 Reem Saleh
Reem Saleh Salman M. Toor
Salman M. Toor Varun Sasidharan Nair
Varun Sasidharan Nair Eyad Elkord
Eyad Elkord- 1Cancer Research Center, Qatar Biomedical Research Institute (QBRI), Hamad Bin Khalifa University (HBKU), Qatar Foundation (QF), Doha, Qatar
- 2Biomedical Research Center, School of Science, Engineering and Environment, University of Salford, Manchester, United Kingdom
A balance between co-inhibitory and co-stimulatory signals in the tumor microenvironment (TME) is critical to suppress tumor development and progression, primarily via maintaining effective immunosurveillance. Aberrant expression of immune checkpoints (ICs), including programmed cell death protein 1 (PD-1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), T cell immunoglobulin and mucin-domain containing-3 (TIM-3), lymphocyte-activation gene 3 (LAG-3) and T cell immunoreceptor with Ig and ITIM domains (TIGIT), can create an immune-subversive environment, which helps tumor cells to evade immune destruction. Recent studies showed that epigenetic modifications play critical roles in regulating the expression of ICs and their ligands in the TME. Reports showed that the promoter regions of genes encoding ICs/IC ligands can undergo inherent epigenetic alterations, such as DNA methylation and histone modifications (acetylation and methylation). These epigenetic aberrations can significantly contribute to the transcriptomic upregulation of ICs and their ligands. Epigenetic therapeutics, including DNA methyltransferase and histone deacetylase inhibitors, can be used to revert these epigenetic anomalies acquired during the progression of disease. These discoveries have established a promising therapeutic modality utilizing the combination of epigenetic and immunotherapeutic agents to restore the physiological epigenetic profile and to re-establish potent host immunosurveillance mechanisms. In this review, we highlight the roles of epigenetic modifications on the upregulation of ICs, focusing on tumor development, and progression. We discuss therapeutic approaches of epigenetic modifiers, including clinical trials in various cancer settings and their impact on current and future anti-cancer therapies.
Introduction
Epigenetics involves heritable and long-term changes in gene expression, which are mediated by various mechanisms, without altering the DNA sequence. In physiological and pathological settings, epigenetics plays profound, and ubiquitous roles in the regulation of gene transcription (1, 2). Epigenetic alterations in genes encoding tumor suppressors, suppressive cytokines and inhibitory immune checkpoints (ICs) can lead to impaired activation of anti-tumor immunity, tumor growth, immune escape and drug resistance, and significantly contribute to cancer development and progression (3, 4). Genetic and epigenetic modifications acquired by the tumor microenvironment (TME) play an indispensable role in tumorigenesis and result in uncontrolled growth of malignant cells (5). As cancer cells divide, they acquire genetic and epigenetic alterations giving rise to new cancer clones with different genetic and epigenetic make-ups, and inheritable traits favoring growth and survival of malignant cells (4).
The contribution of ICs to cancer pathogenesis and progression is well-recognized and has rationalized the development of monoclonal antibodies that target ICs and their ligands for cancer therapy (6, 7). Inhibitory ICs and their ligands are immunomodulatory molecules, and their physiological expression is crucial to maintain immune hemostasis and immunosurveillance to avoid potential risks of autoimmunity (8). Over expression of inhibitory ICs has been recognized as one of the major contributing factors to cancer development and progression, as well as autoimmune/chronic inflammatory diseases. Inhibitory ICs, including cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1), T cell immunoglobulin and mucin-domain containing-3 (TIM-3), lymphocyte-activation gene 3 (LAG-3) and T cell immunoreceptor with Ig and ITIM domains (TIGIT), can negatively influence the activation of antigen-presenting cells (APCs) and T effector cells (Teffs), and enhance the function of T regulatory cells (Tregs) and myeloid-derived suppressive cells (MDSCs) (9).
Immune checkpoints and their ligands are differentially expressed by immune cells (Table 1). The binding of CTLA-4 to CD80 and CD86 causes inhibitory signals toward T cell activation (10). PD-1 is expressed by multiple types of immune cells, including activated T cells. Upon its interaction with its ligands, programmed cell death-ligand 1 or 2 (PD-L1 or PD-L2), inhibitory signals are generated to inhibit T cell activation/proliferation (11, 22, 23). The interaction between TIM-3 and galectin-9 has also been reported to suppress T cell function (14). The binding of LAG-3 to its ligand reduces antigen-specific CD4+ Teff responses and suppresses cytokine production (24–26). The interaction between TIGIT and its ligands inhibits Teff activation (19–21).
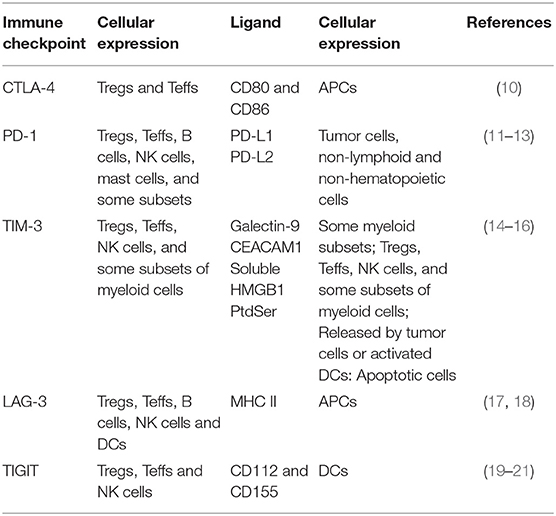
Table 1. Expression of immune checkpoints and their ligands.
Despite the success of ICIs in treating various cancer types, a large proportion of patients show low response rates due to primary or acquired resistance mechanisms. Primary resistance mechanisms are mainly dependent on the existing immune response, while acquired resistance mechanisms are governed by tumor heterogeneity/plasticity, immunosuppressive cells (including Tregs and MDSCs), T cell exhaustion and increased expression of inhibitory ICs (6, 8, 9, 27).
The overexpression ICs and their ligands, within the TME, can be mediated by different forms of epigenetic alterations, including DNA methylation, histone modifications and microRNAs (3, 28, 29). Epigenetic modifiers, including DNA methyltransferase and histone deacetylase inhibitors, can be used to revert the changes acquired during cancer onset or progression (30). The use of combined therapies targeting epigenetic modifications and ICs could serve as a highly promising therapeutic strategy to restore the physiological epigenetic profile and to boost anti-tumor immunity. In this review, we focus on the role of epigenetic modifications regulating IC expression, and promoting cancer development and progression. We discuss the different therapeutic approaches of utilizing epigenetic modifiers, including clinical trials in various cancer settings and their impact on anti-cancer therapies.
Role of Epigenetics in Cancer Development and Progression
Epigenetics controls the transcriptional and post-transcriptional regulations of a vast array of genes, which mediate various cellular processes and functions ranging from proliferation, differentiation, invasion, survival, growth, metabolism, and immune responses (31). The development and progression of many pathological conditions, including cancer, trauma, and infectious and autoimmune diseases can be driven by aberrant epigenetic modifications (1–3, 5). Cancer was initially considered as a “genetic disease” due to gene mutations associated with loss of gene function or gene overexpression; these mutations were initially thought to be the main driving force behind disease pathogenies and progression (32). However, there is emerging evidence implicating a crucial role for epigenetics in carcinogenesis. During tumorigenesis, the epigenome is subjected to various alterations such as global changes in histone modifications, dysregulation in the non-coding RNA networks, global loss of DNA methylation, and regional hypermethylation particularly in the promoter regions of tumor suppressor genes (33). Using whole-genome sequencing, Mack et al. showed very low mutation rates, and no recurrent somatic single nucleotide variants were associated with 47 cases of pediatric brain cancer (hindbrain ependymomas) (34). In addition, the authors showed that poor prognosis of hindbrain ependymomas exhibit a CpG island methylation phenotype, which is known to induce transcriptional silencing of differentiation genes through trimethylation of lysine 27 on histone H3 (H3K27) (34). Moreover, genetic alterations in genes encoding enzymes that regulate DNA methylation and histone modifications are also responsible for predisposing individuals to cancer (35, 36). For instance, mutations in DNA methylation enzyme DNMT3a are found in ~22% of patients with acute myeloid leukemia (AML) and T cell lymphoma have been associated with poor disease outcomes (36–38). Another study showed that mutations in ten-eleven translocation 2 (TET2) methylcytosine dioxygenase, which mediates DNA demethylation, are present in ~15% of myeloid cancers (39). Mutations in genes encoding proteins that facilitate histone demethylation on H3K27, such as ubiquitously transcribed tetratricopeptide repeat, X chromosome (UTX) and additional sex combs like 1 (ASXL1), have been detected in 11% of patients with myelodysplastic syndromes and 43% of chronic myelomonocytic leukemia (40, 41). Furthermore, mutations in histone lysine acetyltransferases (HATs) and histone deacetylases (HDACs) have been associated with hematological malignancies and solid tumors (42).
Global DNA methylation and histone modifications are closely linked with cancer development and progression. The level of DNA methylation (hypo or hyper) and levels of histone methylation or acetylation can vary across different cancer types. For instance, development of breast cancer has been associated with global DNA hypermethylation, while prostate cancer pathology has been linked with DNA hypomethylation and increased active histone methylation (43). Therefore, these findings suggest that DNA methylation could occur in a tissue-specific manner depending on the TME. Certain patterns of DNA methylation, hypermethylation or hypomethylation, targeting specific genes in a particular TME could have a profound impact on cancer development and/or progression (44, 45). Based on this and since DNA hypermethylation is associated with transcriptional silencing (46), it could be anticipated that development of particular cancer types is driven by DNA hypermethylation causing reduced expression of genes related to tumor suppression and activation of anti-tumor immunity (47). On the other hand, transcriptional activation mediated by DNA hypomethylation could result in the overexpression of genes favoring tumor growth, angiogenesis, metastasis and immunosuppression, leading to the development of cancer types, which have been linked with DNA hypomethylation (44, 48).
Role of Immune Checkpoints in Cancer Development and Progression
Increased expression of CTLA-4 and PD-1/PD-L1 and their negative correlations with overall survival (OS) in various cancer cases have been well-established (49–51). Toor et al. reported a positive correlation between tumor-node-metastasis (TNM) staging and increased expression of CTLA-4 in circulating CD4+ T cells of colorectal cancer (CRC) patients (52). Elevated co-expression of LAG-3 and PD-L1 in tumor tissues from triple negative breast cancer (TNBC) patients treated with adjuvant therapy has been associated with poor disease prognosis (53). Zhang et al. demonstrated that TIM-3 expression, in colorectal tumor tissues, was positively correlated with TNM staging, lymph metastasis and shorter OS (54). Furthermore, overexpression of TIM-3 on tumor-infiltrating cells showed a positive correlation with poor prognosis and shorter OS in hepatocellular carcinoma (HCC) patients (55, 56).
More recently, soluble forms of ICs/IC ligands, generated by alternative splicing and circulating in the plasma of cancer patients, have been implicated in cancer development and poor prognosis, and were suggested to serve as prognostic biomarkers. Simon et al. reported that serum soluble CTLA-4 (sCTLA-4) in pediatric patients with acute lymphoblastic leukemia can be used as a prognostic biomarker (57). High levels of sPD-1 in the plasma of patients with hepatitis B virus (HBV) were associated with high viral load and increased risk of hepatocellular carcinoma (HCC) (58). Increased levels of sPD-L1 have also been associated with poor clinical outcomes in various cancers including HCC, diffuse large B-cell lymphoma, renal cell carcinoma, and gastric and lung cancer (59–63). Additionally, poor prognosis and short OS have been linked with elevated levels of sTIM-3 in HCC patients (64). The mechanisms by which ICs and IC ligands (membrane-bound or soluble forms) mediate immunosuppression and promote tumorigenesis have been reviewed elsewhere (7, 9, 65). Collectively, the interactions between ICs and their ligands impair APC function, reduce T cell proliferation and cytokine release, induce T cell apoptosis, and enhance suppressive activity of Tregs and MDSCs (6, 27).
Epigenetic Mechanisms Regulating the Transcription of Immune Checkpoints in Cancer
The epigenetic machinery is mainly comprised of three components: DNA methylation, histone modifications (e.g., acetylation, methylation, phosphorylation, and ubiquitylation) and non-coding RNAs/microRNAs (miRNAs) (66). In this section, we discuss how these mechanisms control the expression of IC and IC ligand genes in the TME of various cancer types.
DNA Methylation
DNA methylation is defined as the covalent transfer of a methyl group to the C-5 position of the cytosine ring of DNA mediated by DNA methyltransferases (DNMTs) (67). DNA methylation patterns are governed by the action of DNMTs: DNMT1, DNMT3a, and DNMT3b (67, 68). Mechanistically, transcriptional silencing is mediated by a methylated cytosine by eliminating components of transcriptional regulation from their target sites (67).
DNA methylation can be passively lost or actively driven by TET family of dioxygenases, which catalyze the oxidation of methylcytosine to hydroxymethylcytosine (69, 70). While transcriptional silencing is driven by the action of DNMT(s) leading to DNA methylation, transcriptional activation is caused by hypomethylation or demethylation facilitated by the action of TET enzyme(s) (46). Indeed, the imbalance between the activity of DNMTs and TETs can affect the expression of many genes favoring transcriptional silencing or activation during many pathological conditions, including cancer (71). For example, the upregulation of TET enzymes and downregulation of DNMTs in the circulation and tumor tissues of breast cancer (BC) and colorectal cancer (CRC) patients could be associated with DNA hypomethylation causing the upregulation of ICs/IC ligands (Figure 1) (28, 29, 72).
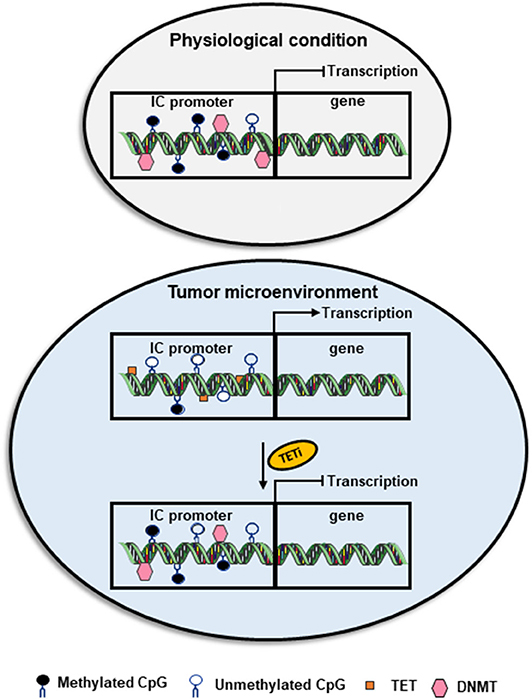
Figure 1. Role of DNA methylation in the transcriptional regulation of immune checkpoint expression. Under physiological conditions, the CpG islands in the promoter region of immune checkpoints are methylated by DNMTs, which leads to the transcriptional repression of ICs. However, in the TME, the activity of DNMTs could be override by the action of TETs causing TET-mediated active demethylation and favoring IC transcription. TET inhibitors could be used as a therapeutic agent to block TET-mediated active demethylation and retain the physiological condition by downregulating the transcription of genes, including ICs.
Restoring normal patterns of DNA methylation, especially in genes related to immune modulation and tumorigenesis, has been recognized as one of the goals for cancer therapy improvement (as described in Section Epigenetic Modifiers Targeting DNA Methylation). Aberrant DNA methylation patterns have been associated with immune evasion in cancer patients. For instance, Jung et al. reported that genomic methylation in lung and melanoma patients correlates with the immune escape signatures, independently of mutation burden and aneuploidy (73). Additionally, authors found significant negative correlations between genomic demethylation, and immunomodulatory-related pathways/immune cell markers (73), suggesting that demethylation could be responsible for silencing the transcription of these genes in patients with lung cancer and melanoma. Interestingly, they reported that global hypomethylation in these cancer patients correlated with poor clinical responses following immunotherapy, indicating that alterations in DNA methylation can be used to predict clinical benefits of immunotherapies (73). In another study, global DNA hypomethylation in human melanoma cell lines was associated with elevated expression of PD-L1, implicating a therapeutic potential for targeting PD-L1 using DNA methylation modifying agents (74).
The role of DNA methylation in regulating the expression of several ICs/IC ligands in the circulation and tumor tissues of BC and CRC patients has been addressed previously. Elashi et al. reported that increased expression of TIM-3, PD-L1, and TIGIT in the peripheral blood of both BC and CRC patients (72). DNA methylation has no role in regulating the expression of TIM-3 in the circulation of BC and CRC patients, while PD-L1 upregulation was found to be mediated by DNA hypomethylation (72). Elevated level of TIGIT in the circulation of CRC patients was mediated by DNA hypomethylation; however, DNA methylation has no role in regulating the expression of TIGIT in the circulation of BC patients (72).
Elevated expression of PD-1, CTLA-4, and TIM-3 genes in breast tumor tissues was found to be mediated by DNA hypomethylation in the CpG islands of their promoter regions (28). In the same study, authors found that the promoter regions of LAG-3 genes were completely hypomethylated in breast tumor tissues, and paired-normal tissue, suggesting that DNA methylation has no role in the upregulation of these genes in BC (28). In another study, it was reported that elevated expression of CTLA-4 and TIGIT genes in human CRC tumor tissues is driven by DNA hypomethylation (29). Additionally, authors demonstrated that DNA methylation plays no role in the overexpression of PD-1, PD-L1, galectin-9, and TIM-3 in colorectal tumor tissues (29).
A study by Marwitz et al. demonstrated that elevated expression of PD-1 and CTLA-4 in tumor tissues of non-small-cell lung cancer (NSCLC) patients is driven by DNA hypomethylation (75). However, increased expression of PD-L1 in NSCLC tumor tissues was not associated with DNA methylation (75). In contrast, elevated level of PD-L1 expression in tumor tissues of head and neck squamous-cell carcinoma (HNSCC) was a resultant of DNA hypomethylation (76). Goltz et al. demonstrated that PD-L1 promoter methylation predicts the survival rate and disease prognosis of various cancer settings, including CRC, HNSCC and AML (77–79). Another study by Rover et al. showed that increased expression of CTLA-4, PD-1, PD-L1, and PD-L2 was associated with DNA hypomethylation in patients with lower-grade gliomas (80). Altogether, these data suggest that DNA hypomethylation is responsible for increasing the expression of ICs/IC ligands in cancers; however, the set of genes regulated by DNA methylation differ from one cancer type to another.
Histone Modifications
Histone Methylation
Histone methylation is another mechanism by which epigenetic modifications occur to cause transcriptional and post-transcriptional alterations in many genes, including those related to cancer development and immune evasion. These alterations affect chromatin compaction/structure, recruitment and binding of transcription factors, initiation and elongation factors with target DNAs, and RNA processing (81). Histone methylation is a dynamic process which takes place on the side-chain nitrogen atoms of lysine (K) residues, mainly on H3 followed by H4 (82). It is controlled by the activity of six major family classes of histone lysine methyltransferase complexes (KMT1, KMT2, KMT3, KMT4, KMT5, and KMT6). Lysine residues can be mono- di-, or tri-methylated by the action of KMTs (83). Lysine methylation can be reversed by lysine demethylases (KDMs), which also comprised of at least six families with distinct and overlapping functions (KDM1, KDM2, KDM3, KDM4, KDM5, and KDM6) (84, 85). The regulation of histone methylation and demethylation is a complex process (86); each KMT or KDM family consists of several enzymes that target a specific lysine residue. Additionally, different methylation states on lysine residues are controlled by different family classes of KMT or KDM, and have a different impact on transcriptional regulation.
Histone methylation on lysine residues appears to be a more stable mark; its loss on histones H3 and H4 causes transcriptional repression or silencing. Mono-, di- or trimethylation of lysine 4 in histone H3 (H3K4me1/2/3) and H3K36me3/me2 correlates with transcriptional activation (87, 88). On the other hand, trimethylation of lysine 9 and 27 in histone H3 (H3K9me3 and H3K27me3) correlates with repression (Figure 2A) (87, 88). The contribution of histone methylation to the regulation of IC transcription in breast and colorectal tumor tissues has been previously demonstrated. We have shown that upregulation of PD-1, CTLA-4 and LAG-3 in breast tumor tissues is associated with low enrichment of repressive histones, H3K9me3 and H3K27me3, in their promoter regions (Figure 2B) (28). In contrast, the expression of TIM-3 gene in breast tumor tissues was associated with low enrichment of H3K27me3 in its promoter region (28). In another study, increased expression of PD-1 and TIGIT in colorectal tumor tissues was shown to be associated with the low abundance of H3K9me3 in their promoter regions (29). Moreover, transcriptional upregulation of CTLA-4 in colorectal tumor tissues was found to be driven by the low abundance of H3K27me3 in its promoter region, while the low abundance of both H3K9me3 and H3K27me3 repressive histones was associated with the upregulation of TIM-3 in colorectal tumor tissues (29). Based on the above findings, it could be anticipated that targeting the activity of enzymes (KDMs) on repressive histones, H3K9me3 and H3K27me3, to maintain their trimethylation can result in the transcriptional repression of IC/IC ligand, thereby offering a therapeutic strategy for cancer treatment. The contribution of some of lysine demethylases (such as KDM3B, KDM4A, and KDM5B) to the development and/or progression of different cancer types, including breast cancer, prostate cancer and AML, have been reported, thus rationalizing the development of drugs targeting the activities of these enzymes [as reviewed in (89)].
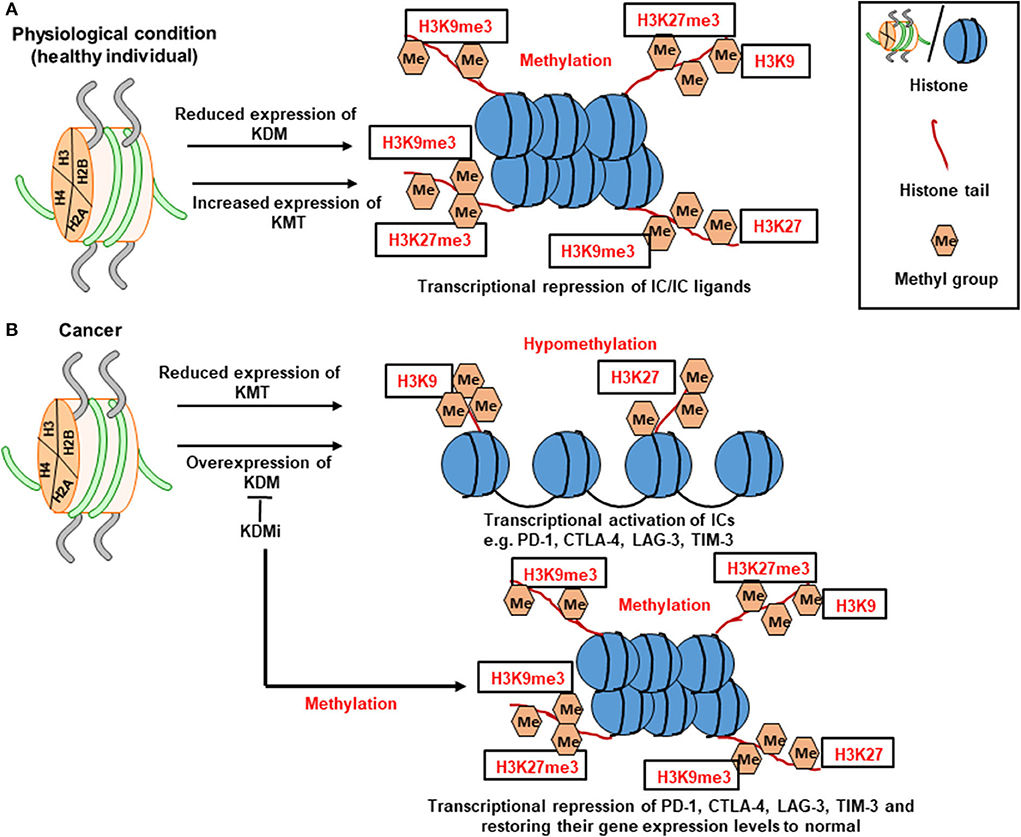
Figure 2. Role of histone methylation in the transcriptional regulation of immune checkpoints. Schematic diagrams simplify the complexity of gene transcription via histone methylation. Histone methylation depends on the interplay between KMTs (lysine methyltransferases) and KDMs (lysine demethylases). KMTs transfer methyl group to the histone tails. Under physiological conditions, histone methylation on the 9th and 27th lysine residues of H3 tail (H3K9me3 and H3K27me3, respectively) leads to transcriptional repression of ICs (A). In tumor conditions, the low abundance of H3K9me3 and H3K27me3 leads to transcriptional activation of ICs such as PD-1, CTLA-4, LAG-3, and TIM-3. Meanwhile, utilization of KDM inhibitor (KDMi) could be beneficial in restoring the normal levels of ICs (B).
Histone Acetylation
The importance of histone acetylation in regulating gene transcription and cellular processes, such as immune response, apoptosis, autophagy, cell cycle arrest, DNA damage repair, and metabolism, has been shown in cancer (86, 90). It is a highly reversible process, which involves the catalytic activity of histone acetyltransferases (HATs) and histone deacetylases (HDACs) (91). Histone acetylation occurs on lysine residues at the N-terminus induced by the activity of HATs, resulting in the removal of the basic charge at unmodified lysine residues, and leading to active transcription (92, 93). HDACs and HATs control histone acetylation act in opposite directions causing an altered structure of the chromatin, and dictate the accessibility of DNA to transcription factors (sequence-specific DNA-binding factors) and other elements of the transcriptional machinery, such as co-activators. Disrupting the equilibrium of histone acetylation or deacetylation is also reported to be associated with tumorigenesis and poor prognosis (94).
HDACs stabilize the nucleosomal DNA-histone interaction causing transcriptional silencing (Figure 3A), while the action of HATs mediates transcriptional activation (Figure 3B) (91). HDACs can be divided into four classes: class I, II, III, and IV (95). The role of HDACs in cancer epigenetics and disease development is receiving an increasing attention, and targeting their activity has recently been postulated as potential therapeutic strategy for cancer treatment. HDACs repress the transcription of genes associated with immune responses and tumor suppression by restricting the accessibility of transcription factors to their binding sites and inducing a closed chromatin confirmation (96). Preclinical models of melanoma and lung adenocarcinoma showed that the expression of PD-L1 and T cell chemokines can be upregulated by HDAC inhibitors to enhance the sensitivity of the immune response to anti-PD-1/PD-L1 therapy and improve clinical outcomes (97, 98). Recently, Fan et al. reported that upregulated levels of HAT1 is associated with poor prognosis of pancreatic cancer (99). Using in vitro and in vivo models, authors also demonstrated that knockdown of HAT1 reduced the proliferation of pancreatic tumor cells, and downregulated PD-L1 expression (99). Furthermore, it was shown that PD-L1 expression positively correlated with HAT1 expression in pancreatic tumor tissues (99). Altogether, these findings suggested that HAT1 transcriptionally regulate PD-L1 expression in cancer settings, and implicated that targeting HAT1 activity could be used as a therapeutic approach for cancer treatment (99) (Figure 3B, i). Alternatively, the use of ICIs targeting PD-1/PD-L1 axis in patients with acquired resistance (97, 100) due to aberrant expressions of HAT1 and PD-L1 (99) could be beneficial in maximizing the anti-tumor immune response, enhancing the sensitivity to ICI, and overcoming resistance (Figure 3B, ii). Collectively, these findings suggest that HDACs act opposite to HATs in terms of IC regulation, and that HDAC inhibition in combination with ICIs could be beneficial in enhancing the therapeutic efficacy of cancer treatment by increasing the sensitivity of the host immune response to ICIs. This particular therapeutic strategy could be favorable for cancer patients who developed acquired resistance to ICIs.
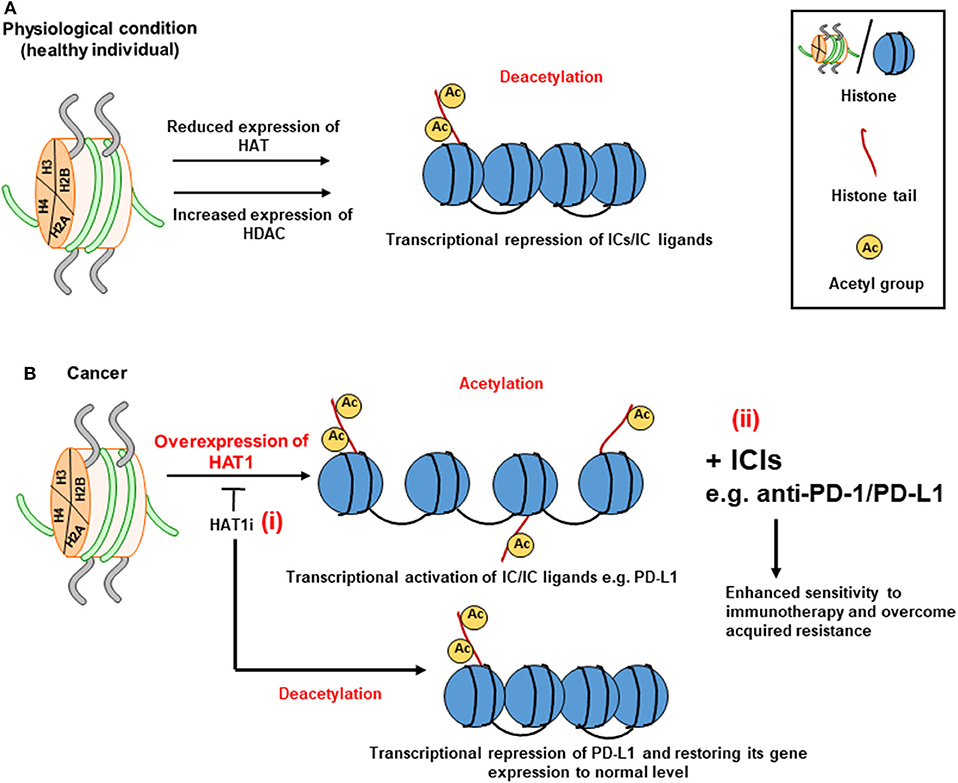
Figure 3. Role of histone acetylation in the transcriptional regulation of immune checkpoints. The transcriptional regulation of ICs by means of acetylation relies on the balance between HATs and HDACs on lysine residues at histone tails. A set of HDACs can keep the heterochromatin structure and downregulate the transcription of ICs in physiological conditions (A). However, via tumor-acquired mechanisms, HAT activity is dominated resulting in the conversion of heterochromatin (closed chromatin) to euchromatin (open chromatin) by transferring acetyl molecules to the histone tails, thereby favoring gene transcription. Overexpression of HAT1 can lead to increased expression of PD-L1 in cancer tissues by enhancing histone acetylation. The use of HAT1 inhibitor (HAT1i) could be useful in restoring the normal expression of PD-L1- (i) (B). Immune checkpoint inhibitors (ICIs) targeting PD-1/PD-L1 axis could be used in patients with aberrant expression of HAT1 and PD-L1 (ii).
Given the complexity of epigenetic regulations and knowing the fact that HATs and HDACs can alter the transcription of multiple target genes, it is crucial to take this into consideration during the development of HAT and HDAC inhibitors and the design of therapeutic protocols. For instance, HDAC inhibitors, valproic acid (VPA; class I HDAC inhibitor) and trichostatin-A (TSA; class I and II inhibitor), could induce apoptosis and alter the acetylation status of p53, on ETS Related Gene (ERG)+ prostate cancer cells (101, 102). In addition, VPA and TSA were able to repress the transcription of ERG, which its overexpression has been associated with poor prognosis and unfavorable clinical outcomes in prostate cancer patients (101).
Long Non-coding RNAs and MicroRNAs
Long non-coding RNAs (lncRNAs) are a series of non-coding RNAs comprised of more than 200 nucleotides. lncRNAs are pointed to as potential candidates to evaluate the prognosis, diagnosis, and development of cancers, even though their capacity of protein-coding is very little (103). MicroRNAs (miRNAs) are small non-coding RNAs (19–25 nucleotides long), which complementary pair with the 3′ untranslated region of target mRNAs, resulting in the repression of transcription and/or the degradation of target mRNAs (104). Studies demonstrated that miRNAs can regulate more than 30% of human genes involved in many cellular processes, including cell cycle arrest and cell growth/proliferation/differentiation/apoptosis (105–107).
miRNAs in various cancers can influence the transcriptional regulation of immunomodulatory genes, including ICs and their ligands (108). Wei et al. showed that transfection of human CD4+ T cells with miR-138 abolished the expression of CTLA-4, PD-1 and FoxP3 expression in glioma mouse models (109). Another miRNA with tumor-suppressive functions is miR-28. Li et al. reported that the expression of miR-28 is downregulated ~30% in exhausted PD-1+ T cells from melanoma patients (110). Authors reported that miR-28 inhibits the expression of the TIM-3 and PD-1 in T cells upon the binding to their respective 3′ UTRs (110). In ovarian carcinoma, signaling pathways mediated by the interactions of CTLA-4 with CD80 and PD-1 with PD-L1 are negatively regulated by mi-R424(322) (111), suggesting the importance of mi-R424 in the downregulating of CTLA-4 and PD-1 signaling pathways. In support of this, it was shown that high levels of mi-R424(322) in tumors are positively correlated with progression-free survival in patients with ovarian carcinoma (111). More recently, Richardsen et al. demonstrated that low levels of miR424-3p in prostate cancer (PC) tissues associated with an aggressive phenotype of PC, poor disease prognosis and low survival rate (112). Authors also reported a negative correlation between CTLA-4 expression and miR424-3p expression in PC tissues (112), highlighting the role of miR424-3p in regulating CTLA-4 expression in PC as it has been reported in other cancer types (111, 113).
A study by Cortez et al. demonstrated that PD-L1 expression in NSCLC is negatively regulated by p53 via miR-34, suggesting that miRNA delivery could serve as a novel therapeutic approach for lung cancer therapy (114). Studies indicated that miRNAs can affect the progression of AML by modulating the expression of target genes such as TIM-3. Based on bioinformatics, it was predicted that miR-330-5p may silence the transcription of TIM-3 in the AML cell line, HL-60 (115). Acquired resistance against anti-PD-1 therapy has been associated with the upregulation of TIM-3 on T cells in lung cancer and HNSCC patients (116, 117). Another study by Oweida et al. showed that response to anti-PD-L1 mAb and radiotherapy was compensated by the increased expression of TIM-3 on CD8+ T cells and Tregs, associated with tumor relapse, poor survival rate in a mouse model of head and neck tumor (118). Collectively, these studies suggest that the use of ICIs in combination with miRNA therapy to target alternative ICs, could be beneficial in preventing the development of acquired resistance in response to anti-PD-1 or anti-PD-L1 therapies.
In lymphoma, the expression of CTLA-4, PD-1, PD-L1, TIM-3 and LAG-3 are negatively regulated by miR-146 (119). The expression of PD-L1 on tumor cells and the suppression of anti-tumor immunity in human lung cancer are negatively regulated by miR-200 (120). Another miRNA with tumor suppressive functions is miR-34a. Its expression is induced by p53, which in turn suppresses the expression of PD-L1 (120). In line with this, it was reported that low levels of miR-34a in AML and NSCLC are positively correlated with the overexpression of membrane–bound PD-L1 (53, 114). Overexpression of PD-L1, and low levels of p53 and miR-34a have been associated with poor clinical outcomes in patients with NSCLC (120). On the other hand, overexpression of miR-34a can dysregulate the activation of PD-1/PD-L1 signaling pathway, causing the reversal of CD8+ T cell exhaustion, and triggering T cell activation and cytokine expression, such as IFN-γ and TNF-α (121). In CRC, low levels of miR138-5p, a tumor suppressive miRNA, positively correlates with advanced disease stages, lymph node metastasis and poor clinical outcomes (122). miR138-5p negatively regulates PD-L1 expression in CRC, which is associated with reduced cell proliferation and cell cycle progression (122).
Collectively, these findings clearly imply the importance of miRNAs in regulating the expression of genes related to tumorigenesis, immune evasion and cancer progression. One miRNA may have several mRNA targets, and therefore could influence the function of many genes, pathways and cellular processes. The overall role of various miRNAs on the regulation of ICs and their ligands are summarized in Figure 4. The above findings also suggest the potential therapeutic benefit of including miRNAs in cancer therapy as it will be discussed below.
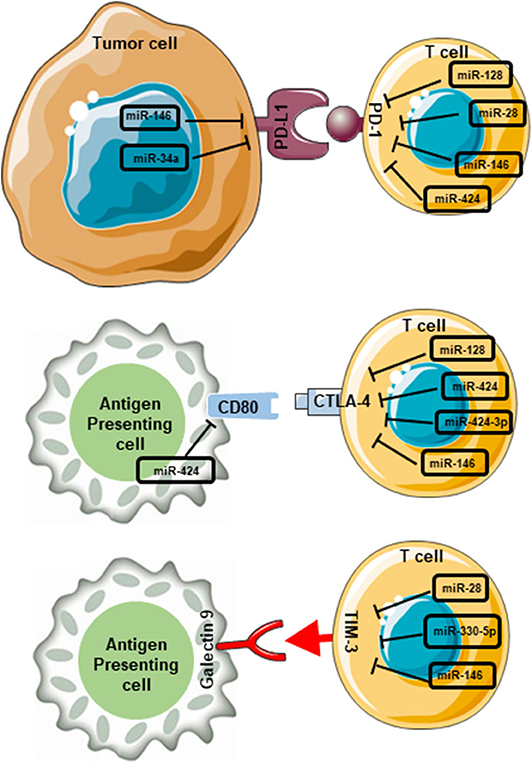
Figure 4. miRNA-mediated interruption of interactions between immune checkpoints and their ligands in the tumor microenvironment. miRNAs which contribute to the blockade of PD-1/PD-L1 interactions are miR-146, miR-34a, miR-128, miR-28, miR-146, and miR-424. miR-146 and miR-34a expressed on tumor cells, and miR-128, miR-28, miR-146, and miR-424 expressed on T cells. Likewise, miR-424 expressed on APCs, and miR-128, miR-424, miR-424-3p, and miR-146 expressed on T cells interfere with CD80/CTLA-4 interactions. Furthermore, miR-28, miR-330-5p, and miR-146 expressed on T cells interfere with TIM-3/galectin 9 interaction. These miRNA-mediated interruptions could lead to the blockade of downstream pathways, which ultimately favor anti-tumor immunity.
Potential Therapeutic Applications of Epigenetic Modifiers for Cancer Treatment
Studies have shown that cancers exploit epigenetic mechanisms mainly in two ways: (1) to delineate the normal transcriptional regulation of gene expression to assist tumor progression; and (2) to deactivate anti-tumor immune responses, and regulate oncogenes and tumor suppressor genes. Dysregulated transcription of co-activators or suppressors of oncogenes/proto-oncogenes and tumor suppressor genes leads to the development of various human cancers. Hypomethylation leads to genomic instability, while hypermethylation may lead to silencing of tumor suppressor genes (123). Therefore, the development and use of epigenetic modifiers aiming to modulate the activity of enzymes involved in these epigenetic pathways, including DNMTs, TETs, HATs and HDACs, may offer therapeutic benefits (96, 124). However, it is important to consider the complexity of epigenetic regulations and take into consideration the tumor type, nature of the TME, and all the target genes that can be altered upon the inhibition of epigenetic mediators (DNA/histone modifiers) during the development of epigenetic drugs and the design of therapeutic protocols. The communication between immune cells and tumor cells via IC/IC ligand interactions results into immunosuppression and tumor progression (Figure 5A). Some epigenetic drugs can be used to enhance anti-tumor immunity by downregulating the expression of ICs/IC ligands (99) (Figure 5B), while others could be used in combination with ICIs to improve the sensitivity of the host response to therapy by upregulating the expression of IC ligands (97, 100) (Figure 5C). This should be useful during the assignment of therapeutic protocols for cancer patients.
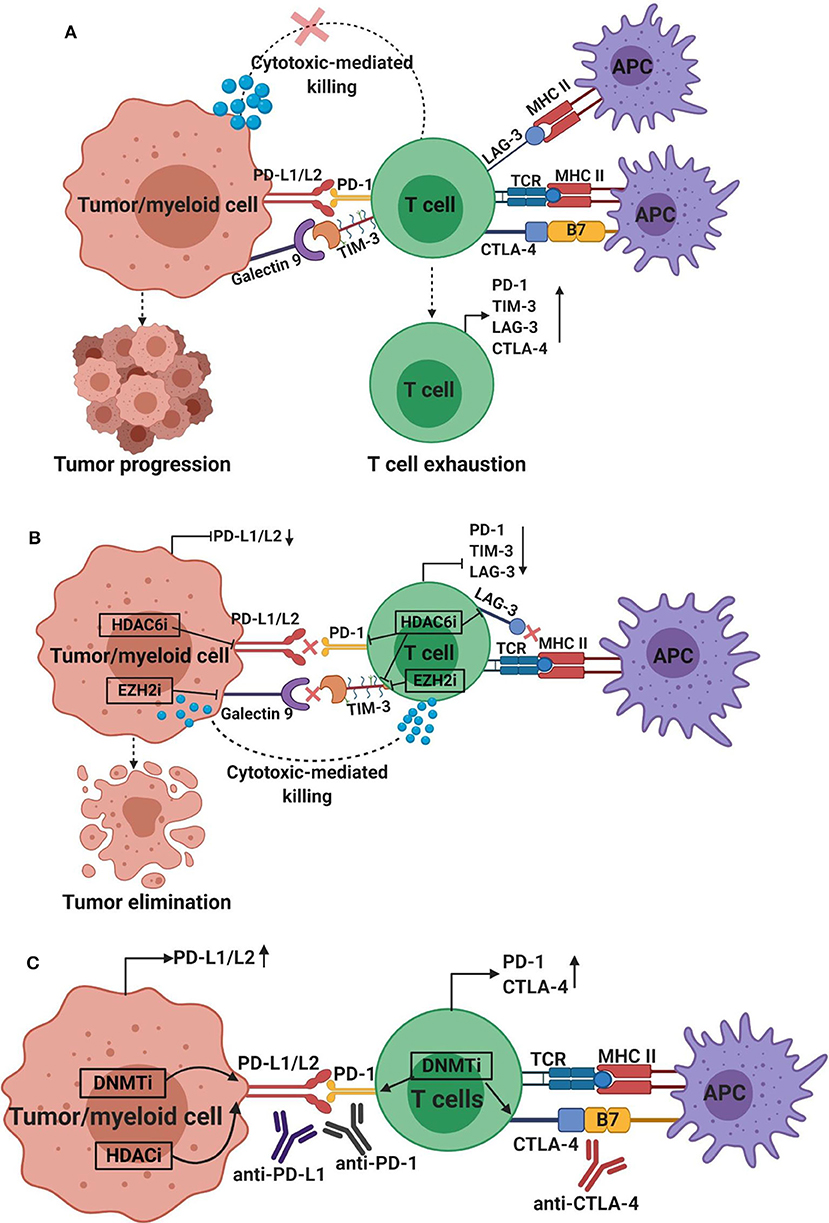
Figure 5. Effect of epigenetic modifiers on the expression of immune checkpoints and their ligands in the tumor microenvironment. The interaction between co-inhibitory immune checkpoints on immune cells and their ligands on tumor cells or myeloid cells results in tumor progression, immunosuppression and T cell exhaustion characterized by increased expression of immune checkpoints, including PD-1, CTLA-4, TIM-3, and LAG-3, and loss of effector functions, such as cytokine release and cell-mediated cytotoxicity. The interactions between PD-1, TIM-3, CTLA-4, and LAG-3 on T cells with their respective ligands PD-L1/PD-L2, galectin-9, B7 ligands or MHC II on tumor cells/myeloid cells or APC, generate signals that inhibit T cell activation/proliferation (A). Depending on the tumor microenvironment and tumor type, the application of epigenetic modifiers can downregulate or upregulate the expression of immune checkpoints and their ligands. The application of HDAC6 inhibitor (HDAC6i) can downregulate the expression of PD-L1/2, PD-1, TIM-3, and LAG-3, and EZH2 inhibitor (EZH2i) can downregulate the expression of galectin-9 and TIM-3 (B), indicating the potential benefits of using these modifiers to enhance anti-tumor immune responses and promote tumor cell killing. On the other hand, application of DNMT inhibitor (DNMTi; azacytidine or decitabine) can upregulate the expression of PD-L1/2, PD-1 and CTLA-4, and HDAC inhibitor (HDACi; vorinostat; or panobinostat) can upregulate the expression of PD-L1/2 (C), suggesting the potential benefit of combining epigenetic modifies with immune checkpoint inhibitors, such as anti-CTLA-4, anti-PD-1 or anti-PD-L1, to increase the sensitivity of the host immune response and promote more potent anti-tumor immunity.
Epigenetic Modifiers Targeting DNA Methylation
DNA methylation may lead to silencing of suppressor genes, such as TP53 and CDKN2A, thereby increasing susceptibility to cancer onset. Inhibition of DNMTs has been shown to correlate with increased expression of tumor suppressor genes and reduction in tumorigenicity (125). Hypomethylating agents, which inhibit DNMT, target the methylation patterns of tumor cells to reinstate normal methylation signatures. DNMT inhibitors (DNMTis), such as 5-azacytidine (AZA/5AC) and decitabine, have been developed to inhibit and degrade DNMTs, reverse hypermethylation and promote transcriptional activation (126). Several signaling pathways such as those related to double-stranded RNA (ds-RNA) response, type I interferon response and apoptosis are induced upon the application of DNMTis. In a preclinical melanoma model, DNMTi treatment was able to increase the sensitivity to anti-CTLA-4 therapy by affecting hypermethylated endogenous retrovirus genes (127). DNMTis, azacytidine and decitabine, have also been shown effective in increasing PD-L1 and PD-L2 levels in melanoma (97) (Figure 5C). Animal studies showed that combining azacytidine or decitabine with anti-CTLA-4 in ovarian cancer and melanoma is beneficial in improving the immune response to anti-CTLA-4 and reducing tumor burden (128, 129). Altogether, these data rationalized the use of DNMTis in combination with ICIs to maximize the therapeutic efficacy and clinical outcomes in cancer patients (129). Azacytidine and decitabine serve as the most commonly used DNMTi in oncology for the treatment of chronic myelomonocytic leukemia (CMML), myelodysplastic syndromes (MDS) and AML (130). Treatment of MDS with decitabine increased the mRNA expression of PD-1, its ligands (PD-L1 and PD-L2) in addition to CTLA-4 (131), rationalizing the potential synergy between DNMTi and ICIs in enhancing the therapeutic efficacy of combined treatment, as hypomethylation may increase the expression of ICs/IC ligands, and subsequently sensitize tumor cells to the ICIs (Figure 5C). Several trials are currently underway to investigate DNMTi use in treating different solid malignancies (132–134). However, DNMTi therapies are frequently associated with severe side effects, no or partial treatment responses and therapy resistance in a significant patient cohort. Therefore, identifying novel, more specific targets against DNMTi are currently being explored.
TET-mediated DNA demethylation contributing toward developmental processes including disease progression and its dysregulation may lead to tumorigenesis (135). As previously discussed in Section DNA Methylation, TET-mediated DNA demethylation could be associated with the overexpression of IC/IC ligand in the circulation and tumor tissues of patients with breast and colorectal cancers (28, 29, 72). Therefore, the inhibition of TET activities can have a therapeutic potential and could be beneficial in restoring the normal transcriptomic expression and methylation patterns of IC/IC ligand. Furthermore, TET mutations have been associated with various hematological malignancies; however, specific TET protein inhibitors have not been tested till present in clinical oncology (124, 136). Nevertheless, upstream targets for TET-associated pathways have been identified in different malignancies and have been the focus of numerous preclinical studies. For instance, mutations in genes encoding isocitrate dehydrogenase 1 and 2 lead to TET1 inactivation in gliomas (137).
Epigenetic Modifiers Targeting Histone Modifications
HATs modify chromatin histones to exert their effects of epigenetic modulation of gene transcription, and are dysregulated in various human diseases including cancers (138). For instance, HAT1 has been implicated in the transcriptional upregulation of PD-L1 in pancreatic cancer (99). Additionally, it has been demonstrated that knockdown of HAT1 reduced the proliferation of pancreatic tumor cells, and expression of PD-L1 (99). These findings suggest that targeting HATs could be beneficial in reducing the expression of IC ligands, and ultimately could have clinical benefits for cancer patients. However, in contrast to HDAC inhibitors (HDACis), HAT inhibitors (HATis) are yet to be explored in preclinical/clinical trials (139). Significance of HATi is mainly overshadowed by the well-established HDACi. However, studies have shown that HATi can be equally potent blockers of tumorigenesis as HDACi (140).
Vorinostat (class I and II) and romidepsin (class I) are FDA-approved HDACi commonly used to treat several malignancies. HDACi promote acetylation of histones and modulate expression of ~2–10% of cellular genes via effects on chromatin structure and transcription factor/cofactor binding, leading to either increase or decrease in expression (141). It has been shown that the use of vorinostat and panobinostat (pan-HDACi) is able to increase the expression of PD-L1 in TNBC, and PD-L1 and PD-L2 in melanoma by altering chromatin compaction on their promoter regions (100, 142) (Figure 5C).
Other non-canonical effects of HDACis on the regulation of immune responses are also evident from numerous studies. Reports have shown that HDACis can inhibit tumor growth and enhance the host immune response against cancer cells via the suppression of Tregs and FoxP3 expression (143), upregulation of NK cell activating ligands, MHC molecules (class I and II), enhancement of NK and CD8+ T cell cytotoxicity and production of pro-inflammatory cytokines (143–145). Class II HDACi (entinostat) in combination with DNMTi (azacytidine), anti-CTLA-4 and anti-PD-1 mAbs improved treatment outcomes, associated with tumor regression and absence of metastasis in murine models of CT26 colorectal tumors and 4T1 metastatic breast cancer (146). The number of tumor-infiltrating FoxP3+ Tregs was significantly reduced upon treatment with epigenetic modulators, compared to ICIs; however, the effect of epigenetic modulators on tumor-infiltrating CD8+ T cell number was similar to that induced by ICIs alone (146). A study by Orillion et al. demonstrated that the use of entinostat (class I HDACi) suppressed the function of MDSC and enhanced the anti-tumor effects of anti-PD-1 therapy in murine models of lung and renal cell carcinoma, suggesting a rationale for combining HDACi and ICIs in clinical trials (147). Other studies showed that using HDACis in combination with anti-PD-1 therapy enhances the anti-tumor immune response, reduces tumor burden and increases survival in murine tumor models (97, 100). Woods et al. demonstrated that the treatment with HDACi increases the expression of PD-L1 in murine melanoma mouse model, thereby enhancing the sensitivity to anti-PD-1 therapy and overcoming resistance to therapy (97). Similarly, Briere et al. showed that class I/IV HDACi increased the expression of PD-L1 in syngeneic tumor models, and demonstrated that the HDACi in combination with anti-PD-L1 enhanced the anti-tumor immune response compared to their use as a monotherapy (148).
Preclinical studies demonstrated that the upregulation of ICs/IC ligands can be epigenetically modulated. Inhibition of HDAC activity has been reported to modulate PD-L1 expression in chronic lymphocytic leukemia (CLL) and melanoma (97, 149). Recently, Knox et al. demonstrated that the use of HDAC6i significantly reduced the upregulation of PD-L1 and PD-L2 (Figure 5B) in SM1 murine melanoma model, increased expression of IFN-γ and IL-2, and improved survival rates (150). Notably, Kim et al. have recently shown that CG-745, a class I and HDAC6i, induced IL-2 and IFN-γ expression, promoted cytotoxic T cell/NK cell proliferation and inhibited Treg proliferation, which consequently promoted effects of anti-PD-1 therapy in syngeneic mouse models (151). Furthermore, Laino et al. showed that HDAC6 inhibition downregulated the expressions of TIM-3, PD-1, and LAG-3 on expanded T cells from the circulation of melanoma patients (152), indicating the potential benefits of blocking HDAC6 activity to alleviate T cell suppression. Additionally, Bae et al. showed that HDAC6 inhibition reduced the expression of PD-L1 on multiple myeloma bone marrow cells and PD-1 expression on CD8+ T cells (153) (Figure 5B).
The regulation of gene transcription by histone methylation/demethylation is a complex process, which is controlled by the activity of different family classes of enzyme complexes, KMTs (83) and KDMs (84, 85). Different classes of KTMs and KDMs act on different lysine residues on histone H3 or H4 and regulate the expression of various target genes. H3K27me3 is known as a transcriptional repressor for many genes including those associated with tumor resistance to therapy (147). Methylation of H3K27me3 is positively regulated by polycomb repressive complex 2 (PRC2), a member of the KMT family, and its enzymatic subunit, enhancer of zeste homolog 2 (EZH2) (147, 154, 155). Together, these data suggest that targeting EZH2 could interfere with the transcriptional repression mediated by H3K27me3, and therefore overcome tumor resistance to therapy and improve disease outcomes. EZH2 has been implicated in various cancers including melanomas, ovarian, prostrate, and breast cancers (136, 156).
Increased expression of EZH2 has been associated with the development of acquired resistance against recombinant IL-2 (rIL2) and anti-CTLA-4 therapies in melanoma mouse model (154). On the other hand, co-inhibition of EZH2 with rIL-2/anti-CTLA-4 immunotherapies resulted in the downregulation of PD-L1 expression in melanoma cells, increased number of intratumoral PD-1lowTIM-3lowLAG-3lowCD8+ T cells expressing high levels of IFN-γ and suppression of tumor growth (154). Using in vitro and in vivo models, EZH2 activity has been reported to be responsible for the progression of hepatocellular carcinoma by enhancing the expression of galectin-9, TIM-3 ligand, via the trimethylation of H3K27 (157), suggesting that inhibition of EZH2 could be useful for targeting galectin-9 and TIM-3 expression (Figure 5B). Collectively, these results suggest the potential therapeutic benefits of targeting EZH2 in cancer to downregulate IC/IC ligand expression and enhance anti-tumor immunity, and rationalized for the development of histone methylase inhibitors targeting EZH2 in cancer (136), which are currently under different clinical trials for treating different malignancies.
Long Non-coding RNAs and microRNAs as Potential Therapeutic Strategies for Cancer
A recent study by Ma et al. showed that lncRNA, lnMX1-215, negatively regulates PD-L1 and galectin-9 in HNCC and its overexpression significantly reduces tumor cell proliferation /metastasis in vitro and in vivo (158). Authors proposed lnMX1-215 as a potential therapeutic target for HNCC by interfering with PD-1/PD-L1 and TIM-3/galectin-9 signaling pathways and restoring anti-tumor immunity (158).
miRNAs are aberrantly expressed in many types of cancer and malignancies; they regulate the expression of tumor suppressor genes, oncogenes, ICs and immune checkpoint ligands (108). miRNAs have a great advantage over other non-coding RNAs, and mRNAs; they are more stable in biopsy specimens and body fluids, allowing their use as biomarkers (159–161). Moreover, miRNA expression profiles are tissue-specific, which is helpful in speeding up the diagnosis of specific cancer types (160, 161). By upregulating the expression of ICs and IC ligands, miRNAs can contribute to cancer development/progression and compromise the anti-tumor immune responses (108, 162). Targeting this regulatory function of miRNAs can be used to improve clinical responses and enhance the sensitivity of cancer patients' response to immune checkpoint inhibitors (ICIs).
The single blockade of IC commonly results in the upregulation of alternative ICs, suggesting the emergence of compensatory mechanisms which ultimately leads to resistance to ICIs (27, 117, 118, 163). Single miRNA can target multiple ICs/IC ligand in multiple cell types in the same tumor tissue. Hence, this will mimic the effect of the treatment with multiple ICIs and could be used as a therapeutic agent. For instance, tumor suppressive miRNA, miR-138, can be used to reduce the expression of PD-1, and CTLA-4, induce tumor cell apoptosis and impair invasion and tumor metastasis (109, 164, 165). Zhao et al. reported SHNG14/ZEB1/miR-5590-3p positive feedback loop in diffuse B cell lymphoma (DBCL) is associated with attenuated CD8+ T cell activation through PD-1/PD-L1 axis, suggesting that targeting SHNG14 holds the promise of enhancing anti-tumor immunity and restrain tumor progression (166). Another therapeutic strategy that could be employed in cancer treatment is targeting the function of tumor promoting miRNAs using anti-miRNAs (167, 168).
The use of miRNA as a monotherapy is not beneficial and may result in adverse immunologic effects, given that each miRNA can act on multiple target genes, including those encoding immune modulatory molecules (169). Therefore, small doses of anti-miRNAs can be used in combination with chemotherapy or immunotherapies to minimize the risk of adverse effects (108). In addition, miRNAs could be more beneficial if used in combination with ICIs. They may increase the sensitivity of the host immune response to a particular ICI and overcome tumor acquired resistance. In other words, this combination therapy would convert non-responder patients into responders. For instance, Li et al. demonstrated that miR-28 induces T cell exhaustion by upregulating the expression of PD-1, TIM-3, and BTLA (110). This potentially suggests that use of miR-28 in addition to ICIs, especially those targeting PD-1 and/or TIM-3 could result in beneficial outcomes and enhance anti-tumor immunity. Studies have shown negative correlations between miR-138-5p and PD-L1 expression (122), miR-138 and PD1/CTLA-4 expression (109), and miR-424 and PD-L1 expression (111), suggesting that targeting these miRNAs increase the expression of ICs. Thus, we could rationalize that targeting particular miRNAs could be useful in upregulating the expression of ICs, which increases the sensitivity and efficacy of ICIs.
Clinical Trials for Combined Therapeutic Strategies of Epigenetic Modifiers and ICIs
Epigenetic modifiers have the potential to increase the sensitivity to ICIs and restore more potent anti-tumor immune responses and enhance the clinical responses in cancer patients. Several preclinical models have supported the rationale for combining epigenetic modifiers with ICIs, and implicated the need to design clinical trials to assess the efficacy of targeting DNA methylation and HDAC activity, in combination with ICIs, in different cancers (details of ongoing clinical trials are listed in Table 2). Results from completed phase II clinical trial of pembrolizumab (anti-PD-1) in combination with azacytidine in microsatellite stable (MSS) metastatic colorectal cancer patients showed that the combined therapy had mild anti-tumor effects associated with some adverse effects such as anemia, leukopenia and constipation (171).
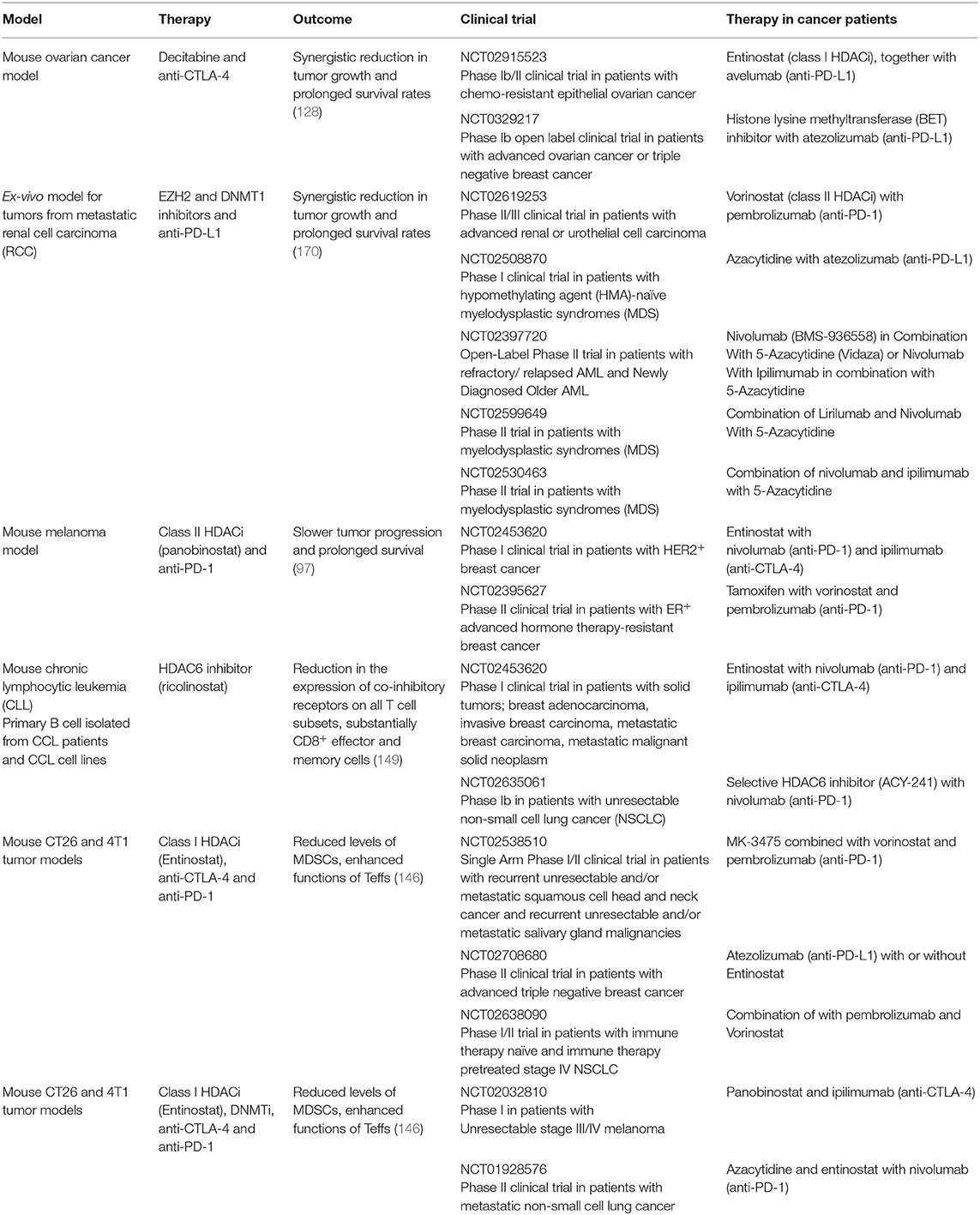
Table 2. Examples of preclinical models and ongoing clinical trials for combination therapies utilizing ICIs and epigenetic modifiers.
Conclusions and Future Directions
Epigenetic modifiers have thus seen important advances in recent years, and currently several are being explored in combination with established ICIs in various clinical trials (172). The rationale for these studies is based on the recent success of ICIs in different cancers, and the unresponsiveness of some cancer patients to current therapies, which is believed to be associated with acquired resistance mechanisms mediated by epigenetic alterations. However, it is noteworthy that several epigenetic enzymes also contribute to cancer progression via other non-epigenetic mechanisms, and ultimately combination therapies to tackle cancer on different fronts with more targeted precision medicine approaches may provide the most effective anti-cancer therapy. It is important to note the complexity of epigenetic regulations while designing epigenetic drugs, and take into consideration all the target genes, which their transcription can be regulated by a specific epigenetic drug. In addition, epigenetic modifiers may have different effects on cancer cells and different types of immune cells, depending on the target genes (173). Further investigations are required to assess the clinical efficacy of using miRNAs in combination with ICIs, and the risk of adverse effects related to toxicity and potential development of autoimmunity.
Author Contributions
RS wrote the manuscript. ST and VS assisted in writing and reviewing the manuscript. EE conceived the topic, wrote, and revised the manuscript. All authors were involved in the final approval of the manuscript.
Funding
This work was supported by a start-up grant [VR04] for Prof. EE from Qatar Biomedical Research Institute, Qatar Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
APC, Antigen-presenting cell; BC, breast cancer; CEACAM-1, carcinoembryonic antigen cell adhesion molecule 1; CRC, colorectal cancer; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; DC, dendritic cell; DNMT, DNA methyltransferase; HAT, histone aceyltransferase; HDAC, histone deacetylase; HMGB1, high-mobility group protein B1; IC, Immune checkpoints; ICI, immune checkpoint inhibitors; LAG-3, Lymphocyte-activation gene 3; MDSC, myeloid-derived suppressor cells; miRNA, micro RNA; NK, natural killer cell; PBMC, peripheral blood mononuclear cells; PC, prostate cancer; PD-1, programmed cell death-1; PD-L1/2, programmed cell death-ligand 1/2; PS, phosphatidylserine; Teff, T effector cell; TET, ten-eleven translocation; TIGIT, T cell immunoreceptor with Ig and ITIM domains; TIM-3, T-cell T cell immunoglobulin and mucin-domain containing-3; TILs, tumor-infiltrating cells; TME, tumor microenvironment; Treg, T regulatory cell.
References
1. Lu Q. The critical importance of epigenetics in autoimmunity. J Autoimmun. (2013) 41:1–5. doi: 10.1016/j.jaut.2013.01.010
2. Suarez-Alvarez B, Baragano Raneros A, Ortega F, Lopez-Larrea C. Epigenetic modulation of the immune function: a potential target for tolerance. Epigenetics. (2013) 8:694–702. doi: 10.4161/epi.25201
3. Ali MA, Matboli M, Tarek M, Reda M, Kamal KM, Nouh M, et al. Epigenetic regulation of immune checkpoints: another target for cancer immunotherapy? Immunotherapy. (2017) 9:99–108. doi: 10.2217/imt-2016-0111
4. Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. (2014) 54:716–27. doi: 10.1016/j.molcel.2014.05.015
5. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. (2010) 31:27–36. doi: 10.1093/carcin/bgp220
6. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. (2018) 50:165. doi: 10.1038/s12276-018-0191-1
7. Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors in cancer therapy: a focus on T-regulatory cells. Immunol Cell Biol. (2018) 96:21–33. doi: 10.1111/imcb.1003
8. Toor SM, Sasidharan Nair V, Decock J, Elkord E. Immune checkpoints in the tumor microenvironment. Semin Cancer Biol. (2019) 19:30123–3. doi: 10.1016/j.semcancer.2019.06.021
9. Saleh R, Elkord E. Treg-mediated acquired resistance to immune checkpoint inhibitors. Cancer Lett. (2019) 457:168–79. doi: 10.1016/j.canlet.2019.05.003
10. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
11. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
12. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
13. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. (2018) 8:86. doi: 10.3389/fonc.2018.00086
14. Anderson AC. Tim-3, a negative regulator of anti-tumor immunity. Curr Opin Immunol. (2012) 24:213–6. doi: 10.1016/j.coi.2011.12.005
15. Anderson AC. Tim-3: an emerging target in the cancer immunotherapy landscape. Cancer Immunol Res. (2014) 2:393–8. doi: 10.1158/2326-6066.CIR-14-0039
16. He Y, Cao J, Zhao C, Li X, Zhou C, Hirsch FR. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. (2018) 11:7005–9. doi: 10.2147/OTT.S170385
17. Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur J Immunol. (1995) 25:2718–21. doi: 10.1002/eji.1830250949
18. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. (1990) 171:1393–405. doi: 10.1084/jem.171.5.1393
19. Tahara-Hanaoka S, Shibuya K, Onoda Y, Zhang H, Yamazaki S, Miyamoto A, et al. Functional characterization of DNAM-1 (CD226) interaction with its ligands PVR (CD155) and nectin-2 (PRR-2/CD112). Int Immunol. (2004) 16:533–8. doi: 10.1093/intimm/dxh059
20. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. (2009) 10:48–57. doi: 10.1038/ni.1674
21. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. (2014) 26:923–37. doi: 10.1016/j.ccell.2014.10.018
22. Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, et al. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci USA. (2004) 101:10691–6. doi: 10.1073/pnas.0307252101
23. Rozali EN, Hato SV, Robinson BW, Lake RA, Lesterhuis WJ. Programmed death ligand 2 in cancer-induced immune suppression. Clin Dev Immunol. (2012) 2012:656340. doi: 10.1155/2012/656340
24. Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, et al. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol. (2010) 184:6545–51. doi: 10.4049/jimmunol.0903879
25. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res. (2015) 3:412–23. doi: 10.1158/2326-6066.CIR-14-0150
26. Workman CJ, Vignali DA. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur J Immunol. (2003) 33:970–9. doi: 10.1002/eji.200323382
27. Saleh R, Elkord E. Acquired resistance to cancer immunotherapy: Role of tumor-mediated immunosuppression. Semin Cancer Biol. (2019) 19:30171–3. doi: 10.1016/j.semcancer.2019.07.017
28. Sasidharan Nair V, El Salhat H, Taha RZ, John A, Ali BR, Elkord E. DNA methylation and repressive H3K9 and H3K27 trimethylation in the promoter regions of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, and PD-L1 genes in human primary breast cancer. Clin Epigenetics. (2018) 10:78. doi: 10.1186/s13148-018-0512-1
29. Sasidharan Nair V, Toor SM, Taha RZ, Shaath H, Elkord E. DNA methylation and repressive histones in the promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and galectin-9 genes in human colorectal cancer. Clin Epigenetics. (2018) 10:104. doi: 10.1186/s13148-018-0539-3
30. Patnaik S, Anupriya. Drugs targeting epigenetic modifications and plausible therapeutic strategies against colorectal cancer. Front Pharmacol. (2019) 10:588. doi: 10.3389/fphar.2019.00588
31. Kagohara LT, Stein-O'Brien GL, Kelley D, Flam E, Wick HC, Danilova LV, et al. Epigenetic regulation of gene expression in cancer: techniques, resources and analysis. Brief Funct Genomics. (2018) 17:49–63. doi: 10.1093/bfgp/elx018
32. Adjiri A. DNA mutations may not be the cause of cancer. Oncol Ther. (2017) 5:85–101. doi: 10.1007/s40487-017-0047-1
33. Roberti A, Valdes AF, Torrecillas R, Fraga MF, Fernandez AF. Epigenetics in cancer therapy and nanomedicine. Clin Epigenetics. (2019) 11:81. doi: 10.1186/s13148-019-0675-4
34. Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stutz AM, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. (2014) 506:445–50.
35. Huidobro C, Fernandez AF, Fraga MF. The role of genetics in the establishment and maintenance of the epigenome. Cell Mol Life Sci. (2013) 70:1543–73. doi: 10.1007/s00018-013-1296-2
36. Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. (2011) 17:330–9. doi: 10.1038/nm.2305
37. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. (2010) 363:2424–33. doi: 10.1056/NEJMoa1005143
38. Haouas H, Haouas S, Uzan G, Hafsia A. Identification of new markers discriminating between myeloid and lymphoid acute leukemia. Hematology. (2010) 15:193–203. doi: 10.1179/102453310X12647083620769
39. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. (2009) 360:2289–301. doi: 10.1056/NEJMoa0810069
40. van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. (2009) 41:521–3. doi: 10.1038/ng.349
41. Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, Carbuccia N, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. (2009) 145:788–800. doi: 10.1111/j.1365-2141.2009.07697.x
42. You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. (2012) 22:9–20. doi: 10.1016/j.ccr.2012.06.008
43. Chen QW, Zhu XY, Li YY, Meng ZQ. Epigenetic regulation and cancer (review). Oncol Rep. (2014) 31:523–32. doi: 10.3892/or.2013.2913
44. Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. (2009) 1:239–59. doi: 10.2217/epi.09.33
45. Lewandowska J, Bartoszek A. DNA methylation in cancer development, diagnosis and therapy–multiple opportunities for genotoxic agents to act as methylome disruptors or remediators. Mutagenesis. (2011) 26:475–87. doi: 10.1093/mutage/ger019
46. Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol. (2014) 6:a019133. doi: 10.1101/cshperspect.a019133
47. Estecio MR, Issa JP. Dissecting DNA hypermethylation in cancer. FEBS Lett. (2011) 585:2078–86. doi: 10.1016/j.febslet.2010.12.001
48. Torano EG, Petrus S, Fernandez AF, Fraga MF. Global DNA hypomethylation in cancer: review of validated methods and clinical significance. Clin Chem Lab Med. (2012) 50:1733–42. doi: 10.1515/cclm-2011-0902
49. Hu P, Liu Q, Deng G, Zhang J, Liang N, Xie J, et al. The prognostic value of cytotoxic T-lymphocyte antigen 4 in cancers: a systematic review and meta-analysis. Sci Rep. (2017) 7:42913. doi: 10.1038/srep42913
50. Le Goux C, Damotte D, Vacher S, Sibony M, Delongchamps NB, Schnitzler A, et al. Correlation between messenger RNA expression and protein expression of immune checkpoint-associated molecules in bladder urothelial carcinoma: a retrospective study. Urol Oncol. (2017) 35:257–63. doi: 10.1016/j.urolonc.2017.01.014
51. Santoni G, Amantini C, Morelli MB, Tomassoni D, Santoni M, Marinelli O, et al. High CTLA-4 expression correlates with poor prognosis in thymoma patients. Oncotarget. (2018) 9:16665–77. doi: 10.18632/oncotarget.24645
52. Toor SM, Murshed K, Al-Dhaheri M, Khawar M, Abu Nada M, Elkord E. Immune checkpoints in circulating and tumor-infiltrating CD4+ T cell subsets in colorectal cancer patients. Front Immunol. (2019) 10:2936. doi: 10.3389/fimmu.2019.02936
53. Wang Y, Dong T, Xuan Q, Zhao H, Qin L, Zhang Q. Lymphocyte-activation gene-3 expression and prognostic value in neoadjuvant-treated triple-negative breast cancer. J Breast Cancer. (2018) 21:124–33. doi: 10.4048/jbc.2018.21.2.124
54. Zhou E, Huang Q, Wang J, Fang C, Yang L, Zhu M, et al. Up-regulation of Tim-3 is associated with poor prognosis of patients with colon cancer. Int J Clin Exp Pathol. (2015) 8:8018–27.
55. Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X, et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology. (2012) 56:1342–51. doi: 10.1002/hep.25777
56. Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, et al. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut. (2015) 64:1593–604. doi: 10.1136/gutjnl-2014-307671
57. Simone R, Tenca C, Fais F, Luciani M, De Rossi G, Pesce G, et al. A soluble form of CTLA-4 is present in paediatric patients with acute lymphoblastic leukaemia and correlates with CD1d+ expression. PLoS ONE. (2012) 7:e44654. doi: 10.1371/journal.pone.0044654
58. Cheng HY, Kang PJ, Chuang YH, Wang YH, Jan MC, Wu CF, et al. Circulating programmed death-1 as a marker for sustained high hepatitis B viral load and risk of hepatocellular carcinoma. PLoS ONE. (2014) 9:e95870. doi: 10.1371/journal.pone.0095870
59. Finkelmeier F, Canli O, Tal A, Pleli T, Trojan J, Schmidt M, et al. High levels of the soluble programmed death-ligand (sPD-L1) identify hepatocellular carcinoma patients with a poor prognosis. Eur J Cancer. (2016) 59:152–9. doi: 10.1016/j.ejca.2016.03.002
60. Rossille D, Gressier M, Damotte D, Maucort-Boulch D, Pangault C, Semana G, et al. High level of soluble programmed cell death ligand 1 in blood impacts overall survival in aggressive diffuse large B-Cell lymphoma: results from a French multicenter clinical trial. Leukemia. (2014) 28:2367–75. doi: 10.1038/leu.2014.137
61. Fukuda T, Kamai T, Masuda A, Nukui A, Abe H, Arai K, et al. Higher preoperative serum levels of PD-L1 and B7-H4 are associated with invasive and metastatic potential and predictable for poor response to VEGF-targeted therapy and unfavorable prognosis of renal cell carcinoma. Cancer Med. (2016) 5:1810–20. doi: 10.1002/cam4.754
62. Takahashi N, Iwasa S, Sasaki Y, Shoji H, Honma Y, Takashima A, et al. Serum levels of soluble programmed cell death ligand 1 as a prognostic factor on the first-line treatment of metastatic or recurrent gastric cancer. J Cancer Res Clin Oncol. (2016) 142:1727–38. doi: 10.1007/s00432-016-2184-6
63. Okuma Y, Hosomi Y, Nakahara Y, Watanabe K, Sagawa Y, Homma S. High plasma levels of soluble programmed cell death ligand 1 are prognostic for reduced survival in advanced lung cancer. Lung Cancer. (2017) 104:1–6. doi: 10.1016/j.lungcan.2016.11.023
64. Li F, Li N, Sang J, Fan X, Deng H, Zhang X, et al. Highly elevated soluble Tim-3 levels correlate with increased hepatocellular carcinoma risk and poor survival of hepatocellular carcinoma patients in chronic hepatitis B virus infection. Cancer Manag Res. (2018) 10:941–51. doi: 10.2147/CMAR.S162478
65. Gu D, Ao X, Yang Y, Chen Z, Xu X. Soluble immune checkpoints in cancer: production, function and biological significance. J Immunother Cancer. (2018) 6:132. doi: 10.1186/s40425-018-0449-0
66. Putiri EL, Robertson KD. Epigenetic mechanisms and genome stability. Clin Epigenetics. (2011) 2:299–314. doi: 10.1007/s13148-010-0017-z
67. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. (1999) 99:247–57. doi: 10.1016/S0092-8674(00)81656-6
68. Hermann A, Goyal R, Jeltsch A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem. (2004) 279:48350–9. doi: 10.1074/jbc.M403427200
69. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. (2009) 324:930–5. doi: 10.1126/science.1170116
70. Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. (2011) 286:35334–8. doi: 10.1074/jbc.C111.284620
71. Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene. (2001) 20:3139–55. doi: 10.1038/sj.onc.1204341
72. Elashi AA, Sasidharan Nair V, Taha RZ, Shaath H, Elkord E. DNA methylation of immune checkpoints in the peripheral blood of breast and colorectal cancer patients. Oncoimmunology. (2019) 8:e1542918. doi: 10.1080/2162402X.2018.1542918
73. Jung H, Kim HS, Kim JY, Sun JM, Ahn JS, Ahn MJ, et al. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat Commun. (2019) 10:4278. doi: 10.1038/s41467-019-12159-9
74. Gowrishankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, Hersey P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kappaB. PLoS One. (2015) 10:e0123410. doi: 10.1371/journal.pone.0123410
75. Marwitz S, Scheufele S, Perner S, Reck M, Ammerpohl O, Goldmann T. Epigenetic modifications of the immune-checkpoint genes CTLA4 and PDCD1 in non-small cell lung cancer results in increased expression. Clin Epigenetics. (2017) 9:51. doi: 10.1186/s13148-017-0354-2
76. Franzen A, Vogt TJ, Muller T, Dietrich J, Schrock A, Golletz C, et al. PD-L1 (CD274) and PD-L2 (PDCD1LG2) promoter methylation is associated with HPV infection and transcriptional repression in head and neck squamous cell carcinomas. Oncotarget. (2018) 9:641–50. doi: 10.18632/oncotarget.23080
77. Goltz D, Gevensleben H, Dietrich J, Dietrich D. PD-L1 (CD274) promoter methylation predicts survival in colorectal cancer patients. Oncoimmunology. (2017) 6:e1257454. doi: 10.1080/2162402X.2016.1257454
78. Goltz D, Gevensleben H, Dietrich J, Schroeck F, de Vos L, Droege F, et al. PDCD1 (PD-1) promoter methylation predicts outcome in head and neck squamous cell carcinoma patients. Oncotarget. (2017) 8:41011–20. doi: 10.18632/oncotarget.17354
79. Goltz D, Gevensleben H, Grunen S, Dietrich J, Kristiansen G, Landsberg J, et al. PD-L1 (CD274) promoter methylation predicts survival in patients with acute myeloid leukemia. Leukemia. (2017) 31:738–43. doi: 10.1038/leu.2016.328
80. Rover LK, Gevensleben H, Dietrich J, Bootz F, Landsberg J, Goltz D, et al. PD-1 (PDCD1) promoter methylation is a prognostic factor in patients with diffuse lower-grade gliomas harboring isocitrate dehydrogenase (IDH) mutations. EBioMedicine. (2018) 28:97–104. doi: 10.1016/j.ebiom.2018.01.016
81. Zhao Z, Shilatifard A. Epigenetic modifications of histones in cancer. Genome Biol. (2019) 20:245. doi: 10.1186/s13059-019-1870-5
82. Herz HM, Garruss A, Shilatifard A. SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem Sci. (2013) 38:621–39. doi: 10.1016/j.tibs.2013.09.004
83. Mohan M, Herz HM, Shilatifard A. SnapShot: Histone lysine methylase complexes. Cell. (2012) 149:498–e1. doi: 10.1016/j.cell.2012.03.025
84. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. (2004) 119:941–53. doi: 10.1016/j.cell.2004.12.012
85. Fang R, Barbera AJ, Xu Y, Rutenberg M, Leonor T, Bi Q, et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol Cell. (2010) 39:222–33. doi: 10.1016/j.molcel.2010.07.008
86. Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harb Perspect Biol. (2016) 8:a019521. doi: 10.1101/cshperspect.a019521
87. Nowak SJ, Corces VG. Phosphorylation of histone H3 correlates with transcriptionally active loci. Genes Dev. (2000) 14:3003–13. doi: 10.1101/gad.848800
88. Trievel RC. Structure and function of histone methyltransferases. Crit Rev Eukaryot Gene Expr. (2004) 14:147–69. doi: 10.1615/CritRevEukaryotGeneExpr.v14.i3.10
89. D'Oto A, Tian QW, Davidoff AM, Yang J. Histone demethylases and their roles in cancer epigenetics. J Med Oncol Ther. (2016) 1:34–40.
90. Sasidharan Nair V, Saleh R, Toor SM, Taha RZ, Ahmed AA, Kurer MA, et al. Transcriptomic profiling disclosed the role of DNA methylation and histone modifications in tumor-infiltrating myeloid-derived suppressor cell subsets in colorectal cancer. Clin Epigenetics. (2020) 12:13. doi: 10.1186/s13148-020-0808-9
91. Yang XJ, Seto E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr Opin Genet Dev. (2003) 13:143–53. doi: 10.1016/S0959-437X(03)00015-7
92. Kouzarides T. Chromatin modifications and their function. Cell. (2007) 128:693–705. doi: 10.1016/j.cell.2007.02.005
93. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. (2009) 325:834–40. doi: 10.1126/science.1175371
94. Seligson DB, Horvath S, McBrian MA, Mah V, Yu H, Tze S, et al. Global levels of histone modifications predict prognosis in different cancers. Am J Pathol. (2009) 174:1619–28. doi: 10.2353/ajpath.2009.080874
95. Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. (2014) 6:a018713. doi: 10.1101/cshperspect.a018713
96. Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther. (2019) 4:62. doi: 10.1038/s41392-019-0095-0
97. Woods DM, Sodre AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC inhibition upregulates pd-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res. (2015) 3:1375–85. doi: 10.1158/2326-6066.CIR-15-0077-T
98. Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, et al. HDAC inhibitors enhance t-cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res. (2016) 22:4119–32. doi: 10.1158/1078-0432.CCR-15-2584
99. Fan P, Zhao J, Meng Z, Wu H, Wang B, Wu H, et al. Overexpressed histone acetyltransferase 1 regulates cancer immunity by increasing programmed death-ligand 1 expression in pancreatic cancer. J Exp Clin Cancer Res. (2019) 38:47–59. doi: 10.1186/s13046-019-1044-z
100. Terranova-Barberio M, Thomas S, Ali N, Pawlowska N, Park J, Krings G, et al. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget. (2017) 8:114156–72. doi: 10.18632/oncotarget.23169
101. Fortson WS, Kayarthodi S, Fujimura Y, Xu H, Matthews R, Grizzle WE, et al. Histone deacetylase inhibitors, valproic acid and trichostatin-A induce apoptosis and affect acetylation status of p53 in ERG-positive prostate cancer cells. Int J Oncol. (2011) 39:111–9. doi: 10.3892/ijo.2011.1014
102. Chen HP, Zhao YT, Zhao TC. Histone deacetylases and mechanisms of regulation of gene expression. Crit Rev Oncog. (2015) 20:35–47. doi: 10.1615/CritRevOncog.2015012997
103. Wang P, Ning S, Zhang Y, Li R, Ye J, Zhao Z, et al. Identification of lncRNA-associated competing triplets reveals global patterns and prognostic markers for cancer. Nucleic Acids Res. (2015) 43:3478–89. doi: 10.1093/nar/gkv233
104. O'Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol. (2018) 9:402. doi: 10.3389/fendo.2018.00402
105. Bueno MJ, Malumbres M. MicroRNAs and the cell cycle. Biochim Biophy Acta. (2011) 1812:592–601. doi: 10.1016/j.bbadis.2011.02.002
106. Otto T, Candido SV, Pilarz MS, Sicinska E, Bronson RT, Bowden M, et al. Cell cycle-targeting microRNAs promote differentiation by enforcing cell-cycle exit. Proc Natl Acad Sci USA. (2017) 114:10660–5. doi: 10.1073/pnas.1702914114
107. Loh HY, Norman BP, Lai KS, Rahman N, Alitheen NBM, Osman MA. The regulatory role of microRNAS in breast cancer. Int J Mol Sci. (2019) 20(19). doi: 10.3390/ijms20194940
108. Smolle MA, Calin HN, Pichler M, Calin GA. Noncoding RNAs and immune checkpoints-clinical implications as cancer therapeutics. FEBS J. (2017) 284:1952–66. doi: 10.1111/febs.14030
109. Wei J, Nduom EK, Kong LY, Hashimoto Y, Xu S, Gabrusiewicz K, et al. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro Oncol. (2016) 18:639–48. doi: 10.1093/neuonc/nov292
110. Li Q, Johnston N, Zheng X, Wang H, Zhang X, Gao D, et al. miR-28 modulates exhaustive differentiation of T cells through silencing programmed cell death-1 and regulating cytokine secretion. Oncotarget. (2016) 7:53735–50. doi: 10.18632/oncotarget.10731
111. Xu S, Tao Z, Hai B, Liang H, Shi Y, Wang T, et al. miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat Commun. (2016) 7:11406. doi: 10.1038/ncomms11406
112. Richardsen E, Andersen S, Al-Saad S, Rakaee M, Nordby Y, Pedersen MI, et al. Low expression of miR-424-3p is highly correlated with clinical failure in prostate cancer. Sci Rep. (2019) 9:10662. doi: 10.1038/s41598-019-47234-0
113. Wu CT, Lin WY, Chang YH, Lin PY, Chen WC, Chen MF. DNMT1-dependent suppression of microRNA424 regulates tumor progression in human bladder cancer. Oncotarget. (2015) 6:24119–31. doi: 10.18632/oncotarget.4431
114. Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, et al. PDL1 regulation by p53 via miR-34. J Natl Cancer Inst. (2016) 108:djv303. doi: 10.1093/jnci/djv303
115. Fooladinezhad H, Khanahmad H, Ganjalikhani-Hakemi M, Doosti A. Negative regulation of TIM-3 expression in AML cell line (HL-60) using miR-330-5p. Br J Biomed Sci. (2016) 73:129–33. doi: 10.1080/09674845.2016.1194564
116. Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology. (2017) 6:e1261779. doi: 10.1080/2162402X.2016.1261779
117. Shayan G, Ferris RL. PD-1 blockade upregulate TIM-3 expression as a compensatory regulation of immune check point receptors in HNSCC TIL. J Immunother Cancer. (2015) 3(Suppl 2):P196–P. doi: 10.1186/2051-1426-3-S2-P196
118. Oweida A, Hararah MK, Phan A, Binder D, Bhatia S, Lennon S, et al. Resistance to radiotherapy and PD-L1 blockade is mediated by TIM-3 upregulation and regulatory T-cell infiltration. Clin Cancer Res. (2018) 24:5368–80. doi: 10.1158/1078-0432.CCR-18-1038
119. Querfeld C, Wu X, Sanchez J, Palmer J, Motevalli A, Zain J, et al. The miRNA profile of cutaneous T cell lymphoma correlates with the dysfunctional immunophenotype of the disease. Blood. (2016) 128:4132. doi: 10.1182/blood.V128.22.4132.4132
120. Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. (2014) 5:5241. doi: 10.1038/ncomms6241
121. Bader AG. miR-34 - a microRNA replacement therapy is headed to the clinic. Front Genet. (2012) 3:120. doi: 10.3389/fgene.2012.00120
122. Zhao L, Yu H, Yi S, Peng X, Su P, Xiao Z, et al. The tumor suppressor miR-138-5p targets PD-L1 in colorectal cancer. Oncotarget. (2016) 7:45370–84. doi: 10.18632/oncotarget.9659
123. Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. (1998) 395:89–93. doi: 10.1038/25779
124. Park JW, Han JW. Targeting epigenetics for cancer therapy. Arch Pharm Res. (2019) 42:159–70. doi: 10.1007/s12272-019-01126-z
125. Suzuki T, Miyata N. Non-hydroxamate histone deacetylase inhibitors. Curr Med Chem. (2005) 12:2867–80. doi: 10.2174/092986705774454706
126. Sorm F, Piskala A, Cihak A, Vesely J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia. (1964). 20:202–3. doi: 10.1007/BF02135399
127. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. (2015) 162:974–86. doi: 10.1016/j.cell.2015.07.011
128. Wang L, Amoozgar Z, Huang J, Saleh MH, Xing D, Orsulic S, et al. Decitabine enhances lymphocyte migration and function and synergizes with CTLA-4 blockade in a murine ovarian cancer model. Cancer Immunol Res. (2015) 3:1030–41. doi: 10.1158/2326-6066.CIR-15-0073
129. Chiappinelli KB, Zahnow CA, Ahuja N, Baylin SB. Combining epigenetic and immunotherapy to combat cancer. Cancer Res. (2016) 76:1683–9. doi: 10.1158/0008-5472.CAN-15-2125
130. Gnyszka A, Jastrzebski Z, Flis S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. (2013) 33:2989–96.
131. Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. (2014) 28:1280–8. doi: 10.1038/leu.2013.355
132. Cowan LA, Talwar S, Yang AS. Will DNA methylation inhibitors work in solid tumors? A review of the clinical experience with azacitidine and decitabine in solid tumors. Epigenomics. (2010) 2:71–86. doi: 10.2217/epi.09.44
133. Fu S, Hu W, Iyer R, Kavanagh JJ, Coleman RL, Levenback CF, et al. Phase 1b-2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer. Cancer. (2011) 117:1661–9. doi: 10.1002/cncr.25701
134. Singal R, Ramachandran K, Gordian E, Quintero C, Zhao W, Reis IM. Phase I/II study of azacitidine, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel-based therapy. Clin Genitourin Cancer. (2015) 13:22–31. doi: 10.1016/j.clgc.2014.07.008
135. Tan L, Shi YG. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development. (2012) 139:1895–902. doi: 10.1242/dev.070771
136. Ning B, Li W, Zhao W, Wang R. Targeting epigenetic regulations in cancer. Acta Biochim Biophys Sin (Shanghai). (2016) 48:97–109. doi: 10.1093/abbs/gmv116
137. Bian EB, Zong G, Xie YS, Meng XM, Huang C, Li J, et al. TET family proteins: new players in gliomas. J Neurooncol. (2014) 116:429–35. doi: 10.1007/s11060-013-1328-7
138. Wapenaar H, Dekker FJ. Histone acetyltransferases: challenges in targeting bi-substrate enzymes. Clin Epigenetics. (2016) 8:59. doi: 10.1186/s13148-016-0225-2
139. Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. (2007) 26:5310–8. doi: 10.1038/sj.onc.1210599
140. Gerrard DL, Boyd JR, Stein GS, Jin VX, Frietze S. Disruption of broad epigenetic domains in PDAC cells by HAT inhibitors. Epigenomes. (2019) 3: doi: 10.3390/epigenomes3020011
141. Tiffon C, Adams J, van der Fits L, Wen S, Townsend P, Ganesan A, et al. The histone deacetylase inhibitors vorinostat and romidepsin downmodulate IL-10 expression in cutaneous T-cell lymphoma cells. Br J Pharmacol. (2011) 162:1590–602. doi: 10.1111/j.1476-5381.2010.01188.x
142. Woods DM, Woan K, Cheng F, Wang H, Perez-Villarroel P, Lee C, et al. The antimelanoma activity of the histone deacetylase inhibitor panobinostat (LBH589) is mediated by direct tumor cytotoxicity and increased tumor immunogenicity. Melanoma Res. (2013) 23:341–8. doi: 10.1097/CMR.0b013e328364c0ed
143. Shen L, Ciesielski M, Ramakrishnan S, Miles KM, Ellis L, Sotomayor P, et al. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PLoS ONE. (2012) 7:e30815. doi: 10.1371/journal.pone.0030815
144. West AC, Smyth MJ, Johnstone RW. The anticancer effects of HDAC inhibitors require the immune system. Oncoimmunology. (2014) 3:e27414. doi: 10.4161/onci.27414
145. Oki Y, Buglio D, Zhang J, Ying Y, Zhou S, Sureda A, et al. Immune regulatory effects of panobinostat in patients with Hodgkin lymphoma through modulation of serum cytokine levels and T-cell PD1 expression. Blood Cancer J. (2014) 4:e236. doi: 10.1038/bcj.2014.58
146. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. (2014) 111:11774–9. doi: 10.1073/pnas.1410626111
147. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. (2011) 469:343–9. doi: 10.1038/nature09784
148. Briere D, Sudhakar N, Woods DM, Hallin J, Engstrom LD, Aranda R, et al. The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy. Cancer Immunol Immunother. (2018) 67:381–92. doi: 10.1007/s00262-017-2091-y
149. Powers JJ, Maharaj KK, Sahakian E, Xing L, PerezVillarroel P, Knox T, et al. Histone deacetylase 6 (HDAC6) as a regulator of immune check-point molecules in chronic lymphocytic leukemia (CLL). Blood. (2014) 124:3311–. doi: 10.1182/blood.V124.21.3311.3311
150. Knox T, Sahakian E, Banik D, Hadley M, Palmer E, Noonepalle S, et al. Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells. Sci Rep. (2019) 9:6136. doi: 10.1038/s41598-019-42237-3
151. Kim YD, Park SM, Ha HC, Lee AR, Won H, Cha H, et al. HDAC Inhibitor, CG-745, enhances the anti-cancer effect of Anti-PD-1 immune checkpoint inhibitor by modulation of the immune microenvironment. J Cancer. (2020) 11:4059–72. doi: 10.7150/jca.44622
152. Laino AS, Betts BC, Veerapathran A, Dolgalev I, Sarnaik A, Quayle SN, et al. HDAC6 selective inhibition of melanoma patient T-cells augments anti-tumor characteristics. J Immunother Cancer. (2019) 7:33. doi: 10.1186/s40425-019-0517-0
153. Bae J, Hideshima T, Tai YT, Song Y, Richardson P, Raje N, et al. Histone deacetylase (HDAC) inhibitor ACY241 enhances anti-tumor activities of antigen-specific central memory cytotoxic T lymphocytes against multiple myeloma and solid tumors. Leukemia. (2018) 32:1932–47. doi: 10.1038/s41375-018-0062-8
154. Zingg D, Arenas-Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J, et al. The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. (2017) 20:854–67. doi: 10.1016/j.celrep.2017.07.007
155. Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. (2012) 8:890–6. doi: 10.1038/nchembio.1084
156. Jones BA, Varambally S, Arend RC. Histone methyltransferase EZH2: a therapeutic target for ovarian cancer. Mol Cancer Ther. (2018) 17:591–602. doi: 10.1158/1535-7163.MCT-17-0437
157. Chen S, Pu J, Bai J, Yin Y, Wu K, Wang J, et al. EZH2 promotes hepatocellular carcinoma progression through modulating miR-22/galectin-9 axis. J Exp Clin Cancer Res. (2018) 37:3. doi: 10.1186/s13046-017-0670-6
158. Ma H, Chang H, Yang W, Lu Y, Hu J, Jin S. A novel IFNalpha-induced long noncoding RNA negatively regulates immunosuppression by interrupting H3K27 acetylation in head and neck squamous cell carcinoma. Mol Cancer. (2020) 19:4. doi: 10.1186/s12943-019-1123-y
159. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. (2006) 6:857–66. doi: 10.1038/nrc1997
160. Blondal T, Jensby Nielsen S, Baker A, Andreasen D, Mouritzen P, Wrang Teilum M, et al. Assessing sample and miRNA profile quality in serum and plasma or other biofluids. Methods. (2013) 59:S1–6. doi: 10.1016/j.ymeth.2012.09.015
161. Pichler M, Calin GA. MicroRNAs in cancer: from developmental genes in worms to their clinical application in patients. Br J Cancer. (2015) 113:569–73. doi: 10.1038/bjc.2015.253
162. Lee HM, Nguyen DT, Lu LF. Progress and challenge of microRNA research in immunity. Front Genet. (2014) 5:178. doi: 10.3389/fgene.2014.00178
163. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. (2016) 7:10501. doi: 10.1038/ncomms10501
164. Liu X, Jiang L, Wang A, Yu J, Shi F, Zhou X. MicroRNA-138 suppresses invasion and promotes apoptosis in head and neck squamous cell carcinoma cell lines. Cancer Lett. (2009) 286:217–22. doi: 10.1016/j.canlet.2009.05.030
165. Yeh YM, Chuang CM, Chao KC, Wang LH. MicroRNA-138 suppresses ovarian cancer cell invasion and metastasis by targeting SOX4 and HIF-1alpha. Int J Cancer. (2013) 133:867–78. doi: 10.1002/ijc.28086
166. Zhao L, Liu Y, Zhang J, Liu Y, Qi Q. LncRNA SNHG14/miR-5590-3p/ZEB1 positive feedback loop promoted diffuse large B cell lymphoma progression and immune evasion through regulating PD-1/PD-L1 checkpoint. Cell Death Dis. (2019) 10:731. doi: 10.1038/s41419-019-1886-5
167. Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. (2005) 438:685–9. doi: 10.1038/nature04303
168. Elmen J, Lindow M, Schutz S, Lawrence M, Petri A, Obad S, et al. LNA-mediated microRNA silencing in non-human primates. Nature. (2008) 452:896–9. doi: 10.1038/nature06783
169. Hanna J, Hossain GS, Kocerha J. The Potential for microRNA Therapeutics and Clinical Research. Front Genet. (2019) 10:478. doi: 10.3389/fgene.2019.00478
170. Panda A, de Cubas AA, Stein M, Riedlinger G, Kra J, Mayer T, et al. Endogenous retrovirus expression is associated with response to immune checkpoint blockade in clear cell renal cell carcinoma. JCI. (2018) 3:e121522. doi: 10.1172/jci.insight.121522
171. Lee JJ, Sun W, Bahary N, Ohr J, Rhee JC, Stoller RG, et al. Phase 2 study of pembrolizumab in combination with azacitidine in subjects with metastatic colorectal cancer. Int J Clin Oncol. (2017) 35:3054. doi: 10.1200/JCO.2017.35.15_suppl.3054
172. Gallagher SJ, Shklovskaya E, Hersey P. Epigenetic modulation in cancer immunotherapy. Curr Opin Pharmacol. (2017) 35:48–56. doi: 10.1016/j.coph.2017.05.006
Keywords: cancer, immune checkpoints, epigenetics, DNA methylation, histone modifications, therapeutic targets
Citation: Saleh R, Toor SM, Sasidharan Nair V and Elkord E (2020) Role of Epigenetic Modifications in Inhibitory Immune Checkpoints in Cancer Development and Progression. Front. Immunol. 11:1469. doi: 10.3389/fimmu.2020.01469
Received: 22 March 2020; Accepted: 05 June 2020;
Published: 14 July 2020.
Edited by:
Lidia Karabon, Polish Academy of Sciences, PolandReviewed by:
Zong Sheng Guo, University of Pittsburgh School of Medicine, United StatesAlejandro Villagra, George Washington University, United States
Copyright © 2020 Saleh, Toor, Sasidharan Nair and Elkord. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eyad Elkord, ZWVsa29yZEBoYmt1LmVkdS5xYQ==; ZS5lbGtvcmRAc2FsZm9yZC5hYy51aw==