 Jefferson Elias-Oliveira1
Jefferson Elias-Oliveira1 Jefferson Antônio Leite1
Jefferson Antônio Leite1 Ítalo Sousa Pereira1
Ítalo Sousa Pereira1 Jhefferson Barbosa Guimarães1
Jhefferson Barbosa Guimarães1 Gabriel Martins da Costa Manso1
Gabriel Martins da Costa Manso1 João Santana Silva1
João Santana Silva1 Rita Cássia Tostes2
Rita Cássia Tostes2 Daniela Carlos1*
Daniela Carlos1*- 1Department of Biochemistry and Immunology, Ribeirão Preto Medical School, University of São Paulo, Ribeirão Preto, Brazil
- 2Department of Pharmacology, Ribeirão Preto Medical School, University of São Paulo, Ribeirão Preto, Brazil
The intestinal microbiome maintains a close relationship with the host immunity. This connection fosters a health state by direct and indirect mechanisms. Direct influences occur mainly through the production of short-chain fatty acids (SCFAs), gastrointestinal hormones and precursors of bioactive molecules. Indirect mechanisms comprise the crosstalk between bacterial products and the host's innate immune system. Conversely, intestinal dysbiosis is a condition found in a large number of chronic intestinal inflammatory diseases, such as ulcerative colitis and Crohn's disease, as well as in diseases associated with low-grade inflammation, such as obesity, type 1 and 2 diabetes mellitus and cardiovascular diseases. NOD-Like receptors (NLRs) are cytoplasmic receptors expressed by adaptive and innate immune cells that form a multiprotein complex, termed the inflammasome, responsible for the release of mature interleukin (IL)-1β and IL-18. NLRs are also involved in the recognition of bacterial components and production of antimicrobial molecules that shape the gut microbiota and maintain the intestinal homeostasis. Recent novel findings show that NLRs may act as positive or negative regulators of inflammation by modulating NF-κB activation. This mini-review presents current and updated evidence on the interplay between NLRs and gut microbiota and their dual role, contributing to progression or conferring protection, in diabetes and other inflammatory diseases.
Introduction
The healthy human intestine is colonized by several microorganisms, including fungi, viruses, and bacteria belonging to different families (1). Studies on the gut microbiome show a high number of bacteria from the Bacteroidaceae, Prevotellaceae, Rikenellaceae, and Ruminococcaceae families in the colon (2). On the other hand, the small intestine is mainly colonized by bacteria from the Lactobacillaceae and Enterobacteriaceae families (3). In recent years, sequencing analysis of the 16S rRNA gene revealed an association between the gut microbiota and inflammatory diseases (4). Changes in the composition of the intestinal microbiota, a process called dysbiosis, play a key role in the pathogenesis of inflammatory diseases, such as rheumatoid arthritis (5), atherosclerosis (6), ulcerative colitis, Crohn's disease (7), and diabetes mellitus type 1 and 2 (8, 9). Accordingly, modulation of the gut microbiota by prebiotics and probiotics, as preventive or therapeutic strategies to mitigate the pathogenesis of inflammatory diseases, has been increasingly investigated (10).
Innate immunity receptors, also called pattern recognition receptors (PRRs), are expressed by several cells and are involved in the recognition of microbial products or endogenous self-molecules. PRRs are key components in the pathogenesis of inflammatory and autoimmune diseases (11, 12). Toll-Like Receptors (TLRs) and NOD-Like Receptors (NLRs) are among the main families that comprise the PRRs superfamily (13). In the process of dysbiosis, the increased pathobiontic bacteria modulates the expression and activation of TLRs, leading to a pro-inflammatory response in the intestine and in extra-intestinal sites (14, 15). On the other hand, NLRs have either beneficial or harmful effects that rely on the antimicrobial factors and pro-inflammatory cytokine profile following gut microbiota activation. This mini review highlights the divergent roles of NLRs in metabolic and inflammatory diseases associated with gut dysbiosis.
Gut Dysbiosis in Inflammatory Diseases
The intestinal microbiota, when in homeostasis, is directly related to the host's health. The intestinal microbiota influences host metabolism (16), immune system (17, 18), gut microbicide mechanisms (19), and maintains the intestinal barrier (20). Many studies show that environmental factors, such as the use of antibiotics (21, 22), diet (23) and stress (24) can alter the intestinal microbiota, increasing pathobiontic bacteria at the expense of commensal bacteria, a process known as dysbiosis (25). Gut dysbiosis contributes to the development of several autoimmune, inflammatory and metabolic diseases, such as rheumatoid arthritis (RA), inflammatory bowel diseases (IBD), and diabetes mellitus (26, 27). However, in many cases, as in IBD for example, it is not yet known whether dysbiosis is the cause or consequence of the disease (28, 29). The exact role of the gut microbiota in the pathogenesis of RA is not fully understood either. However, germ-free (GF) mice exhibit a delay in the development of RA when compared to the control group (30). In the early stages of RA, a decrease in some commensal bacteria, such as those belonging to the Bifidobacteria and Bacteroides genus, and an increase in Escherichia coli and Proteus mirabilis have been reported (31, 32). In addition, RA patients have an increase in Prevotella copri as well as in anti-P. copri IgA and IgG, suggesting that this bacteria may contribute to the pathogenesis of RA (33).
Inflammatory bowel diseases, such as Crohn's disease (CD) and ulcerative colitis (UC), affect ~3 million people in Europe and the USA, with a high and accelerated incidence in developing countries (34, 35). Although the etiology is still unclear, genetic predisposition and environmental factors, such as diet and use of antibiotics, are triggers of these diseases, characterized mainly by chronic intestinal inflammation (34, 36). In addition, disruption of the epithelial barrier and gut dysbiosis are widely reported in patients and in experimental models of gastrointestinal infections (37, 38) including patients with IBD (39, 40). 16S rRNA metagenomic analysis showed that the microbiota present in the feces of mice with UC is very different from microbiota in the feces of healthy mice, mainly by an increase in species of the phylum Verrucomicrobia and a decrease in Tenericutes in mice with colitis, which correlates with a higher disease score (41, 42). An increase in Enterobacteria is observed in fecal samples from patients with CD (43). Escherichia and Shigella abundance is also increased in this condition, when compared to healthy individuals. In addition, a reduction of the Roseburia, Coprococcus, and Ruminococcus genera, which are important butyrate producers, has been reported (44, 45). Analysis of colon biopsies from patients with IBD also shows a decrease in Firmicutes and an increase in Bacteroidetes (46) and patients with IBD exhibit increased biofilm production of strains of Enterococcus when compared to strains from the control group (47).
An imbalanced gut microbiota and changes in the intestinal barrier function are also closely linked to the pathogenesis of diabetes mellitus (DM) (48). DM comprises a group of metabolic diseases characterized mainly by chronic hyperglycemia, resulting from impaired secretion and/or insulin functionality (49). In type 1 diabetes (T1D), also called autoimmune diabetes, autoantibodies are present and autoreactive lymphocytes mediate pancreatic β-cell destruction, leading to complete insulin deficiency (50). The impact of the microbiota on the development of T1D was demonstrated using Myd88-deficient non-obese diabetic (NOD) mice bred in pathogen-free (SPF) or germ-free (GF) conditions. Whereas, SPF NOD.Myd88−/− mice are protected from T1D, mice under GF conditions develop T1D, showing that Myd88 protective effects depend on the presence of gut microbiota (51). In this context, many studies have shown differences in the composition of the microbiota between diabetic and non-diabetic patients, suggesting that these changes are associated with the development and severity of T1D (52, 53). Bacterial proteome studies show high enrichment with Clostridium and Bacteroides proteins in children with T1D, whereas the control group exhibit greater enrichment with Bifidobacterium proteins (54). Furthermore, the decrease in lactate- and butyrate-producing species, such B. adolescentis, is associated with T1D autoimmunity (55).
In models of type 2 diabetes (T2D), gut dysbiosis aggravates the inflammatory process, increases intestinal permeability and also alters the metabolism of short-chain fatty acids, which are important in insulin resistance (56), in addition to accelerating the development of obesity, retinopathy and nephropathy (57). In patients with T2D, excessive intake of carbohydrates and proteins is associated with an imbalance in the gut microbiota, with an increase in the Clostridium genus and a decrease in Bifidobacterium spp. and Lactobacillus, in addition to glucose intolerance (58). Moreover, in experimental models of T2D, the administration of bacteria of the Bifidobacterium genus improves glucose tolerance and confers a protective role in the development of T2D (59, 60). Similarly, the administration of Bacteroides acidifaciens decreases insulin resistance and even prevents obesity (61).
Innate immunity receptors, such as NLRs, have a decisive role in protecting the intestinal barrier against various microorganisms from the environment. These receptors also modulate microbial intestinal composition, being associated with the development of inflammatory diseases (62).
Protective Role of NLRs in Gut Microbiota Homeostasis and IBD
The innate immune system components are the first barrier against infections and recognize cell death, generating a rapid immune response due to the recognition of Pathogen-Associated Molecular Pattern (PAMPs) and Damage-associated molecular patterns (DAMPs), respectively (63). NLRs are part of a variety of innate immunity receptors, located in the intracellular environment, and initiate inflammatory processes. NOD1 and NOD2, central members of NLRs, mainly recognize bacterial peptidoglycan and, thus, induce gene transcription of NF-kB and mitogen-activated protein kinases (MAPKs), activating the expression of pro-inflammatory factors by different cells (13).
The NOD1 receptor detects gamma D-glutamyl-meso-diaminopimelic acid (γ iE-DAP), a peptide found mainly in Gram-negative bacteria, but also in groups of Gram-positive bacteria such as Listeria spp. and Bacillus spp (64–66). In the absence of NOD1, there is expansion of some intestinal bacteria, such as Clostridiales, Bacteroides spp., segmented filamentous bacteria (SFB), and Enterobacteriaceae. The NOD2 receptor detects the muramyl dipeptide (MDP) present in the bacterial peptideoglycan and is the most important receptor in intestinal homeostatic control (67). This receptor controls commensal microbiota and the elimination of pathogenic bacteria in intestinal crypts, minimizing the risk of intestinal inflammation and colorectal cancer (68–70). Interestingly, NOD2 expression depends on the presence of intestinal commensal bacteria, indicating a positive feedback relationship. NOD2 deficiency breaks this homeostatic interaction, resulting in gut dysbiosis, and increased IBD susceptibility (69).
Other NLRs also play an important role in intestinal homeostasis. The activation of NOD-like receptor family-pyrin domain containing 6 (NLRP6), through oligomerization and assembly of proteins–inflammasome complex–activates caspase-1 and leads to the synthesis of IL-1β and IL-18 in the intestinal epithelium (71). The deficiency of NLRP6 in mouse colonic epithelial cells decreases IL-18 levels, promotes gut dysbiosis and increases the risk of colitis (72, 73). IL-18 secreted by epithelial cells stimulates the barrier function and the regeneration of epithelial cells (73). In addition, commensal microbiota itself activates the NLRP6 inflammasome, leading to the production of mucus by goblet cells and antimicrobial peptides, maintaining a healthy composition of the intestinal microbiota (74).
NLRP3, another type of NLRs, is highly expressed in the monocytic lineage (75), and favors a greater production of IL-1β over IL-18, leading to changes in the composition of the intestinal microbiota (76). Under normal conditions, NLRP3 deficient mice exhibit gut dysbiosis associated with an excessive growth of Prevotellaceae and Bacteroidetes (77), whereas the ratio between Firmicutes and Bacterioidetes decreases (78). Unlike other NLRs, NLRP12 has anti-inflammatory effects, inhibiting canonical and non-canonical NF-κB; decreasing the production of inflammatory cytokines, chemokines and tumorigenic factors (79–82), and controlling infection by Gram-negative bacteria (83). NLRP12 deficiency, in a dextran sodium sulfate (DSS)-induced colitis model, promotes colon inflammation, decreases gut microbiota diversity and increases colitogenic bacteria, such Erysipelotrichaceae family, depicting a protective role of NLRP12 in IBD (84).
Divergent Roles of NLRs and AIM2 in T1D Development
In the past few years, several lines of evidence have demonstrated that members of the NLRs family participate in T1D pathogenesis. Recently, we reported that mice lacking NOD2, but not NOD1, are resistant to streptozotocin (STZ)-induced T1D and are unable to induce a Th1 and Th17 immune response in the pancreatic lymph nodes (PLNs) and pancreas. Interestingly, diabetic mice exhibit changes in the composition of the gut microbiota, and this is associated with gut microbiota translocation to PLNs (Figure 1). When these mice are submitted to a broad-spectrum antibiotic treatment, previously to the STZ injections, they do not develop signs of T1D, such as hyperglycemia. Additionally, the administration of the NOD2 ligand, MDP, promotes STZ-induced T1D in antibiotic-treated, STZ-injected wild-type (WT) mice. Our results demonstrate that gut microbiota recognition by NOD2 in the PLNs triggers a proinflammatory response, which induces a Th1 and Th17 cell pathogenic immune response, thus contributing to STZ-induced T1D pathogenesis (Table 1) (85).
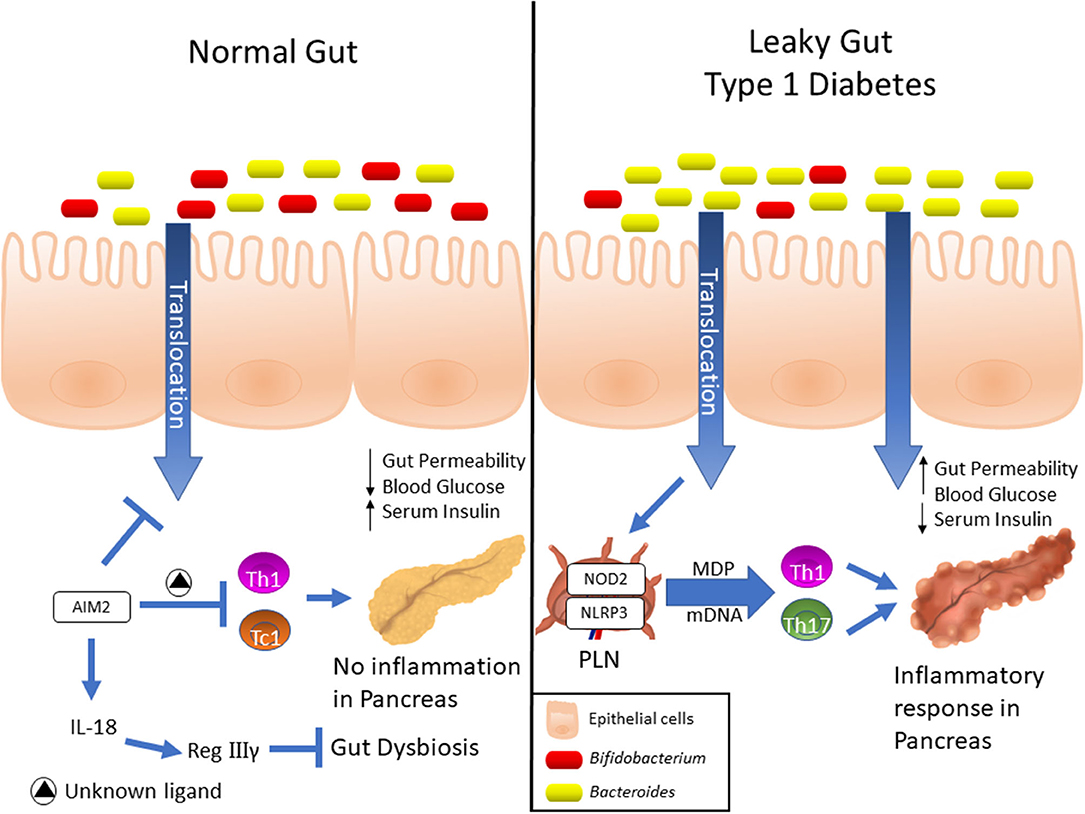
Figure 1. Expression and differential functions of NLRs in Type 1 diabetes development. Elevated AIM2 expression was detected into intestinal mucosa of pre-diabetic mice, and its activation induces the IL-18 release, which in turn, promotes the RegIIIγ production. This mechanism attenuates the gut dysbiosis, reinforces the gut barrier and dampens the Th1 and Tc1 lymphocyte response against insulin-producing β cells, which ultimately protects against T1D. On the other hand, NOD2 recognizes translocated muramyl dipeptide (MDP) from dysbiotic microbiota, and contributes to the activation of Th1 and Th17 lymphocytes in T1D. Finally, upregulation of NLRP3 expression in PLNs was observed in diabetic mice, which is activated in macrophages by recognition of mitochondrial DNA (mDNA), leads to IL-1β production and drives the pathogenic Th17 and Th1 lymphocyte generation, resulting in T1D onset.
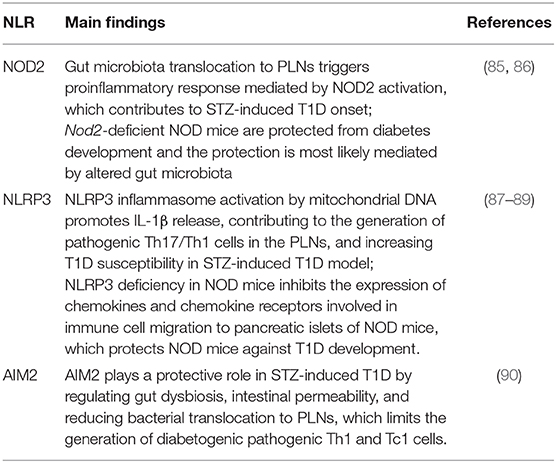
Table 1. Summary of experimental studies about the role of NLRs in T1D.
STZ-injected WT mice display an increase in different bacteria groups in the gut microbiota, such as Bacteroidaceae family and the Bacteroides genus that have been associated with increased susceptibility to T1D in humans (91, 92). These results recapitulate what has been found in type 1 diabetic patients, with the Bacteroidetes phylum, the Bacteroidaceae family, and the Bacteroides genus being more commonly found in autoantibody-positive children than in autoantibody-negative peers (55). Other important observation found among type 1 diabetic patients is decreased microbiota diversity, associated with reduced relative abundance of Bifidobacterium, Roseburia, Faecalibacterium, and Lachnospira (91). These data indicate that gut dysbiosis observed in type 1 diabetic patients may act as an environmental trigger in the development of the disease and that strategies aiming blockade of NOD2 signaling emerge as potential therapies for T1D. Similar results were reported in spontaneous T1D mice model. Non-cohoused NOD.NOD2−/− mice exhibit reduced T1D incidence and a decrease in CD4+ IFN-γ+/CD8+ IFN-γ+ (Th1/Tc1) and CD4+ IL-17+/CD8+ IL-17+ (Th17/Tc17) T cells in PLNs, indicating that NOD2 activation regulates T1D development by altering the composition of gut microbiota and by modulating the adaptive immune response (86).
Other studies also revealed that NLRP3 is required for T1D pathogenesis. NLRP3 deficiency in NOD mice protects against T1D by inhibiting the expression of chemokines and chemokine receptors involved in immune cell migration to pancreatic islets. NLRP3 deficiency in NOD mice reduces the expression of CCR5 and CXRC3 on T cells and also the gene expression of CCL5 and CXCL10 in pancreatic tissue and these processes occur in an IRF1-dependent manner (87). Additionally, our research group demonstrated that NLRP3 inflammasome activation by mitochondrial DNA (mDNA) promotes IL-1β release by macrophages, contributing to the generation of pathogenic Th17/Th1 cells in the PLNs and to T1D susceptibility in STZ-induced T1D model (Figure 1, Table 1) (88). In accordance, an association study in a north-eastern Brazilian population identified two single-nucleotide polymorphisms (SNPs) in NLRP3, rs10754558, and rs358294199, that are associated with T1D in humans, suggesting that variations in NLRP3 may be a predisposing genetic factor for the development of autoimmune T1D (89).
Another innate immune receptor that results in inflammasome assembly upon its activation, is the DNA sensor absent in melanoma 2 (AIM2) (93, 94). The activation of AIM2 is involved in autoimmune and inflammatory diseases (95). In the STZ T1D model, AIM2 is highly expressed in the ileum at early stages of the disease. Interestingly, AIM2−/− STZ-injected mice display increased T1D incidence, augmented intestinal permeability and bacterial translocation to PLNs, which leads to a proinflammatory response mediated by Th1 and Tc1 cells. When the gut microbiota is depleted by a broad-spectrum antibiotic cocktail before STZ-injections, the increased susceptibility to T1D observed in AIM2−/− mice is abrogated (Table 1). The effects induced by AIM2 activation in vivo are mediated by IL-18 release, which favors regenerating islet-derived III gamma (RegIIIγ) production, thus mitigating gut microbiota alterations and reinforcing the intestinal barrier function. Together, our data show that AIM2 activation limits gut microbiota dysbiosis, intestinal permeability and translocation to PLNs, decreasing a proinflammatory response, and conferring protection against T1D (90).
NLRs Role in Obesity, T2D and Comorbidities
Gut dysbiosis can lead to increased permeability of the intestinal barrier, resulting in low-grade systemic inflammation and metabolic disorders such as obesity, T2D and ischemic stroke (96, 97). Receptors of the innate immunity play a role in systemic inflammation caused by obesity. Mice fed a high-fat diet (HFD) exhibit an increase in colonic inflammation and endotoxemia due to elevated intestinal permeability of the colon mucosa (98). Additionally, increased TLR4 signaling in the colon and activation of NF-κB are observed (98). However, female mice lacking TLR4 display higher risk of developing obesity, but also greater protection to insulin resistance, perhaps due to the lack of TLR4 signaling in important organs for metabolic homeostasis (99). In addition, other studies showed that gut dysbiosis promotes a state of metabolic endotoxemia during obesity, resulting in blood LPS accumulation, metainflammation and insulin resistance through CD14/TLR4 pathway (100–102).
T2D is a chronic metabolic inflammatory condition and is the most common type of diabetes in adults worldwide (103). This disease is initiated by the worsening of pancreatic dysfunction, established when insulin production by β-pancreatic cells cannot keep up with the increase in peripheral insulin resistance (104, 105). Low-grade systemic inflammation accompanies diabetes, with high serum levels of C-reactive protein (CRP), tumor necrosis factor (TNF-α), monocyte chemo-attracting protein-1 (MCP-1) and IL-1β (106, 107). In addition, obesity, aging and other conditions that promote low-grade chronic inflammation are linked to increased risk of developing T2D (108–110). Systemically, the high serum concentrations of IL-6, IL-1β, and TNF-α increase insulin resistance and cause endothelial dysfunction, priming the vascular system to the development of diabetes-related diseases, including systemic arterial hypertension (111). Meanwhile, increased pancreatic IL-1β, IL-6, and IL-8 decrease insulin gene expression in β-pancreatic cells, contributing to increased insulin resistance (112).
A fine balance between the activation of innate NOD1 and NOD2 receptors is crucial for maintaining peripheral insulin resistance. Direct activation of NOD1 receptors through intraperitoneal administration of NOD1 ligand in WT mice leads to an increase in peripheral insulin resistance in up to 6 h (113). In this same study, the activation of NOD1 induced small increases in circulating proinflammatory cytokines. In addition, higher concentrations of inflammatory mediators are observed in cultures of 3T3-L1 fibroblasts differentiated into adipocytes and exposed for 18 h to NOD1 ligands (113). Inflammation of peripheral tissues, especially adipose tissue (114), is a hallmark of T2D and directly contributes to its pathogenesis through adipose tissue dysfunction and subsequent complications in energy homeostasis and intermediate metabolism (115). In this context, double knockout NOD1/NOD2 mice are protected against peripheral insulin resistance and peripheral inflammation observed in the obesity (HFD)-induced T2D model (113).
NOD2 is an innate immunity receptor that recognizes peptidoglycan in the cell wall of bacteria and, therefore, constitutes an important link between gut microbiota and immunity (116). Therefore, NOD2 activation profile may be important in metabolic diseases with immune branches and, therefore, may represent the link in the cross-talk between the gut microbiota and these diseases. The deficiency of NOD2 in mice allows greater translocation of bacteria from the intestine (116). In a model of HFD, mice deficient in NOD2 exhibit greater peripheral resistance to insulin, inflammation of visceral adipose tissue, and higher content of bacterial DNA in the liver (117). HFD increases the Firmicutes to Bacteriodetes ratio and NOD2−/− mice submitted to HFD exhibit dysbiosis, represented by an increase in the number of Helicobacter bacteria and in the Peptococcaceae family and reduction of the Clostridium genus, when compared to HFD-fed WT mice (117). Fecal transplantation from obese mice to lean GF mice increases total body mass and adipose-tissue mass. Alternatively, fecal transplantation from lean mice to obese GF mice reduces the adipose-tissue mass (118). In this last experimental setup, there is an increase in the number of Bacteriodetes phylum in the gut, which has been related to the production of microbiota metabolites with host modulatory properties, in particular: SCFAs (118). Interestingly, adequate consumption of dietary fiber favors the secretion of SCFAs by the intestinal microbiota. In addition, the activation of the GPR43 receptor in mice, by SCFAs, in M2 macrophages of adipose tissue leads to increased metabolic activity and favors maintenance and homeostasis of healthy adipose tissue, improving metabolic health (119).
Concluding Remarks
The different categories of NLRs modulate gut dysbiosis-driven extra-intestinal and intestinal inflammatory diseases. The effects of NLRs are diverse and may be either protective or deleterious depending on the immunological context. In the intestine, NLRs regulate gut microbiota composition and translocation by influencing mucus secretion and antimicrobial peptide production, thus playing a key role in the protection against inflammatory bowel diseases, such as ulcerative colitis and Crohn's disease. Alternatively, NLRs also are activated by microbial PAMPs (gut microbiota) or endogenous DAMPs (components from dead or dying cells), which act as negative or positive regulators of the innate and adaptive immunity response and contribute to the susceptibility or resistance to metabolic diseases such as obesity, type 1, type 2 diabetes, and their comorbidities. Thus, the pharmacological modulation of these receptors may represent new therapeutic strategies for these inflammatory and metabolic diseases.
Author Contributions
JE-O, JL, ÍP, JG, and GM equally contributed to the manuscript writing. JS and RT provided scientific assistance and revised it critically. DC coordinated and reviewed the manuscript. All authors approved the submission and publication.
Funding
This study was supported by grants from the São Paulo Research Foundation (FAPESP) (Process numbers: 2012/10395-0; 2018/14815-0).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Dieterich W, Schink M, Zopf Y. Microbiota in the gastrointestinal tract. Med Sci. (2018) 6:116. doi: 10.3390/medsci6040116
2. Hall AB, Tolonen AC, Xavier RJ. Human genetic variation and the gut microbiome in disease. Nat Rev Genet. (2017) 18:690–9. doi: 10.1038/nrg.2017.63
3. Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol. (2016) 14:20–32. doi: 10.1038/nrmicro3552
4. Wang J, Jia H. Metagenome-wide association studies: fine-mining the microbiome. Nat Rev Microbiol. (2016) 14:508–22. doi: 10.1038/nrmicro.2016.83
5. Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med. (2015) 21:895–905. doi: 10.1038/nm.3914
6. Karlsson FH, Fak F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, et al. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun. (2012) 3:1245. doi: 10.1038/ncomms2266
7. Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease. Gut. (2004) 53:1–4. doi: 10.1136/gut.53.1.1
8. Tai N, Wong FS, Wen L. The role of gut microbiota in the development of type 1, type 2 diabetes mellitus and obesity. Rev Endocr Metab Disord. (2015) 16:55–65. doi: 10.1007/s11154-015-9309-0
9. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. (2012) 490:55–60. doi: 10.1038/nature11450
10. Kolodziejczyk AA, Zheng D, Elinav E. Diet–microbiota interactions and personalized nutrition. Nat Rev Microbiol. (2019) 17:742–53. doi: 10.1038/s41579-019-0256-8
11. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
12. Suresh R, Mosser DM. Pattern recognition receptors in innate immunity, host defense, and immunopathology. Adv Physiol Educ. (2013) 37:284–91. doi: 10.1152/advan.00058.2013
13. Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. (2009) 21:317–37. doi: 10.1093/intimm/dxp017
14. Li X, Watanabe K, Kimura I. Gut microbiota dysbiosis drives and implies novel therapeutic strategies for diabetes mellitus and related metabolic diseases. Front Immunol. (2017) 8:1882. doi: 10.3389/fimmu.2017.01882
15. Frosali S, Pagliari D, Gambassi G, Landolfi R, Pandolfi F, Cianci R. How the intricate interaction among toll-like receptors, microbiota, and intestinal immunity can influence gastrointestinal pathology. J Immunol Res. (2015) 2015:489821. doi: 10.1155/2015/489821
16. Rowland I, Gibson G, Heinken A, Scott K, Swann J, Thiele I, et al. Gut microbiota functions : metabolism of nutrients and other food components. Eur J Nutr. (2018) 57:1–24. doi: 10.1007/s00394-017-1445-8
17. Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. (2012) 489:231–41. doi: 10.1038/nature11551
18. Lazar V, Ditu L-M, Pircalabirou GG, Gheorghe I, Curutiu C, Holban AM, et al. Aspects of gut microbiota and immune system interactions in infectious diseases, immunopathology, and cancer. Front Immunol. (2018) 9:1–1830. doi: 10.3389/fimmu.2018.01830
19. Iacob S, Iacob DG, Luminos LM. Intestinal microbiota as a host defense mechanism to infectious threats. Front Microbiol. (2019) 9:3328. doi: 10.3389/fmicb.2018.03328
20. Alam A, Neish A. Role of gut microbiota in intestinal wound healing and barrier function. Tissue Barriers. (2018) 6:1539595. doi: 10.1080/21688370.2018.1539595
21. Zarrinpar A, Chaix A, Xu ZZ, Chang MW, Marotz CA, Saghatelian A, et al. Antibiotic-induced microbiome depletion alters metabolic homeostasis by affecting gut signaling and colonic metabolism. Nat Commun. (2018) 9:2872. doi: 10.1038/s41467-018-05336-9
22. Kim HJ, Lee SH, Hong SJ. Antibiotics-induced dysbiosis of intestinal microbiota aggravates atopic dermatitis in mice by altered short-chain fatty acids. Allergy Asthma Immunol Res. (2020) 12:137–48. doi: 10.4168/aair.2020.12.1.137
23. Alou MT, Lagier J, Raoult D. Diet influence on the gut microbiota and dysbiosis related to nutritional disorders. Hum Microbiome J. (2016) 1:3–11. doi: 10.1016/j.humic.2016.09.001
24. Karl JP, Hatch AM, Arcidiacono SM, Pearce SC, Pantoja-Feliciano IG, Doherty LA, et al. Effects of psychological, environmental and physical stressors on the gut microbiota. Front Microbiol. (2018) 9:2013. doi: 10.3389/fmicb.2018.02013
25. Tilg H, Zmora N, Adolph TE, Elinav E. The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol. (2020) 20:40–54. doi: 10.1038/s41577-019-0198-4
26. Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol. (2016) 13:412–25. doi: 10.1038/nrgastro.2016.85
27. Wilkins LJ, Monga M, Miller AW. Defining dysbiosis for a cluster of chronic diseases. Sci Rep. (2019) 9:12918. doi: 10.1038/s41598-019-49452-y
28. Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol. (2012) 9:599–608. doi: 10.1038/nrgastro.2012.152
29. Buttó LF, Haller D. Dysbiosis in intestinal inflammation: cause or consequence. Int J Med Microbiol. (2016) 306:302–9. doi: 10.1016/j.ijmm.2016.02.010
30. Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ. Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis. (2015) 26:26191. doi: 10.3402/mehd.v26.26191
31. Vaahtovuo J, Munukka E, Korkeamaki M, Luukkainen R, Toivanen P. Fecal microbiota in early rheumatoid arthritis. J Rheumatol. (2008) 35:1500–5.
32. Newkirk MM, Zbar A, Baron M, Manges AR. Distinct bacterial colonization patterns of Escherichia coli subtypes associate with rheumatoid factor status in early inflammatory arthritis. Rheumatology. (2010) 49:1311–6. doi: 10.1093/rheumatology/keq088
33. Pianta A, Arvikar S, Strle K, Drouin EE, Wang Q, Costello CE, et al. Evidence of the immune relevance of prevotella copri, a gut microbe, in patients with rheumatoid arthritis. Arthritis Rheumatol. (2017) 69:964–75. doi: 10.1002/art.40003
34. Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: causation or correlation? Nat Publ Gr. (2017) 14:573–84. doi: 10.1038/nrgastro.2017.88
35. Collaborators G 2017 IBD. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990-2017: a systematic analysis for the Global burden of disease study 2017. Lancet Gastroenterol Hepatol. (2020) 5:17–30. doi: 10.1016/S2468-1253(19)30333-4
36. Mattos BRR, Garcia MPG, Nogueira JB, Paiatto LN, Albuquerque CG, Souza CL, et al. Inflammatory bowel disease : an overview of immune mechanisms and biological treatments. Mediators Inflamm. (2015) 2015:493012. doi: 10.1155/2015/493012
37. Beatty JK, Akierman S V, Motta JP, Muise S, Workentine ML, Harrison JJ, et al. Giardia duodenalis induces pathogenic dysbiosis of human intestinal microbiota biofilms. Int J Parasitol. (2017) 47:311–26. doi: 10.1016/j.ijpara.2016.11.010
38. Yu LCH, Shih YA, Wu LL, Lin YD, Kuo WT, Peng WH, et al. Enteric dysbiosis promotes antibiotic-resistant bacterial infection: systemic dissemination of resistant and commensal bacteria through epithelial transcytosis. Am J Physiol Gastrointest Liver Physiol. (2014) 307:G824–35. doi: 10.1152/ajpgi.00070.2014
39. Prorok-Hamon M, Friswell MK, Alswied A, Roberts CL, Song F, Flanagan PK, et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut. (2014) 63:761–70. doi: 10.1136/gutjnl-2013-304739
40. Chassaing B, Gewirtz AT. Pathobiont hypnotises enterocytes to promote tumour development. Gut. (2014) 63:1837–8. doi: 10.1136/gutjnl-2014-306890
41. Samanta AK, Torok VA, Percy NJ, Abimosleh SM, Howarth GS. Microbial fingerprinting detects unique bacterial communities in the faecal microbiota of rats with experimentally-induced colitis. J Microbiol. (2012) 50:218–25. doi: 10.1007/s12275-012-1362-8
42. Nagalingam NA, Kao JY, Young VB. Microbial ecology of the murine gut associated with the development of dextran sodium sulfate-induced colitis. Inflamm Bowel Dis. (2011) 17:917–26. doi: 10.1002/ibd.21462
43. Seksik P, Rigottier-Gois L, Gramet G, Sutren M, Pochart P, Marteau P, et al. Alterations of the dominant faecal bacterial groups in patients with Crohn's disease of the colon. Gut. (2003) 52:237–42. doi: 10.1136/gut.52.2.237
44. Chen L, Wang W, Zhou R, Ng SC, Li J, Huang M, et al. Characteristics of fecal and mucosa-associated microbiota in Chinese patients with inflammatory bowel disease. Medicine. (2014) 93:e51. doi: 10.1097/MD.0000000000000051
45. Kowalska-Duplaga K, Gosiewski T, Kapusta P, Sroka-Oleksiak A, Wedrychowicz A, Pieczarkowski S, et al. Differences in the intestinal microbiome of healthy children and patients with newly diagnosed Crohn's disease. Sci Rep. (2019) 9:18880. doi: 10.1038/s41598-019-55290-9
46. Walker AW, Sanderson JD, Churcher C, Parkes GC, Hudspith BN, Rayment N, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. (2011) 11:7. doi: 10.1186/1471-2180-11-7
47. Golinska E, Tomusiak A, Gosiewski T, Wiecek G, Machul A, Mikolajczyk D, et al. Virulence factors of Enterococcus strains isolated from patients with inflammatory bowel disease. World J Gastroenterol. (2013) 19:3562–72. doi: 10.3748/wjg.v19.i23.3562
48. Lu J, Ma KL, Ruan XZ. Dysbiosis of gut microbiota contributes to the development of diabetes mellitus. Infect microbes Dis. (2019) 1:43–8. doi: 10.1097/IM9.0000000000000011
49. Kharroubi AT. Diabetes mellitus: the epidemic of the century. World J Diabetes. (2015) 6:850. doi: 10.4239/wjd.v6.i6.850
50. Katsarou A, Gudbjörnsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Prim. (2017) 3:1–17. doi: 10.1038/nrdp.2017.16
51. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. (2008) 455:1109–13. doi: 10.1038/nature07336
52. Paun A, Yau C, Danska JS. The influence of the microbiome on type 1 diabetes. J Immunol. (2017) 198:590–5. doi: 10.4049/jimmunol.1601519
53. Jamshidi P, Hasanzadeh S, Tahvildari A, Farsi Y, Arbabi M, Mota JF, et al. Is there any association between gut microbiota and type 1 diabetes? A systematic review. Gut Pathog. (2019) 11:49. doi: 10.1186/s13099-019-0332-7
54. Pinto E, Anselmo M, Calha M, Bottrill A, Duarte I, Andrew PW, et al. The intestinal proteome of diabetic and control children is enriched with different microbial and host proteins. Microbiology. (2017) 163:161–74. doi: 10.1099/mic.0.000412
55. de Goffau MC, Luopajarvi K, Knip M, Ilonen J, Ruohtula T, Harkonen T, et al. Fecal microbiota composition differs between children with beta-cell autoimmunity and those without. Diabetes. (2013) 62:1238–44. doi: 10.2337/db12-0526
56. Sircana A, Framarin L, Leone N, Berrutti M, Castellino F, Parente R, et al. Altered gut microbiota in type 2 diabetes: just a coincidence? Curr Diab Rep. (2018) 18:98. doi: 10.1007/s11892-018-1057-6
57. Fernandes R, Viana SD, Nunes S, Reis F. Diabetic gut microbiota dysbiosis as an inflammaging and immunosenescence condition that fosters progression of retinopathy and nephropathy. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:1876–97. doi: 10.1016/j.bbadis.2018.09.032
58. Yamaguchi Y, Adachi K, Sugiyama T, Shimozato A, Ebi M, Ogasawara N, et al. Association of intestinal microbiota with metabolic markers and dietary habits in patients with type 2 diabetes. Digestion. (2016) 94:66–72. doi: 10.1159/000447690
59. Le TKC, Hosaka T, Nguyen TT, Kassu A, Dang TO, Tran HB, et al. Bifidobacterium species lower serum glucose, increase expressions of insulin signaling proteins, and improve adipokine profile in diabetic mice. Biomed Res. (2015) 36:63–70. doi: 10.2220/biomedres.36.63
60. Kikuchi K, Ben Othman M, Sakamoto K. Sterilized bifidobacteria suppressed fat accumulation and blood glucose level. Biochem Biophys Res Commun. (2018) 501:1041–7. doi: 10.1016/j.bbrc.2018.05.105
61. Yang JY, Lee YS, Kim Y, Lee SH, Ryu S, Fukuda S, et al. Gut commensal Bacteroides acidifaciens prevents obesity and improves insulin sensitivity in mice. Mucosal Immunol. (2017) 10:104–16. doi: 10.1038/mi.2016.42
62. Lipinski S, Rosenstiel P. Debug your bugs - how NLRs shape intestinal host-microbe interactions. Front Immunol. (2013) 4:479. doi: 10.3389/fimmu.2013.00479
63. Daolin T, Rui K, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. (2012) 249:158–75. doi: 10.1111/j.1600-065X.2012.01146.x
64. Girardin SE, Travassos LH, Hervé M, Blanot D, Boneca IG, Philpott DJ, et al. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem. (2003) 278:41702–8. doi: 10.1074/jbc.M307198200
65. Girardin SE, Boneca IG, Carneiro LAM, Antignac A, Jéhanno M, Viala J, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. (2003) 300:1584–7. doi: 10.1126/science.1084677
66. Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. (2003) 4:702–7. doi: 10.1038/ni945
67. Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2: implications for Crohn's disease. J Biol Chem. (2003) 278:5509–12. doi: 10.1074/jbc.C200673200
68. Rehman A, Sina C, Gavrilova O, Häsler R, Ott S, Baines JF, et al. Nod2 is essential for temporal development of intestinal microbial communities. Gut. (2011) 60:1354–62. doi: 10.1136/gut.2010.216259
69. Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci USA. (2009) 106:15813–8. doi: 10.1073/pnas.0907722106
70. Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. (2013) 123:700–11. doi: 10.1172/JCI62236
71. Henao-Mejia J, Elinav E, Thaiss CA, Flavell RA. Inflammasomes and metabolic disease. Annu Rev Physiol. (2014) 76:57–78. doi: 10.1146/annurev-physiol-021113-170324
72. Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell. (2015) 163:1428–43. doi: 10.1016/j.cell.2015.10.048
73. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. (2011) 145:745–57. doi: 10.1016/j.cell.2011.04.022
74. Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell. (2014) 156:1045–59. doi: 10.1016/j.cell.2014.01.026
75. Awad F, Assrawi E, Jumeau C, Georgin-Lavialle S, Cobret L, Duquesnoy P, et al. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLoS ONE. (2017) 12:e0175336. doi: 10.1371/journal.pone.0175336
76. Yao X, Zhang C, Xing Y, Xue G, Zhang Q, Pan F, et al. Remodelling of the gut microbiota by hyperactive NLRP3 induces regulatory T cells to maintain homeostasis. Nat Commun. (2017) 8:1–17. doi: 10.1038/s41467-017-01917-2
77. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. (2012) 482:179–85. doi: 10.1038/nature10809
78. Ehlers MR, Todd RM. Genesis and maintenance of attentional biases: the role of the locus coeruleus-noradrenaline system. Neural Plast. (2017) 1:2–3. doi: 10.1155/2017/6817349
79. Williams KL, Lich JD, Duncan JA, Reed W, Rallabhandi P, Moore C, et al. The CATERPILLER protein Monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor α-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J Biol Chem. (2005) 280:39914–24. doi: 10.1074/jbc.M502820200
80. Lich JD, Williams KL, Moore CB, Arthur JC, Davis BK, Taxman DJ, et al. Cutting edge: monarch-1 suppresses non-canonical NF-κB activation and p52-dependent chemokine expression in monocytes. J Immunol. (2007) 178:1256–60. doi: 10.4049/jimmunol.178.3.1256
81. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity. (2012) 36:742–54. doi: 10.1016/j.immuni.2012.03.012
82. Zaki MH, Vogel P, Malireddi RKS, Body-Malapel M, Anand PK, Bertin J, et al. The NOD-Like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell. (2011) 20:649–60. doi: 10.1016/j.ccr.2011.10.022
83. Vladimer GI, Weng D, Paquette SWM, Vanaja SK, Rathinam VAK, Aune MH, et al. The NLRP12 inflammasome recognizes yersinia pestis. Immunity. (2012) 37:96–107. doi: 10.1016/j.immuni.2012.07.006
84. Chen L, Wilson JE, Koenigsknecht MJ, Chou WC, Montgomery SA, Truax AD, et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat Immunol. (2017) 18:541–51. doi: 10.1038/ni.3690
85. Costa FRC, Francozo MCS, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al. Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med. (2016) 213:1223–39. doi: 10.1084/jem.20150744
86. Li YY, Pearson JA, Chao C, Peng J, Zhang X, Zhou Z, et al. Nucleotide-binding oligomerization domain-containing protein 2 (Nod2) modulates T1DM susceptibility by gut microbiota. J Autoimmun. (2017) 82:85–95. doi: 10.1016/j.jaut.2017.05.007
87. Hu C, Ding H, Li Y, Pearson JA, Zhang X, Flavell RA, et al. NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets. Proc Natl Acad Sci USA. (2015) 112:11318–23. doi: 10.1073/pnas.1513509112
88. Carlos D, Costa FRC, Pereira CA, Rocha FA, Yaochite JNU, Oliveira GG, et al. Mitochondrial DNA activates the NLRP3 inflammasome and predisposes to type 1 diabetes in murine model. Front Immunol. (2017) 8:164. doi: 10.3389/fimmu.2017.00164
89. Pontillo A, Brandao L, Guimaraes R, Segat L, Araujo J, Crovella S. Two SNPs in NLRP3 gene are involved in the predisposition to type-1 diabetes and celiac disease in a pediatric population from northeast Brazil. Autoimmunity. (2010) 43:583–9. doi: 10.3109/08916930903540432
90. Leite JA, Pessenda G, Guerra-Gomes IC, de Santana AKM, André Pereira C, Ribeiro Campos Costa F, et al. The DNA sensor AIM2 protects against streptozotocin-induced type 1 diabetes by regulating intestinal homeostasis via the IL-18 pathway. Cells. (2020) 9:959. doi: 10.3390/cells9040959
91. Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. (2013) 11:46. doi: 10.1186/1741-7015-11-46
92. Patterson E, Marques TM, O'Sullivan O, Fitzgerald P, Fitzgerald GF, Cotter PD, et al. Streptozotocin-induced type-1-diabetes disease onset in Sprague-Dawley rats is associated with an altered intestinal microbiota composition and decreased diversity. Microbiology. (2015) 161:182–93. doi: 10.1099/mic.0.082610-0
93. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. (2009) 458:514–8. doi: 10.1038/nature07725
94. Rathinam VAK, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. (2010) 11:395–402. doi: 10.1038/ni.1864
95. Man SM, Karki R, Kanneganti T-D. AIM2 inflammasome in infection, cancer, and autoimmunity: role in DNA sensing, inflammation, and innate immunity. Eur J Immunol. (2016) 46:269–80. doi: 10.1002/eji.201545839
96. Yamashiro K, Tanaka R, Urabe T, Ueno Y, Yamashiro Y, Nomoto K, et al. Gut dysbiosis is associated with metabolism and systemic inflammation in patients with ischemic stroke. PLoS ONE. (2017) 12:e0171521. doi: 10.1371/journal.pone.0171521
97. Pasini E, Corsetti G, Assanelli D, Testa C, Romano C, Dioguardi FS, et al. Effects of chronic exercise on gut microbiota and intestinal barrier in human with type 2 diabetes. Minerva Med. (2019) 110:3–11. doi: 10.23736/S0026-4806.18.05589-1
98. Kim KA, Gu W, Lee IA, Joh EH, Kim DH. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE. (2012) 7:e47713. doi: 10.1371/journal.pone.0047713
99. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. (2006) 116:3015–25. doi: 10.1172/JCI28898
100. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. (2007) 56:1761–72. doi: 10.2337/db06-1491
101. Poggi M, Bastelica D, Gual P, Iglesias MA, Gremeaux T, Knauf C, et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia. (2007) 50:1267–76. doi: 10.1007/s00125-007-0654-8
102. Luche E, Cousin B, Garidou L, Serino M, Waget A, Barreau C, et al. Metabolic endotoxemia directly increases the proliferation of adipocyte precursors at the onset of metabolic diseases through a CD14-dependent mechanism. Mol Metab. (2013) 2:281–91. doi: 10.1016/j.molmet.2013.06.005
103. Piper MS, Saad RJ. Diabetes mellitus and the Cclon. Curr Treat Options Gastroenterol. (2017) 15:460–74. doi: 10.1007/s11938-017-0151-1
104. Alarcon C, Boland BB, Uchizono Y, Moore PC, Peterson B, Rajan S, et al. Pancreatic β-cell adaptive plasticity in obesity increases insulin production but adversely affects secretory function. Diabetes. (2016) 65:438–50. doi: 10.2337/db15-0792
105. Boland BB, Rhodes CJ, Grimsby JS. The dynamic plasticity of insulin production in β-cells. Mol Metab. (2017) 6:958–73. doi: 10.1016/j.molmet.2017.04.010
106. Randeria SN, Thomson GJA, Nell TA, Roberts T, Pretorius E. Inflammatory cytokines in type 2 diabetes mellitus as facilitators of hypercoagulation and abnormal clot formation. Cardiovasc Diabetol. (2019) 18:72. doi: 10.1186/s12933-019-0870-9
107. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-α in human obesity and insulin resistance. J Clin Invest. (1995) 95:2409–15. doi: 10.1172/JCI117936
108. Yan ST, Jia JH, Lv XF, Shao YH, Yin SN, Zhang XG, et al. Glycemic control and comprehensive metabolic risk factors control in older adults with type 2 diabetes. Exp Gerontol. (2019) 127:110713. doi: 10.1016/j.exger.2019.110713
109. Maskarinec G, Sadakane A, Sugiyama H, Brenner A, Tatsukawa Y, Grant E. Type 2 diabetes, obesity, and breast cancer risk among Japanese women of the atomic bomb survivor cohort. Cancer Epidemiol. (2019) 60:179–84. doi: 10.1016/j.canep.2019.04.009
110. Lee S, Lacy ME, Jankowich M, Correa A, Wu WC. Association between obesity phenotypes of insulin resistance and risk of type 2 diabetes in African Americans: the Jackson heart study. J Clin Transl Endocrinol. (2020) 19:100210. doi: 10.1016/j.jcte.2019.100210
111. Eckardt K, Görgens SW, Raschke S, Eckel J. Myokines in insulin resistance and type 2 diabetes. Diabetologia. (2014) 57:1087–99. doi: 10.1007/s00125-014-3224-x
112. Hotamisligil GkS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. (1996) 271:665–70. doi: 10.1126/science.271.5249.665
113. Schertzer JD, Tamrakar AK, Magalhães JG, Pereira S, Bilan PJ, Fullerton MD, et al. NOD1 activators link innate immunity to insulin resistance. Diabetes. (2011) 60:2206–15. doi: 10.2337/db11-0004
114. Rakotoarivelo V, Lacraz G, Mayhue M, Brown C, Rottembourg D, Fradette J, et al. Inflammatory cytokine profiles in visceral and subcutaneous adipose tissues of obese patients undergoing bariatric surgery reveal lack of correlation with obesity or diabetes. EBioMedicine. (2018) 30:237–47. doi: 10.1016/j.ebiom.2018.03.004
115. Garg A. Adipose tissue dysfunction in obesity and lipodystrophy. Clin Cornerstone. (2006) 8(Suppl. 4):S7–13. doi: 10.1016/S1098-3597(06)80039-6
116. de Souza PR, Guimarães FR, Sales-Campos H, Bonfá G, Nardini V, Chica JEL, et al. Absence of NOD2 receptor predisposes to intestinal inflammation by a deregulation in the immune response in hosts that are unable to control gut dysbiosis. Immunobiology. (2018) 223:577–85. doi: 10.1016/j.imbio.2018.07.003
117. Denou E, Lolmède K, Garidou L, Pomie C, Chabo C, Lau TC, et al. Defective NOD 2 peptidoglycan sensing promotes diet-induced inflammation, dysbiosis, and insulin resistance. EMBO Mol Med. (2015) 7:259–74. doi: 10.15252/emmm.201404169
118. Muegge BD, Kuczynski J, Knights D, Clemente JC, González A, Fontana L, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. (2011) 332:970–4. doi: 10.1126/science.1198719
Keywords: NLRs, microbiota, gut dysbiosis, diabetes, inflammatory diseases
Citation: Elias-Oliveira J, Leite JA, Pereira ÍS, Guimarães JB, Manso GMdC, Silva JS, Tostes RC and Carlos D (2020) NLR and Intestinal Dysbiosis-Associated Inflammatory Illness: Drivers or Dampers? Front. Immunol. 11:1810. doi: 10.3389/fimmu.2020.01810
Received: 15 May 2020; Accepted: 07 July 2020;
Published: 11 August 2020.
Edited by:
Alessio Fasano, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Charles Kelly, King's College London, United KingdomMegan K. L. MacLeod, University of Glasgow, United Kingdom
Copyright © 2020 Elias-Oliveira, Leite, Pereira, Guimarães, Manso, Silva, Tostes and Carlos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Carlos, ZGFuaWNhciYjeDAwMDQwO3VzcC5icg==