: 10.1089/vim.2019.0032
17. England JT, Abdulla A, Biggs CM, Lee AYY, Hay KA, Hoiland RL, et al. Weathering the COVID-19 storm: lessons from hematologic cytokine syndromes. Blood Rev. (2020) 2020:100707. doi: 10.1016/j.blre.2020.100707
18. Yao X, Ye F, Zhang M, Cui C, Huang B, Niu P, et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin Infect Dis. (2020) ciaa237. doi: 10.1093/cid/ciaa237. [Epub ahead of print].
19. Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. (2020) 30:269–71. doi: 10.1038/s41422-020-0282-0
20. Wu R, Wang L, Kuo HD, Shannar A, Peter R, Chou PJ, et al. An update on current therapeutic drugs treating COVID-19. Curr Pharmacol Rep. (2020) 2020:1–15. doi: 10.1007/s40495-020-00216-7
21. Silva JC, Mariz HA, Rocha LF Jr, Oliveira PS, Dantas AT, et al. Hydroxychloroquine decreases Th17-related cytokines in systemic lupus erythematosus and rheumatoid arthritis patients. Clinics (São Paulo). (2013) 68:766–71. doi: 10.6061/clinics/2013(06)07
22. Ohkoshi M, Fujii S. Effect of the synthetic protease inhibitor [N,N-dimethylcarbamoyl-methyl 4-(4-guanidinobenzoyloxy)-phenylacetate] methanesulfate on carcinogenesis by 3-methylcholanthrene in mouse skin. J Natl Cancer Inst. (1983) 71:1053–7.
23. Ikeda S, Manabe M, Muramatsu T, Takamori K, Ogawa H. Protease inhibitor therapy for recessive dystrophic epidermolysis bullosa. In vitro effect and clinical trial with camostat mesylate. J Am Acad Dermatol. (1988) 18:1246–52. doi: 10.1016/S0190-9622(88)70130-9
24. Sai JK, Suyama M, Kubokawa Y, Matsumura Y, Inami K, Watanabe S. Efficacy of camostat mesilate against dyspepsia associated with non-alcoholic mild pancreatic disease. J Gastroenterol. (2010) 45:335–41. doi: 10.1007/s00535-009-0148-1
25. Yamamoto M, Matsuyama S, Li X, Takeda M, Kawaguchi Y, Inoue JI, et al. Identification of nafamostat as a potent inhibitor of middle east respiratory syndrome coronavirus s protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob Agents Chemother. (2016) 60:6532–9. doi: 10.1128/AAC.01043-16
26. Iwaki M, Ino Y, Motoyoshi A, Ozeki M, Sato T, Kurumi M, et al. Pharmacological studies of FUT-175, nafamostat mesilate. V. Effects on the pancreatic enzymes and experimental acute pancreatitis in rats. Jpn J Pharmacol. (1986) 41:155–62. doi: 10.1254/jjp.41.155
27. Berry JD, Hay K, Rini JM, Yu M, Wang L, Plummer FA, et al. Neutralizing epitopes of the SARS-CoV S-protein cluster independent of repertoire, antigen structure or mAb technology. MAbs. (2010) 2:53–66. doi: 10.4161/mabs.2.1.10788
28. ter Meulen J, van den Brink EN, Poon LL, Marissen WE, Leung CS, Cox F, et al. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med. (2006) 3:e237. doi: 10.1371/journal.pmed.0030237
29. Ng OW, Keng CT, Leung CS, Peiris JS, Poon LL, Tan YJ. Substitution at aspartic acid 1128 in the SARS coronavirus spike glycoprotein mediates escape from a S2 domain-targeting neutralizing monoclonal antibody. PLoS ONE. (2014) 9:e102415. doi: 10.1371/journal.pone.0102415
30. Lip KM, Shen S, Yang X, Keng CT, Zhang A, Oh HL, et al. Monoclonal antibodies targeting the HR2 domain and the region immediately upstream of the HR2 of the S protein neutralize in vitro infection of severe acute respiratory syndrome coronavirus. J Virol. (2006) 80:941–50. doi: 10.1128/JVI.80.2.941-950.2006
31. ter Meulen J, Bakker AB, van den Brink EN, Weverling GJ, Martina BE, Haagmans BL, et al. Human monoclonal antibody as prophylaxis for SARS coronavirus infection in ferrets. Lancet. (2004) 363:2139–41. doi: 10.1016/S0140-6736(04)16506-9
32. van den Brink EN, Ter Meulen J, Cox F, Jongeneelen MA, Thijsse A, Throsby M, et al. Molecular and biological characterization of human monoclonal antibodies binding to the spike and nucleocapsid proteins of severe acute respiratory syndrome coronavirus. J Virol. (2005) 79:1635–44. doi: 10.1128/JVI.79.3.1635-1644.2005
33. Warren TK, Jordan R, Lo MK, Ray AS, Mackman RL, Soloveva V, et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature. (2016) 531:381–5. doi: 10.1038/nature17180
34. Agostini ML, Andres EL, Sims AC, Graham RL, Sheahan TP, Lu X, et al. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. (2018) 9:e00221-18. doi: 10.1128/mBio.00221-18
35. Lim J, Jeon S, Shin HY, Kim MJ, Seong YM, Lee WJ, et al. Case of the index patient who caused tertiary transmission of COVID-19 infection in korea: the application of lopinavir/ritonavir for the treatment of COVID-19 infected pneumonia monitored by quantitative RT-PCR. J Korean Med Sci. (2020) 35:e79. doi: 10.3346/jkms.2020.35.e89
36. Pecheur EI, Borisevich V, Halfmann P, Morrey JD, Smee DF, Prichard M, et al. The synthetic antiviral drug arbidol inhibits globally prevalent pathogenic viruses. J Virol. (2016) 90:3086–92. doi: 10.1128/JVI.02077-15
37. Conti P, Ronconi G, Caraffa A, Gallenga CE, Ross R, Frydas I, et al. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents. (2020) 34:1. doi: 10.23812/CONTI-E
38. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. (2020) 395:507–13. doi: 10.1016/S0140-6736(20)30211-7
39. Gautret P, Lagier JC, Parola P, Hoang VT, Meddeb L, Mailhe M, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents. (2020) 2020:105949. doi: 10.1016/j.ijantimicag.2020.105949
40. Molina JM, Delaugerre C, Le Goff J, Mela-Lima B, Ponscarme D, Goldwirt L, et al. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Med Mal Infect. (2020) 50:384. doi: 10.1016/j.medmal.2020.03.006
41. Russell CD, Millar JE, Baillie JK. Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet. (2020) 395:473–5. doi: 10.1016/S0140-6736(20)30317-2
42. Iba T, Levy JH, Levi M, Connors JM, Thachil J. Coagulopathy of coronavirus disease 2019. Crit Care Med. (2020) doi: 10.1097/CCM.0000000000004458
43. Liu Y, Sun W, Guo Y, Chen L, Zhang L, Zhao S, et al. Association between platelet parameters and mortality in coronavirus disease 2019: retrospective cohort study. Platelets. (2020) 31:490–6. doi: 10.1080/09537104.2020.1754383
44. Shanmugaraj B, Siriwattananon K, Wangkanont K, Phoolcharoen W. Perspectives on monoclonal antibody therapy as potential therapeutic intervention for Coronavirus disease-19 (COVID-19). Asian Pac J Allergy Immunol. (2020) 38:10–8. doi: 10.12932/AP-200220-0773
45. Sui J, Li W, Roberts A, Matthews LJ, Murakami A, Vogel L, et al. Evaluation of human monoclonal antibody 80R for immunoprophylaxis of severe acute respiratory syndrome by an animal study, epitope mapping, and analysis of spike variants. J Virol. (2005) 79:5900–6. doi: 10.1128/JVI.79.10.5900-5906.2005
46. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. (2020) 94:e00127-20. doi: 10.1128/JVI.00127-20
47. Li JL, Li G, Jing XZ, Li YF, Ye QY, Jia HH, et al. Assessment of clinical sepsis-associated biomarkers in a septic mouse model. J Int Med Res. (2018) 46:2410–22. doi: 10.1177/0300060518764717
48. Tanaka T, Narazaki M, Kishimoto T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy. (2016) 8:959–70. doi: 10.2217/imt-2016-0020
49. Pedersen SF, Ho YC. SARS-CoV-2: a storm is raging. J Clin Invest. (2020) 130:2202–5. doi: 10.1172/JCI137647
50. Izumiyama T, Mori Y, Mori S, Mori N, Kodama T, Itoi E. The effect of anti-IL-6 receptor antibody for the treatment of McH-lpr/lpr-RA1 mice that spontaneously developed destructive arthritis and enthesitis. BMC Musculoskelet Disord. (2019) 20:286. doi: 10.1186/s12891-019-2664-3
51. Michot JM, Albiges L, Chaput N, Saada V, Pommeret F, Griscelli F, et al. Tocilizumab, an anti-IL6 receptor antibody, to treat Covid-19-related respiratory failure: a case report. Ann Oncol. (2020) 31:961–4. doi: 10.1016/j.annonc.2020.03.300
52. Velazquez-Salinas L, Verdugo-Rodriguez A, Rodriguez LL, Borca MV. The role of interleukin 6 during viral infections. Front Microbiol. (2019) 10:1057. doi: 10.3389/fmicb.2019.01057
53. Atal S, Fatima Z. IL-6 inhibitors in the treatment of serious COVID-19: a promising therapy? Pharmaceut Med. (2020) 1–9. doi: 10.1007/s40290-020-00342-z. [Epub ahead of print].
54. Feldmann M, Maini RN, Woody JN, Holgate ST, Winter G, Rowland M, et al. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet. (2020) 395:1407–9. doi: 10.1016/S0140-6736(20)30858-8
55. Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. (2006) 7:311–7. doi: 10.1038/ni1309
56. Krafts K, Hempelmann E, Skorska-Stania A. From methylene blue to chloroquine: a brief review of the development of an antimalarial therapy. Parasitol Res. (2012) 111:1–6. doi: 10.1007/s00436-012-2886-x
57. Lamoureux F, Zoubeidi A. Dual inhibition of autophagy and the AKT pathway in prostate cancer. Autophagy. (2013) 9:1119–20. doi: 10.4161/auto.24921
58. Li G, De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat Rev Drug Discov. (2020) 19:149–50. doi: 10.1038/d41573-020-00016-0
59. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. (2010) 140:313–26. doi: 10.1016/j.cell.2010.01.028
60. Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol. (2009) 625:220–33. doi: 10.1016/j.ejphar.2009.06.063
61. Savarino A, Di Trani L, Donatelli I, Cauda R, Cassone A. New insights into the antiviral effects of chloroquine. Lancet Infect Dis. (2006) 6:67–9. doi: 10.1016/S1473-3099(06)70361-9
62. Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J. (2005) 2:69. doi: 10.1186/1743-422X-2-69
63. van den Borne BE, Dijkmans BA, de Rooij HH, le Cessie S, Verweij CL. Chloroquine and hydroxychloroquine equally affect tumor necrosis factor-alpha, interleukin 6, and interferon-gamma production by peripheral blood mononuclear cells. J Rheumatol. (1997) 24:55–60.
64. Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol. (2011) 186:4794–804. doi: 10.4049/jimmunol.1000702
65. Coelho AS, Chagas CEP, de Padua RM, Pianetti GA, Fernandes C. A comprehensive stability-indicating HPLC method for determination of chloroquine in active pharmaceutical ingredient and tablets: Identification of oxidation impurities. J Pharm Biomed Anal. (2017) 145:248–54. doi: 10.1016/j.jpba.2017.06.023
66. Melles RB, Marmor MF. The risk of toxic retinopathy in patients on long-term hydroxychloroquine therapy. JAMA Ophthalmol. (2014) 132:1453–60. doi: 10.1001/jamaophthalmol.2014.3459
67. Zhao X, Jiang Y, Zhao Y, Xi H, Liu C, Qu F, et al. Analysis of the susceptibility to COVID-19 in pregnancy and recommendations on potential drug screening. Eur J Clin Microbiol Infect Dis. (2020) 39:1209–20. doi: 10.1007/s10096-020-03897-6
68. Chen J, Liu D, Liu L, Liu P, Xu Q, Xia L, et al. [A pilot study of hydroxychloroquine in treatment of patients with moderate COVID-19]. Zhejiang Da Xue Xue Bao Yi Xue Ban. (2020) 49:215–9.
69. Spiezia L, Boscolo A, Poletto F, Cerruti L, Tiberio I, Campello E, et al. COVID-19-related severe hypercoagulability in patients admitted to intensive care unit for acute respiratory failure. Thromb Haemost. (2020) 120:998–1000. doi: 10.1055/s-0040-1710018
70. Sheahan TP, Sims AC, Graham RL, Menachery VD, Gralinski LE, Case JB, et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med. (2017) 9:396. doi: 10.1126/scitranslmed.aal3653
71. Ko WC, Rolain JM, Lee NY, Chen PL, Huang CT, Lee PI, et al. Arguments in favour of remdesivir for treating SARS-CoV-2 infections. Int J Antimicrob Agents. (2020) 2020:105933. doi: 10.1016/j.ijantimicag.2020.105933
72. Bhimraj A, Morgan RL, Shumaker AH, Lavergne V, Baden L, Cheng VC, et al. Infectious diseases society of america guidelines on the treatment and management of patients with COVID-19. Clin Infect Dis. (2020) doi: 10.1093/cid/ciaa478. [Epub ahead of print].
73. Wang Y, Zhou F, Zhang D, Zhao J, Du R, Hu Y, et al. Evaluation of the efficacy and safety of intravenous remdesivir in adult patients with severe COVID-19: study protocol for a phase 3 randomized, double-blind, placebo-controlled, multicentre trial. Trials. (2020) 21:422. doi: 10.1186/s13063-020-04352-9
74. Davies M, Osborne V, Lane S, Roy D, Dhanda S, Evans A, et al. Remdesivir in treatment of COVID-19: a systematic benefit-risk assessment. Drug Saf. (2020) 43:645–56. doi: 10.1007/s40264-020-00966-9
75. Chen F, Chan KH, Jiang Y, Kao RY, Lu HT, Fan KW, et al. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. J Clin Virol. (2004) 31:69–75. doi: 10.1016/j.jcv.2004.03.003
76. Morgenstern B, Michaelis M, Baer PC, Doerr HW, Cinatl J Jr. Ribavirin and interferon-beta synergistically inhibit SARS-associated coronavirus replication in animal and human cell lines. Biochem Biophys Res Commun. (2005). 326:905–8. doi: 10.1016/j.bbrc.2004.11.128
77. Booth CM, Matukas LM, Tomlinson GA, Rachlis AR, Rose DB, Dwosh HA, et al. Clinical features and short-term outcomes of 144 patients with SARS in the greater Toronto area. JAMA. (2003) 289:2801–9. doi: 10.1001/jama.289.21.JOC30885
78. Mathurin KS, Martens GW, Kornfeld H, Welsh RM. CD4 T-cell-mediated heterologous immunity between mycobacteria and poxviruses. J Virol. (2009) 83:3528–39. doi: 10.1128/JVI.02393-08
79. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of t cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol. (2020). 11:827. doi: 10.3389/fimmu.2020.00827
80. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Jacobs C, Xavier RJ, et al. BCG-induced trained immunity in NK cells: Role for non-specific protection to infection. Clin Immunol. (2014) 155:213–9. doi: 10.1016/j.clim.2014.10.005
Keywords: SARS-CoV-2, COVID-19, immune therapy, monoclonal antibody, respiratory distress, cytokine treatment
Citation: Benani A and Ben Mkaddem S (2020) Mechanisms Underlying Potential Therapeutic Approaches for COVID-19. Front. Immunol. 11:1841. doi: 10.3389/fimmu.2020.01841
Received: 24 April 2020; Accepted: 08 July 2020;
Published: 21 July 2020.
Edited by:
Zisis Kozlakidis, International Agency for Research on Cancer (IARC), FranceReviewed by:
Talia H. Swartz, Icahn School of Medicine at Mount Sinai, United StatesVijayakumar Velu, Emory University, United States
Copyright © 2020 Benani and Ben Mkaddem. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanae Ben Mkaddem, c2FuYWUuYmVubWthZGRlbSYjeDAwMDQwO2luc2VybS5mcg==