 Oliver Nicolai1†
Oliver Nicolai1† Christian Pötschke1†
Christian Pötschke1† Dina Raafat1,2
Dina Raafat1,2 Julia van der Linde3
Julia van der Linde3 Sandra Quosdorf1
Sandra Quosdorf1 Anna Laqua1†Claus-Dieter Heidecke3
Anna Laqua1†Claus-Dieter Heidecke3 Claudia Berek4
Claudia Berek4 Murthy N. Darisipudi1
Murthy N. Darisipudi1 Christoph J. Binder5
Christoph J. Binder5 Barbara M. Bröker1*
Barbara M. Bröker1*- 1Department of Immunology, Institute of Immunology and Transfusion Medicine, University Medicine Greifswald, Greifswald, Germany
- 2Department of Microbiology and Immunology, Faculty of Pharmacy, Alexandria University, Alexandria, Egypt
- 3Department of General Surgery, Visceral, Thoracic and Vascular Surgery, University Medicine Greifswald, Greifswald, Germany
- 4German Rheumatism Research Centre (DRFZ), Berlin, Germany
- 5Department of Laboratory Medicine, Medical University of Vienna, Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria
In murine abdominal sepsis by colon ascendens stent peritonitis (CASP), a strong increase in serum IgM and IgG antibodies was observed, which reached maximum values 14 days following sepsis induction. The specificity of this antibody response was studied in serum and at the single cell level using a broad panel of bacterial, sepsis-unrelated as well as self-antigens. Whereas an antibacterial IgM/IgG response was rarely observed, studies at the single-cell level revealed that IgM antibodies, in particular, were largely polyreactive. Interestingly, at least 16% of the IgM mAbs and 20% of the IgG mAbs derived from post-septic mice showed specificity for oxidation-specific epitopes (OSEs), which are known targets of the innate/adaptive immune response. This identifies those self-antigens as the main target of B cell responses in sepsis.
Introduction
Sepsis, by definition, is a life-threatening organ dysfunction caused by a dysregulated host response to infection (1, 2), which is associated with high morbidity and mortality (3, 4). Due to an aging population, a steady increase in surgical interventions, and the occurrence of antibiotic resistance, sepsis is still of high clinical relevance (4–6).
B cells have been ascribed a protective function in sepsis which encompasses antibody-dependent as well as -independent mechanisms (7, 8). However, during sepsis, a large number of B cells and other immune cells are lost by apoptosis (9, 10) and it is assumed that B cell responses are severely impaired (11, 12). In fact, Mohr et al. have shown that B cell priming with defined antigens is defective in sepsis (13). By contrast, we and other research groups have shown that sepsis induces high concentrations of serum IgM and IgG antibodies of unknown specificities (13, 14). Since these may be responsible for the observed antibody-mediated protection, we set out to examine their antigen specificities in a mouse model of abdominal sepsis.
During sepsis, the organism is flooded with bacterial antigens as well as self-antigens, which are released by dying host cells. Moreover, there is an abundance of danger signals, both pathogen- and damage-associated molecular patterns (PAMPs and DAMPs, respectively) (15, 16). These may, on the one hand, act as adjuvants in an antigen-driven B cell response and, on the other, trigger a polyclonal B cell reaction (17–19). In addition to these danger signals, inflammation and cell death are accompanied by lipid peroxidation, resulting in the generation of oxidation-specific epitopes (OSEs), which are also recognized by pattern recognition receptors (PRR) of the innate immune system exerting an adjuvant effect (20–22). All of these factors could contribute to the B cell response in sepsis.
During sepsis, antibody production is induced by T cell-dependent (TD) as well as -independent (TI) mechanisms (14). In a TD immune reaction, follicular B cells are activated via the B cell receptor. With the help of activated T cells, they differentiate and form germinal centers, where class switch to all Ig (sub)classes and somatic hypermutation take place. At the end of this process, affinity-matured plasma cells have developed that continuously secrete antibodies (23). TI B cell responses may be triggered in two ways: TI-2 antigens like polysaccharides efficiently crosslink B cell receptors and initiate a strong and long-lasting antigen-specific primary response (24). In contrast, TI-1 antigens like lipopolysaccharide (LPS) and bacterial DNA (CpG) activate B cells polyclonally, i.e., independent of the B cell receptor, via TLR triggering (25–27). Predominantly B-1- and marginal zone (MZ) B cells can rapidly respond to TI-1 antigens (28).
The main reservoir of B-1 cells are the pleural and peritoneal cavities, but a small proportion can be found in all lymphoid organs. Not only are B-1 cells prone to TI responses, but they are also the main producers of natural antibodies (NAbs), defined as antibodies that circulate in normal individuals in the absence of exogenous antigenic stimulation (29). NAbs are considered polyreactive, usually lack somatic hypermutation and are said to use a restricted set of B cell receptor genes (30–32). NAbs are hence at the interface between innate and adaptive immune responses, and can bridge the time gap until the TD response has matured. In terms of their antigen specificity, B-1 cells are selected for a certain strength of self-antigen binding. Remarkably, ~30% of B-1 cell-derived IgM binds to OSEs. B-1 cells are able to switch to all IgG subclasses in vitro, whereas in vivo, they are producers of NAbs, mainly of the IgM, IgG3, and IgA isotype [reviewed extensively in (32–34)].
MZ B cells are located close to the marginal sinus in the spleen, where they have direct access to blood-borne antigens (35). Although they have the capacity to generate TD and TI responses, their main function is the TI response against blood-borne pathogens (36, 37). Very early in the course of an infection, they differentiate to IgM- or IgG-secreting cells (38). Both TD as well as TI processes take place during sepsis, and all B cell populations become activated (8, 14). We have therefore tested sepsis-induced IgM- and IgG-binding to a broad panel of bacterial as well as autoantigens. OSE were identified as the dominant target of the B cell response.
Materials and Methods
Animals and Ethics Statement
All experiments were performed on 8–12 weeks old female C57BL/6 wild type (WT) mice. All animals were housed in a conventional, temperature-controlled animal facility with a 12-h light and dark cycle, and provided with food and water ad-libitum. All experiments were approved by the animal ethics committee of the local animal protection authority (LALLF, State Office for Agriculture, Food Safety and Fisheries Mecklenburg-Western Pomerania; numbers LALLF M-V/TSD/7221.3-1.1-052/07 and LALLF M-V/TSD/7221.3-1.2-013/09). All efforts were made to minimize the suffering of mice.
CASP Surgery
CASP surgery was performed as described before (39, 40). Briefly, mice were anesthetized with Ketamin (Ketanest, Parke-Davis GmbH, Berlin) and xylazin (Rompun, Bayer Health Care, Leverkusen) intraperitoneally (100 mg/10 mg per kg bodyweight, respectively) and a 18G stent (Ohmeda AB, Helsingborg, Sweden) was implanted into their colon ascendens. Mice were monitored every 4 h until recovery.
Hybridoma Generation
Splenocytes from mice 10 or 14 days following CASP or sham surgeries were prepared and fused with X63 AG8.653 myeloma cells using polyethylene glycol (Sigma-Aldrich), following an extensive protocol for fusion and selection as described elsewhere (http://www.umass.edu/vetimm/docs/Wagner_Hybridoma.pdf). Briefly, 10 million X63-cells were fused with 1–3 × 107 splenocytes, and the fusion products were plated in several dilutions into 96-well cell culture plates. Ten days later, plates were screened under a light microscope for hybridoma growth. Only hybridomas from plates where <50% of the wells showed cell growth were taken into account, since this improves the probability of monoclonality to over 85% (41). In addition, wells were observed under a light microscope to select for single clone growth based on morphology.
Immunohistochemical Autoantibody Screening on HEp-2-Cells
Screening of autoantibodies was performed as described before (13). Briefly, serum samples, diluted 1:100 in PBS containing 20% FCS or undiluted hybridoma supernatants were incubated on HEp-2-ANA slides (INOVA Diagnostics, San Diego, CA, USA) overnight at 4°C. Slides were washed with PBS, and bound antibodies were detected either with polyclonal goat anti-mouse IgG or IgM conjugated to FITC (10 μg/mL, Southern Biotech, Atlanta, GA, USA). Slides were then equally exposed, and pictures were taken with a Zeiss Axio imager A.1 fluorescence microscope (Zeiss, Göttingen, Germany) equipped with Spotadvanced software (Diagnostic Instruments, Sterling Heights, MI, USA) using the same settings for all images.
ELISA
Antigens were prepared for ELISA as follows: (i) Bacterial antigens: Bacteria (E. coli, E. faecalis, P. mirabilis, S. aureus 8325-4 Δspa) were grown overnight in tryptic soy broth (TSB) medium as described previously (41). Cells were washed twice in cold PBS (3,340 g for 15 min at 4°C) and diluted to an optical density at 595 nm (OD595) of 1.0 in PBS before being inactivated by irradiation with UV-light for 10 min; (ii) Self-antigens: Histone H2A (calf thymus, 2 μg/mL) and dsDNA (calf thymus, 10 μg/mL), both obtained from Sigma-Aldrich, were diluted in PBS, whereas murine IgG-Fc-fragments (Dianova, 1 μg/mL) were diluted in coating buffer (pH 9.6; Candor Bioscience GmbH, Wangen, Germany); (iii) LPS (E. coli O55:B5, Sigma Aldrich, 10 μg/mL) and sepsis unrelated antigens, including TNP (14)-bovine serum albumin (BSA, Biosearch Technologies, 10 μg/mL) and ovalbumin (OVA, Sigma-Aldrich, 10 μg/mL) were diluted in coating buffer to the indicated concentrations; (iv) OSEs: phosphocholine-conjugated BSA (PC-BSA) was obtained from Biosearch Technologies Inc. Malondialdehyde-acetaldehyde-modified BSA (MAA-BSA) was prepared as described elsewhere (42). Human native LDL was isolated, and Cu2+-oxidized LDL (CuOx-LDL) and Malondialdehyde-modified LDL (MDA-LDL) were prepared as described previously (42).
96-well flat-bottom microplates (Nunc MaxiSorp™) were coated with the respective antigens overnight at 4°C (bacterial antigens, H2A: 100 μL per well), LPS, dsDNA, murine IgG-Fc-fragments and sepsis unrelated antigens: 50 μl per well). The plates were then washed three times with PBS containing 0.05% Tween20, and blocked with PBS containing 10% fetal calf serum (150 μL/°) for 1 h at room temperature (RT). Murine sera (dilution range 1:100–1:2,500) or hybridoma supernatants were added to the wells and incubated for 90 min at RT. After three washing steps, bound antibodies were detected using F(ab)2-fragments of goat anti-mouse IgG or IgM conjugated to POD (Dianova) as previously described (43). Chemiluminescence was measured using a Tecan Sunrise photometer (Tecan Group Ltd., Maennedorf, Switzerland), and the data were expressed as relative light units (RLU) per 100 ms.
RNA Isolation From Hybridoma Cells and Complementary DNA Generation
Total RNA was isolated from about 1 × 105 hybridoma cells using the RNeasy Mini Kit (QIAGEN GmbH, Hilden, Germany) according to manufacturer's instructions. The isolation steps included a DNase-digestion step to eliminate genomic DNA. The quantity of RNA was determined by using the DS-11 Series Spectrophotometer (DeNovix Inc., DE, USA). One microgram of RNA was transcribed to cDNA using oligo DT Primers of the RevertAidTM First Strand cDNA Synthesis Kit (Fermentas) as per the manufacturer's instructions. For the amplification of VH-domain sequences of the immunoglobulins, cDNA was amplified by multiplex PCR using msVHE as universal forward primer and a mixture of the Ig-specific reverse primers (Table 1). The reaction was performed in a total volume of 25 μL using 2 μL of cDNA, 0.2 μL of GoTaq-polymerase (Promega), 2.5 μL of dNTP (1 mM, Roche Diagnostics), 5 μL MgCl2 (25 mM, Promega). Primers were used at a final concentration of 200 nM each, and the PCR was performed for 35 cycles at 95°C 30 s, 65°C 30 s, 72°C 45 s and a final elongation step at 72°C for 10 min. PCR products were analyzed on 1.5% agarose gels, and the DNA bands of around 400 bp size were cut under UV light for extraction of DNA using the Nucleo-Spin® Extract II kit (Machery-Nagel).

Table 1. Primers used for VH-gene sequence amplification.
Cloning and Transformation of Target DNA
The extracted DNA-fragments were cloned into the pCR® 2.1-TOPO® TA vector using the TOPO® TA Cloning® Kit (Life Technologies, Thermo Fisher Scientific, Carlsbad, CA, USA). All steps were done according to manufacturer's instructions. Products were transfected into chemically competent E. coli by the heat-shock method. Transformed E. coli were cultivated on LB agar plates containing 50 μg/mL of each IPTG, x-Gal and Ampicillin, and incubated at 37°C for 12 h. At least three single white colonies were picked and seeded further into single wells of a 96-well plate containing LB agar with 50 μg/mL Ampicillin.
VH-N-DH-N-JH Fragment Sequencing and Analysis
Plasmid isolation and sequencing of the single colonies was performed by GATC Biotech AG, Konstanz, Germany, using the vector-specific M13 forward primer. The resulting sequences were further verified using IMGT/V-Quest database, (https://www.imgt.org/IMGT_vquest/vquest?livret=0&Option=mouseIg).
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 6 (GraphPad software, San Diego, CA, USA). Data were assessed for significant differences using One-Way ANOVA with Bonferroni post test for selected pairs, or using the unpaired Student t-test, whenever appropriate. P < 0.05 were considered to be significant.
Results
A Strong Induction of Total Serum IgG During Sepsis Was Not Due to an Antibacterial IgG Response
We and others have shown that sepsis induced a marked increase in serum IgM levels followed by an even stronger IgG response (11, 13). As we have previously shown, most ASCs reside in the spleen, and the class switch to IgG results from both TI and TD processes (14). Since the antigen-specific T cell response is still fully functional at the onset of sepsis (47), we assumed that the class switched IgG response was mainly directed at the invasive bacteria. To test this hypothesis, we induced sepsis in WT mice using the CASP sepsis model. First, systemically disseminated bacteria were identified by plating sera of septic mice onto agar plates followed by microbiological identification of growing colonies. Enterococcus faecalis (E. faecalis) and Escherichia coli (E. coli), two microbial species of the intestinal flora, were regularly found in the blood of septic mice. Antibody binding to these bacteria as well as to Staphylococcus aureus (S. aureus), which is often observed in the murine intestine, was then measured by ELISA. UV-inactivated washed bacterial cells served as antigens. To avoid non-specific IgG binding to S. aureus, the protein A-deficient mutant strain 8425-Δspa was used. Additionally, LPS of E. coli was included. As shown in Figure 1, sepsis induced a significant increase in IgM-binding to whole bacteria as well as LPS. In contrast, there was no significant anti-bacterial or anti-LPS IgG response in the majority of animals. The absence of an anti-bacterial IgG response in sepsis was in striking contrast to immunization with inactivated bacteria (without adjuvant), where ímmunized animals did not develop disease symptoms, but elaborated high specific antibacterial IgG titres (Supplementary Figure 1). Thus, the very strong serum IgG increase in sepsis was not due to an anti-IgG response to the bacteria tested, namely S. aureus, E. coli and E. faecalis.
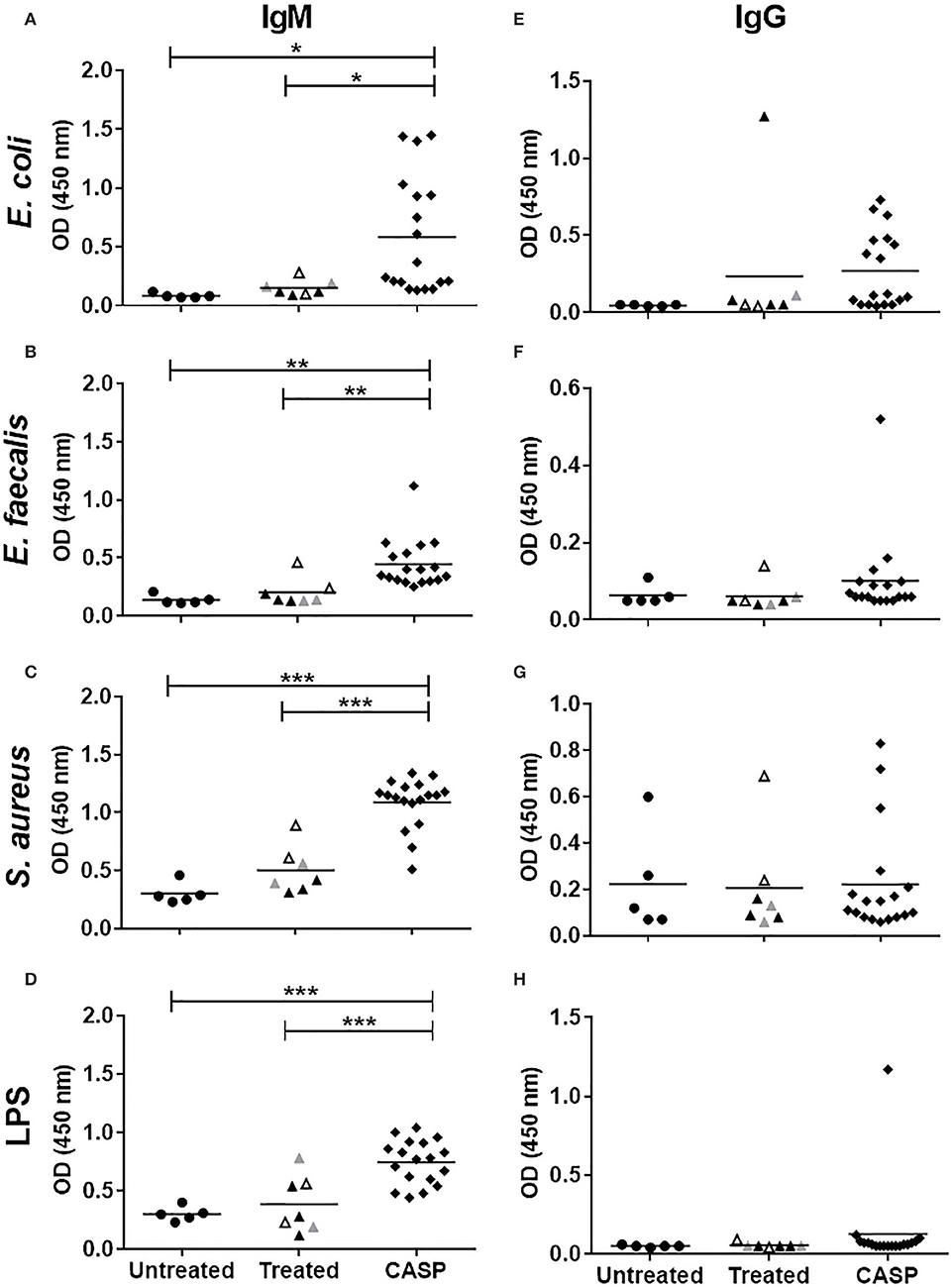
Figure 1. Binding of sera IgM/IgG to bacteria and LPS. The binding of murine serum antibodies, IgM (A-D) or IgG (E-H), to indicated bacteria; E. coli (A,C), E. faecalis (B,F), S. aureus (C,G), and LPS (D,H) was tested by ELISA. Fourteen days after sepsis induction, blood was collected, and sera were diluted 1:100 in blocking buffer. Sera from non-septic animals were used as controls. Control mice were untreated (•), whereas the treated mice were anesthetized only (▴), or additionally received a laparotomy ( ), or sham surgery (▵). Mice that underwent CASP surgery are indicated by a black diamond (♦). Mean values of OD 450 nm are shown. Each symbol represents one animal (N = 5–18). Statistical analysis was done by One-way ANOVA with the Bonferroni post test for selected pairs. *p < 0.05, **p < 0.01, ***p < 0.001.
), or sham surgery (▵). Mice that underwent CASP surgery are indicated by a black diamond (♦). Mean values of OD 450 nm are shown. Each symbol represents one animal (N = 5–18). Statistical analysis was done by One-way ANOVA with the Bonferroni post test for selected pairs. *p < 0.05, **p < 0.01, ***p < 0.001.
Sepsis-Induced Serum Antibodies Are Directed Against Self-Antigens
In sepsis, the strong general IgG response with only a modest reaction to bacterial antigens could be caused by polyclonal activation of B-cells due to the release of large amounts of PAMPs and DAMPs. We reasoned that autoreactive Ig should be increased in this case, because the normal B-cell pool contains numerous self-specificities (48–50). Moreover, the PAMPs and DAMPs could also act as adjuvants in a TD immune response to self-antigens that are released from damaged cells and tissues. Therefore, we tested the sera of septic and control mice for Ig-binding to eukaryotic cellular structures using HEp-2 cells as antigens and FITC-labeled anti-mouse-IgM or IgG antibodies for detection of binding. As shown in Figure 2, the sera of septic mice (data from two representative septic animals are depicted) showed an increase in autoreactive IgM and IgG response, as compared to untreated animals (left panel), 14 days after sepsis induction. The fluorescence patterns observed were mostly diverse, where often more than one cellular structure was stained (Figure 2, arrows). The binding patterns differed between individual septic mice from the same cage. Hence, sepsis, which was probably caused by very similar gut microbiota, induced autoantibodies of different specificities. Remarkably, a similar autoreactive response was observed in the sera of splenectomized animals upon sepsis induction (Supplementary Figure 2).
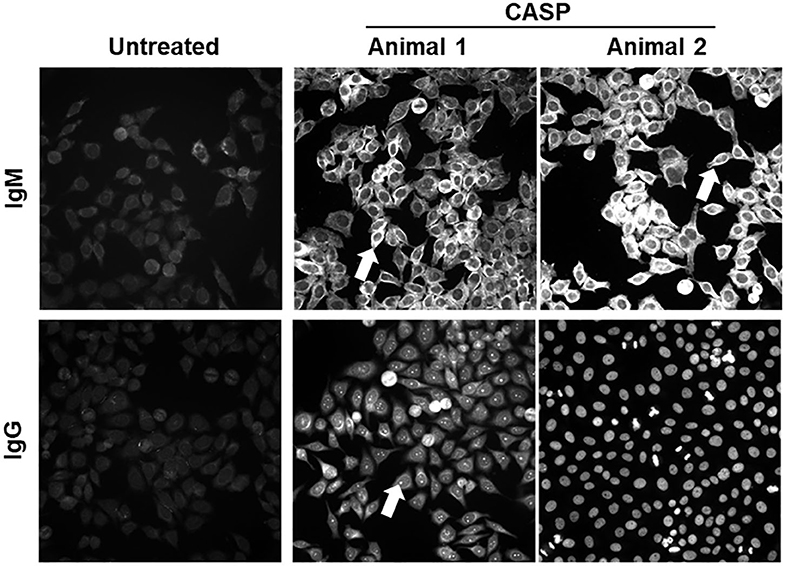
Figure 2. Serum antibodies directed against self-antigens 14 days after sepsis induction. Sepsis was induced in C57BL/6 mice by 18G CASP. Control mice remained untreated. Fourteen days later blood was taken, and sera diluted 1:100 in 20% FCS/PBS were incubated on HEp-2-ANA slides. Bound antibodies were detected by FITC-labeled anti-mouse IgM or IgG antibodies. Out of the tested serum antibodies, 12/13 IgM and 12/13 IgG antibodies were autoreactive. Shown are representative pictures of septic and control mice. N = 8 per group. Serum IgM and IgG binding patterns varied greatly. In most cases more than one cellular structure is stained, albeit at variable intensities.
Sepsis Increased Serum Antibodies Directed Against Sepsis-Unrelated Antigens
An immune response which is largely polyclonal rather than antigen-driven would be expected to show increased antibody binding to antigens unrelated to sepsis. Hence, besides anti-bacterial and self-binding antibodies, we screened for the presence of antibodies binding to such sepsis-unrelated antigens. At first, we investigated the binding of serum antibodies to two antigens the animals had never been exposed to, namely the classical TD protein antigen ovalbumin (OVA) and the hapten 2,4,6-trinitrophenyl (TNP) conjugated to bovine serum albumin (TNP-(14)-BSA). Regarding OVA, only IgM binding increased significantly during sepsis (data not shown), whereas IgM- as well as IgG-binding to TNP-(14)-BSA were both very strongly enhanced (Supplementary Figure 3). Together, these results clearly support the concept of a polyclonal B cell response, which may be dominated by NAb specificities.
Sepsis Induced High Titer IgM and IgG Antibodies Directed Against Oxidation-Specific Epitopes (OSEs)
Since sepsis induced a robust autoreactive-antibody response, we questioned whether these antibodies would be able to bind to OSEs, which are present at high density on apoptotic cells, whose numbers are known to increase during sepsis. OSEs generated during sepsis act as an endogenous DAMPs, and initiate an innate immune response. It has been shown that a large proportion of NAbs, especially those produced by B-1 cells, bind to OSEs (20, 42). Therefore, we tested sera of septic and untreated animals at day 10 and 14 for OSE-specific antibodies by chemiluminescent ELISA. Sepsis elicited a very strong IgM and IgG response to known OSEs in all tested animals (Figure 3 and Supplementary Figure 4), while there were very low levels of OSE-specific antibodies or none at all in untreated animals. Hence, at day 14 post sepsis, the IgM response to both modified LDL and BSA was significantly higher in septic mice than in the control group, where specific IgM was low or even under the limit of detection (Figure 3). On the same day, the IgG/IgM response to MAA-BSA was 4-fold higher than in the untreated group. The IgM response to CuOx-LDL showed a trend toward increased levels, while IgG levels were significantly higher compared to untreated animals (Supplementary Figure 4). Since under other conditions, such as hypercholesterolemia, IgM antibodies dominate the immune response to OSEs (51), the pronounced specific class-switched response in sepsis is remarkable.
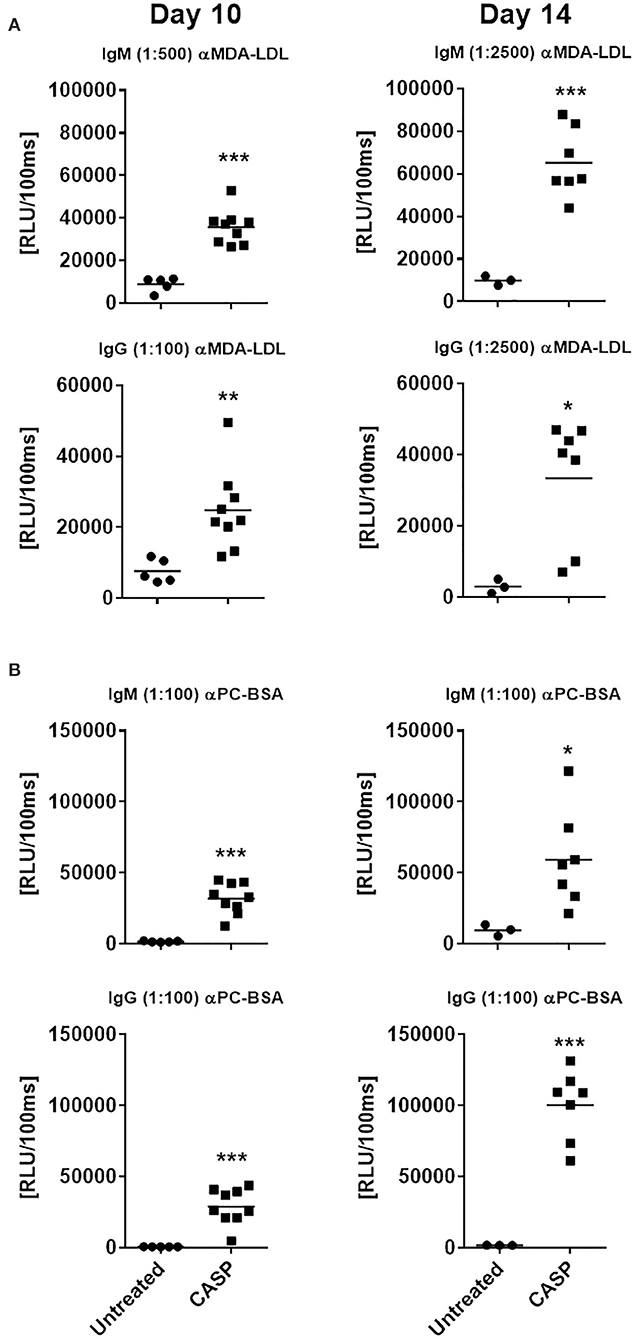
Figure 3. Serum IgM and IgG binding to model oxidation-specific epitopes. C57BL/6 mice were subjected to CASP surgery, while control animals remained untreated. Ten and 14 days after sepsis induction, blood was drawn to determine serum IgM and IgG binding to malondialdehyde-modified LDL [MDA-LDL; (A)] and phosphocholine-conjugated BSA [PC-BSA; (B)], using chemiluminescent ELISA. Serum dilutions used are indicated in brackets. Statistical analysis was performed using the unpaired t-test. N = 3–9 per group. *p < 0.05, **p < 0.01, ***p < 0.001.
Studies at the Single Cell Level Revealed a High Proportion of Autoreactive IgM Antibodies
To study the sepsis-induced antibodies at a single-cell level, monoclonal antibodies (mAbs) were generated. Since we discovered the spleen as the main source of antibody secreting cells (ASCs) in sepsis (14), splenocytes from eight septic mice were obtained on day 10 after sepsis induction and fused. Three quarters (74.3%) of the hybridomas from septic mice produced IgM (Table 2). However, sepsis induced a much stronger increase in serum IgG than IgM, which peaked at day 14 (14). Assuming that the class switch to IgG may not have been completed on day 10 after sepsis induction, additional fusions were performed using splenocytes from six septic mice on day 14. However, choosing this later time point did not change the proportion of IgM- to IgG-producing cells, where 82% of the resulting hybridomas secreted IgM (Table 2). Several attempts to produce hybridomas from non-septic control mice failed, presumably due to the lack of activated B cells and plasmablasts in the spleen, which is a limitation of this study (data not shown). Determination of the mAb specificities confirmed and extended the observations made with serum antibodies, namely that sepsis induces autoreactive antibodies, predominantly IgM.

Table 2. HEp-2-positive IgM- or IgG-secreting hybridomas.
The total of 386 hybridoma supernatants derived from the 14 above mentioned septic mice and containing either IgM or IgG mAbs were screened for autoreactivity. A large proportion of IgM mAbs (31% to 40%) reacted with specific structures of the HEp-2 cells (Table 2). HEp-2-binding IgM mAbs were found in every animal, regardless of whether the spleen cells were obtained 10 or 14 days after sepsis. In contrast, only three out of 93 IgG mAbs showed detectable HEp-2-binding, when using undiluted hybridoma culture supernatants. These auto-reactive IgG hybridomas were obtained from three mice. Figure 4 illustrates the variability of the binding patterns of the mAbs derived from splenic B cells of septic mice 10 or 14 days after CASP induction, which bind several cellular self-antigens. FITC-labeled anti-mouse IgG or IgM antibodies were used to determine the binding of those mAbs and clearly showed that, similar to our previous experiment at the serum level, the binding patterns of the individual antibodies were highly variable.
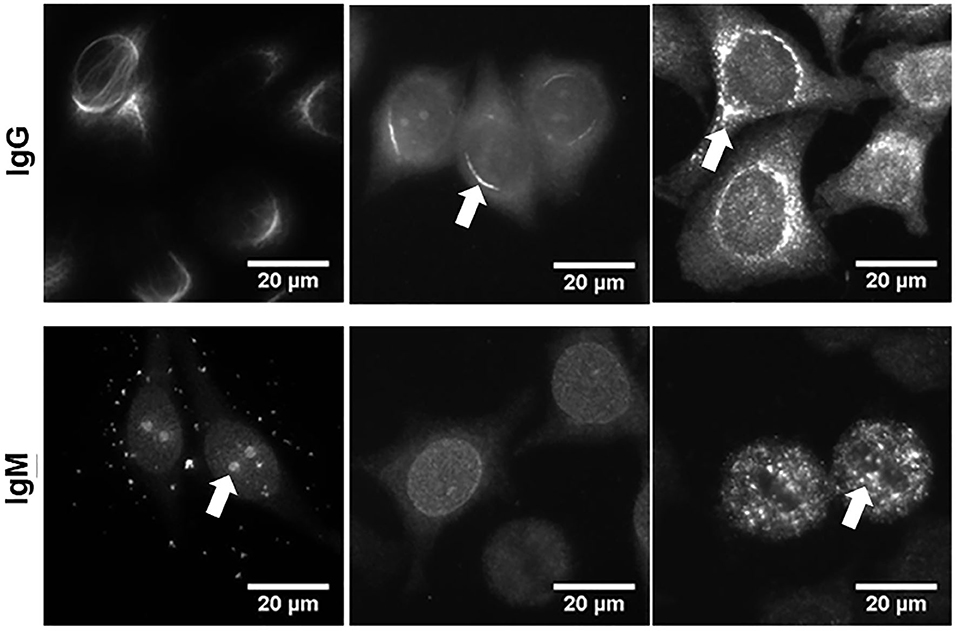
Figure 4. Monoclonal antibodies derived from splenic B cells of septic mice bind several cellular self-antigens. Ten or 14 days after CASP induction, splenocytes were isolated and fused with myeloma cells to obtain hybridomas. Supernatants of monoclonal IgM- or IgG-producing hybridomas were incubated on HEp-2-ANA slides to detect auto-reactivity. Bound IgG or IgM was detected by FITC-labeled anti-mouse IgG or IgM antibodies. Three examples, respectively, of autoreactive monoclonal IgM- and IgG-antibodies are shown. The binding patterns of the various antibodies showed obvious differences, where in most cases more than one cellular structure was stained, albeit at varying intensities (indicated by arrows). For quantified data, regarding the numbers/percent of IgM or IgG hybridoma supernatants from septic mice directed against self-antigens, please refer to Table 2.
Most IgM Antibodies Are Polyreactive
Since we observed high titres of autoantibodies in sera of septic mice at day 10 or 14 (Figure 2), we wondered whether this also applies to the mAbs present in the hybridoma supernatants of six septic mice (generated from splenocytes at day 10 of sepsis).
We tested the hybridoma supernatants for mAb-binding to a broad panel of antigens, including bacterial and self-antigens, as well as two antigens that the animals had never been exposed to, namely TNP and OVA. Binding was observed with 27 out of 120 IgM-supernatants (22.5%) (Table 3) but with none of the 40 tested IgG supernatants (data not shown). The majority of the binding IgM mAbs (85%) were polyreactive, recognizing at least two antigens in the test panel used.
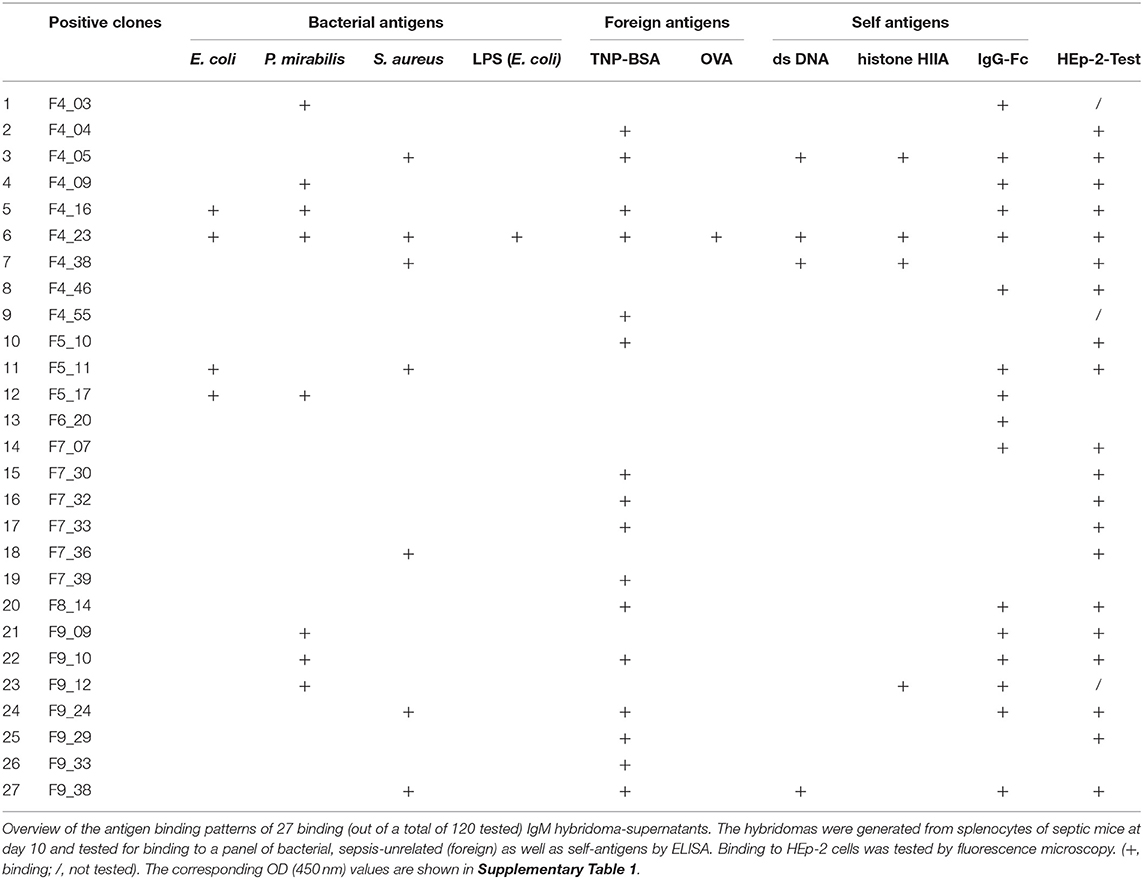
Table 3. Antigen-binding patterns of positively-tested IgM hybridomas.
In Septic Mice, a Large Proportion of mAbs, Both IgM and IgG, Binds to Oxidation Specific Epitopes
Eighteen IgM mAbs that showed reactivity to the antigen panel in Table 3, as well as all available IgG mAbs (N = 20) of so far unknown specificity, were tested for binding to OSEs. Thirteen (72%) IgM and 5 (25%) IgG mAbs showed binding to one or several OSEs in ELISA (Table 4). Examples of IgG mAb titration curves are shown in the Supplementary Figure 5. The five OSE-specific IgG hybridomas were derived from four different mice. Remarkably, with the exception of IgG3, the OSE-specific mAbs encompassed all IgG subclasses. These findings underline that OSEs are prominent targets of the humoral immune response in sepsis.
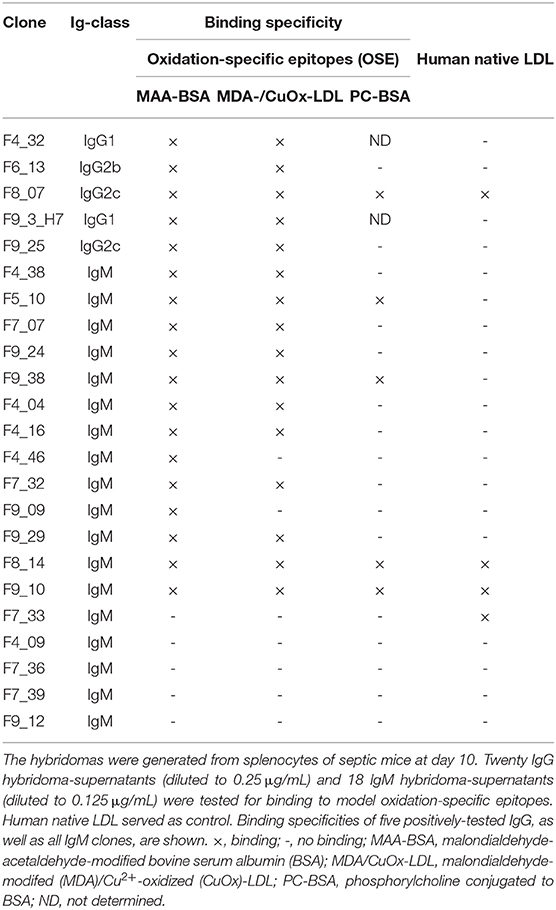
Table 4. Binding pattern of CASP d10 monoclonal IgG and IgM to oxidation-specific epitopes.
Most Sepsis-Induced IgG mAbs Carried Few Somatic Mutations
Many class switched antibody responses are TD and are characterized by somatic hypermutation. Therefore, the IgG mAbs were tested for somatic hypermutation by sequencing the variable domains of the IgG heavy chains. IgG mAbs at day 10 or 14 after sepsis mostly showed no significant differences in mutations (Supplementary Figure 6A). Most sequences were near germline, similar to 12 IgG monoclonal antibodies obtained from an untreated animal, which all had <4 mutations/100 nucleotides (Supplementary Figure 6A). However, four out of the 34 IgG mAbs derived from septic animals had five or more mutations per 100 nucleotides, indicative of somatic hypermutation. As such mAbs were present already at day 10, they were probably derived from memory B cells (52). OSE-specific and –non-specific IgG mAbs showed similar proportions of highly mutated IgG (Figure 5). Thus, most of the IgG antibodies generated during sepsis used near-germline gene sequences. Regarding the CDR3 length, the number of amino acid exchanges per V-region as well as the proportion of non-silent mutations, there were no significant differences between septic and non-septic animals (Supplementary Figures 6, 7).
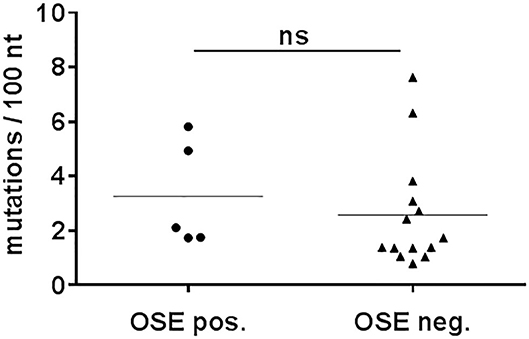
Figure 5. Number of mutations in Vh-genes of monoclonal IgG. CASP surgery was performed on C57BL/6 mice. Hybridomas were generated by fusing splenocytes from 10- days-septic mice. The resultant monoclonal IgG hybridomas were sequenced for Vh-genes. Mutations of monoclonal IgG that showed binding to OSEs (OSE pos.) were compared to IgG that showed no binding (OSE neg.). Statistical analysis was performed using the unpaired t-test. N = 5–14 per group. nt, nucleotides; ns, not significant.
Discussion
Our data clearly show that OSEs are the main targets of the strong humoral immune response observed in sepsis. While Ig binding patterns to other self-epitopes were individual, OSE-specific binding was observed in all tested animals.
The sepsis-induced suppression of the adaptive immune system is well-documented (10, 13, 53). Nevertheless, we and others observed a strong increase in serum IgM and IgG concentrations following sepsis (13, 14). Interestingly, the peak of the IgG serum concentration exceeded that of IgM by a factor of four, and an Ig switching to all IgG subclasses was seen. However, at the single cell level, there were three times as many IgM hybridomas as those producing IgG. Although one could argue that fusion was performed too soon, we found essentially the same proportions on day 14 as on day 10, which rules out time as the causal factor. Although the numbers of hybridomas are in general too low to make a definitive conclusion, a plausible explanation for this observation may be found in the half-lives of IgM vs. IgG antibodies. IgM has a markedly shorter serum half-life than IgG, and the half-life of polyreactive IgM is further reduced by Ag-binding, leading to a relative accumulation of IgG (54, 55).
Since sepsis induces dissemination of endogenous bacteria into the bloodstream (11), a specific antibacterial IgG response would have been expected. However, there was very little serum IgG binding to bacteria in post-septic mice. Since we focussed our investigation on culturable bacteria in the sera of septic mice, it is possible that bacterial species other than the ones examined here were the culprits, since ELISA was performed only for E. coli, S. aureus, and E. faecalis. However, E. coli and E. faecalis originate from the serum of a septic animal. In fact, the bacterial strains used for testing are definitely present in large numbers in the intestine. When animals were injected i.p. with dead bacteria (E. coli, S. aureus, and E. faecalis) without sepsis, they were perfectly able to elaborate a specific IgG response. Depending on the bacterial species, we could show that 107-109 UV-inactivated bacteria trigger a robust specific IgG response (Supplementary Figure 1). Hence, we assume that sepsis interferes with an antibacterial antibody response. Interestingly enough, we rather observed Ig binding to autoantigens, as well as antigens, to which the animals had never been exposed. The production of natural autoantibodies against a spectrum of autoantigens has been described in sepsis patients (56–58). Using a relatively small panel of autoantigens, Burbelo et al. detected autoantibodies in 46% of severe sepsis patients, suggesting that this is a relatively common phenomenon (58). However, the production of autoantibodies is not confined to sepsis, but appears to be a general phenomenon observed with other types of bacterial, viral, and protozoan infections (18, 59–61), in addition to autoimmune disorders. In their recent study, Sakakibara et al. observed that the immune response to murine γ-herpesvirus 68 is accompanied by autoantibody production from polyreactive B cells in the germinal center, whose self-reactivity is generated through somatic hypermutation (61). More interestingly, several reports suggest a role of polyclonal activation in triggering anti-self responses, and hence leading to autoimmune disorders as a consequence of infections (18, 62). For instance, a direct link between infection and autoimmune encephalitis has been experimentally demonstrated in mice, and has been attributed to the production of autoantibodies (62).
It has been demonstrated that sepsis induces massive apoptosis of B and T lymphocytes, as well as DCs in mice and humans, and the release of danger molecules, known as PAMPs and DAMPs (8, 9, 63–67). We confirmed this in our model by TUNEL staining (terminal deoxynucleotidyl transferase dUTP nick end labeling) of the spleens for apoptotic nuclei (data not shown). The observed autoantigen response in sepsis could be explained by a polyclonal B cell activation through PAMPs and DAMPs, which activate B cells independent of the BCR, leading to the production of the so-called NAbs, which are polyreactive (68). To test whether polyreactive (natural) Abs or monoreactive Ig from a large variety of B cells dominate the humoral immune response after sepsis, an analysis at the single cell level was conducted. It revealed that a large proportion of IgM-producing cells were polyreactive, as one would have expected based on the results obtained with serum.
Our results identify OSEs as the dominant target of the B cell response in murine polymicrobial sepsis: First, all post-septic animals had OSE-binding IgM and IgG, whereas antibody binding patterns to other tested autoantigens differed between individuals, even from the same cage. Second, anti-OSE titres were much higher than those of other tested bacterial or self-antigens. Finally, OSE-specific B cells were very frequent. Thirteen out of 18 tested IgM mAbs bound to OSEs even at low concentrations. Since 19% (23/120) of the IgM mAbs were polyreactive, at least 14% of IgM-producing B cells were OSE-specific. This frequency is somewhat similar to that of IgG mAbs, of which 25% were OSE-reactive.
Several points argue for a B-1 cell-driven immune response in the case of IgM. B-1 cells reside in the peritoneum and upon activation migrate to the spleen as well as other lymphatic organs, where they differentiate into ASCs (69, 70). These are the main producers of NAbs, which are polyreactive and utilize germline-near sequences (68). Chou et al. have shown that 20–30% of B-1 cell-derived IgM targets model-OSEs (42). This reflects the frequencies we find in monoclonal IgM and IgG. Moreover, Chang et al. have demonstrated that immunization of mice with apoptotic cells, which are abundant in sepsis, induces OSE-binding IgM (70).
In contrast, it is rather unlikely that the IgG response to OSEs is also dominated by B-1 cells. In B-1 cells, only a switch to IgG3 has been shown in vivo (31, 71). However, none of the five OSE-specific IgG mAbs were IgG3, whereas all other IgG subclasses were represented. Hence, a TD response of follicular B cells appears more likely. Chang et al. were able to induce OSE-binding IgG in mice by immunizing with model-OSE and apoptotic cells simultaneously (70). Moreover, Grasset et al. showed that some follicular B cells can bind fluorescently-labeled oxidized LDL. Repeated immunization with apoptotic cells induced OSE-binding IgG (72).
We suggest the following model: In sepsis, numerous immune cells undergo apoptosis. PAMPs and DAMPs polyclonally activate peritoneal B-1 cells to migrate to the spleen and differentiate into ASCs. These produce NAbs, explaining the increase in serum IgM targeting OSEs as well as other autoantigens. NAbs, including OSE-targeting IgM antibodies, may act as scavengers, promoting the clearance of apoptotic cells and debris, which would be relevant in sepsis (22, 42). In this study, however, we observed that OSE-specific antibodies had switched to IgG as well, presumably in a TD response. It may be speculated that the dominant response to OSEs restricts a specific antibody response to the sepsis-causing bacteria, which the organism would mount in the absence of apoptosis. Besides this, massive apoptosis has been shown to be tolerogenic, which explains the well-known fact that sepsis does not induce strong protective immune memory (11). The pathophysiological function of OSEs and OSE-specific IgM and IgG in sepsis should now be investigated, to determine whether OSE-specific antibodies could have therapeutic potential.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by The Animal Ethics Committee of the local animal protection authority (LALLF, State Office for Agriculture, Food Safety, and Fisheries Mecklenburg-Western Pomerania).
Author Contributions
ON, CP, CB, and BB: conceptualization and project design. ON, CP, JL, SQ, and AL: methodology and performance of experiments. ON and CP: data evaluation. ON, CP, DR, C-DH, MD, and BB: interpretation of data. ON, DR, MD, CJB, and BB: writing—original draft preparation. All authors critically reviewed the manuscript and approved the submitted version.
Funding
This work was financially supported by the German Research Foundation (RTG 840; grants to ON, CP, and JL). We acknowledge support for the Article Processing Charge from the DFG (German Research Foundation, 393148499) and the Open Access Publication Fund of the University of Greifswald.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Maria Ozsvar Kozma for excellent technical support. The authors would also like to thank Juliane Moritz for her contribution to this work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01570/full#supplementary-material
References
1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
2. Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. (2013) 369:840–51. doi: 10.1056/NEJMra1208623
3. Fleischmann C, Hartmann M, Hartog CS, Welte T, Heublein S, Thomas-Rueddel D, et al. Epidemiology of sepsis in Germany: incidence, mortality and associated costs of care 2007-2013. Intensive Care Med Exp. (2015) 3(Suppl. 1):A50. doi: 10.1186/2197-425X-3-S1-A50
4. La Suarez De Rica A, Gilsanz F, Maseda E. Epidemiologic trends of sepsis in western countries. Ann Transl Med. (2016) 4:325. doi: 10.21037/atm.2016.08.59
5. Fitzpatrick F, Tarrant C, Hamilton V, Kiernan FM, Jenkins D, Krockow EM. Sepsis and antimicrobial stewardship: two sides of the same coin. BMJ Qual Saf. (2019) 28:758–61. doi: 10.1136/bmjqs-2019-009445
6. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the global burden of disease study. Lancet. (2020) 395:200–11. doi: 10.1016/S0140-6736(19)32989-7
7. Boes M, Prodeus AP, Schmidt T, Carroll MC, Chen J. A critical role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J Exp Med. (1998) 188:2381–6. doi: 10.1084/jem.188.12.2381
8. Kelly-Scumpia KM, Scumpia PO, Weinstein JS, Delano MJ, Cuenca AG, Nacionales DC, et al. B cells enhance early innate immune responses during bacterial sepsis. J Exp Med. (2011) 208:1673–82. doi: 10.1084/jem.20101715
9. Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE Jr, Hui JJ, Chang KC, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol. (2001) 166:6952–63. doi: 10.4049/jimmunol.166.11.6952
10. Inoue S, Unsinger J, Davis CG, Muenzer JT, Ferguson TA, Chang K, et al. IL-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J Immunol. (2010) 184:1401–9. doi: 10.4049/jimmunol.0902307
11. Pötschke C, Kessler W, Maier S, Heidecke C-D, Bröker BM. Experimental sepsis impairs humoral memory in mice. PLoS ONE. (2013) 8:e81752. doi: 10.1371/journal.pone.0081752
12. Sjaastad FV, Condotta SA, Kotov JA, Pape KA, Dail C, Danahy DB, et al. Polymicrobial sepsis chronic immunoparalysis is defined by diminished Ag-specific T cell-dependent B cell responses. Front Immunol. (2018) 9:2532. doi: 10.3389/fimmu.2018.02532
13. Mohr A, Polz J, Martin EM, Griessl S, Kammler A, Potschke C, et al. Sepsis leads to a reduced antigen-specific primary antibody response. Eur J Immunol. (2012) 42:341–52. doi: 10.1002/eji.201141692
14. Nicolai O, Pötschke C, Schmoeckel K, Darisipudi MN, van der Linde J, Raafat D, et al. Antibody production in murine polymicrobial sepsis-kinetics and key players. Front Immunol. (2020) 11:828. doi: 10.3389/fimmu.2020.00828
15. Gentile LF, Moldawer LL. DAMPs, PAMPs, and the origins of SIRS in bacterial sepsis. Shock. (2013) 39:113–4. doi: 10.1097/SHK.0b013e318277109c
16. Sursal T, Stearns-Kurosawa DJ, Itagaki K, Oh S-Y, Sun S, Kurosawa S, et al. Plasma bacterial and mitochondrial DNA distinguish bacterial sepsis from sterile systemic inflammatory response syndrome and quantify inflammatory tissue injury in nonhuman primates. Shock. (2013) 39:55–62. doi: 10.1097/SHK.0b013e318276f4ca
17. Suthers AN, Sarantopoulos S. TLR7/TLR9- and B cell receptor-signaling crosstalk: promotion of potentially dangerous B cells. Front Immunol. (2017) 8:775. doi: 10.3389/fimmu.2017.00775
18. Montes CL, Acosta-Rodríguez EV, Merino MC, Bermejo DA, Gruppi A. Polyclonal B cell activation in infections: infectious agents' devilry or defense mechanism of the host? J Leuk Biol. (2007) 82:1027–32. doi: 10.1189/jlb.0407214
19. Lanzavecchia A, Bernasconi N, Traggiai E, Ruprecht CR, Corti D, Sallusto F. Understanding and making use of human memory B cells. Immunol Rev. (2006) 211:303–9. doi: 10.1111/j.0105-2896.2006.00403.x
20. Miller YI, Choi S-H, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. (2011) 108:235–48. doi: 10.1161/CIRCRESAHA.110.223875
21. Weismann D, Binder CJ. The innate immune response to products of phospholipid peroxidation. Biochim Biophys Acta. (2012) 1818:2465–75. doi: 10.1016/j.bbamem.2012.01.018
22. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. (2016) 16:485–97. doi: 10.1038/nri.2016.63
23. Shlomchik MJ, Weisel F. Germinal center selection and the development of memory B and plasma cells. Immunol Rev. (2012) 247:52–63. doi: 10.1111/j.1600-065X.2012.01124.x
24. Garcia De Vinuesa C, O'Leary P, Sze DM, Toellner KM, MacLennan IC. T-independent type 2 antigens induce B cell proliferation in multiple splenic sites, but exponential growth is confined to extrafollicular foci. Eur J Immunol. (1999) 29:1314–23. doi: 10.1002/(SICI)1521-4141(199904)29:04<1314::AID-IMMU1314>3.0.CO;2-4
25. Coutinho A, Gronowicz E, Bullock WW, Möller G. Mechanism of thymus-independent immunocyte triggering. Mitogenic activation of B cells results in specific immune responses. J Exp Med. (1974) 139:74–92. doi: 10.1084/jem.139.1.74
26. Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. (1995) 374:546–9. doi: 10.1038/374546a0
27. Pasare C, Medzhitov R. Control of B-cell responses by Toll-like receptors. Nature. (2005) 438:364–8. doi: 10.1038/nature04267
28. Genestier L, Taillardet M, Mondiere P, Gheit H, Bella C, Defrance T. TLR agonists selectively promote terminal plasma cell differentiation of B cell subsets specialized in thymus-independent responses. J Immunol. (2007) 178:7779–86. doi: 10.4049/jimmunol.178.12.7779
29. Ehrenstein MR, Notley CA. The importance of natural IgM: scavenger, protector and regulator. Nat Rev Immunol. (2010) 10:778–86. doi: 10.1038/nri2849
30. Cerutti A, Cols M, Puga I. Marginal zone B cells: virtues of innate-like antibody-producing lymphocytes. Nat Rev Immunol. (2013) 13:118–32. doi: 10.1038/nri3383
31. Panda S, Ding JL. Natural antibodies bridge innate and adaptive immunity. J Immunol. (2015) 194:13–20. doi: 10.4049/jimmunol.1400844
32. Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol. (2011) 11:34–46. doi: 10.1038/nri2901
33. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. (2015) 15:160–71. doi: 10.1038/nri3795
34. Savage HP, Baumgarth N. Characteristics of natural antibody-secreting cells. Ann N Y Acad Sci. (2015) 23:12799. doi: 10.1111/nyas.12799
35. Martin F, Kearney JF. Marginal-zone B cells. Nat Rev Immunol. (2002) 2:323–35. doi: 10.1038/nri799
36. Zouali M, Richard Y. Marginal zone B-cells, a gatekeeper of innate immunity. Front Immunol. (2011) 2:63. doi: 10.3389/fimmu.2011.00063
37. MacLennan IC, Toellner KM, Cunningham AF, Serre K, Sze DM, Zuniga E, et al. Extrafollicular antibody responses. Immunol Rev. (2003) 194:8–18. doi: 10.1034/j.1600-065X.2003.00058.x
38. Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. (2001) 14:617–29. doi: 10.1016/S1074-7613(01)00129-7
39. Maier S, Traeger T, Entleutner M, Westerholt A, Kleist B, Hüser N, et al. Cecal ligation and puncture versus colon ascendens stent peritonitis: two distinct animal models for polymicrobial sepsis: two distinct animal models for polymicrobial sepsis. Shock. (2004) 21:505–11. doi: 10.1097/01.shk.0000126906.52367.dd
40. Zantl N, Uebe A, Neumann B, Wagner H, Siewert JR, Holzmann B, et al. Essential role of gamma interferon in survival of colon ascendens stent peritonitis, a novel murine model of abdominal sepsis. Infect Immun. (1998) 66:2300–9. doi: 10.1128/IAI.66.5.2300-2309.1998
41. Holtfreter S, Nguyen TT, Wertheim H, Steil L, Kusch H, Truong QP, et al. Human immune proteome in experimental colonization with Staphylococcus aureus. Clin Vaccine Immunol. (2009) 16:1607–14. doi: 10.1128/CVI.00263-09
42. Chou M-Y, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest. (2009) 119:1335–49. doi: 10.1172/JCI36800
43. Binder CJ, Hörkkö S, Dewan A, Chang M-K, Kieu EP, Goodyear CS, et al. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med. (2003) 9:736–43. doi: 10.1038/nm876
44. Kantor AB, Merrill CE, Herzenberg LA, Hillson JL. An unbiased analysis of V(H)-D-J(H) sequences from B-1a, B-1b, and conventional B cells. J Immunol. (1997) 158:1175–86.
45. Tiller T, Busse CE, Wardemann H. Cloning and expression of murine Ig genes from single B cells. J Immunol Methods. (2009) 350:183–93. doi: 10.1016/j.jim.2009.08.009
46. Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med. (2006) 203:553–61. doi: 10.1084/jem.20052438
47. Schmoeckel K, Traffehn S, Eger C, Potschke C, Broker BM. Full activation of CD4+ T cells early during sepsis requires specific antigen. Shock. (2015) 43:192–200. doi: 10.1097/SHK.0000000000000267
48. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. (2003) 301:1374–7. doi: 10.1126/science.1086907
49. Tiller T, Tsuiji M, Yurasov S, Velinzon K, Nussenzweig MC, Wardemann H. Autoreactivity in human IgG+ memory B cells. Immunity. (2007) 26:205–13. doi: 10.1016/j.immuni.2007.01.009
50. Tiller T, Kofer J, Kreschel C, Busse CE, Riebel S, Wickert S, et al. Development of self-reactive germinal center B cells and plasma cells in autoimmune Fc gammaRIIB-deficient mice. J Exp Med. (2010) 207:2767–78. doi: 10.1084/jem.20100171
51. Palinski W, Hörkkö S, Miller E, Steinbrecher UP, Powell HC, Curtiss LK, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest. (1996) 98:800–14. doi: 10.1172/JCI118853
52. Berek C, Milstein C. Mutation drift and repertoire shift in the maturation of the immune response. Immunol Rev. (1987) 96:23–41. doi: 10.1111/j.1600-065X.1987.tb00507.x
53. Cabrera-Perez J, Condotta SA, James BR, Kashem SW, Brincks EL, Rai D, et al. Alterations in antigen-specific naive CD4 T cell precursors after sepsis impairs their responsiveness to pathogen challenge. J Immunol. (2015) 194:1609–20. doi: 10.4049/jimmunol.1401711
54. Vieira P, Rajewsky K. The half-lives of serum immunoglobulins in adult mice. Eur J Immunol. (1988) 18:313–6. doi: 10.1002/eji.1830180221
55. Sigounas G, Harindranath N, Donadel G, Notkins AL. Half-life of polyreactive antibodies. J Clin Immunol. (1994) 14:134–40. doi: 10.1007/BF01541346
56. Barnay-Verdier S, Fattoum L, Borde C, Kaveri S, Gibot S, Maréchal V. Emergence of autoantibodies to HMGB1 is associated with survival in patients with septic shock. Intensive care Med. (2011) 37:957–62. doi: 10.1007/s00134-011-2192-6
57. Malfussi H, Santana IV, Gasparotto J, Righy C, Tomasi CD, Gelain DP, et al. Anti-NMDA receptor autoantibody is an independent predictor of hospital mortality but not brain dysfunction in septic patients. Front Neurol. (2019) 10:221. doi: 10.3389/fneur.2019.00221
58. Burbelo PD, Seam N, Groot S, Ching KH, Han BL, Meduri GU, et al. Rapid induction of autoantibodies during ARDS and septic shock. J Transl Med. (2010) 8:97. doi: 10.1186/1479-5876-8-97
59. Rivera-Correa J, Rodriguez A. Divergent roles of antiself antibodies during infection. Trends Immunol. (2018) 39:515–22. doi: 10.1016/j.it.2018.04.003
60. Portocala R, Spyrou N, Lambropoulou V, Pateraki E. The presence of both antivirus and antiself antibodies in sera from patients with adenovirus and influenza B. Virologie. (1988) 39:207–16.
61. Sakakibara S, Yasui T, Jinzai H, O'donnell K, Tsai C-Y, Minamitani T, et al. Self-reactive and polyreactive B cells are generated and selected in the germinal center during γ-herpesvirus infection. Int Immunol. (2020) 32:27–38. doi: 10.1093/intimm/dxz057
62. Joubert B, Dalmau J. The role of infections in autoimmune encephalitides. Rev Neurol. (2019) 175:420–6. doi: 10.1016/j.neurol.2019.07.004
63. Hotchkiss RS, Tinsley KW, Swanson PE, Grayson MH, Osborne DF, Wagner TH, et al. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol. (2002) 168:2493–500. doi: 10.4049/jimmunol.168.5.2493
64. Peck-Palmer OM, Unsinger J, Chang KC, Davis CG, McDunn JE, Hotchkiss RS. Deletion of MyD88 markedly attenuates sepsis-induced T and B lymphocyte apoptosis but worsens survival. J Leuk Biol. (2008) 83:1009–18. doi: 10.1189/jlb.0807528
65. Tinsley KW, Grayson MH, Swanson PE, Drewry AM, Chang KC, Karl IE, et al. Sepsis induces apoptosis and profound depletion of splenic interdigitating and follicular dendritic cells. J Immunol. (2003) 171:909–14. doi: 10.4049/jimmunol.171.2.909
66. Wang SD, Huang KJ, Lin YS, Lei HY. Sepsis-induced apoptosis of the thymocytes in mice. J Immunol. (1994) 152:5014–21.
67. Busse M, Traeger T, Potschke C, Billing A, Dummer A, Friebe E, et al. Detrimental role for CD4+ T lymphocytes in murine diffuse peritonitis due to inhibition of local bacterial elimination. Gut. (2008) 57:188–95. doi: 10.1136/gut.2007.121616
68. Palma J, Tokarz-Deptuła B, Deptuła J, Deptuła W. Natural antibodies – facts known and unknown. Cent Eur J Immunol. (2018) 43:466–75. doi: 10.5114/ceji.2018.81354
69. Yang Y, Tung JW, Ghosn EE, Herzenberg LA. Division and differentiation of natural antibody-producing cells in mouse spleen. Proc Natl Acad Sci USA. (2007) 104:4542–6. doi: 10.1073/pnas.0700001104
70. Chang M-K, Binder CJ, Miller YI, Subbanagounder G, Silverman GJ, Berliner JA, et al. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med. (2004) 200:1359–70. doi: 10.1084/jem.20031763
71. Panda S, Zhang J, Tan NS, Ho B, Ding JL. Natural IgG antibodies provide innate protection against ficolin-opsonized bacteria. EMBO J. (2013) 32:2905–19. doi: 10.1038/emboj.2013.199
Keywords: CASP, polymicrobial sepsis, B cell response, polyreactive antibodies, oxidation-specific epitopes
Citation: Nicolai O, Pötschke C, Raafat D, van der Linde J, Quosdorf S, Laqua A, Heidecke C-D, Berek C, Darisipudi MN, Binder CJ and Bröker BM (2020) Oxidation-Specific Epitopes (OSEs) Dominate the B Cell Response in Murine Polymicrobial Sepsis. Front. Immunol. 11:1570. doi: 10.3389/fimmu.2020.01570
Received: 14 February 2020; Accepted: 15 June 2020;
Published: 31 July 2020.
Edited by:
Florian Uhle, Heidelberg University Hospital, GermanyReviewed by:
Huan Yang, Feinstein Institute for Medical Research, United StatesPrasad Srikakulapu, University of Virginia, United States
Xuchu Que, University of California, San Diego, United States
Copyright © 2020 Nicolai, Pötschke, Raafat, van der Linde, Quosdorf, Laqua, Heidecke, Berek, Darisipudi, Binder and Bröker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara M. Bröker, YnJvZWtlckB1bmktZ3JlaWZzd2FsZC5kZQ==
†Present address: Oliver Nicolai, Z.A.S. Zentral Archiv Service GmbH, Neubrandenburg, Germany
Christian Pötschke, Salutas Pharma GmbH, Barleben, Germany
Anna Laqua, Department of Hematology, University Hospital Schleswig-Holstein, Kiel, Germany