 Yuhui Yang
Yuhui Yang Chunyan Li
Chunyan Li Tao Liu
Tao Liu Xiaofang Dai
Xiaofang Dai Alexandr V. Bazhin
Alexandr V. Bazhin- 1Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Department of Nuclear Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Hubei Province Key Lab of Molecular Imaging, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 4Department of General, Visceral, and Transplant Surgery, Ludwig-Maximilians-University Munich, Munich, Germany
Among the various immunological and non-immunological tumor-promoting activities of myeloid-derived suppressor cells (MDSCs), their immunosuppressive capacity remains a key hallmark. Effort in the past decade has provided us with a clearer view of the suppressive nature of MDSCs. More suppressive pathways have been identified, and their recognized targets have been expanded from T cells and natural killer (NK) cells to other immune cells. These novel mechanisms and targets afford MDSCs versatility in suppressing both innate and adaptive immunity. On the other hand, a better understanding of the regulation of their development and function has been unveiled. This intricate regulatory network, consisting of tumor cells, stromal cells, soluble mediators, and hostile physical conditions, reveals bi-directional crosstalk between MDSCs and the tumor microenvironment. In this article, we will review available information on how MDSCs exert their immunosuppressive function and how they are regulated in the tumor milieu. As MDSCs are a well-established obstacle to anti-tumor immunity, new insights in the potential synergistic combination of MDSC-targeted therapy and immunotherapy will be discussed.
Introduction
Myeloid cells are a group of highly diverse cells that are essential for the normal functioning of innate and adaptive immunity. Mononuclear myeloid cells include monocytes, macrophages, and dendritic cells (DCs), and granulocytic myeloid cells include neutrophils, eosinophils, basophils, and mast cells. In steady state, myelopoiesis is under tight control and remains predominantly quiescent. A wide range of pathological stimuli, such as infectious microorganisms, tissue damage, and malignantly transformed cells, induce emergency myelopoiesis that largely leads to robust expansion of activated monocytes and neutrophils to eliminate potential threats. If these conditions terminate in time, the homeostasis of myeloid cells will be restored, leaving no negative consequence to the host; conversely, the persistent presence of low-strength stimuli leads to the accumulation of immature myeloid cells characterized by powerful immunosuppressive capacity, which may serve as a protective mechanism to prevent excessive tissue damage caused by unresolved immune response (1).
Studies since the 1970s have highlighted a group of systematically expanded and pathologically activated immature myeloid cells in tumor-bearing hosts. Based on their myeloid origin and immunosuppressive potency, these cells were termed myeloid-derived suppressor cells (MDSCs) in 2007 (2). In addition to cancer, MDSCs are implicated in other diseases, such as chronic inflammation or infection, autoimmune disorder, trauma, and graft-versus-host disease (2). MDSCs are a heterogeneous population consisting of myeloid progenitor cells and immature myeloid cells, characterized by the lack of surface markers associated with fully differentiated myeloid cells and by their morphological resemblance to granulocytic and monocytic cells (3).
MDSCs are generally divided into two main subsets: polymorphonuclear MDSCs (PMN-MDSCs, also known as granulocytic MDSCs) and monocytic MDSCs (M-MDSCs), which morphologically and phenotypically resemble neutrophils and monocytes, respectively. In tumor-bearing mice, MDSCs are generally defined as positive for myeloid lineage differentiation markers CD11b and Gr-1, with PMN-MDSCs being Ly6G+Ly6Clow and M-MDSCs being Ly6G−Ly6Chigh (4). On the other hand, their counterparts in cancer patients are less definite, since studies on human MDSCs have been hampered by cellular diversity and a lack of unequivocal markers. Nonetheless, human PMN-MDSCs are now commonly defined as CD11b+CD14−CD15+ or CD11b+CD14−CD66b+ and M-MDSCs as CD11b+CD14+HLA-DR−/lowCD15− (4). Another population of immature MDSCs has recently been identified. These LIN− (including CD3, CD14, CD15, CD19, and CD56) HLA-DR−CD33+ cells contain mixed groups of MDSCs comprising more immature progenitors and have been defined as “early-stage MDSCs (e-MDSCs)” (4). However, the murine equivalent of these e-MDSCs has not yet been defined.
Activated MDSCs actively participate in multiple aspects of tumor progression, including immune evasion, angiogenesis, pre-metastatic niche formation, and epithelial-mesenchymal transition (EMT) (5–7). Among these tumor-promoting activities, suppression of immune cells is the defining feature of MDSCs. Since the aforementioned surface markers are not exclusive to MDSCs and some are shared by other myeloid cells, phenotyping together with suppressive function assessment has been proven to be the optimal strategy for identifying bona fide MDSCs (4). Studies in the past decade have provided us with a clearer view of the immunosuppressive nature of MDSCs. In this work, we intend to thoroughly review the ever-expanding list of suppressive machineries and cell targets of MDSCs (Figure 1). The nature of MDSC-mediated immune suppression will be discussed in detail, highlighting the antigen specificity of suppression and the regulatory role of the tumor microenvironment.
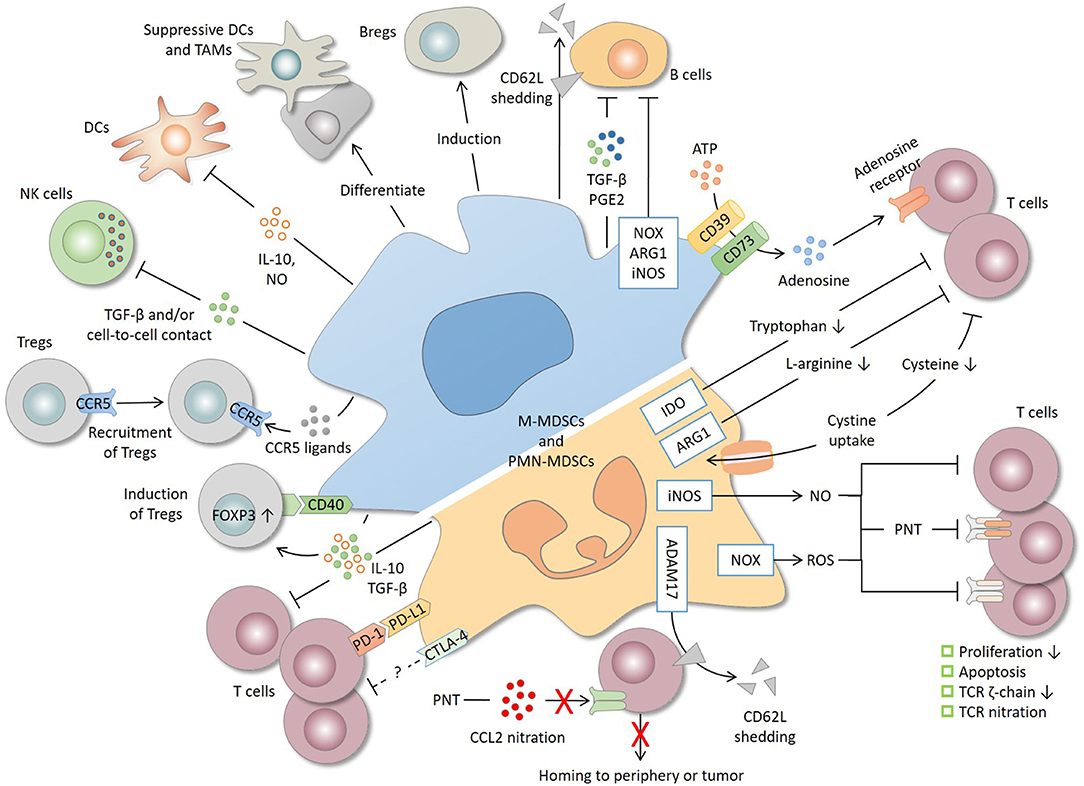
Figure 1. Immunosuppressive mechanisms and cell targets of MDSCs. T cells are the primary target of MDSCs. MDSCs produce a high level of nitric oxide (NO), reactive oxygen species (ROS), and peroxynitrite (PNT), which suppress T cells by inhibiting proliferation, inducing apoptosis, decreasing the TCR ζ-chain and nitrating the TCR complex. MDSCs deplete amino acids essential for T-cell response. For instance, MDSCs decrease L-arginine and tryptophan level through arginase 1 (ARG1) and indoleamine 2, 3-dioxygenase (IDO), respectively, and reduce the cysteine availability through cystine uptake. CD39/CD73 expression by MDSCs produces adenosine that inhibits T cells through adenosine receptors. By shedding CD62L (L-selectin) off the T-cell surface or by nitrating CCL2, MDSCs interrupt T-cell trafficking to the periphery or tumor site. MDSCs express both programmed cell death-ligand 1 (PD-L1), which inhibits T cells through interaction with programmed cell death protein 1 (PD-1), and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), whose precise role remains unclear. IL-10 and TGF-β, two major immunosuppressive cytokines produced by MDSCs, are implicated in T-cell suppression and regulatory T cell (Treg) induction. MDSCs also induce Tregs through CD40 in a contact-dependent manner and recruit Tregs through the production of various chemokines. In addition to T cells, MDSCs suppress natural killer (NK) cells and dendritic cells (DCs), inhibit B cells, and induce regulatory B cells (Bregs). Lastly, tumor-infiltrating MDSCs, mostly M-MDSCs, may differentiate into suppressive DCs and tumor-associated macrophages (TAMs). ADAM17, a disintegrin and metalloproteinase 17; iNOS, inducible nitric oxide synthase; NOX2, NADPH oxidase 2.
Suppressive Mechanisms and Cell Targets of MDSCs
Nitric Oxide, Reactive Oxygen Species, and Peroxynitrite
It is well-established that MDSCs are capable of inhibiting T-cell function. MDSCs express a high level of inducible nitric oxide synthase (iNOS), which produces nitric oxide (NO) (8–11). It is reported that NO suppresses T-cell proliferation, probably directly by inhibiting the Jak/STAT5 pathway or indirectly by inhibiting the antigen presentation from DCs (11, 12). Meanwhile, NO induces apoptosis of T cells (13). On the other hand, MDSCs produce a high amount of reactive oxygen species (ROS) via NADPH oxidase (NOX2) (8, 14). The inhibitory effect of ROS on T-cell function is well-described (15). For MDSCs, this suppression is caused by decreased expression of T-cell receptor (TCR) ζ-chain and is abrogated by inhibiting ROS production (14).
Studies have identified peroxynitrite (PNT), a potent oxidant produced by reaction between NO and superoxide anion (), as a crucial effector molecule of MDSCs. Local production of PNT in the tumor microenvironment is responsible for the non-responsiveness of tumor-infiltrating cytotoxic T lymphocytes (CTLs), and consistently, these CTLs are associated with a high level of nitrotyrosine, a marker of PNT activity (16). PNT suppresses T cells by nitrating the TCR complex, leading to loss of response to specific antigen presented by MDSCs (see below) (17). In addition to the TCR complex, it has recently been shown that MDSCs inhibit T-cell activation by nitrating Tyr394 of lymphocyte-specific protein tyrosine kinase (LCK), an initiating tyrosine kinase in the TCR-mediated signaling cascade (18).
Interference With the Trafficking of T Cells
MDSCs impede the access of T cells to target sites by interfering with their trafficking (19). Expression of a disintegrin and metalloproteinase 17 (ADAM17), a major sheddase of L-selectin (CD62L), by MDSCs cleaves the ectodomain of L-selectin and consequently reduces L-selectin on the surface of naïve CD4+ and CD8+ T cells, therefore limiting their homing to peripheral lymph nodes and tumor sites (20). In another study, this MDSC-mediated decreased L-selectin level on T cells is regulated by high mobility group box protein 1 (HMGB1) in the tumor microenvironment (21).
Besides directly interfering with T-cell trafficking, MDSC-derived NO reduces E-selectin expression on endothelial cells, and PNT causes nitration and inactivation of CCL2 chemokine, both of which indirectly hamper the migration of T cells to the tumor site (22, 23).
Depletion of Amino Acids Necessary for T-Cell Response
MDSCs are able to deplete amino acids required for T-cell activation and proliferation. A high level of arginase 1 (ARG1) expression by MDSCs depletes L-arginine in the tumor microenvironment, leading to downregulation of the CD3 ζ-chain of the TCR complex and proliferative arrest of T cells (24).
On the other hand, MDSCs deprive T cells of cysteine, an essential amino acid for T-cell activation, by uptaking cystine and not exporting cysteine. Since T cells depend on exogenously generated cysteine, the decreased availability of cysteine in the tumor milieu results in impaired T-cell activation (25). Furthermore, it is also reported that indoleamine 2, 3-dioxygenase (IDO) expression is upregulated in MDSCs isolated from fresh breast cancer tissue and is responsible for MDSC-mediated inhibition on T-cell proliferation and Th1 polarization (26).
Adenosine and Adenosine Receptors
Recent studies have identified adenosine, a purine nucleoside, as a novel effector molecule of MDSCs. Extracellular ATP or ADP is hydrolyzed by CD39 (nucleoside triphosphate diphosphohydrolase) into AMP, which is in turn cleaved by CD73 (ecto-5'-nucleotidase) into adenosine (27). Both CD39 and CD73 are expressed by MDSCs from tumor-bearing mice and cancer patients, suggesting that MDSCs are capable of producing adenosine (28–30). TGF-β promotes the differentiation of MDSCs into CD39+CD73+ terminally differentiated myeloid cells with high adenosine production in tumor-bearing mice (31). Consistently, another recent study has demonstrated that tumor-derived TGF-β induces CD39/CD73 expression on MDSCs from lung cancer patients through the mammalian target of rapamycin (mTOR)-hypoxia-inducible factor 1α (HIF-1α) pathway, and these CD39+CD73+ MDSCs represent a distinct subpopulation that expresses higher levels of HIF-1α, cyclooxygenase 2 (COX2), IL-10, tumor necrosis factor (TNF)-α, and TGF-β as compared to their counterparts (32).
It is well-studied that adenosine inhibits the activation and effector function of T cells, which signals primarily through A2A and A3 adenosine receptors (33). In the presence of CD73 substrate 5′-AMP, the inhibition of PMN-MDSCs on anti-CD3/CD28-induced T-cell proliferation is potentiated (28). On the contrary, CD73−/− MDSCs or MDSCs whose CD39 or CD73 enzymatic activity is inhibited show reduced capacity to suppress T cells and natural killer (NK) cells (30, 32, 34). Furthermore, it is reported that MDSCs promote chemoresistance through the activity of CD39 and CD73 (32). Metformin, a biguanide used for type 2 diabetes, reduces the expression and activity of CD39 and CD73 on MDSCs, which leads to reduced MDSC-mediated suppression of CD8+ T cells in vitro and in vivo, and may partially account for the survival benefit seen in diabetic ovarian cancer patients treated with metformin (30).
The adenosine receptors expressed on MDSCs contribute indirectly to the adenosine-induced immune suppression. Stimulation of A2B receptors preferentially expands PMN-MDSCs (28). In mice with melanoma, blockade of A2B receptors reduces IL-10, monocyte chemoattractant protein 1 (MCP-1), and MDSCs in the tumor site, which is associated with increased frequency of intratumoral CD8+ T cells, elevated levels of TNF-α and IFN-γ, and delayed tumor growth (35). In another murine melanoma model, selective deletion of A2A receptors in myeloid cells leads to significantly reduced IL-10 production by MDSCs, an increase in activated CD8+ T cells and NK cells, and delayed primary tumor growth and metastasis (36).
CD39 and CD73 are also expressed on tumor cells, regulatory T cells (Tregs), effector T cells, Th17 cells, and other stromal cells (33). Ectonucleotidases are supposed to prevent excessive T cell-mediated immune response and to regulate the balance between pro-inflammatory ATP and immunosuppressive adenosine. However, tumor hijacks this network to facilitate immune evasion. In line with the abovementioned findings, Umansky et al. have proposed two modes of adenosine signaling. Firstly, MDSCs, Tregs, and tumor cells may produce extracellular adenosine to suppress T-cell function in a paracrine manner. Secondly, adenosine produced by ectonucleotidase on tumor-infiltrating lymphocytes suppresses their own function in an autocrine manner; the upregulated CD39 and CD73 expression by MDSCs and Tregs also enables autocrine adenosine signaling and potentiates their expansion and/or suppressive activity (33).
MDSC-Derived IL-10
MDSCs are a major source of IL-10 in tumor-bearing host (37–40), and consistently, the frequency of MDSCs is correlated with the IL-10 level in peripheral blood of cancer patients (41). It is becoming clear that IL-10 serves as a non-redundant suppressive mechanism of MDSCs, and accordingly, blockade of IL-10 signaling or neutralization of IL-10 leads to alleviated T-cell suppression, delayed tumor progression, and improved therapeutic efficacy (37, 42). In addition to T-cell inhibition, MDSC-derived IL-10 is implicated in the induction of Tregs and the suppression of DCs (see below).
Recent studies are unraveling the regulation on IL-10 production by MDSCs, which involves cellular and non-cellular participants. For instance, hypoxia significantly upregulates IL-10 secreted by MDSCs (43). Exposure to lipopolysaccharide (LPS), a Toll-like receptor (TLR) ligand, increases IL-10 production by MDSCs, which may require the MyD88 signaling pathway (44). Transmembrane TNF-α (tmTNF-α), but not the secretory form, activates MDSCs to upregulate IL-10 and other immunosuppressive effector molecules through TNFR2 (45). The level of interferon regulatory factor 4 (IRF4), an essential transcription factor required for lymphoid and myeloid cell differentiation, reduces remarkably during the development of MDSCs and modulates the suppression of T cells through IL-10 and ROS production (46). Tumor cells, not surprisingly, participate in the MDSC-derived IL-10 regulation. For instance, knockdown of semaphorin 4D, a pro-angiogenic factor overexpressed in many malignancies, in tumor cells reduces the IL-10 production by MDSCs (47). Glioma stem cell-derived exosomes induce systemic T-cell suppression by polarizing CD14+ monocytes toward M-MDSC phenotype with heightened IL-10 level (48). In another study, the NKG2D ligand RAE-1ε expressed on tumor cells facilitated the expansion and activation of MDSCs that display pronounced ARG1 activity and IL-10 production (49).
Similarly, MDSCs developed in the settings of microbial infection are also capable of producing IL-10 (50–52). In patients with chronic hepatitis B, IL-10 induced by programmed cell death protein 1 (PD-1) signaling is responsible for T-cell suppression by MDSCs (50). In patients with chronic hepatitis C virus infection, M-MDSCs have higher levels of phosphorylated STAT3 and IL-10, while blocking STAT3 signaling reduces hepatitis C virus (HCV)-mediated M-MDSC expansion and IL-10 expression (51).
TGF-β
TGF-β is another well-documented immunosuppressive cytokine secreted by MDSCs in tumor-bearing host (22, 43, 53). MDSCs developed in non-cancer settings are also capable of producing TGF-β (52, 54). Evidence for the regulation of MDSC-derived TGF-β remains elusive. It was shown previously that TGF-β produced by MDSCs is induced in vivo by IL-13 and CD1d-restricted T cells that are most likely natural killer T (NKT) cells (55). Recent studies have shown that TGF-β production by MDSCs is regulated by tmTNF-α, ribosomal protein S19, and semaphorin 4D (45, 47, 56). On the contrary, CD14+HLA-DR−/low MDSCs from patients with liver cancer show no TGF-β secretion (57). These findings suggest that TGF-β production by MDSCs may be context-dependent.
MDSC-derived TGF-β contributes to T-cell suppression, although it is probably not the principal mechanism (53). CD14+HLA-DR−/low MDSCs isolated from melanoma patients inhibit T cells via TGF-β with no involvement of ARG1 and iNOS (58). Song et al. have shown that transfer of tumor-derived MDSCs to asthmatic mice leads to reduced pulmonary recruitment of inflammatory cells, suppressed Th2 response, and decreased IgE production in a TGF-β1-dependent manner (59). Furthermore, TGF-β is essential in Treg induction by MDSCs (see below).
Other immune cells are also inhibited by MDSC-derived TGF-β. For instance, in a murine model of AIDS, M-MDSCs suppressed B-cell response by superoxide, nitric oxide, PNT, and TGF-β (54). CD14+HLA-DR−/low MDSCs from melanoma patients inhibit NK cells primarily through TGF-β that is stimulated by tumor-derived PGE2 (60). In addition to soluble TGF-β, MDSCs expanded in tumor-bearing mice express and utilize membrane-bound TGF-β to suppress NK cells and NKT cells in a contact-dependent manner (61, 62).
In addition to immune suppression, TGF-β has been implicated in the regulation of tumor metastasis facilitated by MDSCs. A portion of tumor cells undergoes EMT to disseminate, invade surrounding tissue, and metastasize. In a spontaneous murine model of melanoma, Toh and colleagues have shown for the first time that MDSCs use TGF-β, epidermal growth factor, and hepatocyte growth factor to induce EMT and that depletion of MDSCs results in reduced EMT and fewer metastases (63). In another study, anti-TGF-β treatment in a murine model of mammary tumor inhibited tumor growth and lung metastasis, and depletion of MDSCs diminished this beneficial effect of TGF-β neutralization (64). Another study from the same group later demonstrated that specific deletion of gene encoding TGF-β receptor II in myeloid cells significantly reduces metastasis, which is mediated by decreased TGF-β1 and type 2 cytokine production and by reduced ARG1 and iNOS expression. This effect was largely ascribed to the CD11b+Ly6G+ myeloid subset (65).
PD-L1 and CTLA-4 Expression by MDSCs
Immune checkpoint pathways act as negative regulators and prevent excessive immune response. MDSCs assist tumor to hijack this mechanism in order to promote T-cell anergy, which signals mostly through the PD-1/programmed cell death-ligand 1 (PD-L1) pathway (66). MDSCs express PD-L1 in various tumor models (43, 67–73). Meanwhile, numerous studies have found PD-L1 expression in MDSCs from cancer patients (29, 42, 53, 72, 74–76). In liver cancer patients, the percentage of PD-L1+ MDSCs in peripheral blood correlates with disease stage and correlates inversely with clinical outcome (76). On the other hand, MDSCs developed during microbial infection also express PD-L1 (77, 78).
PD-L1 is implicated in MDSC-mediated T-cell suppression. PD-L1 blockade reduces the suppressive capacity of MDSCs on T cells (29, 42, 53, 68, 73, 74, 77–79). In addition to conventional T cells, in a murine model of liver metastasis, PD-L1 expression by MDSCs impairs the proliferation of chimeric antigen receptor cells, while MDSC depletion or PD-L1 blockade improves their therapeutic efficacy (80). Blocking PD-L1 relieves inhibition on DCs by MDSCs as well (81).
Several studies have shown that tumor-infiltrating MDSCs express a higher level of PD-L1 than their peripheral counterparts, suggesting microenvironmental regulation of PD-L1 expression (43, 68, 72, 73, 75). For instance, tumor cells upregulate the PD-L1 expression in MDSCs by interfering with their arachidonic acid metabolism (82). Tumor-derived soluble mediators are also responsible for PD-L1 induction in intratumoral MDSCs (76, 80). Other microenvironmental signals that regulate PD-L1 expression by MDSCs, such as hypoxia, cytokines, and stromal cells, will be discussed in detail in the following sections.
On the other hand, it is reported that MDSCs express cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) (43, 71). However, unlike PD-L1, the precise role and regulation of CTLA-4 is less well-studied in MDSCs. It is reported that blocking or silencing CTLA-4 reduces the frequency and ARG1 activity of MDSCs (83).
Induction and Recruitment of Regulatory T Cells
MDSCs inhibit effector T cells not only by themselves but also by inducing and recruiting Tregs. The proliferation of Tregs is relatively insensitive to suppression by MDSCs as compared with effector T cells (84). Intratumoral accumulation of Tregs occurs later than that of MDSCs, while depletion of MDSCs reduces infiltrating Tregs, suggesting that MDSCs may facilitate the development of Tregs (85). In non-cancer settings, co-culturing CD4+ T cells with MDSCs from HIV+ individuals or chronic hepatitis C patients significantly increases the differentiation of Foxp3+ Tregs (51, 86).
The mechanism(s) for Treg induction by MDSCs is not fully understood. During tumor progression, a subset of DCs with an immature myeloid phenotype is licensed by tumor cells to promote proliferation of Tregs by producing TGF-β (87). Huang and colleagues have shown that MDSCs induce Tregs both in vitro and in vivo, which requires activation of T cells and is dependent on IFN-γ and IL-10. The authors speculated that, in response to IFN-γ produced by activated T cells, MDSCs secret TGF-β and IL-10, both of which participate in the development of Tregs (88). Another study from this same group later demonstrated that CD40 expression on MDSCs is required for Treg induction, since adoptive transfer of CD40-deficient MDSCs or administration of anti-CD40 antibodies fails to induce Tregs (89). Treg induction by MDSCs is attenuated in the Transwell system that separates the two cell types, suggesting the requirement of direct cell-to-cell contact (90). In a murine model of B-cell lymphoma, MDSCs promoted the expansion of Tregs from pre-existing natural Tregs but not conversion from naïve T cells. In that study, MDSCs induced tumor-specific Tregs via antigen uptake, processing, and presentation, which requires ARG1 but not TGF-β (91).
In addition, MDSCs may promote the recruitment of Tregs to the tumor milieu. Tumor-infiltrating M-MDSCs produce CCR5 ligands CCL3, CCL4, and CCL5, and meanwhile, Tregs exhibit high surface expression of CCR5 and are recruited to tumor tissue by CCL4 and CCL5. Accordingly, Tregs from CCR5 knockout mice almost completely lost their ability to migrate toward M-MDSCs in vitro (92). In a murine model of glioblastoma multiforme, both M-MDSCs and Tregs were recruited by CCL2 produced by tumor-associated macrophages (TAMs) and microglia (93). A recent study revealed a closed loop between mast cells, MDSCs, and Tregs in the tumor microenvironment. Mast cells induce infiltration of MDSCs to tumor and induce their IL-17 secretion; MDSC-derived IL-17 attracts Tregs indirectly and potentiates their suppressive activity and IL-9 production; IL-9 in turn promotes the survival and tumor-promoting function of mast cells. In that study, IL-17 promoted Treg recruitment by increasing the level of CCL17 and CCL22 in the tumor microenvironment (94).
Studies on the relation between MDSCs and Tregs in cancer patients are relatively limited. A positive correlation between MDSCs and Tregs in peripheral blood and tumor site has been detected in cancer patients (40, 95). Hoechst and colleagues have shown that CD14+HLA-DR−/low M-MDSCs from hepatocellular carcinoma patients induce suppressive CD4+CD25+Foxp3+ Tregs in a contact-dependent manner when co-cultured with autologous CD3/CD28-stimulated CD4+ T cells (57). In addition, to induce Tregs from CD4+ T cells, a study from the same group has shown that CD14+HLA-DR−/low M-MDSCs are able to convert Th17 cells to Foxp3+ Tregs, which is dependent on MDSC-derived TGF-β and retinoic acid (96). Jitschin et al. have shown that M-MDSCs from chronic lymphocytic leukemia (CLL) patients suppress T-cell activation and promote Treg induction, which is partly dependent on IDO activity (95). Furthermore, the authors have also demonstrated that after co-culture with CLL cells, monocytes from healthy donors resemble the phenotypic, suppressive, and Treg-inducing characteristics of M-MDSCs from CLL patients (95). In patients with lung cancer, a novel tumor-infiltrating B7-H3+CD14+HLA-DR−/low subset of MDSCs is reported to induce Tregs in vitro, which is partly dependent upon IL-10 (40).
Interestingly, there are also reports revealing no clear association between MDSCs and Tregs. In mice bearing T-cell lymphoma, the percentage of intratumoral Tregs is invariably high throughout tumor growth and does not relate to the accumulation kinetics of MDSCs (9). In another study, the T-cell non-responsiveness induced by adoptive transfer of MDSCs was not caused by Treg induction (97). Furthermore, in contrast to the abovementioned Treg-inducing action of M-MDSCs, it is reported that PMN-MDSCs impair TGF-β-mediated generation of inducible Tregs (iTregs) from naïve T cells and inhibit proliferation of naturally occurring Tregs (nTregs) without affecting Foxp3 expression (98). These discrepancies need to be clarified by further study.
Suppression of Natural Killer Cells
NK cells are another major target of MDSCs. The reduced number and impaired function of NK cells in tumor-bearing mice are inversely correlated with the increased level of MDSCs and are restored by depletion of MDSCs (61, 99). A similar inverse correlation is also observed in patients with non-Hodgkin lymphoma (39). It is shown that the enhanced lactate production by tumor cells inhibits NK cells not only directly by inhibiting their cytotoxicity but also indirectly by increasing the number of MDSCs (100). Interestingly, a recent study has demonstrated that a portion of immature NK cells is converted into MDSCs in the presence of GM-CSF and that this conversion is abolished by IL-2 exposure (101). This novel developmental pathway of MDSCs may account, at least partially, for the reduced level of NK cells in tumor-bearing host.
In murine models, the cytotoxicity, NKG2D expression, and IFN-γ production of NK cells are inhibited by MDSCs both in vitro and in vivo (61, 102). This suppression is contact-dependent and requires membrane-bound TGF-β1 on MDSCs (61, 102). In a recent study, Elkabets et al. identified a novel subset of Gr-1high PMN-MDSCs that is induced by IL-1β and lacks Ly6C expression (Ly6Cneg). These Ly6Cneg MDSCs produce higher levels of iNOS and ROS than Ly6Clow MDSCs and, correspondingly, exhibit stronger suppression of T cells and NK cells (103). The MDSC-mediated NK cell suppression is associated with increased metastasis in mice during gestation (104). In tumor-bearing mice treated with medroxyprogesterone acetate, which is commonly used as hormone replacement therapy and as a contraceptive, MDSCs exhibit higher suppression of NK cells as compared with MDSCs from control mice, implying a potential mechanism for increased breast cancer incidence associated with prolonged medroxyprogesterone acetate administration (105).
In patients with liver cancer or advanced melanoma, CD14+HLA-DR−/low MDSCs suppress autologous NK-cell cytotoxicity and IFN-γ production (60, 106). This suppression is independent of ARG1 and iNOS but requires cell-to-cell contact through NK-activating receptor NKp30 on NK cells, suggesting expression of NKp30 ligand(s) by MDSCs (106). In addition, TGF-β produced by MDSCs from melanoma patients, which is stimulated by PGE2, also serves as a major mechanism for NK-cell suppression (60). In addition, MDSCs from cancer patients inhibit Fc receptor-mediated signal transduction and downstream effector function of NK cells, including antibody-dependent cellular cytotoxicity and cytokine production, probably through NO production (107).
As an essential defensive mechanism of the innate immune system, it is not surprising that NK cells are suppressed by MDSCs generated in microbial infection. It is shown that polymorphonuclear neutrophils and PMN-MDSCs dampen the activation and cytotoxic activity of NK cells toward Aspergillus fumigatus (108). In another study with mice infected by vaccinia virus, PMN-MDSCs negatively regulated the proliferation, activation, and function of NK cells, which helped to contain excessive NK cell activity (109). In HCV infection, CD33+CD11blowHLA-DRlow MDSCs suppress the IFN-γ production of NK cells by depleting L-arginine via ARG1 (110). Interestingly, CD66b+CD33b+HLA-DRlow PMN-MDSCs increase strikingly in the cord blood of neonates when compared with peripheral blood of healthy children and adults. These cord blood PMN-MDSCs are able to inhibit the function of T cells and NK cells, which may be responsible for the impaired host defense of neonates (111).
Conversely, there are studies showing NK cell activation by MDSCs. For instance, Nausch et al. have found that MDSCs from tumor-bearing mice express NKG2D ligand RAE-1 and activate NK cells to produce IFN-γ, which is partially contact-dependent and requires signaling through NKG2D (112). In mice bearing NK-sensitive tumor, poly I:C treatment allows MDSCs to prime NK cells and consequently leads to delayed tumor growth. MDSC-derived IFN-α after poly I:C administration activates NK cells, which drives CD69 expression and IFN-γ production but does not induce cytotoxic activity of NK cells (113). A recent study has shown that M-MDSCs infiltrate in the tumor microenvironment prior to NK cells and are required for the tumoricidal activity of NK cells to eradicate galectin-1-deficient GL26 glioma (114). Taken together, the seemingly contradictory findings mentioned above suggest that the effect of MDSCs on NK cells, either inhibitory or stimulatory, is most likely context-dependent.
Impaired Function of Dendritic Cells by MDSCs
Relatively less information is available on the direct impact of MDSCs on DCs. Accumulation of MDSCs in tumor-bearing mice and cancer patients is associated with impaired differentiation and accumulation of DCs (115–117). Unfortunately, the underlying mechanism(s) is not fully understood. In a murine model of allergic airway inflammation, LPS exposure promoted the development of a group of myeloid cells in the lung that resembled MDSCs phenotypically and functionally. These cells inhibited the reactivation of primed Th2 cells by DCs (118). In mice with hepatocellular carcinoma, MDSC-derived IL-10 was found to be responsible for the impaired TLR ligand-induced IL-12 production and T-cell stimulatory activity of DCs (116). Recently, it was shown that MDSC-mediated suppression of antigen presentation from DCs to CD4+ T cells depends on NO, which may cause nitration of STAT1, a key mediator for antigen presentation, and, consistently, this suppression is abrogated by iNOS inhibitors (11). In another recent study, Notch and STAT3 signals were found to be required by MDSCs to suppress the differentiation, maturation, and antigen presentation ability of DCs in vitro and in vivo (119).
Due to their superior antigen presentation and T-cell activation properties, DCs are utilized as cancer vaccines to prompt immunity against malignant cells. DC vaccines loaded with tumor antigens through various approaches aim to induce and potentiate tumor antigen-specific T-cell response. In line with MDSC-mediated suppression of DCs, favorable therapeutic efficacy of DC vaccination is associated with a reduced level of MDSCs in tumor-bearing mice (120, 121). In cancer patients, when monocyte-derived DCs are used as vaccines, the presence of CD14+HLA-DR−/low MDSCs in the starting monocyte population causes impairment of DC maturation, antigen uptake, migration, and T-cell stimulation capacity (122). Therefore, it is reasonable to apply DC-based vaccines in combination with agents that target MDSCs. These agents include chemotherapeutics (e.g., all-trans retinoic acid, gemcitabine, and cyclophosphamide) (123, 124), tyrosine kinase inhibitors (e.g., sunitinib, axitinib, and dasatinib) (125–127), lenalidomide (128), and anti-Gr-1 antibody (120), and these combinations have shown reduced levels of MDSCs and improved efficacy in pre-clinical studies. The initiation of immune response by DC vaccines involves interaction between multiple immune cell types. Therefore, to overcome the immunosuppression mediated by MDSCs and maximize efficacy, further research is still needed to accurately define the action of MDSCs and other immune cells in DC vaccine-induced anti-tumor immunity.
B Cells
In recent years, B cells have emerged as a novel target of MDSCs. In an in vitro model of B lymphopoiesis, MDSCs induced by adipocyte-derived factors inhibited B-cell development through IL-1 production (129). PMN-MDSCs inhibited the recruitment, proliferation, and cytokine secretion of B cells in the central nervous system of mice with experimental autoimmune encephalomyelitis (130). In the settings of retroviral infection and autoimmune disease, several animal studies have revealed that MDSCs impair B cell response by many of the mechanisms utilized in T-cell suppression, such as ROS, iNOS, ARG1, TGF-β, and PGE2 (54, 131). MDSCs from mice infected with retrovirus express V-domain Ig-containing suppressor of T-cell activation (VISTA), a negative checkpoint regulator that is homologous to PD-L1 and inhibits T-cell response, and VISTA deficiency in MDSCs or neutralization of VISTA by blocking antibody partially rescues the impaired B-cell proliferation (132). Both contact-dependent and contact-independent inhibition have been implicated in these studies (54, 131).
Whether these suppressive mechanisms are used by MDSCs in cancer settings is less well-elucidated. ROS, ARG1, iNOS PGE2, and TGF-β have recently been suggested to exert suppressive effects on B-cell proliferation and antibody production by tumor-induced MDSCs (133). In a murine model of lung cancer, the impeded B cell differentiation was associated with tumor progression and MDSC infiltration; mechanistically, MDSCs inhibit B cell response by TGF-β-mediated modulation of IL-7 and downstream STAT5 signaling, which are both essential in B-cell differentiation and function (133). In another study, Ku et al. showed that tumor-induced MDSCs reduce L-selectin on naïve CD4+ and CD8+ T cells and that even moderate L-selectin reduction is sufficient to profoundly disrupt homing of T cells to distant lymph nodes. Interestingly, the loss of L-selectin has also been found in B cells. In the study concerned, the shedding of L-selectin from naïve T cells and B cells was contact-dependent and was independent of major L-selectin sheddase ADAM17. Since the trafficking of both naïve B cells and CD4+ precursors of follicular helper T cells was hindered, the authors suggested that the T cell-dependent antibody production in lymph nodes may have been severely impaired (134).
Regulatory B cells (Bregs) are immunosuppressive and inhibit the expansion of pathogenic T cells and other pro-inflammatory lymphocytes through the production of IL-10, IL-35, and TGF-β. In consistence with these properties, Bregs have been shown to suppress anti-tumor immunity and promote tumor growth. In patients with colorectal cancer, the level of Bregs positively correlates with disease stage and with the frequency of MDSCs (135). In a murine model of breast cancer, Shen et al. showed that MDSCs upregulate PD-L1 expression on B cells and dampen their anti-tumor response; more interestingly, MDSCs may transform B cells into a novel subtype of Bregs that possesses higher inhibitory capability on T cells as compared with other subsets of Bregs (136). In another study, MDSCs induced the expansion of IL-10-producing Bregs, probably through iNOS, and ameliorated autoimmunity in mice with systemic lupus erythematosus (137). Conversely, in mice infected with retrovirus, M-MDSCs inhibited the proliferation of IL-10-producing Bregs in response to LPS stimulation (54).
Antigen-Specific and Non-specific Suppression of CD8+ and CD4+ T Cells
Among the various cell targets, suppression of T cells remains the characteristic necessary to define bona fide MDSCs, provided that the phenotypic criteria are met. With the abovementioned mechanisms, MDSCs are capable of suppressing both antigen-specific and non-specific T-cell response (Figure 2). It is now generally accepted that ROS, and PNT in particular, are responsible for antigen-specific suppression, provided that MDSCs and T cells are in close contact, since these substances are unstable and short-lived, while iNOS, ARG1, and immunosuppressive cytokines are responsible for antigen-non-specific suppression, since effector molecules of these mechanisms have relatively longer half-lives and require cellular proximity, but not close interaction, to exert inhibition (1).
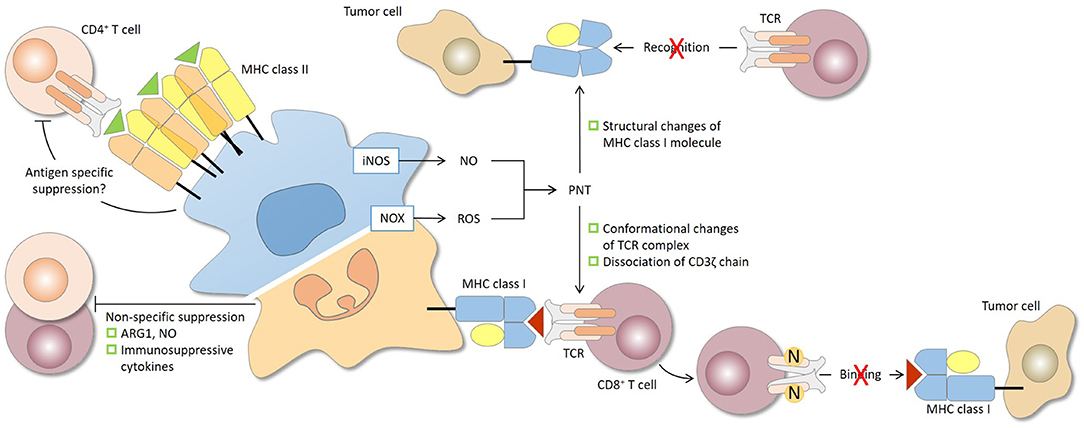
Figure 2. Antigen specificity of MDSC-mediated suppression of CD4+ and CD8+ T cells. The antigen specificity of T-cell suppression by MDSCs is determined largely by the characteristics of the effector molecules involved. The short-lived reactive oxygen species (ROS) and peroxynitrite (PNT) are responsible for antigen-specific suppression, provided that MDSCs and T cells are in close contact, while arginase 1 (ARG1), nitric oxide (NO), and immunosuppressive cytokines, which have relatively longer half-lives, mediate antigen-non-specific suppression. During the close and prolonged interaction between MDSCs and CD8+ T cells in antigen recognition, PNT causes nitration and conformational changes of the TCR complex and dissociation of CD3ζ molecules. CD8+ T cells consequently lose their binding ability to peptide-MHC class I complex and are rendered non-responsive to specific peptide presented by tumor cells. PNT may also induce structural changes of MHC class I on tumor cells, leading to reduced antigenic peptide binding. In this case, antigen-specific CD8+ T cells, even if functional, fail to recognize tumor cells. For CD4+ T cells, antigen-specific suppression by MDSCs has been reported and may require sufficient MHC class II expression by MDSCs. iNOS, nitric oxide synthase; NOX2, NADPH oxidase 2.
Early studies have shown that Gr-1+ immature myeloid cells isolated from tumor-bearing mice are able to uptake and process soluble proteins and present the antigenic epitopes on their surface (97). Their suppression of antigen-specific CD8+ T cells requires antigen presentation via MHC class I and ROS production (14, 138). Studies in the last decade have revealed that MDSC-induced antigen-specific T-cell tolerance results from post-translational modification of the TCR complex. MDSCs from gp91phox−/− mice produce little ROS and fail to inhibit CD8+ T cells, and neutralization of PNT abrogates the suppressive activity of MDSCs on T cells (17). Nagaraj et al. demonstrated that the close and prolonged cell-to-cell contact during antigen recognition allows MDSC-derived PNT to cause nitration of tyrosines in the TCR-CD8 complex, which induces conformational changes in these molecules and leads to loss of binding ability to peptide-MHC complex (17). Consistently, using double TCR transgenic CD8+ T cells, the same group later showed that MDSCs induce CD8+ T-cell tolerance only against the peptide presented by themselves, while they do not affect T-cell response to peptide specific for other TCR that is not presented by MDSCs (139). In accordance with previous findings, the authors showed that nitration of surface molecules of T cells is localized to the site of physical interaction between MDSCs and T cells, which may lead to dissociation between TCR and CD3ζ molecules, and consequently, nitrotyrosine positive CD8+ T cells are rendered non-responsive to specific peptide (139). In another study, however, ROS were found not to be involved in antigen-specific T-cell suppression by MDSCs, and MDSCs deficient in MHC class I showed no impairment in antigen-specific suppression, which excludes the necessity of antigen presentation (9).
Interestingly, PNT produced by MDSCs can facilitate immune evasion of tumor cells even in the presence of normal functioning T cells. PNT induces nitration and structural changes of MHC class I molecules on tumor cells, which hampers their capacity to bind antigenic peptide and subsequently impairs the recognition by CTLs, therefore affording tumor cells resistance to antigen-specific CTLs (140). These findings collectively suggest the involvement of multiple mechanisms in antigen-specific CD8+ T-cell suppression by MDSCs.
On the other hand, evidence for MDSC-mediated antigen-specific suppression of CD4+ T cells remains elusive, and different results have been reported. It was previously indicated that MDSCs fail to suppress antigen-specific CD4+ T-cell proliferation, which may be due to the low MHC class II expression on MDSCs, which precludes them from forming close contact with CD4+ T cells (91, 138). However, MDSC-mediated suppression of the proliferation of CD4+ T cells exposed to a specific peptide has been reported, which is at least partially due to cysteine deprivation by MDSCs (25, 88). Interestingly, Nagaraj and colleagues have shown that MDSCs are able to suppress antigen-specific CD4+ T-cell response in vitro and in vivo, as long as their MHC class II expression reaches a sufficient level (141). In different experimental systems, MDSCs are able to blunt IFN-γ production of both tumor-specific CD8+ and CD4+ T cells in the spleen of tumor-bearing mice in vivo (142). In patients with liver cancer, depletion of CD14+HLA-DR−/low M-MDSCs enhances IFN-γ secreting CD4+ T cells specific to α-fetoprotein (57). These discrepancies might be explained, in part, by the varied MHC class II level of MDSCs that has been described in different tumor models and human studies, and under some experimental conditions, MDSCs could inhibit the proliferation of T cells without affecting the IFN-γ production and vice versa (3, 4).
Regulation on the Suppressive Nature of MDSCs
In most studies, immunosuppressive activity is detected only in MDSCs derived from tumor-bearing host but not in their control counterparts from tumor-free host, suggesting a tight control over MDSCs by tumor. MDSCs carry out immune suppression principally in the tumor microenvironment, which is a highly dynamic complex and plays a crucial role in tumor development. The constant bi-directional communication between MDSCs and the ever-changing microenvironment shapes the phenotype and function of MDSCs (Figure 3). For instance, tumor-derived M-MDSCs show higher suppression of T cells than spleen- or bone marrow-derived M-MDSCs from the same mice. Several cellular and non-cellular components of the tumor microenvironment, including the subset composition of MDSCs, tumor cells, stromal cells, cytokines, metabolic state, and hypoxia, regulate the suppressive nature of MDSCs.
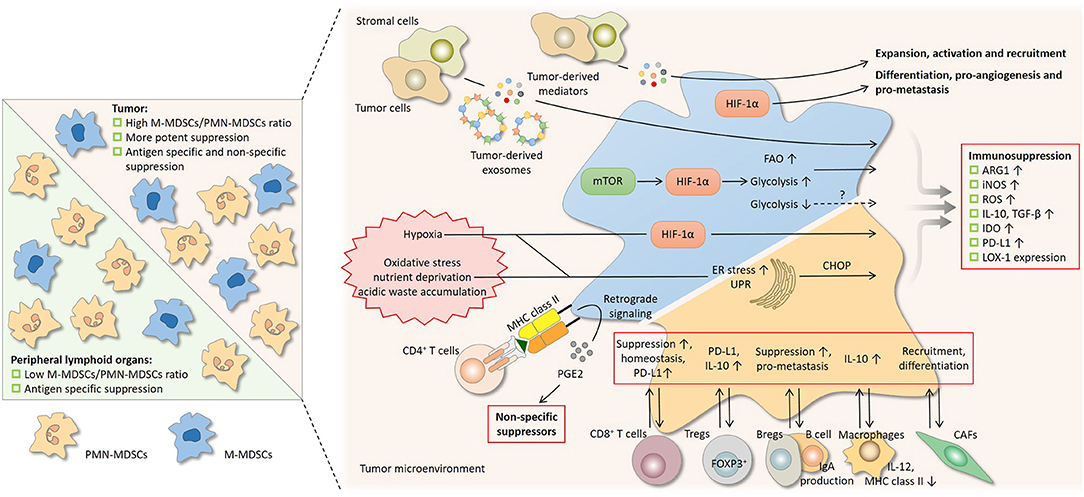
Figure 3. Regulation on the suppressive nature of MDSCs by subset composition and the tumor microenvironment. Differential suppressive capacity and mechanism(s) between PMN-MDSCs and M-MDSCs influence the suppression by MDSCs as a whole population (left). In peripheral lymphoid organs where PMN-MDSCs predominate, suppression by MDSCs is mainly antigen-specific, since the activity of PMN-MDSCs depends largely on reactive oxygen species (ROS) and peroxynitrite (PNT). In tumor where the proportion of M-MDSCs increases, suppression is more potent and is both antigen-specific and non-specific, since M-MDSCs are more suppressive and mainly rely on arginase 1 (ARG1), nitric oxide (NO), and immunosuppressive cytokines. A network of cytokines, hostile physical conditions, and cells in the tumor microenvironment regulates MDSCs in multiple aspects (right). Soluble mediators derived from tumor regulate the suppressive activity of MDSCs and also drive their development. After being taken by MDSCs, the contents of tumor-derived exosomes, which act as intercellular messengers, promote the expansion and potentiate the suppressive capacity of MDSCs. Like tumor cells, MDSCs undergo metabolic reprogramming to adapt to varying surroundings. Hypoxia-inducible factor 1α (HIF-1α) induced by the mammalian target of rapamycin (mTOR) pathway enhances glycolysis and may potentiate suppression by MDSCs, whereas glycolysis has also been reported to be a negative regulator. The heightened fatty acid oxidation (FAO) is associated with upregulated ARG1 and increased NO and PNT production. Hypoxic signaling, primarily through HIF-1α, is another central regulator. HIF-1α promotes many non-immunological activities of MDSCs, including differentiation, pro-angiogenesis, and pro-metastasis. HIF-1α augments MDSC-mediated suppression by upregulating several effector molecules. The hostile conditions in the tumor milieu, such as oxidative stress, nutrient deprivation, and acidic waste accumulation, causes ER stress and induce unfolded protein response (UPR) in MDSCs. ER stress response marker C/EBP homologous protein (CHOP) regulates ARG1, superoxide, and PNT production by MDSCs. The bidirectional communication with stromal cells fine-tunes the induction, homeostasis, differentiation, and suppressive function of MDSCs. Bregs, regulator B cells; CAFs, cancer-associated fibroblasts; DCs, dendritic cells; iNOS, nitric oxide synthase; LOX-1, lectin-type oxidized LDL receptor-1; NK, natural killer; PD-L1, programmed cell death-ligand 1; Tregs, regulatory T cells.
Subset Composition and Antigen Specificity and Capacity of MDSC-Mediated Suppression
It is now clear that the suppressive machineries of MDSCs do not act simultaneously, and subsets of MDSCs use different mechanisms for T-cell suppression (9, 10, 92, 143). For instance, M-MDSCs, whose activity mainly relies on ARG1, NO, and immunosuppressive cytokines, inhibit both antigen-specific and non-specific T-cell response (8, 10, 19, 92, 143, 144), while PMN-MDSCs, whose activity largely depends on high ROS and PNT production, inhibit T cells in an antigen-specific manner (10, 92). In one study, only M-MDSCs, but not PMN-MDSCs, were able to augment the activation-induced Fas upregulation of CD8+ T cells through NO production and sensitize them to Fas-mediated apoptosis and were able to impede the differentiation of mature CTLs (143). Therefore, the suppressive nature of MDSCs is influenced by their subset composition.
PMN-MDSCs is commonly the predominant subpopulation in peripheral lymphoid organs in many murine tumor models, and accordingly, antigen-specific T-cell tolerance is detected at these sites (145, 146). This peripheral antigen-dependent T-cell inhibition may partially explain the findings in some studies that T cells in the periphery retain their responsiveness to other non-specific stimuli (3, 17, 97). On the other hand, the proportion of M-MDSCs is substantially higher in the tumor milieu (144, 145), and in spite of the common findings that PMN-MDSCs may still be the prevalent subpopulation, M-MDSCs are more suppressive than PMN-MDSCs on a per-cell basis (9, 119). As a consequence, tumor-infiltrating MDSCs demonstrate higher immunosuppressive capacity than their peripheral counterparts and are able to inhibit both antigen-specific and non-specific T-cell function (19, 147, 148).
In spite of these findings, it is noteworthy to point out that similar or even stronger inhibitory capacity of peripheral MDSCs has also been reported (140, 149, 150) and that non-specific T-cell suppression is not uncommon in MDSCs derived from peripheral lymphoid organs (149, 151).
It is common that the ratio between subgroups of MDSCs varies in different tumor models. Unfortunately, many of these studies have not addressed the subset composition of intratumoral or peripheral MDSCs in detail, nor have they assessed the suppressive capacity of PMN-MDSCs and M-MDSCs separately. Therefore, the discrepancies on antigen specificity and capacity of MDSC-mediated suppression, on the one hand, should be interpreted with care, and on the other hand, may suggest that subset composition of MDSCs is not likely the sole nor a major determinant that influences their suppressive nature.
Tumor-Derived Mediators
The generation of MDSCs includes two phases. Firstly, aberrant myelopoiesis and blocked differentiation of immature myeloid cells lead to the expansion of MDSCs, mainly driven by various growth factors; secondly, these MDSCs are activated to be fully functional, primarily promoted by pro-inflammatory factors. This two-signal model of expansion and activation may answer the question of why MDSCs are not generated under normal physiological settings or during acute inflammation. In steady state, growth factors stimulate normal hematopoiesis without generating MDSCs due to the absence of pro-inflammatory factors, whereas during acute inflammation, in the absence of sustained growth factors, pro-inflammatory factors alone do not lead to MDSC generation either, since immature myeloid cells may rapidly differentiate into mature myeloid cells.
As discussed in the previous sections, many of the tumor-derived mediators actively regulate the suppressive function of MDSCs. In a murine model of tissue-specific inflammatory response, MDSCs from inflammatory or tumor site are more suppressive than MDSCs from spleen, and splenic MDSCs from inflamed mice are more suppressive than splenic MDSCs from naïve mice (148). Further study from the same group has shown that MDSCs exposed to IFN-γ, IL-13, and GM-CSF in vitro or MDSCs localized in inflammatory or tumor site in vivo have elevated L-arginine transporter cationic amino acid transporter 2 expression, which parallels the expression of ARG1 and iNOS and is required for optimal suppressive activity of MDSCs (146). These findings suggest a priming effect of tumor-derived pro-inflammatory cytokines. In a more recent study, tumor cells upregulate tumor necrosis factor-α-induced protein 8-like 2 (TIPE2) in MDSCs through ROS, which in turn controls the polarization of MDSCs by increasing pro-tumoral and inhibiting anti-tumoral mediator expression (152).
Several pro-inflammatory factors are reported to enhance the suppressive potency of MDSCs. For instance, PGE2 generated by COX2 in tumor cells upregulates ARG1 expression of MDSCs through the EP4 receptor (153). PGE2 promotes hypermethylation and repression of a cluster of myeloid genes, which is in contrast to the profile from DCs generated in vitro or CD11b+ cells from healthy controls. This MDSC-specific gain of methylation requires the upregulation of DNA methyltransferase 3A, while its downregulation abolishes the immunosuppressive properties of MDSCs (154). It another study, PGE2 potentiates the suppressive function of human M-MDSCs induced by GM-CSF/IL-6 from peripheral blood mononuclear cells (155). However, whether these actions of PGE2 occur in vivo remains to be determined. IL-17 not only enhances tumor-infiltrating MDSCs, probably by increasing CXCL1 and CXCL5 secretion by tumor cells, but also potentiates their inhibition on T cells through upregulation of ARG1 and IDO (156). The calcium-binding pro-inflammatory proteins S100A8 and S100A9, which are ubiquitously present in the tumor microenvironment, drive the accumulation of MDSCs through increased recruitment to primary tumor and pre-metastatic niche (150). It was recently reported that S100A8 enhances T-cell suppression by MDSCs (157) and that S100A9 induces IL-6 and IL-10 release by MDSCs (158). Furthermore, MDSCs also express and secret S100A8/A9, thus forming a positive feedback loop that helps to maintain suppressive MDSCs in the tumor microenvironment (150).
Both type I and II interferons upregulate PD-L1 expression in MDSCs. It is well-documented that IFN-γ functions as a master regulator of PD-L1 expression in tumor. IFN-γ neutralization reduces tumor-infiltrating PD-L1+ MDSCs in vivo, and mechanistically, IFN-γ upregulates IRF1, which in turn binds to IRF-binding sequence in cd274 promoter and activates PD-L1 expression (72). The IFN-γ level in the tumor microenvironment may be reduced due to MDSC-mediated suppression of T cells and NK cells, which are important sources of IFN-γ. As a compensatory mechanism, MDSCs may maintain their PD-L1 expression by secreting IFN-α and IFN-β, which bind to IFN receptor type I and upregulate PD-L1 in an autocrine manner (159).
It is noteworthy to point out that many of the tumor-derived mediators influence more than one aspect of MDSCs. For instance, in addition to promoting expansion, GM-CSF alone is able to promote immunosuppression by MDSCs (160). GM-CSF increases IL-4Rα expression on MDSCs, which leads to IL-13-induced ARG1 upregulation (161), and GM-CSF drives PD-L1 and IDO expression of MDSCs through STAT3 activation (69, 80). Tumor-derived migration inhibitory factor has been reported to promote the differentiation, recruitment, and suppressive activity of MDSCs (162, 163). These pleiotropic and redundant effects further complicate the regulatory network of MDSC development.
Tumor-Derived Exosomes
Exosomes are small extracellular vesicles released by nearly all cells and are present in most body fluids. These membrane-bound vesicles contain proteins, DNA, mRNA, and miRNA and act as intercellular messengers (164). Tumor constantly produces and secrets exosomes. Upon contact with target cells, tumor derived-exosomes are able to alter the phenotypic and functional characters of the recipients, reprogramming them into participants in tumor progression. In the early phase of tumor growth, exosomes derived from immune cells in the tumor microenvironment may facilitate anti-tumor response, while in more advanced disease, tumor derived-exosomes promote immune suppression by interfering with the differentiation, maturation, and anti-tumor activity of immune cells (164). Several recent studies have shown that MDSCs also produce exosomes, whose contents are implicated in their own chemotaxis, survival, pro-metastatic, and immunosuppressive activity (165).
Studies have shown that tumor-derived exosomes promote the expansion of MDSCs. Administration of tumor-derived exosomes to healthy mice leads to increased frequency of immature myeloid cells that acquire the phenotypic and functional characters of MDSCs (166). Tumor derived-exosomes induce accumulation of splenic and intratumoral MDSCs that are able to promote tumor growth, which is dependent on exosomal PGE2 and TGF-β (167). In multiple myeloma, exosomes derived from both tumor cells and stromal cells expand MDSCs (168, 169). In addition, tumor derived-exosomes may contribute to metastasis by inducing accumulation of MDSCs, PMN-MDSCs in particular, in the pre-metastatic niche (170, 171).
Many of the suppressive machineries of MDSCs can be potentiated by tumor derived-exosomes, including expression of ARG1 and iNOS, and production of IL-10 and VEGF (48, 167, 169, 172). The suppressive capacity of MDSCs on T cells is accordingly heightened (48, 169). STAT3 is implicated in this exosomal regulation on MDSCs (169). Chalmin et al. have shown that HSP72 expressed on tumor derived-exosomes induces suppressive activity of MDSCs, which activates STAT3 in a TLR2/MyD88-dependent manner through autocrine production of IL-6 (142). Similarly, in another study, MDSCs were expanded and activated by exosomal HSP70, which induced phosphorylation of STAT3 through the TLR2/MyD88 pathway (172). In consistence with these findings, T-cell proliferation is inhibited by MDSCs isolated from mice treated with tumor derived-exosomes but not by MDSCs isolated from MyD88 knockout mice treated with tumor derived-exosomes (171). Furthermore, stromal cell-derived exosomes are also reported to enhance T-cell suppression by MDSCs, probably through the STAT3 pathway as well (168).
Tumor-derived exosomes are able to mediate RNA transfer from tumor cells to recipient cells. Ridder and colleagues have shown that MDSCs are the major recombined cells in the tumor microenvironment after the uptake of exosomes and their RNA content and that MDSCs recombined with exosomal RNA display enhanced ARG1, TGF-β, and PD-L1 expression as compared to the non-recombined counterparts (173). In a recent study, hypoxia increases exosome secretion by glioma cells. Moreover, hypoxia upregulates miR-10a and miR-21 in glioma-derived exosomes, which in turn potentiates the suppressive function of MDSCs (174).
Metabolic Reprogramming of MDSCs
Along with disease progression, malignant cells undergo dramatic alteration in their energy metabolism to meet the demand for rapid tumor growth and to adapt to the varying microenvironment. Meanwhile, it was recently demonstrated that tumor-associated immune cells also experience metabolic changes that help to shape their pro- and/or anti-tumor response (175). In this regard, metabolic reprogramming is emerging as a regulator of MDSCs. Using MSC-1 cells, an immortalized murine MDSC cell line, early in vitro studies have revealed two distinct bioenergetic states that coincide with the exponential and stationary growth phases of MSC-1 cells (176) and that their maturation and suppressive potential are accompanied by an increase in the central carbon metabolism activity (177).
MDSCs exhibit a high glycolytic rate (175). The enhanced glycolysis of MDSCs helps to keep their ROS level within a safe range and promotes their survival and accumulation in tumor-bearing host (178). mTOR-mediated HIF-1α induction is essential in glycolytic activation (175). Inhibiting the mTOR pathway blocks the differentiation of M-MDSCs from precursors by impairing glycolysis. Consistently, 2-deoxyglucose, which inhibits glycolysis, blocks the differentiation of M-MDSCs, while metformin, which promotes glycolysis, rescues the reduction in M-MDSCs caused by mTOR inhibition (179). On the other hand, glycolysis in tumor cells also contributes to the expansion of MDSCs, which is mediated by increased production of G-CSF, GM-CSF, and lactate (100, 180).
In addition to promoting expansion, glycolysis regulates the function of MDSCs. A recent study has found that enhanced glycolysis mediated by the mTOR pathway leads to stronger suppressive capacity of tumor-infiltrating M-MDSCs as compared with splenic M-MDSCs and that mTOR inhibition by rapamycin reduces the glycolysis, intratumoral level, and suppressive activity of M-MDSCs (181). Attenuated iNOS and ARG1 may be responsible for the impaired function caused by rapamycin-mediated glycolysis inhibition (179).
On the contrary, glycolysis as a negative regulator of MDSCs has also been reported. It is shown that mTOR- and HIF-1α-induced glycolytic activation is required for differentiation of MDSCs to a less suppressive M1 phenotype (182). In the settings of transplantation and autoimmune disorder, dexamethasone expands MDSCs and strengthens their function. In a model of immunological hepatic injury, dexamethasone inhibits HIF-1α-dependent glycolysis in MDSCs and promotes their suppressive activity to protect against inflammatory injury (183). In addition, there are studies showing that mTOR inhibition by rapamycin potentiates the suppressive activity of MDSCs, which protects against acute graft-versus-host disease and acute kidney injury (184, 185); yet, unfortunately, the glycolytic or other metabolic characteristics of MDSCs were not determined in these studies. These seemingly conflicting results indicate the complexity and the possibly context-dependent manner in which glycolytic rate determines the function of MDSCs.
Recently, it is shown that tumor-infiltrating MDSCs have increased fatty acid oxidation (FAO), which is accompanied by upregulated ARG1, increased NO, and PNT production, and that FAO inhibition impairs the suppressive activity of MDSCs in vitro and in vivo (186). Only intratumoral MDSCs, and not splenic MDSCs, have increased FAO, suggesting that the microenvironment is responsible for this metabolic alteration (186). Consistently, a further study from the same group demonstrated that tumor-derived cytokines, such as G-CSF and GM-CSF, induce the expression of lipid transport receptors in intratumoral MDSCs through the activation of STAT3 and STAT5, which leads to increased uptake of lipids that are present at high concentrations in the tumor microenvironment; intracellular accumulation of lipids in turn increases the oxidative metabolism and suppressive activity of MDSCs (187).
Hypoxia and HIF-1α
Hypoxia caused by excessive oxygen consumption by tumor cells and aberrant organization of tumor vasculature is a common feature of the tumor microenvironment and plays a central role in tumor progression, primarily through HIF-dependent signalings. Multiple activities of MDSCs are regulated by hypoxia. For instance, hypoxia facilitates the recruitment of MDSCs to tumor site (188, 189). Intratumoral MDSCs preferentially localize in poorly perfused and hypoxic regions, and their pro-angiogenic capacity is generally enhanced by hypoxia (6). The homeostasis of tumor-infiltrating MDSCs is fine-tuned by the hypoxic microenvironment, since hypoxia promotes the differentiation of intratumoral MDSCs to TAMs (190), while it is also reported that hypoxia promotes the maintenance of MDSCs by upregulating ectonucleoside triphosphate diphosphohydrolas 2 in tumor cells, which forms a 5′-AMP-rich microenvironment and prevents differentiation of MDSCs (191).
It is now generally accepted that microenvironmental hypoxia directly augments the suppressive function of MDSCs (1). In a tumor model with similar PMN-MDSC to M-MDSC ratios in spleen and tumor site, Corzo et al. found that the inhibition on T cells is antigen-specific by splenic MDSCs, which display higher ROS production, while it is both antigen-specific and non-specific by tumor-infiltrating MDSCs, which exhibit upregulated ARG1 and iNOS. Exposure of splenic MDSCs to hypoxia leads to non-specific T-cell suppression, suggesting that the hypoxic microenvironment may convert MDSCs into non-specific suppressors. This conversion is mediated by HIF-1α (190). A similar difference in suppressive mechanisms and antigen specificity is detected in MDSCs obtained from peripheral blood and tumor tissue of patients with head and neck cancer (190). Similarly, it was recently reported that HIF-1α potentiates the immunosuppressive activity of splenic MDSCs in a murine model of chronic Leishmania infection (192).
Noman et al. have shown that the PD-L1 level is higher on intratumoral MDSCs than on splenic MDSCs and that hypoxic stress upregulates PD-L1 on splenic MDSCs through HIF-1α. More importantly, hypoxia potentiates the ability of splenic MDSCs to suppress both specific and non-specific stimuli-mediated T-cell proliferation, while PD-L1 blockade abrogates the enhanced suppression under hypoxia, in part by decreasing the production of suppressive cytokines, particularly IL-6 and IL-10, in hypoxic MDSCs (43). The authors have also found that hypoxia increases the secretion of IL-6, IL10, and TGF-β from MDSCs (43). In another study from the same group, tumor-infiltrating MDSCs expressed an increased level of miR-210 as compared with splenic MDSCs, and hypoxia induced miR-210 in splenic MDSCs via HIF-1α. MiR-210 in turn enhanced the suppressive capacity of splenic MDSCs by increasing their ARG1 activity and NO production without affecting ROS, IL-6, or IL-10 production or PD-L1 expression (67). In a more recent study, HIF-1α acted as a transcriptional activator of VISTA, a negative checkpoint regulator in the B7 family, in MDSCs and consistently, antibody blockade or genetic ablation of VISTA abolished MDSC-mediated suppression of T cells under hypoxic but not normoxic conditions (193). These findings suggest that hypoxia regulates MDSC-mediated suppression through multiple pathways.
MDSCs actively participate in tumor metastasis by inducing EMT, increasing the invasiveness and stemness of tumor cells, and stimulating angiogenesis (5, 6). Unfortunately, the precise roles of hypoxia and hypoxic signalings in these MDSC-driven steps of metastatic cascade are not well-defined. On the other hand, MDSCs actively participate in pre-metastatic niche formation. MDSCs, especially the granulocytic subset, reach the pre-metastatic site prior to the arrival of disseminated tumor cells, which is regulated indirectly by hypoxia in the primary tumor. In a murine mammary tumor model, tumor that grows in pre-irradiated mammary tissue has decreased vascular density and is more hypoxic and metastatic, recapitulating the clinical features of locally relapsed breast cancer after radiation therapy; HIF-1-dependent Kit ligand expression by hypoxic tumor cells mobilizes c-Kit+ PMN-MDSCs to home to pre-metastatic lungs to promote metastasis (194). In other studies, PMN-MDSCs are recruited by MCP-1 or G-CSF derived from hypoxic tumor cells to pre-metastatic lungs, where they may inhibit the cytotoxicity of NK cells (195, 196).
Endoplasmic Reticulum Stress and Unfolded Protein Response
In homeostatic settings, the endoplasmic reticulum (ER) readily handles the folding of secretory and transmembrane proteins. The hostile conditions in the tumor milieu, such as hypoxia, oxidative stress, nutrient deprivation, and acidic waste accumulation, impair the protein-folding capacity of ER, thus provoking a cellular state of ER stress. When the misfolded proteins exceed a tolerable level, PKR-like ER-resident kinase (PERK), inositol-requiring enzyme 1α (IRE1α), and activating transcription factor 6α (ATF6α) detect the presence of ER stress and trigger unfolded protein response (UPR) to improve the folding efficiency in ER (197). These ER-localized sensors are held inactive by chaperone BiP in steady state, while upon ER stress, the dissociation of BiP activates all three sensors: PERK phosphorylates the translation initiation factor eIF2α, which restricts cap-dependent translation and in turn upregulates activating transcription factor 4 (ATF4) and its downstream target C/EBP homologous protein (CHOP); IRE1α cleaves the X-box-binding protein 1 (XBP1) mRNA, and the spliced mRNA is re-ligated to produce highly active XBP1s that regulates gene expression involved in protein folding; ATF6α fine-tunes UPR by regulating the transcription of ER chaperone genes (197).
Unresolvable ER stress often leads to cell death, while tolerable defect in protein-folding capacity may fuel tumor cell survival, metastasis, angiogenesis, and therapeutic resistance. The immunosuppressive effect of ER stress is receiving growing attention (197). Mahadevan et al. have shown that stressed tumor cells actively regulate the function of myeloid cells. For instance, tumor cells undergoing ER stress release yet unidentified soluble mediators that lead to upregulated UPR markers and pro-inflammatory cytokines in responder macrophages (198). This transmissible ER stress also imprints bone marrow-derived DCs with increased ARG1 and decreased ability to cross-present antigen to CD8+ T cells (199). On the other hand, intrinsic ER stress regulates the myeloid cell activity as well. STAT3 synergizes with STAT6 in macrophages to promote cathepsin secretion and tumor invasion through the IRE1α pathway (200). ER stress and XBP1 activation in tumor-infiltrating DCs lead to abnormal lipid accumulation, which impairs their antigen presentation capacity (201).
In line with macrophages and DCs, MDSCs exhibit clear signs of ER stress and UPR. MDSCs isolated from tumor-bearing host have a higher level of ER stress response markers as compared with monocytes and neutrophils from the same host or healthy control (202). Furthermore, the CHOP level in tumor-infiltrating MDSCs is higher than in splenic MDSCs or other tumor-infiltrating immune cells (203).
Recent studies have demonstrated that ER stress response regulates the homeostasis and suppressive function of MDSCs. ER stress induces apoptosis of MDSCs through upregulation of TRAIL-R or through the eIF2α-ATF4-CHOP pathway; though the lifespan of MDSCs is shortened by ER stress, it may stimulate myelopoiesis and the turnover of MDSCs in tumor-bearing host (202, 203). Administration of the ER stress inducer thapsigargin promotes infiltration of MDSCs in tumor and enhances their suppressive capacity through upregulation of ARG1, iNOS, and NOX2 (204).
Thevenot et al. have elaborately shown that the suppressive activity of MDSCs is regulated by ER stress response marker CHOP (203). In CHOP-deficient mice, tumor growth is significantly retarded, while it is partially restored by depletion of MDSCs, suggesting a reversal of the tumor-promoting activity of MDSCs. Consistently, functional assessment of tumor-infiltrating CHOP−/− MDSCs reveals reduced suppression of T cells, which is associated with decreased ARG1, superoxide, and PNT; furthermore, these CHOP−/− MDSCs acquire a DC-like phenotype and are able to stimulate immune response.
In another study, ER stress-related genes were found to be among the most upregulated in PMN-MDSCs, as compared with neutrophils from the same cancer patient or a healthy individual (205). Surface expression of lectin-type oxidized LDL receptor-1 (LOX-1), which is regulated by ER stress, effectively distinguishes immunosuppressive PMN-MDSCs from neutrophils in cancer patients (205). ER stress induced by thapsigargin promotes LOX-1 upregulation in human neutrophils and converts them into immunosuppressive cells, which is prevented by inhibiting the IRE1α-XBP1s pathway (205, 206). However, whether downstream signaling through LOX-1 is responsible for the acquisition of suppressive activity by neutrophils remains undetermined.
Crosstalk Between MDSCs and Stromal Cells in the Tumor Microenvironment
Many of the tumor-promoting roles of MDSCs, such as immune suppression, pro-angiogenesis, and pro-metastasis, are regulated by the surrounding cells in the tumor microenvironment. How tumor cells regulate the immunosuppressive function of MDSCs has been discussed in previous sections. The induction and suppressive capacity of MDSCs are also fine-tuned during the dynamic and mutualistic communication with the non-malignant stromal cells in the tumor milieu. Many of these cells are not merely targets but also regulators of MDSCs.
MDSCs primarily inhibit T-cell response, and on the other way round, T cells influence the suppressive nature of MDSCs. The antigen-specific CD4+ T cells, and not CD8+ T cells, enhance the immunosuppressive capacity of MDSCs by turning them into non-specific suppressors in vitro and in vivo. Mechanistically, this effect requires cross-linking of MHC class II on MDSCs during cell-to-cell contact with activated CD4+ T cells, and the subsequent retrograde signaling in MDSCs upregulates COX2 and PGE2 expression (141). In a recent study, IFN-γ produced by T cells was found to be critical in regulating the enhanced suppressive activity of MDSCs induced by TLR2 ligand, which promoted differentiation of MDSCs into iNOS+ macrophages (207).
In addition to immunosuppression, T cells regulate the induction of MDSCs. It has been reported that FasL+-activated T cells may regulate the homeostasis of MDSCs through Fas-FasL interaction, which induces apoptosis of MDSCs (208). In human colorectal cancer, γδT cells promote the recruitment, proliferation, and survival of PMN-MDSCs through secretion of large amounts of IL-17 and other cytokines, including IL-8, GM-CSF, and TNF-α (209). It has been shown in different murine tumor models that TNF-α secreted by CD4+ T cells, and partially by CD8+ T cells, induces myelopoiesis, which increases the frequency of MDSCs (210).
PD-L1 expression on MDSCs is upregulated upon co-culture with T cells (79), and MDSCs are able to induce PD-1 expression on T cells through TGF-β (75, 211). In melanoma-bearing mice receiving IL-2- and TNF-α-coding adenovirus in combination with adoptive T-cell therapy, PD-L1 was upregulated in intratumoral MDSCs, and the frequency of PD-1+ CD8+ T cells correlated with the PD-L1 expression level on MDSCs in tumor site (70).
Not only do MDSCs promote Treg induction and recruitment: their suppressive function is also modified by Tregs. An earlier study reported that CD80 expression is required for MDSC-mediated antigen-specific T-cell suppression, which is dependent on CD4+CD25+ Tregs and CTLA-4 and that depletion of CD4+CD25+ Tregs diminishes the suppression mediated by MDSCs (212). In a more recent study, Treg depletion decreased PD-L1 expression and IL-10 production by MDSCs (73). In a murine model of melanoma, the expansion, recruitment, and activation of MDSCs occurred in a Treg-dependent manner and required the expression of IDO (213). Therefore, it is likely that MDSCs and Tregs do not act separately but rather cooperate reciprocally in immune suppression.
Crosstalk between MDSCs and B cells has been found recently. In one study, MDSCs that accumulated around the germinal center co-localized with B cells in the spleen of tumor-bearing mice, and cell-to-cell interaction through TNFR2 on MDSCs and membranous TNF on B cells promoted the proliferation and differentiation of B cells into IgA-producing plasma cells (214). Both IL-10 and TGF-β are crucial for this MDSC-mediated IgA response. In another study, Bregs from tumor-bearing mice increased the immunosuppressive and pro-metastatic function of MDSCs, partially through the TGF-β type I/II receptor signaling axis (215).
IL-10 is implicated in the interaction between MDSCs and other immune cells. Through cell-to-cell contact, MDSCs produce IL-10 to downregulate IL-12 by macrophages, and macrophages in turn stimulate IL-10 upregulation by MDSCs (216). The increased IL-10 level and reduced IL-12 level consequently skew the immunity toward a tumor-promoting type 2 response. In another recent study, MDSC-derived IL-10 decreased IL-6 and TNF-α while increasing NO produced by macrophages (217). Meanwhile, IL-10 produced by MDSCs may reduce MHC class II molecule expression on macrophages, leading to diminished antigen-presentation capacity (218). This bi-directional crosstalk between MDSCs and macrophages is accentuated by the inflammatory microenvironment. MDSCs isolated from tumor with a heightened IL-1β level produce more IL-10 and downregulate IL-12 by macrophages to a greater degree as compared with MDSCs from less inflammatory tumors (38). This IL-10 elevation by MDSCs requires IL-6 from macrophages and signaling through TLR4 on MDSCs and macrophages (38, 218). This action of inflammation is further corroborated by the findings that pro-inflammatory mediators PGE2 and HMGB1 upregulate IL-10 in MDSCs in the presence of macrophages (21, 218).
It is reported that mast cells not only induce the recruitment but also promote the suppressive function of MDSCs, probably through CD40L-CD40 interaction (219, 220).
Cancer-associated fibroblasts (CAFs) are a heterogeneous group of activated fibroblasts that play pleiotropic roles in tumor development and are able to modulate anti-tumor immunity on various levels. Through secretion of CCL2 and CXCL12, CAFs facilitate the recruitment of MDSCs (221). Meanwhile, pancreatic CAFs produce multiple MDSC-promoting soluble mediators, IL-6 in particular, and favor the differentiation of MDSCs (222). CAFs from hepatic cancer attract monocytes to the tumor microenvironment by CXCL12 and induce their differentiation into MDSCs through IL-6-mediated STAT3 activation (223). The MDSC-promoting effect of CAFs in breast cancer involves epigenetic regulation by histone deacetylase 6 (224). Consistently, inhibiting CAFs leads to reduced in vivo induction and intratumoral level of MDSCs (225, 226). On the other hand, MDSCs promote activation and migration of CAFs, suggesting a positive feedback loop that amplifies interaction between them. To further complicate the issue, in recent studies, CAFs show similar phenotypic and immunosuppressive characteristics to the circulating fibrocytes that may arise from MDSCs and may represent a novel MDSC subset (227).
Conclusions and Perspectives
Among the multiple tumor-promoting characteristics of MDSCs, the capacity to suppress T-cell response remains a key hallmark. Given the complexity of the tumor immune microenvironment, it is not surprising that MDSCs are more than a T-cell suppressor and that their function is regulated on multiple levels. With the advances in phenotyping and functional assessment in recent years, a clearer view of the immunosuppressive nature of MDSCs has been achieved. Firstly, several novel suppressive mechanisms have been identified, which makes MDSCs a versatile suppressor. Secondly, the antigenicity of MDSC-mediated T-cell inhibition depends largely on the properties of the effector molecules utilized, since a different level and duration of intercellular contact is required; furthermore, differential suppressive potency and preferential mechanisms between subsets of MDSCs compartmentalize T-cell suppression in tumor-bearing host: immunosuppression is relatively weak and is antigen-specific in the periphery, while it is strong and is both antigen-specific and non-specific in the tumor milieu. Thirdly, the recognized targets of MDSCs have been extended from T cells to other immune cells, such as NK cells, DCs, and B cells, which broadens their suppressive spectrum and makes them suppressive in both innate and adaptive immunity. Lastly, in addition to clarification of their expansion and activation in the presence of tumor, the development and function of MDSCs are fine-tuned by several microenvironmental factors. With these characteristics unraveled, a pivotal role of MDSCs in the intricate network of immune suppression within the tumor microenvironment has been unveiled.
As a competent accomplice in carcinogenesis and disease progression, the correlation between MDSCs and tumor burden and disease stage is well-documented. For instance, a recent meta-analysis has shown for the first time that a high level of MDSCs is associated with shorter survival outcomes in patients with solid tumors and hematologic malignancies (228). This notion has two therapeutic implications. On the one hand, MDSCs have been regarded as an attractive target in cancer therapy. Various pre-clinical and clinical studies have shown promising benefits by targeting MDSCs, which can be achieved by depleting their quantity, blocking their trafficking, or inhibiting their immunosuppressive activity (5). On the other hand, due to their potent immunosuppressive capacity, MDSCs act as a major obstacle to natural anti-tumor immunity, hinder the efficacy of immunotherapy, and constitute an important mechanism for resistance. Accordingly, a high level of MDSCs predicts poor response to immune checkpoint inhibitor ipilimumab in metastatic melanoma patients (66). More importantly, it is rational to target MDSCs in combination with immunotherapy, which may yield a synergistic effect and delay, or even reverse, the occurrence of resistance (66). For instance, as compared to monotherapy, the efficacy of an immune checkpoint inhibitor or cancer vaccine is enhanced by combining with MDSC-targeted therapy in pre-clinical studies and clinical trials (66), and T cell-based immunotherapy efficacy is enhanced by inhibiting the trafficking of MDSCs (229). These benefits are associated with improved T cell-mediated immune response against tumor or increased antigen presentation capacity of DCs, probably due to the relieved inhibition imposed by MDSCs.
However, approaches to combat with MDSCs are still in their infancy, and there are several conundrums to be addressed. Considering their versatility and the complexity of microenvironmental regulation, the suppressive machineries of MDSCs are not likely to act simultaneously, but most probably function in a context-dependent manner. As a consequence, when we target the suppressive function MDSCs, it would be difficult to identify the most relevant target(s). Meanwhile, taking into account the indispensability of myelopoiesis in physiological processes and the phenotypic similarity between MDSCs and normal myeloid cells, it would be challenging to target MDSCs accurately without affecting the normal myeloid compartment.
Since they were firstly reported in the late 1970s and consensus on their nomenclature was reached in 2007, MDSCs as a group of suppressive myeloid cells have received increasing attention, and research on MDSCs is booming. Their roles in malignant and non-malignant settings are becoming clarified. With the effort in the past decade, an algorithm that includes phenotypic and functional, and, if possible, molecular criteria necessary to identify MDSCs was proposed in 2016 (4). This step-by-step approach aims to minimize ambiguity and helps to dissect the function of MDSCs in future studies. For instance, it may help us to better distinguish MDSCs from normal myeloid cells in the same host. Determining how to target MDSCs more precisely and efficiently relies, hopefully, on a better understanding of the development and suppressive nature of MDSCs. Lastly, more clinical trials are needed to validate the synergistic effect of MDSC-targeted therapy and cancer immunotherapy.
Author Contributions
YY conceptualized the study, wrote the manuscript, and approved the final draft for publication. YY and CL prepared the figures. TL, XD, and AB reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China under grant numbers 81402553 to YY, 81801760 to CL, and 81602696 to TL.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. (2016) 37:208–20. doi: 10.1016/j.it.2016.01.004
2. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. (2007) 67:425; author reply 6. doi: 10.1158/0008-5472.CAN-06-3037
3. Nagaraj S, Gabrilovich DI. Regulation of suppressive function of myeloid-derived suppressor cells by CD4+ T cells. Semin Cancer Biol. (2012) 22:282–8. doi: 10.1016/j.semcancer.2012.01.010
4. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
5. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front Immunol. (2018) 9:398. doi: 10.3389/fimmu.2018.00398
6. Li C, Liu T, Bazhin AV, Yang Y. The sabotaging role of myeloid cells in anti-angiogenic therapy: coordination of angiogenesis and immune suppression by hypoxia. J Cell Physiol. (2017) 232:2312–22. doi: 10.1002/jcp.25726
7. Karakhanova S, Link J, Heinrich M, Shevchenko I, Yang Y, Hassenpflug M, et al. Characterization of myeloid leukocytes and soluble mediators in pancreatic cancer: importance of myeloid-derived suppressor cells. Oncoimmunology. (2015) 4:e998519. doi: 10.1080/2162402X.2014.998519
8. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. (2008) 181:5791–802. doi: 10.4049/jimmunol.181.8.5791
9. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. (2008) 111:4233–44. doi: 10.1182/blood-2007-07-099226
10. Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer. (2014) 134:2853–64. doi: 10.1002/ijc.28622
11. Markowitz J, Wang J, Vangundy Z, You J, Yildiz V, Yu L, et al. Nitric oxide mediated inhibition of antigen presentation from DCs to CD4(+) T cells in cancer and measurement of STAT1 nitration. Sci Rep. (2017) 7:15424. doi: 10.1038/s41598-017-14970-0
12. Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol. (1998) 160:5729–34.
13. Wang Z, Jiang J, Li Z, Zhang J, Wang H, Qin Z. A myeloid cell population induced by Freund adjuvant suppresses T-cell-mediated antitumor immunity. J Immunother. (2010) 33:167–77. doi: 10.1097/CJI.0b013e3181bed2ba
14. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. (2004) 172:989–99. doi: 10.4049/jimmunol.172.2.989
15. Yang Y, Bazhin AV, Werner J, Karakhanova S. Reactive oxygen species in the immune system. Int Rev Immunol. (2013) 32:249–70. doi: 10.3109/08830185.2012.755176
16. Bronte V, Kasic T, Gri G, Gallana K, Borsellino G, Marigo I, et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med. (2005) 201:1257–68. doi: 10.1084/jem.20042028
17. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. (2007) 13:828–35. doi: 10.1038/nm1609
18. Feng S, Cheng X, Zhang L, Lu X, Chaudhary S, Teng R, et al. Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proc Natl Acad Sci USA. (2018) 115:10094–9. doi: 10.1073/pnas.1800695115
19. Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. (2012) 72:876–86. doi: 10.1158/0008-5472.CAN-11-1792
20. Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S. Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J Immunol. (2009) 183:937–44. doi: 10.4049/jimmunol.0804253
21. Parker KH, Sinha P, Horn LA, Clements VK, Yang H, Li J, et al. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res. (2014) 74:5723–33. doi: 10.1158/0008-5472.CAN-13-2347
22. Gehad AE, Lichtman MK, Schmults CD, Teague JE, Calarese AW, Jiang Y, et al. Nitric oxide-producing myeloid-derived suppressor cells inhibit vascular E-selectin expression in human squamous cell carcinomas. J Invest Dermatol. (2012) 132:2642–51. doi: 10.1038/jid.2012.190
23. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. (2011) 208:1949–62. doi: 10.1084/jem.20101956
24. Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. (2008) 222:180–91. doi: 10.1111/j.1600-065X.2008.00608.x
25. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. (2010) 70:68–77. doi: 10.1158/0008-5472.CAN-09-2587
26. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. (2013) 190:3783–97. doi: 10.4049/jimmunol.1201449
27. Adam T, Mathes A, Isayev O, Philippov PP, Werner J, Karakhanova S, et al. In vivo immunological effects of CD73 deficiency. Cell Physiol Biochem. (2019) 52:1192–202. doi: 10.33594/000000081
28. Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol. (2011) 187:6120–9. doi: 10.4049/jimmunol.1101225
29. Limagne E, Euvrard R, Thibaudin M, Rebe C, Derangere V, Chevriaux A, et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res. (2016) 76:5241–52. doi: 10.1158/0008-5472.CAN-15-3164
30. Li L, Wang L, Li J, Fan Z, Yang L, Zhang Z, et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. (2018) 78:1779–91. doi: 10.1158/0008-5472.CAN-17-2460
31. Ryzhov SV, Pickup MW, Chytil A, Gorska AE, Zhang Q, Owens P, et al. Role of TGF-beta signaling in generation of CD39+CD73+ myeloid cells in tumors. J Immunol. (2014) 193:3155–64. doi: 10.4049/jimmunol.1400578
32. Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-beta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology. (2017) 6:e1320011. doi: 10.1080/2162402X.2017.1320011
33. Umansky V, Shevchenko I, Bazhin AV, Utikal J. Extracellular adenosine metabolism in immune cells in melanoma. Cancer Immunol Immunother. (2014) 63:1073–80. doi: 10.1007/s00262-014-1553-8
34. Ye C, Geng Z, Dominguez D, Chen S, Fan J, Qin L, et al. Targeting ornithine decarboxylase by alpha-difluoromethylornithine inhibits tumor growth by impairing myeloid-derived suppressor cells. J Immunol. (2016) 196:915–23. doi: 10.4049/jimmunol.1500729
35. Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Blockade of A2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia. (2013) 15:1400–9. doi: 10.1593/neo.131748
36. Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. (2014) 74:7250–9. doi: 10.1158/0008-5472.CAN-13-3583
37. Hart KM, Byrne KT, Molloy MJ, Usherwood EM, Berwin B. IL-10 immunomodulation of myeloid cells regulates a murine model of ovarian cancer. Front Immunol. (2011) 2:29. doi: 10.3389/fimmu.2011.00029
38. Bunt SK, Clements VK, Hanson EM, Sinha P, Ostrand-Rosenberg S. Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J Leukoc Biol. (2009) 85:996–1004. doi: 10.1189/jlb.0708446
39. Sato Y, Shimizu K, Shinga J, Hidaka M, Kawano F, Kakimi K, et al. Characterization of the myeloid-derived suppressor cell subset regulated by NK cells in malignant lymphoma. Oncoimmunology. (2015) 4:e995541. doi: 10.1080/2162402X.2014.995541
40. Zhang G, Huang H, Zhu Y, Yu G, Gao X, Xu Y, et al. A novel subset of B7-H3(+)CD14(+)HLA-DR(-/low) myeloid-derived suppressor cells are associated with progression of human NSCLC. Oncoimmunology. (2015) 4:e977164. doi: 10.4161/2162402X.2014.977164
41. Arihara F, Mizukoshi E, Kitahara M, Takata Y, Arai K, Yamashita T, et al. Increase in CD14+HLA-DR -/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol Immunother. (2013) 62:1421–30. doi: 10.1007/s00262-013-1447-1
42. Azzaoui I, Uhel F, Rossille D, Pangault C, Dulong J, Le Priol J, et al. T-cell defect in diffuse large B-cell lymphomas involves expansion of myeloid-derived suppressor cells. Blood. (2016) 128:1081–92. doi: 10.1182/blood-2015-08-662783
43. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
44. Koike Y, Kanai T, Saeki K, Nakamura Y, Nakano M, Mikami Y, et al. MyD88-dependent interleukin-10 production from regulatory CD11b(+)Gr-1(high) cells suppresses development of acute cerulein pancreatitis in mice. Immunol Lett. (2012) 148:172–7. doi: 10.1016/j.imlet.2012.08.008
45. Hu X, Li B, Li X, Zhao X, Wan L, Lin G, et al. Transmembrane TNF-alpha promotes suppressive activities of myeloid-derived suppressor cells via TNFR2. J Immunol. (2014) 192:1320–31. doi: 10.4049/jimmunol.1203195
46. Nam S, Kang K, Cha JS, Kim JW, Lee HG, Kim Y, et al. Interferon regulatory factor 4 (IRF4) controls myeloid-derived suppressor cell (MDSC) differentiation and function. J Leukoc Biol. (2016) 100:1273–84. doi: 10.1189/jlb.1A0215-068RR
47. Younis RH, Han KL, Webb TJ. Human head and neck squamous cell carcinoma-associated semaphorin 4D induces expansion of myeloid-derived suppressor cells. J Immunol. (2016) 196:1419–29. doi: 10.4049/jimmunol.1501293
48. Domenis R, Cesselli D, Toffoletto B, Bourkoula E, Caponnetto F, Manini I, et al. Systemic T Cells immunosuppression of glioma stem cell-derived exosomes is mediated by monocytic myeloid-derived suppressor cells. PLoS ONE. (2017) 12:e0169932. doi: 10.1371/journal.pone.0169932
49. Qian L, Liu Y, Wang S, Gong W, Jia X, Liu L, et al. NKG2D ligand RAE1epsilon induces generation and enhances the inhibitor function of myeloid-derived suppressor cells in mice. J Cell Mol Med. (2017) 21:2046–54. doi: 10.1111/jcmm.13124
50. Huang A, Zhang B, Yan W, Wang B, Wei H, Zhang F, et al. Myeloid-derived suppressor cells regulate immune response in patients with chronic hepatitis B virus infection through PD-1-induced IL-10. J Immunol. (2014) 193:5461–9. doi: 10.4049/jimmunol.1400849
51. Ren JP, Zhao J, Dai J, Griffin JW, Wang L, Wu XY, et al. Hepatitis C virus-induced myeloid-derived suppressor cells regulate T-cell differentiation and function via the signal transducer and activator of transcription 3 pathway. Immunology. (2016) 148:377–86. doi: 10.1111/imm.12616
52. Brudecki L, Ferguson DA, McCall CE, El Gazzar M. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect Immun. (2012) 80:2026–34. doi: 10.1128/IAI.00239-12
53. Chikamatsu K, Sakakura K, Toyoda M, Takahashi K, Yamamoto T, Masuyama K. Immunosuppressive activity of CD14+ HLA-DR- cells in squamous cell carcinoma of the head and neck. Cancer Sci. (2012) 103:976–83. doi: 10.1111/j.1349-7006.2012.02248.x
54. Rastad JL, Green WR. Myeloid-derived suppressor cells in murine AIDS inhibit B-cell responses in part via soluble mediators including reactive oxygen and nitrogen species, and TGF-beta. Virology. (2016) 499:9–22. doi: 10.1016/j.virol.2016.08.031
55. Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med. (2003) 198:1741–52. doi: 10.1084/jem.20022227
56. Markiewski MM, Vadrevu SK, Sharma SK, Chintala NK, Ghouse S, Cho JH, et al. The ribosomal protein s19 suppresses antitumor immune responses via the complement C5a receptor 1. J Immunol. (2017) 198:2989–99. doi: 10.4049/jimmunol.1602057
57. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. (2008) 135:234–43. doi: 10.1053/j.gastro.2008.03.020
58. Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol. (2007) 25:2546–53. doi: 10.1200/JCO.2006.08.5829
59. Song C, Yuan Y, Wang XM, Li D, Zhang GM, Huang B, et al. Passive transfer of tumour-derived MDSCs inhibits asthma-related airway inflammation. Scand J Immunol. (2014) 79:98–104. doi: 10.1111/sji.12140
60. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res. (2014) 20:4096–106. doi: 10.1158/1078-0432.CCR-14-0635
61. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
62. Zhang H, Li Z, Wang L, Tian G, Tian J, Yang Z, et al. Critical role of myeloid-derived suppressor cells in tumor-induced liver immune suppression through inhibition of NKT cell function. Front Immunol. (2017) 8:129. doi: 10.3389/fimmu.2017.00129
63. Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong WC, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. (2011) 9:e1001162. doi: 10.1371/journal.pbio.1001162
64. Li Z, Pang Y, Gara SK, Achyut BR, Heger C, Goldsmith PK, et al. Gr-1+CD11b+ cells are responsible for tumor promoting effect of TGF-beta in breast cancer progression. Int J Cancer. (2012) 131:2584–95. doi: 10.1002/ijc.27572
65. Pang Y, Gara SK, Achyut BR, Li Z, Yan HH, Day CP, et al. TGF-beta signaling in myeloid cells is required for tumor metastasis. Cancer Discov. (2013) 3:936–51. doi: 10.1158/2159-8290.CD-12-0527
66. Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol. (2018) 9:1310. doi: 10.3389/fimmu.2018.01310
67. Noman MZ, Janji B, Hu S, Wu JC, Martelli F, Bronte V, et al. Tumor-promoting effects of myeloid-derived suppressor cells are potentiated by hypoxia-induced expression of miR-210. Cancer Res. (2015) 75:3771–87. doi: 10.1158/0008-5472.CAN-15-0405
68. Fuse H, Tomihara K, Heshiki W, Yamazaki M, Akyu-Takei R, Tachinami H, et al. Enhanced expression of PD-L1 in oral squamous cell carcinoma-derived CD11b(+)Gr-1(+) cells and its contribution to immunosuppressive activity. Oral Oncol. (2016) 59:20–9. doi: 10.1016/j.oraloncology.2016.05.012
69. Thorn M, Guha P, Cunetta M, Espat NJ, Miller G, Junghans RP, et al. Tumor-associated GM-CSF overexpression induces immunoinhibitory molecules via STAT3 in myeloid-suppressor cells infiltrating liver metastases. Cancer Gene Ther. (2016) 23:188–98. doi: 10.1038/cgt.2016.19
70. Tahtinen S, Blattner C, Vaha-Koskela M, Saha D, Siurala M, Parviainen S, et al. T-cell therapy enabling adenoviruses coding for IL2 and TNFα induce systemic immunomodulation in mice with spontaneous melanoma. J Immunother. (2016) 39:343–54. doi: 10.1097/CJI.0000000000000144
71. Liu Y, Zeng B, Zhang Z, Zhang Y, Yang R. B7-H1 on myeloid-derived suppressor cells in immune suppression by a mouse model of ovarian cancer. Clin Immunol. (2008) 129:471–81. doi: 10.1016/j.clim.2008.07.030
72. Lu C, Redd PS, Lee JR, Savage N, Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology. (2016) 5:e1247135. doi: 10.1080/2162402X.2016.1247135
73. Fujimura T, Ring S, Umansky V, Mahnke K, Enk AH. Regulatory T cells stimulate B7-H1 expression in myeloid-derived suppressor cells in ret melanomas. J Invest Dermatol. (2012) 132:1239–46. doi: 10.1038/jid.2011.416
74. Gorgun G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, et al. Lenalidomide enhances immune checkpoint blockade-induced immune response in multiple myeloma. Clin Cancer Res. (2015) 21:4607–18. doi: 10.1158/1078-0432.CCR-15-0200
75. Dubinski D, Wolfer J, Hasselblatt M, Schneider-Hohendorf T, Bogdahn U, Stummer W, et al. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro Oncol. (2016) 18:807–18. doi: 10.1093/neuonc/nov280
76. Iwata T, Kondo Y, Kimura O, Morosawa T, Fujisaka Y, Umetsu T, et al. PD-L1(+)MDSCs are increased in HCC patients and induced by soluble factor in the tumor microenvironment. Sci Rep. (2016) 6:39296. doi: 10.1038/srep39296
77. Drabczyk-Pluta M, Werner T, Hoffmann D, Leng Q, Chen L, Dittmer U, et al. Granulocytic myeloid-derived suppressor cells suppress virus-specific CD8(+) T cell responses during acute Friend retrovirus infection. Retrovirology. (2017) 14:42. doi: 10.1186/s12977-017-0364-3
78. Zhang ZN, Yi N, Zhang TW, Zhang LL, Wu X, Liu M, et al. Myeloid-derived suppressor cells associated with disease progression in primary HIV infection: PD-L1 blockade attenuates inhibition. J Acquir Immune Defic Syndr. (2017) 76:200–8. doi: 10.1097/QAI.0000000000001471
79. Ballbach M, Dannert A, Singh A, Siegmund DM, Handgretinger R, Piali L, et al. Expression of checkpoint molecules on myeloid-derived suppressor cells. Immunol Lett. (2017) 192:1–6. doi: 10.1016/j.imlet.2017.10.001
80. Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA, et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother. (2015) 64:817–29. doi: 10.1007/s00262-015-1692-6
81. Heine A, Held SAE, Schulte-Schrepping J, Wolff JFA, Klee K, Ulas T, et al. Generation and functional characterization of MDSC-like cells. Oncoimmunology. (2017) 6:e1295203. doi: 10.1080/2162402X.2017.1295203
82. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci USA. (2017) 114:1117–22. doi: 10.1073/pnas.1612920114
83. Pico de Coana Y, Poschke I, Gentilcore G, Mao Y, Nystrom M, Hansson J, et al. Ipilimumab treatment results in an early decrease in the frequency of circulating granulocytic myeloid-derived suppressor cells as well as their Arginase1 production. Cancer Immunol Res. (2013) 1:158–62. doi: 10.1158/2326-6066.CIR-13-0016
84. Dugast AS, Haudebourg T, Coulon F, Heslan M, Haspot F, Poirier N, et al. Myeloid-derived suppressor cells accumulate in kidney allograft tolerance and specifically suppress effector T cell expansion. J Immunol. (2008) 180:7898–906. doi: 10.4049/jimmunol.180.12.7898
85. Zhang Y, Liu Q, Zhang M, Yu Y, Liu X, Cao X. Fas signal promotes lung cancer growth by recruiting myeloid-derived suppressor cells via cancer cell-derived PGE2. J Immunol. (2009) 182:3801–8. doi: 10.4049/jimmunol.0801548
86. Wang L, Zhao J, Ren JP, Wu XY, Morrison ZD, Elgazzar MA, et al. Expansion of myeloid-derived suppressor cells promotes differentiation of regulatory T cells in HIV-1+ individuals. AIDS. (2016) 30:1521–31. doi: 10.1097/QAD.0000000000001083
87. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med. (2005) 202:919–29. doi: 10.1084/jem.20050463
88. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. (2006) 66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299
89. Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. (2010) 70:99–108. doi: 10.1158/0008-5472.CAN-09-1882
90. Siret C, Collignon A, Silvy F, Robert S, Cheyrol T, André P, et al. Deciphering the crosstalk between myeloid-derived suppressor cells and regulatory T cells in pancreatic ductal adenocarcinoma. Front Immunol. (2020) 10:3070. doi: 10.3389/fimmu.2019.03070
91. Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. (2008) 68:5439–49. doi: 10.1158/0008-5472.CAN-07-6621
92. Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol. (2012) 189:5602–11. doi: 10.4049/jimmunol.1201018
93. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. (2016) 76:5671–82. doi: 10.1158/0008-5472.CAN-16-0144
94. Yang Z, Zhang B, Li D, Lv M, Huang C, Shen GX, et al. Mast cells mobilize myeloid-derived suppressor cells and Treg cells in tumor microenvironment via IL-17 pathway in murine hepatocarcinoma model. PLoS ONE. (2010) 5:e8922. doi: 10.1371/journal.pone.0008922
95. Jitschin R, Braun M, Buttner M, Dettmer-Wilde K, Bricks J, Berger J, et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood. (2014) 124:750–60. doi: 10.1182/blood-2013-12-546416
96. Hoechst B, Gamrekelashvili J, Manns MP, Greten TF, Korangy F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood. (2011) 117:6532–41. doi: 10.1182/blood-2010-11-317321
97. Kusmartsev S, Nagaraj S, Gabrilovich DI. Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J Immunol. (2005) 175:4583–92. doi: 10.4049/jimmunol.175.7.4583
98. Centuori SM, Trad M, LaCasse CJ, Alizadeh D, Larmonier CB, Hanke NT, et al. Myeloid-derived suppressor cells from tumor-bearing mice impair TGF-beta-induced differentiation of CD4+CD25+FoxP3+ Tregs from CD4+CD25-FoxP3- T cells. J Leukoc Biol. (2012) 92:987–97. doi: 10.1189/jlb.0911465
99. Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, et al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. (2014) 74:104–18. doi: 10.1158/0008-5472.CAN-13-1545
100. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. (2013) 191:1486–95. doi: 10.4049/jimmunol.1202702
101. Park YJ, Song B, Kim YS, Kim EK, Lee JM, Lee GE, et al. Tumor microenvironmental conversion of natural killer cells into myeloid-derived suppressor cells. Cancer Res. (2013) 73:5669–81. doi: 10.1158/0008-5472.CAN-13-0545
102. Liu C, Yu S, Kappes J, Wang J, Grizzle WE, Zinn KR, et al. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood. (2007) 109:4336–42. doi: 10.1182/blood-2006-09-046201
103. Elkabets M, Ribeiro VS, Dinarello CA, Ostrand-Rosenberg S, Di Santo JP, Apte RN, et al. IL-1beta regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol. (2010) 40:3347–57. doi: 10.1002/eji.201041037
104. Mauti LA, Le Bitoux MA, Baumer K, Stehle JC, Golshayan D, Provero P, et al. Myeloid-derived suppressor cells are implicated in regulating permissiveness for tumor metastasis during mouse gestation. J Clin Invest. (2011) 121:2794–807. doi: 10.1172/JCI41936
105. Spallanzani RG, Dalotto-Moreno T, Raffo Iraolagoitia XL, Ziblat A, Domaica CI, Avila DE, et al. Expansion of CD11b(+)Ly6G (+)Ly6C (int) cells driven by medroxyprogesterone acetate in mice bearing breast tumors restrains NK cell effector functions. Cancer Immunol Immunother. (2013) 62:1781–95. doi: 10.1007/s00262-013-1483-x
106. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. (2009) 50:799–807. doi: 10.1002/hep.23054
107. Stiff A, Trikha P, Mundy-Bosse B, McMichael E, Mace TA, Benner B, et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing Fc receptor-mediated natural killer cell function. Clin Cancer Res. (2018) 24:1891–904. doi: 10.1158/1078-0432.CCR-17-0691
108. Mueller-Leisse J, Brueggemann S, Bouzani M, Schmitt AL, Einsele H, Loeffler J. Polymorphonuclear neutrophils and granulocytic myeloid-derived suppressor cells inhibit natural killer cell activity toward Aspergillus fumigatus. Med Mycol. (2015) 53:622–9. doi: 10.1093/mmy/myv030
109. Fortin C, Huang X, Yang Y. NK cell response to vaccinia virus is regulated by myeloid-derived suppressor cells. J Immunol. (2012) 189:1843–9. doi: 10.4049/jimmunol.1200584
110. Goh CC, Roggerson KM, Lee HC, Golden-Mason L, Rosen HR, Hahn YS. Hepatitis C virus-induced myeloid-derived suppressor cells suppress NK Cell IFN-gamma production by altering cellular metabolism via arginase-1. J Immunol. (2016) 196:2283–92. doi: 10.4049/jimmunol.1501881
111. Rieber N, Gille C, Kostlin N, Schafer I, Spring B, Ost M, et al. Neutrophilic myeloid-derived suppressor cells in cord blood modulate innate and adaptive immune responses. Clin Exp Immunol. (2013) 174:45–52. doi: 10.1111/cei.12143
112. Nausch N, Galani IE, Schlecker E, Cerwenka A. Mononuclear myeloid-derived “suppressor” cells express RAE-1 and activate natural killer cells. Blood. (2008) 112:4080–9. doi: 10.1182/blood-2008-03-143776
113. Shime H, Kojima A, Maruyama A, Saito Y, Oshiumi H, Matsumoto M, et al. Myeloid-derived suppressor cells confer tumor-suppressive functions on natural killer cells via polyinosinic:polycytidylic acid treatment in mouse tumor models. J Innate Immun. (2014) 6:293–305. doi: 10.1159/000355126
114. Baker GJ, Chockley P, Zamler D, Castro MG, Lowenstein PR. Natural killer cells require monocytic Gr-1(+)/CD11b(+) myeloid cells to eradicate orthotopically engrafted glioma cells. Oncoimmunology. (2016) 5:e1163461. doi: 10.1080/2162402X.2016.1163461
115. Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. (2008) 205:2235–49. doi: 10.1084/jem.20080132
116. Hu CE, Gan J, Zhang RD, Cheng YR, Huang GJ. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand J Gastroenterol. (2011) 46:156–64. doi: 10.3109/00365521.2010.516450
117. Brimnes MK, Vangsted AJ, Knudsen LM, Gimsing P, Gang AO, Johnsen HE, et al. Increased level of both CD4+FOXP3+ regulatory T cells and CD14+HLA-DR(-)/low myeloid-derived suppressor cells and decreased level of dendritic cells in patients with multiple myeloma. Scand J Immunol. (2010) 72:540–7. doi: 10.1111/j.1365-3083.2010.02463.x
118. Arora M, Poe SL, Oriss TB, Krishnamoorthy N, Yarlagadda M, Wenzel SE, et al. TLR4/MyD88-induced CD11b+Gr-1 int F4/80+ non-migratory myeloid cells suppress Th2 effector function in the lung. Mucosal Immunol. (2010) 3:578–93. doi: 10.1038/mi.2010.41
119. Wang SH, Lu QY, Guo YH, Song YY, Liu PJ, Wang YC. The blockage of Notch signalling promoted the generation of polymorphonuclear myeloid-derived suppressor cells with lower immunosuppression. Eur J Cancer. (2016) 68:90–105. doi: 10.1016/j.ejca.2016.08.019
120. van Deventer HW, Burgents JE, Wu QP, Woodford RM, Brickey WJ, Allen IC, et al. The inflammasome component NLRP3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res. (2010) 70:10161–9. doi: 10.1158/0008-5472.CAN-10-1921
121. Vandenberk L, Garg AD, Verschuere T, Koks C, Belmans J, Beullens M, et al. Irradiation of necrotic cancer cells, employed for pulsing dendritic cells (DCs), potentiates DC vaccine-induced antitumor immunity against high-grade glioma. Oncoimmunology. (2016) 5:e1083669. doi: 10.1080/2162402X.2015.1083669
122. Poschke I, Mao Y, Adamson L, Salazar-Onfray F, Masucci G, Kiessling R. Myeloid-derived suppressor cells impair the quality of dendritic cell vaccines. Cancer Immunol Immunother. (2012) 61:827–38. doi: 10.1007/s00262-011-1143-y
123. Ghansah T, Vohra N, Kinney K, Weber A, Kodumudi K, Springett G, et al. Dendritic cell immunotherapy combined with gemcitabine chemotherapy enhances survival in a murine model of pancreatic carcinoma. Cancer Immunol Immunother. (2013) 62:1083–91. doi: 10.1007/s00262-013-1407-9
124. Rossowska J, Pajtasz-Piasecka E, Anger N, Wojas-Turek J, Kicielinska J, Piasecki E, et al. Cyclophosphamide and IL-12-transduced DCs enhance the antitumor activity of tumor antigen-stimulated DCs and reduce Tregs and MDSCs number. J Immunother. (2014) 37:427–39. doi: 10.1097/CJI.0000000000000054
125. Bose A, Taylor JL, Alber S, Watkins SC, Garcia JA, Rini BI, et al. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int J Cancer. (2011) 129:2158–70. doi: 10.1002/ijc.25863
126. Bose A, Lowe DB, Rao A, Storkus WJ. Combined vaccine+axitinib therapy yields superior antitumor efficacy in a murine melanoma model. Melanoma Res. (2012) 22:236–43. doi: 10.1097/CMR.0b013e3283538293
127. Lowe DB, Bose A, Taylor JL, Tawbi H, Lin Y, Kirkwood JM, et al. Dasatinib promotes the expansion of a therapeutically superior T-cell repertoire in response to dendritic cell vaccination against melanoma. Oncoimmunology. (2014) 3:e27589. doi: 10.4161/onci.27589
128. Nguyen-Pham TN, Jung SH, Vo MC, Thanh-Tran HT, Lee YK, Lee HJ, et al. Lenalidomide Synergistically Enhances the Effect of Dendritic Cell Vaccination in a Model of Murine Multiple Myeloma. J Immunother. (2015) 38:330–9. doi: 10.1097/CJI.0000000000000097
129. Kennedy DE, Knight KL. Inhibition of B lymphopoiesis by adipocytes and IL-1-producing myeloid-derived suppressor cells. J Immunol. (2015) 195:2666–74. doi: 10.4049/jimmunol.1500957
130. Knier B, Hiltensperger M, Sie C, Aly L, Lepennetier G, Engleitner T, et al. Myeloid-derived suppressor cells control B cell accumulation in the central nervous system during autoimmunity. Nat Immunol. (2018) 19:1341–51. doi: 10.1038/s41590-018-0237-5
131. Crook KR, Jin M, Weeks MF, Rampersad RR, Baldi RM, Glekas AS, et al. Myeloid-derived suppressor cells regulate T cell and B cell responses during autoimmune disease. J Leukoc Biol. (2015) 97:573–82. doi: 10.1189/jlb.4A0314-139R
132. Green KA, Wang L, Noelle RJ, Green WR. Selective involvement of the checkpoint regulator VISTA in suppression of B-Cell, but Not T-Cell, responsiveness by monocytic myeloid-derived suppressor cells from mice infected with an immunodeficiency-causing retrovirus. J Virol. (2015) 89:9693–8. doi: 10.1128/JVI.00888-15
133. Wang Y, Schafer CC, Hough KP, Tousif S, Duncan SR, Kearney JF, et al. Myeloid-derived suppressor cells impair B cell responses in lung cancer through IL-7 and STAT5. J Immunol. (2018) 201:278–95. doi: 10.4049/jimmunol.1701069
134. Ku AW, Muhitch JB, Powers CA, Diehl M, Kim M, Fisher DT, et al. Tumor-induced MDSC act via remote control to inhibit L-selectin-dependent adaptive immunity in lymph nodes. Elife. (2016) 5:e17375. doi: 10.7554/eLife.17375
135. Wang K, Gong H, Chai R, Yuan H, Chen Y, Liu J. Aberrant frequency of IL-35 producing B cells in colorectal cancer patients. Cytokine. (2018) 102:206–10. doi: 10.1016/j.cyto.2017.10.011
136. Shen M, Wang J, Yu W, Zhang C, Liu M, Wang K, et al. A novel MDSC-induced PD-1(-)PD-L1(+) B-cell subset in breast tumor microenvironment possesses immuno-suppressive properties. Oncoimmunology. (2018) 7:e1413520. doi: 10.1080/2162402X.2017.1413520
137. Park MJ, Lee SH, Kim EK, Lee EJ, Park SH, Kwok SK, et al. Myeloid-derived suppressor cells induce the expansion of regulatory B cells and ameliorate autoimmunity in the sanroque mouse model of systemic lupus erythematosus. Arthritis Rheumatol. (2016) 68:2717–27. doi: 10.1002/art.39767
138. Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. (2001) 166:5398–406. doi: 10.4049/jimmunol.166.9.5398
139. Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. (2010) 184:3106–16. doi: 10.4049/jimmunol.0902661
140. Lu T, Ramakrishnan R, Altiok S, Youn JI, Cheng P, Celis E, et al. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest. (2011) 121:4015–29. doi: 10.1172/JCI45862
141. Nagaraj S, Nelson A, Youn JI, Cheng P, Quiceno D, Gabrilovich DI. Antigen-specific CD4(+) T cells regulate function of myeloid-derived suppressor cells in cancer via retrograde MHC class II signaling. Cancer Res. (2012) 72:928–38. doi: 10.1158/0008-5472.CAN-11-2863
142. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest. (2010) 120:457–71. doi: 10.1172/JCI40483
143. Schouppe E, Mommer C, Movahedi K, Laoui D, Morias Y, Gysemans C, et al. Tumor-induced myeloid-derived suppressor cell subsets exert either inhibitory or stimulatory effects on distinct CD8+ T-cell activation events. Eur J Immunol. (2013) 43:2930–42. doi: 10.1002/eji.201343349
144. Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX, Moughon DL, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. (2010) 115:1461–71. doi: 10.1182/blood-2009-08-237412
145. Younos I, Donkor M, Hoke T, Dafferner A, Samson H, Westphal S, et al. Tumor- and organ-dependent infiltration by myeloid-derived suppressor cells. Int Immunopharmacol. (2011) 11:816–26. doi: 10.1016/j.intimp.2011.02.021
146. Cimen Bozkus C, Elzey BD, Crist SA, Ellies LG, Ratliff TL. Expression of cationic amino acid transporter 2 is required for myeloid-derived suppressor cell-mediated control of T cell immunity. J Immunol. (2015) 195:5237–50. doi: 10.4049/jimmunol.1500959
147. Maenhout SK, Van Lint S, Emeagi PU, Thielemans K, Aerts JL. Enhanced suppressive capacity of tumor-infiltrating myeloid-derived suppressor cells compared with their peripheral counterparts. Int J Cancer. (2014) 134:1077–90. doi: 10.1002/ijc.28449
148. Haverkamp JM, Crist SA, Elzey BD, Cimen C, Ratliff TL. In vivo suppressive function of myeloid-derived suppressor cells is limited to the inflammatory site. Eur J Immunol. (2011) 41:749–59. doi: 10.1002/eji.201041069
149. Connolly MK, Mallen-St Clair J, Bedrosian AS, Malhotra A, Vera V, Ibrahim J, et al. Distinct populations of metastases-enabling myeloid cells expand in the liver of mice harboring invasive and preinvasive intra-abdominal tumor. J Leukoc Biol. (2010) 87:713–25. doi: 10.1189/jlb.0909607
150. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. (2008) 181:4666–75. doi: 10.4049/jimmunol.181.7.4666
151. Watanabe S, Deguchi K, Zheng R, Tamai H, Wang LX, Cohen PA, et al. Tumor-induced CD11b+Gr-1+ myeloid cells suppress T cell sensitization in tumor-draining lymph nodes. J Immunol. (2008) 181:3291–300. doi: 10.4049/jimmunol.181.5.3291
152. Yan D, Wang J, Sun H, Zamani A, Zhang H, Chen W, et al. TIPE2 specifies the functional polarization of myeloid-derived suppressor cells during tumorigenesis. J Exp Med. (2020) 217:e20182005. doi: 10.1084/jem.20182005
153. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. (2005) 202:931–9. doi: 10.1084/jem.20050715
154. Rodriguez-Ubreva J, Catala-Moll F, Obermajer N, Alvarez-Errico D, Ramirez RN, Company C, et al. Prostaglandin E2 leads to the acquisition of DNMT3A-dependent tolerogenic functions in human myeloid-derived suppressor cells. Cell Rep. (2017) 21:154–67. doi: 10.1016/j.celrep.2017.09.018
155. Tomic S, Joksimovic B, Bekic M, Vasiljevic M, Milanovic M, Colic M, et al. Prostaglanin-E2 potentiates the suppressive functions of human mononuclear myeloid-derived suppressor cells and increases their capacity to expand IL-10-producing regulatory T cell subsets. Front Immunol. (2019) 10:475. doi: 10.3389/fimmu.2019.00475
156. Novitskiy SV, Pickup MW, Gorska AE, Owens P, Chytil A, Aakre M, et al. TGF-beta receptor II loss promotes mammary carcinoma progression by Th17 dependent mechanisms. Cancer Discov. (2011) 1:430–41. doi: 10.1158/2159-8290.CD-11-0100
157. Sido JM, Yang X, Nagarkatti PS, Nagarkatti M. Delta9-Tetrahydrocannabinol-mediated epigenetic modifications elicit myeloid-derived suppressor cell activation via STAT3/S100A8. J Leukoc Biol. (2015) 97:677–88. doi: 10.1189/jlb.1A1014-479R
158. De Veirman K, De Beule N, Maes K, Menu E, De Bruyne E, De Raeve H, et al. Extracellular S100A9 Protein in Bone Marrow Supports Multiple Myeloma Survival by Stimulating Angiogenesis and Cytokine Secretion. Cancer Immunol Res. (2017) 5:839–46. doi: 10.1158/2326-6066.CIR-17-0192
159. Xiao W, Klement JD, Lu C, Ibrahim ML, Liu K. IFNAR1 Controls Autocrine Type I IFN Regulation of PD-L1 Expression in Myeloid-Derived Suppressor Cells. J Immunol. (2018) 201:264–77. doi: 10.4049/jimmunol.1800129
160. Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. (2010) 185:2273–84. doi: 10.4049/jimmunol.1000901
161. Kohanbash G, McKaveney K, Sakaki M, Ueda R, Mintz AH, Amankulor N, et al. GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-alpha. Cancer Res. (2013) 73:6413–23. doi: 10.1158/0008-5472.CAN-12-4124
162. Zhang H, Ye YL, Li MX, Ye SB, Huang WR, Cai TT, et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene. (2017) 36:2095–104. doi: 10.1038/onc.2016.367
163. Yaddanapudi K, Rendon BE, Lamont G, Kim EJ, Al Rayyan N, Richie J, et al. MIF is necessary for late-stage melanoma patient MDSC immune suppression and differentiation. Cancer Immunol Res. (2016) 4:101–12. doi: 10.1158/2326-6066.CIR-15-0070-T
164. Whiteside TL. Exosomes and tumor-mediated immune suppression. J Clin Invest. (2016) 126:1216–23. doi: 10.1172/JCI81136
165. Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. (2018) 200:422–31. doi: 10.4049/jimmunol.1701019
166. Altevogt P, Bretz NP, Ridinger J, Utikal J, Umansky V. Novel insights into exosome-induced, tumor-associated inflammation and immunomodulation. Semin Cancer Biol. (2014) 28:51–7. doi: 10.1016/j.semcancer.2014.04.008
167. Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer. (2009) 124:2621–33. doi: 10.1002/ijc.24249
168. Wang J, De Veirman K, De Beule N, Maes K, De Bruyne E, Van Valckenborgh E, et al. The bone marrow microenvironment enhances multiple myeloma progression by exosome-mediated activation of myeloid-derived suppressor cells. Oncotarget. (2015) 6:43992–4004. doi: 10.18632/oncotarget.6083
169. Wang J, De Veirman K, Faict S, Frassanito MA, Ribatti D, Vacca A, et al. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J Pathol. (2016) 239:162–73. doi: 10.1002/path.4712
170. Wen SW, Sceneay J, Lima LG, Wong CS, Becker M, Krumeich S, et al. The biodistribution and immune suppressive effects of breast cancer-derived exosomes. Cancer Res. (2016) 76:6816–27. doi: 10.1158/0008-5472.CAN-16-0868
171. Liu Y, Xiang X, Zhuang X, Zhang S, Liu C, Cheng Z, et al. Contribution of MyD88 to the tumor exosome-mediated induction of myeloid derived suppressor cells. Am J Pathol. (2010) 176:2490–9. doi: 10.2353/ajpath.2010.090777
172. Diao J, Yang X, Song X, Chen S, He Y, Wang Q, et al. Exosomal Hsp70 mediates immunosuppressive activity of the myeloid-derived suppressor cells via phosphorylation of Stat3. Med Oncol. (2015) 32:453. doi: 10.1007/s12032-014-0453-2
173. Ridder K, Sevko A, Heide J, Dams M, Rupp AK, Macas J, et al. Extracellular vesicle-mediated transfer of functional RNA in the tumor microenvironment. Oncoimmunology. (2015) 4:e1008371. doi: 10.1080/2162402X.2015.1008371
174. Guo X, Qiu W, Liu Q, Qian M, Wang S, Zhang Z, et al. Immunosuppressive effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten Pathways. Oncogene. (2018) 37:4239–59. doi: 10.1038/s41388-018-0261-9
175. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity. (2015) 43:435–49. doi: 10.1016/j.immuni.2015.09.001
176. Hammami I, Chen J, Bronte V, De Crescenzo G, Jolicoeur M. Myeloid-derived suppressor cells exhibit two bioenergetic steady-states in vitro. J Biotechnol. (2011) 152:43–8. doi: 10.1016/j.jbiotec.2011.01.009
177. Hammami I, Chen J, Murschel F, Bronte V, De Crescenzo G, Jolicoeur M. Immunosuppressive activity enhances central carbon metabolism and bioenergetics in myeloid-derived suppressor cells in vitro models. BMC Cell Biol. (2012) 13:18. doi: 10.1186/1471-2121-13-18
178. Jian SL, Chen WW, Su YC, Su YW, Chuang TH, Hsu SC, et al. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ROS-mediated apoptosis. Cell Death Dis. (2017) 8:e2779. doi: 10.1038/cddis.2017.192
179. Wu T, Zhao Y, Wang H, Li Y, Shao L, Wang R, et al. mTOR masters monocytic myeloid-derived suppressor cells in mice with allografts or tumors. Sci Rep. (2016) 6:20250. doi: 10.1038/srep20250
180. Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab. (2018) 28:87–103 e6. doi: 10.1016/j.cmet.2018.04.022
181. Deng Y, Yang J, Luo F, Qian J, Liu R, Zhang D, et al. mTOR-mediated glycolysis contributes to the enhanced suppressive function of murine tumor-infiltrating monocytic myeloid-derived suppressor cells. Cancer Immunol Immunother. (2018) 67:1355–64. doi: 10.1007/s00262-018-2177-1
182. Liu G, Bi Y, Shen B, Yang H, Zhang Y, Wang X, et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1alpha-dependent glycolysis. Cancer Res. (2014) 74:727–37. doi: 10.1158/0008-5472.CAN-13-2584
183. Lu Y, Liu H, Bi Y, Yang H, Li Y, Wang J, et al. Glucocorticoid receptor promotes the function of myeloid-derived suppressor cells by suppressing HIF1alpha-dependent glycolysis. Cell Mol Immunol. (2018) 15:618–29. doi: 10.1038/cmi.2017.5
184. Lin Y, Wang B, Shan W, Tan Y, Feng J, Xu L, et al. mTOR inhibitor rapamycin induce polymorphonuclear myeloid-derived suppressor cells mobilization and function in protecting against acute graft-versus-host disease after bone marrow transplantation. Clin Immunol. (2018) 187:122–31. doi: 10.1016/j.clim.2017.11.005
185. Zhang C, Wang S, Li J, Zhang W, Zheng L, Yang C, et al. The mTOR signal regulates myeloid-derived suppressor cells differentiation and immunosuppressive function in acute kidney injury. Cell Death Dis. (2017) 8:e2695. doi: 10.1038/cddis.2017.86
186. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. (2015) 3:1236–47. doi: 10.1158/2326-6066.CIR-15-0036
187. Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology. (2017) 6:e1344804. doi: 10.1080/2162402X.2017.1344804
188. Chiu DK, Xu IM, Lai RK, Tse AP, Wei LL, Koh HY, et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology. (2016) 64:797–813. doi: 10.1002/hep.28655
189. Hong J, Tobin NP, Rundqvist H, Li T, Lavergne M, Garcia-Ibanez Y, et al. Role of tumor pericytes in the recruitment of myeloid-derived suppressor cells. J Natl Cancer Inst. (2015) 107:djv209. doi: 10.1093/jnci/djv209
190. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. (2010) 207:2439–53. doi: 10.1084/jem.20100587
191. Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK, Li LL, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. (2017) 8:517. doi: 10.1038/s41467-017-00530-7
192. Hammami A, Abidin BM, Charpentier T, Fabie A, Duguay AP, Heinonen KM, et al. HIF-1alpha is a key regulator in potentiating suppressor activity and limiting the microbicidal capacity of MDSC-like cells during visceral leishmaniasis. PLoS Pathog. (2017) 13:e1006616. doi: 10.1371/journal.ppat.1006616
193. Deng J, Li J, Sarde A, Lines JL, Lee YC, Qian DC, et al. Hypoxia-induced VISTA promotes the suppressive function of myeloid-derived suppressor cells in the tumor microenvironment. Cancer Immunol Res. (2019) 7:1079–90. doi: 10.1158/2326-6066.CIR-18-0507
194. Kuonen F, Laurent J, Secondini C, Lorusso G, Stehle JC, Rausch T, et al. Inhibition of the Kit ligand/c-Kit axis attenuates metastasis in a mouse model mimicking local breast cancer relapse after radiotherapy. Clin Cancer Res. (2012) 18:4365–74. doi: 10.1158/1078-0432.CCR-11-3028
195. Chafe SC, Lou Y, Sceneay J, Vallejo M, Hamilton MJ, McDonald PC, et al. Carbonic anhydrase IX promotes myeloid-derived suppressor cell mobilization and establishment of a metastatic niche by stimulating G-CSF production. Cancer Res. (2015) 75:996–1008. doi: 10.1158/0008-5472.CAN-14-3000
196. Sceneay J, Chow MT, Chen A, Halse HM, Wong CS, Andrews DM, et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. (2012) 72:3906–11. doi: 10.1158/0008-5472.CAN-11-3873
197. Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. (2017) 168:692–706. doi: 10.1016/j.cell.2016.12.004
198. Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc Natl Acad Sci USA. (2011) 108:6561–6. doi: 10.1073/pnas.1008942108
199. Mahadevan NR, Anufreichik V, Rodvold JJ, Chiu KT, Sepulveda H, Zanetti M. Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8(+) T cell priming. PLoS One. (2012) 7:e51845. doi: 10.1371/journal.pone.0051845
200. Yan D, Wang HW, Bowman RL, Joyce JA. STAT3 and STAT6 signaling pathways synergize to promote cathepsin secretion from macrophages via IRE1alpha activation. Cell Rep. (2016) 16:2914–27. doi: 10.1016/j.celrep.2016.08.035
201. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER Stress Sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. (2015) 161:1527–38. doi: 10.1016/j.cell.2015.05.025
202. Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest. (2014) 124:2626–39. doi: 10.1172/JCI74056
203. Thevenot PT, Sierra RA, Raber PL, Al-Khami AA, Trillo-Tinoco J, Zarreii P, et al. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity. (2014) 41:389–401. doi: 10.1016/j.immuni.2014.08.015
204. Lee BR, Chang SY, Hong EH, Kwon BE, Kim HM, Kim YJ, et al. Elevated endoplasmic reticulum stress reinforced immunosuppression in the tumor microenvironment via myeloid-derived suppressor cells. Oncotarget. (2014) 5:12331–45. doi: 10.18632/oncotarget.2589
205. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. (2016) 1:aaf8943. doi: 10.1126/sciimmunol.aaf8943
206. Nan J, Xing YF, Hu B, Tang JX, Dong HM, He YM, et al. Endoplasmic reticulum stress induced LOX-1(+) CD15(+) polymorphonuclear myeloid-derived suppressor cells in hepatocellular carcinoma. Immunology. (2018) 154:144–55. doi: 10.1111/imm.12876
207. Shime H, Maruyama A, Yoshida S, Takeda Y, Matsumoto M, Seya T. Toll-like receptor 2 ligand and interferon-gamma suppress anti-tumor T cell responses by enhancing the immunosuppressive activity of monocytic myeloid-derived suppressor cells. Oncoimmunology. (2017) 7:e1373231. doi: 10.1080/2162402X.2017.1373231
208. Sinha P, Chornoguz O, Clements VK, Artemenko KA, Zubarev RA, Ostrand-Rosenberg S. Myeloid-derived suppressor cells express the death receptor Fas and apoptose in response to T cell-expressed FasL. Blood. (2011) 117:5381–90. doi: 10.1182/blood-2010-11-321752
209. Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, et al. gammadeltaT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity. (2014) 40:785–800. doi: 10.1016/j.immuni.2014.03.013
210. Al Sayed MF, Amrein MA, Buhrer ED, Huguenin AL, Radpour R, Riether C, et al. T-cell-Secreted TNFalpha induces emergency myelopoiesis and myeloid-derived suppressor cell differentiation in cancer. Cancer Res. (2019) 79:346–59. doi: 10.1158/0008-5472.CAN-17-3026
211. Chen X, Wang L, Li P, Song M, Qin G, Gao Q, et al. Dual TGF-beta and PD-1 blockade synergistically enhances MAGE-A3-specific CD8(+) T cell response in esophageal squamous cell carcinoma. Int J Cancer. (2018) 143:2561–74. doi: 10.1002/ijc.31730
212. Yang R, Cai Z, Zhang Y, Yutzy WHt, Roby KF, Roden RB. CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1+CD11b+ myeloid cells. Cancer Res. (2006) 66:6807–15. doi: 10.1158/0008-5472.CAN-05-3755
213. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-expressed IDO recruits and activates MDSCs in a Treg-dependent manner. Cell Rep. (2015) 13:412–24. doi: 10.1016/j.celrep.2015.08.077
214. Xu X, Meng Q, Erben U, Wang P, Glauben R, Kuhl AA, et al. Myeloid-derived suppressor cells promote B-cell production of IgA in a TNFR2-dependent manner. Cell Mol Immunol. (2017) 14:597–606. doi: 10.1038/cmi.2015.103
215. Bodogai M, Moritoh K, Lee-Chang C, Hollander CM, Sherman-Baust CA, Wersto RP, et al. Immunosuppressive and Prometastatic functions of myeloid-derived suppressive cells rely upon education from tumor-associated B cells. Cancer Res. (2015) 75:3456–65. doi: 10.1158/0008-5472.CAN-14-3077
216. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. (2007) 179:977–83. doi: 10.4049/jimmunol.179.2.977
217. Beury DW, Parker KH, Nyandjo M, Sinha P, Carter KA, Ostrand-Rosenberg S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J Leukoc Biol. (2014) 96:1109–18. doi: 10.1189/jlb.3A0414-210R
218. Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol. (2012) 22:275–81. doi: 10.1016/j.semcancer.2012.01.011
219. Danelli L, Frossi B, Gri G, Mion F, Guarnotta C, Bongiovanni L, et al. Mast cells boost myeloid-derived suppressor cell activity and contribute to the development of tumor-favoring microenvironment. Cancer Immunol Res. (2015) 3:85–95. doi: 10.1158/2326-6066.CIR-14-0102
220. Jachetti E, Cancila V, Rigoni A, Bongiovanni L, Cappetti B, Belmonte B, et al. Cross-talk between myeloid-derived suppressor cells and mast cells mediates tumor-specific immunosuppression in prostate cancer. Cancer Immunol Res. (2018) 6:552–65. doi: 10.1158/2326-6066.CIR-17-0385
221. Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. (2016) 76:4124–35. doi: 10.1158/0008-5472.CAN-15-2973
222. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. (2013) 73:3007–18. doi: 10.1158/0008-5472.CAN-12-4601
223. Deng Y, Cheng J, Fu B, Liu W, Chen G, Zhang Q, et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene. (2017) 36:1090–101. doi: 10.1038/onc.2016.273
224. Li A, Chen P, Leng Y, Kang J. Histone deacetylase 6 regulates the immunosuppressive properties of cancer-associated fibroblasts in breast cancer through the STAT3-COX2-dependent pathway. Oncogene. (2018) 37:5952–66. doi: 10.1038/s41388-018-0379-9
225. Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell. (2017) 32:654–68 e5. doi: 10.1016/j.ccell.2017.10.005
226. Ohshio Y, Teramoto K, Hanaoka J, Tezuka N, Itoh Y, Asai T, et al. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci. (2015) 106:134–42. doi: 10.1111/cas.12584
227. Gunaydin G, Kesikli SA, Guc D. Cancer associated fibroblasts have phenotypic and functional characteristics similar to the fibrocytes that represent a novel MDSC subset. Oncoimmunology. (2015) 4:e1034918. doi: 10.1080/2162402X.2015.1034918
228. Ai L, Mu S, Wang Y, Wang H, Cai L, Li W, et al. Prognostic role of myeloid-derived suppressor cells in cancers: a systematic review and meta-analysis. BMC Cancer. (2018) 18:1220. doi: 10.1186/s12885-018-5086-y
Keywords: myeloid-derived suppressor cells, immune suppression, tumor microenvironment, immunotherapy, endoplasmic reticulum stress
Citation: Yang Y, Li C, Liu T, Dai X and Bazhin AV (2020) Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 11:1371. doi: 10.3389/fimmu.2020.01371
Received: 19 March 2020; Accepted: 28 May 2020;
Published: 22 July 2020.
Edited by:
Joao P. B. Viola, National Cancer Institute (INCA), BrazilReviewed by:
Fabian Flores-Borja, King's College London, United KingdomJohn P. Vasilakos, 3M, United States
Copyright © 2020 Yang, Li, Liu, Dai and Bazhin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuhui Yang, yuhui_yang@hust.edu.cn