 Hiroyuki Oshiumi
Hiroyuki Oshiumi- Department of Immunology, Faculty of Life Sciences, Graduate School of Medical Sciences, Kumamoto University, Kumamoto, Japan
RIG-I and MDA5 are cytoplasmic viral RNA sensors and are essential for antiviral innate immune responses, such as type I interferon production. Post-translational modification is critical for the activation and inactivation of RIG-I and MDA5. At least seven ubiquitin ligases have been reported to be involved in either K63- or K48-linked polyubiquitination of RIG-I and MDA5, and these ubiquitin ligases are further regulated by other factors. TRIM25 is an E3 ubiquitin ligase that delivers a K63-linked polyubiquitin moiety to the caspase activation and recruitment domains (CARDs) of RIG-I, thereby activating the antiviral innate immune response. Recent studies have shown that NDR2, ZCCHC3, and Lnczc3h7a promote TRIM25-mediated RIG-I activation. Riplet is another ubiquitin ligase that mediates the K63-linked polyubiquitination of the C-terminal domain (CTD) of RIG-I; however, it was also reported that Riplet delivers the K63-linked polyubiquitin moiety to the CARDs of RIG-I as well as to the CTD, thereby activating RIG-I. Further, there are several factors that attenuate the activation of RIG-I and MDA5. RNF125, TRIM40, and c-Cbl mediate K48-linked polyubiquitination and induce degradation of RIG-I and/or MDA5. USP21 and CYLD remove the K63-linked polyubiquitin chain from RIG-I, and NLRP12 inhibits polyubiquitin-mediated RIG-I activation. Although these new regulators have been reported, their distinctive roles and functional differences remain elusive, and in some cases, studies on the topic are contradictory to each other. In the present review, recent studies related to post-translational modifications of RIG-I and MDA5 are summarized, and several controversies and unanswered questions in this field are discussed.
Introduction
Cytoplasmic viral double-stranded RNA (dsRNA) is recognized by RIG-I-like receptors (RLRs), which include RIG-I, MDA5, and LGP2 (1, 2). 5′ tri- and di-phosphate of dsRNA are crucial for the recognition of viral RNA by RIG-I, whereas 5′phosphate of dsRNA is dispensable for MDA5 activation (3–5). The expression of RLRs is induced after viral infection in various types of cells. Dendritic cells and macrophages as well as epithelial cells utilize RIG-I and MDA5 to sense viral RNA and to produce type I interferon (IFN) at an early phase of viral infection (6, 7). An accumulating body of evidence has shown that RLRs play a crucial role in antiviral innate immune responses (5, 8).
After recognition of viral dsRNA, RIG-I and MDA5 trigger the signals that induce the expression of type I IFN and other pro-inflammatory cytokines via a MAVS adaptor molecule, which is also called IPS-1, CARDIF, and VISA (9–12). While RIG-I recognizes relatively short dsRNA molecule (<1 kbp), MDA5 recognizes longer dsRNAs (>1 kbp) (13); thus, these proteins exhibit different specificities to viral infections (7). For instance, influenza A virus, hepatitis C virus, and respiratory syncytial virus are preferentially recognized by RIG-I, whereas encephalomyocarditis virus and Mengovirus are recognized by MDA5 (14).
The RIG-I and MDA5 proteins carry two caspase activation and recruitment domains (CARDs) at their N-terminal regions, which are essential for MAVS activation (8). In contrast, LGP2 does not possess any CARDs and plays only a regulatory role in the activation of RIG-I and MDA5 (8). The C-terminal domains (CTDs) of RIG-I and MDA5 recognize viral RNA, and the proteins assemble along dsRNA to make a nucleoprotein filament (15, 16). The formation of the nucleoprotein filament leads to inter-molecular association of RIG-I CARDs, which results in the formation of a 2CARD tetramer structure (17). This 2CARD tetramer structure functions as a core of MAVS oligomerization, resulting in MAVS prion-like fiber formation (18). The MAVS fibers on mitochondria are essential for activating downstream protein kinases, such as TBK1 and IKK-ε (18). Activated protein kinases phosphorylate an IRF3 transcription factor to induce type I IFN expression (19). Recently, we identified a scaffold protein, zyxin, that stabilizes the interaction between MAVS, RIG-I, and MDA5 (20). Considering that the zyxin protein localizes to focal adhesions and regulates actin filament assembly (21, 22), it might function as the scaffold required to start the assembly and filament formation of MAVS as well as actin.
There are many regulators of RIG-I and MDA5, such as ubiquitin ligases and protein kinases. Further, those ubiquitin ligases are regulated by several host and viral proteins. In the present review, we summarize recent findings related to the ubiquitin-mediated regulation of RLRs.
Ubiquitin Ligases That Mediate K63-Linked Polyubiquitination OF RIG-I
The RIG-I protein harbors several post-translational modifications, and polyubiquitin modification is crucial for RIG-I activation and degradation. Gack et al. first reported that RIG-I harbors K63-linked polyubiquitination, leading to the activation of downstream signaling (23) (Figure 1). The K172 residue of the RIG-I CARDs is ubiquitinated, which is essential for the induction of type I IFN expression (23). Later studies showed that both covalent binding and non-covalent binding of the K63-linked polyubiquitin chain to the K172 residue are sufficient to induce the RIG-I activation (17, 24). The K63-linked polyubiquitin chains stabilize the RIG-I 2CARD tetramer (17). TRIM25 was first reported to be responsible for delivering the polyubiquitin moiety to RIG-I CARDs (23, 24).
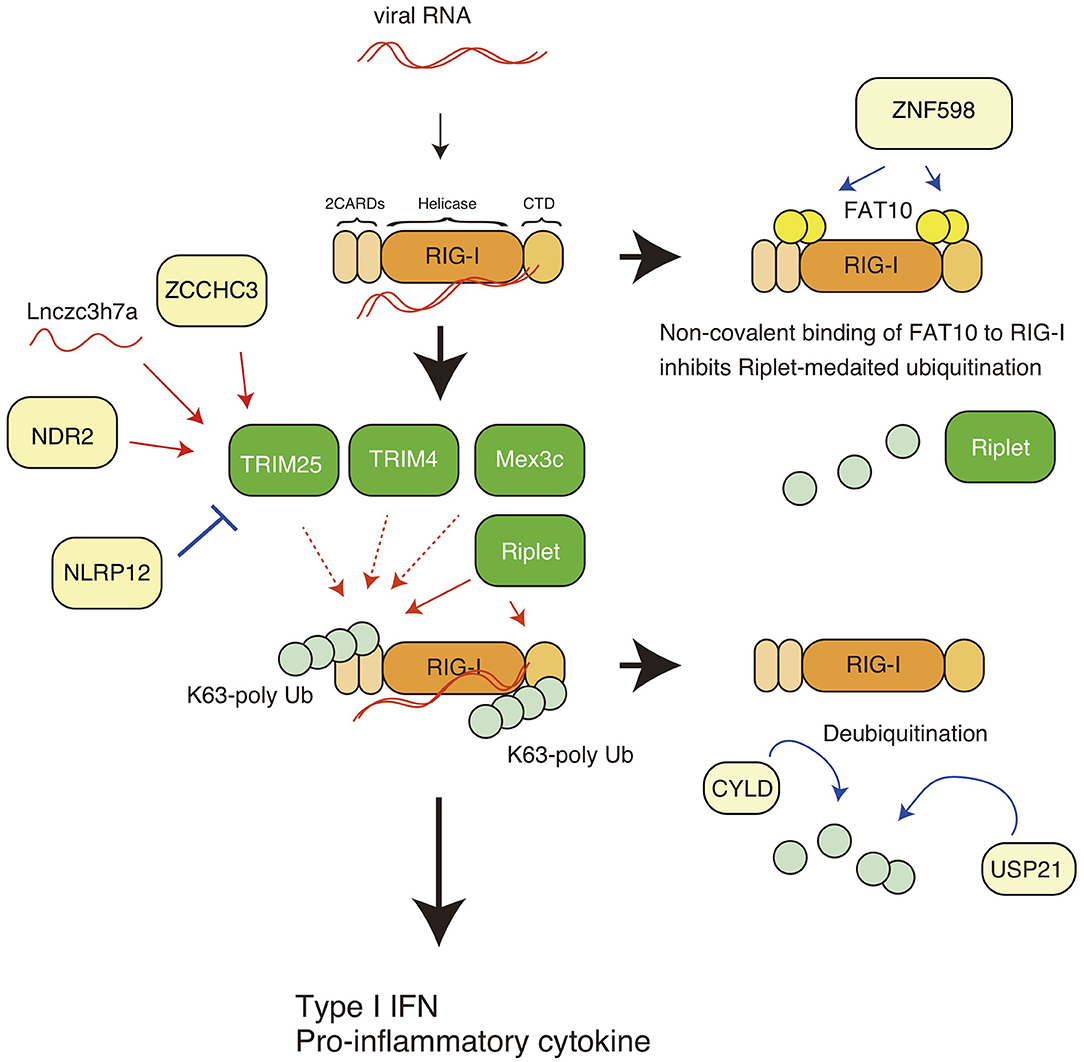
Figure 1. K63-linked polyubiquitin chain-mediated regulation of RIG-I. The RIG-I protein comprises the N-terminal caspase activation and recruitment domains (CARDs), an RNA helicase domain, and the C-terminal domain (CTD). K63-linked polyubiquitination leads to activation of RIG-I, leading to the expression of type I IFN and other pro-inflammatory cytokines. TRIM25, Riplet, Mex3c, and TRIM4 are reported to mediate K63-linked polyubiquitination of the CARDs, and Riplet mediates the polyubiquitination of both the CARDs and the CTD. ZNF598 promotes the binding of FAT10 to RIG-I, thereby inhibiting Riplet-mediated ubiquitination of RIG-I. CYLD and USP remove the polyubiquitin chain from RIG-I. The ZCCHC3 and NDR2 protein promote TRIM25-medaited polyubiquitination of RIG-I. A non-coding long RNA, Lnczc3h7a, promotes TRIM25-medated RIG-I activation.
Riplet (also called RNF135 and Reul) is another ubiquitin ligase (25). Since knockout (KO) of Riplet results in severe defects in the in vivo antiviral innate immune response, such as impaired type I IFN production and decreased survival of virally infected mice (26), Riplet-mediated RIG-I ubiquitination is crucial for in vivo antiviral innate immune responses. The Riplet protein preferentially binds to the RIG-I CTD and mediates K63-linked polyubiquitination at K788, K849, K851, K888, K907, and K909 (27, 28). It has been shown that Riplet promotes the binding of TRIM25 to RIG-I, thereby augmenting TRIM25-mediated RIG-I activation (28). Thus, a sequential ubiquitination model has been postulated (25, 28). However, recent studies have reported that Riplet ubiquitinated multiple sites of RIG-I, including the 2CARDs, and produced unanchored polyubiquitin chains, thereby promoting RIG-I activation (29, 30).
Many studies have reported that TRIM25 is responsible for RIG-I ubiquitination and activation, whereas several recent studies have shown that Riplet, but not TRIM25 is essential for RIG-I activation (30, 31). In addition, there are several reports illustrating the RIG-I-independent antiviral activities of TRIM25 (32, 33). On the other hand, it has been reported that Riplet can activates IRF3 transcription factor in a RIG-I-independent manner (34). Since there are differences in the experimental conditions between recent studies, and other possible explanations which have been recently discussed in more detail (35), the apparent contradictions should be reconciled by further studies. Despite these recent contradictory results, many studies have identified regulatory factors (proteins and lncRNAs) that regulate TRIM25-medaited RIG-I K63-linked polyubiquitination and activation, and over the past several years, many viral antagonists of TRIM25 have been discovered which function to inhibit the K63-linked polyubiquitination and signaling of RIG-I (as described below). In addition to TRIM25 and Riplet, Mex3c and TRIM4 ubiquitin ligases have been reported to be involved in the K63-linked polyubiquitination and activation of RIG-I (36, 37). Amino acid substitution at K45, K99, and K169 attenuated Mex3c-mediated polyubiquitination of RIG-I (36), and TRIM4 targeted K164 and K172 of RIG-I (37) (Figure 1). However, Shi et al. reported that knockout of Mex3c and TRIM4 did not affect the activation of MAVS upon stimulation (38). These apparent discrepancies should be addressed in further studies to determine why RIG-I requires four ubiquitin ligases and why previous studies apparently contradict to each other.
Ubiquitin Ligases That Mediate K48-Linked Polyubiquitination OF RIG-I
The RIG-I protein is also regulated by K48-linked polyubiquitination, which is well-known to lead to protein degradation in a proteasome-dependent manner (Figure 2). RNF125 is a ubiquitin ligase that mediates the K48-linked polyubiquitination of RIG-I, thereby promoting the degradation of the protein via proteasomes (39). RNF125 attenuates RIG-I-induced IFN-β production (39). c-Cbl is an E3 ubiquitin ligase and is recruited to the cytoplasmic tail of Siglec-G together with SHP2 (40). c-Cbl mediates K48-lined polyubiquitination of RIG-I at the K813 residue and thereby promotes RIG-I degradation (40). TRIM40 is a member of the TRIM family and binds to the CARDs of RIG-I (41). TRIM40 promotes K27- and K48-linked polyubiquitination of RIG-I (41). Although the in vivo roles of RNF125 and c-Cbl in the RIG-I-mediated innate immune response remain unclear, it has been shown that knockout of TRIM40 enhances RIG-I-mediated type I IFN production and improves the survival of mice infected with vesicular stomatitis virus, which is recognized by RIG-I (41). Since excessive activation of RIG-I leads to autoimmune disorders (42), these ubiquitin ligases are expected to be important in preventing excessive RIG-I activation.
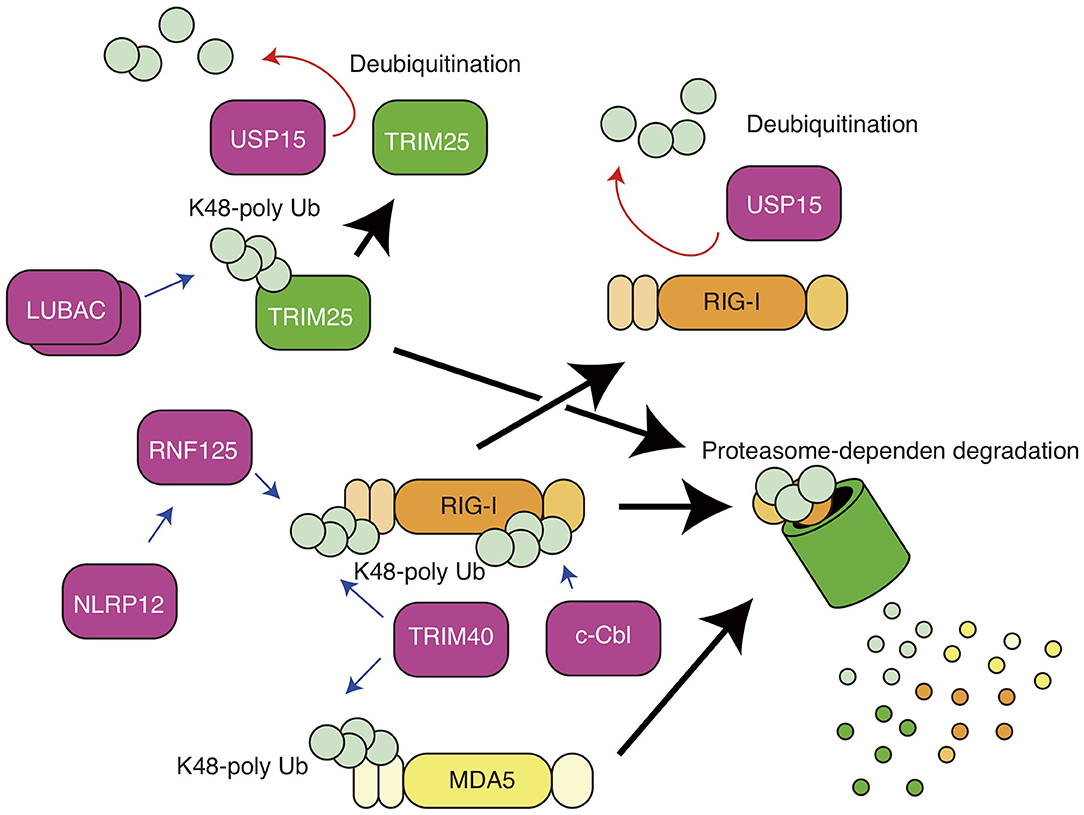
Figure 2. K48-linked polyubiquitin chain-mediated regulation of RIG-I and MDA5. K48-linked polyubiquitination of RIG-I and TRIM25 leads to proteasomal degradation of the proteins. RNF125 mediates K48-linked polyubiquitination of RIG-I, and NLRP12 promotes TRIM25-mediated ubiquitination. TRIM40 conjugates K48-linked polyubiquitin chain to both RIG-I and MDA5. c-Cbl mediates K48-linked polyubiquitination of RIG-I. USP15 removes RNF125-mediated polyubiquitin chain.
Regulators of Ubiquitin Ligase Targeting RIG-I
FAT10 is a ubiquitin-like modifier and is also involved in the control of RIG-I activation (43, 44). In general, covalent binding of FAT10 to a target protein leads to degradation of the target protein via proteasome (45, 46). A previous study reported that non-covalent binding of FAT10 to RIG-I decreased the solubility of the RIG-I protein, resulting in attenuation of RIG-I-mediated cytokine expression (44).
The ZNF598 protein is known to exhibit E3 ubiquitin ligase activity and to be required for the ubiquitination of ribosome components for ribosome quality control (47, 48). DiGiuseppe et al. first reported that ZNF598 decreases the expression of antiviral genes, such as PKR, ISG56, and MxA (49). Recently, we showed that ZNF598 promoted non-covalent binding of FAT10 to RIG-I, thereby inhibiting Riplet-mediated K63-linked polyubiquitination of RIG-I (50).
There are several other mechanisms that inhibit K63-linked polyubiquitin chain-mediated RIG-I activation. USP21 is a deubiquitinating enzyme that removes the K63-linked polyubiquitin chain from RIG-I, resulting in the attenuation of innate immune responses (51). CYLD is another deubiquitinating enzyme that also removes the K63-linked polyubiquitin chain from RIG-I, and thus functions as a negative regulatory factor (52). Those activities might be promoted by FAT10, because the binding of FAT10 to RIG-I leads to reduced polyubiquitination of RIG-I (50).
Previous studies have reported several factors that regulate the activities of RIG-I via TRIM25. TRIM25 ubiquitin ligase itself is also regulated by ubiquitination. Liner ubiquitin assembly complex (LUBAC), comprising the HOIL-1L and HOIP proteins, conjugates K48-linked polyubiquitin chains to TRIM25, resulting in proteasome-dependent degradation of TRIM25 (53). USP15 is a ubiquitin-specific protease and cleaves the K48-linked polyubiquitin chain of TRIM25 (54).
NLRP12, a member of the Nod-like receptor (NLR) family, is expressed in dendritic cells and neutrophils (55) and recognizes Yersinia pestis infection, leading to inflammasome-mediated IL-1β processing and the production of mature IL-1β (56). NLRP12 also has inflammasome-independent functions that regulates RIG-I. The nucleotide-binding domain of NLRP12 physically interacts with TRIM25 and attenuates TRIM25-mediated K63-linked polyubiquitination (57). In addition, NLRP12 enhances the RNF125-mediated K48-linked polyubiquitination of RIG-I; thus, NLRP12 KO augments serum type I IFN levels and reduces viral titers in brain after intranasal inoculation with vesicular stomatitis virus (57).
NDR2 is a protein kinase and is involved in a variety of biological processes, such as apoptosis, tumorigenesis, and cell division (58). NDR2 deficiency decreases RIG-I-mediated type I IFN production, resulting in increased susceptibility to vesicular stomatitis virus and influenza A virus infection (59). The NDR2 protein associate with RIG-I and TRIM25 and promotes the TRIM25-mediated K63-linked polyubiquitination of RIG-I (59). ZCCHC3 is a zinc-finger protein and functions as a co-sensor of cGAS, which is a cytosolic DNA sensor. The protein binds to cytoplasmic viral RNA and acts as a co-receptor of RIG-I. Biochemical studies have shown that ZCCHC3 recruits TRIM25 to RIG-I, thereby facilitating K63-linked polyubiquitination (60). In addition, long non-coding RNA (lncRNA) also regulates RIG-I activation via TRIM25. For example, the lncRNA Lnczc3h7a binds TRIM25 (61). The association between lncRNA and TRIM25 occurs even in resting cells and promotes TRIM25-mediated K63-linked polyubiquitination of RIG-I during viral infection (61). The roles of those factors are summarized in Figures 1, 2. Since there are contradictory reports related to TRIM25 function (30, 31), further studies of those RIG-I regulators, such as FAT10, NDR2, ZCCHC3, and NLRP12, might reconcile the apparent contradictions.
Post-Translational Modification OF MDA5
The MDA5 protein is regulated by phosphorylation (62). In resting cells, the 2CARDs and the CTD of MDA5 are phosphorylated to suppress abnormal activation (63). After recognition of a viral RNA, phosphatase 1 (PP1) dephosphorylates MDA5, resulting in the activation of its downstream signaling (63). Since excessive activation of MDA5 causes systemic lupus erythematosus-like autoimmune diseases (64), its phosphorylation is thought to be important for the prevention of harmful MDA5 activation in non-infected cells. RIOK3, a protein kinase, phosphorylates the CTD of MDA5 and enhanced MDA5-mediated cytokine expression (65). However, the CARDs of MDA5 are not phosphorylated by RIOK3, and thus it is expected that there is another protein kinase that phosphorylates the CARDs of MDA5.
The MDA5 protein is also polyubiquitinated. As in RIG-I protein, the CARDs of MDA5 bind to K63-linked polyubiquitin chains. The K174 residue of the MDA5 CARDs is essential for this binding; a ubiquitination-defective K174A mutant of MDA5 failed to induce type I IFN expression (66). However, which ubiquitin ligase is responsible for K63-polyubiquitination remains unclear. Zhao et al. reported that TRIM40 binds to the CARDs of MDA5 and promotes K27- and K48-linked polyubiquitination of MDA5, leading to proteasomal degradation of the protein (41). Further studies are required to fully reveal the role of ubiquitin in the MDA5 activation.
Viral Proteins Targeting Ubiquitin Ligases
Several viral proteins target ubiquitin ligases to escape RIG-I-mediated innate immune responses. The NS1 protein of influenza A virus attenuates the IRF3 activation that is required for type I IFN expression (67). Gack et al. have shown that NS1 binds to TRIM25 and Riplet and attenuates the K63-linked polyubiquitination of RIG-I (68, 69). Hepatitis C virus persistently infects the host liver for several decades and can evade innate immune responses. Viral NS3-4A proteases cleaves viral poly-protein to produce mature viral proteins, and it also cleaves the MAVS adaptor, resulting in the release of MAVS from the mitochondrial outer membrane and thus inhibition of RIG-I-mediated type I IFN expression (10). The hepatitis C virus NS3-4A protein also binds to Riplet and inhibits Riplet-dependent antiviral activities (28, 34). Although NS3-4A cleaves and degrade Riplet in a protease activity-dependent manner, it has bee also postulated that there is a RIG-I-independent Riplet antiviral activity, attenuated by NS3-4A, plays a role (28, 34). RIG-I is able to recognize DNA virus infection (70, 71), and it has been shown that RIG-I senses HSV-1 infection via RNA5SP141 (72). Human papilloma virus E6 oncoprotein targets USP15 and TRIM25, thereby inhibiting RIG-I-mediated antiviral activities (73). In addition, it is also reported that viral proteins of EB virus antagonizes TRIM25 function (74). Considering that viruses have evolved to escape host innate immune responses (75), these observations support the notion that Riplet and TRIM25 are crucial for antiviral innate immune responses.
Perspective
An accumulating body of evidence has shown that ubiquitination is a key post-translational modification for RIG-I (62). At least four ubiquitin ligases are reported to be required for K63-linked polyubiquitination of RIG-I. Among these ligases, the antiviral activities of Riplet and TRIM25 have been confirmed by several groups. Recent studies have reported that TRIM25 is dispensable for RIG-I ubiquitination (30, 31, 38); however, it is still possible that other ubiquitin ligases, such as Mex3c and TRIM4, play redundant roles and thus compensate for a lack of TRIM25. To test this possibility, it will be necessary to uncover the underlying molecular mechanism via analysis of double or triple KO of TRIM25, Mex3c, and TRIM4. It is also possible that Riplet and TRIM25 regulate RIG-I in a cell-type specific manner. Another possibility is that the expression levels of ZCCHC3, NDR2, and NLRP12 affect the dependency on TRIM25. Further studies are required to reconcile the apparent discrepancy. On the other hand, the question of why there are so many factors that regulate RIG-I is yet to be answered. ZNF598, RNF125, c-Cbl, and TRIM40 are negative regulators of RIG-I, but their distinct roles remain elusive. Their redundancy and functional differences should be further investigated to provide greater insight into the regulation of RIG-I.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported in part by Grants-in-Aid from the Japan Agency for Medical Research and Development (AMED), the Ministry of Education, Science, and Technology (MEXT), and Japan Society of the Promotion of Science (JSPS).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank our laboratory members for their helpful discussion and Enago for English language editing.
References
1. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. (2004) 5:730–7. doi: 10.1038/ni1087
2. Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. (2005) 175:2851–8. doi: 10.4049/jimmunol.175.5.2851
3. Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, et al. Recognition of 5' triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. (2009) 31:25–34. doi: 10.1016/j.immuni.2009.05.008
4. Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5'-diphosphates. Nature. (2014) 514:372–5. doi: 10.1038/nature13590
5. Hur S. Double-stranded RNA sensors and modulators in innate immunity. Annu Rev Immunol. (2019) 37:349–75. doi: 10.1146/annurev-immunol-042718-041356
6. Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity. (2005) 23:19–28. doi: 10.1016/j.immuni.2005.04.010
7. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. (2006) 441:101–5. doi: 10.1038/nature04734
8. Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol. (2020). doi: 10.1038/s41577-020-0288-3. [Epub ahead of print].
9. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. (2005) 6:981–8. doi: 10.1038/ni1243
10. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. (2005) 437:1167–72. doi: 10.1038/nature04193
11. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. (2005) 122:669–82. doi: 10.1016/j.cell.2005.08.012
12. Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. (2005) 19:727–40. doi: 10.1016/j.molcel.2005.08.014
13. Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. (2008) 205:1601–10. doi: 10.1084/jem.20080091
14. Loo YM, Gale MJr. Immune signaling by RIG-I-like receptors. Immunity. (2011) 34:680–92. doi: 10.1016/j.immuni.2011.05.003
15. Peisley A, Wu B, Yao H, Walz T, Hur S. RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol Cell. (2013) 51:573–83. doi: 10.1016/j.molcel.2013.07.024
16. Wu B, Peisley A, Richards C, Yao H, Zeng X, Lin C, et al. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. (2013) 152:276–89. doi: 10.1016/j.cell.2012.11.048
17. Peisley A, Wu B, Xu H, Chen ZJ, Hur S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature. (2014) 509:110–4. doi: 10.1038/nature13140
18. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. (2011) 146:448–61. doi: 10.1016/j.cell.2011.06.041
19. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. (2003) 4:491–6. doi: 10.1038/ni921
20. Kouwaki T, Okamoto M, Tsukamoto H, Fukushima Y, Matsumoto M, Seya T, et al. Zyxin stabilizes RIG-I and MAVS interactions and promotes type I interferon response. Sci Rep. (2017) 7:11905. doi: 10.1038/s41598-017-12224-7
21. Beckerle MC. Zyxin: zinc fingers at sites of cell adhesion. Bioessays. (1997) 19:949–57. doi: 10.1002/bies.950191104
22. Hirota T, Morisaki T, Nishiyama Y, Marumoto T, Tada K, Hara T, et al. Zyxin, a regulator of actin filament assembly, targets the mitotic apparatus by interacting with h-warts/LATS1 tumor suppressor. J Cell Biol. (2000) 149:1073–86. doi: 10.1083/jcb.149.5.1073
23. Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. (2007) 446:916–20. doi: 10.1038/nature05732
24. Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, et al. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. (2010) 141:315–30. doi: 10.1016/j.cell.2010.03.029
25. Okamoto M, Kouwaki T, Fukushima Y, Oshiumi H. Regulation of RIG-I activation by K63-linked polyubiquitination. Front Immunol. (2017) 8:1942. doi: 10.3389/fimmu.2017.01942
26. Oshiumi H, Miyashita M, Inoue N, Okabe M, Matsumoto M, Seya T. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe. (2010) 8:496–509. doi: 10.1016/j.chom.2010.11.008
27. Oshiumi H, Matsumoto M, Hatakeyama S, Seya T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J Biol Chem. (2009) 284:807–17. doi: 10.1074/jbc.M804259200
28. Oshiumi H, Miyashita M, Matsumoto M, Seya T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. (2013) 9:e1003533. doi: 10.1371/journal.ppat.1003533
29. Gao D, Yang YK, Wang RP, Zhou X, Diao FC, Li MD, et al. REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS ONE. (2009) 4:e5760. doi: 10.1371/journal.pone.0005760
30. Cadena C, Ahmad S, Xavier A, Willemsen J, Park S, Park JW, et al. Ubiquitin-dependent and -independent roles of E3 ligase RIPLET in innate immunity. Cell. (2019) 177:1187–200. doi: 10.1016/j.cell.2019.03.017
31. Hayman TJ, Hsu AC, Kolesnik TB, Dagley LF, Willemsen J, Tate MD, et al. RIPLET, and not TRIM25, is required for endogenous RIG-I-dependent antiviral responses. Immunol Cell Biol. (2019) 97:840–52. doi: 10.1111/imcb.12284
32. Li MM, Lau Z, Cheung P, Aguilar EG, Schneider WM, Bozzacco L, et al. TRIM25 enhances the antiviral action of zinc-finger antiviral protein (ZAP). PLoS Pathog. (2017) 13:e1006145. doi: 10.1371/journal.ppat.1006145
33. Meyerson NR, Zhou L, Guo YR, Zhao C, Tao YJ, Krug RM, et al. Nuclear TRIM25 Specifically Targets Influenza Virus Ribonucleoproteins to Block the Onset of RNA Chain Elongation. Cell Host Microbe. (2017) 22:627–38 e627. doi: 10.1016/j.chom.2017.10.003
34. Vazquez C, Tan CY, Horner SM. Hepatitis C virus infection is inhibited by a noncanonical antiviral signaling pathway targeted by NS3-NS4A. J Virol. (2019) 93:e00725–19. doi: 10.1128/JVI.00725-19
35. Hage A, Rajsbaum R. To TRIM or not to TRIM: the balance of host-virus interactions mediated by the ubiquitin system. J Gen Virol. (2019) 100:1641–62. doi: 10.1099/jgv.0.001341
36. Kuniyoshi K, Takeuchi O, Pandey S, Satoh T, Iwasaki H, Akira S, et al. Pivotal role of RNA-binding E3 ubiquitin ligase MEX3C in RIG-I-mediated antiviral innate immunity. Proc Natl Acad Sci USA. (2014) 111:5646–51. doi: 10.1073/pnas.1401674111
37. Yan J, Li Q, Mao AP, Hu MM, Shu HB. TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J Mol Cell Biol. (2014) 6:154–63. doi: 10.1093/jmcb/mju005
38. Shi Y, Yuan B, Zhu W, Zhang R, Li L, Hao X, et al. Ube2D3 and Ube2N are essential for RIG-I-mediated MAVS aggregation in antiviral innate immunity. Nat Commun. (2017) 8:15138. doi: 10.1038/ncomms15138
39. Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci USA. (2007) 104:7500–5. doi: 10.1073/pnas.0611551104
40. Chen W, Han C, Xie B, Hu X, Yu Q, Shi L, et al. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell. (2013) 152:467–78. doi: 10.1016/j.cell.2013.01.011
41. Zhao C, Jia M, Song H, Yu Z, Wang W, Li Q, et al. The E3 ubiquitin ligase TRIM40 attenuates antiviral immune responses by targeting MDA5 and RIG-I. Cell Rep. (2017) 21:1613–23. doi: 10.1016/j.celrep.2017.10.020
42. Kato H, Fujita T. RIG-I-like receptors and autoimmune diseases. Curr Opin Immunol. (2015) 37:40–5. doi: 10.1016/j.coi.2015.10.002
43. Liu YC, Pan J, Zhang C, Fan W, Collinge M, Bender JR, et al. A MHC-encoded ubiquitin-like protein (FAT10) binds noncovalently to the spindle assembly checkpoint protein MAD2. Proc Natl Acad Sci USA. (1999) 96:4313–8. doi: 10.1073/pnas.96.8.4313
44. Nguyen NT, Now H, Kim WJ, Kim N, Yoo JY. Ubiquitin-like modifier FAT10 attenuates RIG-I mediated antiviral signaling by segregating activated RIG-I from its signaling platform. Sci Rep. (2016) 6:23377. doi: 10.1038/srep23377
45. Schmidtke G, Aichem A, Groettrup M. FAT10ylation as a signal for proteasomal degradation. Biochim Biophys Acta. (2014) 1843:97–102. doi: 10.1016/j.bbamcr.2013.01.009
46. Aichem A, Groettrup M. The ubiquitin-like modifier FAT10 in cancer development. Int J Biochem Cell Biol. (2016) 79:451–61. doi: 10.1016/j.biocel.2016.07.001
47. Garzia A, Jafarnejad SM, Meyer C, Chapat C, Gogakos T, Morozov P, et al. The E3 ubiquitin ligase and RNA-binding protein ZNF598 orchestrates ribosome quality control of premature polyadenylated mRNAs. Nat Commun. (2017) 8:16056. doi: 10.1038/ncomms16056
48. Sundaramoorthy E, Leonard M, Mak R, Liao J, Fulzele A, Bennett EJ. ZNF598 and RACK1 regulate mammalian ribosome-associated quality control function by mediating regulatory 40S ribosomal ubiquitylation. Mol Cell. (2017) 65:751–60 e754. doi: 10.1016/j.molcel.2016.12.026
49. DiGiuseppe S, Rollins MG, Bartom ET, Walsh D. ZNF598 plays distinct roles in interferon-stimulated gene expression and poxvirus protein synthesis. Cell Rep. (2018) 23:1249–58. doi: 10.1016/j.celrep.2018.03.132
50. Wang G, Kouwaki T, Okamoto M, Oshiumi H. Attenuation of the innate immune response against viral infection due to ZNF598-promoted binding of FAT10 to RIG-I. Cell Rep. (2019) 28:1961–70 e1964. doi: 10.1016/j.celrep.2019.07.081
51. Fan Y, Mao R, Yu Y, Liu S, Shi Z, Cheng J, et al. USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J Exp Med. (2014) 211:313–28. doi: 10.1084/jem.20122844
52. Friedman CS, O'Donnell MA, Legarda-Addison D, Ng A, Cardenas WB, Yount JS, et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. (2008) 9:930–6. doi: 10.1038/embor.2008.136
53. Inn KS, Gack MU, Tokunaga F, Shi M, Wong LY, Iwai K, et al. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol Cell. (2011) 41:354–65. doi: 10.1016/j.molcel.2010.12.029
54. Pauli EK, Chan YK, Davis ME, Gableske S, Wang MK, Feister KF, et al. The ubiquitin-specific protease USP15 promotes RIG-I-mediated antiviral signaling by deubiquitylating TRIM25. Sci Signal. (2014) 7:ra3. doi: 10.1126/scisignal.2004577
55. Arthur JC, Lich JD, Ye Z, Allen IC, Gris D, Wilson JE, et al. Cutting edge: NLRP12 controls dendritic and myeloid cell migration to affect contact hypersensitivity. J Immunol. (2010) 185:4515–9. doi: 10.4049/jimmunol.1002227
56. Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity. (2012) 37:96–107. doi: 10.1016/j.immuni.2012.07.006
57. Chen ST, Chen L, Lin DS, Chen SY, Tsao YP, Guo H, et al. NLRP12 Regulates Anti-viral RIG-I Activation via Interaction with TRIM25. Cell Host Microbe. (2019) 25:602–16 e607. doi: 10.1016/j.chom.2019.02.013
58. Cornils H, Kohler RS, Hergovich A, Hemmings BA. Downstream of human NDR kinases: impacting on c-myc and p21 protein stability to control cell cycle progression. Cell Cycle. (2011) 10:1897–904. doi: 10.4161/cc.10.12.15826
59. Liu Z, Wu C, Pan Y, Liu H, Wang X, Yang Y, et al. NDR2 promotes the antiviral immune response via facilitating TRIM25-mediated RIG-I activation in macrophages. Sci Adv. (2019) 5:eaav0163. doi: 10.1126/sciadv.aav0163
60. Lian H, Zang R, Wei J, Ye W, Hu MM, Chen YD, et al. The zinc-finger protein ZCCHC3 binds RNA and facilitates viral RNA sensing and activation of the RIG-I-like receptors. Immunity. (2018) 49:438–48 e435. doi: 10.1016/j.immuni.2018.08.014
61. Lin H, Jiang M, Liu L, Yang Z, Ma Z, Liu S, et al. The long noncoding RNA Lnczc3h7a promotes a TRIM25-mediated RIG-I antiviral innate immune response. Nat Immunol. (2019) 20:812–23. doi: 10.1038/s41590-019-0379-0
62. Chiang C, Gack MU. Post-translational control of intracellular pathogen sensing pathways. Trends Immunol. (2017) 38:39–52. doi: 10.1016/j.it.2016.10.008
63. Wies E, Wang MK, Maharaj NP, Chen K, Zhou S, Finberg RW, et al. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity. (2013) 38:437–49. doi: 10.1016/j.immuni.2012.11.018
64. Funabiki M, Kato H, Miyachi Y, Toki H, Motegi H, Inoue M, et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity. (2014) 40:199–212. doi: 10.1016/j.immuni.2013.12.014
65. Takashima K, Oshiumi H, Takaki H, Matsumoto M, Seya T. RIOK3-Mediated PHOSPHORYLATION of MDA5 interferes with its assembly and attenuates the innate immune response. Cell Rep. (2015) 11:192–200. doi: 10.1016/j.celrep.2015.03.027
66. Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, Grishin NV, et al. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity. (2012) 36:959–73. doi: 10.1016/j.immuni.2012.03.022
67. Talon J, Horvath CM, Polley R, Basler CF, Muster T, Palese P, et al. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol. (2000) 74:7989–96. doi: 10.1128/JVI.74.17.7989-7996.2000
68. Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. (2009) 5:439–49. doi: 10.1016/j.chom.2009.04.006
69. Rajsbaum R, Albrecht RA, Wang MK, Maharaj NP, Versteeg GA, Nistal-Villan E, et al. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. (2012) 8:e1003059. doi: 10.1371/journal.ppat.1003059
70. Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol. (2009) 10:1065–72. doi: 10.1038/ni.1779
71. Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. (2009) 138:576–91. doi: 10.1016/j.cell.2009.06.015
72. Chiang JJ, Sparrer KMJ, van Gent M, Lassig C, Huang T, Osterrieder N, et al. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat Immunol. (2018) 19:53–62. doi: 10.1038/s41590-017-0005-y
73. Chiang C, Pauli EK, Biryukov J, Feister KF, Meng M, White EA, et al. The human papillomavirus E6 oncoprotein targets USP15 and TRIM25 to suppress RIG-I-mediated innate immune signaling. J Virol. (2018) 92:e01737–17. doi: 10.1128/JVI.01737-17
74. Gupta S, Yla-Anttila P, Callegari S, Tsai MH, Delecluse HJ, Masucci MG. Herpesvirus deconjugases inhibit the IFN response by promoting TRIM25 autoubiquitination and functional inactivation of the RIG-I signalosome. PLoS Pathog. (2018) 14:e1006852. doi: 10.1371/journal.ppat.1006852
Keywords: RIG-I, MDA5, innate immunity, ubiquitin, virus, type I interferon
Citation: Oshiumi H (2020) Recent Advances and Contradictions in the Study of the Individual Roles of Ubiquitin Ligases That Regulate RIG-I-Like Receptor-Mediated Antiviral Innate Immune Responses. Front. Immunol. 11:1296. doi: 10.3389/fimmu.2020.01296
Received: 06 April 2020; Accepted: 22 May 2020;
Published: 24 June 2020.
Edited by:
Thomas A. Kufer, University of Hohenheim, GermanyReviewed by:
Michaela Gack, University of Chicago, United StatesRicardo Rajsbaum, University of Texas Medical Branch at Galveston, United States
Katrin Rittinger, Francis Crick Institute, United Kingdom
Copyright © 2020 Oshiumi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroyuki Oshiumi, oshiumi@kumamoto-u.ac.jp