 Sergio Dellepiane
Sergio Dellepiane Jeremy S. Leventhal2
Jeremy S. Leventhal2 Paolo Cravedi
Paolo Cravedi- 1Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 2Division of Nephrology, White Plains Hospital, White Plains, NY, United States
Acute Kidney Injury (AKI) complicates up to 10% of hospital admissions substantially increasing patient morbidity and mortality. Experimental evidence supports that AKI initiation and maintenance results from immune-mediated damage. Exogenous injury sources directly damage renal cells which produce pro-inflammatory mediators recruiting immune cells and furthering kidney injury. Many AKI studies focus on activation of innate immunity; major components include complement pathways, neutrophils, and monocytes. Recently, growing evidence emphasizes T lymphocytes role in affecting AKI pathogenesis and magnitude. In particular, T helper 17 lymphocytes enhance tissue injury by recruiting neutrophils and other inflammatory cells, while regulatory T cells conversely reduce renal injury and facilitate repair. Intriguingly, evidence supports local parenchymal-T cell interactions as essential to producing T cell phenotypic changes affecting long-term kidney and patient survival. Herein, we review T cells effects on AKI and patient outcomes and discuss related new therapeutic approaches to improve outcomes of affected individuals.
Acute kidney injury (AKI) is clinically defined by rapid renal function decline indicated by serum creatine rise ≥0.3 mg/dl (or >50% from baseline) and/or urine output ≤500 ml/day (1). It is classified as pre-renal, post-renal or parenchymal (also known as intrinsic) depending on the primary site of injury. Pre-renal and post-renal AKI are consequences of altered renal perfusion or urinary tract obstruction, respectively; thus, they represent extrinsic disorders. However, if pre/post-renal injuries persist, AKI will eventually evolve to cellular damage and intrinsic kidney disease. Pathophysiologically, AKI represents complex interactions of exogenous injury and host responses culminating in decreased glomerular filtration.
In the last decade, new approaches focused on more specific nomenclature across types of parenchymal AKI (2). Indeed, while pre- and post-renal AKI are frequently reversible and minimally impactful on patient survival (3), parenchymal AKI is an emerging global health concern, increases patient morbidity/mortality risk, and rose in incidence over the last 30 years (4). In industrialized countries, AKI affects 5–10% of hospitalized patients and 25–50% of those in intensive care units (ICU) (4, 5). A 2013 meta-analysis estimated that mortality rates for hospital-acquired AKI is ~23% and rises to 50% in subsets requiring dialysis (5). Similarly, a large registry study on >190,000 patients reported 90-day AKI mortality rates of 37% (vs. 7% in non-AKI group). In the same cohort 2 years post-discharge, AKI survivors' combined risk of death, end stage renal disease (ESRD) or chronic kidney disease (CKD) was >30%, more than double of the cohort without AKI (6, 7).
Taken together, clinical data and experimental animal AKI models, concur that AKI associates or contributes to lung, heart, liver, brain, or gut damage (8) that produces long-term sequelae in affected organs (9). Importantly, immune system function is tightly linked to AKI with bidirectional influence. While sepsis is a recognized leading cause of hospital-associated kidney injury (4), AKI also associates with increased infection risk even after full recovery of renal function (10, 11). The first studies about immune cell activation during AKI focus on innate immune response; more recently research shows adaptive immunity activation during AKI contributing to renal and extra-renal outcomes. Herein, we will review both adaptive immune contributions to AKI and immune function changes related to AKI.
Etiologies of Hospital Related Acute Kidney Injury
AKI encompasses a broad spectrum of renal insults causing decreased filtration. In the last decade, multiple classifications were proposed to identify and study underlying conditions (2). From an epidemiological point of view, an important difference exists between community acquired vs. hospital related AKI. Community-AKI is more likely pre-renal and usually occurs in older or medically compromised patients from dehydration or from drugs that limit glomerular perfusion (e.g., non-steroidal anti-inflammatory drugs or inhibitors of the renin angiotensin aldosterone axis) (3). Conversely, hospital-acquired AKI is more often intrinsic and more likely to be severe. Another classification identifies major clinical syndromes and procedures that have a strong causative link with AKI (e.g., sepsis related-AKI, post-cardiac surgery AKI etc.); the definition of these clinical settings may guide clinicians in the diagnostic and therapeutic approach. From an etiologic point of view, these AKI types share a large part of the underlying mechanisms (2) (Figure 1).
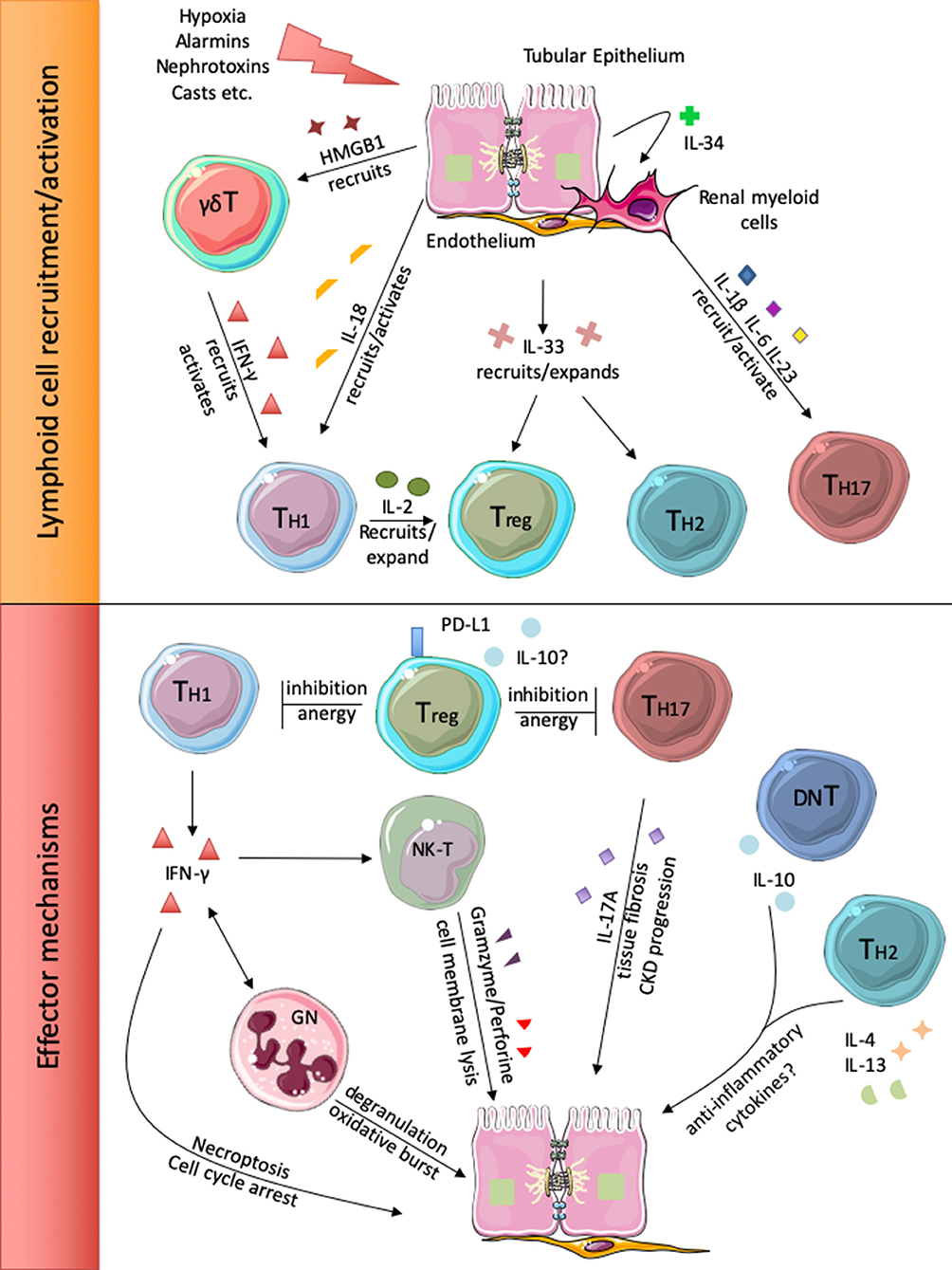
Figure 1. Injured renal cells release different alarm signals that recruit and activate local and circulating lymphoid cells (upper panel). Subsequently, the different lymphocyte subsets participate to renal injury perpetration or inhibition (lower panel).
Sepsis is a leading cause of in-hospital AKI accounting for 30–50% of cases (4). During sepsis, microbial and released host products act as alarm signals (or alarmins) targeting pattern recognition receptors (PRR) (12). Renal endothelium, tubular epithelial cells (TEC) and immune cells express PRR that sense a wide variety injury related molecular motifs. PRR activation produces pro-inflammatory phenotypes in renal cells which also activate programmed cell death pathways. Immune cells migrate to the site of alarmin release and contribute to local inflammation. In addition to infection consequences, patient courses are further complicated by nephrotoxic drugs (e.g., aminoglycosides) and sepsis-related ischemic injury, discussed in more depth below.
Ischemia-reperfusion injury (IRI) is probably the most studied experimental AKI model. Clinically, it occurs from severe renal hypoperfusion caused by blood loss, septic shock, and other anatomical abnormalities of renal blood supply. Some authors classify post-surgical AKI as a distinct entity due to predictable complicating factors of alarmin release (as consequence of bacterial contamination or tissue damage) or nephrotoxic drug administration (e.g., contrast dye) (13). In animal models, protracted IRI induces acute tubular necrosis (ATN), a lesion characterized by the extensive necrosis of the proximal tubular segments at the corticomedullary junction. However, renal biopsies from patients with ischemic AKI show mild parenchymal damage, despite severe organ dysfunction; thus, since 2000, more studies hypothesize microvascular failure and forms of cellular dysfunction (e.g., loss of polarity, epithelial leaking, organelle injury etc.) being predominant features in humans (14).
Multiple interacting etiologies contribute to cancer-related AKI. Oncologic patients suffer AKI from combinations of tumor lysis syndrome, infections, procedural complications, neoplastic renal invasion, paraproteinemia (mostly related to plasma-cell cancers) and drug toxicity. Traditionally, alkylating agents most frequently caused ATN (e.g., platin compounds). In the last decade new agents, most prominently immune checkpoint blockade inhibitors, are increasingly used. Multiple studies showed how immune checkpoint inhibitors can trigger intra-renal inflammation and autoimmune renal damage (15).
AKI frequently occurs in hospitalized patients but is rarely the cause for admission and frequently occurs from distant organ injury/dysfunction. In hepatorenal syndrome, for example, vasoactive aminic metabolites accumulate in liver failure and produce renal circulatory failure (8). Subtypes of acute cardiorenal syndromes involve renal injury resulting from decreased cardiac output or venous congestion, while others involve cytokine release and neurohormonal changes. Other relevant detrimental cross-talk are mediated by lung (hypoxia, cytokine release), brain (natriuretic peptides, cytokines), or intestine (leaking of bacteria and toxic metabolites) (12). Finally, massive muscle cell lysis seen in crush injuries cause injury. Cell lysis byproducts produce electrolyte unbalance, alarmin release, increased circulating waste products, and myoglobin precipitation in tubular lumen (cast nephropathy) that can combine with dehydration and profoundly injury kidneys (16).
Role of the Innate Inflammatory Response in AKI Pathogenesis
Innate immune cells perpetrate AKI damage directly (e.g., neutrophil degranulation) or by recruiting the adaptive immune cells. At baseline, renal tissue hosts mainly macrophages and dendritic cells (DC), while immature monocytes and neutrophils migrate to the kidney in response to the alarm signal.
Neutrophils
Neutrophils are the most abundant leucocytes infiltrating the kidney immediately after IRI (17) and multiple studies have shown that in-hospital risk of AKI positively correlates with the percentage of circulating neutrophils, suggesting an involvement of these cells in AKI pathogenesis (18). In response to stress, tubular cells acutely release IL-18 which recruits and activates neutrophils (19). Activated renal endothelial cells express E-selectins that bind neutrophil β-integrins and initiate the diapedesis process (20). Neutrophils damage renal tissue by degranulation, IFN-γ release and by recruiting of NK T-cells (21).
Macrophages
Macrophages are the most abundant kidney resident immune cells. During AKI, TEC acutely release IL-34 that activates resident macrophages and promotes tubular injury (22). TEC damage induces also the recruitment and the activation of circulating monocytes (23), which perpetrate the injury by releasing pro-inflammatory cytokines as IL-1β, TNF-α, IL-6, and IL-12 (24). Subsequently, both resident and infiltrating macrophages switch to the regulatory phenotype and promote tissue repair. In particular, resident macrophages develop an embryonic-like gene signature soon after IRI and secrete WNT4, which in turn activates β-catenin pathway in TEC and promotes their proliferation (17, 23).
Dendritic Cells and NK Cells
Resident renal dendritic cells (DC) are the predominant source of TNF-α during AKI and promote TH1 activation by secreting IL-12p40 (25, 26); moreover, they release TH17 inducing cytokines as IL-1 and IL-23 (27).
Natural killer (NK) cells migrate to the kidney in inflammatory conditions and induce TEC damage by releasing perforin (28). A subset of NK expresses an invariant form of T cell receptor (NKT or invariant NK) and substantially contributes to interferon production after renal IRI (21).
Complement System
Complement cascade is acutely activated during AKI and contributes to renal damage, as indicated by data from C3 knock-out mice that are protected from IRI (29). Human and murine data have shown that complement get activated during AKI trough the alternative (30) and the mannose binding leptin pathways (31). Additionally, C3 can be activated in kidney parenchyma after binding ammonia (amidic-C3) (32). Complement activation injuries perpetrates renal injury trough the generation of membrane attack complex, the recruitment of immune cells and the activation of C3b and C5b receptors on tubular and endothelial cells (33).
Adaptive Immune Response
Most studies investigating the role of T cells as AKI mediators focus on CD4 (i.e., T helper cells) while CD8 (cytotoxic) involvement is controversial (see below). Depending on inflammatory context, naïve CD4 cells differentiate to T-helper (TH) 1, 2, 17, or regulatory T cells (Treg). After AKI, TEC primarily release TH1 inducing signals and renal myeloid cells polarize toward TH17; TH2 activation during AKI is less understood.
TH1 Cells
TH1 responses are orchestrated by master regulator transcription factor T-bet and characterized by expression of surface marker CXCR3. TH1 are classically associated with IFN-γ secretion and responses to intracellular pathogens. IFN-γ is a cytokine with a ubiquitous receptor promoting MHC expression, autophagy, reducing cell proliferation, and activating inflammatory death pathways (e.g., necroptosis and pyroptosis) (34). TH1 differentiation and activity are promoted by IL-12, IL-18, and IFN-γ itself. Upon injury, TEC release IL-18, leading to conversion of naïve CD4 T cells into TH1 (35). Rapid intrarenal migration of IFN-γ+ CD4 cells after LPS injection is prevented in IL-18 receptor deficient mice that, in turn, limits kidney injury (36). Cytokines alone cause cell injury, but TH1 cells also recruit other immune cells. Li et al. reported TH1 rapidly travel to ischemic kidneys and promote neutrophil and NK chemotaxis (that peaks after 3 h) (21). Additionally, IFN-γ alters TEC making them apro-inflammatory via expression of costimulatory molecules ICOS-L and B7-1 (37), preventing TEC proliferation (38), and promoting their death by necroptosis (39).
Although T cells promote inflammatory transformation of TEC, cytokine dependent damage appears predominantly based on TH1 responses. Day et al. observed that infiltrating lymphocytes were the main source of IFN-γ, while the cytokine fraction released by TEC was dispensable (40) in experimental models. Human data is corroboratory: in a multicenter prospective study on 1,400 patients undergoing cardiac surgery, post-operatory increase in serum IFN-γ significantly and directly associated with AKI incidence and 1-year mortality (41). Rather than non-specific responses to damage associated molecular patterns (DAMP) experimental evidence points to antigen-specific T cell function. Renal injury was worse after IRI in T-cell depleted mice reconstituted with heterogeneous CD4 cells compared to those given monoclonal ones (42).
TH2 Cells
Little is known regarding T helper 2 (TH2) cells during AKI. TH2 constitutively express transcription factor GATA3 and surface marker Crth2 (CCR4 in mice): canonically, these cells orchestrate the anti-parasitic immune response via IL-4 and IL-13 secretion and are associated with asthma and allergic diseases. Yokota and colleagues induced IRI in mice lacking the transcription factors STAT4 or 6; the first is essential for TH1 response while STAT6 induces TH2 phenotype (43). While STAT4 deficient mice were protected from AKI, STAT6 knock down associated with worse outcomes. Increased tubular injury in IL-4 deficient mice further supports a renoprotective designation for TH2 responses (43). Conversely, human data from post-cardiac surgery patients showed direct relationships between IL-4 and IL-13 serum concentrations, AKI incidence, and 1-year mortality (41). Clearly, further experimental and clinical evidence are required to understand how TH2 responses affect AKI.
TH17 Cells
T helper 17 cells (TH17) are a subset of CD4 lymphocytes characterized by expression of IL-17A, IL-17F, and the transcription factor ROR-γt. TH17 are frequently identified by surface marker CCR6, with variable expression of IL-23R, CCR4, and CCR2 (humans) or CCR7 (mice) (27). In both humans and mice, naïve CD4 experimental polarization is driven by TGF-β, IL-6, IL-21, and IL-23. Other stimuli contribute to mature TH17 activation: angiotensin II, salt excess and IL-1β (27, 44). TH17 cells are particularly abundant in barrier epithelia (e.g., skin, gut) and respond primarily to fungal and extracellular bacterial infections by recruiting neutrophils and activating epithelium via IL-17. TH17 are linked to various autoimmune diseases; the prototypical TH17 disease is psoriasis and it is effectively treated with anti-IL-17 therapies (45).
More recently, TH17 cells emerged as main players in AKI pathophysiology. Different groups demonstrated that TH17 are the most abundant kidney infiltrating lymphocytes infiltrating following AKI in mice (27, 44). Pindjakova et al. observed that resident dendritic cells and TEC release IL-1 (α and β), IL-23, and IL-6 to promote intrarenal IL-17 migration and activation after AKI from ureteral obstruction. IL-1 signaling dominates the phenomenon and its suppression pushes lymphocytes phenotypes to TH1 (27). Mehrota and colleagues demonstrated that TH17 cells expressing calcium channel Orai1 are solely responsible for IL-17 production after IRI and, ultimately, for renal injury (46). A 10-fold increase in circulating Orai1+ TH17 cells are found in ICU patients with AKI compared to those without (46). In vivo, intra-renal expression of Orai1 persisted for days after AKI resolution and its inhibition prevented the transition to chronic kidney disease (CKD) (46). Intriguingly, the kidney also possesses mechanisms to counteract TH17 cell activation. Our group observed that erythropoietin (EPO), a kidney produced hormone, prevents TH17 induction (47) and ameliorates renal injury in a murine model of Balkan nephropathy. Together with studies demonstrating EPO prevents IRI, current experimental evidence strongly support inhibition of dominant TH17 responses are feasible to prevent AKI and related progression to CKD.
T Regulatory Cells
CD4 regulatory cells (Treg) are immunosuppressive T cells characterized phenotypically by constitutively high levels of IL-2 receptor (CD25) expression and maintained functionally via transcription factor FOXP3.
Research suggests that Treg attenuate AKI (48). Jaworska and colleagues observed IRI amelioration after Treg transfer, an effect that was dependent on programmed death ligand 1 and 2 (PD-L1/2) expression by Treg. The relevance of PDL to renal inflammation is supported by experimental demonstration of PD-1 expression by tubular cells (49) and clinical observations of renal adverse events in patients treated with immune checkpoint inhibitors targeting PD-1/PD-L1 axis (50).
After injury, renal Treg inhibit inflammation in multiple ways; they release TGFβ and IL-10, halt production of IL-1β, TNF-α, and IFN-γ, and reduce overall CD4 proliferation (51). It is unclear which between intra-renal or circulating Treg represent the active pool during AKI, though evidence exists supporting both central and peripheral sources. Investigators observed that DC heat shock protein 70 (HSP70) production increased splenic Tregs that migrated to the kidney and attenuated IRI (52), while another recent paper pointed out the role of renal resident Treg during IRI (53). The authors observed a progressive increase in CD3+ T cells after ischemia that paralleled the development of tissue fibrosis. Among the most expanded subsets there was a resident Treg population that was characterized by the expression of IL-33 receptor, a marker usually associated with TH2 phenotype. The administration of IL2 and IL33 at the time of IRI activated this population, promoted rapid recovery, and prevented tissue fibrosis. Of note, a previous study demonstrated that IL-33 is released by renal endothelium after cisplatin administration (54). Given these conflicting results, the answer may be model dependent.
Other T Cells (Gamma-Delta, Double Negative, CD8)
Nomenclature of T cells is receptor based: αβ T cells constitute the majority (i.e., TH1, T2, TH17, and Treg subsets) while γδ T cells, resident in skin and the gut epithelia, constitute <1% of peripheral T cells. Their effector responses, based on still undefined antigens, include release of IL-17, IFN-γ, and TNF-α. In murine IRI, γδ lymphocytes rapidly infiltrate kidney tissue and promote subsequent migration of αβ cells; of note, γδ depletion delays but does not prevent injury, while αβ T cell ablation is protective (55). This suggests γδ T cells affect kinetics of kidney injury but are not necessary for AKI. In a clinical study of 20 patients undergoing abdominal aortic repair, magnitude of γδ T cells disappearance from the circulation was proportional to kidney injury markers (56). The same pattern was seen experimentally in mice where TEC HMGB1 release induced γδ T cell kidney migration, supporting the paradigm that early γδ T cell AKI responses facilitate αβ T cell recruitment.
CD8 T cells role in AKI is less defined, if it exists. A 2001 study found no significant pathogenic role for CD8 cells in IRI (57). A subsequent paper reported that CD8 deficient mice were mildly protected from cisplatin induced AKI, but less than those with CD4 depletion (58). Finally, in a study on acute aristolochic acid nephropathy, authors reported both CD4 and CD8 depletions were detrimental. In particular, absence of CD8 cells was associated to higher intra-renal TNF-α production and reduction of anti-inflammatory macrophages (59). More work is required to more clearly define how, and if, CD8 cells affect AKI.
Double negative (DN) T cells represent an early stage of T cell maturation lacking CD4 and CD8 expression. DN T cells are ubiquitous and some authors hypothesize they are an independent differentiated population rather than a maturation stage (60). These lymphocytes constitute more than 30% of kidney T cells at baseline and rapidly proliferate after tubular damage. Martina et al. reported that DN cells secrete IL-10 after IRI thus being anti-inflammatory (61) and, ostensibly, protective.
B Cells
Experimental B-cells work has not definitively defined their role in AKI. One IRI study with B-cell deficient mice showed decreased AKI. Serum from control mice having restored AKI; these results suggests an antibody-mediated mechanism (62). Renner et al. observed the opposite effect; B-cell deficient mice had less intra-renal IL-10 production and a worse renal outcome (63). Of note, the same study reported a harmful B-cell subset; natural-IgM from peritoneal lymphocytes precipitated on the glomerular basal membrane and activated the complement alternative pathway. Conversely, Lobo and Okusa reported that infusion of natural-IgM was actually protective from IRI by inducing B-regulatory cells (64). These conflicting studies are emblematic of ongoing conflict regarding B cells and AKI.
Larger consensus has been reached about the B-cell role in post-AKI renal fibrosis (65). It has been shown that fibroblasts increase their collagen production in tissues with a higher B cell signature (66). Consistently, B-cell depleting therapy (anti-CD20) prevented kidney interstitial fibrosis after ureteral obstruction (67).
Immune Therapies in AKI
The important effects adaptive immune responses have in AKI pathogenesis suggest, that immune modulatory therapies might effectively achieve clinically desirable results. Pechman and coworkers observed that mycophenolate mofetil (an immunosuppressive agent that inhibits purine synthesis in lymphocytes) prevented AKI long-term sequalae as renal fibrosis and salt-sensitive hypertension (68). Experimental models further link T cells, MMF, and CKD; a murine study modeling AKI transitions to CKD, showed an inverse association between mycophenolate treatment vs. TH17 proliferation and CKD (69). Clinically relevant approaches preventing TH17 responses (i.e., EPO receptor agonism and Orai1 inhibition) effectively halted kidney disease in murine models. Moreover, targeting TH17 effector molecule, IL-17, prevented calcineurin inhibitor related renal fibrosis (70). Taken together, these findings justify further studies targeting TH17 responses to prevent CKD.
A promising approach to treat AKI is the promotion of endogenous Treg expansion. A known strategy promoting Treg expansion involves IL-2 function. The IL-2/anti-IL2 complex is a mixture of IL-2 with an antibody that prevents IL-2 binding to the β-chain of its receptor (CD25); this complex strongly induced Treg proliferation and attenuated IRI by reducing neutrophil and macrophage migration in renal tissue (71). Other authors generated a fusion IL-2/IL-33 cytokine that expanded intra-renal IL-33R+ Treg, halted CD4 effector cell proliferation and prevented 100% of the observed mortality in a murine IRI model (72). Dimethylsphingosine (DMS) promotes CD4 migration to kidneys at baseline and after ischemia; Lai et al. demonstrated that FOXP3+ lymphocytes were proportionally more abundant in the renal tissue after DMS treatment and prevented IRI in mice (73).
Treg adoptive transfer effectively downregulates IRI and other types of renal injury in pre-clinical models (48); however, its clinical application remains challenging. Therapeutic cell products need specific cell factories, are temperature and time-sensitive (thus needing complicated stock and transport procedures), are prone to contamination, may degenerate in neoplastic disease and can trigger the host immune response toward allo-antigens (73).
Stem cell therapies have been proposed for the management of AKI; in particular, mesenchymal stromal cells (MSC) have been successfully used in different preclinical models and are currently under investigation in clinical trials (74). MSC are immune-regulatory; importantly, MSC infusion expands intra-renal Treg after IRI. Consequent reductions in circulating IL-6, TNF-α and IFN-γ levels are Treg dependent (75). Treg strategies, therefore, intersect with other established experimental protocols.
However, cell infusion poses previously mentioned challenges, even if MSC are relatively easy to expand and not immunogenic. An interesting alternative comes from the observation that MSC conditioned medium is as effective as MSC infusion in promoting tissue regeneration (76). Indeed, MSC beneficial effects in AKI are not contact-mediated and MSC do not differentiate in any mature kidney cell. If future efforts identify substances inducing intra-renal Treg expansion therapies would avoid cell infusion complications.
Conclusions
Inflammation produces AKI via reciprocal interactions between renal parenchyma, resident immune cells, and recruited immune cells. Increasing recent evidence indicates a dominant role of the adaptive immune response, and T cells in particular, as prominent pathogenic elements as well as mitigating factors. Myriad AKI etiologies frequently condense into recurrent identifiable immune patterns associated with tubular injury and T cells. In particular, CD4 and γδ T cells are initial immune effectors migrating to kidneys and orchestrating activation of innate cells. Early injury phases are characterized by a strong IFN-γ response, possibly produced by TH1 cells. In later phases, TH17 perpetuate injury and tissue fibrosis. Conversely, Treg and possibly TH2 exert opposing anti-inflammatory roles and limit or prevent injury. Pre-clinical and observational studies provide strong bases of feasibility for future pharmacological interventions targeting lymphocyte function to prevent and limit AKI as well as subsequent renal fibrosis.
Author Contributions
SD wrote the first draft. JL reviewed it. PC had the original idea and reviewed the manuscript. All authors approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Khwaja A, Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract. (2012) 120:c179–c84. doi: 10.1159/000339789
2. Kellum JA, Lameire N. The definition of acute kidney injury. Lancet Lond Engl. (2018) 391:202–3. doi: 10.1016/S0140-6736(17)31630-6
3. Kaufman J, Dhakal M, Patel B, Hamburger R. Community-acquired acute renal failure. Am J Kidney Dis. (1991) 17:191–8. doi: 10.1016/s0272-6386(12)81128-0
4. Bellomo R. The epidemiology of acute renal failure: 1975 versus 2005. Curr Opin Crit Care. (2006) 12:557–60. doi: 10.1097/01.ccx.0000247443.86628.68
5. Susantitaphong P, Cruz DN, Cerda J, Abulfaraj M, Alqahtani F, Koulouridis I, et al. World incidence of AKI: a meta-analysis. Clin J Am Soc Nephrol. (2013) 8:1482–93. doi: 10.2215/CJN.00710113
6. Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR. Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis. (2009) 53:961–73. doi: 10.1053/j.ajkd.2008.11.034
7. Pannu N, James M, Hemmelgarn B, Klarenbach S, Alberta Kidney Disease Network. Association between AKI, recovery of renal function, and long-term outcomes after hospital discharge. Clin J Am Soc Nephrol. (2013) 8:194–202. doi: 10.2215/CJN.06480612
8. Lee SA, Cozzi M, Bush EL, Rabb H. Distant organ dysfunction in acute kidney injury: a review. Am J Kidney Dis. (2018) 72:846–56. doi: 10.1053/j.ajkd.2018.03.028
9. Vieira JM, Castro I, Curvello-Neto A, Demarzo S, Caruso P, Pastore L, et al. Effect of acute kidney injury on weaning from mechanical ventilation in critically ill patients. Crit Care Med. (2007) 35:184–91. doi: 10.1097/01.CCM.0000249828.81705.65
10. Mehta RL, Bouchard J, Soroko SB, Ikizler TA, Paganini EP, Chertow GM, et al. Sepsis as a cause and consequence of acute kidney injury: program to improve care in acute renal disease. Intensive Care Med. (2011) 37:241–8. doi: 10.1007/s00134-010-2089-9
11. Lai T-S, Wang C-Y, Pan S-C, Huang T-M, Lin M-C, Lai C-F, et al. Risk of developing severe sepsis after acute kidney injury: a population-based cohort study. Crit Care Lond Engl. (2013) 17:R231. doi: 10.1186/cc13054
12. Cantaluppi V, Quercia AD, Dellepiane S, Ferrario S, Camussi G, Biancone L. Interaction between systemic inflammation and renal tubular epithelial cells. Nephrol Dial Transplant. (2014) 29:2004–11. doi: 10.1093/ndt/gfu046
13. Wang Y, Bellomo R. Cardiac surgery-associated acute kidney injury: risk factors, pathophysiology and treatment. Nat Rev Nephrol. (2017) 13:697–711. doi: 10.1038/nrneph.2017.119
14. Kellum JA. Acute kidney injury. Crit Care Med. (2008) 36:S141–5. doi: 10.1097/CCM.0b013e318168c4a4
15. Murakami N, Motwani S, Riella LV. Renal complications of immune checkpoint blockade. Curr Probl Cancer. (2017) 41:100–10. doi: 10.1016/j.currproblcancer.2016.12.004
16. Sever MS, Lameire N, Van Biesen W, Vanholder R. Disaster nephrology: a new concept for an old problem. Clin Kidney J. (2015) 8:300–9. doi: 10.1093/ckj/sfv024
17. Lever JM, Hull TD, Boddu R, Pepin ME, Black LM, Adedoyin OO, et al. Resident macrophages reprogram toward a developmental state after acute kidney injury. JCI Insight. (2019) 4:e125503. doi: 10.1172/jci.insight.125503
18. Gameiro J, Fonseca JA, Dias JM, Milho J, Rosa R, Jorge S, et al. Neutrophil, lymphocyte and platelet ratio as a predictor of postoperative acute kidney injury in major abdominal surgery. BMC Nephrol. (2018) 19:320. doi: 10.1186/s12882-018-1073-4
19. Leung BP, Culshaw S, Gracie JA, Hunter D, Canetti CA, Campbell C, et al. A role for IL-18 in neutrophil activation. J Immunol. (2001) 167:2879–86. doi: 10.4049/jimmunol.167.5.2879
20. Block H, Herter JM, Rossaint J, Stadtmann A, Kliche S, Lowell CA, et al. Crucial role of SLP-76 and ADAP for neutrophil recruitment in mouse kidney ischemia-reperfusion injury. J Exp Med. (2012) 209:407–21. doi: 10.1084/jem.20111493
21. Li L, Huang L, Sung SJ, Lobo PI, Brown MG, Gregg RK, et al. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol. (2007) 178:5899–911. doi: 10.4049/jimmunol.178.9.5899
22. Zhang M-Z, Yao B, Yang S, Jiang L, Wang S, Fan X, et al. CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest. (2012) 122:4519–32. doi: 10.1172/JCI60363
23. Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi B-S, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. (2011) 22:317–26. doi: 10.1681/ASN.2009060615
24. Duque AG, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. (2014) 5:491. doi: 10.3389/fimmu.2014.00491
25. Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. (2007) 71:619–28. doi: 10.1038/sj.ki.5002132
26. Bajwa A, Huang L, Ye H, Dondeti K, Song S, Rosin DL, et al. Dendritic cell sphingosine 1-phosphate receptor-3 regulates Th1-Th2 polarity in kidney ischemia-reperfusion injury. J Immunol. (2012) 189:2584–96. doi: 10.4049/jimmunol.1200999
27. Pindjakova J, Hanley SA, Duffy MM, Sutton CE, Weidhofer GA, Miller MN, et al. Interleukin-1 accounts for intrarenal Th17 cell activation during ureteral obstruction. Kidney Int. (2012) 81:379–90. doi: 10.1038/ki.2011.348
28. Zhang Z-X, Wang S, Huang X, Min W-P, Sun H, Liu W, et al. NK cells induce apoptosis in tubular epithelial cells and contribute to renal ischemia-reperfusion injury. J Immunol. (2008) 181:7489–98. doi: 10.4049/jimmunol.181.11.7489
29. Farrar CA, Zhou W, Lin T, Sacks SH. Local extravascular pool of C3 is a determinant of postischemic acute renal failure. FASEB J. (2006) 20:217–26. doi: 10.1096/fj.05-4747com
30. Thurman JM, Ljubanovic D, Edelstein CL, Gilkeson GS, Holers VM. Lack of a functional alternative complement pathway ameliorates ischemic acute renal failure in mice. J Immunol. (2003) 170:1517–23. doi: 10.4049/jimmunol.170.3.1517
31. De Vries B, Walter SJ, Peutz-Kootstra CJ, Wolfs TGAM, van Heurn LWE, Buurman WA. The mannose-binding lectin-pathway is involved in complement activation in the course of renal ischemia-reperfusion injury. Am J Pathol. (2004) 165:1677–88. doi: 10.1016/S0002-9440(10)63424-4
32. Nath KA, Hostetter MK, Hostetter TH. Pathophysiology of chronic tubulo-interstitial disease in rats. Interactions of dietary acid load, ammonia, and complement component C3. J Clin Invest. (1985) 76:667–75. doi: 10.1172/JCI112020
33. Castellano G, Franzin R, Sallustio F, Stasi A, Banelli B, Romani M, et al. Complement component C5a induces aberrant epigenetic modifications in renal tubular epithelial cells accelerating senescence by Wnt4/βcatenin signaling after ischemia/reperfusion injury. Aging. (2019) 11:4382–406. doi: 10.18632/aging.102059
34. Bhat MY, Solanki HS, Advani J, Khan AA, Keshava Prasad TS, Gowda H, et al. Comprehensive network map of interferon gamma signaling. J Cell Commun Signal. (2018) 12:745–51. doi: 10.1007/s12079-018-0486-y
35. Xu D, Trajkovic V, Hunter D, Leung BP, Schulz K, Gracie JA, et al. IL-18 induces the differentiation of Th1 or Th2 cells depending upon cytokine milieu and genetic background. Eur J Immunol. (2000) 30:3147–56. doi: 10.1002/1521-4141(200011)30:11<3147::AID-IMMU3147>3.0.CO;2-J
36. Nozaki Y, Hino S, Ri J, Sakai K, Nagare Y, Kawanishi M, et al. Lipopolysaccharide-Induced acute kidney injury is dependent on an IL-18 receptor signaling pathway. Int J Mol Sci. (2017) 18:2777. doi: 10.3390/ijms18122777
37. De Haij S, Woltman AM, Trouw LA, Bakker AC, Kamerling SW, van der Kooij SW, et al. Renal tubular epithelial cells modulate T-cell responses via ICOS-L and B7-H1. Kidney Int. (2005) 68:2091–102. doi: 10.1111/j.1523-1755.2005.00665.x
38. García-Sánchez O, López-Novoa JM, López-Hernández FJ. Interferon-γ reduces the proliferation of primed human renal tubular cells. Nephron Extra. (2014) 4:1–7. doi: 10.1159/000353587
39. Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, et al. A role for tubular necroptosis in cisplatin-induced AKI. J Am Soc Nephrol. (2015) 26:2647–58. doi: 10.1681/ASN.2014080741
40. Day Y-J, Huang L, Ye H, Li L, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: the role of CD4+ T cells and IFN-gamma. J Immunol. (2006) 176:3108–14. doi: 10.4049/jimmunol.176.5.3108
41. Moledina DG, Mansour SG, Jia Y, Obeid W, Thiessen-Philbrook H, Koyner JL, et al. Association of T cell-derived inflammatory cytokines with acute kidney injury and mortality after cardiac surgery. Kidney Int Rep. (2019) 4:1689–97. doi: 10.1016/j.ekir.2019.09.003
42. Satpute SR, Park JM, Jang HR, Agreda P, Liu M, Gandolfo MT, et al. The role for T cell repertoire/antigen-specific interactions in experimental kidney ischemia reperfusion injury. J Immunol. (2009) 183:984–92. doi: 10.4049/jimmunol.0801928
43. Yokota N, Burne-Taney M, Racusen L, Rabb H. Contrasting roles for STAT4 and STAT6 signal transduction pathways in murine renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. (2003) 285:F319–25. doi: 10.1152/ajprenal.00432.2002
44. Mehrotra P, Patel JB, Ivancic CM, Collett JA, Basile DP. Th-17 cell activation in response to high salt following acute kidney injury is associated with progressive fibrosis and attenuated by AT-1R antagonism. Kidney Int. (2015) 88:776–84. doi: 10.1038/ki.2015.200
45. Balato A, Scala E, Balato N, Caiazzo G, Di Caprio R, Monfrecola G, et al. Biologics that inhibit the Th17 pathway and related cytokines to treat inflammatory disorders. Expert Opin Biol Ther. (2017) 17:1363–74. doi: 10.1080/14712598.2017.1363884
46. Mehrotra P, Sturek M, Neyra JA, Basile DP. Calcium channel Orai1 promotes lymphocyte IL-17 expression and progressive kidney injury. J Clin Invest. (2019) 129:4951–61. doi: 10.1172/JCI126108
47. Donadei C, Angeletti A, Cantarelli C, D'Agati VD, La Manna G, Fiaccadori E, et al. Erythropoietin inhibits SGK1-dependent TH17 induction and TH17-dependent kidney disease. JCI Insight. (2019) 5:e127428. doi: 10.1172/jci.insight.127428
48. Lee H, Nho D, Chung H-S, Lee H, Shin M-K, Kim S-H, et al. CD4+CD25+ regulatory T cells attenuate cisplatin-induced nephrotoxicity in mice. Kidney Int. (2010) 78:1100–9. doi: 10.1038/ki.2010.139
49. Jaworska K, Ratajczak J, Huang L, Whalen K, Yang M, Stevens BK, et al. Both PD-1 ligands protect the kidney from ischemia reperfusion injury. J Immunol. (2015) 194:325–33. doi: 10.4049/jimmunol.1400497
50. Wanchoo R, Karam S, Uppal NN, Barta VS, Deray G, Devoe C, et al. Adverse renal effects of immune checkpoint inhibitors: a narrative review. Am J Nephrol. (2017) 45:160–9. doi: 10.1159/000455014
51. Chen G, Dong Z, Liu H, Liu Y, Duan S, Liu Y, et al. mTOR signaling regulates protective activity of transferred CD4+Foxp3+ T cells in repair of acute kidney injury. J Immunol. (2016) 197:3917–26. doi: 10.4049/jimmunol.1601251
52. Kim M-G, Jung Cho E, Won Lee J, Sook Ko Y, Young Lee H, Jo S-K, et al. The heat-shock protein-70-induced renoprotective effect is partially mediated by CD4+ CD25+ Foxp3 + regulatory T cells in ischemia/reperfusion-induced acute kidney injury. Kidney Int. (2014) 85:62–71. doi: 10.1038/ki.2013.277
53. Do Valle Duraes F, Lafont A, Beibel M, Martin K, Darribat K, Cuttat R, et al. Immune cell landscaping reveals a protective role for regulatory T cells during kidney injury and fibrosis. JCI Insight. (2020) 5:e130651. doi: 10.1172/jci.insight.130651
54. Akcay A, Nguyen Q, He Z, Turkmen K, Won Lee D, Hernando AA, et al. IL-33 exacerbates acute kidney injury. J Am Soc Nephrol. (2011) 22:2057–67. doi: 10.1681/ASN.2010091011
55. Hochegger K, Schätz T, Eller P, Tagwerker A, Heininger D, Mayer G, et al. Role of alpha/beta and gamma/delta T cells in renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. (2007) 293:F741–7. doi: 10.1152/ajprenal.00486.2006
56. Göcze I, Ehehalt K, Zeman F, Riquelme P, Pfister K, Graf BM, et al. Postoperative cellular stress in the kidney is associated with an early systemic γδ T-cell immune cell response. Crit Care Lond Engl. (2018) 22:168. doi: 10.1186/s13054-018-2094-x
57. Burne MJ, Daniels F, El Ghandour A, Mauiyyedi S, Colvin RB, O'Donnell MP, et al. Identification of the CD4(+) T cell as a major pathogenic factor in ischemic acute renal failure. J Clin Invest. (2001) 108:1283–90. doi: 10.1172/JCI12080
58. Liu M, Chien C-C, Burne-Taney M, Molls RR, Racusen LC, Colvin RB, et al. A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J Am Soc Nephrol. (2006) 17:765–74. doi: 10.1681/ASN.2005010102
59. Baudoux T, Husson C, De Prez E, Jadot I, Antoine M-H, Nortier JL, et al. CD4+ and CD8+ T cells exert regulatory properties during experimental acute aristolochic acid nephropathy. Sci Rep. (2018) 8:5334. doi: 10.1038/s41598-018-23565-2
60. Ascon DB, Ascon M, Satpute S, Lopez-Briones S, Racusen L, Colvin RB, et al. Normal mouse kidneys contain activated and CD3+CD4- CD8- double-negative T lymphocytes with a distinct TCR repertoire. J Leukoc Biol. (2008) 84:1400–9. doi: 10.1189/jlb.0907651
61. Martina MN, Noel S, Saxena A, Bandapalle S, Majithia R, Jie C, et al. Double-Negative αβ T cells are early responders to AKI and are found in human kidney. J Am Soc Nephrol. (2016) 27:1113–23. doi: 10.1681/ASN.2014121214
62. Burne-Taney MJ, Ascon DB, Daniels F, Racusen L, Baldwin W, Rabb H. B cell deficiency confers protection from renal ischemia reperfusion injury. J Immunol. (2003) 171:3210–5. doi: 10.4049/jimmunol.171.6.3210
63. Renner B, Strassheim D, Amura CR, Kulik L, Ljubanovic D, Glogowska MJ, et al. B cell subsets contribute to renal injury and renal protection after ischemia/reperfusion. J Immunol. (2010) 185:4393–400. doi: 10.4049/jimmunol.0903239
64. Lobo PI, Okusa MD. Role of natural IgM and IgM induced bregs in preventing ischemia induced innate inflammation and acute kidney injury. Nephron. (2019) 143:166–9. doi: 10.1159/000501639
65. Zhu F, Bai X, Chen X. B lymphocytes in renal interstitial fibrosis. J Cell Commun Signal. (2017) 11:213–8. doi: 10.1007/s12079-017-0382-x
66. Whitfield ML, Finlay DR, Murray JI, Troyanskaya OG, Chi J-T, Pergamenschikov A, et al. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci USA. (2003) 100:12319–24. doi: 10.1073/pnas.1635114100
67. Han H, Zhu J, Wang Y, Zhu Z, Chen Y, Lu L, et al. Renal recruitment of B lymphocytes exacerbates tubulointerstitial fibrosis by promoting monocyte mobilization and infiltration after unilateral ureteral obstruction. J Pathol. (2017) 241:80–90. doi: 10.1002/path.4831
68. Pechman KR, Basile DP, Lund H, Mattson DL. Immune suppression blocks sodium-sensitive hypertension following recovery from ischemic acute renal failure. Am J Physiol Regul Integr Comp Physiol. (2008) 294:R1234–9. doi: 10.1152/ajpregu.00821.2007
69. Mehrotra P, Collett JA, McKinney SD, Stevens J, Ivancic CM, Basile DP. IL-17 mediates neutrophil infiltration and renal fibrosis following recovery from ischemia reperfusion: compensatory role of natural killer cells in athymic rats. Am J Physiol Renal Physiol. (2017) 312:F385–F97. doi: 10.1152/ajprenal.00462.2016
70. Chiasson VL, Pakanati AR, Hernandez M, Young KJ, Bounds KR, Mitchell BM. Regulatory T-cell augmentation or interleukin-17 inhibition prevents calcineurin inhibitor-induced hypertension in mice. Hypertension. (2017) 70:183–91. doi: 10.1161/HYPERTENSIONAHA.117.09374
71. Kim M-G, Koo TY, Yan J-J, Lee E, Han KH, Jeong JC, et al. IL-2/anti-IL-2 complex attenuates renal ischemia-reperfusion injury through expansion of regulatory T cells. J Am Soc Nephrol. (2013) 24:1529–36. doi: 10.1681/ASN.2012080784
72. Stremska ME, Jose S, Sabapathy V, Huang L, Bajwa A, Kinsey GR, et al. IL233, a novel IL-2 and IL-33 hybrid cytokine, ameliorates renal injury. J Am Soc Nephrol. (2017) 28:2681–93. doi: 10.1681/ASN.2016121272
73. Lai LW, Yong KC, Lien YH. Pharmacologic recruitment of regulatory T cells as a therapy for ischemic acute kidney injury. Kidney Int. (2012) 81:983–92. doi: 10.1038/ki.2011.412
74. Dellepiane S, Medica D, Quercia AD, Cantaluppi V. The exciting “bench to bedside” journey of cell therapies for acute kidney injury and renal transplantation. J Nephrol. (2017) 30:319–36. doi: 10.1007/s40620-017-0384-z
75. Bai M, Zhang L, Fu B, Bai J, Zhang Y, Cai G, et al. IL-17A improves the efficacy of mesenchymal stem cells in ischemic-reperfusion renal injury by increasing treg percentages by the COX-2/PGE2 pathway. Kidney Int. (2018) 93:814–25. doi: 10.1016/j.kint.2017.08.030
Keywords: AKI, IRI, regulatory T cell, Treg, TH1, TH2, TH17
Citation: Dellepiane S, Leventhal JS and Cravedi P (2020) T Cells and Acute Kidney Injury: A Two-Way Relationship. Front. Immunol. 11:1546. doi: 10.3389/fimmu.2020.01546
Received: 06 May 2020; Accepted: 11 June 2020;
Published: 17 July 2020.
Edited by:
Giuseppe Castellano, University of Bari Aldo Moro, ItalyReviewed by:
Ankit Saxena, National Institutes of Health (NIH), United StatesRui Li, Perelman School of Medicine at the University of Pennsylvania, United States
Copyright © 2020 Dellepiane, Leventhal and Cravedi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Cravedi, cGFvbG8uY3JhdmVkaUBtc3NtLmVkdQ==