 Chao Peng
Chao Peng Yaobin Ouyang
Yaobin Ouyang Nonghua Lu
Nonghua Lu Nianshuang Li
Nianshuang Li- 1Department of Gastroenterology, The First Affiliated Hospital of Nanchang University, Nanchang, China
- 2Institute of Digestive Disease, The First Affiliated Hospital of Nanchang University, Nanchang, China
Gastrointestinal (GI) cancers, especially gastric cancer and colorectal cancer (CRC), represent a major global health burden. A large population of microorganisms residing in the GI tract regulate physiological processes, such as the immune response, metabolic balance, and homeostasis. Accumulating evidence has revealed the alteration of microbial communities in GI tumorigenesis. Experimental studies in cell lines and animal models showed the functional roles and molecular mechanisms of several bacteria in GI cancers, including Helicobacter pylori in gastric cancer as well as Fusobacterium nucleatum, Escherichia coli, Peptostreptococcus anaerobius, and Bacteroides fragilis in CRC. The transcriptional factor NF-κB plays a crucial role in the host response to microbial infection through orchestrating innate and adaptive immune functions. Moreover, NF-κB activity is linked to GI cancer initiation and development through its induction of chronic inflammation, cellular transformation and proliferation. Here, we provide an overview and discussion of modulation of the NF-κB signaling pathway by microbiota, especially infectious bacteria, in GI tumorigenesis, with a major focus on gastric cancer and CRC.
Introduction
Cancer is the second leading cause of death globally behind cardiovascular disease, according to statistical data from the World Health Organization (1). Gastrointestinal carcinoma remains the main cause of cancer-related morbidity and mortality worldwide, particularly in East Asian countries (2). The roles of genetic risk factors in cancer development have been well-studied. Germline mutation in CDH1 (E-cadherin) is widely detected in gastric cancer (3). The genetic mutation of adenomatous polyposis coli (APC) is associated with a higher risk of familial adenomatous polyposis and colorectal cancer (4). In addition, abundant and diverse microbes reside in the human body. These microorganisms include bacteria, fungi, archaea, and viruses. Approximately 100 trillion of microorganisms exist in the human gastrointestinal tract (5, 6). The activities of complex microbial communities orchestrate many aspects of human health, such as immune responses, metabolic balance, and homeostasis. Recently, accumulating evidence suggests that disruption of the microbiota is involved in diverse human diseases, including gastrointestinal disorders, obesity, inflammatory bowel disease (IBD), and depression (7, 8). Data at different levels from animal models and cell lines indicate that microbial pathogens exert oncogenic properties during gastrointestinal tumorigenesis (9, 10). It has been well-established that infection with the gram-negative bacterium Helicobacter pylori (H. pylori) significantly increases the risk of gastric cancer. The presence of Fusobacterium nucleatum (F. nucleatum), a gram-negative obligate anaerobic bacterium, can contribute to intestinal tumorigenesis (11). The NF-κB signaling pathway can be activated to modulate host cellular events after exposure to different microbial pathogens or microbial products, such as lipopolysaccharide (LPS) and pathogen-associated molecular patterns (PAMPs) (12). Cytoplasmic NF-κB is transferred to the nucleus, where it induces antimicrobial inflammatory cytokine expression, which functions as a rapid defense mechanism against microbes, including infectious bacteria. However, prolonged chronic inflammation due to the activation of NF-κB proteins may result in tissue damage, further contributing to tumorigenesis by changing the genetic and epigenetic states of damaged tissues and the host microenvironment (13). In this review, we provide an update on recent advances in our understanding of the modulation of the NF-κB signaling pathway by microbes, particularly infectious bacteria in gastrointestinal tumorigenesis, with a major focus on stomach and intestinal cancers.
Signal Transduction of the NF-κB Pathway
Activation of NF-κB Signaling
The NF-κB family of transcriptional factors regulates a large number of genes involved in different cellular processes, such as cell proliferation, differentiation, genome stability, and the innate immune and adaptive immune responses (14). The NF-κB family consists of five members that interact with each other to homodimerize or heterodimerize: NF-κB1 (also named p50), NF-κB1 (also named p52), RelA (also named p65), RelB, and c-Rel (15). Activation of the NF-κB signaling pathway can occur through canonical and non-canonical (or alternative) pathways (16). The IKK kinase complex, including the catalytic subunits IKKα, IKKβ, and a regulatory subunit NF-κB essential modulator (NEMO), is the core component of the NF-κB signaling cascade (17). Under normal physiological conditions, NF-κB dimers in an inactive form are sequestered to the cytoplasm through their interaction with IKB-inhibitory proteins (IκBα, IκBβ, and IκBε). Upon stimulation with diverse bacteria, various immune receptors, such as Toll-like receptors (TLRs) and TNF receptors (TNFRs), can be activated to mediate the NF-κB signaling pathway. The primary mechanism of canonical NF-κB activation is the degradation of IκBα. In this process, IKK phosphorylates IκBα and leads to its ubiquitination through the SCFβTrCP ligase-dependent proteasome degradation machinery. As a result, NF-κB is released and translocated from the cytoplasm to the nucleus, where it binds DNA and regulates downstream gene transcription (18–20). The alternative NF-κB signaling is mainly dependent on the activation of NF-κB2 (p100)/ RelB complex, which specifically responds to a subset of receptors, including BAFF (B-cell activating factor belonging to TNF family) receptor (BAFFR), CD40, and receptor activator for NF-κB (RANK) (21). NF-κB-inducing kinase (NIK) is a core component of the non-canonical pathway. Ikkα is activated by NIK and then phosphorylates p100 (22). Then, p100 is processed to its active form, p52, which forms a heterodimer with RelB that translates to the nucleus (21, 23–25). Both the canonical and non-canonical pathways can be mediated to orchestrate host inflammation in response to microbial pathogen infection (Figure 1).
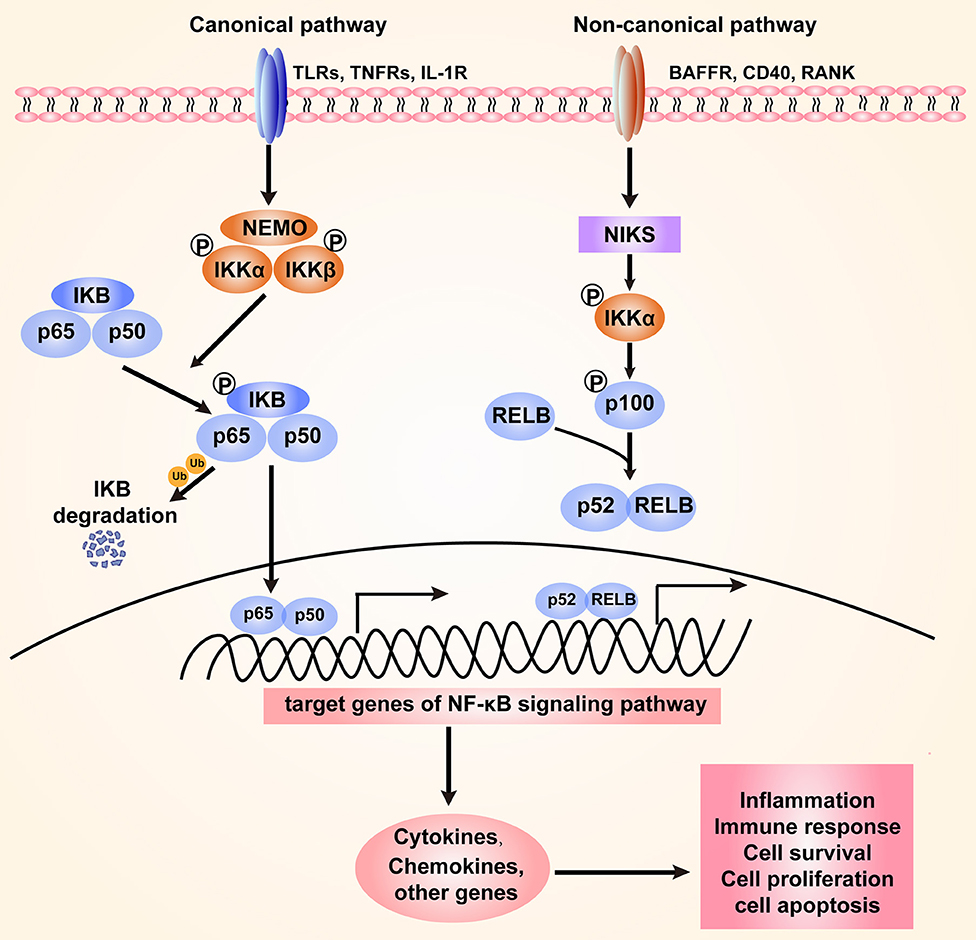
Figure 1. The canonical and non-canonical NF-κB signaling pathway. The canonical pathway is induced by TLRs, TNFRs, and IL-1R. Activation of this cascade leads to the phosphorylation and degradation of inhibitory protein IκB. NF-κB is activated by release from the IκB-containing complex, then translocating into nucleus. The non-canonical pathway is dependent on the activation of NF-κB2 (p100)/ RelB complex by BAFFR, CD40, and RANK. This cascade induces phosphorylation of NIK, which subsequently phosphorylates IKKα. Then p52-RelB heterodimer is activated and translocate to the nucleus. The activation of NF-κB signaling regulates various cellular processes through targeting the expression of cytokines, chemokines and other genes.
NF-κB Activation, Inflammation and Cancer
Activation of the NF-κB cascade is a central regulator of host responses to microbial infection. The innate immune response, a first line of host defense against different microorganisms, is modulated by the NF-κB signaling pathway, which in turn promotes the expression of target genes (12, 26). Most importantly, NF-κB acts as a central regulator of the immune response and inflammation by upregulating many chemokines (CXCL1, CXCL2, CXCL3, etc.) and cytokines (TNFα, IL-1β, IL-6, IL-8, etc.) (27). Activated NF-κB also affects cellular proliferation and apoptosis by targeting Bcl2, IAPs, and cyclins. In addition, NF-κB is essential for the induction of antimicrobial effectors that can effectively eliminate pathogenic microbes, such as antimicrobial peptides (AMPs) (28).
NF-κB, a critical regulator, has been linked to inflammation and cancer at multiple levels (13, 29). On the one hand, inflammation is a host-protective response to microbial pathogens or tissue damage. Upon stimulation by diverse bacterial species (H. pylori, F. nucleatum, etc.), NF-κB is highly activated at the site of infection for its antimicrobial activity and maintenance of tissue homeostasis (30). On the other hand, the strong involvement of the NF-κB pathway in the adaptive immune response, through either B or T cells, increases the severity and extent of inflammation (31). Constitutive chronic inflammation may lead to damaged tissues, autoimmune diseases and cancer initiation by increased cellular stresses and the accumulation of DNA damage. The alteration of genetic stability and epigenetic states at the site of damaged tissues contributes to generating a pro-tumorigenic microenvironment (32). Elevated NF-κB activity and the increased expression of proinflammatory cytokines have been documented in various tumorous tissues (33, 34). Constitutive activation of NF-κB turns on the transcription of genes that promote cell proliferation, cell survival, and genomic instability and thereby contributes to oncogenic mutations. There is strong evidence that the inducible activation of the NF-κB cascade promotes cell proliferation by targeting cyclin D1 expression and inhibits cell apoptosis by targeting BCl2 expression (35, 36). NF-κB can be activated following DNA damage. The activation of NF-κB triggers acute and chronic inflammation, which in turn is linked to decreased genomic stability and genetic mutations in cancer initiation and progression (37). As a result, the NF-κB signaling pathway is believed to play an important role in the pathogenesis and carcinogenesis of microbial infection. Here, we focus on the bacteria that can cause gastrointestinal cancer by modulating the NF-κB signaling pathway.
Bacterial Pathogens Linked to Gastrointestinal Tumorigenesis
The Gastric Microbiota and Gastric Cancer
No bacterium was known to specialize in colonization of the human stomach, with its unique acid environment, until the discovery of the gram-negative bacterium H. pylori, which was first reported in the stomachs of patients with peptic ulcers in 1982 (38). To survive in acidic conditions, H. pylori produces a large amount of the enzyme urease, which catalyzes the hydrolysis of urea to ammonia, thereby neutralizing gastric acid (39). Approximately half of the world's population is infected with H. pylori, mainly in developing countries (40). H. pylori infection has been extensively studied and found to be associated with an increased risk of gastric adenocarcinoma. Long-term infection with H. pylori is an inducible factor leading to gastric atrophic gastritis, intestinal metaplasia, dysplasia, and ultimately gastric cancer, a sequence also called the Correa cascade of multistep gastric carcinogenesis (41). Accumulating data from clinical follow-up studies suggest that eradication of H. pylori significantly reduces the risk of gastric cancer (42, 43). This is illustrated by the finding that patients have a lower incidence of metachronous gastric cancer following treatment to eradicate H. pylori (44). Additionally, in patients with H. pylori infection who had a family history of gastric carcinoma in their first-degree relatives, H. pylori eradication significantly decreased gastric cancer risk (45).
Some heterogeneity exists between different H. pylori strains. High prevalence of H. pylori infection, but low prevalence of GC incidence, was found in many African countries (46). Multiple mechanisms are involved in the interaction between the host and pathogenic H. pylori. Both bacterial and host genetic factors contribute to H. pylori infection-induced chronic inflammation, metaplasia and gastric tumorigenesis (47). From the perspective of bacteria, the virulence factors of H. pylori have been demonstrated to influence this microorganism's pathogenicity. Cytotoxin-associated gene A (CagA) and vacuolating cytotoxin A (VacA), the most intensively investigated virulence factors, play significant roles in gastric adenocarcinoma induced by H. pylori infection. The bacterium utilizes a type IV secretion system (T4SS) to inject CagA into host gastric epithelial cells. As a result, CagA is responsible for the dysregulation of cellular proliferation and apoptosis through disturbing the PI3K/AKT, MEK/ERK, and Wnt/β-catenin signaling pathways (48). Additionally, it has been indicated that CagA induces an inflammatory response via activation of the NF-κB pathway (49). In addition, the VacA toxin of H. pylori can rapidly cause vacuolation in gastric epithelial cells (50). From the perspective of host genetics, gene polymorphisms can increase the risk of gastric cancer in patients with H. pylori infection. Polymorphisms in the IL-1β gene increase the risk of gastric carcinogenesis in H. pylori-positive populations (51).
With the development of sequencing platforms and bioinformatics tools, 16S rRNA gene sequencing based on hypervariable regions was performed to study the profile of the human microbiome, including mainly the microbiotas of the stomach and intestine (52). As a result, many gastric bacteria species other than H. pylori have been identified. The stomachs of H. pylori-positive and H. pylori-negative individuals exhibit significantly different bacterial communities. Among H. pylori-positive patients, H. pylori is the most dominant bacterium in the stomach. In contrast, the gastric microbiota of H. pylori-negative individuals is more diverse and consists mainly of Firmicutes, Proteobacteria, Bacteroidetes, Fusobacteria, and Actinobacteria (53). Recently, gastric bacterial communities were shown to be associated with gastric malignancies. Ferreira et al. showed that Firmicutes and Actinobacteria are over-represented in the gastric carcinoma microbiota compared with the chronic gastritis microbiota. Furthermore, gastric cancer samples exhibit a significant reduction in the abundance of H. pylori (54). Coker et al. identified differences in mucosal bacterial interactions across stages of gastric carcinogenesis, from superficial gastritis to atrophic gastritis, intestinal metaplasia, and GC. The significant enrichment and central network locations of five microbes (Peptostreptococcus stomatis, Streptococcus anginosus, Parvimonas micra, Slackia exigua, and Dialister pneumosintes) suggest their important role in GC progression (55).
The effect of non-H. pylori bacteria on gastric pathology is further supported by animal model systems. In transgenic INS-GAS mice with high circulating gastrin levels, colonization of H. pylori led to a significant increase in Firmicutes and reduction in Bacteroidetes (41). Other Helicobacter species, such as Helicobacter felis (H. felis), commonly colonize animals. Mongolian gerbils infected with H. felis developed premalignant gastric lesions (56). Moreover, germ-free transgenic INS-GAS mice supplemented with normal intestinal flora (IF) or 3 species of commensal bacteria (rASF; ASF356 Clostridium species, ASF361 Lactobacillus murinus, and ASF519 Bacteroides species) developed more severe gastric lesions and elevated levels of proinflammatory genes than H. pylori-monocolonized INS-GAS mice (57). These findings suggest that microbial diversity contributes to gastric cancer risk. Long-term H. pylori infection causes gastric atrophy, which leads to achlorhydria and decreased acid secretion. Notably, H. pylori infection and alteration of the acidity of the gastric environment may result in alterations in the gastric microbiota (58). However, due to the difficulty in bacterial isolation and culture, the functional role and pathogenic mechanisms of microbial communities in gastric neoplasia remain unclear.
There are some genetic, environmental, dietary, and lifestyle factors that influence microbiome system. Genetic mutation such as CDH1 and TP53, lifestyles including smoking, low fruits and vegetables consumption, high salts, nitrates, and pickled foods intake and overweight are also found to be associated with increased GC risk (59, 60). He et al. reported that 12 week high-fat diet lead to the dysbiosis of gastrointestinal microbiota in C57BL/6 mice, what's more, the alterations of microbiota in stomach was earlier than that in gut and the dysbiosis of gastrointestinal microbiota may related with the metabolic disorders of mice (61). Arita et al. showed that high-fat diet leads to severe dysbiosis of gastric microbiota and increased gastric leptin, which lead to the development of gastric intestinal metaplasia in C57 mice (62). A recent large scale blinded randomized placebo controlled trial in China showed that both H. pylori treatment and vitamin supplementation lead to a significant reduced incidence of GC, and H. pylori treatment, vitamin or garlic supplementation lead to a significant reduction of GC mortality (42).
Imbalance Between Gut Microbiota and Intestinal Cancer
Changes in the gut microbiota, referred to as dysbiosis, are involved in the development of multiple diseases, including cancer, metabolic diseases, cardiovascular diseases and depression (63, 64). Colorectal cancer (CRC) is the third most common cancer worldwide (2, 65). Effects of the fecal microbiota transplant of human fecal samples to germ-free mice suggested the effect of gut microbiota on colorectal carcinogenesis. Germ-free mice gavaged with stool from patients with CRC showed increased levels of proinflammatory genes, including the chemokines CXCR1 and CXRC2 as well as the cytokines IL-17A, IL-22, and IL-23A (66). Sequencing studies have revealed discrepancies in the gut microbiomes of patients with colorectal cancer and healthy individuals. 16S rRNA sequencing data from stool samples from CRC patients suggested that several genera, including Fusobacterium, Porphyromonas, Enterococcus, Escherichia, Klebsiella, Streptococcus, and Parvimonas, are linked to CRC (67–69). Hibberd et al. analyzed the intestinal microbiota from tumor tissues and the normal mucosa. Several taxa, including Fusobacterium, Selenomonas and Peptostreptococcus, were selectively enriched in patients with colon cancer compared to control individuals. Probiotic intervention significantly altered the microbial composition (70). Furthermore, significant differences in microbial communities were observed across the stages of colorectal carcinogenesis. Nakatsu et al. compared the mucosal microbiotas of normal tissues, adenomatous polyps and carcinomas. Fusobacterium, Parvimonas, Gemella, and Leptotrichia were significantly increased in patients with adenoma, which is an early-stage CRC, whereas the abundance of bacterial communities, including Bacteroides, Blautia, and F. prausnitzii, was decreased. In late-stage CRC, neither of these changes was significant. Furthermore, Peptostreptococcus and Parvimonas showed a strongly positive relationship in colonic carcinoma and carcinoma-adjacent mucosa (71). In addition, Warren et al. compared the CRC tissues and matched normal control tissues. Co-occurrence network analysis was performed to identify microbe-microbe and host-microbe associations specific to tumors. The authors confirmed tumor over-representation of Fusobacterium species and observed significant co-occurrence within individual tumors of Fusobacterium, Leptotrichia, and Campylobacter species (72). As a corollary, these studies may identify novel CRC-associated microbial markers, such as Fusobacterium, Peptostreptococcus, Leptotrichia, and Parvimonas.
Recently, an increasing number of studies have elucidated the functional roles and molecular mechanisms of several specific bacterial species in CRC carcinogenesis, including Fusobacterium nucleatum (F. nucleatum), Escherichia coli (E. coli), Peptostreptococcus anaerobius (P. anaerobius), and Bacteroides fragilis (B. fragilis) (Figure 3). F. nucleatum is highly increased in CRC patients compared to healthy individuals, and its abundance is closely related to a worse survival rate (73, 74). Biological informatic, functional and mechanistic studies in human samples, cell lines and animal models have revealed the potential role of F. nucleatum in CRC chemotherapy. Specifically, these bacteria could promote CRC resistance to chemotherapy through the activation of autophagy mediated by TLR4/MYD88 and miRNAs (75). These studies suggest that F. nucleatum serves as a candidate prognostic biomarker in CRC. Although E. coli is a commensal bacterium that colonizes the human GI tract, several studies indicate a link between some pathogenic E. coli strains and CRC risk. E. coli are divided into 4 phylotypes, among which specific E. coli strains from each phylotype have been associated with IBD, which is a known risk factor for CRC (76, 77). Infection with pathogenic E. coli in multiple intestinal neoplasia (Min) mice significantly increased colonic polys compared with those in control groups (78). Pathogenic E. coli produces various virulence factors. Colibactin, a hybrid polyketide-peptide encoded by the pks genomic island, has been shown to induce DNA double-strand breaks and genomic instability in human cells (79), contributing to mutational signatures in CRC (80). In addition, the anaerobic bacterium P. anaerobius, which is enriched in the fecal and mucosal samples of CRC patients, was recently demonstrated to promote CRC carcinogenesis through in vitro and in vivo studies (81). The gram-negative bacterium B. fragilis is a normal commensal bacterial species that colonizes the colon. A subset of B. fragilis termed enterotoxigenic B. fragilis produces the B. fragilis toxin (BFT) and has been found to play an important role in CRC tumorigenesis and development. Sears et al. revealed the increased abundance of the BFT gene in mucosal biopsies from patients with CRC compared to normal individuals. Furthermore, increased bft positivity was found in early to progressive CRC patients (82). This research team then investigated the role of the gut microbiota in the development of familial adenomatous polyposis (FAP), which is an autosomal dominant disease caused by the APC gene in which many colorectal adenomas can develop (83). The genes colibactin from E. coli and bft from B. fragilis are more highly expressed in patients with FAP compared to healthy individuals. Mechanistically, the co-colonization of E. coli and B. fragilis in the colon promotes the expression of inflammatory cytokines and accelerates DNA damage (84). As a result, modulation of the gut microbiota may be an effective strategy for the prevention or treatment of CRC.
Some genetic, diet, lifestyle and other environmental factor have been shown to modulate gut microbiota, further affect host metabolism, immune response and promote colorectal carcinogenesis. Liang et al. reported that APC mutation was closely related to the alteration of gut microbiota that plays an important role in the development of CRC from intestinal adenomatous polyps (85). The increasing incidence of CRC in western countries is partly attributed to the increasing adoption of western lifestyles and overweight. High fat diet is a well-known factor to influence the gut microbiota and promote CRC development in animal model (86). Interestingly, it has been demonstrated that Mediterranean Diet (MD) could counteract CRC that caused by high-fat diet by modulate apoptosis and gut microbiota in mice (87).
Modulation of the NF-κB Signaling Pathway in Response to Bacterial Infection in Gastric Tumorigenesis
Commensal microbes in the GI tract are essential for the maintenance of GI functions, including development, immune responses and homeostasis. It is becoming increasingly clear that disruption of the microbiota contributes to gastric and intestinal tumorigenesis. In particular, upon infection with intestinal bacteria, host cells rapidly employ the intracellular NF-κB signaling pathway to activate antibacterial immunity and maintain intestinal barrier integrity (30). A number of studies have reported that during bacterial infection, the NF-κB pathway controls multiple cellular processes, including inflammation, proliferation and apoptosis, by regulating the expression of a network of downstream effectors. As a result, the NF-κB signaling pathway is strongly involved in microbiota-associated gastric (Figure 2) and colorectal tumorigenesis (Figure 3). Here, we focus on the bacteria that modulate the NF-κB pathway in gastrointestinal tumorigenesis.
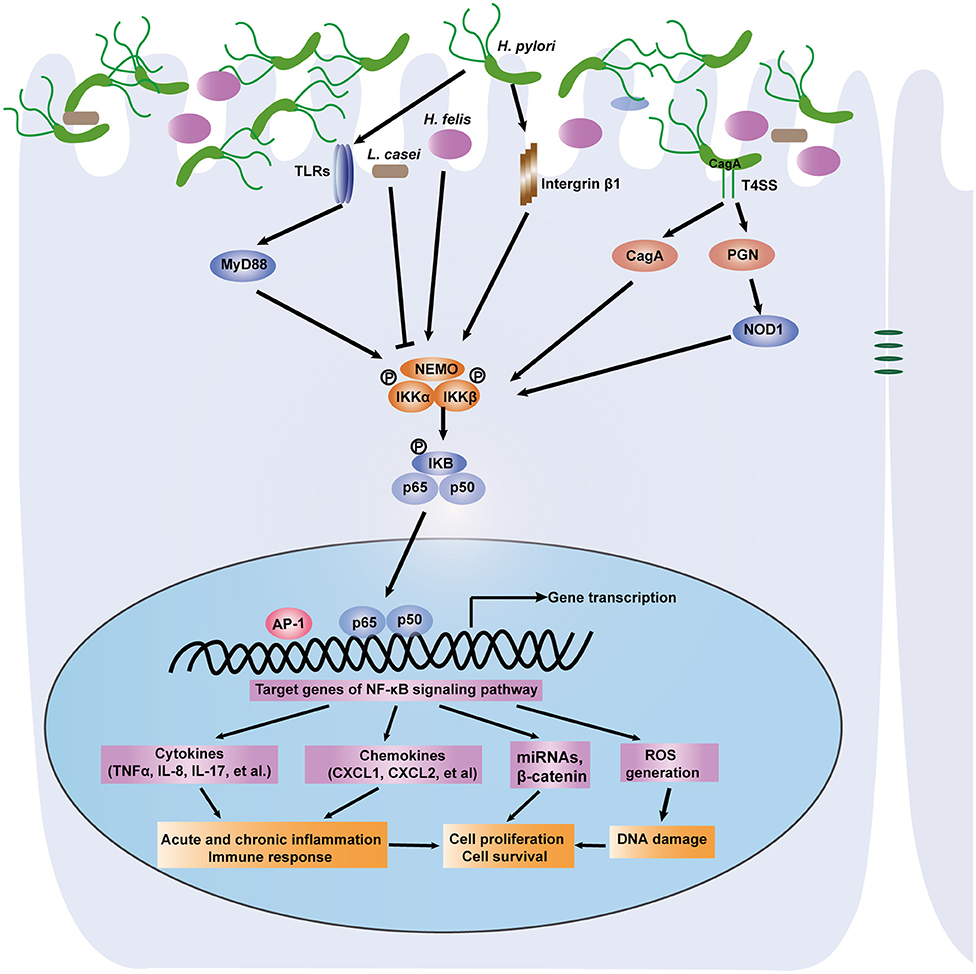
Figure 2. A depiction of the modulation of NF-κB signaling pathway by gastric microbiota, especially H. pylori. Gastric epithelial cells PRRS (NOD1, TLRs) specifically recognize the virulence factors such as CagA and peptidoglycan that are delivered by the H. pylori cag pathogenicity island. Then the NF-κB pathway is activated through IKK-mediated phosphorylation of IκB. Active NF-κB translocate to nucleus and induce downstream genes expression. NF-κB activation promotes acute, chronic inflammation and immune response via induction of cytokines and chemokines. The accumulation of DNA damage is associated with activation of the NF-κB signaling pathway. Several miRNAs and other genes can be activated. As a result, H. pylori infection induces gastric carcinogenesis via regulation of cell proliferation and survival. Other microorganisms including L. casei and H. felis link the NF-κB pathway to gastric inflammation and tumorigenesis.
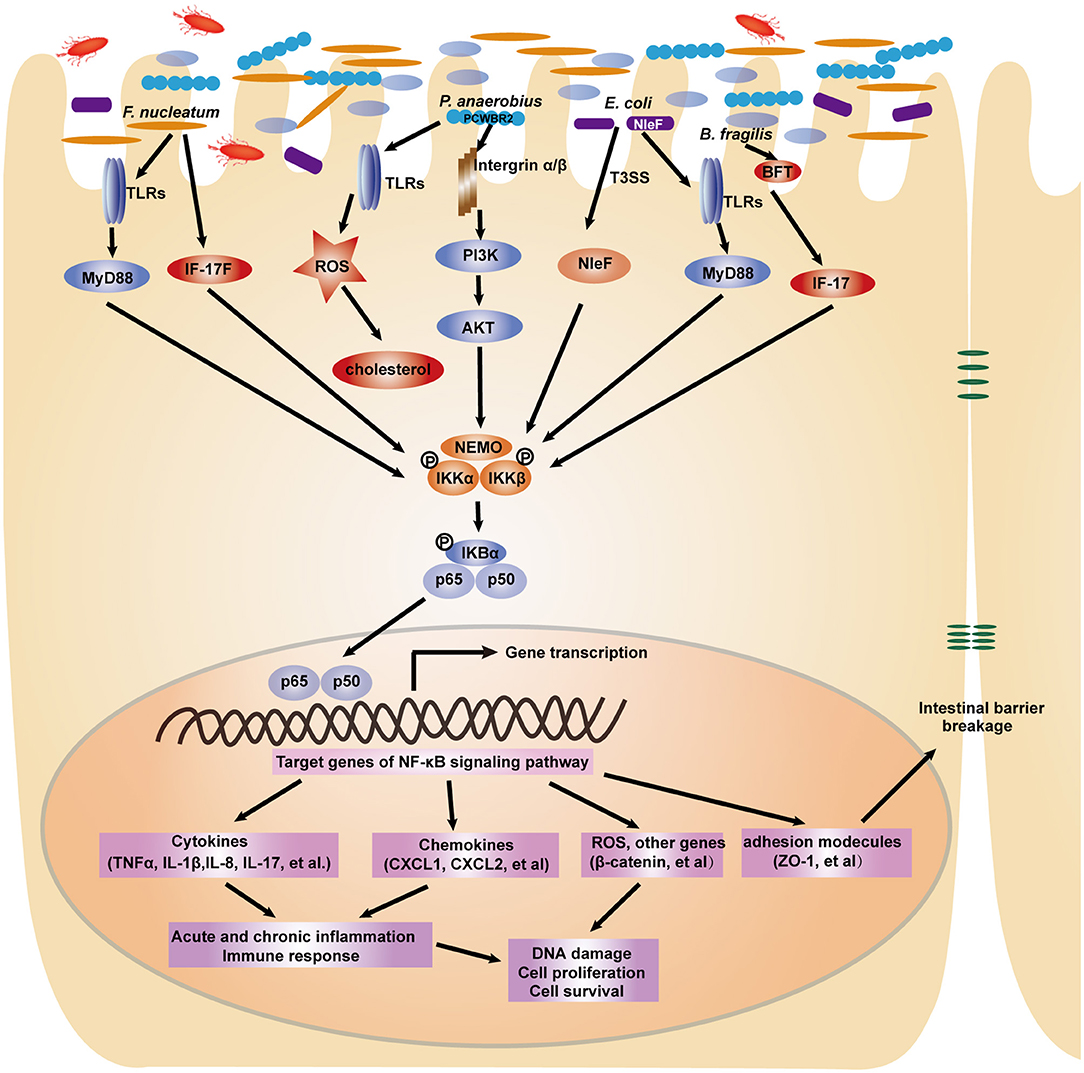
Figure 3. A depiction of the modulation of NF-κB signaling pathway by gut microbiota in colorectal tumorigenesis. In response to intestinal bacterial pathogens including F. nucleatum, P. anaerobius, E. coli, B. fragilis, intestinal epithelial cells employ several receptors such as TLRs and integrins to recognize distinct microbial components. The NF-κB signaling pathway is subsequently activated to induce the expression of pro-inflammatory cytokines, chemokines, adhesion molecules and other genes, which ultimately lead to the cellular processes changes and contribute to colorectal carcinogenesis.
H. pylori Infection, the NF-κB Signaling Pathway, and Gastric Carcinogenesis
H. pylori-infected individuals develop acute or chronic gastritis, and in a subset of subjects, gastritis may progress to peptic ulcer disease, in particular, intestinal metaplasia and gastric carcinomas (88). There is very compelling evidence for the involvement of the NF-κB pathway in H. pylori-associated gastric tumorigenesis. The activity of NF-κB was shown to be markedly increased in the lamina propria and epithelium of the antral mucosa of H. pylori-infected adults compared to those of uninfected controls, suggesting that neutrophil infiltration in the gastric mucosa of H. pylori-infected gastritis patients is attributed to activation of the NF-κB pathway (89). Twenty years ago, it was discovered that H. pylori infection increased NF-κB activity and the nuclear translocation of NF-κB p50/p65 heterodimers and p50 homodimers in transformed gastric epithelial AGS cells. Furthermore, activation of the NF-κB pathway by H. pylori infection could lead to the induction of proinflammatory cytokines such as IL-8 and IL-17 (90, 91). Consistently, the importance of NF-κB in H. pylori infection-induced gastric neoplasia was confirmed in animal models (92).
Upon sensing pathogenic H. pylori, cellular pattern recognition receptors (PRRs) induce intracellular signaling pathways in the innate immune response. TLRs and NOD1 are common PRRs involved in activation of the NF-κB signaling pathway (93). It was reported that infection with H. pylori strain 26695 increased NF-κB activity and chemokine gene expression in HEK293 and gastric epithelial MKN45 cells transfected with TLR2 and TLR5 but not TLR4 (94). NOD1 specifically recognizes peptidoglycan (PGN) delivered by the H. pylori cag pathogenicity island. Experimental studies, both in vivo and in vitro, have indicated that NOD1 induces NF-κB activity in response to pathogenic H. pylori, which is implicated in gastric inflammation and malignant lesions (95, 96). The virulence factor CagA has been identified to be responsible for the NF-κB-induced response subsequent to NOD1 activation (97).
Gastric chronic inflammation caused by the NF-κB pathway in response to H. pylori infection finally contributes to gastric carcinogenesis through accumulated DNA damage and abnormal cell polarity and proliferation. H. pylori infection induces DNA damage to exert genotoxic effects (98). Hartung et al. reported that the induction of DNA double-strand breaks (DSBs) is associated with activation of the NF-κB signaling pathway. DSBs were greatly reduced in AGS cells treated with p65/RelA RNAi or an NF-κB inhibitor. Similarly, the loss of β1 integrin resulted in decreased DSBs and inhibited IL-8 secretion after infection with H. pylori. Intriguingly, H. pylori-induced DNA damage can promote NF-κB target gene transactivation and host cell survival (99).
Moreover, NF-κB drives cell proliferation, which promotes metaplastic hyperplasia and neoplasia in the stomach. H. pylori infection has been shown to induce the activity of NF-κB and AP-1, which in turn promotes the expression of oncogenes (β-catenin, c-myc) and mediates the hyperproliferation of gastric cells (100). DARPP-32 has been identified as a transcriptional target gene of the NF-κB pathway that is significantly upregulated following H. pylori infection. Consequently, induction of DARPP-32 counteracted H. pylori-induced cell death and promoted gastric cell survival through the activation of AKT (101). Small RNAs such as miRNA-223-3p have been documented to link NF-κB, cellular proliferation and gastric carcinogenesis (102). As a result, H. pylori infection rapidly leads to activation of the NF-κB pathway, which triggers various molecular mechanisms to affect gastric neoplastic lesions.
In addition to H. pylori, several other microorganisms exhibit distinct functions in the GI tract. Many probiotics have been shown to inhibit the development of gastric diseases. Hwang et al. reported that Lactobacillus casei (L. casei) extract suppressed the NF-κB pathway by decreasing the expression of NF-κB p65 and IκB, which in turn induced apoptosis and inhibited the growth of gastric cancer cells (103). Infection with the non-pylori Helicobacter species H. felis links the IKKβ/NF-κB pathway to gastric inflammation and tumorigenesis. Shibata et al. generated mice in which IKKβ was conditionally deleted in gastric epithelial cells and myeloid cells to determine the role of IKKβ/NF-κB signaling in H. felis infection-associated gastric neoplasia. They found that deletion of IKKβ in gastric epithelial cells resulted in increased apoptosis, the accumulation of ROS and DNA damage, severe inflammation and more rapid progression to gastric preneoplasia (104).
Modulation of the NF-κB Signaling Pathway by the Intestinal Microbiota in CRC
F. nucleatum and the NF-κB Signaling Pathway
Multiple studies over the past few years have definitively addressed the carcinogenic properties of F. nucleatum in the initiation and development of CRC. It has become clear that F. nucleatum infection modulates the NF-κB signaling pathway, targeting downstream genes that regulate various cellular processes, such as the inflammatory response, cell proliferation, and cell migration, and finally affecting tumorigenesis (105–107). In the APCMin/+ mouse model, F. nucleatum treatment accelerated small intestinal and colonic tumorigenesis. Increased nuclear translocation of the p65 NF-κB subunit was observed in tumors (106). F. nucleatum infection can rapidly induce the host innate immune response, which activates the NF-κB signaling pathway. Several studies have shown that the common TLR4/MYD88 innate immune signaling pathway is activated following F. nucleatum infection in CRC cells (75). Yang et al. found that infection with F. nucleatum significantly activated the TLR4/MyD88 pathway to upregulate the activity of NF-κB p65 and p50 in HCT116 CRC cells. Then, miR-21, an oncogenic target miRNA in cancer, was found to be increased by F. nucleatum through binding to NF-κB, which thereby promoted cell survival and invasion. Inhibition of NF-κB impaired F. nucleatum-induced CRC cell proliferation and cell invasion. Hyperactivation of NF-κB was found in CRC tissues with high amounts of F. nucleatum (107). NLRX1 is a member of the NLR family that plays an important role in host innate immunity. NLRX1 was shown to negatively modulate inflammatory cytokine IL-8 expression via the transcriptional factor NF-κB in response to F. nucleatum infection (108). Additionally, induction of NF-κB by F. nucleatum infection facilitated ROS generation and production of the proinflammatory cytokines TNF-α and IL-1β (109).
Ulcerative colitis (UC) is a major type of IBD and a known risk factor for CRC (108). F. nucleatum was more highly enriched in human UC tissues than in normal tissues. Mechanistically, experimental studies from cells and animal models have shown that F. nucleatum infection activates the canonical NF-κB pathway through increasing phosphorylation levels of the NF-κB subunits p65 and IκB-α. The intestinal epithelial barrier marker ZO-1, a downstream target gene of the NF-κB pathway, was significantly decreased following F. nucleatum infection. Furthermore, pretreatment with human anti-IL-17F antibody attenuated F. nucleatum-induced NF-κB activity and intestinal inflammation, which suggests that F. nucleatum activates the NF-κB pathway via IL-17F (110). In addition, F. nucleatum was found to be more abundant in the patients with Crohn's diseases, which is another common type of IBD. F. nucleatum infection could promote intestinal mucosal barrier destruction during the development of Crohn's diseases (111). F. nucleatum is a heterogeneous species with five proposed subspecies (ssp.), i.e., ssp. animalis, ssp. fusiforme, ssp. nucleatum, ssp. polymorphum, and ssp. vincentii, which show the pathogenic differences. Among the five subspecies, ssp. fusiforme and ssp. vincentii are more frequently associated with health while ssp. nucleatum associated with diseases (112, 113). Adherence and invasion are essential mechanisms for antimicrobial host defense mechanism and induction of inflammatory response. The invasiveness of F. nucleatum varies widely among different strains, and has been shown as directly related to IBD disease status (114).
E. coli and the NF-κB Signaling Pathway
E. coli is one of the most common bacterial species that colonizes the human GI tract. Numerous studies have identified E. coli as a risk factor for the development of Crohn's disease, ulcerative colitis, and CRC. Enteropathogenic and enterohemorrhagic E. coli use a type III secretion system (T3SS) to transport dozens of effector proteins into host cells; these effector proteins in turn manipulate the host inflammatory response through activation of the NF-κB pathway (115). This bacterial pathogen has developed various mechanisms to regulate the activation of NF-κB. Pallett et al. found that the highly conserved non-LEE (locus of enterocyte effacement)-encoded effector (NleF) is responsible for nuclear translocation of the NF-κB p65 subunit and IL-8 production (116). Sahu et al. reported that non-pathogenic E. coli has emerged as a tumor promoter that enhances oncogenicity through enrichment of the cancer stem cell population. Mechanistically, internalization of E. coli leads to the activation of NF-κB through increased phosphorylation of the NF-κB subunit RelA/p65 and IKKα, inactivation of IκBα, and induction of the Wnt/β-catenin pathway through the upregulation of β-catenin and its downstream genes. Then, NF-κB and Wnt/β-catenin synergistically promote tumorigenic stemness traits (117). In addition, high NF-κB expression has been demonstrated in E. coli-associated IBD patients. E. coli strains isolated from IBD patients were found to induce the NF-κB and TNF-α promoters in HT-29 human colonic cells (118). Karrasch et al. determined the role of the TLR/NF-κB signaling pathway in bacteria-induced colitis using animal models—IL-10-deficient mice and NF-κB knock-in mice. As a result, coinfection with the commensal bacterial strain Enterococcus faecalis led to experimental colitis through activation of the TLR/NF-κB signaling pathway (119). Taken together, these results show that some E. coli strains, in which the NF-κB pathway induces chronic inflammation, play a crucial role in colorectal carcinogenesis.
P. anaerobius and the NF-κB Signaling Pathway
P. anaerobius is a gram-positive anaerobic bacterium that was recently identified to be especially enriched in the stool samples of CRC patients. Jun Yu et al. from the Chinese University of Hong Kong recently undertook research to determine the role of P. anaerobius and its molecular mechanism in colorectal carcinogenesis. P. anaerobius was found to induce the production of intracellular ROS through its interaction with TLR2 and TLR4. As a result, total cholesterol synthesis and the proliferation of intestinal epithelial cells were significantly enhanced (81). Similar to other bacteria, P. anaerobius adheres to the intestinal mucosa and encodes a virulence factor to induce the host immune response. Putative cell wall binding repeat 2 (PCWBR2), a P. anaerobius surface protein, interacts with the corresponding cell surface receptor integrin α2/β1. Subsequently, experimental data from CRC cells and APCMin/+ mice indicated that P. anaerobius challenge activated the PI3K/AKT and NF-κB signaling pathways via phosphorylation of the AKT and p65 NF-κB subunits. As a result, P. anaerobius modulates the tumor immune microenvironment, including the expansion of myeloid-derived suppressor cells, tumor-associated macrophages and granulocytic tumor-associated neutrophils. Cell proliferation and the proinflammatory response were significantly increased by P. anaerobius infection, further accelerating colorectal tumorigenesis (120).
Enterotoxigenic B. fragilis and the NF-κB Signaling Pathway
Enterotoxigenic B. fragilis, but not non-toxigenic B. fragilis, is associated with the development of intestinal diseases, such as human inflammatory diarrhea and colorectal carcinogenesis. Enterotoxigenic B. fragilis targets intestinal epithelial cells by producing BFT. As a result, cells develop molecular mechanisms to activate the NF-κB signaling pathway. To explore the role of BFT in enterotoxigenic B. fragilis-triggered tumorigenesis. Chung et al. constructed ApcMin mice colonized with an enterotoxigenic B. fragilis strain possessing an in-frame chromosomal deletion of bft gene. The results showed that B. fragilis stimulated intracellular IL-17 secretion to activate the NF-κB pathway, which in turn induced the expression of chemokines (CXCL1, CXCL2, and CXCL5) that collectively contributed to colonic carcinogenesis (121, 122). The cytokine IL-8, a key downstream target gene of NF-κB, was also significantly increased in intestinal epithelial cells treated with active enterotoxigenic B. fragilis (123). The β-catenin and GSK3β cellular signaling pathways are involved in NF-κB activity and IL-8 expression in B. fragilis-infected cells (124). Host tissues recruit inflammatory cells to induce sustained inflammation through activation of the NF-κB signaling pathway in pathological processes and increase the risk of CRC through aberrant regulation of other cellular processes, such as cell proliferation and angiogenesis.
Concluding Remarks
The gastrointestinal microbiota plays an important role in maintaining host physiological processes. Aberration of the microbiota might ultimately result in various diseases such as metabolic, cardiovascular, immune, mental, and gastrointestinal diseases or even cancer. The causal relationship between gastrointestinal cancer and the microbiota has been gradually validated. Indeed, multiple studies suggest that utilizing the microbiota, especially specific bacteria, may provide novel microbial biomarkers for prevention, diagnosis and treatment. Currently, some clinical trials based on these microbes (e.g., F. nucleatum)are ongoing. Moreover, the activated NF-κB signaling pathway is considered an important line of defense against microbial pathogens. Abnormal and sustained activation of NF-κB signaling contributes to malignant transformation from inflammation to cancer. Therefore, selectively targeting molecules of the NF-κB signaling pathway to block the association between pathogens and NF-κB shows therapeutic potential and benefit in cancer treatment. For example, specific NF-κB inhibitors targeting the IKK complex have shown promise as anticancer therapeutics (12). However, beyond the involvement of NF-κB, the relationship between the microbiota and gastrointestinal carcinogenesis is very complex. Moving forward, there is a need to explore and more deeply understand the underlying mechanisms that link the microbiota and host response.
In addition, molecular pathological epidemiology (MPE) that investigate the interactive effect of some specific molecular features and environmental factors on disease prognosis and clinical outcome has been widely applied to cancer research (125, 126). The host genetic, diet, lifestyle and other environmental factors, which have been closely linked with gut microbiota, are critical for the prevention of gastrointestinal cancer. Modifications of the western lifestyles such as high fat diet that resulting in overweight or obesity could substantially reduce the CRC incidence (127). Eating less salted or pickled foods is considered to be important for prevention of GC (128). This promising direction may help to gain new insights into the pathogenic process, prevention and treatment of gastrointestinal cancer.
Author Contributions
NLi, CP, and NLu discussed the contents. NLi, CP, and YO wrote and edited this manuscript. NLi supervised and oversaw the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81900500, 81670507, and 81870395), the Education Department of Jiangxi Province (GJJ180115) and Jiangxi Provincial Department of Science & Technology (20192BAB215006).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Global Burden of Disease Cancer Collaboration, Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol. (2017) 3:524–48. doi: 10.1001/jamaoncol.2016.5688
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
3. Hansford S, Kaurah P, Li-Chang H, Woo M, Senz J, Pinheiro H, et al. Hereditary diffuse gastric cancer syndrome: CDH1 mutations and beyond. JAMA Oncol. (2015) 1:23–32. doi: 10.1001/jamaoncol.2014.168
4. Zhang L, Theodoropoulos PC, Eskiocak U, Wang W, Moon YA, Posner B, et al. Selective targeting of mutant adenomatous polyposis coli (APC) in colorectal cancer. Sci Transl Med. (2016) 8:361ra140. doi: 10.1126/scitranslmed.aaf8127
5. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. (2016) 14:e1002533. doi: 10.1371/journal.pbio.1002533
6. Thursby E, Juge N. Introduction to the human gut microbiota. Biochem J. (2017) 474:1823–36. doi: 10.1042/BCJ20160510
7. Valdes AM, Walter J, Segal E, Spector TD. Role of the gut microbiota in nutrition and health. BMJ. (2018) 361:k2179. doi: 10.1136/bmj.k2179
8. Kahrstrom CT, Pariente N, Weiss U. Intestinal microbiota in health and disease. Nature. (2016) 535:47. doi: 10.1038/535047a
9. Wroblewski LE, Peek RM Jr, Coburn LA. The role of the microbiome in gastrointestinal cancer. Gastroenterol Clin North Am. (2016) 45:543–56. doi: 10.1016/j.gtc.2016.04.010
10. Wong SH, Yu J. Gut microbiota in colorectal cancer: mechanisms of action and clinical applications. Nat Rev Gastroenterol Hepatol. (2019) 16:690–704. doi: 10.1038/s41575-019-0209-8
11. Ng C, Li H, Wu WKK, Wong SH, Yu J. Genomics and metagenomics of colorectal cancer. J Gastrointest Oncol. (2019) 10:1164–70. doi: 10.21037/jgo.2019.06.04
12. Rahman MM, McFadden G. Modulation of NF-κB signalling by microbial pathogens. Nat Rev Microbiol. (2011) 9:291–306. doi: 10.1038/nrmicro2539
13. Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. (2018) 18:309–24. doi: 10.1038/nri.2017.142
14. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
15. Napetschnig J, Wu H. Molecular basis of NF-κB signaling. Annu Rev Biophys. (2013) 42:443–68. doi: 10.1146/annurev-biophys-083012-130338
16. Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. (2013) 12:86. doi: 10.1186/1476-4598-12-86
17. Israel A. The IKK complex, a central regulator of NF-κB activation. Cold Spring Harb Perspect Biol. (2010) 2:a000158. doi: 10.1101/cshperspect.a000158
18. Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBalpha and beta-catenin and stimulates IκBalpha ubiquitination in vitro. Genes Dev. (1999) 13:270–83. doi: 10.1101/gad.13.3.270
19. Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I κ B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. (1995) 267:1485–8. doi: 10.1126/science.7878466
20. Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. (2011) 12:695–708. doi: 10.1038/ni.2065
21. Cildir G, Low KC, Tergaonkar V. Non-canonical NF-κB signaling in health and disease. Trends Mol Med. (2016) 22:414–29. doi: 10.1016/j.molmed.2016.03.002
22. Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G et al. Activation by IKKalpha of a second, evolutionary conserved, NF-κ B signaling pathway. Science. (2001) 293:1495–9. doi: 10.1126/science.1062677
23. Maruyama T, Fukushima H, Nakao K, Shin M, Yasuda H, Weih F et al. Processing of the NF-κ B2 precursor p100 to p52 is critical for RANKL-induced osteoclast differentiation. J Bone Miner Res. (2010) 25:1058–67. doi: 10.1359/jbmr.091032
24. Sun SC. The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol. (2017) 17:545–58. doi: 10.1038/nri.2017.52
25. Sun SC. The non-canonical NF-κB pathway. Immunol Rev. (2012) 246:125–40. doi: 10.1111/j.1600-065X.2011.01088.x
26. Li C, Wang S, He J. The two NF-κB pathways regulating bacterial and WSSV infection of shrimp. Front Immunol. (2019) 10:1785. doi: 10.3389/fimmu.2019.01785
28. Ganesan S, Aggarwal K, Paquette N, Silverman N. NF-κB/Rel proteins and the humoral immune responses of Drosophila melanogaster. Curr Top Microbiol Immunol. (2011) 349:25–60. doi: 10.1007/82_2010_107
29. DiDonato JA, Mercurio F, Karin M. NF-κB and the link between inflammation and cancer. Immunol Rev. (2012) 246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x
30. Zhai Z, Boquete JP, Lemaitre B. Cell-specific Imd-NF-κB responses enable simultaneous antibacterial immunity and intestinal epithelial cell shedding upon bacterial infection. Immunity. (2018) 48:897–910.e897. doi: 10.1016/j.immuni.2018.04.010
31. Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell. (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
32. Xia Y, Shen S, Verma IM. NF-κB, an active player in human cancers. Cancer Immunol Res. (2014) 2:823–30. doi: 10.1158/2326-6066.CIR-14-0112
33. Dai W, Wu J, Wang D, Wang J. Cancer gene therapy by NF-κB-activated cancer cell-specific expression of CRISPR/Cas9 targeting telomeres. Gene Ther. (2020). doi: 10.1038/s41434-020-0128-x. [Epub ahead of print].
34. Inoue S, Ide H, Mizushima T, Jiang G, Netto GJ, Gotoh M, et al. Nuclear factor-κB promotes urothelial tumorigenesis and cancer progression via cooperation with androgen receptor signaling. Mol Cancer Ther. (2018) 17:1303–14. doi: 10.1158/1535-7163.MCT-17-0786
35. Liu JR, Xu GM, Shi XM, Zhang GJ. Low temperature plasma promoting fibroblast proliferation by activating the NF-κB pathway and increasing cyclinD1 expression. Sci Rep. (2017) 7:11698. doi: 10.1038/s41598-017-12043-w
36. Buchholz TA, Garg AK, Chakravarti N, Aggarwal BB, Esteva FJ, Kuerer HM, et al. The nuclear transcription factor κB/bcl-2 pathway correlates with pathologic complete response to doxorubicin-based neoadjuvant chemotherapy in human breast cancer. Clin Cancer Res. (2005) 11:8398–402. doi: 10.1158/1078-0432.CCR-05-0885
37. Ji Z, He L, Regev A, Struhl K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc Natl Acad Sci USA. (2019) 116:9453–62. doi: 10.1073/pnas.1821068116
38. Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. (1984) 1:1311–5. doi: 10.1016/S0140-6736(84)91816-6
39. Jones MD, Li Y, Zamble DB. Acid-responsive activity of the Helicobacter pylori metalloregulator NikR. Proc Natl Acad Sci USA. (2018) 115:8966–71. doi: 10.1073/pnas.1808393115
40. Hooi JKY, Lai WY, Ng WK, Suen MMY, Underwood FE, Tanyingoh D, et al. Global prevalence of helicobacter pylori infection: systematic review and meta-analysis. Gastroenterology. (2017) 153:420–9. doi: 10.1053/j.gastro.2017.04.022
41. Correa P, Piazuelo MB. Helicobacter pylori infection and gastric adenocarcinoma. US Gastroenterol Hepatol Rev. (2011) 7:59–64.
42. Li WQ, Zhang JY, Ma JL, Li ZX, Zhang L, Zhang Y, et al. Effects of Helicobacter pylori treatment and vitamin and garlic supplementation on gastric cancer incidence and mortality: follow-up of a randomized intervention trial. BMJ. (2019) 366:l5016. doi: 10.1136/bmj.l5016
43. Leung WK, Wong IOL, Cheung KS, Yeung KF, Chan EW, Wong AYS, et al. Effects of helicobacter pylori treatment on incidence of gastric cancer in older individuals. Gastroenterology. (2018) 155:67–75. doi: 10.1053/j.gastro.2018.03.028
44. Choi IJ, Kook MC, Kim YI, Cho SJ, Lee JY, Kim CG, et al. Helicobacter pylori therapy for the prevention of metachronous gastric cancer. N Engl J Med. (2018) 378:1085–95. doi: 10.1056/NEJMoa1708423
45. Choi IJ, Kim CG, Lee JY, Kim YI, Kook MC, Park B, et al. Family history of gastric cancer and helicobacter pylori treatment. N Engl J Med. (2020) 382:427–36. doi: 10.1056/NEJMoa1909666
46. Holcombe C. Helicobacter pylori: the African enigma. Gut. (1992) 33:429–31. doi: 10.1136/gut.33.4.429
47. Peng C, Li NS, Hu Y, Lu NH. Impact factors that modulate gastric cancer risk in Helicobacter pylori-infected rodent models. Helicobacter. (2019) 24:e12580. doi: 10.1111/hel.12580
48. Hatakeyama M. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe. (2014) 15:306–16. doi: 10.1016/j.chom.2014.02.008
49. Brandt S, Kwok T, Hartig R, Konig W, Backert S. NF-κB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci USA. (2005) 102:9300–5. doi: 10.1073/pnas.0409873102
50. McClain MS, Beckett AC, Cover TL. Helicobacter pylori vacuolating toxin and gastric cancer. Toxins (Basel). (2017) 9:316. doi: 10.3390/toxins9100316
51. El-Omar EM, Rabkin CS, Gammon MD, Vaughan TL, Risch HA, Schoenberg JB, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. (2003) 124:1193–201. doi: 10.1016/S0016-5085(03)00157-4
52. Ames NJ, Ranucci A, Moriyama B, Wallen GR. The human microbiome and understanding the 16S rRNA gene in translational nursing science. Nurs Res. (2017) 66:184–97. doi: 10.1097/NNR.0000000000000212
53. Noto JM, Peek RM Jr. The gastric microbiome, its interaction with Helicobacter pylori, and its potential role in the progression to stomach cancer. PLoS Pathog. (2017) 13:e1006573. doi: 10.1371/journal.ppat.1006573
54. Ferreira RM, Pereira-Marques J, Pinto-Ribeiro I, Costa JL, Carneiro F, Machado JC, et al. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut. (2018) 67:226–36. doi: 10.1136/gutjnl-2017-314205
55. Coker OO, Dai Z, Nie Y, Zhao G, Cao L, Nakatsu G, et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut. (2018) 67:1024–32. doi: 10.1136/gutjnl-2017-314281
56. Burkitt MD, Duckworth CA, Williams JM, Pritchard DM. Helicobacter pylori-induced gastric pathology: insights from in vivo and ex vivo models. Dis Model Mech. (2017) 10:89–104. doi: 10.1242/dmm.027649
57. Lertpiriyapong K, Whary MT, Muthupalani S, Lofgren JL, Gamazon ER, Feng Y, et al. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut. (2014) 63:54–63. doi: 10.1136/gutjnl-2013-305178
58. Espinoza JL, Matsumoto A, Tanaka H, Matsumura I. Gastric microbiota: an emerging player in Helicobacter pylori-induced gastric malignancies. Cancer Lett. (2018) 414:147–52. doi: 10.1016/j.canlet.2017.11.009
59. Pharoah PD, Guilford P, Caldas C, International Gastric Cancer Linkage C. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology. (2001) 121:1348–53. doi: 10.1053/gast.2001.29611
60. Yang P, Zhou Y, Chen B, Wan HW, Jia GQ, Bai HL, et al. Overweight, obesity and gastric cancer risk: results from a meta-analysis of cohort studies. Eur J Cancer. (2009) 45:2867–73. doi: 10.1016/j.ejca.2009.04.019
61. He C, Cheng D, Peng C, Li Y, Zhu Y, Lu N. High-fat diet induces dysbiosis of gastric microbiota prior to gut microbiota in association with metabolic disorders in mice. Front Microbiol. (2018) 9:639. doi: 10.3389/fmicb.2018.00639
62. Arita S, Inagaki-Ohara K. High-fat-diet-induced modulations of leptin signaling and gastric microbiota drive precancerous lesions in the stomach. Nutrition. (2019) 67–68:110556. doi: 10.1016/j.nut.2019.110556
63. Kho ZY, Lal SK. The human gut microbiome - a potential controller of wellness and disease. Front Microbiol. (2018) 9:1835. doi: 10.3389/fmicb.2018.01835
64. Cani PD. Human gut microbiome: hopes, threats and promises. Gut. (2018) 67:1716–25. doi: 10.1136/gutjnl-2018-316723
65. Rawla P, Sunkara T, Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol. (2019) 14:89–103. doi: 10.5114/pg.2018.81072
66. Wong SH, Zhao L, Zhang X, Nakatsu G, Han J, Xu W, et al. Gavage of fecal samples from patients with colorectal cancer promotes intestinal carcinogenesis in germ-free and conventional mice. Gastroenterology. (2017) 153:1621–33.e6. doi: 10.1053/j.gastro.2017.08.022
67. Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X, et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. (2012) 6:320–9. doi: 10.1038/ismej.2011.109
68. Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, et al. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst. (2013) 105:1907–11. doi: 10.1093/jnci/djt300
69. Yu J, Feng Q, Wong SH, Zhang D, Liang QY, Qin Y, et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. (2017) 66:70–8. doi: 10.1136/gutjnl-2015-309800
70. Hibberd AA, Lyra A, Ouwehand AC, Rolny P, Lindegren H, Cedgard L, et al. Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterol. (2017) 4:e000145. doi: 10.1136/bmjgast-2017-000145
71. Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WK, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun. (2015) 6:8727. doi: 10.1038/ncomms9727
72. Warren RL, Freeman DJ, Pleasance S, Watson P, Moore RA, Cochrane K, et al. Co-occurrence of anaerobic bacteria in colorectal carcinomas. Microbiome. (2013) 1:16. doi: 10.1186/2049-2618-1-16
73. Mima K, Nishihara R, Qian ZR, Cao Y, Sukawa Y, Nowak JA, et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut. (2016) 65:1973–1980. doi: 10.1136/gutjnl-2015-310101
74. Brennan CA, Garrett WS. Fusobacterium nucleatum - symbiont, opportunist and oncobacterium. Nat Rev Microbiol. (2019) 17:156–66. doi: 10.1038/s41579-018-0129-6
75. Yu T, Guo F, Yu Y, Sun T, Ma D, Han J et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell. (2017) 170:548–63.e16. doi: 10.1016/j.cell.2017.07.008
76. Wassenaar TM. E. coli and colorectal cancer: a complex relationship that deserves a critical mindset. Crit Rev Microbiol. (2018) 44:619–32. doi: 10.1080/1040841X.2018.1481013
77. Mirsepasi-Lauridsen HC, Vallance BA, Krogfelt KA, Petersen AM. Escherichia coli pathobionts associated with inflammatory bowel disease. Clin Microbiol Rev. (2019) 32:e00060–18. doi: 10.1128/CMR.00060-18
78. Bonnet M, Buc E, Sauvanet P, Darcha C, Dubois D, Pereira B, et al. Colonization of the human gut by E. coli and colorectal cancer risk. Clin Cancer Res. (2014) 20:859–67. doi: 10.1158/1078-0432.CCR-13-1343
79. Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrede JP. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci USA. (2010) 107:11537–42. doi: 10.1073/pnas.1001261107
80. Pleguezuelos-Manzano C, Puschhof J, Huber AR, van Hoeck A, Wood HM, Nomburg J et al. Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature. (2020) 580:269–73. doi: 10.1038/s41586-020-2080-8
81. Tsoi H, Chu ESH, Zhang X, Sheng J, Nakatsu G, Ng SC, et al. Peptostreptococcus anaerobius induces intracellular cholesterol biosynthesis in colon cells to induce proliferation and causes dysplasia in mice. Gastroenterology. (2017) 152:1419–33.e5. doi: 10.1053/j.gastro.2017.01.009
82. Boleij A, Hechenbleikner EM, Goodwin AC, Badani R, Stein EM, Lazarev MG, et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin Infect Dis. (2015) 60:208–15. doi: 10.1093/cid/ciu787
83. Li J, Wang R, Zhou X, Wang W, Gao S, Mao Y, et al. Genomic and transcriptomic profiling of carcinogenesis in patients with familial adenomatous polyposis. Gut. (2019) 69:1283–93. doi: 10.1136/gutjnl-2019-319438
84. Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science. (2018) 359:592–7. doi: 10.1126/science.aah3648
85. Liang S, Mao Y, Liao M, Xu Y, Chen Y, Huang X, et al. Gut microbiome associated with APC gene mutation in patients with intestinal adenomatous polyps. Int J Biol Sci. (2020) 16:135–46. doi: 10.7150/ijbs.37399
86. Zhang Y, Kang C, Wang XL, Zhou M, Chen MT, Zhu XH, et al. Dietary factors modulate colonic tumorigenesis through the interaction of gut microbiota and host chloride channels. Mol Nutr Food Res. (2018) 62:554. doi: 10.1002/mnfr.201700554
87. Piazzi G, Prossomariti A, Baldassarre M, Montagna C, Vitaglione P, Fogliano V, et al. A mediterranean diet mix has chemopreventive effects in a murine model of colorectal cancer modulating apoptosis and the gut microbiota. Front Oncol. (2019) 9:140. doi: 10.3389/fonc.2019.00140
88. Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. (2006) 19:449–90. doi: 10.1128/CMR.00054-05
89. Bontems P, Aksoy E, Burette A, Segers V, Deprez C, Mascart F, et al. NF-κB activation and severity of gastritis in Helicobacter pylori-infected children and adults. Helicobacter. (2014) 19:157–67. doi: 10.1111/hel.12118
90. Keates S, Hitti YS, Upton M, Kelly CP. Helicobacter pylori infection activates NF-κ B in gastric epithelial cells. Gastroenterology. (1997) 113:1099–109. doi: 10.1053/gast.1997.v113.pm9322504
91. Luzza F, Parrello T, Monteleone G, Sebkova L, Romano M, Zarrilli R, et al. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J Immunol. (2000) 165:5332–7. doi: 10.4049/jimmunol.165.9.5332
92. Ferrero RL, Ave P, Ndiaye D, Bambou JC, Huerre MR, Philpott DJ, et al. NF-κB activation during acute Helicobacter pylori infection in mice. Infect Immun. (2008) 76:551–61. doi: 10.1128/IAI.01107-07
93. Hu Y, Liu JP, Zhu Y, Lu NH. The importance of toll-like receptors in NF-κB signaling pathway activation by helicobacter pylori infection and the regulators of this response. Helicobacter. (2016) 21:428–40. doi: 10.1111/hel.12292
94. Smith MF Jr, Mitchell A, Li G, Ding S, Fitzmaurice AM, Ryan K, et al. Toll-like receptor (TLR) 2 and TLR5, but not TLR4, are required for Helicobacter pylori-induced NF-κ B activation and chemokine expression by epithelial cells. J Biol Chem. (2003) 278:32552–60. doi: 10.1074/jbc.M305536200
95. Suarez G, Romero-Gallo J, Piazuelo MB, Wang G, Maier RJ, Forsberg LS, et al. Modification of Helicobacter pylori peptidoglycan Enhances NOD1 activation and promotes cancer of the stomach. Cancer Res. (2015) 75:1749–59. doi: 10.1158/0008-5472.CAN-14-2291
96. Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. (2004) 5:1166–74. doi: 10.1038/ni1131
97. Gall A, Gaudet RG, Gray-Owen SD, Salama NR. TIFA signaling in gastric epithelial cells initiates the cag Type 4 secretion system-dependent innate immune response to helicobacter pylori infection. mBio. (2017) 8:e01168–17. doi: 10.1128/mBio.01168-17
98. Koeppel M, Garcia-Alcalde F, Glowinski F, Schlaermann P, Meyer TF. Helicobacter pylori infection causes characteristic DNA damage patterns in human cells. Cell Rep. (2015) 11:1703–13. doi: 10.1016/j.celrep.2015.05.030
99. Hartung ML, Gruber DC, Koch KN, Gruter L, Rehrauer H, Tegtmeyer N, et al. H. pylori-induced DNA strand breaks are introduced by nucleotide excision repair endonucleases and promote NF-κB target gene expression. Cell Rep. (2015) 13:70–9. doi: 10.1016/j.celrep.2015.08.074
100. Byun E, Park B, Lim JW, Kim H. Activation of NF-κB and AP-1 mediates hyperproliferation by inducing beta-Catenin and c-Myc in Helicobacter pylori-Infected gastric epithelial cells. Yonsei Med J. (2016) 57:647–51. doi: 10.3349/ymj.2016.57.3.647
101. Zhu S, Soutto M, Chen Z, Peng D, Romero-Gallo J, Krishna US, et al. Helicobacter pylori-induced cell death is counteracted by NF-κB-mediated transcription of DARPP-32. Gut. (2017) 66:761–2. doi: 10.1136/gutjnl-2016-312141
102. Yang F, Xu Y, Liu C, Ma C, Zou S, Xu X, et al. NF-κB/miR-223-3p/ARID1A axis is involved in Helicobacter pylori CagA-induced gastric carcinogenesis and progression. Cell Death Dis. (2018) 9:12. doi: 10.1038/s41419-017-0020-9
103. Hwang JW, Baek YM, Yang KE, Yoo HS, Cho CK, Lee YW, et al. Lactobacillus casei extract induces apoptosis in gastric cancer by inhibiting NF-κB and mTOR-mediated signaling. Integr Cancer Ther. (2013) 12:165–73. doi: 10.1177/1534735412442380
104. Shibata W, Takaishi S, Muthupalani S, Pritchard DM, Whary MT, Rogers AB et al. Conditional deletion of IκB-kinase-beta accelerates helicobacter-dependent gastric apoptosis, proliferation, and preneoplasia. Gastroenterology. (2010) 138:1022–34.e1021–10. doi: 10.1053/j.gastro.2009.11.054
105. Chen S, Su T, Zhang Y, Lee A, He J, Ge Q, et al. Fusobacterium nucleatum promotes colorectal cancer metastasis by modulating KRT7-AS/KRT7. Gut Microbes. (2020) 7:1–15. doi: 10.1080/19490976.2019.1695494
106. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. (2013) 14:207–15. doi: 10.1016/j.chom.2013.07.007
107. Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y, et al. Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear factor-κB, and up-regulating expression of microRNA-21. Gastroenterology. (2017) 152:851–66.e24. doi: 10.1053/j.gastro.2016.11.018
108. Hung SC, Huang PR, Almeida-da-Silva CLC, Atanasova KR, Yilmaz O, Ojcius DM. NLRX1 modulates differentially NLRP3 inflammasome activation and NF-κB signaling during Fusobacterium nucleatum infection. Microbes Infect. (2018) 20:615–25. doi: 10.1016/j.micinf.2017.09.014
109. Kang W, Jia Z, Tang D, Zhang Z, Gao H, He K, et al. Fusobacterium nucleatum facilitates apoptosis, ros generation, and inflammatory cytokine production by activating AKT/MAPK and NF-κB signaling pathways in human gingival fibroblasts. Oxid Med Cell Longev. (2019) 2019:1681972. doi: 10.1155/2019/1681972
110. Chen Y, Chen Y, Cao P, Su W, Zhan N, Dong W. Fusobacterium nucleatum facilitates ulcerative colitis through activating IL-17F signaling to NF-κB via the upregulation of CARD3 expression. J Pathol. (2020) 250:170–82. doi: 10.1002/path.5358
111. Cao P, Chen Y, Chen Y, Su W, Zhan N, Dong W. Fusobacterium nucleatum activates endoplasmic reticulum stress to promote crohn's disease development via the upregulation of CARD3 expression. Front Pharmacol. (2020) 11:106. doi: 10.3389/fphar.2020.00106
112. Lourenco TG, Heller D, Silva-Boghossian CM, Cotton SL, Paster BJ, Colombo AP. Microbial signature profiles of periodontally healthy and diseased patients. J Clin Periodontol. (2014) 41:1027–36. doi: 10.1111/jcpe.12302
113. Han YW. Fusobacterium nucleatum: a commensal-turned pathogen. Curr Opin Microbiol. (2015) 23:141–7. doi: 10.1016/j.mib.2014.11.013
114. Strauss J, Kaplan GG, Beck PL, Rioux K, Panaccione R, Devinney R, et al. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm Bowel Dis. (2011) 17:1971–8. doi: 10.1002/ibd.21606
115. Litvak Y, Sharon S, Hyams M, Zhang L, Kobi S, Katsowich N, et al. Epithelial cells detect functional type III secretion system of enteropathogenic Escherichia coli through a novel NF-κB signaling pathway. PLoS Pathog. (2017) 13:e1006472. doi: 10.1371/journal.ppat.1006472
116. Pallett MA, Berger CN, Pearson JS, Hartland EL, Frankel G. The type III secretion effector NleF of enteropathogenic Escherichia coli activates NF-κB early during infection. Infect Immun. (2014) 82:4878–88. doi: 10.1128/IAI.02131-14
117. Sahu U, Choudhury A, Parvez S, Biswas S, Kar S. Induction of intestinal stemness and tumorigenicity by aberrant internalization of commensal non-pathogenic E. coli. Cell Death Dis. (2017) 8:e2667. doi: 10.1038/cddis.2017.27
118. La Ferla K, Seegert D, Schreiber S. Activation of NF-κB in intestinal epithelial cells by E. coli strains isolated from the colonic mucosa of IBD patients. Int J Colorectal Dis. (2004) 19:334–42. doi: 10.1007/s00384-004-0583-7
119. Karrasch T, Kim JS, Muhlbauer M, Magness ST, Jobin C. Gnotobiotic IL-10-/-;NF-κ B(EGFP) mice reveal the critical role of TLR/NF-κ B signaling in commensal bacteria-induced colitis. J Immunol. (2007) 178:6522–32. doi: 10.4049/jimmunol.178.10.6522
120. Long X, Wong CC, Tong L, Chu ESH, Ho Szeto C, Go MYY, et al. Peptostreptococcus anaerobius promotes colorectal carcinogenesis and modulates tumour immunity. Nat Microbiol. (2019) 4:2319–30. doi: 10.1038/s41564-019-0541-3
121. Chung L, Thiele Orberg E, Geis AL, Chan JL, Fu K, DeStefano Shields CE, et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe. (2018) 23:421. doi: 10.1016/j.chom.2018.01.007
122. Kim JM, Cho SJ, Oh YK, Jung HY, Kim YJ, Kim N. Nuclear factor-κ B activation pathway in intestinal epithelial cells is a major regulator of chemokine gene expression and neutrophil migration induced by Bacteroides fragilis enterotoxin. Clin Exp Immunol. (2002) 130:59–66. doi: 10.1046/j.1365-2249.2002.01921.x
123. Wu S, Powell J, Mathioudakis N, Kane S, Fernandez E, Sears CL. Bacteroides fragilis enterotoxin induces intestinal epithelial cell secretion of interleukin-8 through mitogen-activated protein kinases and a tyrosine kinase-regulated nuclear factor-κB pathway. Infect Immun. (2004) 72:5832–39. doi: 10.1128/IAI.72.10.5832-5839.2004
124. Jeon JI, Ko SH, Kim JM. Intestinal epithelial cells exposed to bacteroides fragilis enterotoxin regulate NF-κB activation and inflammatory responses through beta-catenin expression. Infect Immun. (2019) 87:e00312–19. doi: 10.1128/IAI.00312-19
125. Ogino S, Nowak JA, Hamada T, Milner DA Jr, Nishihara R. Insights into pathogenic interactions among environment, host, and tumor at the crossroads of molecular pathology and epidemiology. Annu Rev Pathol. (2019) 14:83–103. doi: 10.1146/annurev-pathmechdis-012418-012818
126. Hamada T, Nowak JA, Milner DA Jr, Song M, Ogino S. Integration of microbiology, molecular pathology, and epidemiology: a new paradigm to explore the pathogenesis of microbiome-driven neoplasms. J Pathol. (2019) 247:615–28. doi: 10.1002/path.5236
127. Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol. (2019) 16:713–32. doi: 10.1038/s41575-019-0189-8
Keywords: NF-κB signaling pathway, Helicobacter pylori, microbiota, gastric cancer, colorectal cancer
Citation: Peng C, Ouyang Y, Lu N and Li N (2020) The NF-κB Signaling Pathway, the Microbiota, and Gastrointestinal Tumorigenesis: Recent Advances. Front. Immunol. 11:1387. doi: 10.3389/fimmu.2020.01387
Received: 20 March 2020; Accepted: 29 May 2020;
Published: 30 June 2020.
Edited by:
Paul Laszlo Bollyky, Stanford University, United StatesReviewed by:
Shuji Ogino, Dana–Farber Cancer Institute, United StatesEmma Allen-Vercoe, University of Guelph, Canada
Copyright © 2020 Peng, Ouyang, Lu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nianshuang Li, enl5YWxucyYjeDAwMDQwOzEyNi5jb20=
†These authors have contributed equally to this work