 Landon W. Locke
Landon W. Locke Larry S. Schlesinger
Larry S. Schlesinger Elliott D. Crouser3*
Elliott D. Crouser3*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 August 2020
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01719
This article is part of the Research Topic Novel Insights into The Immunology of Pulmonary Granulomatous Diseases View all 11 articles
The inability to effectively model sarcoidosis in the laboratory or in animals continues to hinder the discovery and translation of new, targeted treatments. The granuloma is the signature pathological hallmark of sarcoidosis, yet there are significant knowledge gaps that exist with regard to how granulomas form. Significant progress toward improved therapeutic and prognostic strategies in sarcoidosis hinges on tractable experimental models that recapitulate the process of granuloma formation in sarcoidosis and allow for mechanistic insights into the molecular events involved. Through its inherent representation of the complex genetics underpinning immune cell dysregulation in sarcoidosis, a recently developed in vitro human granuloma model holds promise in providing detailed mechanistic insight into sarcoidosis–specific disease regulating pathways at play during early stages of granuloma formation. The purpose of this review is to critically evaluate current sarcoidosis models and assess their potential to progress the field toward the goal of improved therapies in this disease. We conclude with the potential integrated use of preclinical models to accelerate progress toward identifying and testing new drugs and drug combinations that can be rapidly brought to clinical trials.
Sarcoidosis is a complex immune disease with genetic susceptibility and environmental factors playing important roles. The disease occurs in about 15–40 per 100,000 people in the United States (1). In terms of both frequency and severity, the disease disproportionately affects women, people of African American and Northern European descent, and economically disadvantaged populations (1, 2). Severe clinical phenotypes are associated with progressive lung, heart, or brain damage and are debilitating and potentially fatal (2). The failure to effectively model sarcoidosis in the laboratory or in animals has been a roadblock to gaining crucial insights into the triggers and underlying cellular and molecular mechanisms driving the disease pathogenesis. This has manifested not only in terms of hindering the establishment of disease-specific biomarkers for diagnostic and prognostic purposes, but lack of mechanistic insight into disease regulating targets also impairs our capacity to treat this disease. In order to identify disease-specific treatments, the field needs a tractable model that recapitulates the essential elements of the disease pathogenesis. The granuloma is the signature pathological hallmark of sarcoidosis. Importantly, the architecture and immunology of granuloma formation differ significantly from one granulomatous disorder to the next. These differences, which are dictated by a highly complex and coordinated interplay of many diverse immune cell populations, are evident even during the early stages of granuloma formation, and they are further amplified as the structures evolve. However, a significant knowledge gap exists with regard to how granulomas form due to the fact that local factors, such as cytokines, chemokines, and direct cell-cell interactions among various immune cells, strongly influence the function of each immune cell during granuloma formation. Significant progress toward improved therapeutic and prognostic strategies in sarcoidosis hinges on tractable experimental models that recapitulate the process of granuloma formation in sarcoidosis and allow for mechanistic insights into the molecular events involved. The purpose of this review is to critically evaluate current sarcoidosis models and assess their potential to progress the field toward these goals. Selected sarcoidosis models described in the following sections have been summarized in Table 1.
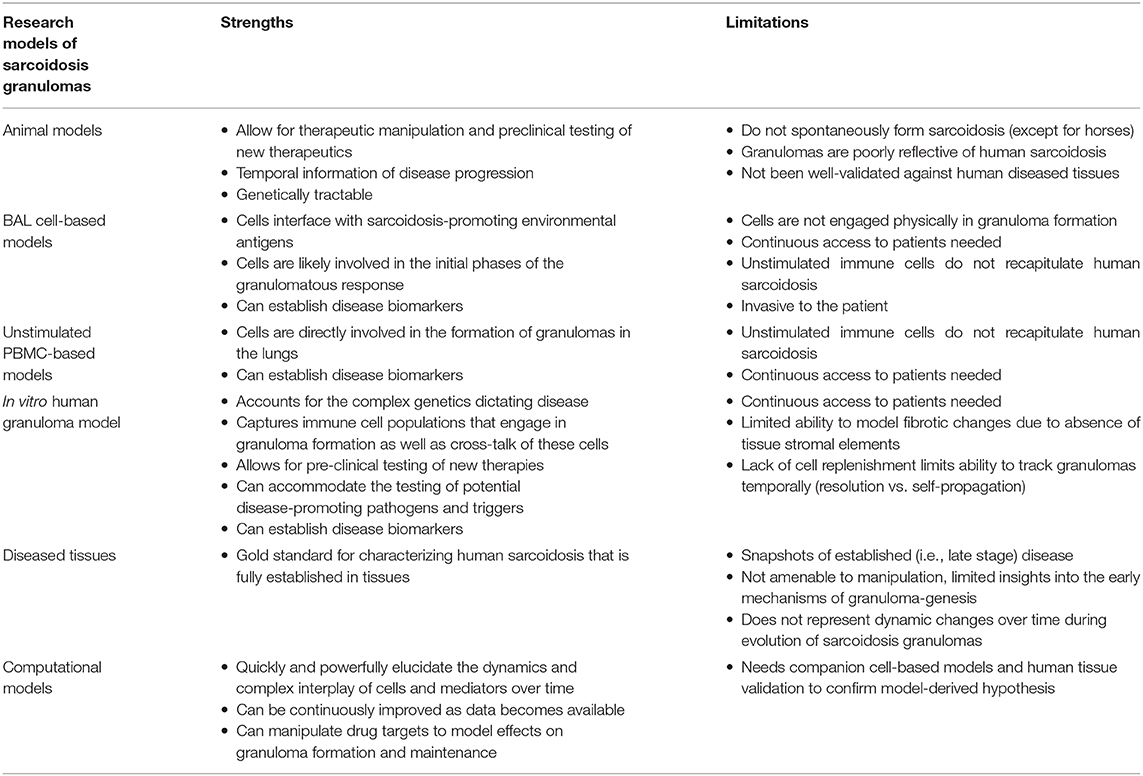
Table 1. The strengths and limitations of current sarcoidosis research models.
Studies of diseased patient biopsy tissue are the foundation of sarcoidosis research. The original description of sarcoidosis in 1877 was based on the characteristic appearance of biopsied skin lesions and histological features that discriminated sarcoidosis from tuberculosis (TB) and other infectious granulomatous disorders (3). Despite the legitimacy of conducting research based on human sarcoidosis tissue samples, such research has yielded limited information regarding disease pathogenesis over the past 143 years. The information that can be obtained from sarcoidosis tissues is limited to a snapshot view of the disease in the form of established granulomas, a perspective that provides very limited insights into the critical and dynamic mechanisms underpinning the pathogenic formation of granulomas.
Sarcoidosis granuloma formation is considered to be environmental antigen-mediated (4), and the lungs are the primary interface between environmental antigens and the host's immune surveillance system. This premise has led many researchers to investigate cells derived from bronchoalveolar lavage (BAL) fluid, particularly alveolar macrophages (AMs) and lymphocytes, to gain insight into sarcoidosis pathogenesis. Such studies indicated that sarcoidosis AMs exhibit enhanced antigen processing capacity and promote greater T cell activation (5), and as such, AMs likely play an important role during the initiation of the granulomatous response in the lungs. While it is likely that AMs interface with sarcoidosis-promoting environmental antigens and may be involved in the initial phases of the granulomatous response, they are not the primary source of macrophages involved in granuloma formation and are therefore insufficient for modeling their complexities. Cells in the peripheral blood, on the other hand, are directly involved in the formation of granulomas in the lungs (6) (Figure 1), and laboratory models based on peripheral blood mononuclear cells (PBMCs) have successfully replicated many of the histological and molecular features of human granulomatous diseases, including TB (7, 8), leprosy (9), and sarcoidosis (10). Along with tissue resident antigen-presenting cells (APCs) such as dendritic cells or AMs, blood monocytes act as sentinels for the detection of potential threats to the host and exit the vascular space to “patrol” within the interstitial space of the lungs (6). These monocytes will migrate back into the vascular space if no danger is sensed. When activated by a danger signal (e.g., bacterial antigens) (Figure 1A), the monocytes establish residency in the lung tissue (6, 11). The subsequent presentation of antigens by monocytes or tissue resident APCs to T cells (Figure 1B) leads to a localized immune response that drives the recruitment and activation of circulating monocytes and lymphocytes into the tissue (12, 13) (Figure 1C). The early granuloma begins to get assembled by these infiltrated cells along with tissue-resident cells to wall off the pathogen and protect the host (Figure 1D). Thus, PBMCs are essential for granuloma formation in the lungs and in other tissues as well.
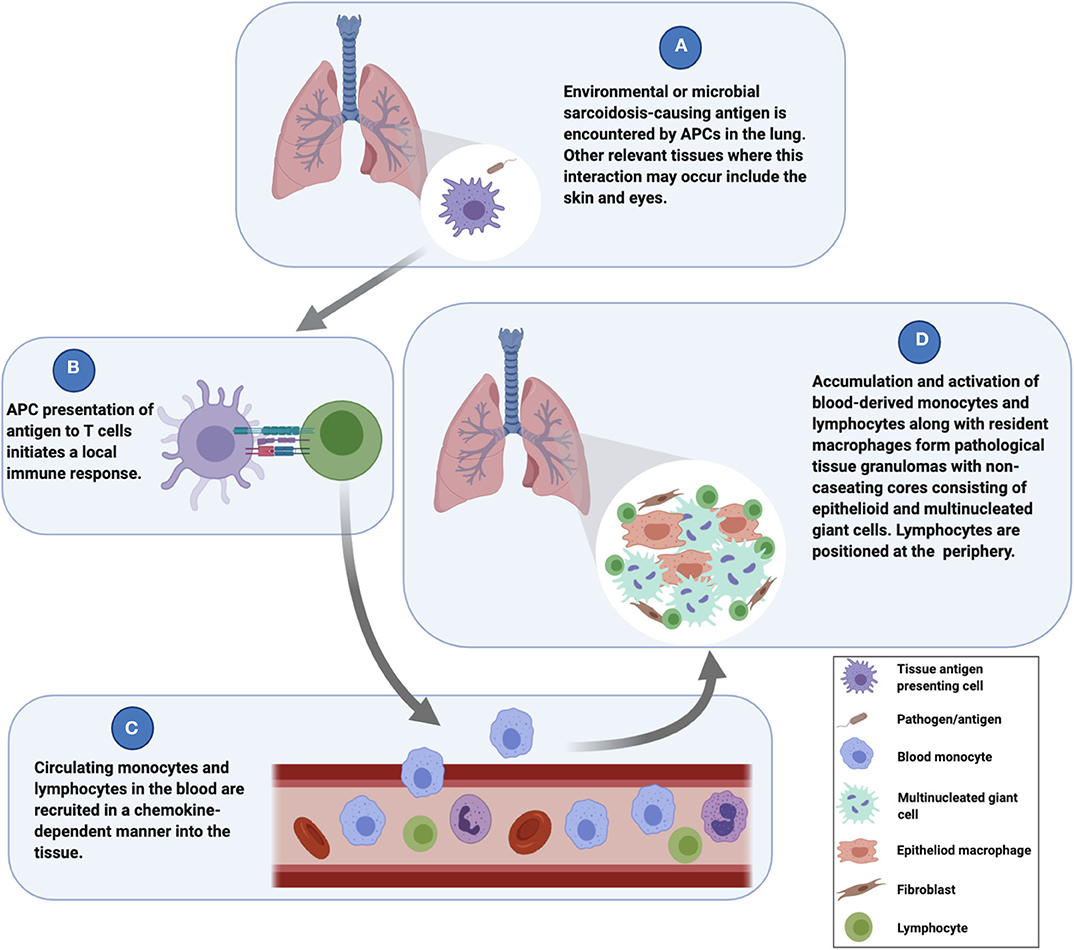
Figure 1. Mechanisms underlying the initiation and evolution of sarcoidosis granuloma formation.
Animal models are commonly relied upon to understand human disease mechanisms, and they offer potential strengths related to therapeutic manipulation and temporal information pertaining to disease progression. Currently there is no universally accepted animal model of human sarcoidosis, largely because animals [other than horses (14)] do not develop sarcoidosis and the link between human gene polymorphisms and disease prevalence has not been established to the degree that it can be recapitulated in the genetic manipulation of animals (e.g., mice) (15). While pulmonary granuloma formation can be achieved in rodents via pre-sensitization to putative antigens followed by pulmonary administration of the same antigens (i.e., a hypersensitivity response), injection and pulmonary embolization of pathogen-coated beads, or bronchial administration of foreign body particles, the granuloma cellular composition, morphology, local inflammatory milieu, and sustainability of these granulomas are poorly reflective of human sarcoidosis. For example, pulmonary granulomas that form in response to repeated exposures to the bacterium Propionibacterium acnes are consistent with hypersensitivity pneumonitis in that, unlike sarcoidosis granulomas, they are poorly formed, transient, and surrounded by prominent lymphocytic pneumonitis (16). Other models that instill carbon nanotubes into the airways of mice induce granulomas with features distinct from sarcoidosis, including foamy lipid-laden macrophages and a paucity of surrounding lymphocytes (17). The immunology of foreign body granulomas fundamentally differs from sarcoidosis because foreign bodies do not promenently feature adaptive immune responses (18, 19). Given the limited ability to recapitulate critical aspects of human sarcoidosis granulomas, animal models have not yielded major breakthroughs in the field of sarcoidosis, albeit with one exception.
A serendipitous link to human sarcoidosis granuloma formation was recently discovered based on a murine Tsc2 knockout model (20). The Tsc2 gene encodes for tuberous sclerosis protein-2 (TSC2), which normally represses the activity of mammalian target of rapamycin complex 1 (mTORc1) (21). The Tsc2 knockout mice exhibit spontaneous systemic tissue granuloma formation with features resembling those of sarcoidosis patients with active disease progression, including epithelioid-like macrophages, M2 macrophage polarization, and mTORc1 pathway activation (20). However, genome wide association studies have not identified variations of the Tsc2 gene as a risk factor for sarcoidosis, and the upstream regulators of mTORc1 in humans with sarcoidosis are unknown. Additionally, it is not known if abnormal regulation of mTORc1 alone is sufficient to cause disease in humans. In this regard, rapamycin, an mTORc1 inhibitor, is currently under investigation as a treatment for sarcoidosis and these studies may provide a more definitive answer to this question. It is reasonable to conclude based on available evidence that the murine Tsc2 knockout model identifies an important immunological pathway that contributes to sarcoidosis disease progression. As such, the model could be leveraged for pre-clinical testing of new sarcoidosis therapeutics targeting mTORc1 regulation pathways.
A major barrier to developing a viable sarcoidosis model is the inability to identify a specific sarcoidosis disease-causing antigen. However, pulmonary sarcoidosis is clinically and histologically very similar to berylliosis, a human disease linked to genetic variability of T cell receptors for divalent beryllium molecules (22). Given the clinical similarities, it has been proposed that sarcoidosis and berylliosis may have similar mechanistic underpinnings (23, 24). Indeed, the berylliosis animal model produces multicellular aggregates of mononuclear cells in the lung consistent with human sarcoidosis (22). However, sarcoidosis patients do not exhibit abnormal T cell-mediated responses to beryllium (25). New data, discussed below, suggest that abnormal macrophage antigen presentation, as opposed to abnormal T cell-mediated antigen recognition, plays a primary role in sarcoidosis pathogenesis.
Given that the polygenic nature of sarcoidosis renders it difficult to model using conventional murine knockout approaches, the field continues to rely on human subjects to conduct meaningful mechanistic research to understand sarcoidosis disease pathogenesis. There have been several studies that have examined single cell populations from BAL or blood of sarcoidosis patients, yielding new insights into the disease and potential disease trajectories. Drake et al. showed that CD4+ T cells derived from sarcoidosis BAL samples induced fibroblasts to produce increased amounts of collagen in a STAT3-IL17A dependent manner, thus clarifying signaling events upstream of fibrotic tissue changes (26). Given its ability to modify immune cell function, Yang et al. sought to identify DNA methylation changes in BAL cells from sarcoidosis patients ranging from remitting to severe phenotypes (27). While this study demonstrated an increased variability in DNA methylation in pooled sarcoidosis BAL cells, further insights into DNA methylation patterns associated with different sarcoidosis phenotypes are likely to emerge in the future from a larger patient sample size. In order to better clarify inciting antigens in sarcoidosis, Grunewald et al. used elegant molecular simulations based on CD4+ T cells derived from BAL to demonstrate a potential role for an autoantigen in triggering sarcoidosis (28). Other studies have similarly implicated an autoantigen in sarcoidosis. Chen et al. showed increased levels of serum amyloid A, an amyloid precursor protein, in biopsied sarcoidosis tissues, specifically showing it to be localized in macrophages and giant cells in granulomas (29). The authors postulated that this autoantigen might act to sustain granulomatous inflammation even if the inciting antigen was microbial in nature. In all, studies that have focused on single cell populations or diseased tissues have revealed factors that are likely important modifiers of disease but possess shortcomings with respect to determining the complex and multi-faceted mechanisms underlying pathogenic granuloma formation.
Considering the limitations of current laboratory models, we sought to develop an improved lab model of human sarcoidosis, an in vitro human granuloma model (Figure 2), with the purpose of gaining mechanistic insights into the molecular events specifically involved in pathological granuloma formation. We posit that there are three key criteria of a viable model to recapitulate the process of granuloma formation in sarcoidosis. First, the model must account for the complex genetics and related unique immune features of sarcoidosis patients. Second, the model must allow for antigen-immune cell interplay, capturing the currently poorly defined immune cell populations that become dysregulated and engaged in granuloma formation. Third, the model must account for the immune cell microenvironment consisting of cytokines and chemokines that promote antigen processing, presentation, and immune cell phenotype changes that occur during the evolution of granulomas. The use of a stimulating antigen is crucial as there is a wealth of evidence supporting an evolution from active granulomatous infection phase to sterile sarcoidosis granulomas (30–32) often harboring non-viable microbial remnants of infectious organisms (33, 34). Recent in vitro granuloma models for TB have been developed that appear to exhibit many of these features (35).
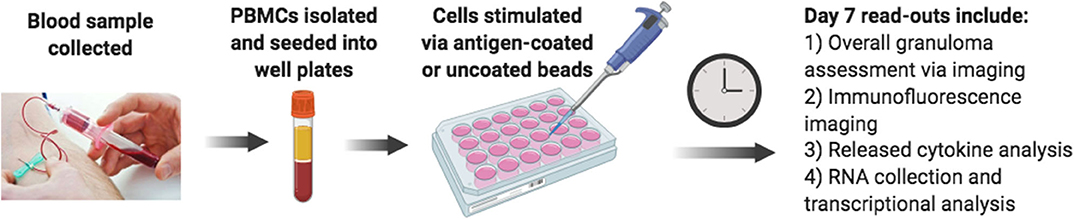
Figure 2. Schematic diagram of the in vitro human granuloma model.
The importance of the criteria described above, including accounting for the complex genetics, immune cell cross-talk, and antigen-immune cell interplay dictating granuloma formation was demonstrated in the in vitro granuloma model by comparing PBMC responses between Mycobacterium tuberculosis (M.tb) naïve sarcoidosis patients and healthy controls. No difference in PBMC gene expression profiles was observed between M.tb naïve sarcoidosis patients and healthy controls at baseline (Figure 3A) (i.e., in the absence of antigen stimulation) in the model, but a vastly divergent response was noted in sarcoidosis PBMCs following challenge with immunogenic M.tb antigens (purified protein derivative, PPD) as reflected by >1,000 differentially expressed (DE) genes (Figure 3B). While the lack of DE gene expression observed in the absence of antigen likely speaks to variability of gene expression among humans, it provides confidence that the DE expression of >1,000 genes observed after antigen stimulation is not simply due to differences that were already apparent at baseline. Compared to healthy controls, M.tb antigen-stimulated sarcoidosis PBMCs also formed more robust granuloma-like aggregates composed of CD11b+ macrophages and CD3+ lymphocytes physically arranged in a manner typical of sarcoidosis granulomas (Figure 4) and produced a very different cytokine profile (10). These results challenge the relevance of studying immune cells in an unstimulated fashion and are consistent with the notion that sarcoidosis is a disease incited by exposure to infectious antigen (36).
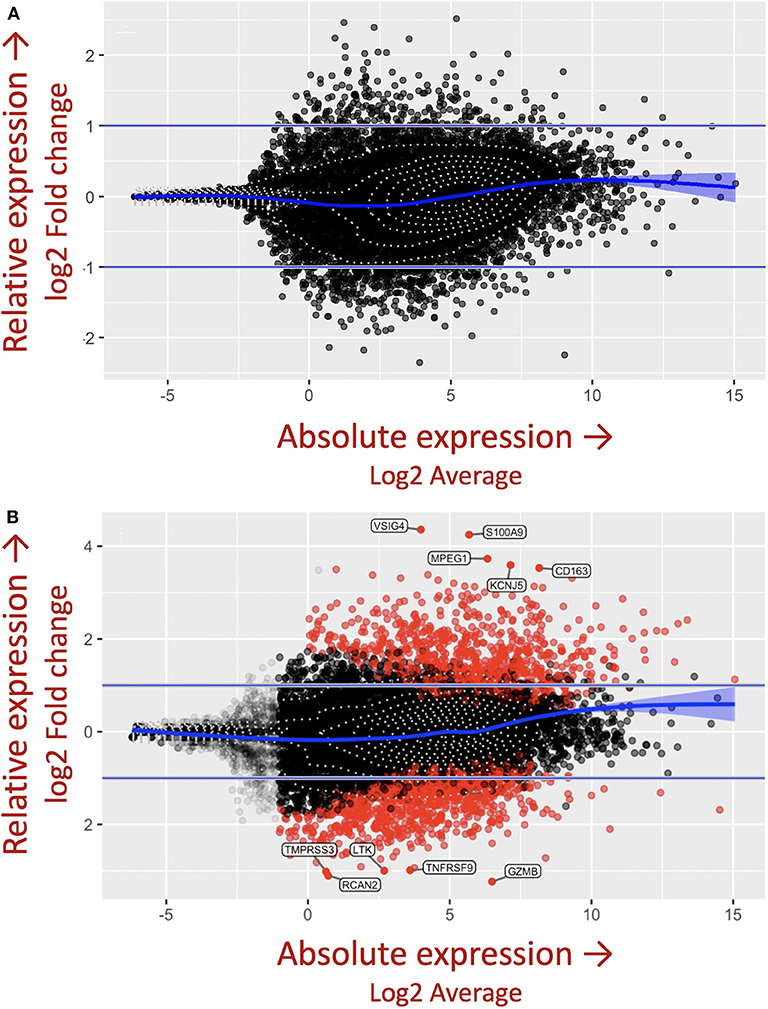
Figure 3. M.tb antigen stimulation induces a unique and divergent transcriptional response in sarcoidosis. Differential gene expression in PBMCs derived from patients with sarcoidosis and healthy control subjects after (A) uncoated and (B) M.tb antigen-coated bead stimulation shown as mva style plots. The x-axis is the log2 average of the gene expression level. All genes with an adjusted P-value of 0.05 and at least a log2-fold change in the magnitude of gene expression (indicated by the two horizontal blue lines) between M.tb antigen and uncoated beads are shaded red.
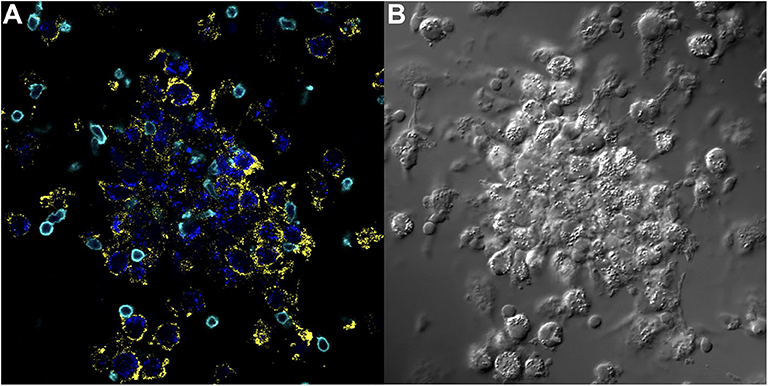
Figure 4. Key immune cell populations are represented in the in vitro human granuloma model. (A) Immunofluorescence microscopy image showing CD11b+ macrophages and CD3+ lymphocytes are present in a representative granuloma-like structure formed by M.tb. antigen-simulated PBMCs from a M.tb. naïve sarcoidosis patient. The image is a composite of 3 fluorescent channels: blue, yellow, and cyan channels represent the M.tb. antigen-coated beads, CD11b staining, and CD3 staining, respectively. (B) A differential interference contrast image of the same granuloma-like structure.
Through its inherent representation of the complex genetics underpinning immune cell dysregulation, the in vitro granuloma model holds promise in providing detailed mechanistic insight into sarcoidosis–specific disease regulating pathways at play during early stages of granuloma formation. This was demonstrated through the common stimulation of PBMCs derived from M.tb naïve sarcoidosis patients and individuals with latent TB infection (LTBI) with M.tb-derived antigens. Both groups produced granuloma-like structures following stimulation, albeit the structures derived from LTBI individuals had a denser lymphocytic cuff. However, gene expression differences were vast between the groups revealing divergent mechanisms of granuloma formation that once again were dependent on antigen stimulation (37). Specifically, we found >5,000 unique DE genes in sarcoidosis compared to LTBI, with many of these DE genes being associated with macrophages (37). Ingenuity Pathway Analysis showed that one distinct pathway in sarcoidosis involves enhanced and prolonged antigen uptake, processing, and presentation (37). Featured in this pathway is the increased expression of vacuolar H+-ATPase (V-ATPase), a multisubunit proton pump that actively acidifies lysosomal compartments. In addition to this role, V-ATPase forms a signaling complex with mTORc1 that promotes its activation when phagolysosomes are acidified (38). This finding in the in vitro granuloma model links macrophage antimicrobial activities with abnormally sustained mTORc1 signaling shown to lead to unchecked granuloma formation in the Tsc2 knockout mouse model and raises the speculation that evolution of sarcoidosis involves unchecked development of a granulomatous response as a result of enhanced microbial killing. The abnormal immune response in sarcoidosis featuring enhanced intracellular microbial killing via phagolysosomes is unlikely to be confined to M.tb and its antigens produced, as other intracellular pathogens, such as Histoplasmosis capsulatum and P. acnes have been incriminated in the pathogenesis of sarcoidosis (39).
Given that macrophages are the dominant cell type observed in sarcoidosis granulomas, there have been many different efforts aimed at modeling the macrophage response in sarcoidosis. Macrophage polarization is thought to play a major role in inflammatory diseases including diseases infectious in nature (7), and a growing body of evidence points to a functional imbalance in sarcoidosis in favor of a Th2-biased immune response. Macrophages display enormous plasticity in their phenotypes and appear along a continuum (40). One polar subset identified early on by defined agonists is the so-called M2-type macrophage that takes on immunoregulatory and tissue maintenance and reparative properties (41). Elevated expression of M2 macrophage-associated markers have been noted in diseased sarcoidosis tissues, including CD206 and CD163, and their expression correlated with disease severity (42–44). Macrophage features observed in our in vitro human granuloma model strongly align with this theory, including a significant increase in >50 different M2-like macrophage gene transcripts and elevated expression of CD163 in sarcoidosis granulomas at the protein level (Figure 5). Pathway analysis predicted the cytokine IL-13 as being an important upstream regulator of the observed gene expression changes in sarcoidosis compared to healthy controls after antigen stimulation, and we verified this gene network to be highly overrepresented in human sarcoidosis lung and lymph node tissues (45) (Figure 6). IL-13 signaling is associated with a Th2-biased immune response and is a known promoter of alternative or M2-like macrophage activation (46, 47). Furthermore, characteristic features of sarcoidosis such as multinucleated giant cell formation (48, 49) and fibrotic tissue changes (8, 39) associated with severe disease phenotypes are signature behaviors of M2-like macrophages.
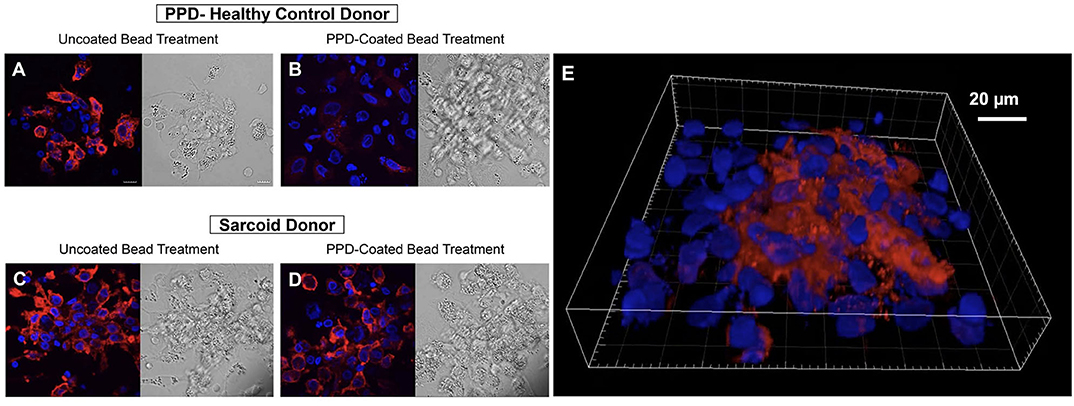
Figure 5. CD163 expression is increased in granuloma-like structures formed by antigen-stimulated sarcoidosis PBMCs in the in vitro human granuloma model. Immunofluorescence microscopy imaging demonstrating macrophage CD163 expression upon stimulating healthy control PBMCs with uncoated beads (A) with dramatic loss of expression following 7-days M.tb antigen-stimulation (B). In contrast, abundant CD163 expression was observed on sarcoidosis macrophages after uncoated bead treatment (C) that persisted after M.tb antigen-stimulation (D). A 3D rendered volume of a sarcoidosis granuloma-like structure 7 days after M.tb antigen-stimulation showing centrally clustered CD163-expressing macrophages (E). DAPI and CD163 staining shown in blue and red, respectively.
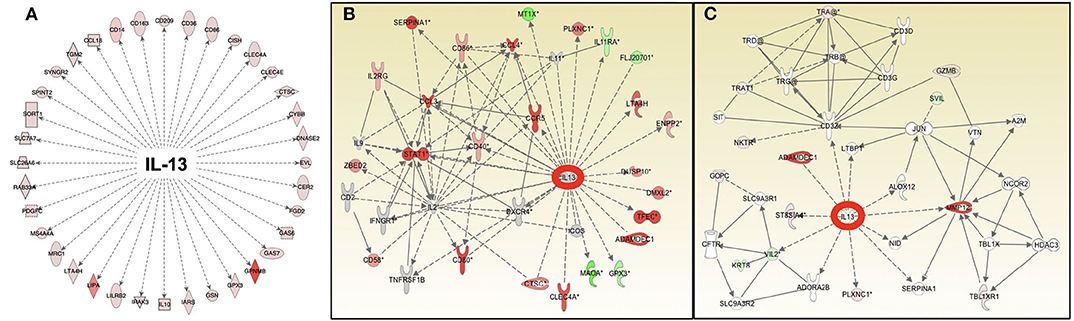
Figure 6. Pathway analysis of gene expression identifies IL-13 as an important and common upstream mediator of the antigen-dependent sarcoidosis granulomatous response in the in vitro granuloma model (A), human sarcoidosis lymph node tissue (B), and lung biopsies (C).
Sarcoidosis and TB appear to have similar triggers, potentially occupying extremes of a common disease spectrum. Histologically, important differences between sarcoidosis and TB granulomas have been described with a subset of TB granulomas featuring lipid-containing, necrotic macrophages leading to caseation while sarcoidosis granulomas do not present with necrosis or caseation. CD163-expressing macrophages, which were recently shown to be highly abundant in sarcoidosis pulmonary granulomas but not in TB granulomas (43), do not accumulate lipids (50) and therefore their presence in sarcoidosis may explain why sarcoidosis granulomas are “non-caseating.” Examination of TB granulomas from infected macaques similarly revealed an absence of CD163 expression in caseating central cores of these structures containing lipid-filled macrophages (51). There is undoubtedly some overlap, however, in granuloma phenotypes between sarcoidosis and TB which is expected given the heterogeneity of these diseases. Ultimately, the connection between macrophage polarization, granuloma characteristics, and antimicrobial capacities including the restriction of M.tb growth remains incompletely understood, with the classic picture of M1 macrophages associated with improved protection likely an oversimplification.
Other data from the model that point to a functional imbalance in favor of Th2-immune signaling in sarcoidosis was the relatively blunted INF-γ production associated with sarcoidosis following M.tb antigen stimulation and the significant inhibition of granuloma formation in sarcoidosis through pharmacologic signal transducer and activator of transcription 6 (STAT6) inhibition (45). IL-13–mediated activation of STAT6 is a well-established pathway for M2-like macrophage polarization (52). It has also been shown that pharmacologic inhibition of STAT3 via upstream Janus kinase (JAK) inhibition leads to complete resolution of skin lesions in sarcoidosis patients (53). Activation of STAT3 is associated with M2-like macrophage polarization (54). In the in vitro human granuloma model we also observed a subset of STAT1-regulated genes that were DE between sarcoidosis and LTBI following M.tb antigen stimulation, suggesting a more complex signaling landscape. Indeed, the observed co-existence of both non-CD163-expressing macrophages and CD163-expressing macrophages in the granuloma-like structures (Figure 5) supports heterogeneous signaling at the level of the granuloma. Collectively, data from the model with respect to transcriptome-based network characteristics, immune signaling dysfunction, and macrophage immunohistological protein expression patterns reflective of diseased tissues indicate that antigen-stimulated PBMCs derived from patients with sarcoidosis has the ability to manifest the complex and, as yet, undefined genetic features that predispose to sarcoidosis.
As cell-based laboratory models continue to advance, the use of mathematical models will act in concert with them to accelerate the pace of discovery (55, 56). A computational model of sarcoidosis has been established and is capable of modeling physical measurements and parameters based on input from companion in vitro models and human studies to model the interconnectedness of the cytokine, chemokine, and growth factor proteins contributing to granuloma formation over time (57). We envision that the mathematical model can be used to rapidly interrogate the complex interplay of cells and inflammatory mediators on the evolution of granuloma formation over time through the manipulation of a variable (cytokine for example) or combination of variables, thus informing on specific druggable targets in sarcoidosis. Targets identified in this manner can be rapidly tested in the in vitro model for efficacy and thus be combined in a synergistic fashion to bring new therapies (mono or in combo with another) into human trials to effectively inhibit the cycle of pathological granuloma formation (Figure 7).
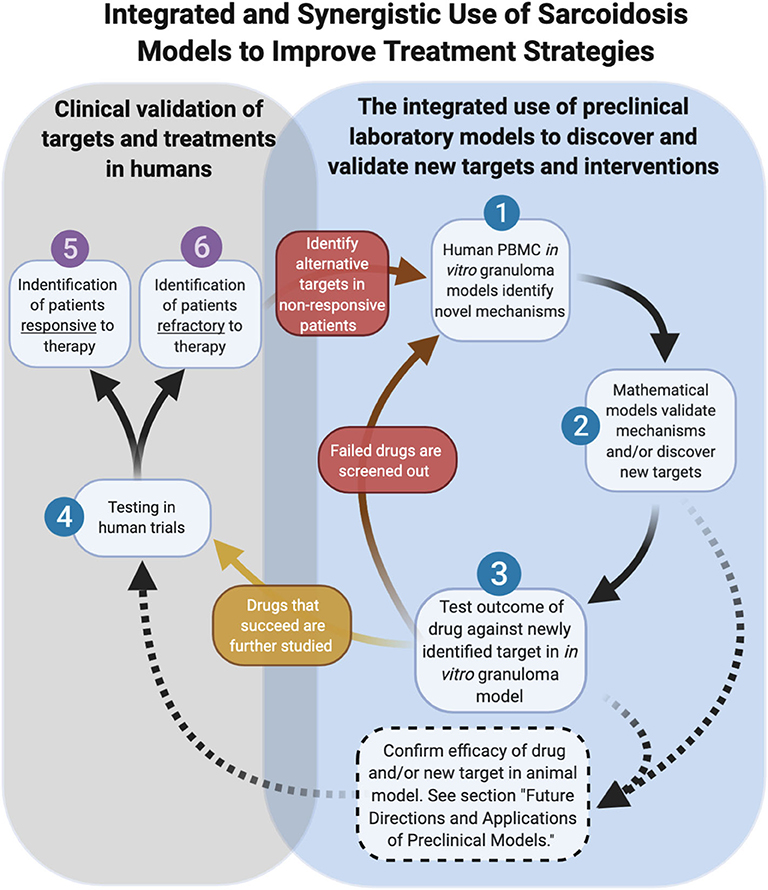
Figure 7. Integrated and synergistic use of sarcoidosis models. Dashed arrows represent future pathways dependent on animal model advancements.
As we look toward the future of therapeutic development in sarcoidosis, the role of animal models is currently unclear. Undoubtedly animal models have proven valuable for defining new aspects of disease pathogenesis and pre-clinical testing of new therapeutics. Relevant to sarcoidosis, novel imaging techniques can be performed in mice to enable powerful visual insights into the interplay of immune cells and tissue stroma (58). The future development of new transgenic animal models of sarcoidosis, however, will be contingent upon identifying disease-causing genetic factors in humans. An alternative approach would be to “humanize” animals, a process involving the replacement of the native bone marrow with stem cells from human sarcoidosis donors. There are inherent limitations of this approach, including impaired adaptive immunity in the humanized mice (59), which likely hinder these models from accurately modeling the full cellular and molecular determinants of granuloma formation in sarcoidosis. A humanized model of TB demonstrated pathology that closely modeled human disease, lending promise to this approach in the future (60). We envision a future role of humanized animal models of sarcoidosis that are explored in parallel with in vitro human granuloma models as complementary platforms for testing new therapies and evaluating pharmacokinetic and toxicity aspects ahead of human trials (Figure 7).
In addition to the providing greater insights into granuloma formation in sarcoidosis, the in vitro human granuloma model could be leveraged to screen novel therapies and determine the contribution of specific genes to granuloma formation using siRNA or CRISPR technology (Figure 7). Because the model accounts for the inherent genetic variables that predispose to disease, it could be used to gain insights into the ethnic/racial and gender disparities characteristic of this disease. Given that approximately one-third of patients have a progressive form of the disease (61), the model may be able to clarify the unique mechanisms driving granuloma formation in this severe phenotype. An additional strength of the in vitro human granuloma model is that it can be used to better understand the dynamic, multicellular mechanisms underpinning distinct granulomatous immune responses that occur in humans and is not restricted to sarcoidosis (Figure 8). For example, the model can be used to explore mechanisms driving early granulomatous responses to etiologic factors other than TB antigens, including exposures to foreign bodies.
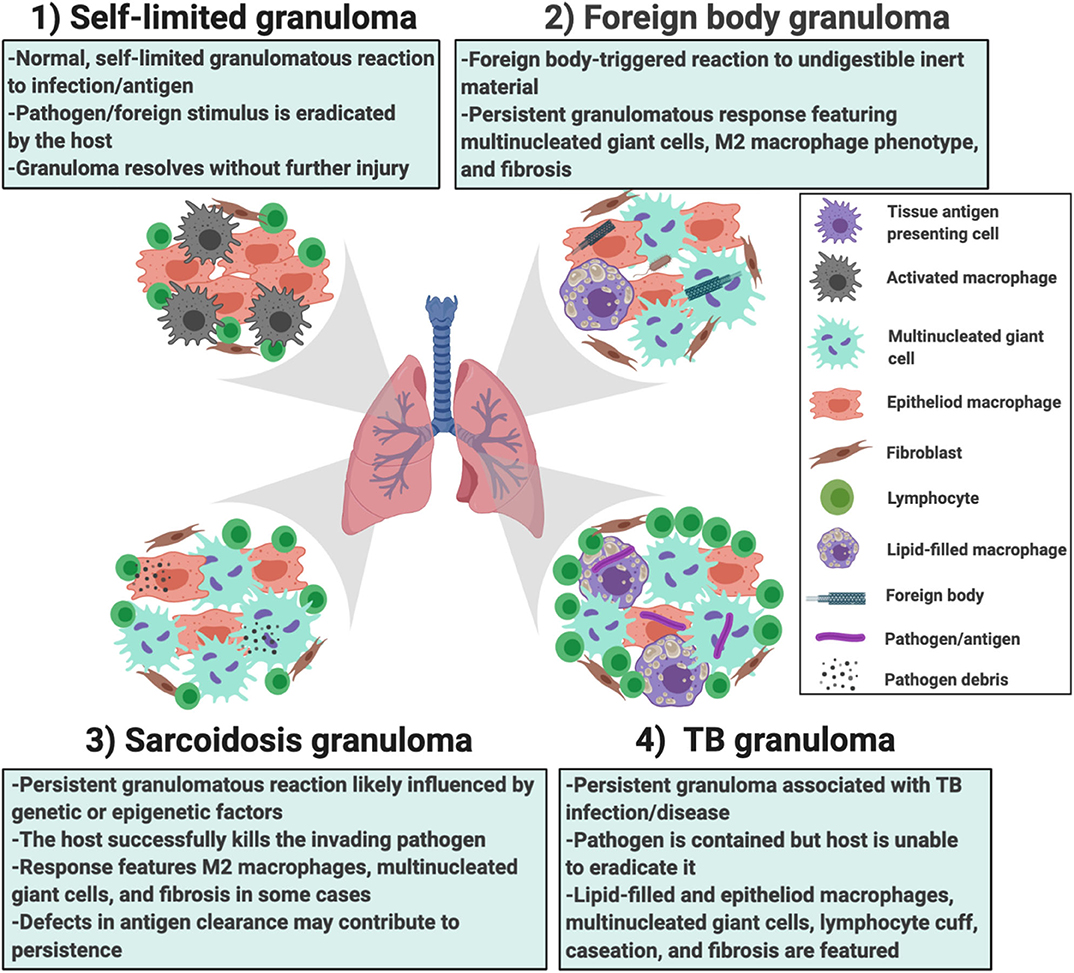
Figure 8. Different scenarios in which the in vitro human granuloma model can be used to better understand the dynamic, multicellular mechanisms underpinning distinct granulomatous immune responses that occur in humans ranging from (1) self-limited immune reactions; (2) foreign body immune reactions; (3) sarcoidosis granulomas; and (4) TB granulomas. The granuloma model can be used to test mechanistic hypotheses as well as further our understanding of the importance of environmental (e.g., the type of antigen used to promote granulomas) and host factors (e.g., genetics/race, epigenetic factors/sex, etc.) in these reactions. They can also be used for throughput testing of new potential therapies.
There are some caveats of current in vitro cell culture models to address. For example, the in vitro human granuloma model is likely not capturing the entire spectrum of disease due to the absence of a tissue-mimicking environment. Recent advances in organ-on-a-chip technology are beginning to provide new opportunities in in vitro cell culture models such as the modeling of more complex cell interactions involving extracellular matrix components and stromal cells, although to date most of these models lack immune cells (62). Enabling the cross-talk between immune cells and matrix cells such as fibroblasts may allow for mechanisms driving fibrotic changes to be modeled in sarcoidosis, a signature feature of severe disease phenotypes (63). Organ-on-a-chip technology also allows for the continuous perfusion of cells, such as PBMCs, through a vascular-like system, thus potentially modeling cellular recruitment processes and enabling a more prolonged study of the eventual fate of granulomas (i.e., self-perpetuating vs. disaggregating). As with any modeling endeavor, it is challenging to know when a model is “good enough.” In the case of sarcoidosis, any modeling advancement must be weighed against the added cost, throughput potential, and the burden of validation.
The failure to effectively model sarcoidosis in the laboratory or in animals continues to hinder the discovery and translation of new, targeted treatments. Preclinical models that are capable of clarifying the significant knowledge gap surrounding the triggers and mechanisms driving granuloma formation and self-perpetuation in sarcoidosis will be key to this cause. Compared to modeling approaches that study only a single cell type, the patient-derived PBMC-based in vitro granuloma model allows for the interrogation of antigen-immune cell interplay and related cell-cell interactions driving the various stages of granuloma formation in sarcoidosis and underpinned by the complex and, as yet, undefined genetic features of this disease. The in vitro human granuloma model provides a powerful approach to addressing critical knowledge gaps in sarcoidosis, and we envision its complementary use with other pre-clinical models will likely accelerate progress toward identifying and testing new drugs and drug combinations that can be rapidly brought to clinical trials.
EC, LS, and LL wrote the article. EC and LL contributed to designing the figures and table for the article. All authors contributed to the article and approved the submitted version.
This work was supported by the following grants: NIH through The Ohio State University Center for Clinical and Translational Science (Clinical and Translational Science Award UL1TR001070 [EC and LS]), ATS Foundation/Mallinckrodt Pharmaceuticals, Inc. Research Fellowship, and a Sarcoidosis Disease Model Grant from the Foundation for Sarcoidosis Research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AM, alveolar macrophage; BAL, bronchoalveolar lavage; PBMC, peripheral blood mononuclear cells; JAK, Janus kinase; STAT6, signal transducer and activator of transcription 6; V-ATPase, vacuolar H+-ATPase; M.tb, Mycobacterium tuberculosis; mTORc1, mammalian target of rapamycin complex 1; Tsc2, tuberous sclerosis-2 gene; DE, differentially expressed; TB, tuberculosis; LTBI, individuals with latent TB infection; PPD, purified protein derivative.
1. Baughman RP, Field S, Costabel U, Crystal RG, Culver DA, Drent M, et al. Sarcoidosis in America. Analysis based on health care use. Ann Am Thorac Soc. (2016) 13:1244–52. doi: 10.1513/AnnalsATS.201511-760OC
2. Gerke AK, Judson MA, Cozier YC, Culver DA, Koth LL. Disease burden and variability in sarcoidosis. Ann Am Thorac Soc. (2017) 14:S421–8. doi: 10.1513/AnnalsATS.201707-564OT
3. James DG. Centenary commemoration of sarcoidosis and of Jonathan Hutchinson. Br Med J. (1969) 2:109–10. doi: 10.1136/bmj.2.5649.109
4. Chen ES, Moller DR. Etiology of sarcoidosis. Clin Chest Med. (2008) 29:365–77. doi: 10.1016/j.ccm.2008.03.011
5. Venet A, Hance AJ, Saltini C, Robinson BW, Crystal RG. Enhanced alveolar macrophage-mediated antigen-induced T-lymphocyte proliferation in sarcoidosis. J Clin Invest. (1985) 75:293–301. doi: 10.1172/JCI111688
6. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. (2013) 39:599–610. doi: 10.1016/j.immuni.2013.08.007
7. Huang Z, Luo Q, Guo Y, Chen J, Xiong G, Peng Y, et al. Mycobacterium tuberculosis-induced polarization of human macrophage orchestrates the formation and development of tuberculous granulomas in vitro. PLoS ONE. (2015) 10:e0129744. doi: 10.1371/journal.pone.0129744
8. Guirado E, Mbawuike U, Keiser TL, Arcos J, Azad AK, Wang SH, et al. Characterization of host and microbial determinants in individuals with latent tuberculosis infection using a human granuloma model. MBio. (2015) 6:e02537–14. doi: 10.1128/mBio.02537-14
9. Wang H, Maeda Y, Fukutomi Y, Makino M. An in vitro model of Mycobacterium leprae induced granuloma formation. BMC Infect Dis. (2013) 13:279. doi: 10.1186/1471-2334-13-279
10. Crouser ED, White P, Caceres EG, Julian MW, Papp AC, Locke LW, et al. A novel in vitro human granuloma model of sarcoidosis and latent tuberculosis infection. Am J Respir Cell Mol Biol. (2017) 57:487–98. doi: 10.1165/rcmb.2016-0321OC
11. Doherty DE, Downey GP, Worthen GS, Haslett C, Henson PM. Monocyte retention and migration in pulmonary inflammation. Requirement for neutrophils. Lab Invest. (1988) 59:200–13.
12. Ichiyasu H, Suga M, Matsukawa A, Iyonaga K, Mizobe T, Takahashi T, et al. Functional roles of MCP-1 in Propionibacterium acnes-induced, T cell-mediated pulmonary granulomatosis in rabbits. J Leukoc Biol. (1999) 65:482–91. doi: 10.1002/jlb.65.4.482
13. Maus UA, Wellmann S, Hampl C, Kuziel WA, Srivastava M, Mack M, et al. CCR2-positive monocytes recruited to inflamed lungs downregulate local CCL2 chemokine levels. Am J Physiol Lung Cell Mol Physiol. (2005) 288:L350–8. doi: 10.1152/ajplung.00061.2004
14. Reijererk EP, Veldhuis Kroeze EJ, van Sloet Oldruitenborgh-Oosterbaan M. Equine sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. (2009) 26:20–3. doi: 10.2746/095777308X307815
15. Hu Y. Yibrehu B, Zabini D, Kuebler WM. Animal models of sarcoidosis. Cell Tissue Res. (2017) 367:651–61. doi: 10.1007/s00441-016-2526-3
16. Werner JL, Escolero SG, Hewlett JT, Mak TN, Williams BP, Eishi Y, et al. Induction of pulmonary granuloma formation by propionibacterium acnes is regulated by MyD88 and Nox2. Am J Respir Cell Mol Biol. (2017) 56:121–30. doi: 10.1165/rcmb.2016-0035OC
17. Huizar I, Malur A, Midgette YA, Kukoly C, Chen P, Ke PC, et al. Novel murine model of chronic granulomatous lung inflammation elicited by carbon nanotubes. Am J Respir Cell Mol Biol. (2011) 45:858–66. doi: 10.1165/rcmb.2010-0401OC
18. Pagan AJ, Ramakrishnan L. The formation and function of granulomas. Annu Rev Immunol. (2018) 36:639–65. doi: 10.1146/annurev-immunol-032712-100022
19. Ohtani H. Granuloma cells in chronic inflammation express CD205 (DEC205) antigen and harbor proliferating T lymphocytes: similarity to antigen-presenting cells. Pathol Int. (2013) 63:85–93. doi: 10.1111/pin.12036
20. Linke M, Pham HT, Katholnig K, Schnoller T, Miller A, Demel F, et al. Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat Immunol. (2017) 18:293–302. doi: 10.1038/ni.3655
21. Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and −2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci USA. (2002) 99:13571–6. doi: 10.1073/pnas.202476899
22. Fontenot AP, Falta MT, Kappler JW, Dai S, McKee AS. Beryllium-induced hypersensitivity: genetic susceptibility and neoantigen generation. J Immunol. (2016) 196:22–7. doi: 10.4049/jimmunol.1502011
23. Rossman MD. Chronic beryllium disease: diagnosis and management. Environ Health Perspect. (1996) 104(Suppl.5):945–7. doi: 10.1289/ehp.96104s5945
24. Li L, Silveira LJ, Hamzeh N, Gillespie M, Mroz PM, Mayer AS, et al. Beryllium-induced lung disease exhibits expression profiles similar to sarcoidosis. Eur Respir J. (2016) 47:1797–808. doi: 10.1183/13993003.01469-2015
25. Muller-Quernheim J, Gaede KI, Fireman E, Zissel G. Diagnoses of chronic beryllium disease within cohorts of sarcoidosis patients. Eur Respir J. (2006) 27:1190–5. doi: 10.1183/09031936.06.00112205
26. Celada LJ, Kropski JA, Herazo-Maya JD, Luo W, Creecy A, Abad AT, et al. PD-1 up-regulation on CD4(+) T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-beta1 production. Sci Transl Med. (2018) 10:aar8356. doi: 10.1126/scitranslmed.aar8356
27. Yang IV, Konigsberg I, MacPhail K, Li L, Davidson EJ, Mroz PM, et al. DNA methylation changes in lung immune cells are associated with granulomatous lung disease. Am J Respir Cell Mol Biol. (2019) 60:96–105. doi: 10.1165/rcmb.2018-0177OC
28. Grunewald J, Kaiser Y, Ostadkarampour M, Rivera NV, Vezzi F, Lotstedt B, et al. T-cell receptor-HLA-DRB1 associations suggest specific antigens in pulmonary sarcoidosis. Eur Respir J. (2016) 47:898–909. doi: 10.1183/13993003.01209-2015
29. Chen ES, Song Z, Willett MH, Heine S, Yung RC, Liu MC, et al. Serum amyloid A regulates granulomatous inflammation in sarcoidosis through Toll-like receptor-2. Am J Respir Crit Care Med. (2010) 181:360–73. doi: 10.1164/rccm.200905-0696OC
30. Freemer M, King TE Jr. The ACCESS study: characterization of sarcoidosis in the United States. Am J Respir Crit Care Med. (2001) 164:1754–5. doi: 10.1164/ajrccm.164.10.2109111b
31. Kuberski T, Yourison I. Coccidioidomycosis A cause of sarcoidosis. Open Forum Infect Dis. (2017) 4:ofw117. doi: 10.1093/ofid/ofw117
32. Wheat LJ, French ML, Wass JL. Sarcoidlike manifestations of histoplasmosis. Arch Intern Med. (1989) 149:2421–6. doi: 10.1001/archinte.149.11.2421
33. Yamamoto K, Uchida K, Furukawa A, Tamura T, Ishige Y, Negi M, et al. Catalase expression of propionibacterium acnes may contribute to intracellular persistence of the bacterium in sinus macrophages of lymph nodes affected by sarcoidosis. Immunol Res. (2019) 67:182–93. doi: 10.1007/s12026-019-09077-9
34. Song Z, Marzilli L, Greenlee BM, Chen ES, Silver RF, Askin FB, et al. Mycobacterial catalase-peroxidase is a tissue antigen and target of the adaptive immune response in systemic sarcoidosis. J Exp Med. (2005) 201:755–67. doi: 10.1084/jem.20040429
35. Elkington P, Lerm M, Kapoor N, Mahon R, Pienaar E, Huh D, et al. In vitro granuloma models of tuberculosis: potential and challenges. J Infect Dis. (2019) 219:1858–66. doi: 10.1093/infdis/jiz020
36. Celada LJ, Hawkins C, Drake WP. The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clin Chest Med. (2015) 36:561–8. doi: 10.1016/j.ccm.2015.08.001
37. Locke L, White P, Julian M, Bicer S, Papp A, Sadee W, et al. Human sarcoidosis vs TB: all granulomas are not created equally. C31. Transl Stud Sarcoidosis. (2019) A4500. doi: 10.1164/ajrccm-conference.2019.199.1_MeetingAbstracts.A4500
38. Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. (2011) 334:678–83. doi: 10.1126/science.1207056
39. Bennett D, Bargagli E, Refini RM, Rottoli P. New concepts in the pathogenesis of sarcoidosis. Expert Rev Respir Med. (2019) 13:981–91. doi: 10.1080/17476348.2019.1655401
40. Murray PJ. Macrophage polarization. Annu Rev Physiol. (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
41. Gordon S. Alternative activation of macrophages. Nat Rev Immunol. (2003) 3:23–35. doi: 10.1038/nri978
42. Prokop S, Heppner FL, Goebel HH, Stenzel W. M2 polarized macrophages and giant cells contribute to myofibrosis in neuromuscular sarcoidosis. Am J Pathol. (2011) 178:1279–86. doi: 10.1016/j.ajpath.2010.11.065
43. Shamaei M, Mortaz E, Pourabdollah M, Garssen J, Tabarsi P, Velayati A, et al. Evidence for M2 macrophages in granulomas from pulmonary sarcoidosis: a new aspect of macrophage heterogeneity. Hum Immunol. (2018) 79:63–9. doi: 10.1016/j.humimm.2017.10.009
44. Isohisa T, Asai J, Kanemaru M, Arita T, Tsutsumi M, Kaneko Y, et al. CD163-positive macrophage infiltration predicts systemic involvement in sarcoidosis. J Cutan Pathol. (2020) 47:584–91. doi: 10.1111/cup.13675
45. Locke LW, Crouser ED, White P, Julian MW, Guirado Caceres E, Papp AC, et al. IL-13-regulated macrophage polarization during granuloma formation in an in vitro human sarcoidosis model. Am J Respir Cell Mol Biol. (2018) 60:84–95. doi: 10.1165/rcmb.2018-0053OC
46. Martinez-Nunez RT, Louafi F, Sanchez-Elsner T. The interleukin 13 (IL-13) pathway in human macrophages is modulated by microRNA-155 via direct targeting of interleukin 13 receptor alpha1 (IL13Ralpha1). J Biol Chem. (2011) 286:1786–94. doi: 10.1074/jbc.M110.169367
47. Standiford TJ. Macrophage polarization in sarcoidosis: an unexpected accomplice? Am J Respir Cell Mol Biol. (2019) 60:9–10. doi: 10.1165/rcmb.2018-0298ED
48. van Maarsseveen TCMT, Vos W, van Diest PJ. Giant cell formation in sarcoidosis: cell fusion or proliferation with non-division? Clin Exp Immunol. (2009) 155:476–86. doi: 10.1111/j.1365-2249.2008.03841.x
49. Helming L, Gordon S. Macrophage fusion induced by IL-4 alternative activation is a multistage process involving multiple target molecules. Eur J Immunol. (2007) 37:33–42. doi: 10.1002/eji.200636788
50. Finn AV, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao X, et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol. (2012) 59:166–77. doi: 10.1016/j.jacc.2011.10.852
51. Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, et al. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol. (2013) 191:773–84. doi: 10.4049/jimmunol.1300113
52. Rahal OM, Wolfe AR, Mandal PK, Larson R, Tin S, Jimenez C, et al. Blocking interleukin (IL)4- and IL13-mediated phosphorylation of STAT6 (Tyr641) decreases M2 polarization of macrophages and protects against macrophage-mediated radioresistance of inflammatory breast cancer. Int J Radiat Oncol Biol Phys. (2018) 100:1034–43. doi: 10.1016/j.ijrobp.2017.11.043
53. Damsky W, Thakral D, McGeary MK, Leventhal J, Galan A, King B. Janus kinase inhibition induces disease remission in cutaneous sarcoidosis and granuloma annulare. J Am Acad Dermatol. (2020) 82:612–21. doi: 10.1016/j.jaad.2019.05.098
54. Tang L, Zhang H, Wang C, Li H, Zhang Q, Bai J. M2A and M2C macrophage subsets ameliorate inflammation and fibroproliferation in acute lung injury through interleukin 10 pathway. Shock. (2017) 48:119–29. doi: 10.1097/SHK.0000000000000820
55. Day J, Friedman A, Schlesinger LS. Modeling the immune rheostat of macrophages in the lung in response to infection. Proc Natl Acad Sci USA. (2009) 106:11246–51. doi: 10.1073/pnas.0904846106
56. Marino S, Linderman JJ, Kirschner DE. A multifaceted approach to modeling the immune response in tuberculosis. Wiley Interdiscip Rev Syst Biol Med. (2011) 3:479–89. doi: 10.1002/wsbm.131
57. Hao W, Crouser ED, Friedman A. Mathematical model of sarcoidosis. Proc Natl Acad Sci USA. (2014) 111:16065–70. doi: 10.1073/pnas.1417789111
58. Laschke MW, Vollmar B, Menger MD. The dorsal skinfold chamber: window into the dynamic interaction of biomaterials with their surrounding host tissue. Eur Cell Mater. (2011) 22:147–64. doi: 10.22203/eCM.v022a12
59. Skelton JK, Ortega-Prieto AM, Dorner M. A Hitchhiker's guide to humanized mice: new pathways to studying viral infections. Immunology. (2018) 154:50–61. doi: 10.1111/imm.12906
60. Calderon VE, Valbuena G, Goez Y, Judy BM, Huante MB, Sutjita P, et al. A humanized mouse model of tuberculosis. PLoS ONE. (2013) 8:e63331. doi: 10.1371/journal.pone.0063331
61. Lockstone HE, Sanderson S, Kulakova N, Baban D, Leonard A, Kok WL, et al. Gene set analysis of lung samples provides insight into pathogenesis of progressive, fibrotic pulmonary sarcoidosis. Am J Respir Crit Care Med. (2010) 181:1367–75. doi: 10.1164/rccm.200912-1855OC
62. Esch EW, Bahinski A, Huh D. Organs-on-chips at the frontiers of drug discovery. Nat Rev Drug Discov. (2015) 14:248–60. doi: 10.1038/nrd4539
Keywords: sarcoidosis, granuloma, modeling, macrophages, lung
Citation: Locke LW, Schlesinger LS and Crouser ED (2020) Current Sarcoidosis Models and the Importance of Focusing on the Granuloma. Front. Immunol. 11:1719. doi: 10.3389/fimmu.2020.01719
Received: 08 April 2020; Accepted: 29 June 2020;
Published: 04 August 2020.
Edited by:
Mary Jane Thomassen, East Carolina University, United StatesReviewed by:
Matyas Sandor, University of Wisconsin-Madison, United StatesCopyright © 2020 Locke, Schlesinger and Crouser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elliott D. Crouser, RWxsaW90dC5jcm91c2VyQG9zdW1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.