 Tiantian Guo
Tiantian Guo Liqing Zou1,2†
Liqing Zou1,2† Jianjiao Ni
Jianjiao Ni Yue Zhou
Yue Zhou Xi Yang
Xi Yang Zhengfei Zhu
Zhengfei Zhu- 1Department of Radiation Oncology, Fudan University Shanghai Cancer Center, Shanghai, China
- 2Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, China
Regulatory T cells (Tregs), which have long been recognized as essential regulators of both inflammation and autoimmunity, also impede effective antitumor immune response due to their immunosuppressive properties. Combined radiotherapy and immunotherapeutic interventions focusing on the removal of Tregs have recently garnered interest as a promising strategy to reverse immunosuppression. Meanwhile, Tregs are emerging as a key player in the pathogenesis of radiation-induced lung injury (RILI), a frequent and potentially life-threatening complication of thoracic radiotherapy. Recognition of the critical role of Tregs in RILI raises the important question of whether radiotherapy combined with Treg-targeting immunotherapy offers any beneficial effects in the protection of normal lung tissue. This present review focuses on the contributions of Tregs to RILI, with particular emphasis on the suspected differential role of Tregs in the pneumonitic phase and fibrotic phase of RILI. We also introduce recent progress on the potential mechanisms by which Tregs modulate RILI and the crosstalk among Tregs, other infiltrating T cells, fibrocytes, and resident epithelial cells driving disease pathogenesis. Finally, we discuss whether Tregs also hold promise as a potential target for immunotherapeutic interventions for RILI.
Introduction
Radiotherapy has a well-established role in the management of thoracic neoplasms (1). However, the lung is a radiosensitive organ, and radiation-induced lung injury (RILI), consisting of radiation-induced pneumonitis (RP) and pulmonary fibrosis (RPF), severely limits the efficacy of radiotherapy and impairs the quality of life of cancer patients, making it a paramount concern for radiation oncologists (2). A variety of factors may affect the risk of RILI, such as the radiation technique used, the volume of normal lung irradiated, dose fractionation regimen, and concurrent chemotherapy (1). From published data to date, the incidence of symptomatic RP is highest for lung cancer (5–50%), followed by mediastinal lymphatics (5–10%) and breast cancer (1–5%) (3, 4); the reported incidence of RPF is in the range of 1–51% in patients receiving thoracic radiotherapy (5–7) and 70–80% in high-dose regions (8).
Regulatory T cells (Tregs), a subset of CD4+ T cells, have long been recognized as essential regulators of inflammation and autoimmunity (9). Recently, Tregs have emerged as a key player in the suppression of antitumor immunity (10). The combination of radiotherapy and Treg-targeting immunotherapy has garnered interest as a promising strategy to reverse immunosuppression, albeit it may also carry potential risks of inducing treatment-related toxicities (11). Meanwhile, recent studies have expanded our understanding on how Tregs modulate fibrogenesis. Intriguingly, Tregs have been shown to be both a friend and a foe of fibrosis; depending on disease stages and their interplay with other immune cells, Tregs may exert anti- or pro-fibrotic effects (12–15). Notably, growing evidence highlights the involvement of Tregs in RPF (16–23), raising the important question of whether radiotherapy combined with Treg-targeting immunotherapy offers any beneficial effects in the protection of normal lung tissue.
In this review, we summarize the current knowledge on the contributions of Tregs to RILI, with particular emphasis on the suspected differential roles of Tregs in the pneumonitic phase and fibrotic phase of RILI. The ultimate goal is to evaluate whether Tregs hold promise as a potential target for immunotherapeutic interventions for RILI.
RILI: A Disturbed Balance Between Inflammation and Tissue Repair
RILI can be generally divided into two distinct, yet tightly connected phases. One is an acute pneumonitic phase, termed RP, which manifests with dyspnea, non-productive cough, and fever within the first 6 months of radiotherapy (1). The second is RPF, a late, irreversible phase (>1 year following therapy) characterized by fibroblast activation, extracellular matrix (ECM) accumulation, and aberrant tissue remodeling (1). Patients with RPF may present with symptoms including dyspnea and respiratory insufficiency (24).
Radiation-induced damage to the resident cells (e.g., epithelial and endothelial cells) triggers a cascade of molecular events, including reactive oxygen species production and DNA damage, which results in an early immune response aimed at initiating tissue repair (20). The subsequent release of damage-associated molecular patterns (DAMPs) induces secretion of pro-inflammatory mediators such as interleukin (IL)−1 and tumor necrosis factor-α (TNF-α) through the activation of the nuclear factor kappa-B (NF-κB) signaling pathway (25), allowing activation of resident immune cells and recruitment of inflammatory cells (26), which, in turn, amplifies the ongoing inflammatory response (2, 26). Notably, accumulating evidence has pointed to the involvement of lymphocytes in radiation-induced early lung inflammation (17, 22, 27). For example, Cappuccini et al. described elevated levels of Th17-associated cytokines (e.g., IL-17 and IL-23) in the bronchoalveolar lavage fluid of irradiated mice at 21 days post thorax irradiation with 15 Gy (22), suggesting the contribution of pro-inflammatory Th17 responses to the development of RILI. Current treatment options for RP primarily focus on the control of excessive inflammatory responses with glucocorticoids, potent anti-inflammatory agents that are known to inhibit the expression of pro-inflammatory cytokines and abrogate the activity of NF- κB (28, 29).
In some cases, RP resolves after treatment with corticosteroids; in other cases, it evolves into an uncontrolled, fibrotic process, namely RPF. In contrast to the normal wound healing process, which involves a series of finely orchestrated biological events, RPF enters a “never-ending spiral,” in which tissue hypoxia, epithelial-mesenchymal transition (EMT), and fibroblast activation ultimately lead to the destruction of the normal lung architecture (2, 18, 30, 31). Transforming growth factor-β (TGF-β) has been recognized as a key signaling molecule in the fibrotic process (26), which stimulates fibroblast proliferation, EMT, and ECM production (26, 32). Furthermore, recent research has deepened our understanding of how lymphocytes - and, in particular, Tregs - are involved in the fibrotic process (16–23, 33). We speculate that a disturbed balance between inflammation and tissue regeneration is a central issue in RPF and that the interaction among Tregs, other infiltrating T cells, epithelial cells, and fibrocytes is instrumental in shifting the local environment toward one favoring fibrosis.
Tregs and Immunoregulatory Cytokines
Two major subgroups of Tregs have been defined as follows, based on their differential developmental origins: (I) natural Tregs arising in the thymus (34) (II) inducible Tregs which can be induced from naïve CD4+ T cells by antigen exposure and cytokines such as TGF-β in the periphery (34–36). Mouse thymus-derived natural Tregs are characterized by their high expression of CD25 and constitutive expression of transcription factor forkhead box P3 (FoxP3) (37). In contrast to mouse Foxp3+ Tregs, human Foxp3+ T cells are more heterogeneous in function (38). For example, human naïve CD4+ T cells can transiently express Foxp3 after in vitro T-cell receptor stimulation without possessing suppressive functions (39–42). Therefore, Foxp3 per se may not represent a sufficient marker for functional Tregs, particularly in humans. In addition, Foxp3 is expressed at low levels or transiently after activation in some subsets of human Tregs (43, 44). Thus, identification of human Tregs is even more complex. At present, additional markers, such as CD127 and CD45RA, are also used to delineate Tregs in humans (45, 46).
Tregs in mice have been shown to exert suppression by cell-cell contact and humoral mechanisms with involvement of a variety of molecules, including surface molecules (e.g., cytotoxic T lymphocyte antigen-4 (CTLA-4), CD39, and CD73), immunoregulatory cytokines (e.g., TGF-β, IL-10, and IL-35), and secreted molecules (e.g., granzyme) (47). Several potential mechanisms of suppression by human Tregs have also been proposed, based on in vitro suppression assays, including cell-contact dependent mechanisms (e.g., Fas/FasL-mediated apoptosis of CD8+ responder cells and lysis of target cells through a perforin/granzyme-dependent pathway) and humoral factor-mediated mechanisms (e.g., secretion of inhibitory cytokines such as IL-10) (48–51). Notably, the mechanisms of human Treg-mediated suppression are similar, but not identical to, mechanisms of regulation by mouse Tregs. For example, IL-35 has been reported to be constitutively expressed by mouse Tregs and contribute to the suppressive activity of Tregs (52), whereas humans Tregs do not constitutively express IL-35 (53). In relations to the lungs, in vivo mouse studies have demonstrated IL-10 mediated Treg effects in allergic conditions (54, 55). In acute lung injury (ALI), Tregs were found to promote the resolution of inflammation through modulation of the alveolar inflammatory milieu in a mouse model of ALI (56). Furthermore, inhibition of TGF-β with neutralizing antibodies abrogated Treg-mediated resolution of inflammation in ALI (56), indicating a potential contribution of TGF-β-dependent mechanisms in the suppressive effects of Tregs. Additionally, recent observations demonstrate that mouse CD4+Foxp3+ Tregs in the lungs can also express the IL-33 receptor suppression of tumorigenicity 2 (ST2) (57, 58). IL-33 is an alarmin cytokine that can be released from damaged epithelial cells to alert the immune system after tissue injury (59). Intriguingly, after exposure to IL-33, mouse Tregs were found to increase the expression levels of GATA-3 as well as ST2 and secrete Th2 cytokines (57). Moreover, it has been reported that mouse ST2+ Tregs secrete high levels of amphiregulin, IL-10, and TGF-β in vitro in the presence of IL-33 (58, 60). Thus, IL-33-responsive Tregs may have a role in tissue repair and fibrotic responses in the lungs, which needs to be confirmed in further studies.
Tregs in Radiation-Induced Pneumonitis
As stated above, Tregs have been implicated in the resolution of inflammation in ALI and allergic conditions, and one can speculate that Tregs may also be beneficial during the pneumonitic phase of RILI in that they dampen excessive pro-inflammatory responses, thereby ameliorating inflammation-associated tissue damage.
Supporting this hypothesis, Wirsdorfer et al. found that thorax irradiation caused a transient accumulation of Tregs in irradiated lungs at 21 days after irradiation. Moreover, the accumulation of Tregs was associated with increased surface expression of immunoregulatory molecules (e.g., CTLA-4, CD73) on CD4+ T cells (17). It is tempting to speculate that Tregs play a protective role in the pneumonitic phase via their suppressive action on pro-inflammatory T cells, such as Th17 cells (22, 61, 62). Another murine study from Liu et al. demonstrated a transient increase in the number of Tregs expressing CTLA-4 in hilar lymph nodes and the spleen at 7 days after the silica exposure (12). Further, depletion of Tregs by intraperitoneal injection of anti-CD25 mAb 1 day before the silica exposure and every 7 days thereafter led to enhanced early lung inflammation with significantly increased infiltration of neutrophils at 7 days after the silica exposure (12), which lends further credence to the protective role of Tregs in the early inflammatory response to lung injury. Moreover, a recent clinical study reported that an imbalance of the Th17/Treg ratio toward Th17 cells was a predictor of RP in patients receiving thoracic radiotherapy (63).
There are several potential explanations for the accumulation of Tregs in irradiated lungs during the pneumonitic phase. First, the increased Treg fraction may be partially attributed to enhanced radioresistance and survival of Tregs compared with other lymphocyte subpopulations (10). Second, given the crucial role of TGF-β in the development of Tregs (34, 36), radiation-induced changes in the cytokine milieu (e.g., increased levels of TGF-β) may favor Treg accumulation in the lung tissue. However, it is noteworthy that radiation induces TGF-β production in a dose-dependent, time-dependent, and tissue-specific manner (64), and the precise role of TGF-β in the accumulation of Tregs in the lung tissue requires further exploration. Additionally, in irradiated macrophages, radiation-induced DNA damage results in potent induction of pro-inflammatory cytokines and chemokines (65). Tregs express a repertoire of inflammatory chemokines receptors (e.g., CCR2, CCR4, and CCR5) (66), suggesting that inflammatory chemokines may mediates the recruitment of Tregs at inflammatory sites through binding to their receptors on Tregs. For example, CCL2 has been shown to have a crucial role in the recruitment of Tregs to an inflamed site (67–69).
Current evidence indicates that Tregs can, via diverse mechanisms, dampen the inflammatory response after tissue injury and promote efficient repair of damaged tissue, which has been elegantly reviewed by Li et al. (70). Further studies are warranted to uncover the precise mechanisms whereby Tregs modulate inflammation in RILI.
Tregs in Radiation-Induced Pulmonary Fibrosis
Current evidence indicates that Tregs exert a pro-fibrotic role in RPF (18, 21). Wirsdörfer et al. demonstrated in a mouse model that CD4+ FoxP3+ Tregs accumulated in irradiated lungs during the pneumonitic and fibrotic phase. Moreover, Tregs generally expressed CD73 (16). Of further interest are the findings by Xiong et al. demonstrating that long-term (6 months) depletion of Tregs by intraperitoneal injection of anti-CD25 mAb 2 h after thorax irradiation and every 7 days therefore effectively attenuated RPF in mice (18, 21). Notably, in these studies, the ablation of Tregs covered both the early phase and the late chronic phase. Therefore, it is plausible to speculate that Tregs may predominantly play a pro-fibrotic role in RILI. Tregs have been shown to contribute to RPF through several mechanisms, including promotion of fibrocyte accumulation, promotion of EMT, modulation of Th1/Th2 balance, and suppression of Th17 responses (shown in Figure 1) (18, 21).
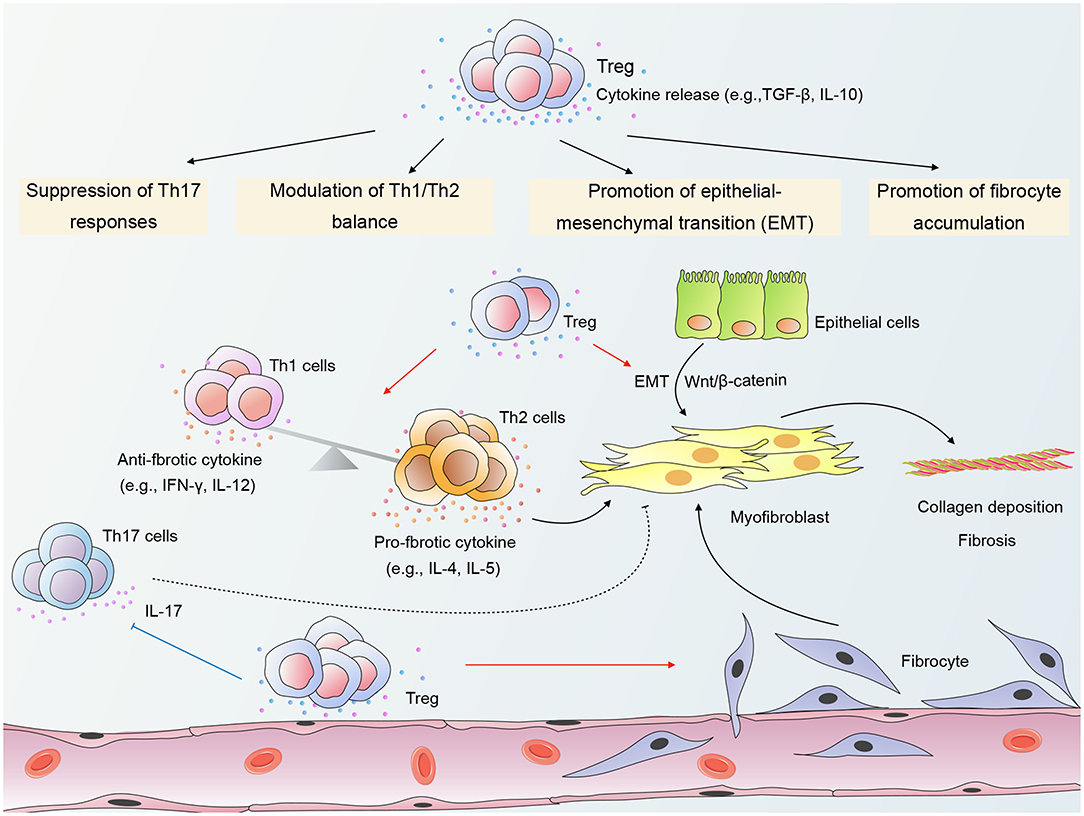
Figure 1. A schematic of mechanisms by which Regulatory T cells (Tregs) modulate radiation-induced pulmonary fibrosis (RPF). Tregs promote the accumulation of fibrocytes in irradiated lungs and β-catenin-mediated epithelial-mesenchymal transition (EMT) in epithelial cells. In addition, Tregs shift the Th1/Th2 cytokine balance toward Th2 dominance, thereby providing a cytokine milieu that favors fibrosis. Tregs may also modulate RPF through suppression of Th17 responses. The crosstalk among Tregs, other infiltrating T cells, epithelial cells, and fibrocytes contributes to the activation of myofibroblasts and collagen deposition, ultimately leading to the destruction of the normal lung architecture. “→”: transform or promote; “⊥”: inhibit; “- - -”: may play a role.
Promotion of Fibrocyte Accumulation
Fibrocytes are bone marrow-derived mesenchymal stem cells that can differentiate into myofibroblasts (71). The accumulation and differentiation of fibrocytes are key processes with massive implications for the pathogenesis of fibroproliferative diseases (72). Regarding RPF, Xiong et al. reported an increased number of fibrocytes in the lung tissue of mice during both early and chronic phase of RILI (21). Interestingly, Treg depletion reduced fibrocyte accumulation and attenuated lung fibrosis (21), indicating that Tregs may contribute to RPF by promoting fibrocyte accumulation in irradiated lungs.
Although the underlying mechanisms have not yet been clarified, Tregs may interact with fibrocytes through secretion of TGF-β, which is known to promote the proliferation, differentiation, and collagen production of fibrocytes (71, 73, 74). Additionally, the differentiation of Gr1+ monocytes into fibrocytes has been demonstrated to be largely under the control of CD4+ T cells (75). As a critical subset of CD4+ T cells, Tregs may also have a role in the development of fibrocytes. The interplay between Tregs and fibrocytes in RILI warrants further investigations.
Promotion of Emt in Epithelial Cells
EMT is a process involving the transdifferentiation of epithelial cells with a progressive loss of apicobasal polarity and cell-cell contacts, as well as an acquisition of a mesenchymal phenotype (32). Growing evidence has demonstrated a key role of EMT in the pathogenesis of RPF (18, 76). Intriguingly, Treg depletion suppressed EMT and substantially attenuated pulmonary fibrosis in irradiated mice (18), revealing a detrimental role for Tregs in RPF through the promotion of EMT.
Furthermore, a study by Boveda-Ruiz et al. has shed light on the interaction between Tregs and epithelial cells, demonstrating that the co-culture of lung epithelial cells with Tregs resulted in increased protein and mRNA expression of TGF-β1 in epithelial cells (13). Thus, stimulation of TGF-β secretion from epithelial cells is a possible mechanism whereby Tregs accelerate RPF. Xiong et al. co-cultured irradiated lung epithelial cells with Tregs and found that Tregs promoted the EMT process and β-catenin expression in epithelial cells (18). Moreover, the EMT-promoting effect of Tregs was partially impaired by β-catenin silencing in vitro (18), suggesting that β-catenin-mediated EMT is a potential mechanism whereby Tregs modulate RPF.
Modulation of Th1/Th2 Balance
Another possible mechanism whereby Tregs accelerate RPF is by shifting the Th1/Th2 cytokine balance toward Th2 dominance. Th2 signature cytokines such as IL-4 have been recognized as potent pro-fibrotic mediators due to their capacity to promote fibroblast activation and collagen production. In contrast to Th2 cytokines, Th1 signature cytokines such as interferon-γ (IFN-γ) exert suppressive effects on fibroblasts and reduce excessive collagen production (77). Therefore, Th2-dominant cytokine milieu may play a critical role in driving fibrosis.
In a mouse model of RPF, Treg depletion skewed the balance toward Th1 dominance with a cytokine profile of increased IFN-γ and IL-12 and decreased IL-4 and IL-5 (21). This observation is reminiscent of a study by Liu et al., demonstrating that Treg depletion in a mouse model of silica-induced lung fibrosis resulted in increased levels of Th1 cytokines and decreased Th2 cytokines (7). One possible mechanism whereby Tregs modulate the Th1/Th2 balance is via promoting the induction of alternatively activated (M2) macrophages (78), which are known to participate in the promotion of Th2 responses and tissue remodeling (79). Additionally, it has been demonstrated that Tregs can undergo molecular differentiation by modifying their expression of Th lineage-specific transcription factors such as T-box expressed in T cells (T-bet) and interferon regulatory factor-4 (IRF-4), thereby enabling them to specifically control Th1 or Th2 responses (80, 81). Thus, in RPF, Tregs may undergo molecular specialization in response to the local milieu that enables them to suppress Th1 responses, which ultimately drives a shift toward a Th2-dominant cytokine milieu.
Suppression of Th17 Responses
To date, the contribution of Th17 cells to fibrotic disorders remains obscure. Animal studies revealed that antibody-mediated neutralization of IL-17A, a hallmark cytokine for Th17 cells, attenuated fibrosis in several damage-associated pulmonary diseases (82, 83). Moreover, in murine models of bleomycin-induced lung injury, IL-17 receptor A- or IL-17A-deficient mice displayed substantially reduced pulmonary fibrosis in contrast to wild-type mice (83, 84). By contrast, Lo Re et al. demonstrated that IL-17A was dispensable for the fibrotic response and that silica-induced pulmonary fibrosis was not attenuated upon treatment with anti-IL-17A antibody or in IL-17R- deficient mice (85).
Evidence suggests that the imbalance of Th17/Treg is a possible mechanism whereby Tregs modulate RPF. According to a preclinical study by Xiong et al. Treg depletion attenuated lung fibrosis, which was accompanied by a significant increase in the number of Th17 cells from month 3 to 6 after irradiation (21), suggesting that Tregs may accelerate RPF through suppression of Th17 responses and that Th17 cells may exert an anti-fibrotic function in RPF. By contrast, Paun et al. demonstrated that the IL-17A-deficient mice were protected from RPF, indicating a pathogenic role of Th17 cells in RPF (86).
Tregs have been shown to restrain Th17 responses in a signal transducers and activators of transcription 3 (Stat3)-dependent manner (87). Meanwhile, other studies have reported that Tregs can promote Th17 differentiation and augment IL-17A induction (88–90). These conflicting findings reflect the complexity of the interplay between Tregs and Th17 cells. The interaction between Tregs and Th17 cells in RPF requires further exploration.
Therapeutic Implications and Future Directions
Notably, the interaction of Tregs with other immune cells (e.g., macrophages) and damaged resident cells (e.g., endothelial cells) may also impact the development of RILI. Tregs can profoundly regulate macrophage phenotype and function (79). For example, co-culture of human monocytes with Tregs steers monocyte differentiation toward M2 macrophages (78), which are known to participate in the resolution of inflammation and tissue remodeling (79). Additionally, macrophages co-cultured with Tregs produce increased levels of TGF-β and decreased levels of TNF-α in response to lipopolysaccharide (56). Thus, we speculate that the interaction between Tregs and macrophages may be involved in tissue repair and pulmonary fibrosis induced by radiation, which warrants further investigation. Additionally, the interaction between Tregs and endothelial cells (ECs) has attracted special attention due to its potential involvement in the regulation of inflammation (91–93). Human umbilical vein endothelial cells co-cultured with Tregs show a diminished capacity to respond to lipopolysaccharide in terms of adhesion molecule expression and pro-inflammatory cytokine production (e.g., monocyte chemoattractant protein-1 and IL-6), suggesting a protective role of Tregs during inflammation (93). In turn, ECs can also affect Treg function, as suggested by Bedke et al., who showed that activated murine lung ECs stimulated the release of IL-10 and TGF-β from Tregs and triggered upregulation of programmed death-1 (PD-1) by Tregs (91). Further investigation on the role of EC–Treg interaction in RILI may provide new insights into the pathogenesis of RILI.
Recognition of the critical role of Tregs in RPF has prompted interest in targeting Tregs to prevent or treat RPF. The observation that Treg depletion attenuated RPF is of particular interest in the era of cancer immunotherapy, as Treg depletion has been shown to have potential for reversing immunosuppression when combined with radiotherapy (11). Nevertheless, targeting Tregs for the treatment of RPF is still limited to preclinical models, and its translation into clinical application remains challenging. One key challenge is to determine the optimal timing for removal of Tregs, given that Tregs may have a requisite role in protecting normal tissue against excessive inflammation-induced damage during the early stage of RILI.
Recently, the finding that CD73 potentiated RPF has prompted interest in targeting CD73 to limit radiation-induced lung toxicity (16). Up to now, although CD73+CD4+Foxp3+ Tregs have been shown to accumulate in irradiated lungs during the pneumonitic and fibrotic phase (16), the role of CD73+CD4+Foxp3+ Tregs in radiation-induced pneumopathy is poorly defined. We speculate that CD73-targeted therapies may limit potential pro-fibrotic actions of Tregs in pulmonary fibrosis induced by radiation. Moreover, given the crucial role of CD73 expression on Tregs in the suppression of antitumor immunity (94, 95), it is intriguing to speculate that CD73-targeted therapy may provide therapeutic benefits by promoting antitumor immunity and reducing late radiation toxicity. Additionally, emerging evidence highlights a novel contribution of the IL-33/ST2 pathway to fibrotic disorders (59). Regarding pulmonary fibrosis, Li et al. reported that bleomycin-induced lung fibrosis was attenuated in ST2-deficient mice or upon treatment with anti–IL-33 antibody (96). Moreover, exogenous administration of mature recombinant IL-33 exacerbated bleomycin-induced fibrosis in mice (96), indicating a critical role of IL-33 in the fibrotic response to lung injury. To date, the mechanisms, especially involving Tregs, whereby IL-33 contributes to pulmonary fibrotic disorders remain underexplored, although some studies have demonstrated strong immunosuppressive properties and Th2-like character of IL-33 activated Tregs in vitro (57, 58, 60). A recent study in mice has revealed a protective role for IL-33 in ALI, demonstrating that IL-33-mediated control of inflammation involves the stimulation of IL-13 secretion by ST2+ Tregs, which reduces the infiltration of inflammatory monocytes and local inflammatory cytokines, such as IL-6 and granulocyte-colony stimulating factor (G-CSF) (97). It is possible to envision that the sustained increase in epithelial-derived IL-33 and chronic activation of ST2+ Tregs triggered by irradiation may eventually shift the local environment toward one favoring fibrosis. Further investigation on the role of IL-33 activated Tregs in radiation-induced pneumopathy is warranted and may provide insight into the therapeutic potential of IL-33/ST2 in RILI.
Concluding Remarks
Accumulating evidence has demonstrated a profound, yet complex role of Tregs in RILI. During the pneumonitic phase, Tregs may play a role in counterbalancing exaggerated pro-inflammatory responses and preventing the worsening of radiation-induced injury. On the other hand, emerging data point to the crucial involvement of Tregs in RPF. The role of Tregs in RPF is likely to be multi-factorial – related to promotion of fibrocyte accumulation, promotion of EMT, and modulation of Th1/Th2 balance.
Although growing interest has been focused on the role of Tregs in RILI and that Treg depletion has been shown to attenuate RILI in mice, from a therapeutic viewpoint, this field is still in its infancy. Further studies that probe into unanswered questions, such as the interaction of Tregs with other cells in RILI, represent the next steps forward in developing effective therapeutic strategies. Given the critical role of Tregs in both tumor-induced immune suppression and radiation-induced fibrotic response, we envision that Tregs represent a promising target for future treatment options. A deeper understanding of the mechanisms whereby Tregs modulate RILI will offer new avenues for the efficient management of RILI.
Author Contributions
TG and LZ drafted the manuscript. JN, YZ, and LY reviewed and edited the manuscript. XY and ZZ conceived the topic and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by National Natural Science Foundation of China (Nos. 81903253, 81872461).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ALI, Acute lung injury; CTLA-4, Cytotoxic T lymphocyte antigen-4; DAMPs, Damage-associated molecular patterns; ECs, Endothelial cells; ECM, Extracellular matrix; EMT, Epithelial-mesenchymal transition; FoxP3, Forkhead box P3; G-CSF, Granulocyte-colony stimulating factor; IFN-γ, Interferon-γ; IL, Interleukin; IRF-4, Interferon regulatory factor-4; M2 macrophage, Alternatively activated macrophage NF-κB, Nuclear factor kappa-B; PD-1, Programmed death-1; RILI, Radiation-induced lung injury; RP, Radiation-induced pneumonitis; RPF, Radiation-induced pulmonary fibrosis; ST2, Suppression of tumorigenicity 2; Stat3, Signal transducers and activators of transcription 3; T-bet, T-box expressed in T cells; TGF-β, Transforming growth factor-β; TNF-α, Tumor necrosis factor-α; Tregs, Regulatory T cells.
References
1. Hanania AN, Mainwaring W, Ghebre YT, Hanania NA, Ludwig M. Radiation-induced lung injury: assessment and management. Chest. (2019) 156:150–62. doi: 10.1016/j.chest.2019.03.033
2. Tsoutsou PG, Koukourakis MI. Radiation pneumonitis and fibrosis: mechanisms underlying its pathogenesis and implications for future research. Int J Radiat Oncol Biol Phys. (2006) 66:1281–93. doi: 10.1016/j.ijrobp.2006.08.058
3. Zhao J, Yorke ED, Li L, Kavanagh BD, Li XA, Das S, et al. Simple factors associated with radiation-induced lung toxicity after stereotactic body radiation therapy of the thorax: a pooled analysis of 88 studies. Int J Radiat Oncol Biol Phys. (2016) 95:1357–66. doi: 10.1016/j.ijrobp.2016.03.024
4. Marks LB, Bentzen SM, Deasy JO, Kong FM, Bradley JD, Vogelius IS, et al. Radiation dose-volume effects in the lung. Int J Radiat Oncol Biol Phys. (2010) 76:S70–6. doi: 10.1016/j.ijrobp.2009.06.091
5. Ding NH, Li JJ, Sun LQ. Molecular mechanisms and treatment of radiation-induced lung fibrosis. Curr Drug Targets. (2013) 14:1347–56. doi: 10.2174/13894501113149990198
6. Mazeron R, Etienne-Mastroianni B, Pérol D, Arpin D, Vincent M, Falchero L, et al. Predictive factors of late radiation fibrosis: a prospective study in non-small cell lung cancer. Int J Radiat Oncol Biol Phys. (2010) 77:38–43. doi: 10.1016/j.ijrobp.2009.04.019
7. Cella L, Liuzzi R, D'Avino V, Conson M, Di Biase A, Picardi M, et al. Pulmonary damage in Hodgkin's lymphoma patients treated with sequential chemo-radiotherapy: Predictors of radiation-induced lung injury. Acta Oncol. (2014) 53:613–9. doi: 10.3109/0284186X.2013.850739
8. Takeda A, Kunieda E, Takeda T, Tanaka M, Sanuki N, Fujii H, et al. Possible misinterpretation of demarcated solid patterns of radiation fibrosis on CT scans as tumor recurrence in patients receiving hypofractionated stereotactic radiotherapy for lung cancer. Int J Radiat Oncol Biol Phys. (2008) 70:1057–65. doi: 10.1016/j.ijrobp.2007.07.2383
9. Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. (2009) 30:636–45. doi: 10.1016/j.immuni.2009.04.010
10. Persa E, Balogh A, Safrany G, Lumniczky K. The effect of ionizing radiation on regulatory T cells in health and disease. Cancer Lett. (2015) 368:252–61. doi: 10.1016/j.canlet.2015.03.003
11. Byrne WL, Mills KH, Lederer JA, O'Sullivan GC. Targeting regulatory T cells in cancer. Cancer Res. (2011) 71:6915–20. doi: 10.1158/0008-5472.Can-11-1156
12. Liu F, Liu J, Weng D, Chen Y, Song L, He Q, et al. CD4+CD25+Foxp3+ regulatory T cells depletion may attenuate the development of silica-induced lung fibrosis in mice. PLoS ONE. (2010) 5:e15404. doi: 10.1371/journal.pone.0015404
13. Boveda-Ruiz D, D'Alessandro-Gabazza CN, Toda M, Takagi T, Naito M, Matsushima Y, et al. Differential role of regulatory T cells in early and late stages of pulmonary fibrosis. Immunobiology. (2013) 218:245–54. doi: 10.1016/j.imbio.2012.05.020
14. Kotsianidis I, Nakou E, Bouchliou I, Tzouvelekis A, Spanoudakis E, Steiropoulos P, et al. Global impairment of CD4+CD25+FOXP3+ regulatory T cells in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2009) 179:1121–30. doi: 10.1164/rccm.200812-1936OC
15. Garibaldi BT, D'Alessio FR, Mock JR, Files DC, Chau E, Eto Y, et al. Regulatory T cells reduce acute lung injury fibroproliferation by decreasing fibrocyte recruitment. Am J Respir Cell Mol Biol. (2013) 48:35–43. doi: 10.1165/rcmb.2012-0198OC
16. Wirsdorfer F, de Leve S, Cappuccini F, Eldh T, Meyer AV, Gau E, et al. Extracellular adenosine production by ecto-5'-Nucleotidase (CD73) enhances radiation-induced lung fibrosis. Cancer Res. (2016) 76:3045–56. doi: 10.1158/0008-5472.Can-15-2310
17. Wirsdorfer F, Cappuccini F, Niazman M, de Leve S, Westendorf AM, Ludemann L, et al. Thorax irradiation triggers a local and systemic accumulation of immunosuppressive CD4+ FoxP3+ regulatory T cells. Radiat Oncol. (2014) 9:98. doi: 10.1186/1748-717x-9-98
18. Xiong S, Pan X, Xu L, Yang Z, Guo R, Gu Y, et al. Regulatory T cells promote beta-catenin–mediated epithelium-to-mesenchyme transition during radiation-induced pulmonary fibrosis. Int J Radiat Oncol Biol Phys. (2015) 93:425–35. doi: 10.1016/j.ijrobp.2015.05.043
19. Xu L, Xiong S, Guo R, Yang Z, Wang Q, Xiao F, et al. Transforming growth factor beta3 attenuates the development of radiation-induced pulmonary fibrosis in mice by decreasing fibrocyte recruitment and regulating IFN-gamma/IL-4 balance. Immunol Lett. (2014) 162:27–33. doi: 10.1016/j.imlet.2014.06.010
20. Wirsdorfer F, Jendrossek V. The role of lymphocytes in radiotherapy-induced adverse late effects in the lung. Front Immunol. (2016) 7:591. doi: 10.3389/fimmu.2016.00591
21. Xiong S, Guo R, Yang Z, Xu L, Du L, Li R, et al. Treg depletion attenuates irradiation-induced pulmonary fibrosis by reducing fibrocyte accumulation, inducing Th17 response, and shifting IFN-gamma, IL-12/IL-4, IL-5 balance. Immunobiology. (2015) 220:1284–91. doi: 10.1016/j.imbio.2015.07.001
22. Cappuccini F, Eldh T, Bruder D, Gereke M, Jastrow H, Schulze-Osthoff K, et al. New insights into the molecular pathology of radiation-induced pneumopathy. Radiother Oncol. (2011) 101:86–92. doi: 10.1016/j.radonc.2011.05.064
23. Liu D, Kong F, Yuan Y, Seth P, Xu W, Wang H, et al. Decorin-modified umbilical cord mesenchymal stem cells (MSCS) attenuate radiation-induced lung injuries via regulating inflammation, fibrotic factors, and immune responses. Int J Radiat Oncol Biol Phys. (2018) 101:945–56. doi: 10.1016/j.ijrobp.2018.04.007
24. Graves PR, Siddiqui F, Anscher MS, Movsas B. Radiation pulmonary toxicity: from mechanisms to management. Semin Radiat Oncol. (2010) 20:201–7. doi: 10.1016/j.semradonc.2010.01.010
25. Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Media Inflamm. (2010) 2010:672395. doi: 10.1155/2010/672395
26. Sprung CN, Forrester HB, Siva S, Martin OA. Immunological markers that predict radiation toxicity. Cancer Lett. (2015) 368:191–7. doi: 10.1016/j.canlet.2015.01.045
27. Paun A, Kunwar A, Haston CK. Acute adaptive immune response correlates with late radiation-induced pulmonary fibrosis in mice. Radiat Oncol. (2015) 10:45. doi: 10.1186/s13014-015-0359-y
28. Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. (1995) 270:286–90. doi: 10.1126/science.270.5234.286
29. Hong JH, Chiang CS, Tsao CY, Lin PY, Wu CJ, McBride WH. Can short-term administration of dexamethasone abrogate radiation-induced acute cytokine gene response in lung and modify subsequent molecular responses? Int J Radiat Oncol Biol Phys. (2001) 51:296–303. doi: 10.1016/s0360-3016(01)01702-3
30. Vujaskovic Z, Anscher MS, Feng QF, Rabbani ZN, Amin K, Samulski TS, et al. Radiation-induced hypoxia may perpetuate late normal tissue injury. Int J Radiat Oncol Biol Phys. (2001) 50:851–5. doi: 10.1016/s0360-3016(01)01593-0
31. Rodemann HP, Bamberg M. Cellular basis of radiation-induced fibrosis. Radiother Oncol. (1995) 35:83–90. doi: 10.1016/0167-8140(95)01540-w
32. Scott LE, Weinberg SH, Lemmon CA. Mechanochemical signaling of the extracellular matrix in epithelial-mesenchymal transition. Front Cell Dev Biol. (2019) 7:135. doi: 10.3389/fcell.2019.00135
33. Li Y, Zou L, Yang X, Chu L, Ni J, Chu X, et al. Identification of lncRNA, MicroRNA, and mRNA-associated CeRNA network of radiation-induced lung injury in a mice model. Dose Res. (2019) 17:1559325819891012. doi: 10.1177/1559325819891012
34. Bluestone JA, Abbas AK. Natural versus adaptive regulatory T cells. Nat Rev Immunol. (2003) 3:253–7. doi: 10.1038/nri1032
35. Karim M, Kingsley CI, Bushell AR, Sawitzki BS, Wood KJ. Alloantigen-induced CD25+CD4+ regulatory T cells can develop in vivo from CD25-CD4+ precursors in a thymus-independent process. J Immunol. (2004) 172:923–8. doi: 10.4049/jimmunol.172.2.923
36. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. (2003) 198:1875–86. doi: 10.1084/jem.20030152
37. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
38. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. (2010) 10:490–500. doi: 10.1038/nri2785
39. Gavin MA, Torgerson TR, Houston E, DeRoos P, Ho WY, Stray-Pedersen A, et al. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci USA. (2006) 103:6659–64. doi: 10.1073/pnas.0509484103
40. Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, et al. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. (2007) 19:345–54. doi: 10.1093/intimm/dxm014
41. Wang J, Ioan-Facsinay A, van der Voort EI, Huizinga TW, Toes RE. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur J Immunol. (2007) 37:129–38. doi: 10.1002/eji.200636435
42. Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. (2007) 110:2983–90. doi: 10.1182/blood-2007-06-094656
43. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med. (2013) 19:739–46. doi: 10.1038/nm.3179
44. Roncarolo MG, Gregori S, Lucarelli B, Ciceri F, Bacchetta R. Clinical tolerance in allogeneic hematopoietic stem cell transplantation. Immunol Rev. (2011) 241:145–63. doi: 10.1111/j.1600-065X.2011.01010.x
45. Hartigan-O'Connor DJ, Poon C, Sinclair E, McCune JM. Human CD4+ regulatory T cells express lower levels of the IL-7 receptor alpha chain (CD127), allowing consistent identification and sorting of live cells. J Immunol Methods. (2007) 319:41–52. doi: 10.1016/j.jim.2006.10.008
46. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. (2009) 30:899–911. doi: 10.1016/j.immuni.2009.03.019
47. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol. (2012) 3:51. doi: 10.3389/fimmu.2012.00051
48. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. (2004) 21:589–601. doi: 10.1016/j.immuni.2004.09.002
49. Strauss L, Bergmann C, Whiteside TL. Human circulating CD4+CD25highFoxp3+ regulatory T cells kill autologous CD8+ but not CD4+ responder cells by Fas-mediated apoptosis. J Immunol. (2009) 182:1469–80. doi: 10.4049/jimmunol.182.3.1469
50. Ito T, Hanabuchi S, Wang YH, Park WR, Arima K, Bover L, et al. Two functional subsets of FOXP3+ regulatory T cells in human thymus and periphery. Immunity. (2008) 28:870–80. doi: 10.1016/j.immuni.2008.03.018
51. Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev. (2006) 212:28–50. doi: 10.1111/j.0105-2896.2006.00420.x
52. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. (2007) 450:566–9. doi: 10.1038/nature06306
53. Bardel E, Larousserie F, Charlot-Rabiega P, Coulomb-L'Herminé A, Devergne O. Human CD4+ CD25+ Foxp3+ regulatory T cells do not constitutively express IL-35. J Immunol. (2008) 181:6898–905. doi: 10.4049/jimmunol.181.10.6898
54. Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. (2008) 28:546–58. doi: 10.1016/j.immuni.2008.02.017
55. Kearley J, Barker JE, Robinson DS, Lloyd CM. Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. J Exp Med. (2005) 202:1539–47. doi: 10.1084/jem.20051166
56. D'Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest. (2009) 119:2898–913. doi: 10.1172/jci36498
57. Chen CC, Kobayashi T, Iijima K, Hsu FC, Kita H. IL-33 dysregulates regulatory T cells and impairs established immunologic tolerance in the lungs. J Allergy Clin Immunol. (2017) 140:1351–63.e7. doi: 10.1016/j.jaci.2017.01.015
58. Siede J, Frohlich A, Datsi A, Hegazy AN, Varga DV, Holecska V, et al. IL-33 receptor-expressing regulatory T cells are highly activated, Th2 biased and suppress CD4 T cell proliferation through IL-10 and TGFbeta release. PLoS ONE. (2016) 11:e0161507. doi: 10.1371/journal.pone.0161507
59. Kotsiou OS, Gourgoulianis KI, Zarogiannis SG. IL-33/ST2 axis in organ fibrosis. Front Immunol. (2018) 9:2432. doi: 10.3389/fimmu.2018.02432
60. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell. (2015) 162:1078–89. doi: 10.1016/j.cell.2015.08.021
61. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. (2011) 34:566–78. doi: 10.1016/j.immuni.2011.03.018
62. MacConmara MP, Tajima G, O'Leary F, Delisle AJ, McKenna AM, Stallwood CG, et al. Regulatory T cells suppress antigen-driven CD4 T cell reactivity following injury. J Leukoc Biol. (2011) 89:137–47. doi: 10.1189/jlb.0210082
63. Wang Y, Xu G, Wang J, Li XH, Sun P, Zhang W, et al. Relationship of Th17/Treg cells and radiation pneumonia in locally advanced esophageal carcinoma. Anticancer Res. (2017) 37:4643–7. doi: 10.21873/anticanres.11866
64. Martin M, Lefaix J, Delanian S. TGF-beta1 and radiation fibrosis: a master switch and a specific therapeutic target? Int J Radiat Oncol Biol Phys. (2000) 47:277–90. doi: 10.1016/s0360-3016(00)00435-1
65. Candéias SM, Testard I. The many interactions between the innate immune system and the response to radiation. Cancer Lett. (2015) 368:173–8. doi: 10.1016/j.canlet.2015.02.007
66. Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med. (2004) 199:303–13. doi: 10.1084/jem.20031562
67. Zhang N, Schröppel B, Lal G, Jakubzick C, Mao X, Chen D, et al. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity. (2009) 30:458–69. doi: 10.1016/j.immuni.2008.12.022
68. Brühl H, Cihak J, Schneider MA, Plachý J, Rupp T, Wenzel I, et al. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J Immunol. (2004) 172:890–8. doi: 10.4049/jimmunol.172.2.890
69. Hamano R, Baba T, Sasaki S, Tomaru U, Ishizu A, Kawano M, et al. Ag and IL-2 immune complexes efficiently expand Ag-specific Treg cells that migrate in response to chemokines and reduce localized immune responses. Eur J Immunol. (2014) 44:1005–15. doi: 10.1002/eji.201343434
70. Li J, Tan J, Martino MM, Lui KO. Regulatory T-cells: potential regulator of tissue repair and regeneration. Front Immunol. (2018) 9:585. doi: 10.3389/fimmu.2018.00585
71. Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. (2001) 166:7556–62. doi: 10.4049/jimmunol.166.12.7556
72. Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. (2004) 114:438–46. doi: 10.1172/jci20997
73. Yang L, Scott PG, Giuffre J, Shankowsky HA, Ghahary A, Tredget EE. Peripheral blood fibrocytes from burn patients: identification and quantification of fibrocytes in adherent cells cultured from peripheral blood mononuclear cells. Lab Invest. (2002) 82:1183–92. doi: 10.1097/01.lab.0000027841.50269.61
74. Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol. (2003) 171:380–9. doi: 10.4049/jimmunol.171.1.380
75. Niedermeier M, Reich B, Rodriguez Gomez M, Denzel A, Schmidbauer K, Gobel N, et al. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci USA. (2009) 106:17892–7. doi: 10.1073/pnas.0906070106
76. Balli D, Ustiyan V, Zhang Y, Wang IC, Masino AJ, Ren X, et al. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. Embo J. (2013) 32:231–44. doi: 10.1038/emboj.2012.336
77. Shao DD, Suresh R, Vakil V, Gomer RH, Pilling D. Pivotal Advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. J Leukoc Biol. (2008) 83:1323–33. doi: 10.1189/jlb.1107782
78. Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci USA. (2007) 104:19446–51. doi: 10.1073/pnas.0706832104
79. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. (2010) 11:889–96. doi: 10.1038/ni.1937
80. Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. (2009) 10:595–602. doi: 10.1038/ni.1731
81. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. (2009) 458:351–6. doi: 10.1038/nature07674
82. Mi S, Li Z, Yang HZ, Liu H, Wang JP, Ma YG, et al. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. J Immunol. (2011) 187:3003–14. doi: 10.4049/jimmunol.1004081
83. Gasse P, Riteau N, Vacher R, Michel ML, Fautrel A, di Padova F, et al. IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PLoS ONE. (2011) 6:e23185. doi: 10.1371/journal.pone.0023185
84. Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, et al. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med. (2010) 207:535–52. doi: 10.1084/jem.20092121
85. Lo Re S, Dumoutier L, Couillin I, Van Vyve C, Yakoub Y, Uwambayinema F, et al. IL-17A-producing gammadelta T and Th17 lymphocytes mediate lung inflammation but not fibrosis in experimental silicosis. J Immunol. (2010) 184:6367–77. doi: 10.4049/jimmunol.0900459
86. Paun A, Bergeron ME, Haston CK. The Th1/Th17 balance dictates the fibrosis response in murine radiation-induced lung disease. Sci Rep. (2017) 7:11586. doi: 10.1038/s41598-017-11656-5
87. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. (2009) 326:986–91. doi: 10.1126/science.1172702
88. Pandiyan P, Conti HR, Zheng L, Peterson AC, Mathern DR, Hernandez-Santos N, et al. CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse Candida albicans Th17 cell infection model. Immunity. (2011) 34:422–34. doi: 10.1016/j.immuni.2011.03.002
89. Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. (2007) 178:6725–9. doi: 10.4049/jimmunol.178.11.6725
90. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. (2006) 24:179–89. doi: 10.1016/j.immuni.2006.01.001
91. Bedke T, Pretsch L, Karakhanova S, Enk AH, Mahnke K. Endothelial cells augment the suppressive function of CD4+ CD25+ Foxp3+ regulatory T cells: involvement of programmed death-1 and IL-10. J Immunol. (2010) 184:5562–70. doi: 10.4049/jimmunol.0902458
92. Lim WC, Olding M, Healy E, Millar TM. Human endothelial cells modulate CD4(+) T cell populations and enhance regulatory T cell suppressive capacity. Front Immunol. (2018) 9:565. doi: 10.3389/fimmu.2018.00565
93. He S, Li M, Ma X, Lin J, Li D. CD4+CD25+Foxp3+ regulatory T cells protect the proinflammatory activation of human umbilical vein endothelial cells. Arterioscler Thromb Vasc Biol. (2010) 30:2621–30. doi: 10.1161/atvbaha.110.210492
94. Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, et al. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. (2011) 71:2892–900. doi: 10.1158/0008-5472.Can-10-4246
95. Sitkovsky MV. T regulatory cells: hypoxia-adenosinergic suppression and re-direction of the immune response. Trends Immunol. (2009) 30:102–8. doi: 10.1016/j.it.2008.12.002
96. Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol. (2014) 134:1422–32.e11. doi: 10.1016/j.jaci.2014.05.011
Keywords: Treg, radiotherapy, lung, pneumonitis, fibrosis
Citation: Guo T, Zou L, Ni J, Zhou Y, Ye L, Yang X and Zhu Z (2020) Regulatory T Cells: An Emerging Player in Radiation-Induced Lung Injury. Front. Immunol. 11:1769. doi: 10.3389/fimmu.2020.01769
Received: 18 April 2020; Accepted: 01 July 2020;
Published: 04 August 2020.
Edited by:
Heiko Mühl, Goethe University Frankfurt, GermanyReviewed by:
Jacob Finkelstein, University of Rochester Medical Center, United StatesFlorian Wirsdörfer, Essen University Hospital, Germany
Copyright © 2020 Guo, Zou, Ni, Zhou, Ye, Yang and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xi Yang, ntgeorge@qq.com; Zhengfei Zhu, fuscczzf@163.com
†These authors have contributed equally to this work