
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 09 September 2020
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01717
 Qi Wang1,2Zhen-Fen Chi3Di Wei3
Qi Wang1,2Zhen-Fen Chi3Di Wei3 Zhen-Ao Zhao1,2
Zhen-Ao Zhao1,2 Hong Zhang1,2
Hong Zhang1,2 Li-Min Zhang1,2Yan-Xu Liu1,2An-Ling Kang1,2Meng Zhao1,2
Li-Min Zhang1,2Yan-Xu Liu1,2An-Ling Kang1,2Meng Zhao1,2 Peng Wang1,2Ling-Hu Nie3*
Peng Wang1,2Ling-Hu Nie3* Chun-Yu Niu4,5*
Chun-Yu Niu4,5* Zi-Gang Zhao1,2,5*
Zi-Gang Zhao1,2,5*Vascular endothelial injury caused by post-hemorrhagic shock mesenteric lymph (PHSML) return is an important manifestation during refractory hemorrhagic shock. Using human umbilical vein endothelial cells (HUVECs) and transcriptome analysis, this study sought to investigate the molecular mechanism underlying the adverse effect of PHSML on vascular endothelium. Post-hemorrhagic shock mesenteric lymph was collected from male rats after they underwent hemorrhagic shock and following resuscitation, while normal mesenteric lymph (NML) was harvested from sham rats. Human umbilical vein endothelial cells were incubated with the culture medium containing either 10% phosphate buffered saline (Control), NML, or PHSML for 3 h, and then were harvested for RNA sequencing. In comparison with NML treated cells, 37 genes were differentially expressed in PHSML-treated HUVECs, including 32 upregulated genes and five downregulated genes. These differentially expressed genes were mainly enriched in inflammatory pathways, including signaling pathways for activation of the NOD-like receptors, NF-κB, and TNF. Furthermore, we found that C–C motif chemokine ligand 2 (CCL2) was increased significantly after PHSML treatment, and Bindarit, a CCL2 production inhibitor, attenuated the damage of HUVECs induced by PHSML. The results provide molecular evidence on vascular endothelium damage caused by PHSML. C–C motif chemokine ligand 2 might represent a new target for reducing vascular injury after severe hemorrhagic shock.
Ischemic hypoxia, hyper-inflammation, and oxidative stress induced by hemorrhagic shock lead to the injury of vascular endothelial cells, which is the main cause of aggravating microcirculation dysfunction and organ injury following refractory hemorrhagic shock. Numerous studies have shown that post-hemorrhagic shock mesenteric lymph (PHSML) return is the main cause of uncontrolled inflammation and acute distant organ damage (1–3). It is reported that PHSML can directly induce the injury of rat or human pulmonary microvascular endothelial cells and human umbilical vein endothelial cells (HUVECs), resulting in increased permeability of monolayer cells in vitro (4–6). However, precise molecular mechanisms have not been elucidated. Second generation sequencing technology is the most effective way to analyze the transcriptional profile (7). Through differential transcriptome analysis, one can study the pathophysiological mechanisms of cell injury and identify new targets for treatment and prevention of hemorrhagic shock. Human umbilical vein endothelial cell was mostly used to study vascular permeability and endothelial barrier function (8). In this study, we sought to explore the mechanism of PHSML injury to vascular endothelium, HUVECs were incubated with PHSML and the differential mRNA expression was investigated using RNA sequencing (RNA-seq) technology. Subsequently, differentially expressed genes were analyzed and verified using the bioinformatic and RT-PCR methods. As a representative gene among differentially expressed genes, C–C motif chemokine ligand 2 (CCL2) and its inhibitor, Bindarit, were used to further confirm the roles of these genes in mediating HUVEC injury.
All experimental protocols involving animals were approved by the Laboratory Animal Research Committee of Hebei North University. Adult male Wistar rats (280–320 g) were purchased from the Experimental Animal Center of the Chinese Academy of Military Medical Sciences and were randomly divided into a sham group and a hemorrhagic shock group, with n = 12 for each group.
Animal models were established according to the routine method of a previous report (9). The rats received an inhalation anesthetic induction with isoflurane and a general anesthetization with 1% pentobarbital sodium (50 mg/kg, Merck, Germany). Then, bilateral inguinal region surgery was performed to separate the femoral artery and vein. Heparin sodium (1 ml/kg, 500 U/kg) was injected into the right femoral vein to prevent blood coagulation during the experiment. The right femoral artery was intubated using polyethylene tubing, which was connected to the PowerLab biological signal acquisition system (ADInstruments, Bella Vista NSW, Australia) for blood pressure monitoring. The left femoral vein was intubated and connected to the syringe for fluid resuscitation. The incision (about 4 cm) was cut along the abdominal white line. After the operation, the animals were kept stable for 10 min. The blood pressure was maintained at 40 ± 2 mmHg for 10 min by drawing blood using an automatic withdrawal-infusion machine (NE-1000, New Era Pump Systems Inc., Farmingdale, NY). The collected blood was mixed with an equal volume of Ringer’s solution and then slowly injected into the left femoral vein within 30 min for resuscitation. In the sham group, all the above operations were performed except bleeding or resuscitation, and the operation time was the same as that of the hemorrhagic shock model.
To prepare the lymph samples, we opened the abdomen along the incision, lifted the right intestine, and found the mesenteric lymphatic duct accompanying the superior mesenteric artery. The infusion needle was adjusted to a suitable bend and inserted into the mesenteric lymphatic duct. The lymph was then drained and extracted by a vacuum tube. After centrifugation at 850 g at 4°C for 10 min, the supernatant was collected at −80°C for cell experiment and ELISA analysis.
Human umbilical vein endothelial cells (purchased from Cobioer Biosciences Co., Ltd, Nanjing, China, Catalog #: CBP60340) were cultured in BEGMTM (Bronchial Epithelial Cell Growth Medium; CC-3170, Lonza) containing 10% FBS at 37°C and 5% CO2. When the confluence of cells reached about 70%, the medium was changed into a culture medium containing 10% phosphate buffered saline (PBS), normal mesenteric lymph (NML), or PHSML. These cells were further cultured for 3 h for the RNA extraction.
Total RNA was extracted using Trizol Reagent, and the integrity of the RNA was analyzed by Agilent 2100 (Agilent Technologies, CA). A library was constructed using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (New England Biolabs, Lspawich, MA, United States) according to instructions. RNA-seq was performed in Novogene Co., Ltd. The sequencing data description was shown in Table 1. Subsequently, TopHat2 software was used to analyze the data obtained with the reference genome. HTSeq v0.6.1 was used to analyze differentially expressed genes in different samples. Log2 (Fold Change) > 1 and Q-value < 0.005 were used as screening criteria (10). Fold Change represents the ratio of expression between two samples.
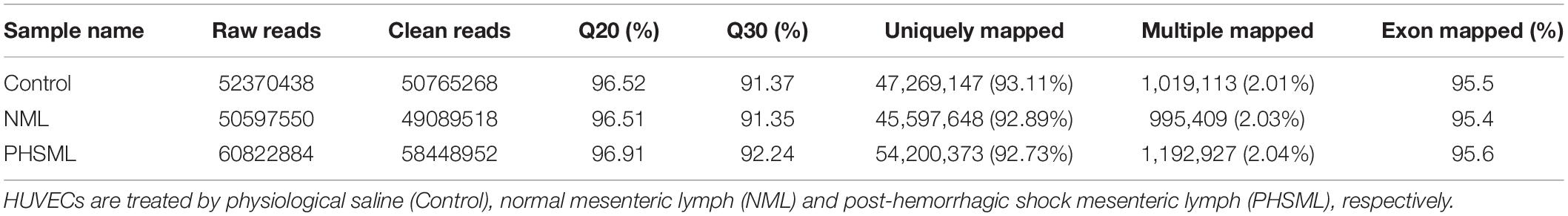
Table 1. Description of RNA-sequencing data.
Gene Ontology (GO) enrichment of differentially expressed genes was analyzed by the GOseq R package with P-value less than 0.05 after the correction was considered to be significantly enriched (11). We used KOBAS v2.0 software to enrich and analyze the differentially expressed genes in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
Total RNA was extracted using Invitrogen TRIzol reagent (Thermo Fisher Scientific). After DNase, I digestion, an aliquot of total RNA (500 ng) was reverse transcribed with Takara PrimeScript RT Reagent Kit (RR037A, Takara). Real-time PCR was performed using a SYBR® Premix Ex TaqTM (DRR420A; Takara) on the Applied Biosystems StepOnePlus Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA). GAPDH was used as an internal reference. The 2–ΔΔCt method was used for relative expression analysis (12). All primer sequences were listed in Table 2.
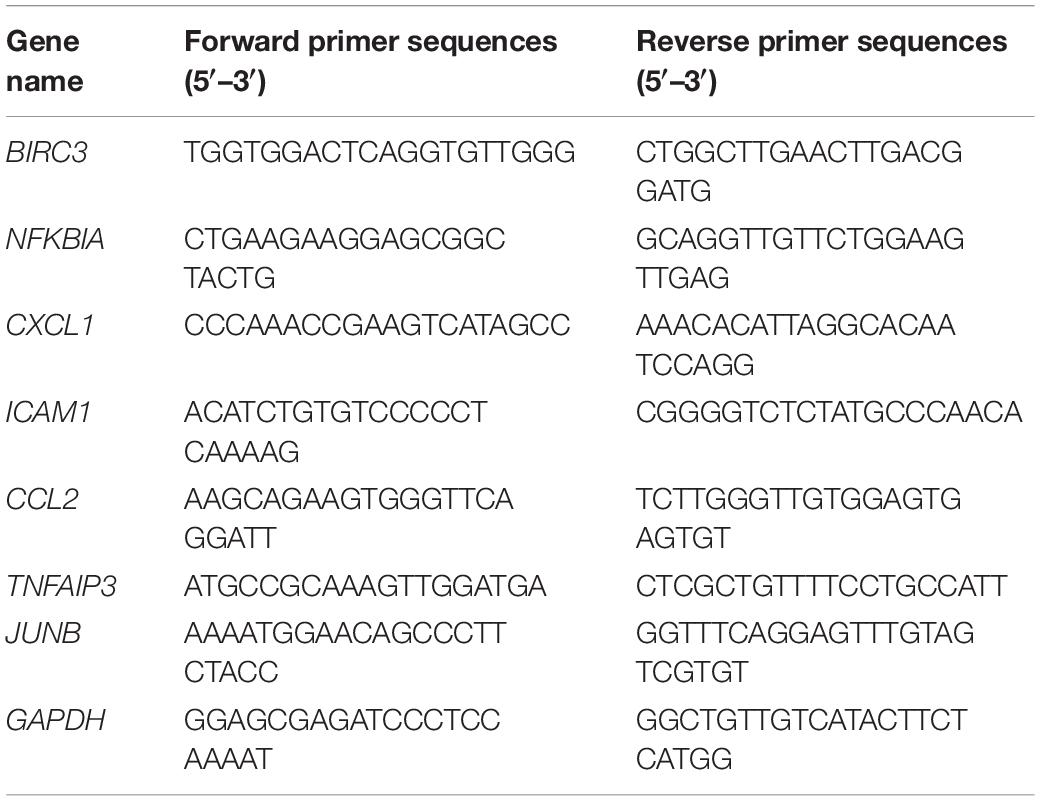
Table 2. Sequences of primers for qRT-PCR.
Thirty thousand cells were planted in each well of a 96-well plate, and the culture medium was removed after 24 h. The cells were further cultured for 24 h by adding a culture medium containing lymph and/or drugs. For CCK8 assay, medium and CCK8 were mixed at a ratio of 10:1 and added into each well for a 4-h incubation. The absorbance at 450 nm was detected by Microplate Reader (SpectraMax® M3, Molecular Devices, Sunnyvale, CA).
Protein samples were extracted from HUVECs with RIPA lysis buffer and sonicated briefly. Equal amounts (30 μg) of protein from each group were separated on a 15% SDS–polyacrylamide gel (SDS-PAGE), and then transferred onto PVDF membranes (Millipore). The membranes were blocked with 5% milk in TBS-T for 1 h at room temperature and then incubated with primary antibody against CCL2 (66272-1-lg, Proteintech Group) at 4°C overnight. After three washes in TBS-T, the membranes were incubated with goat anti-mouse IgG HRP secondary antibody (Applygen) for 1 h at room temperature. The ImageQuant LAS 4000 imager was used for the detection of the protein expression, and the signal density was analyzed by Quantity One software.
The CCL2 concentration in NML and PHSML was measured by rat-specific ELISA kits (CD35498, Wuhan Chundu Biotechnology Co., Ltd, Wuhan, China) in accordance with the manufacturer’s protocols after manufacturing standard curves. The optical density at 450 nm was read using the Microplate Reader.
In this study, data of HUVEC viability and CCL2 concentration were presented as mean ± standard deviation (SD), and the data of mRNA expressions were presented as mean ± standard error (SE). Statistical analysis was performed using SPSS 16.0 (Polar Engineering and Consulting Inc., Chicago, IL, United States). One-way analysis of variance was used between multiple groups, and a Student’s t-test was used between two groups. P < 0.05 was considered to indicate a statistically significant difference.
Between NML and PBS groups, there were only two differentially expressed genes, which were slightly down-regulated in the NML group (Figure 1A). Between the PHSML and PBS groups, 24 genes were up-regulated, with 3 genes down-regulated in the PHSML group (Figure 1B). Compared with the NML group, 32 genes were up-regulated and 5 genes were down-regulated in the PHSML group (Figure 1C). The differentially expressed genes are listed in Tables 3–5. These results indicated that NML had little effect on HUVECs, but PHSML induced a significant change in gene expression pattern. According to the FPKM value of differentially expressed genes, hierarchical clustering analysis further proved the above results from the Tables 3–5 (Figure 2), which show the consistency of differentially expressed genes. NML or PHSML can simulate the physiological or pathological processes of vascular endothelium after lymph return into the circulation system, meaning we need to further analyze the differences between the PHSML and NML groups in the following analysis.
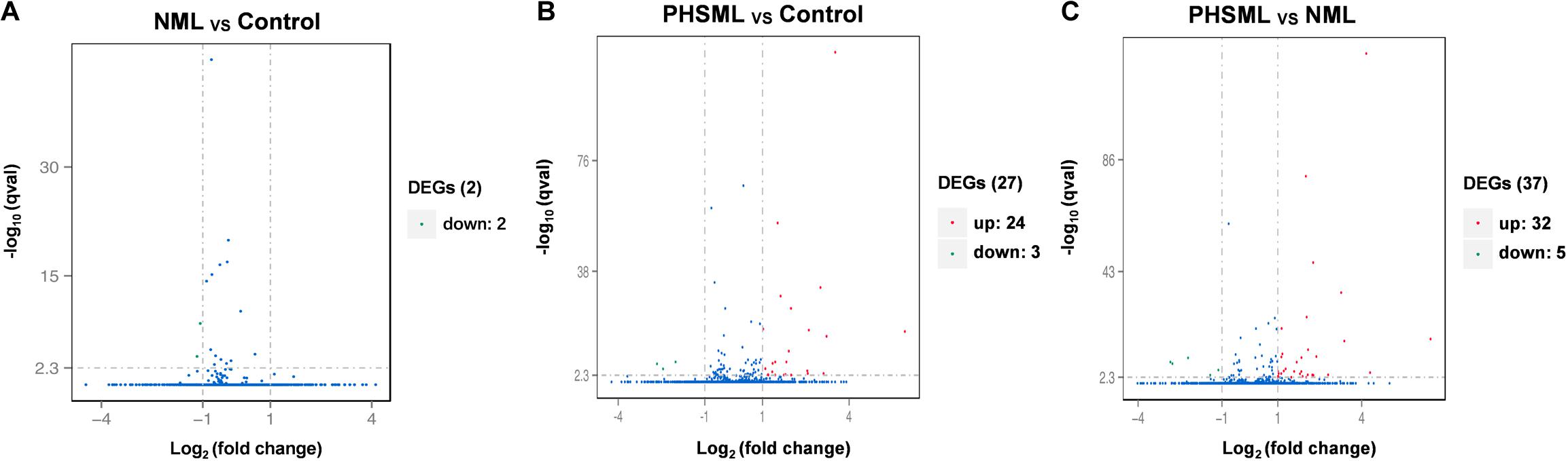
Figure 1. Volcano map of differentially expressed genes. HUVECs are treated by physiological saline (Control), normal mesenteric lymph (NML) and post-hemorrhagic shock mesenteric lymph (PHSML), respectively. (A) Distribution of differentially expressed genes between NML and Control groups. (B) Distribution of differentially expressed genes between PHSML and Control groups. (C) Distribution of differentially expressed genes between PHSML and NML groups. Green: down-regulated; Red: up-regulated, Blue: unchanged.

Table 3. Differentially expressed genes in NML compared to control.
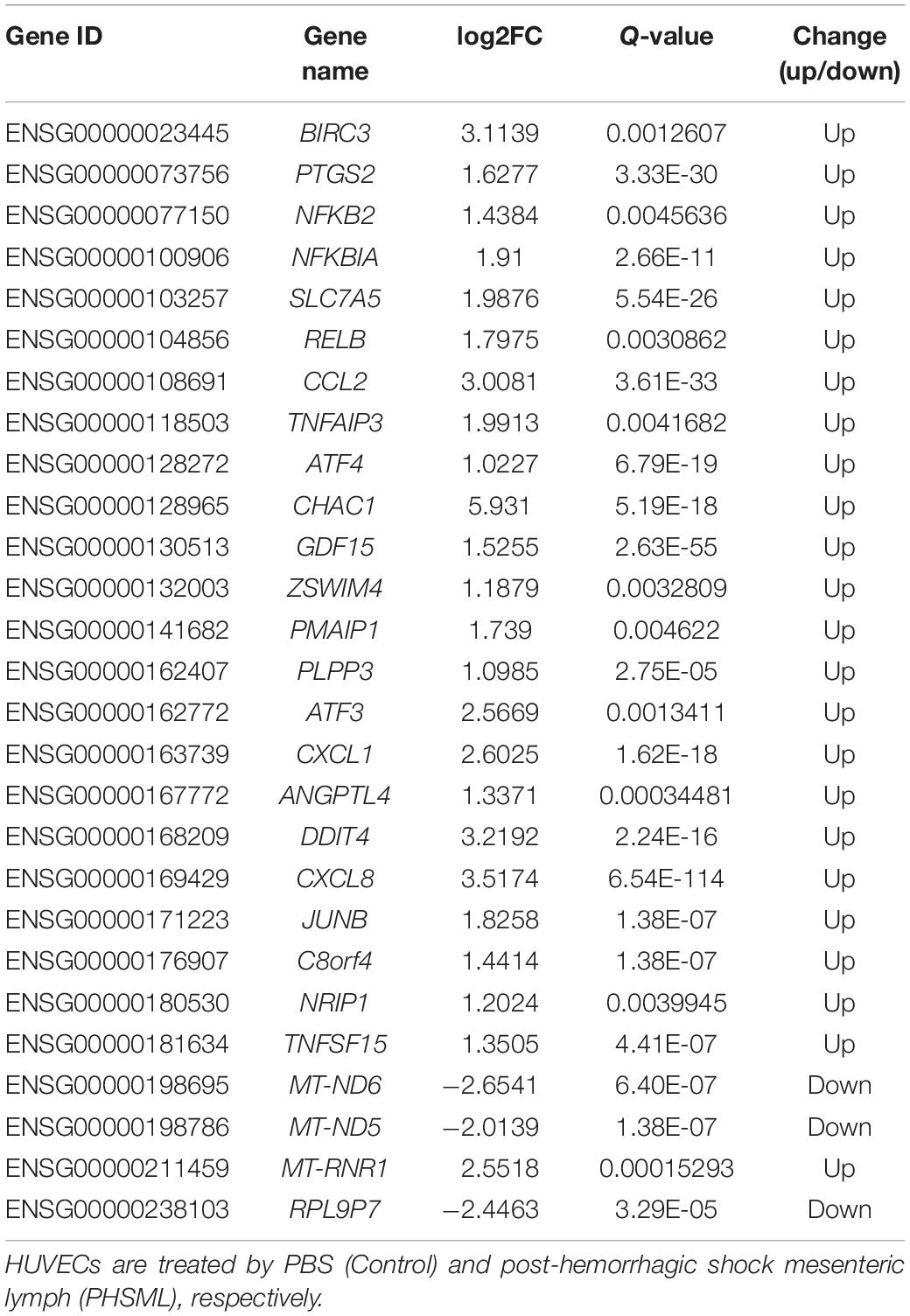
Table 4. Differentially expressed genes in PHSML compared to control.

Table 5. Differentially expressed genes in PHSML compared to NML.
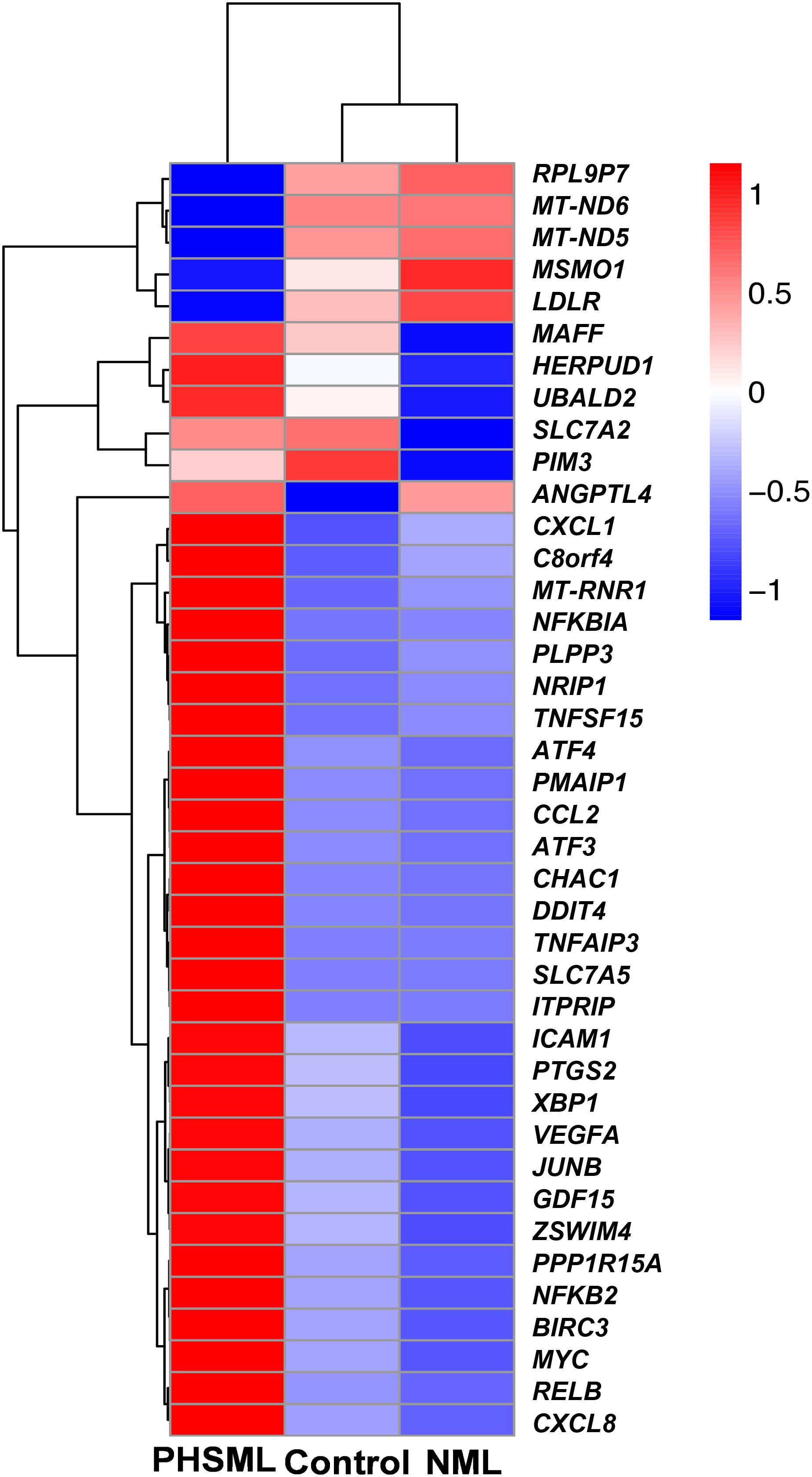
Figure 2. Heatmap of differentially expressed genes in various experimental groups. HUVECs are treated by physiological saline (Control), normal mesenteric lymph (NML), and post-hemorrhagic shock mesenteric lymph (PHSML), respectively.
Firstly, we used GOSeq software for GO enrichment analysis (11). The differentially expressed genes were enriched in 2378 biological processes. We selected 30 GO terms with the most significant enrichment for further analysis (Figure 3). These differentially expressed genes were mainly involved in cell death, response to stress, endoplasmic reticulum nuclear signaling pathway, organic reaction, positive regulation of macromolecule metabolism, adhesion signal, and inflammatory response. It was evident that PHSML may affect the survival of HUVECs.
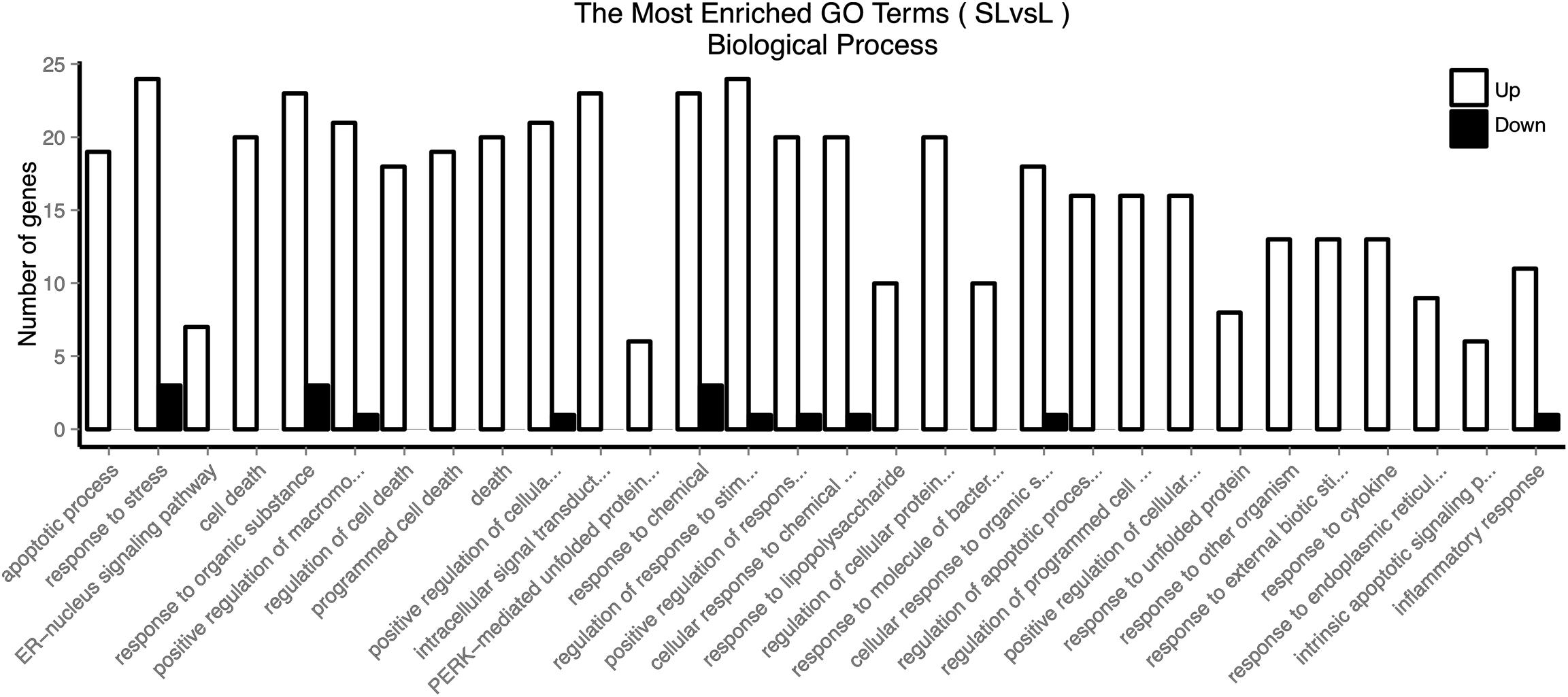
Figure 3. GO enriched biological processes of differentially expressed genes in HUVECs treated with NML and PHSML, respectively.
KEGG collects a large amount of signal pathway information, which can be used to analyze the network of intermolecular interactions (13). The differentially expressed genes between PHSML and NML enriched 105 signaling pathways in the KEGG database. Among the top 20 pathways, the nucleotide-binding oligomerization domain NOD-like receptor signaling pathway, nuclear factor (NF)-κB signaling pathway, and Tumor necrosis factor (TNF) signaling pathway each showed the highest degree of enrichment (Figure 4) respectively. Six differentially expressed genes (BIRC3, TNFAIP3, NFKBIA, CXCL8, CXCL1, and CCL2) were enriched into NOD-like receptor signaling pathway (Figure 5A). Eight differentially expressed genes (BIRC3, NFKBIA, NFKB2, RELB, CXCL8, TNFAIP3, PTGS2, and ICAM1) were enriched into the NF-κB signaling pathway (Figure 5B). Nine differentially expressed genes (BIRC3, NFKBIA, ATF4, CCL2, CXCL1, TNFAIP3, JUNB, ICAM1, and PTGS2) were enriched into the TNF signaling pathway (Figure 5C). Among them, cIAP1 and cIAP2 as regulators of innate immune signaling in mammals, induce robust RIP2 ubiquitination. Moreover, cIAP1 and cIAP2 function as K63 ubiquitin ligases for RIP1, which allows RIP1 to activate the prosurvival NF-kB pathway while preventing it from binding to caspase-8 and inducing apoptosis (14). TNFAIP3 (A20) leads to the ubiquitination of TRAF6 and the deubiquitination of RIP to inhibit the activation of the NF-κB signaling pathway that is mediated by NOD1 and NOD2 (15). Moreover, the genes for the chemokines (CXCL8, CXCL1, and CCL2) have inherent antimicrobial properties. These chemokines play an important role in neutrophil attraction and have been implicated in the prevention of infection in the lung epithelium. Interestingly, these three signaling pathways and the differentially expressed genes are mostly related to inflammation. Thus, excessive inflammation during hemorrhagic shock might further cause tissue damage. To validate the above analysis, seven genes (BIRC3, NFKBIA, CXCL1, ICAM1, CCL2, TNFAIP3, and JUNB) were randomly selected for quantitative real-time polymerase chain reaction (qRT-PCR) verification. The qRT-PCR results demonstrated that our sequencing data were reliable (Figure 6).
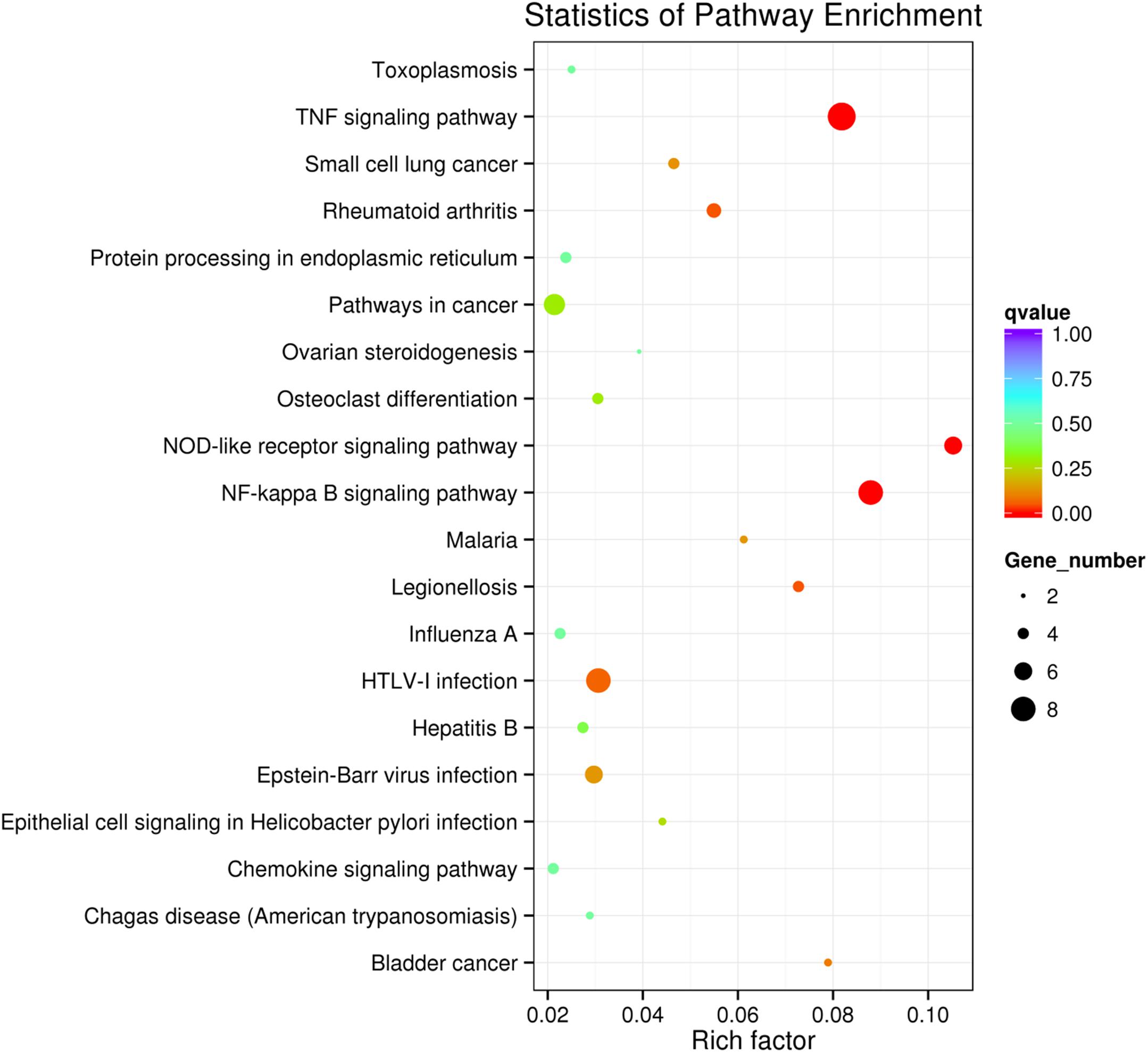
Figure 4. Scatter plot showing the enrichment of KEGG pathways (PHSML vs NML). HUVECs are treated by NML and PHSML, respectively. The size of dots represents the number of differentially expressed genes in this pathway. The point’s color represents the range of the q-value (the closer to zero, the more significant the enrichment is).
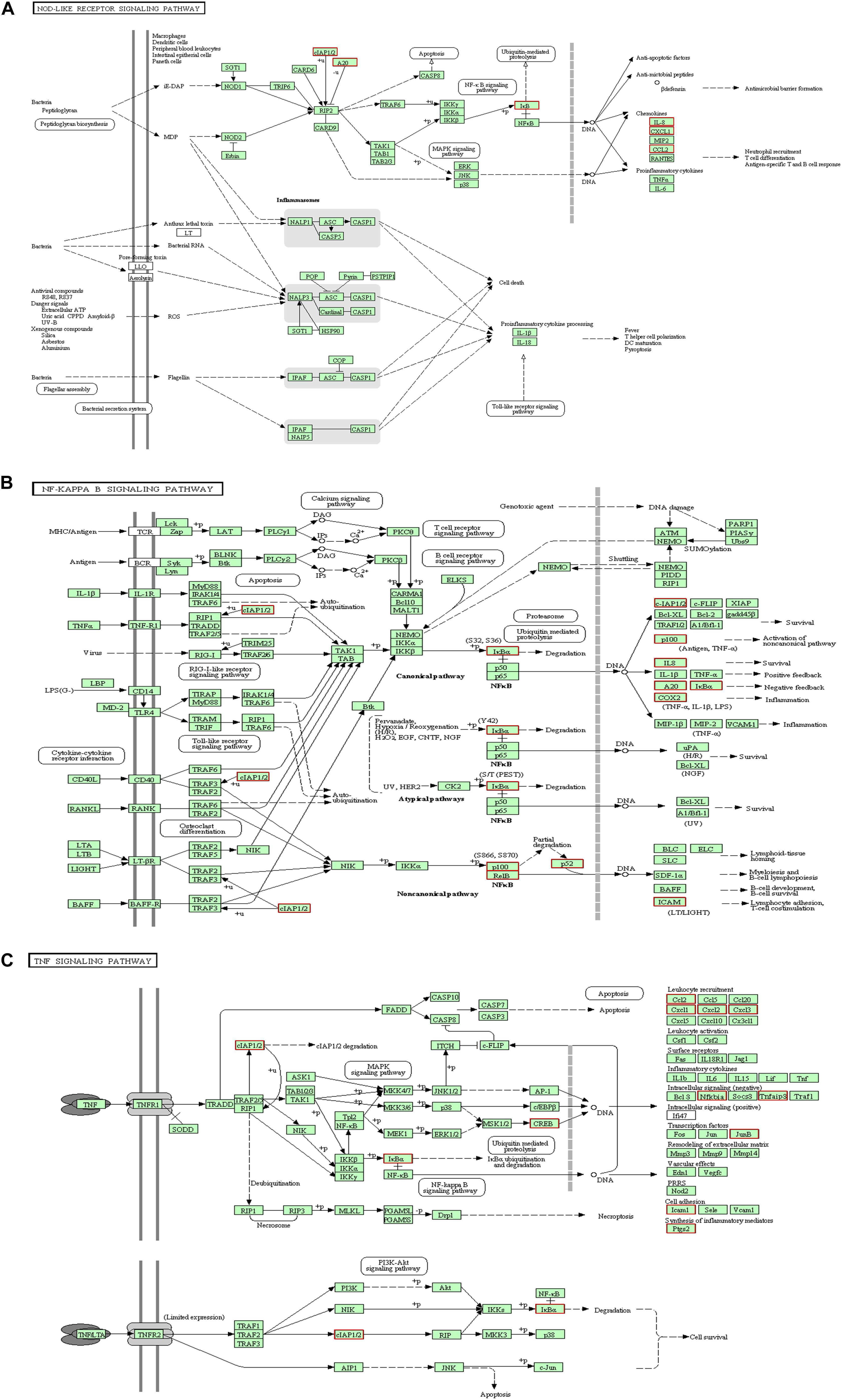
Figure 5. KEGG analysis for signaling pathways. The red box indicates that the up-regulated genes (PHSML vs NML). (A) NOD like receptor signaling pathway. (B) NF-κB signaling pathway. (C) TNF signaling pathway.
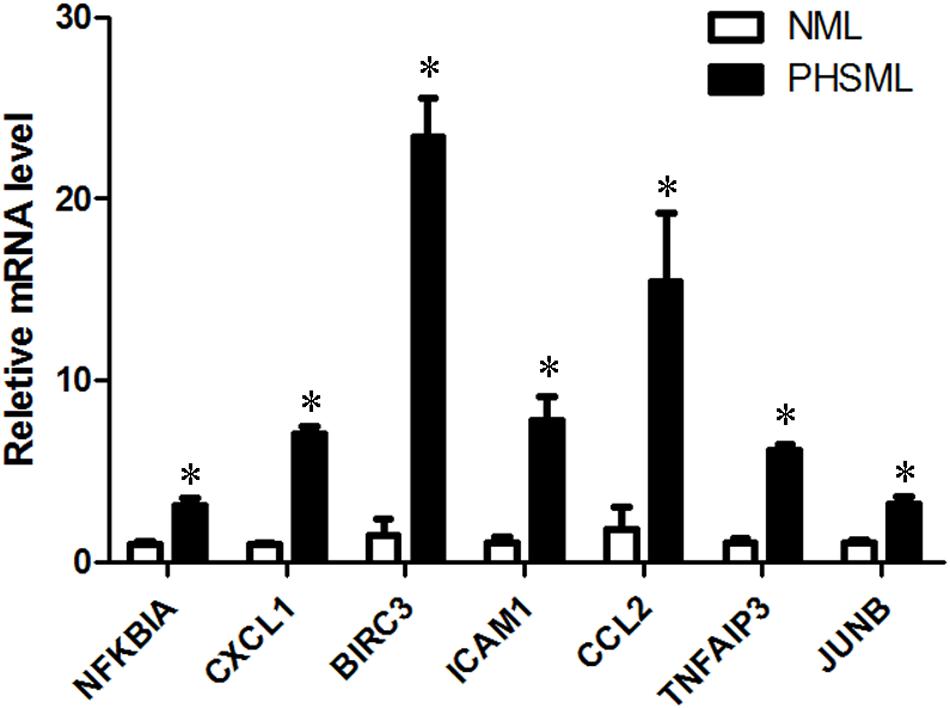
Figure 6. The verification of up-regulated genes (PHSML vs NML) by qRT-PCR. The data were shown as fold change, and expressed as mean ± SE (n = 3), ∗P < 0.05. All the original values of these seven genes in NML group were normalized to 1, respectively.
The above results of RNA-seq and qRT-PCR showed that CCL2 was significantly increased after PHSML treatment, indicating it may play an important role in PHSML stimulated HUVEC injury. Therefore, CCL2 production inhibitor Bindarit might attenuate the cell injury by PHSML, and detected the effects of Bindarit on cell morphology and viability.
Compared with control cells, PHSML treated cells shrank obviously after 24-h incubation, with the appearance of cell debris (Figure 7A). Thus, PHSML treatment for 24 h induced the morphological injury and decreased viability of HUVECs significantly. We found the morphology of HUVECs was partially improved with the increase of Bindarit concentration (Figure 7A). Meanwhile, the results were further confirmed using a CCK8 assay to detect cell viability. When the concentration of Bindarit reached 450 and 600 μM, cell viability was significantly increased (Figure 7B). Quantitative PCR showed the mRNA expression of CCL2 was significantly decreased by Bindarit treatment (Figure 7C). Furthermore, Western blot results confirmed that the PHSML-induced CCL2 expression was inhibited by Bindarit, indicating the inhibitor was effective (Figures 7D,E). Therefore, Bindarit, an inhibitor of CCL2 production, could alleviate the damage of HUVECs by PHSML.
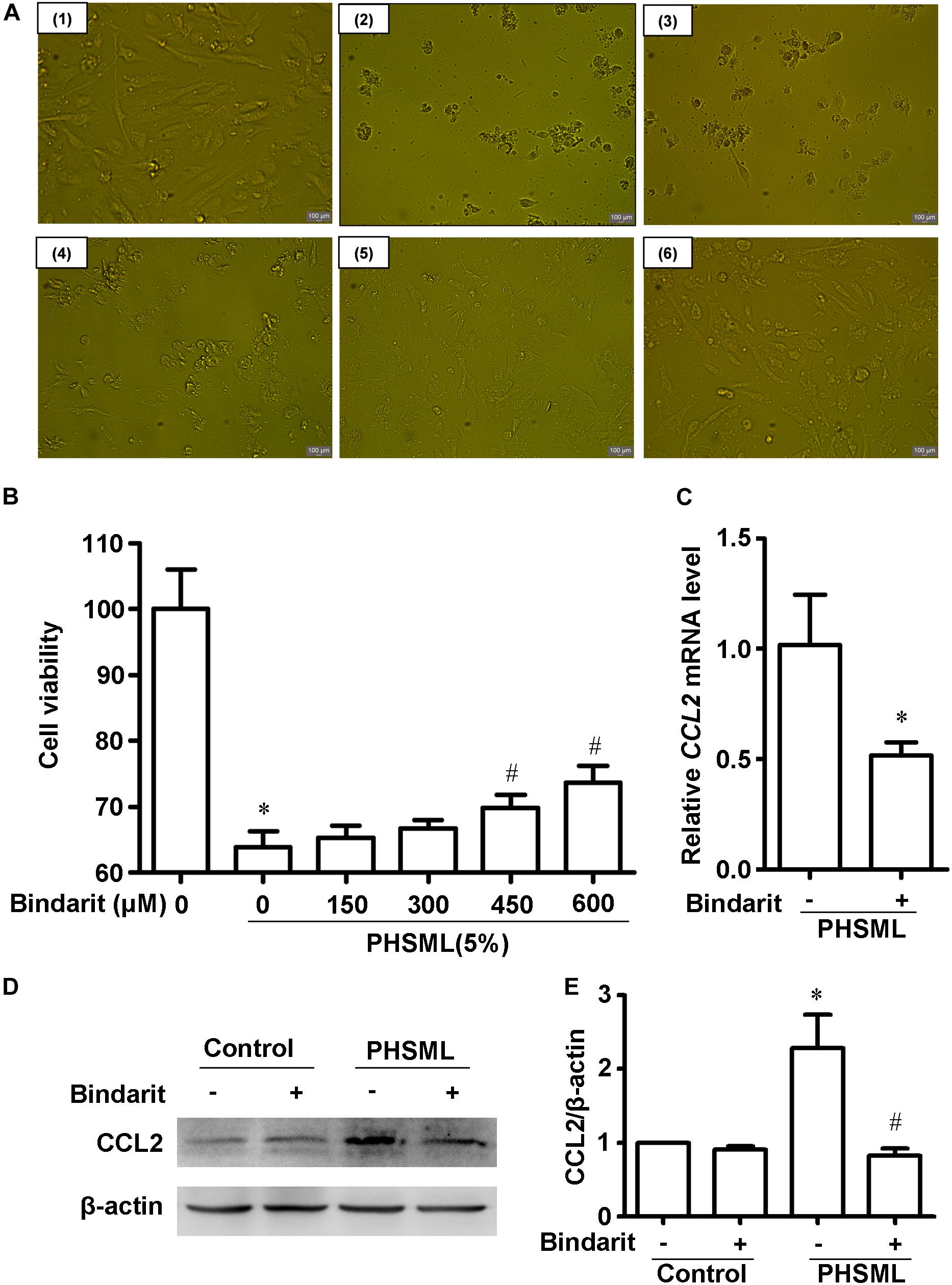
Figure 7. Effect of Bindarit on PHSML-induced HUVECs injury. (A) The effect of Bindarit on the morphology of HUVECs treated by PHSML for 24 h. (1) Control; (2) 5% PHSML; (3) 5% PHSML + 150 μM Bindarit; (4) 5% PHSML + 300 μM Bindarit; (5) 5% PHSML + 450 μM Bindarit; (6) 5% PHSML + 600 μM Bindarit. (B) The effect of Bindarit on the viability of HUVECs treated by PHSML for 24 h. The data is expressed as mean ± SD (n = 10). ∗P < 0.05, compared with the control group; #P < 0.05, compared with the PHSML group. (C) The effect of Bindarit (300 μM) on CCL2 mRNA expression in HUVECs treated with PHSML. The expression was calculated by 2–ΔΔCt method. The data is expressed as mean ± SE (n = 3), ∗P < 0.05, compared with PHSML group. (D) Western blot analysis showed decreased expression of CCL2 protein after Bindarit treatment. (E) Quantitative analysis of Western blot results from panel (D). The data is expressed as mean ± SE (n = 3). ∗P < 0.05, compared with the control group; #P < 0.05, compared with the PHSML group.
The result from the ELISA assay showed that there were no statistical differences in CCL2 level between the PHSML (84.65 ± 5.16 ng/ml) and NML (81.42 ± 6.83 ng/ml).
Hemorrhagic shock increases vascular permeability and induces organ damage, which is related to mesenteric lymph return to the blood circulation system (16). Plasma constituents and blood cells can pass through the vascular endothelial barrier through cellular and paracellular pathways (17). It has been shown that PHSML treatment increased the permeability of HUVECs when the concentration of shock lymph reached 10% in a culture medium, while shock venous serum had no effects (4, 5). In order to dissect the molecular mechanism of PHSML injury on endothelial cells, we performed RNA-seq on HUVECs incubated with 10% PBS, 10% NML, and 10% PHSML, respectively. Furthermore, we verified the mediating role of CCL2, a representative gene among differentially expressed genes, in the impairment of HUVECs induced by PHSML.
According to the data of the RNA-seq, PHSML-treated HUVECs exhibited significant changes in 37 gene expression, while expressed genes in PBS and NML group showed a similar pattern with no changes. We found that these differentially expressed genes are highly enriched in the NOD-like receptor signaling pathway, the NF-κB signaling pathway, and the TNF signaling pathway in the KEGG database. These pathways are closely related to inflammation and immune response, and crosstalk among the three pathways exists. NOD-like receptors are cytoplasmic pattern recognition receptors that contribute to the activation of inflammatory caspases and have been shown to link to inflammatory responses, immune defense, and tissue injury in models of hemorrhagic shock (18–20). Moreover, NOD-like receptors can activate the NF-κB signaling pathway when they recognize danger-related signaling molecules and pathogen-related signaling molecules (21, 22), following hemorrhagic shock (20). TNF is a multifunctional cytokine family that plays an important role in immunity, inflammation, cell proliferation, and differentiation (23). It has been demonstrated that TNF-α is a major cause of lung endothelial injury after lung ischemia-reperfusion (21, 24), which is consistent with the lung injury induced by hemorrhagic shock (25). Furthermore, TNF-α is an important mediator of endothelial dysfunction through the activation of NF-κB and upregulation of NF-κB components (26, 27). Therefore, both the NOD-like receptor signaling pathway and the TNF signaling pathway are involved in NF-κB pathway activation. When cells are in a state of stress and injury, NF-κB, as a coordinator, participates in the expression of various cytokines and adhesion molecules and enhances the immune response. Importantly, the NOD-like receptor signaling pathway (28, 29), the NF-κB signaling pathway (30–32), and TNF signaling pathway (33–35) are all involved in endothelial cell injury. Thus, the inter-related three pathways may form a complicated network that synergistically induces inflammatory injury.
PHSML return plays an important role in the pathogenesis of hemorrhagic shock-induced orange injuries and inflammation. Previous studies have demonstrated that PHSML treatment causes NF-κB activation and I-κB degradation and induces human pulmonary endothelial cells damage (36), intravenous injection of PHSML significantly increased the levels of TNF-α in the plasma of the lung, kidney, etc. (37). The activation of the NOD-like receptor pyrin domain containing-3 (NLRP3) inflammasome is involved in hemorrhagic shock-induced acute lung injury and lung endothelial cells damage through inflammatory response and cell pyroptosis (19, 38, 39). All these factors are related to PHSML effects that need to be determined in future research. Previous studies have demonstrated that NLRP3 siRNA transfection abolished palmitate-induced IL-1β release in HUVECs (40). Blockade of NF-κB activation with a specific inhibitor, BAY 11-7085, partially overthrown gamma radiation-induced cellular hyper-permeability, decreased viability, and excessive inflammation in HUVECs (41). Inhibition of TNF-α with adalimumab also attenuated inflammatory responses in long-term HUVEC cultures (42). These results have confirmed that all NOD-like receptors, NF-κB, and TNF pathways participate in the HUVECs damage induced by various pathogenic factors.
Interestingly, in three groups of differentially expressed genes related to the aforementioned three pathways, several genes that can inhibit inflammation were also altered after PHSML treatment. BIRC3 is a member of the inhibitor of apoptosis family and can bind to TNF receptors TRAF1 and TRAF2 (43). TNFAIP3 is a zinc-containing protein. When some innate immune receptors, such as NOD-like receptors and TNFR, activate the NF-κB signaling pathway, the OTU domain of TNFAIP3 can induce deubiquitination of many related factors such as TRAF6 of the NF-κB signaling pathway, and inhibit the inflammatory response. It has been reported that after renal ischemia-reperfusion, TNFAIP3 can alleviate renal tissue injury by inhibiting the NF-κB signaling pathway and anti-inflammatory reaction (44). To date there are no reports on the role of TNFAIP3 in the inflammatory response after hemorrhagic shock. It can be speculated that the increased TNFAIP3 might be a compensatory response in HUVECs following PHSML treatment. Similarly, NFKBIA, which is considered an anti-inflammatory protein because it inhibits NF-κB (45), was also upregulated. Therefore, further investigation of these anti-inflammatory genes and how they are altered during PHSML treatment warrants further attention in future studies.
Chemokines are small inflammatory proteins that can induce leukocytes to express integrins, recruit leukocytes, and produce superoxide anions. These functions are mainly the first defense against microbial invasion and tissue damage (46). However, when the homeostasis of these molecules is unbalanced and maintains a high level, numerous inflammatory cells will be recruited to the injured site, which expands the inflammatory response and causes more severe tissue damage. According to the location of N-terminal cysteine residues, these factors are divided into four categories: C, CC, CXC, and CX3C (47). In our study, CXCL1, CXCL8, and CCL2 are significantly increased after PHSML treatment. These chemokines play an important role in the development of inflammation. After hepatic hemorrhage and reperfusion, CXCL1 expression was increased, and a large number of neutrophils were recruited to the liver, resulting in aggravation of liver injury (48). Repertaxin, an inhibitor of the CXCL8 receptor, alleviates this inflammatory response, and intestinal ischemia-reperfusion injury (49). After hepatic hemorrhage and reperfusion, the inflammatory response of CCL2-deficient mice was inhibited and the liver was protected (50). In light of these results, it is reasonable to conceive that PHSML may cause tissue damage by inducing pro-inflammatory chemokines of an endothelial cell.
As a pro-inflammatory chemokine, CCL2 is regulated by NF-kB in human aortic endothelial cells (51). Our results of RNA-seq have shown that CCL2 is upregulated by PHSML in HUVECs. Since PHSML-treated HUVECs showed enriched NF-kB signaling genes, CCL2 may be conservatively regulated by NF-κB in endothelial cells. Furthermore, we found that Bindarit, an inhibitor of CCL2 production (52), could protect HUVECs in survival activity and cytomorphology. Therefore, targeting differentially expressed genes can alleviate the damage of PHSML on HUVECs.
PHSML is cytotoxic to endothelial cells, including increased HUVEC permeability and decreased cell viability (53). In our study, we further identified three inflammatory pathways using RNA-seq and found that CCL2 participated in PHSML-induced endothelial cell injury. This result suggests a new strategy for the treatment of PHSML-induced organ injury by targeting the CCL2. However, the role of CCL2 on the secretory or barrier function of endothelial cells treated by PHSML needs to be investigated in the future.
In the present study, to clarify whether CCL2 in PHSML causes HUVECs damage, we performed an animal experiment to collect the mesenteric lymph and tested the CCL2 levels in NML and PHSML using the ELISA method. The result showed that there was no difference in the levels of CCL2 between NML and PHSML, suggesting that the HUVECs damage was not induced by CCL2 payload in PHSML. The component in PHSML that induces cellular injury requires further investigation. It should be noted that many scholars have determined the biologically active component analysis in PHSML that leads to cell damage and shock deterioration using various experimental technologies, for the potential therapeutic interventions of hemorrhagic shock targeting PHSML. Although these studies have identified a number of components carried in PHSML, further studies should be done, targeting the active factors and toxic substances contained in PHSML, a long and hard process.
It should be pointed out that there were certain limitations in the current study. Firstly, the present study demonstrated the role of Bindarit treatment in reducing the expression of CCL2 and in enhancing the viability of HUVECS. However, the effects of Bindarit treatment on the expressions of primary signaling molecules in the NOD-like receptor, NF-κB, and TNF signaling pathways were not determined. Secondly, the present study did not observe the roles of CCL2 inhibitors in in vivo animals following hemorrhagic shock. These issues warrant further investigation and could provide fundamental information for expanding clinical practice and targeting CCL2 in the treatment of hemorrhagic shock in the future.
Taken together, the findings of the present study have demonstrated that the altered genes in HUVECs treated with PHSML were involved in many pathways, including the NOD-like receptor signaling pathway, NF-κB signaling pathway, and TNF signaling pathway, which were most enriched. These pathways and the differentially expressed genes are closely related to inflammation, while inhibition of pro-inflammatory CCL2 can effectively alleviate the damage of PHSML to HUVECs. These results indicate a novel molecular mechanism responsible for the adverse effect of PHSML on vascular endothelium, which may represent a potential target in the treatment of hemorrhagic shock.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA602776/.
The animal study was reviewed and approved by Animal Ethics Committee of Hebei North University.
Z-GZ, C-YN, and L-HN conceived and designed the work. QW, Z-FC, DW, Z-AZ, and HZ acquired and analyzed data for the work. L-MZ, Y-XL, A-LK, MZ, and PW participated in the animal experiments. QW, Z-AZ, and Z-GZ wrote the manuscript. Z-GZ and C-YN revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (Nos. 81670446 and 31371346) and the Natural Science Foundation of Hebei Province (H2019405093).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Deitch EA. Role of the gut lymphatic system in multiple organ failure. Curr Opin Crit Care. (2001) 7:92–8. doi: 10.1097/00075198-200104000-00007
2. Moore EE. Mesenteric lymph: the critical bridge between dysfunctional gut and multiple organ failure. Shock. (1998) 10:415–6.
3. Deitch EA. Gut-origin sepsis: evolution of a concept. Surgeon. (2012) 10:350–6. doi: 10.1016/j.surge.2012.03.003
4. Sun GX, Guo YX, Zhang YP, Zhang LM, Zhao ZG, Niu CY. Posthemorrhagic shock mesenteric lymph enhances monolayer permeability via F-actin and VE-cadherin. J Surg Res. (2016) 203:47–55. doi: 10.1016/j.jss.2016.01.034
5. Deitch EA, Adams CA, Lu Q, Xu DZ. Mesenteric lymph from rats subjected to trauma-hemorrhagic shock are injurious to rat pulmonary microvascular endothelial cells as well as human umbilical vein endothelial cells. Shock. (2001) 16:290–3. doi: 10.1097/00024382-200116040-00010
6. Gonzalez RJ, Moore EE, Ciesla DJ, Nieto JR, Johnson JL, Silliman CC. Post-hemorrhagic shock mesenteric lymph activates human pulmonary microvascular endothelium for in vitro neutrophil-mediated injury: the role of intercellular adhesion molecule-1. J Trauma. (2003) 54:219–23. doi: 10.1097/01.TA.0000047807.12644.95
7. Zhao M, Liu D, Qu H. Systematic review of next-generation sequencing simulators: computational tools, features and perspectives. Brief Funct Genomics. (2017) 16:121–8. doi: 10.1093/bfgp/elw012
8. Ong SP, Ng ML, Chu JJ. Differential regulation of angiopoietin 1 and angiopoietin 2 during dengue virus infection of human umbilical vein endothelial cells: implications for endothelial hyperpermeability. Med Microbiol Immunol. (2013) 202:437–52. doi: 10.1007/s00430-013-0310-5
9. Watts JA, Grattan RM II, Whitlow BS, Kline JA. Activation of poly(ADP-ribose) polymerase in severe hemorrhagic shock and resuscitation. Am J Physiol Gastrointest Liver Physiol. (2001) 281:G498–506. doi: 10.1152/ajpgi.2001.281.2.G498
10. Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. (2010) 26:136–8. doi: 10.1093/bioinformatics/btp612
11. Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. (2010) 11:R14. doi: 10.1186/gb-2010-11-2-r14
12. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. (2001) 25:402–8. doi: 10.1006/meth.2001.1262
13. Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. (2004) 32:D277–80. doi: 10.1093/nar/gkh063
14. Bertrand MJ, Doiron K, Labbe K, Korneluk RG, Barker PA, Saleh M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity. (2009) 30:789–801. doi: 10.1016/j.immuni.2009.04.011
15. Ma D, Liu Q, Zhang M, Feng J, Li X, Zhou Y, et al. iTRAQ-based quantitative proteomics analysis of the spleen reveals innate immunity and cell death pathways associated with heat stress in broilers (Gallus gallus). J Proteomics. (2019) 196:11–21. doi: 10.1016/j.jprot.2019.01.012
16. Deitch EA. Gut lymph and lymphatics: a source of factors leading to organ injury and dysfunction. Ann N Y Acad Sci. (2010) 1207(Suppl. 1):E103–11. doi: 10.1111/j.1749-6632.2010.05713.x
17. Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. (2008) 121:2115–22. doi: 10.1242/jcs.017897
18. Xu P, Wen Z, Shi X, Li Y, Fan L, Xiang M, et al. Hemorrhagic shock augments Nlrp3 inflammasome activation in the lung through impaired pyrin induction. J Immunol. (2013) 190:5247–55. doi: 10.4049/jimmunol.1203182
19. Yang J, Zhao Y, Zhang P, Li Y, Yang Y, Yang Y, et al. Hemorrhagic shock primes for lung vascular endothelial cell pyroptosis: role in pulmonary inflammation following LPS. Cell Death Dis. (2016) 7:e2363. doi: 10.1038/cddis.2016.274
20. Wen Z, Fan L, Li Y, Zou Z, Scott MJ, Xiao G, et al. Neutrophils counteract autophagy-mediated anti-inflammatory mechanisms in alveolar macrophage: role in posthemorrhagic shock acute lung inflammation. J Immunol. (2014) 193:4623–33. doi: 10.4049/jimmunol.1400899
21. Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. (2009) 227:106–28. doi: 10.1111/j.1600-065X.2008.00734.x
22. Kufer TA, Banks DJ, Philpott DJ. Innate immune sensing of microbes by Nod proteins. Ann N Y Acad Sci. (2006) 1072:19–27. doi: 10.1196/annals.1326.020
23. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. (2003) 114:181–90. doi: 10.1016/s0092-8674(03)00521-x
24. Khimenko PL, Bagby GJ, Fuseler J, Taylor AE. Tumor necrosis factor-alpha in ischemia and reperfusion injury in rat lungs. J Appl Physiol. (1998) 85:2005–11. doi: 10.1152/jappl.1998.85.6.2005
25. Cagliani J, Yang WL, McGinn JT, Wang Z, Wang P. Anti-interferon-alpha receptor 1 antibodies attenuate inflammation and organ injury following hemorrhagic shock. J Trauma Acute Care Surg. (2019) 86:881–90. doi: 10.1097/TA.0000000000002214
26. Zhang LN, Gong WD, Luo J, Yu YJ, Qi SH, Yue ZY. Exogenous ghrelin ameliorates acute lung injury by modulating the nuclear factor kappaB inhibitor kinase/nuclear factor kappaB inhibitor/nuclear factor kappaB pathway after hemorrhagic shock. Int Immunopharmacol. (2019) 69:95–102. doi: 10.1016/j.intimp.2019.01.019
27. Wang J, Yang G, Li M, Zhou X. Transient receptor potential melastatin 8 (TRPM8)-based mechanisms underlie both the cold temperature-induced inflammatory reactions and the synergistic effect of cigarette smoke in human bronchial epithelial (16HBE) cells. Front Physiol. (2019) 10:285. doi: 10.3389/fphys.2019.00285
28. Nagyoszi P, Nyul-Toth A, Fazakas C, Wilhelm I, Kozma M, Molnar J, et al. Regulation of NOD-like receptors and inflammasome activation in cerebral endothelial cells. J Neurochem. (2015) 135:551–64. doi: 10.1111/jnc.13197
29. Chen Z, Martin M, Li Z, Shyy JY. Endothelial dysfunction: the role of sterol regulatory element-binding protein-induced NOD-like receptor family pyrin domain-containing protein 3 inflammasome in atherosclerosis. Curr Opin Lipidol. (2014) 25:339–49. doi: 10.1097/MOL.0000000000000107
30. Sehnert B, Burkhardt H, Wessels JT, Schroder A, May MJ, Vestweber D, et al. NF-kappaB inhibitor targeted to activated endothelium demonstrates a critical role of endothelial NF-kappaB in immune-mediated diseases. Proc Natl Acad Sci USA. (2013) 110:16556–61. doi: 10.1073/pnas.1218219110
31. Kempe S, Kestler H, Lasar A, Wirth T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. (2005) 33:5308–19. doi: 10.1093/nar/gki836
32. Rahman A, Fazal F. Blocking NF-kappaB: an inflammatory issue. Proc Am Thorac Soc. (2011) 8:497–503. doi: 10.1513/pats.201101-009MW
33. Zhou P, Lu S, Luo Y, Wang S, Yang K, Zhai Y, et al. Attenuation of TNF-alpha-induced inflammatory injury in endothelial cells by ginsenoside Rb1 via inhibiting NF-kappaB, JNK and p38 signaling pathways. Front Pharmacol. (2017) 8:464. doi: 10.3389/fphar.2017.00464
34. Sawant DA, Tharakan B, Wilson RL, Stagg HW, Hunter FA, Childs, EW. Regulation of tumor necrosis factor-alpha-induced microvascular endothelial cell hyperpermeability by recombinant B-cell lymphoma-extra large. J Surg Res. (2013) 184:628–37. doi: 10.1016/j.jss.2013.04.079
35. Rastogi S, Rizwani W, Joshi B, Kunigal S, Chellappan SP. TNF-alpha response of vascular endothelial and vascular smooth muscle cells involve differential utilization of ASK1 kinase and p73. Cell Death Differ. (2012) 19:274–83. doi: 10.1038/cdd.2011.93
36. Damle SS, Moore EE, Nydam TL, Banerjee M, Gamboni-Robertson F, Su X, et al. Postshock mesenteric lymph induces endothelial NF-kappaB activation. J Surg Res. (2007) 143:136–40. doi: 10.1016/j.jss.2007.04.016
37. Zhao Y, Zhang L, Han R, Si Y, Zhao Z. Intravenous injection of post-hemorrhagic shock mesenteric lymph induces multiple organ injury in rats. Exp Ther Med. (2019) 17:1449–55. doi: 10.3892/etm.2018.7048
38. Xiang M, Shi X, Li Y, Xu J, Yin L, Xiao G, et al. Hemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cells. J Immunol. (2011) 187:4809–17. doi: 10.4049/jimmunol.1102093
39. Bortolotti P, Faure E, Kipnis E. Inflammasomes in tissue damages and immune disorders after trauma. Front Immunol. (2018) 9:1900. doi: 10.3389/fimmu.2018.01900
40. Xing JH, Li R, Gao YQ, Wang MY, Liu YZ, Hong J, et al. NLRP3 inflammasome mediate palmitate-induced endothelial dysfunction. Life Sci. (2019) 239:116882. doi: 10.1016/j.lfs.2019.116882
41. Wang H, Segaran RC, Chan LY, Aladresi AAM, Chinnathambi A, Alharbi SA, et al. Gamma radiation-induced disruption of cellular junctions in HUVECs is mediated through affecting MAPK/NF-kappaB inflammatory pathways. Oxid Med Cell Longev. (2019) 2019:1486232. doi: 10.1155/2019/1486232
42. Prattichizzo F, Giuliani A, Recchioni R, Bonafe M, Marcheselli F, De Carolis S, et al. Anti-TNF-alpha treatment modulates SASP and SASP-related microRNAs in endothelial cells and in circulating angiogenic cells. Oncotarget. (2016) 7:11945–58. doi: 10.18632/oncotarget.7858
43. Yanai A, Hirata Y, Mitsuno Y, Maeda S, Shibata W, Akanuma M, et al. Helicobacter pylori induces antiapoptosis through buclear factor-kappaB activation. J Infect Dis. (2003) 188:1741–51. doi: 10.1086/379629
44. Wei L, Zhang X, Ye Q, Yang Y, Chen X. The transfection of A20 gene prevents kidney from ischemia reperfusion injury in rats. Mol Med Rep. (2017) 16:1486–92. doi: 10.3892/mmr.2017.6725
45. You R, Liu YP, Lin DC, Li Q, Yu T, Zou X, et al. Clonal mutations activate the NF-kappaB pathway to promote recurrence of nasopharyngeal carcinoma. Cancer Res. (2019) 79:5930–43. doi: 10.1158/0008-5472.CAN-18-3845
46. Chen K, Bao Z, Tang P, Gong W, Yoshimura T, Wang JM. Chemokines in homeostasis and diseases. Cell Mol Immunol. (2018) 15:324–34. doi: 10.1038/cmi.2017.134
47. van der Vorst EP, Döring Y, Weber C. Chemokines and their receptors in Atherosclerosis. J Mol Med. (2015) 93:963–71. doi: 10.1007/s00109-015-1317-8
48. Wang S, Yang FJ, Wang X, Zhou Y, Dai B, Han B, et al. PARP-1 promotes tumor recurrence after warm ischemic liver graft transplantation via neutrophil recruitment and polarization. Oncotarget. (2017) 8:88918–33. doi: 10.18632/oncotarget.21493
49. Souza DG, Bertini R, Vieira AT, Cunha FQ, Poole S, Allegretti M, et al. Repertaxin, a novel inhibitor of rat CXCR2 function, inhibits inflammatory responses that follow intestinal ischaemia and reperfusion injury. Br J Pharmacol. (2004) 143:132–42. doi: 10.1038/sj.bjp.0705862
50. Song P, Zhang J, Zhang Y, Shu Z, Xu P, He L, et al. Hepatic recruitment of CD11b+Ly6C+ inflammatory monocytes promotes hepatic ischemia/reperfusion injury. Int J Mol Med. (2018) 41:935–45. doi: 10.3892/ijmm.2017.3315
51. Aljada A, Ghanim H, Saadeh R, Dandona P. Insulin inhibits NFkappaB and MCP-1 expression in human aortic endothelial cells. J Clin Endocrinol Metab. (2001) 86:450–3. doi: 10.1210/jcem.86.1.7278
52. Paccosi S, Musilli C, Mangano G, Guglielmotti A, Parenti A. The monocyte chemotactic protein synthesis inhibitor bindarit prevents mesangial cell proliferation and extracellular matrix remodeling. Pharmacol Res. (2012) 66:526–35. doi: 10.1016/j.phrs.2012.09.006
Keywords: hemorrhagic shock, mesenteric lymph, endothelial cell, inflammation, C–C motif chemokine ligand 2
Citation: Wang Q, Chi Z-F, Wei D, Zhao Z-A, Zhang H, Zhang L-M, Liu Y-X, Kang A-L, Zhao M, Wang P, Nie L-H, Niu C-Y and Zhao Z-G (2020) Transcriptome Analysis Revealed Inflammation Is Involved in the Impairment of Human Umbilical Vein Endothelial Cells Induced by Post-hemorrhagic Shock Mesenteric Lymph. Front. Immunol. 11:1717. doi: 10.3389/fimmu.2020.01717
Received: 17 April 2020; Accepted: 29 June 2020;
Published: 09 September 2020.
Edited by:
Haichao Wang, Feinstein Institute for Medical Research, United StatesReviewed by:
Yong Ming Yao, First Affiliated Hospital of Chinese PLA General Hospital, ChinaCopyright © 2020 Wang, Chi, Wei, Zhao, Zhang, Zhang, Liu, Kang, Zhao, Wang, Nie, Niu and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chun-Yu Niu, bmN5bHhmQDEyNi5jb20=; Zi-Gang Zhao, emdoeWxAMTI2LmNvbQ==; Ling-Hu Nie, NzYyMjIwOEBxcS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.