 Stefano A. P. Colombo
Stefano A. P. Colombo Richard K. Grencis
Richard K. Grencis- 1Department of Tropical Disease Biology, Liverpool School of Tropical Medicine, Liverpool, United Kingdom
- 2Division of Infection, Immunity and Respiratory Medicine, Wellcome Trust Centre for Cell Matrix Research, Lydia Becker Institute for Immunology and Inflammation, The University of Manchester, Manchester, United Kingdom
Infection with soil-transmitted helminths (STH) remains a major burden on global health and agriculture. Our understanding of the immunological mechanisms that govern whether an individual is resistant or susceptible to infection is derived primarily from model infections in rodents. Typically, experimental infections employ an artificially high, single bolus of parasites that leads to rapid expulsion of the primary infection and robust immunity to subsequent challenges. However, immunity in natura is generated slowly, and is only partially effective, with individuals in endemic areas retaining low-level infections throughout their lives. Therefore, there is a gap between traditional model STH systems and observations in the field. Here, we review the immune response to traditional model STH infections in the laboratory. We compare these data to studies of natural infection in humans and rodents in endemic areas, highlighting crucial differences between experimental and natural infection. We then detail the literature to date on the use of “trickle” infections to experimentally model the kinetics of natural infection.
Introduction
Soil-transmitted helminths (STH) are a highly diverse group of parasites present across the globe. Chronic life-long infection with at least one species of STH is common for most vertebrates (1). This includes humans and livestock in low to middle income countries (LMIC). The morbidity and reduced fitness associated with infection make STH helminthiases a major concern both for global health and for agriculture in endemic areas (2, 3). The infectious stages of these parasites are abundant in the environment and, due to their robustness against environmental insult, can persist there for long periods. The longevity of these parasites is compounded by their capacity to act as potent immunomodulators of their hosts (4).
A key determinant in the relationship between a host and STH parasite is the host's immune response. The host must balance an effective response to the parasite with limiting potentially detrimental immunopathology and exhausting vital resources (5). Similarly, the parasite must promote an immune response in the host that supports its own survival but that also protects the host from excessive pathology and infection by other potential pathogens. Given this, it is highly likely that anti-parasite immune responses have evolved to limit parasite burden and promote wound repair rather than to cause rapid and total parasite expulsion.
The majority of studies on immune responses to STHs are performed using rodent-specific STHs that have been adapted to the laboratory setting. These include the gastrointestinal (GI) nematodes, Trichuris muris, Heligmosomoides sp. (Formally Nematospiroides dubius. Herein we refer to the laboratory strain as H. bakeri and those identified in wild rodents as H. polygyrus. It should be noted, however, in the literature to date these names have been used interchangeably for experimental infections), Trichinella spiralis and Nippostrongylus brasiliensis. Additionally, in some cases, human-specific species can be experimentally modeled in rodents, for example Necator americanus (6–9) and Ancylostoma ceylanicum (10, 11) albeit with limited success. Further, the larval migration that occurs during ascariasis, and hookworm infection, can be modeled in mice using the porcine STH, Ascaris suum (12).
Traditional experimental infections using well-established models typically rely on infecting mice with a single, artificially high dose of parasites. This is in contrast to the natural scenario in which frequent low-level exposures are likely to be more common. There is also a clear difference between the kinetics of traditional experimental infections and those seen in naturally infected populations. Thus, should we wish to fully understand the nuances of STH infection, there is a need to ensure that we are accurately modeling the natural situation.
Infection in the Laboratory
Whilst there are species-specific responses based on the model STH used, many aspects of the immune response to experimental high-dose infections can be generalized. Upon invasion of the host, and often throughout infection, STHs cause considerable damage to tissue surrounding the site of infection. Migration of N. brasiliensis through the lung causes gross changes in tissue architecture and long term damage (13). Likewise, invasion of the gut epithelium and lamina propria, by T. muris and H. bakeri, respectively, causes considerable remodeling of the intestinal environment (14, 15).
Breaches by STHs at these barrier sites are associated with the release of alarmins, particularly interleukin (IL)-25, IL-33, and thymic stromal lymphopoietin (TSLP) (16–19). These cytokines trigger innate responses and prime the induction of an adaptive type-2 (Th2) immune response (16, 20, 21). They have also been established as essential to protection against infection with a number of model STHs (22–25). Epithelial cells themselves are potent reservoirs of these cytokines (26–28). Of recent interest is the role tuft cells play in sensing and responding to STH infection. Tuft cells are an epithelial cell subset that exist at low frequency during homeostasis but rapidly proliferate following STH infection (28–30). They sense the presence of STHs and intestinal microbes via taste-chemosensory receptors such as TRPM5 (29, 31) and secrete IL-25 and cysteinyl leukotrienes (CysLTs) to support the establishment of a Th2 mucosal response (28, 32).
Among the first lymphoid responders are the type-2 innate lymphoid cells (ILC2s). ILC2s have been shown to expand during STH infection and act as early sources of IL-4, IL-5 and IL-13 (33–35). Their depletion results in the delayed induction of Th2 immunity (33), although a non-redundant role for these cells in parasite expulsion has only been demonstrated for N. brasiliensis (36, 37). A broad role for alternative activation of macrophages (M2) has also been shown in most model STHs. M2s are required for the trapping and killing of the larval stages of H. bakeri and N. brasiliensis (38–40), this function is dependent on the production of arginase-1 (Arg-1) (41) and can be regulated by the expression level of resistin-like molecule (RELM)-α (40). Whilst expansion of other innate cells – including neutrophils, eosinophils, basophils, and mastocytes—at sites of infection is well-documented (42–44) a functional role for these cells in parasite expulsion has been harder to define and in some cases may be species-specific. For example, depletion of basophils is sufficient to trigger susceptibility to T. muris infection (45) but has no impact on resistance to H. bakeri (46). Similarly, mast cells and eosinophils have been linked to resistance to H. bakeri and T. spiralis (46, 47) but are redundant for expulsion of T. muris (48). Further, neutrophilia has been linked with expulsion of N. brasiliensis and H. bakeri (41, 42), via the release of neutrophil extracellular traps (NETs) (49) and support of M2 polarization (50). However, in cases where ablation of a given cell type does not result in a failure to attenuate infection, these cells may instead function to repair tissue damage once the infection has been resolved (51), or to moderate ongoing responses (40, 52). Alternatively, they may act to prime distal mucosal sites against future infection with other STH species, for example ILC2s primed by T. spiralis infection in the gut migrate to the lung and contribute to protection against a subsequent N. brasiliensis infection (53). Similarly, infection with H. bakeri results in protection against N. brasiliensis infection via IL-33-dependent induction of IL-5+CD4+ T cells capable of recruiting activated eosinophils to the lung (54).
Central to the expulsion of STHs is the CD4+ T cell. This can be inferred form studies of athymic nude mice which sustain long term high dose infections, compared to WT mice which readily expel parasites (55, 56). Depletion or ablation of CD4+ cells is enough to induce to susceptibility to infection in otherwise resistant mouse strains (15, 57). Further, adoptive transfer of CD4+ T cells to T and B cell deficient mice is sufficient to confer protection against infection (58). It is noteworthy that T cell deficient mouse strains such as athymic mice or recombinase 1 or 2 deficient mice still have a functional ILC2 compartment (36, 59, 60). A key function of CD4+ T cells is to provide Th2 cytokines—over and above those produced by ILC2s—in particular IL-4 and IL-13 which signal through IL-4 Receptor α (IL-4Rα) (61). IL-4Rα signaling drives a broad array of down-stream responses that are essential for the expulsion of STHs. These include; hyperproliferation of goblet cells (62); increased expression and secretion of mucins and anti-parasitic peptides, such as Muc5ac and RELM-β (63–66); increased turnover of epithelial cells (67, 68); enhanced gut contractility (69); immunoglobulin (Ig) class-switching to generate parasite-specific IgG1 (46, 70); and polarization of macrophages to an M2 phenotype (41, 71). The CD4+ T cell is also likely to be key to adaptive immune memory to STH infections. Under laboratory conditions, in immunocompetent mice, in response to a high-dose infection, these responses are robustly generated and lead to relatively rapid expulsion of the infection; although the kinetics differ based on genetic background of the host (39, 72–74).
In laboratory models of STH infection, as well as driving parasite expulsion, the immune response to a primary high-dose infection is sufficient to generate immunity to subsequent challenge infections (75–78). During secondary challenges the rate of expulsion is significantly accelerated. Depending on the species this may be a result of enhanced larval trapping mediated by parasite-specific IgG1 (46), priming of localized immune cells (50), or via expeditious induction of mucin secretion or mast cell activity. Regardless of mechanism, high-dose experimental infections produce robust sterilizing immunity to secondary infection.
Natural STH Infection in Mammals
The artificially high doses given during experimental infections have proven a reliable system in which to investigate fundamental mechanisms of resistance to STH infection. However, this regime fails to reflect infection in natura. Not only is a single high-dose of parasites unrealistic in the wild, laboratory rodents are housed in pathogen free environments, with an abundance of resources, and a significant limit to stressors such as predators.
Experimental high-dose infections present a scenario in which primary STH infection is limited in duration, characterized by an immediate and potent Th2-polarized immune response, and generates sterilizing immunity to subsequent challenges. However, epidemiological evaluation of STH burden in human populations shows that in endemic areas, infected individuals suffer chronic parasitism throughout their lives (79–82). This holds true for non-human primates (83–85), livestock (86, 87), and wild rodent populations (88, 89). In humans, infection burden correlates strongly with age following one of two patterns: (i) parasite burden builds rapidly during early childhood but peaks shortly before adolescence, burden then declines and plateaus at a low level throughout adulthood e.g., Trichuris trichiura, and Ascaris lumbricoides (79, 90, 91); or (ii) STH burden builds consistently throughout childhood and early adolescence but plateaus at a moderate level prior to adulthood e.g., Necator americanus and Ancylostoma duodenale (82). Both patterns indicate that protective immunity to infection develops with age. However, they also suggest that this protection is incomplete and is preceded by a sustained period of susceptibility.
Our understanding of immune responses to STH infection in natura is limited. Much of what is known is founded on inferences drawn from blood samples taken from individuals living in endemic regions. As such, these data are caveated by an array of confounding factors. What is clear is that, in humans, up-regulation of the Th2 immune response is associated with STH infection. Importantly, Th2 associated markers, such as IgE and Th2 cytokines, show a clear negative correlation with worm burden (90, 91). Further, a strong IL-5 response in peripheral blood mononuclear cells (PBMC) isolated from infected individuals was shown to be predictive of resistance to reinfection following anthelminthic treatment (92, 93). A recent study utilizing mass cytometry to profile the Th2 and regulatory compartments before and after deworming in an Indonesian cohort confirmed a clear link between infection status and the expansion of ILC2s and Th2 cells, and reaffirmed the role of these cells in production of Th2 cytokines (94). Thus, a functional role for Th2 immunity in resistance to STH infection in natura is likely. What is interesting is that the development of this response is age-associated, with observed increases in anti-parasite IgE levels, and IL-13 and IL-4 production, in older individuals within the same cohort concurrent with a decrease in Th1-associated cytokines (90, 91, 95, 96). However, a complete polarization to Th2 immunity is rare, with most individuals maintaining a mixed Th1/Th2 response. The inability to generate a fully polarized protective response may be, in part, a consequence of STH-mediated immunosuppressive mechanisms. In one human cohort, deworming with the anthelmintic Albendazole resulted in an increase in STH-specific cytokine responses, and correlated with CD4+ T cells decreasing expression of the inhibitory molecule cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) (97). A role for STH-controlled immune suppression in humans is supported by evidence that peptides derived from human-infecting hookworms (Necator americanus and Ancylostoma duodenale) are able to induce IL-10 and TGF-β signaling, and suppress IL-13 secretion in rodent models of colitis and allergy (98–100).
Age-associated, slowly developed resistance to infection is not unique to humans. Both feral and domestic sheep show progressive decreases in STH infection prevalence and fecal egg burden with age (101–103), as do domestic cattle (104). Wood mice (Apodemus sylvaticus) show an age-associated plateau in infection intensity of H. polygyrus akin to that seen in human hookworm infection (88). Further, non-human primates demonstrate slow acquisition of immunity to STHs following long periods of susceptibility, with the infection intensity kinetics of T. trichiura and other STHs paralleling those seen in humans (84, 85). Unfortunately, whilst the kinetics of these infections are broadly characterized, there is little in the way of immunological data accompanying these parasitological findings. However, it has been observed in wood mice that H. polygyrus-specific IgG1 titers increase with age and that treatment with the anthelminthics Ivermectin and Pyrantel was more effective in older mice relative to younger animals (105). This is consistent with a role for IgG1 in host-protection against Heligmosomoides (70).
Of significant importance to the outcome of infection is the overlapping geographical distribution of these parasites; not only with one another but also with other pathogens. STH-STH co-infections are highly common, and have been shown in a number of human cohorts to occur more frequently than single STH infections (106–108). In cases of STH-STH co-infection, infected individuals exhibit higher levels of infection for each individual species relative to individuals with a single-species infection (107, 108). From field data it remains unclear as to whether this is a correlative effect—i.e., an individual susceptible to infection with one species is simply more likely to be susceptible to infection with other STHs—or if STHs act synergistically by activating mechanisms that increase host susceptibility to infection. Experimental co-infection with H. bakeri and T. muris/T. spiralis has demonstrated that mice normally resistant to infection with T. muris or T. spiralis are rendered susceptible to infection when concurrently infected with H. bakeri (109, 110), although the mechanism through which this is mediated remains unresolved. Conversely, existing infection in the gut with H. bakeri or T. spiralis is protective against subsequent N. brasiliensis infection via priming of host-protective responses (53, 54). STH coinfection is also highly common with protist, bacterial and viral pathogens important to human health including malaria, tuberculosis and HIV. The primary focus of research into coinfections of this nature has been the effect STH coinfection has on the outcome of immune responses targeted the other pathogen. The overarching hypothesis being that as potent, and chronic, inducers of Th2/T-regulatory immunity STH infection will suppress the required Th1 immunity that targets single-cell/viral pathogens and thus increase susceptibility to infection and/or impact the efficacy of vaccines (111–113). However, it is also likely that infection with such pathogens will feedback onto the immune response against the STH. Therefore, it is important to bear in mind that in natura STH infections do not exist in isolation and have evolved in the context of a host immune system responding to a complex mix of co-infecting pathogens that elicit a diverse range of responses. Developing model systems with which to interrogate this reality presents exciting and challenging opportunities.
Modeling Natural Infection
Given the difference in lifestyles experienced by laboratory rodents and their wild counterparts, it is perhaps unsurprising that there are considerable differences in their immune systems. Wild mice appear to exist in a state of higher immune activation with a more diverse repertoire of effector/effector-memory cells (114, 115) likely due to greater antigenic exposure (116). This may, in part, explain differences in observations between laboratory experiments, and natural exposure to STHs.
Few studies have attempted to experimentally mimic a “natural” setting for STH infection. Co-housing different mouse strains in large in-door enclosures and allowing for “natural” infection of H. bakeri (through contact with larvae in the enclosure as opposed to controlled oral administration) removed strain-specific resistance to infection resulting in longer-lived infections in BALB/c mice (117). Given the time period in which this experiment was conducted, the means through which this change in immune response occurred was not investigated. It could be speculated that co-housing mice on different backgrounds resulted in a change in the composition of the microbiome rendering previously resistant mice more susceptible to infection. It has previously been shown that strain specific resistance to infection by the enteric bacterial pathogen Citrobacter rodentium can be imposed on normally susceptible mice via fecal transfer from a resistant mouse strain; this effect was mediated by induction of host innate responses including IL-22-stimulated production of antimicrobial peptides (118, 119). However, whether a similar effect can be achieved with model STHs has yet to be shown.
More recently, C57BL/6 mice housed in controlled outdoor enclosures (a process known as “rewilding”) were shown to become susceptible to high-dose T. muris infection and exhibited impaired IL-13 production (120). Similar to observations in humans, higher worm burdens and biomass were correlated with reduced numbers of IL-13+CD4+ cells and increased frequency of IFNγ+CD4+ cells. The authors also found rewilding resulted in a marked increase in fecal microbial diversity. It will be of great interest to define the precise relationship between this increase in community diversity and the outcome of infection. In a subsequent analysis comparing uninfected mice housed in specific pathogen free (SPF) to those that were rewilded, it was shown that overall composition of blood and mesenteric lymph node immune cells was dramatically altered by the rewilding process including increases in central and effector memory T cells (121, 122). Interestingly, germ-free mice reconstituted with the caecal content of rewilded mice showed a significant increase in the proportion of granulocytes—in particular neutrophils—in the peripheral blood relative to mice reconstituted with caecal contents from SPF mice (121). Thus, rewilding has a profound and complex effect on immune cell composition, in part regulated by the microbiome, that may be responsible for impaired resistance to STH infection.
Together, these data suggest that inbred laboratory mice are not simply innately more resistant to STH infection than their wild counterparts, but that that environmental context is a major influence over the outcome of STH infection. Whilst studies that seek to recreate a more natural setting are valuable in bridging the gap between the laboratory and the field, they require an abundance of space and specialized facilities. They also re-introduce a myriad of confounding variables that reductionist laboratory model systems aim to nullify. Thus, the challenge is to develop an infection regime that is easily applicable to a traditional laboratory setting, recapitulates the dynamics observed in natural infections, and that limits the introduction of confounding factors.
Trickle Infection
One factor that is easy to manipulate in a controlled fashion is the dose of parasites administered. In the T. muris system altering single infection dose within a single inbred strain of mouse is sufficient to change both resistance phenotype and the polarization of the immune response (123) with a high dose infection generating a Th2 response and acute infection and a single low dose, chronicity through the generation of a Th1 response (76). However, a single low-dose T. muris infection, in which a chronic infection characterized by a regulated Th1 response is established, also does not recapitulate the dynamic shift from susceptibility to a partial resistance, the observation generally seen in the field. The concept of “threshold” and “subthreshold” levels of infection associated with resistance or susceptibility is not new [see review by Behnke (124)]. Indeed, several observations from both natural and experimental STH infections in ruminants suggest that for some parasites—such as Ostertagia ostertagia (125), Nematodirus battus (126) and Trichostrongylus sp (127, 128)—“lower levels” of infection are consistent with longer survival of parasite burdens.
Historically, there has been interest in so-called “trickle” infections. In principal the trickle infection seeks to mimic natural exposure to a parasite by infecting animals with frequent low-doses of a given STH rather than a single high-dose.
Early on it was observed that, in rats, daily doses of five, third larval stage (L3) N. brasiliensis over either a 12 or 16 week period resulted in steady increase in worm numbers and egg output during the observation periods. Adult worm numbers at the end of the experiments were ≤ 30% of the total number of L3 administered. This data is, therefore, suggestive of partial immunity developing particularly in the latter stages of infection with stunted female worms containing reduced eggs numbers and the presence of few pre-adult/larval stages present. Indeed, if a large (50–1000 L3) single dose challenge infection was then administered to “trickled” rats, clear resistance was evident. When the trickle infection regime was increased to 50 L3 per day over 16 weeks, a rapid increase in parasite burden was observed that peaked at 2 weeks post infection, followed by a steady decline in infection intensity and an increase in the proportion of “stunted” adult female worms for the duration of experiment (129). The kinetics of this infection regime suggested that, in contrast to a high-dose infection in which a robust immune response would drive rapid parasite expulsion, the immune response developed slowly with repeated exposure and was only partially effective i.e., it limited subsequent infections but did not prevent their establishment. Although levels of infection differed, similar observations were made by Ovington (130). The immune response during trickle infections of N. brasiliensis has received little attention. With regards to peripheral antibody responses to parasite surface antigens, there were few differences between single dose and trickle infections (131). Ferens et al. (132, 133) using a shorter trickle of 10 infections of 25 L3 over a 4 week period, followed by a single large challenge, observed that trickle infections primed for a much more robust lung inflammation during the migratory phase of the infection through the lungs, than a single large dose priming infection. Bronchiolar lavage showed that trickle infection generated a marked elevation in eosinophils and alveolar macrophages. This may be indicative that trickle can effectively prime for robust immune mechanisms operating against pre-intestinal larval stages.
In concert with these observations, twice weekly trickle infection of 10–50 H. bakeri L3 in mice (134) or 30–50 Ancylostoma ceylaniucum larvae in hamsters (135) showed similar increases in worm burden followed by a steady decline. This slow expulsion of infection for both species was inhibited by treatment with cortisone (134, 135) suggesting immune control and induction of at least partial immunity by trickle infections. Again, in both systems, a high dose challenge after trickle was largely expelled although some worms still remained in the intestine.
A short-term trickle infection using T. muris demonstrated that infecting C57BL/6 mice on alternate days over the first 35 days of infection resulted an accumulation of parasites. Cytokine and serological analysis at the experimental end-point suggested that the trickle infected mice had an immunophenotype that was intermediate between mice that were infected with a Th2 polarizing high-dose infection and mice that had received a single low-dose infection known to drive a Th1 response (76). This intermediate phenotype parallels the immune-status of individuals in endemic regions that show a mixed Th1/Th2 response as opposed to the strong Th2 polarized responses seen in traditional experimental STH infections in rodents. However, trickle infection of Balb/K mice, a strain that is markedly more resistant than C57BL/6 to a single high dose infection, indicated that although trickle does lead to maintenance of worms within the intestine, lower levels of trickle were required to achieve this and this strain generated stronger Th2 response to the infection.
Similarly, CBA mice susceptible to H. bakeri infection had a blunted immune response during trickle infection and failed to initiate parasite expulsion compared to resistant SWR mice which were able to reduce their parasite burdens (136). Thus, as is evident with single-dose infections, genetic background can influence the progression of trickle infections.
Experimental trickle infections of Trichuris suis have also been undertaken in pigs. Pedersen and Saeed (137) used a trickle regime of 250 eggs twice weekly for 4 weeks and showed that substantial numbers of worms could be found in the gut at week 4 post infection although numbers were considerably reduced by week 14 (137). Trickled animals challenged at this point were significantly immune to a single high dose challenge infection. Nejsum et al. (138) used a more intense trickle regime administering at least 100 eggs per day over a 4, 8, or 14-week period, i.e., cumulative infections of ~ 4,000, 11,000, and 30,000 eggs, respectively. Significant numbers of worms (hundreds) were observed in the intestines at weeks 4 and 8, much lower than the numbest of eggs received. By week 14 few parasites were found in the intestine. Taken together, the data indicates immunity to T. suis can be built up after trickle infections over time and that the dynamics can be affected by the specific conditions of the trickle infection regime used.
We have recently reported a detailed characterization of a long-term trickle infection with T. muris (139). By performing weekly infections of 20 embryonated T. muris eggs in C57BL/6 mice we observed infection kinetics that closely mimicked those seen in human T. trichiura infection. Worm burden rose steadily with subsequent infections for 9 weeks, however, at 11 weeks we observed a decrease in worm burden and an absence of very early larval stages in the caeca of infected mice. This apparent acquisition of immunity correlated with an increase in Th2-associated immune responses including goblet cell hyperplasia, Muc5ac expression, and accelerated epithelial turnover (139). Importantly, depletion of CD4+ cells during the period of expulsion after trickle, removes protection. Given that these responses have been linked to resistance in previous studies using high-dose infection (66, 67, 140) it is reasonable to conclude that the modes by which resistance to T. muris are mediated are similar if not identical between single-dose and trickle infection. What is different between these modes of infection is the environment in which the initial response develops. In single high-dose infection immunologically naïve mice are a blank slate in which Th2 immunity can be rapidly generated. However, during trickle infection a Th2 response must develop in the context of an on-going Th1 response. Given that these types of immunity are mutually antagonistic, understanding the processes through which an on-going Th1 response transitions into a Th2-dominated state will be undoubtedly illuminating on fundamental mechanisms of immune regulation. Thus, a number of interesting questions present themselves.
Is There a Set Threshold of Worm Burden That Can Be Tolerated Before Mechanisms of Expulsion Are Generated?
During trickle infection a transition occurs between susceptibility to infection and subsequent resistance to future challenges. In the case of T. muris this transition is dependent upon the number of doses, with fewer doses being insufficient to generate a protective immune response given the same exposure time (139). This would suggest that it is parasite burden, not the length of exposure that is essential in driving a shift in immune response. A dose-dependency in response to STHs can clearly be seen when comparing the outcome of single high-dose (acute, Th2-dominated) and low-dose (chronic, Th1 dominated) T. muris infection (123). However, what drives this polarization remains unclear. It is possible that there is a genetically set threshold of STH burden, influenced by local environment, that may be tolerated by the host beyond which point the cost exerted by the parasite becomes too great and must be reduced. One explanation could be that there exists a balance between the host response and STH-mediated immune-regulation designed to suppress Th2 immunity (141–143) and the effect of tissue damage that necessitates Th2-dependent wound repair (144). Were this the case, when the damaged caused by the STH becomes greater than its ability to immune-modulate the host, protective immunity is induced. These processes would be dynamic and change as number of infection events alter and the host responds. Recent work using single-dose T. muris infections has implicated B cells as important regulators of the balance between Th1/Th2 immunity. In BALB/c mice, which produce a potent Th2 response to a single high dose T. muris, infection antibody depletion of B cells had no effect. However, in C57BL/6 mice, which initially show a mixed Th1/Th2 response following infection, depletion of B cells resulted in an increase in Th1 cytokines, enhanced IFNγ-associated gene expression, and susceptibility to infection (145). This effect was antibody-independent and places B cells as potential regulators of IFNγ signaling during mixed Th responses. It is exciting to speculate that B cells, whilst previously thought to be largely dispensable for protection against T. muris (56, 57, 146), may play a role in tuning the Th response during trickle infection.
With each subsequent dose of parasite, as parasite burden increases, the relative concentration of available antigen is likely to rise proportionally. The effect of antigen load on T cell receptor (TCR) activation in STH infection remains poorly understood. Based on in vitro studies, using recombinant peptides not derived from STHs, it is canonically thought that a high level of TCR signaling, stimulated by higher antigen concentration or “quality,” favors Th1 differentiation whereas weaker signals allow for Th2 polarization (147). These observations were also mirrored in vivo (148) where it was speculated that paradoxically, large pathogens such as STH do not release amounts of antigen that readily gain access to antigen processing pathways, unlike rapidly dividing microbes. This may act in concert with immunomodulatory mechanisms employed by helminths. Antigen released from the eggs of the blood fluke Schistosoma mansoni actively reduces dendritic cell-T cell interactions lowering activation signal strength and directly biasing toward Th2 differentiation (149). This proactive induction of Th2 immunity by S. mansoni is thought to protect the host against severe pathology caused egg passage (150).
Is There a Role for Tissue Damage in the Induction of Th2 Responses During Trickle Infection?
There is a well-established link between tissue damage and the type-2 immune response. As large macroparasites, STHs cause considerable damage upon invasion of the host and during the course of infection. Indeed, it has been argued that the responses to tissue damage and STH infection co-evolved so that mechanisms that facilitate parasite expulsion also mediate wound repair (144). Tissue damage results in epithelial, mesenchymal and innate-derived cytokines, capable of inducing Th2 responses, being released; these include TSLP, IL-25 and IL-33 (Figure 1) (25, 28, 151). Subsequent expansion of ILC2s, eosinophils, basophils, M2s and Th2 cells promotes/regulates both parasite expulsion and wound healing. As these cytokines are often produced as a result of cell damage, their concentrations present during infection are likely to reflect the magnitude of damage caused. Indeed, a role for IL-25 has been posited in late-stage expulsion of H. bakeri functioning as a key inducer of effector responses against adult-stage parasites (152). Therefore, during a trickle infection the concentration of these alarmins may increase with each subsequent challenge until a threshold concentration is reached that is sufficient to drive a protective response. This notion is consistent with the slow but progressive increase in Th2-associated responses observed during T. muris trickle (139). Further, damage-inducing microparticles have been previously shown to act as potent adjuvants capable of driving innate and antigen-specific Th2 immune responses in vivo comparable in efficacy to Alum (153, 154), as does mechanical abrasion (155). Epithelial derived micro(mi)RNAs are also known to influence resistance to STHs. Epithelial specific deletion of Dicer, a key gene encoding an RNAase involved in miRNA action, can change resistance to T. muris to susceptibility. MiR-375 was identified as an important miRNA in epithelial cells and deletion of MiR-375 in mice phenocopies the Dicer null response to T. muris (156). Little is known of the miRNA response to T. muris after trickle infections. A combination of the repeated release of alarmins, miRNAs and the Th2-specific adjuvant effect of tissue damage resulting from regular repeat infection may facilitate protective immunity.
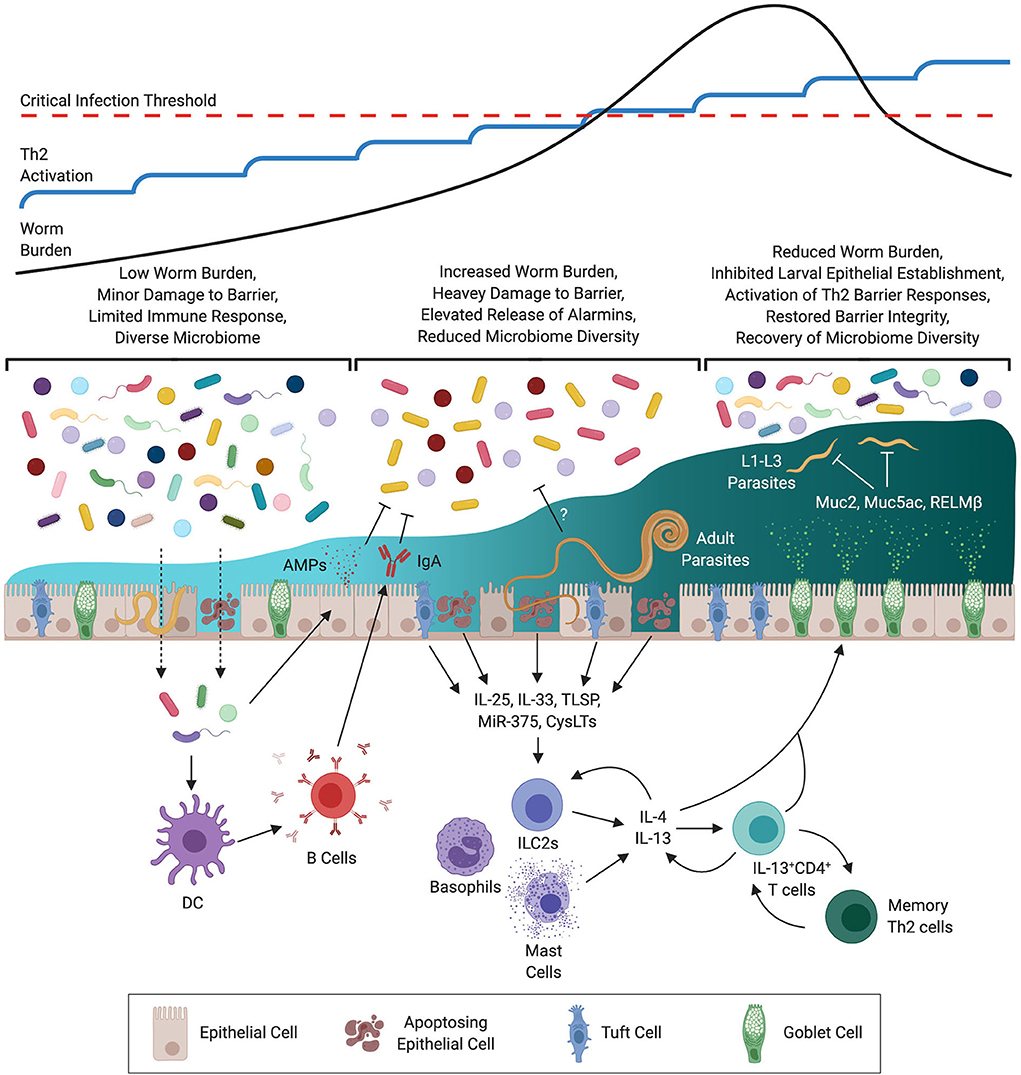
Figure 1. Development of Immunity During Trichuris muris Trickle Infection. At the outset of infection the low level of worm burden results in minor damage to the epithelial barrier. Whilst insufficient to drive a protective Th2 response this minor damage may be sufficient to allow for opportunistic invasion by commensal bacteria triggering the release of antimicrobial peptides (AMPs) and IgA. With repeated infections the level of barrier damage is exacerbated resulting in increased release of alarmins, micro RNAs (MiRs), and cysteinyl leukotrienes (CysLTs) from epithelial, mesenchymal, and innate cells. During this time a decrease in diversity of the microbiome is observed, this may be a result of immune-mediated regulation to prevent invasion by opportunistic pathogenic bacteria, or via STH-mediated remodeling. Activation of innate cells by type-2 signals results in the release of type-2 cytokines (IL-4 & IL-13) resulting in polarization of CD4+ T cells to a Th2 phenotype. Th2 cells then amplify the level of IL-4 & IL-13 signaling to activate host-protective responses at the epithelial barrier including goblet cell hyperproliferation, production of mucins such as Muc5ac, and heightened epithelial cell turnover. These responses operate primarily on early larval stages (L1–3) limiting the establishment of juvenile parasites within the epithelium. As a consequence barrier integrity is restored and intestinal microbial communities recover.
How Is an Effective Memory Response Generated During Trickle Infection?
Consistent with single-dose infection models there is an essential role for CD4+ T cells in immunity during trickle infection (139). These cells likely act as the dominant source of IL-13 that drives anti-parasite effector mechanisms. During infection, homing of T cells to the site of infection is essential for effective parasite expulsion (58, 157, 158). Following single high-dose STH infection, the memory T cells generated persist in the mucosa long after parasite expulsion and are sufficient to facilitate protection against subsequent challenge (159–161). However, the memory T cells generated following high-dose infection were primed in the context of a potent Th2 response, whereas memory cells generated during the early stages of a T. muris trickle infection will likely have been polarized to a Th1 phenotype. Moreover, the activity of the potentially pathogenic Th1 cells is regulated by IL-10 (162, 163) and thus regulation of ongoing responses also accompanies trickle infections. This scenario raises the question as to whether the CD4+ cells required for protective immunity in T. muris trickle infection arise from early memory T cells whose phenotype is plastic and informed by de novo production of Th2 cytokines by innate cells/environment, or if new CD4+ T cells are recruited later in infection? Repolarization of Th1 effector T cells into Th2 cells has previously been shown as a result of STH infection, OVA-specific Th1 cells transferred into naïve mice adopted a Th2 phenotype during N. brasiliensis infection (164), however, this was not in the context of an ongoing Th1 response. Instead, if naïve CD4+ T cells are recruited that differentiate into Th2 cells it will be interesting to determine if their TCR repertoire differs from Th1 cells generated early in infection. Identification of the specific antigens recognized by CD4+ T cells following the development of resistance under a trickle infection may provide a fertile avenue for the discovery of novel vaccine candidates.
What Effect Does the Microbiome Have on the Outcome of Trickle Infection?
There exists an evident relationship between the immune system and the microbiome, especially in the gut where it is required for both the development and maintenance of the mucosal barrier (165), with loss of community diversity associated with inflammatory bowel disease (IBD) (166, 167). Whilst there are relatively few field studies that have investigated the relationship between STH infection and the microbiome in humans, it does appear that infection can affect microbial composition (168, 169). This is consistent with laboratory studies of chronic STH infection that have consistently shown that STHs alter the microbiome (170–172) and that these changes in composition can be reversed following expulsion of the infection (173). Chronic STH infection has been associated with expansion of bacterial genera with the capacity to promote the T regulatory response such as Lactobacillus (174–176) which may contribute to chronicity by suppressing the induction of a Th2 response. During the Th1-dominated susceptible phase of T. muris trickle infection there is a strong reduction of microbial diversity and an expansion of genera associated with chronic STH infection. Interestingly, the reduction in microbial diversity during the susceptible phase leads to a reduced efficiency of egg hatching (177) which is heavily dependent upon the intestinal microbiota (178). It can be speculated that this would have the net effect of keeping successive infection levels low, reducing the induction of protective immunity. Coinciding with the development of resistance during T. muris trickle infection the microbiome appears to partially recover with an increase in diversity and recovery of genera that had been lost earlier in infection (139). The nature of this relationship requires further assessment as several possibilities present themselves: (i) the development of a Th2 response actively promotes a homeostatic microbiota making the recovery in diversity a direct consequence of acquired immunity to STHs; (ii) recovery of the microbiome following loss of diversity occurs independently of host-driven mechanisms, but subsequently facilitates resistance by directly promoting a Th2 response; (iii) STHs produce antimicrobial peptides that restructure the microbiome to suit their own physiology, and when their numbers are reduced this effect is lost and the microbiome recovers as an indirect consequence of host-protective immunity (Figure 1).
Concluding Remarks
Investigation of resistance and susceptibility to intestinal nematode parasites and their underlying immune mechanisms has not only informed on immunity to these particular infectious agents, but has identified novel and fundamental new information on how immunity works. This has no doubt arisen in part from the fact that infection by STHs present a particular set of challenges to the host immune system not seen in other pathogen infections.
The available evidence from the field and from experimental trickle infections of STH has led to a number of generally consistent core observations. Infection from exposure to a low number of infectious stages in any one infection event is more likely to lead to parasite patency than exposure to a “high” number of infectious stages in any one infection event (where the parasites are more often than not expelled, even if not completely). Thus, there appears to be a threshold for an infection event, below which the parasites do not get immunologically expelled and above which they do. This is not only influenced by host genetics, but also the local intestinal environment. It will also vary between different parasite species and the life cycle strategy that they have evolved. It is also clear that as long as the individual infection event remains below a certain level, increases in parasite load are tolerated up to a “critical point.” Again, number of infection events and interval between them will influence the ultimate success of the infection and the speed with which the “critical” point is reached. Ultimately, host protective immunity does begin to operate, although it is generally only partial and not sterilizing immunity. Adult parasites often remain for extended periods, although parasite fecundity eventually drops and new juvenile stages do not appear to be able to complete their development effectively i.e., are expelled. Thus, trickle infections are exemplars of concomitant immunity (179, 180). Protective immunological memory does occur with resistance to both high and low dose infection events, although some level of existing infection generally persists.
The single/challenge high dose infection approach to study experimental immunity to STH has been and continues to be spectacularly informative. Nevertheless, bearing in mind the way in which infections are acquired naturally, the trickle infection approach is set to further inform and refine our understanding of how protective immunity is generated, how it is regulated and, importantly, how it can be improved upon, especially for hosts that are naturally, chronically infected.
Author Contributions
Both authors have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
IC160132 The Royal Society, United Kingdom, 103132/Z/13/Z/WT Wellcome Trust, United Kingdom, Z10661/Z/18/Z/WT Wellcome Trust, United Kingdom, and 203128/Z/16/Z/WT Wellcome Trust, United Kingdom.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Dobson A, Lafferty KD, Kuris AM, Hechinger RF, Jetz W. Homage to Linnaeus: how many parasites? How many hosts? Proc Natl Acad Sci USA. (2008) 105(Suppl. 1): 1482–9. doi: 10.1073/pnas.0803232105
2. Hotez PJ, Alvarado M, Basanez MG, Bolliger I, Bourne R, Boussinesq M, et al. The global burden of disease study 2010: interpretation and implications for the neglected tropical diseases. PLoS Negl Trop Dis. (2014) 8:e2865. doi: 10.1371/journal.pntd.0002865
3. Pullan RL, Smith JL, Jasrasaria R, Brooker SJ. Global numbers of infection and disease burden of soil transmitted helminth infections in 2010. Parasit Vectors. (2014) 7:1–19. doi: 10.1186/1756-3305-7-37
4. Maizels RM, Smits HH, McSorley HJ. Modulation of host immunity by helminths: the expanding repertoire of parasite effector molecules. Immunity. (2018) 49:801–18. doi: 10.1016/j.immuni.2018.10.016
5. Graham AL, Allen JE, Read AF. Evolutionary causes and consequences of immunopathology. Annu Rev Ecol Evol Syst. (2005) 36:373–97. doi: 10.1146/annurev.ecolsys.36.102003.152622
6. Girod N, Brown A, Pritchard DI, Billett EE. Successful vaccination of BALB/c mice against human hookworm (Necator americanus): the immunological phenotype of the protective response. Int J Parasitol. (2003) 33:71–80. doi: 10.1016/S0020-7519(02)00248-5
7. Wilkinson MJ, Wells C, Behnke JM. Necator americanus in the mouse: histopathological changes associated with the passage of larvae through the lungs of mice exposed to primary and secondary infection. Parasitol Res. (1990) 76:386–92. doi: 10.1007/BF00933545
8. Wells C, Behnke JM. Acquired resistance to the human hookworm Necator americanus in mice. Parasite Immunol. (1988) 10:493–505. doi: 10.1111/j.1365-3024.1988.tb00238.x
9. Behnke JM, Wells C, Brown J. An improved technique for experimental infections with skin penetrating nematode larvae (Necator americanus). Int J Parasitol. (1986) 16:461–4. doi: 10.1016/0020-7519(86)90080-9
10. Bungiro RD Jr, Anderson BR, Cappello M. Oral Transfer of adult Ancylostoma ceylanicum hookworms into permissive and nonpermissive host species. Infect Immun. (2003) 71: 1880–6. doi: 10.1128/IAI.71.4.1880-1886.2003
11. Garside P, Behnke JM. Ancylostoma ceylanicum in the hamster: observations on the host—parasite relationship during primary infection. Parasitology. (1989) 98:283–9. doi: 10.1017/S003118200006220X
12. Gazzinelli-Guimaraes PH, de Queiroz Prado R, Ricciardi A, Bonne-Année S, Sciurba J, Karmele EP, et al. Allergen presensitization drives an eosinophil-dependent arrest in lung-specific helminth development. J Clin Invest. (2019) 130:3686–701. doi: 10.1172/JCI127963
13. Marsland BJ, Kurrer M, Reissmann R, Harris NL, Kopf M. Nippostrongylus brasiliensis infection leads to the development of emphysema associated with the induction of alternatively activated macrophages. Eur J Immunol. (2008) 38:479–88. doi: 10.1002/eji.200737827
14. Cliffe LJ, Grencis RK. The Trichuris muris System: a Paradigm of Resistance and Susceptibility to Intestinal Nematode Infection. Adv Parasitol. (2004) 57:255–307. doi: 10.1016/S0065-308X(04)57004-5
15. Hashimoto K, Uchikawa R, Tegoshi T, Takeda K, Yamada M, Arizono N. Depleted intestinal goblet cells and severe pathological changes in SCID mice infected with Heligmosomoides polygyrus. Parasite Immunol. (2009) 31:457–65. doi: 10.1111/j.1365-3024.2009.01123.x
16. Ziegler SF, Artis D. Sensing the outside world: TSLP regulates barrier immunity. Nat Immunol. (2010) 11:289–93. doi: 10.1038/ni.1852
17. Zhao A, Urban JF Jr, Sun R, Stiltz J, Morimoto M, Notari L, et al. Critical role of IL-25 in nematode infection-induced alterations in intestinal function. J Immunol. (2010) 185:6921–9. doi: 10.4049/jimmunol.1000450
18. Pei C, Zhao C, Wang AJ, Fan AX, Grinchuk V, Smith A, et al. Critical role for interleukin-25 in host protective Th2 memory response against heligmosomoides polygyrus bakeri. Infect Immun. (2016) 84:3328–37. doi: 10.1128/IAI.00180-16
19. Angkasekwinai P, Srimanote P, Wang YH, Pootong A, Sakolvaree Y, Pattanapanyasat K, et al. Interleukin-25 (IL-25) promotes efficient protective immunity against Trichinella spiralis infection by enhancing the antigen-specific IL-9 response. Infect Immun. (2013) 81:3731–41. doi: 10.1128/IAI.00646-13
20. Angkasekwinai P, Park H, Wang YH, Wang YH, Chang SH, Corry DB, et al. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. (2007) 204:1509–17. doi: 10.1084/jem.20061675
21. Cayrol C, Girard JP. Interleukin-33 (IL-33): a nuclear cytokine from the IL-1 family. Immunol Rev. (2018) 281:154–68. doi: 10.1111/imr.12619
22. Owyang AM, Zaph C, Wilson EH, Guild KJ, McClanahan T, Miller HRP, et al. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J Exp Med. (2006) 203:843–9. doi: 10.1084/jem.20051496
23. Zaiss MM, Maslowski KM, Mosconi I, Guenat N, Marsland BJ, Harris NL. IL-1β suppresses innate IL-25 and IL-33 production and maintains helminth chronicity. PLoS Pathog. (2013) 9:e1003531. doi: 10.1371/journal.ppat.1003531
24. Hung LY, Lewkowich IP, Dawson LA, Downey J, Yang Y, Smith DE, et al. IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proc Natl Acad Sci USA. (2013) 110:282–7. doi: 10.1073/pnas.1206587110
25. Taylor BC, Zaph C, Troy AE, Du Y, Guild KJ, Comeau MR, et al. TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. J Exp Med. (2009) 206:655–67. doi: 10.1084/jem.20081499
26. Liu YJ, Soumelis V, Watanabe N, Ito T, Wang YH, de Waal Malefyt R, et al. TSLP: an epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu Rev Immunol. (2007) 25:193–219. doi: 10.1146/annurev.immunol.25.022106.141718
27. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an Interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
28. Von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature. (2016) 529:221–5. doi: 10.1038/nature16161
29. Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science. (2016) 351: 1329–33. doi: 10.1126/science.aaf1648
30. Gerbe F, Sidot E, Smyth DJ, Ohmoto M, Matsumoto I, Dardalhon V, et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature. (2016) 529:226–30. doi: 10.1038/nature16527
31. Luo XC, Chen ZH, Xue JB, Zhao DX, Lu C, Li YH, et al. Infection by the parasitic helminth Trichinella spiralis activates a Tas2r-mediated signaling pathway in intestinal tuft cells. Proc Natl Acad Sci USA. (2019) 116: 5564–9. doi: 10.1073/pnas.1812901116
32. McGinty JW, Ting HA, Billipp TE, Nadjsombati MS, Khan DM, Barrett NA, et al. Tuft-Cell-derived leukotrienes drive rapid anti-helminth immunity in the small intestine but are dispensable for anti-protist immunity. Immunity. (2020) 52:528–41.e7. doi: 10.1016/j.immuni.2020.02.005
33. Pelly VS, Kannan Y, Coomes SM, Entwistle LJ, Rückerl D, Seddon B, et al. IL-4-producing ILC2s are required for the differentiation of TH2 cells following Heligmosomoides polygyrus infection. Mucosal Immunol. (2016) 9:1407–17. doi: 10.1038/mi.2016.4
34. Mackley EC, Houston S, Marriott CL, Halford EE, Lucas B, Cerovic V, et al. CCR7-dependent trafficking of RORγ+ ILCs creates a unique microenvironment within mucosal draining lymph nodes. Nat Commun. (2015) 6:5862. doi: 10.1038/ncomms6862
35. Löser S, Smith KA, Maizels RM. Innate lymphoid cells in helminth infections—obligatory or accessory? Front Immunol. (2019) 10:620. doi: 10.3389/fimmu.2019.00620
36. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TKA, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. (2010) 464:1367–70. doi: 10.1038/nature08900
37. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4 + T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity. (2014) 41:283–95. doi: 10.1016/j.immuni.2014.06.016
38. Esser-von Bieren J, Mosconi I, Guiet R, Piersgilli A, Volpe B, Chen F, et al. Antibodies trap tissue migrating helminth larvae and prevent tissue damage by driving IL-4Rα-independent alternative differentiation of macrophages. PLOS Pathog. (2013) 9:1–15. doi: 10.1371/journal.ppat.1003771
39. Reynolds LA, Filbey KJ, Maizels RM. Immunity to the model intestinal helminth parasite Heligmosomoides polygyrus. Semin Immunopathol. (2012) 34:829–46. doi: 10.1007/s00281-012-0347-3
40. Batugedara HM, Li J, Chen G, Lu D, Patel JJ, Jang JC, et al. Hematopoietic cell-derived RELMα regulates hookworm immunity through effects on macrophages. J Leukoc Biol. (2018) 104:855–69. doi: 10.1002/JLB.4A0917-369RR
41. Anthony RM, Urban JF Jr, Alem F, Hamed HA, Rozo CT, Boucher JL, et al. Memory T(H)2 cells induce alternatively activated macrophages to mediate protection against nematode parasites. Nat Med. (2006) 12:955–60. doi: 10.1038/nm1451
42. Sutherland TE, Logan N, Rückerl D, Humbles AA, Allan SM, Papayannopoulos V, et al. Chitinase-like proteins promote IL-17-mediated neutrophilia in a tradeoff between nematode killing and host damage. Nat Immunol. (2014) 15:1116–25. doi: 10.1038/ni.3023
43. Faulkner H, Renauld JC, Van Snick J, Grencis RK. Interleukin-9 enhances resistance to the intestinal nematode Trichuris muris. Infect Immun. (1998) 66:3832–40. doi: 10.1128/IAI.66.8.3832-3840.1998
44. Morimoto M, Morimoto M, Whitmire J, Xiao S, Anthony RM, Mirakami H, et al. Peripheral CD4 T cells rapidly accumulate at the host:parasite interface during an inflammatory Th2 memory response. J Immunol. (2004) 172: 2424–30. doi: 10.4049/jimmunol.172.4.2424
45. Perrigoue JG, Saenz SA, Siracusa MC, Allenspach EJ, Taylor BC, Giacomin PR, et al. MHC class II–dependent basophil–CD4+ T cell interactions promote TH2 cytokine–dependent immunity. Nat Immunol. (2009) 10:697–705. doi: 10.1038/ni.1740
46. Hewitson JP, Filbey KJ, Esser-von Bieren J, Camberis M, Schwartz C, Murray J, et al. Concerted activity of IgG1 antibodies and IL-4/IL-25-dependent effector cells trap helminth larvae in the tissues following vaccination with defined secreted antigens, providing sterile immunity to challenge infection. PLOS Pathog. (2015) 11:e1004676. doi: 10.1371/journal.ppat.1004676
47. McDermott JR, Bartram RE, Knight PA, Miller HRP, Garrod DR, Grencis RK. Mast cells disrupt epithelial barrier function during enteric nematode infection. Proc Natl Acad Sci USA. (2003) 100: 7761–6. doi: 10.1073/pnas.1231488100
48. Betts CJ, Else KJ. Mast cells, eosinophils and antibody-mediated cellular cytotoxicity are not critical in resistance to Trichuris muris. Parasite Immunol. (1999) 21:45–52. doi: 10.1046/j.1365-3024.1999.00200.x
49. Bouchery T, Moyat M, Sotillo J, Silverstein S, Volpe B, Coakley G, et al. Hookworms evade host immunity by secreting a deoxyribonuclease to degrade neutrophil extracellular traps. Cell Host Microbe. (2020) 27:277–89.e6. doi: 10.1016/j.chom.2020.01.011
50. Chen F, Wu W, Millman A, Craft JF, Chen E, Patel N, et al. Neutrophils prime a long-lived effector macrophage phenotype that mediates accelerated helminth expulsion. Nat Immunol. (2014) 15:938–46. doi: 10.1038/ni.2984
51. Chen F, Liu Z, Wu W, Rozo C, Bowdridge S, Millman A, et al. An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat Med. (2012) 18:260–6. doi: 10.1038/nm.2628
52. Strandmark J, Steinfelder S, Berek C, Kühl AA, Rausch S, Hartmann S. Eosinophils are required to suppress Th2 responses in Peyer's patches during intestinal infection by nematodes. Mucosal Immunol. (2017) 10:661–72. doi: 10.1038/mi.2016.93
53. Campbell L, Hepworth MR, Whittingham-Dowd J, Thompson S, Bancroft AJ, Hayes KS, et al. ILC2s mediate systemic innate protection by priming mucus production at distal mucosal sites. J Exp Med. (2019) 216:2714–23. doi: 10.1084/jem.20180610
54. Filbey KJ, Camberis M, Chandler J, Turner R, Kettle AJ, Eichenberger RM, et al. Intestinal helminth infection promotes IL-5- and CD4+ T cell-dependent immunity in the lung against migrating parasites. Mucosal Immunol. (2019) 12:352–62. doi: 10.1038/s41385-018-0102-8
55. Jacobson RH, Reed ND. The immune response of congenitally athymic (Nude) mice to the intestinal nematode nippostrongylus brasiliensis. Proc Soc Exp Biol Med. (1974) 147:667–70. doi: 10.3181/00379727-147-38412
56. Yoichi I. The absence of resistance in congenitally athymic nude mice toward infection with the intestinal nematode, Trichuris muris: resistance restored by lymphoid cell transfer. Int J Parasitol. (1991) 21:65–9. doi: 10.1016/0020-7519(91)90121-M
57. Koyama K, Tamauchi H, Ito Y. The role of CD4+ and CD8+ T cells in protective immunity to the murine nematode parasite Trichuris muris. Parasite Immunol. (1995) 17:161–5. doi: 10.1111/j.1365-3024.1995.tb01018.x
58. Betts J, deSchoolmeester ML, Else KJ. Trichuris muris: CD4+ T cell-mediated protection in reconstituted SCID mice. Parasitology. (2000) 121(Pt 6):631–7. doi: 10.1017/S0031182000006740
59. Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, et al. Transcription factor RORα is critical for nuocyte development. Nat Immunol. (2012) 13:229–36. doi: 10.1038/ni.2208
60. Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat Immunol. (2013) 14:564–73. doi: 10.1038/ni.2584
61. Else KJ, Finkelman FD, Maliszewski CR, Grencis RK. Cytokine-mediated regulation of chronic intestinal helminth infection. J Exp Med. (1994) 179:347–51. doi: 10.1084/jem.179.1.347
62. Turner JE, Stockinger B, Helmby H. IL-22 mediates goblet cell hyperplasia and worm expulsion in intestinal helminth infection. PLOS Pathog. (2013) 9:e1003698. doi: 10.1371/journal.ppat.1003698
63. Artis D, Wang ML, Keilbaugh SA, He W, Brenes M, Swain GP, et al. RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc Natl Acad Sci USA. (2004) 101:13596–600. doi: 10.1073/pnas.0404034101
64. Herbert DR, Yang JQ, Hogan SP, Groschwitz K, Khodoun M, Munitz A, et al. Intestinal epithelial cell secretion of RELM-beta protects against gastrointestinal worm infection. J Exp Med. (2009) 206:2947–57. doi: 10.1084/jem.20091268
65. Hasnain SZ, Wang H, Ghia JE, Haq N, Deng Y, Velcich A, et al. Mucin gene deficiency in mice impairs host resistance to an enteric parasitic infection. Gastroenterology. (2010) 138:1763–71.e5. doi: 10.1053/j.gastro.2010.01.045
66. Hasnain SZ, Evans CM, Roy M, Gallagher AL, Kindrachuk KN, Barron L, et al. Muc5ac: a critical component mediating the rejection of enteric nematodes. J Exp Med. (2011) 208:893–900. doi: 10.1084/jem.20102057
67. Cliffe LJ, Humphreys NE, Lane TE, Potten CS, Booth C, Grencis RK. Accelerated intestinal epithelial cell turnover: a new mechanism of parasite expulsion author(s): Laura J. Cliffe, Neil E. Humphreys, Thomas E. Lane, Chris S. Potten, Cath Booth and Richard K. Grencis Source: Science. (2005) 308:1463–5. doi: 10.1126/science.1108661
68. Zaiss DM, Yang L, Shah PR, Kobie JJ, Urban JF, Mosmann TR. Amphiregulin, a T H 2 cytokine enhancing resistance to nematodes. Science. (2006) 314:1746. doi: 10.1126/science.1133715
69. Khan WI, Richard M, Akiho H, Blennerhasset PA, Humphreys NE, Grencis RK, et al. Modulation of intestinal muscle contraction by interleukin-9 (IL-9) or IL-9 neutralization: correlation with worm expulsion in murine nematode infections. Infect Immun. (2003) 71:2430–8. doi: 10.1128/IAI.71.5.2430-2438.2003
70. Pritchard DI, Williams DJ, Behnke JM, Lee TD. The role of IgG1 hypergammaglobulinaemia in immunity to the gastrointestinal nematode Nematospiroides dubius. The immunochemical purification, antigen-specificity and in vivo anti-parasite effect of IgG1 from immune serum. Immunology. (1983) 49:353–65.
71. Filbey KJ, Grainger JR, Smith KA, Boon L, van Rooijen N, Harcus Y, et al. Innate and adaptive type 2 immune cell responses in genetically controlled resistance to intestinal helminth infection. Immunol Cell Biol. (2014) 92:436–48. doi: 10.1038/icb.2013.109
72. Alizadeh H, Wakelin D. Genetic factors controlling the intestinal mast cell response in mice infected with Trichinella spiralis. Clin Exp Immunol. (1982) 49:331–7.
73. Wakelin P. Genetic control of immune responses to parasites: immunity to Trichuris muris in inbred and random-bred strains of mice. Parasitology. (1975) 71:51–60. doi: 10.1017/S0031182000053142
74. Else K, Wakelin D. The effects of H-2 and non-H-2 genes on the expulsion of the nematode Trichuris muris from inbred and congenic mice. Parasitology. (1988) 96:543–50. doi: 10.1017/S0031182000080173
75. Urban JF, Katona IM, Finkelman FD. Heligmosomoides polygyrus: CD4+ but not CD8+ T cells regulate the IgE response and protective immunity in mice. Exp Parasitol. (1991) 73:500–11. doi: 10.1016/0014-4894(91)90074-7
76. Bancroft AJ, Else KJ, Humphreys NE, Grencis RK. The effect of challenge and trickle Trichuris muris infections on the polarisation of the immune response. Int J Parasitol. (2001) 31:1627–37. doi: 10.1016/S0020-7519(01)00281-8
77. Miller HR, Huntley JF, Wallace GR. Immune exclusion and mucus trapping during the rapid expulsion of Nippostrongylus brasiliensis from primed rats. Immunology. (1981) 44:419–29.
78. Wakelin D, Lloyd M. Immunity to primary and challenge infections of Trichinella spiralis in mice: a re-examination of conventional parameters. Parasitology. (1976) 72:173–82. doi: 10.1017/S0031182000048472
79. Needham C, Thi Kim H, Viet Hoa N, Dinh Cong L, Michael E, Drake L, et al. Epidemiology of soil-transmitted nematode infections in Ha Nam Province, Vietnam. Trop Med Int Heal. (1998) 3:904–12. doi: 10.1046/j.1365-3156.1998.00324.x
80. Brooker S, Clements ACA, Bundy DAP. Global epidemiology, ecology and control of soil-transmitted helminth infections. Adv Parasitol. (2006) 62:221–61. doi: 10.1016/S0065-308X(05)62007-6
81. Bundy DAP, Kan SP, Rose R. Age-related prevalence, intensity and frequency distribution of gastrointestinal helminth infection in urban slum children from Kuala Lumpur, Malaysia. Trans R Soc Trop Med Hyg. (1988) 82:289–94. doi: 10.1016/0035-9203(88)90450-6
82. Brooker S, Jardim-Botelho A, Quinnell RJ, Geiger SM, Caldas IR, Fleming F, et al. Age-related changes in hookworm infection, anaemia and iron deficiency in an area of high Necator americanus hookworm transmission in south-eastern Brazil. Trans R Soc Trop Med Hyg. (2007) 101:146–54. doi: 10.1016/j.trstmh.2006.05.012
83. Gesquiere LR, Habig B, Hansen C, Li A, Freid K, Learn NH, et al. Noninvasive measurement of mucosal immunity in a free-ranging baboon population. Am J Primatol. (2020) 82:e23093. doi: 10.1002/ajp.23093
84. Müller-Graf CDM, Collins DA, Woolhouse MEJ. Intestinal parasite burden in five troops of olive baboons (Papio cynocephalus anubis) in Gombe Stream National Park, Tanzania. Parasitology. (1996) 112:489–97. doi: 10.1017/S0031182000076952
85. MacIntosh AJJ, Hernandez AD, Huffman MA. Host age, sex, and reproductive seasonality affect nematode parasitism in wild Japanese macaques. Primates. (2010) 51:353–64. doi: 10.1007/s10329-010-0211-9
86. McRae KM, Stear MJ, Good B, Keane OM. The host immune response to gastrointestinal nematode infection in sheep. Parasite Immunol. (2015) 37:605–13. doi: 10.1111/pim.12290
87. Armour J. The epidemiology of helminth disease in farm animals. Vet Parasitol. (1980) 6:7–46. doi: 10.1016/0304-4017(80)90037-0
88. Gregory RD, Montgomery SSJ, Montgomery WI. Population biology of Heligmosomoides polygyrus (Nematoda) in the wood mouse. J Anim Ecol. (1992) 61:749–57. doi: 10.2307/5628
89. Fisher MC, Viney ME. The population genetic structure of the facultatively sexual parasitic nematode Strongyloides ratti in wild rats. Proceedings Biol Sci. (1998) 265:703–9. doi: 10.1098/rspb.1998.0350
90. Turner JD, Faulkner H, Kamgno J, Cormont F, Van Snick J, Else KJ, et al. Th2 Cytokines are associated with reduced worm burdens in a human intestinal helminth infection. J Infect Dis. (2003) 188:1768–75. doi: 10.1086/379370
91. Faulkner H, Turner J, Kamgno J, Pion SD, Boussinesq M, Bradley JE. Age- and infection intensity-dependent cytokine and antibody production in human trichuriasis: the importance of IgE. J Infect Dis. (2002) 185:665–74. doi: 10.1086/339005
92. Quinnell RJ, Pritchard DI, Raiko A, Brown AP, Shaw MA. Immune responses in human necatoriasis: association between Interleukin-5 responses and resistance to reinfection. J Infect Dis. (2004) 190:430–8. doi: 10.1086/422256
93. Jackson JA, Turner JD, Rentoul L, Faulkner H, Behnke JM, Hoyle M, et al. T Helper cell type 2 responsiveness predicts future susceptibility to gastrointestinal nematodes in humans. J Infect Dis. (2004) 190:1804–11. doi: 10.1086/425014
94. de Ruiter K, Jochems SP, Tahapary DL, Stam KA, König M, van Unen V, et al. Helminth infections drive heterogeneity in human type 2 and regulatory cells. Sci Transl Med. (2020) 12:eaaw3703. doi: 10.1126/scitranslmed.aaw3703
95. Hagan P, Blumenthal UJ, Dunn D, Simpson AJG, Wilkins HA. Human IgE, IgG4 and resistance to reinfection with Schistosoma haematobium. Nature. (1991) 349:243–5. doi: 10.1038/349243a0
96. Milner T, Reilly L, Nausch N, Midzi N, Mduluza T, Maizels R, et al. Circulating cytokine levels and antibody responses to human Schistosoma haematobium: IL-5 and IL-10 levels depend upon age and infection status. Parasite Immunol. (2010) 32:710–21. doi: 10.1111/j.1365-3024.2010.01235.x
97. Wammes LJ, Hamid F, Wiria AE, May L, Kaisar MMM, Prasetyani-Gieseler MA, et al. Community deworming alleviates geohelminth-induced immune hyporesponsiveness. Proc Natl Acad Sci USA. (2016) 113:12526–31. doi: 10.1073/pnas.1604570113
98. Coronado S, Barrios L, Zakzuk J, Regino R, Ahumada V, Franco L, et al. A recombinant cystatin from Ascaris lumbricoides attenuates inflammation of DSS-induced colitis. Parasite Immunol. (2017) 39:e12425. doi: 10.1111/pim.12425
99. Navarro S, Pickering DA, Ferreira IB, Jones L, Ryan S, Troy S, et al. Hookworm recombinant protein promotes regulatory T cell responses that suppress experimental asthma. Sci Transl Med. (2016) 8:362ra143. doi: 10.1126/scitranslmed.aaf8807
100. Ferreira IB, Pickering DA, Troy S, Croese J, Loukas A, Navarro S. Suppression of inflammation and tissue damage by a hookworm recombinant protein in experimental colitis. Clin Transl Immunol. (2017) 6:e157. doi: 10.1038/cti.2017.42
101. Craig BH, Pilkington JG, Pemberton JM. Gastrointestinal nematode species burdens and host mortality in a feral sheep population. Parasitology. (2006) 133:485–96. doi: 10.1017/S0031182006000618
102. Gulland FMD, Fox M. Epidemiology of nematode infections of Soay sheep (Ovis aries L.) on St. Kilda. Parasitology. (1992) 105:481–92. doi: 10.1017/S0031182000074667
103. Miller JE, Horohov DW. Immunological aspects of nematode parasite control in sheep1. J Anim Sci. (2006) 84(Suppl. 13):E124–32. doi: 10.2527/2006.8413_supplE124x
104. Waruiru RM, Kyvsgaard NC, Thamsborg SM, Nansen P, Bøgh HO, Munyua WK, et al. The Prevalence and intensity of helminth and coccidial infections in dairy cattle in central Kenya. Vet Res Commun. (2000) 24:39–53. doi: 10.1023/A:1006325405239
105. Clerc M, Babayan SA, Fenton A, Pedersen AB. Age affects antibody levels and anthelmintic treatment efficacy in a wild rodent. Int J Parasitol Parasites Wildl. (2019) 8:240–7. doi: 10.1016/j.ijppaw.2019.03.004
106. Kan SP. Soil-transmitted helminthiasis in Selangor, Malaysia. Med J Malaysia. (1982) 37:180–90.
107. Al-Delaimy AK, Al-Mekhlafi HM, Nasr NA, Sady H, Atroosh WM, Nashiry M, et al. Epidemiology of intestinal polyparasitism among orang asli school children in rural malaysia. PLoS Negl Trop Dis. (2014) 8:e3074. doi: 10.1371/journal.pntd.0003074
108. Brooker S, Miguel EA, Moulin S, Luoba AI, Bundy DA, Kremer M. Epidemiology of single and multiple species of helminth infections among school children in Busia District, Kenya. East Afr Med J. (2000) 77:157–61. doi: 10.4314/eamj.v77i3.46613
109. Jenkins SN, Behnke JM. Impairment of primary expulsion of trichuris muris in mice concurrently infected with nematospiroides dubius. Parasitology. (1977) 75:71–8. doi: 10.1017/S0031182000048332
110. Behnke JM, Wakelin D, Wilson MM. Trichinella spiralis: delayed rejection in mice concurrently infected with Nematospiroides dubius. Exp Parasitol. (1978) 46:121–30. doi: 10.1016/0014-4894(78)90162-5
111. Hartgers FC, Yazdanbakhsh M. Co-infection of helminths and malaria: modulation of the immune responses to malaria. Parasite Immunol. (2006) 28:497–506. doi: 10.1111/j.1365-3024.2006.00901.x
112. Babu S, Nutman TB. Helminth-Tuberculosis co-infection: an immunologic perspective. Trends Immunol. (2016) 37:597–607. doi: 10.1016/j.it.2016.07.005
113. Borkow G, Bentwich Z. HIV and helminth co-infection: is deworming necessary? Parasite Immunol. (2006) 28:605–12. doi: 10.1186/1742-4690-3-S1-S79
114. Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. (2016) 532:512–6. doi: 10.1038/nature17655
115. Abolins S, King EC, Lazarou L, Weldon L, Hughes L, Drescher P, et al. The comparative immunology of wild and laboratory mice, Mus musculus domesticus. Nat Commun. (2017) 8:14811. doi: 10.1038/ncomms14811
116. Viney M, Riley EM. The immunology of wild rodents: current status and future prospects. Front Immunol. (2017) 8:1481. doi: 10.3389/fimmu.2017.01481
117. Scott ME. Heligmosomoides polygyrus (Nematoda): susceptible and resistant strains of mice are indistinguishable following natural infection. Parasitology. (1991) 103:429–38. doi: 10.1017/S0031182000059953
118. Willing BP, Vacharaksa A, Croxen M, Thanachayanont T, Finlay BB. Altering Host Resistance to Infections through Microbial Transplantation. PLoS ONE. (2011) 6:e26988. doi: 10.1371/journal.pone.0026988
119. Ghosh S, Dai C, Brown K, Rajendiran E, Makarenko S, Baker J, et al. Colonic microbiota alters host susceptibility to infectious colitis by modulating inflammation, redox status, and ion transporter gene expression. Am J Physiol Gastrointest Liver Physiol. (2011) 301:G39–49. doi: 10.1152/ajpgi.00509.2010
120. Leung JM, Budischak SA, Chung The H, Hansen C, Bowcutt R, Neill R, et al. Rapid environmental effects on gut nematode susceptibility in rewilded mice. PLOS Biol. (2018) 16:1–28. doi: 10.1371/journal.pbio.2004108
121. Yeung F, Chen YH, Lin JD, Leung JM, McCauley C, Devlin JC, et al. Altered immunity of laboratory mice in the natural environment is associated with fungal colonization. Cell Host Microbe. (2020) 27: 809–22.e6. doi: 10.1016/j.chom.2020.02.015
122. Lin JD, Devlin JC, Yeung F, McCauley C, Leung JM, Chen YH, et al. Rewilding Nod2 and Atg16l1 mutant mice uncovers genetic and environmental contributions to microbial responses and immune cell composition. Cell Host Microbe. (2020) 27:830–40.e4 doi: 10.1016/j.chom.2020.03.001
123. Bancroft AJ, Else KJ, Grencis RK. Low-level infection with Trichuris muris significantly affects the polarization of the CD4 response. Eur J Immunol. (1994) 24:3113–8. doi: 10.1002/eji.1830241230
124. Behnke JM. Evasion of immunity by nematode parasites causing chronic infections. Adv Parasitol. (1987) 26:1–71. doi: 10.1016/s0065-308x(08)60294-8
125. Michel JF, Lancaster MB, Hong C. The length of Ostertagia ostertagi in populations of uniform age. Int J Parasitol. (1978) 8:437–41. doi: 10.1016/0020-7519(78)90060-7
126. Martin J, Lee DL. Observations on crystals found in the intestine of Nematodirus battus during the development of immunity to this nematode in lambs. Parasitology. (1976) 72:75–80. doi: 10.1017/S0031182000043201
127. Jackson F, Angus KW, Coop RL. Development of morphological changes in the small intestine of lambs continuously infected with Trichostrongylus vitrinus. Res Vet Sci. (1983) 34:301–4. doi: 10.1016/S0034-5288(18)32227-6
128. Dobson RJ, Waller PJ, Donald AD. Population dynamics of Trichostrongylus colubriformis in sheep: the effect of infection rate on the establishment of infective larvae and parasite fecundity. Int J Parasitol. (1990) 20:347–52. doi: 10.1016/0020-7519(90)90150-L
129. Jenkins DC, Phillipson RF. The kinetics of repeated low-level infections of Nippostrongylus brasiliensis in the laboratory rat. Parasitology. (1971) 62:457–65. doi: 10.1017/S003118200007760X
130. Ovington KS. Trickle infections of Nippostrongylus brasiliensis in rats. Z Parasitenkd. (1986) 72:851–3. doi: 10.1007/BF00925109
131. Maizels RM, Meghji M, Ogilvie BM. Restricted sets of parasite antigens from the surface of different stages and sexes of the nematode parasite Nippostrongylus brasiliensis. Immunology. (1983) 48:107–21.
132. Ferens WA, Arai HP, Befus AD. Trickle infections with Nippostrongylus brasiliensis in rats: larval migration through the lungs. J Parasitol. (1990) 76:684–9. doi: 10.2307/3282983
133. Ferens WA, Arai HP, Befus AD. Bronchoalveolar leucocyte responses to trickle infections with Nippostrongylus brasiliensis in rats. J Parasitol. (1994) 80:654–6. doi: 10.2307/3283207
134. Brailsford TJ, Behnke JM. The dynamics of trickle infections with Heligmosomoides polygyrus in syngeneic strains of mice. Int J Parasitol. (1992) 22:351–9. doi: 10.1016/S0020-7519(05)80013-X
135. Brailsford TJ, Behnke JM. The dynamics of trickle infections with Ancylostoma ceylanicum in inbred hamsters. Parasitology. (1992) 105 (Pt 2):247–53. doi: 10.1017/S0031182000074175
136. Behnke JM, Lowe A, Clifford S, Wakelin D. Cellular and serological responses in resistant and susceptible mice exposed to repeated infection with Heligmosomoides polygyrus bakeri. Parasite Immunol. (2003) 25:333–40. doi: 10.1046/j.1365-3024.2003.00639.x
137. Pedersen S, Saeed I. Acquired immunity to Trichuris suis infection in pigs. Parasitology. (2001) 123:95–101. doi: 10.1017/S0031182001007934
138. Nejsum P, Thamsborg SM, Petersen HH, Kringel H, Fredholm M, Roepstorff A. Population dynamics of Trichuris suis in trickle-infected pigs. Parasitology. (2009) 136:691–7. doi: 10.1017/S0031182009005976
139. Glover M, Colombo SAP, Thornton DJ, Grencis RK. Trickle infection and immunity to Trichuris muris. PLOS Pathog. (2019) 15:1–27. doi: 10.1371/journal.ppat.1007926
140. Bancroft AJ, McKenzie AN, Grencis RK. A critical role for IL-13 in resistance to intestinal nematode infection. J Immunol. (1998) 160:3453–61.
141. Bancroft AJ, Levy CW, Jowitt TA, Hayes KS, Thompson S, Mckenzie EA, et al. The major secreted protein of the whipworm parasite tethers to matrix and inhibits interleukin-13 function. Nat Commun. (2019) 10:2344. doi: 10.1038/s41467-019-09996-z
142. Osbourn M, Soares DC, Vacca F, Cohen ES, Scott IC, Gregory WF, et al. HpARI protein secreted by a helminth parasite suppresses Interleukin-33. Immunity. (2017) 47:739–51.e5. doi: 10.1016/j.immuni.2017.09.015
143. Johnston CJC, Smyth DJ, Kodali RB, White MPJ, Harcus Y, Filbey KJ, et al. A structurally distinct TGF-β mimic from an intestinal helminth parasite potently induces regulatory T cells. Nat Commun. (2017) 8:1741. doi: 10.1038/s41467-017-01886-6
144. Gause WC, Wynn TA, Allen JE. Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat Rev Immunol. (2013) 13:607–14. doi: 10.1038/nri3476
145. Sahputra R, Ruckerl D, Couper KN, Muller W, Else KJ. The essential role played by B cells in supporting protective immunity against Trichuris muris infection is by controlling the Th1/Th2 balance in the mesenteric lymph nodes and depends on host genetic background. Front Immunol. (2019) 10:2842. doi: 10.3389/fimmu.2019.02842
146. Else KJ, Grencis RK. Antibody-independent effector mechanisms in resistance to the intestinal nematode parasite Trichuris muris. Infect Immun. (1996) 64:2950–4. doi: 10.1128/IAI.64.8.2950-2954.1996
147. Yamane H, Paul WE. Early signaling events that underlie fate decisions of naive CD4(+) T cells toward distinct T-helper cell subsets. Immunol Rev. (2013) 252:12–23. doi: 10.1111/imr.12032
148. van Panhuys N, Klauschen F, Germain RN. T-cell-receptor-dependent signal intensity dominantly controls CD4(+) T cell polarization in vivo. Immunity. (2014) 41:63–74. doi: 10.1016/j.immuni.2014.06.003
149. Steinfelder S, Andersen JF, Cannons JL, Feng CG, Joshi M, Dwyer D, et al. The major component in schistosome eggs responsible for conditioning dendritic cells for Th2 polarization is a T2 ribonuclease (omega-1). J Exp Med. (2009) 206:1681–90. doi: 10.1084/jem.20082462
150. Costain AH, MacDonald AS, Smits HH. Schistosome egg migration: mechanisms, pathogenesis and host immune responses. Front Immunol. (2018) 9:3042. doi: 10.3389/fimmu.2018.03042
151. Humphreys NE, Xu D, Hepworth MR, Liew FY, Grencis RK. IL-33, a Potent inducer of adaptive immunity to intestinal nematodes. J Immunol. (2008) 180: 2443–9. doi: 10.4049/jimmunol.180.4.2443
152. Smith KA, Löser S, Varyani F, Harcus Y, McSorley HJ, McKenzie ANJ, et al. Concerted IL-25R and IL-4Rα signaling drive innate type 2 effector immunity for optimal helminth expulsion. Elife. (2018) 7:e38269. doi: 10.7554/eLife.38269.011
153. Mishra PK, Wu W, Rozo C, Hallab NJ, Benevenia J, Gause WC. Micrometer-sized titanium particles can induce potent Th2-type responses through TLR4-independent pathways. J Immunol. (2011) 187: 6491–8. doi: 10.4049/jimmunol.1101392
154. Kuroda E, Ishii KJ, Uematsu S, Ohata K, Coban C, Akira S, et al. Silica crystals and aluminum salts regulate the production of prostaglandin in macrophages via NALP3 inflammasome-independent mechanisms. Immunity. (2011) 34:514–26. doi: 10.1016/j.immuni.2011.03.019
155. Strid J, Sobolev O, Zafirova B, Polic B, Hayday A. The Intraepithelial T cell response to NKG2D-ligands links lymphoid stress surveillance to atopy. Science. (2011) 334: 1293–7. doi: 10.1126/science.1211250
156. Biton M, Levin A, Slyper M, Alkalay I, Horwitz E, Mor H, et al. Epithelial microRNAs regulate gut mucosal immunity via epithelium-T cell crosstalk. Nat Immunol. (2011) 12:239–46. doi: 10.1038/ni.1994
157. Bell LV, Else KJ. Mechanisms of leucocyte recruitment to the inflamed large intestine: redundancy in integrin and addressin usage. Parasite Immunol. (2008) 30:163–70. doi: 10.1111/j.1365-3024.2007.01017.x
158. Artis D, Humphreys NE, Potten CS, Wagner N, Müller W, McDermott JR, et al. β7 integrin-deficient mice: delayed leukocyte recruitment and attenuated protective immunity in the small intestine during enteric helminth infection. Eur J Immunol. (2000) 30:1656–64. doi: 10.1002/1521-4141(200006)30:6<1656::AID-IMMU1656>3.0.CO;2-Z
159. Zaph C, Rook KA, Goldschmidt M, Mohrs M, Scott P, Artis D. Persistence and function of central and effector memory CD4+ T cells following infection with a gastrointestinal helminth. J Immunol. (2006) 177:511–8. doi: 10.4049/jimmunol.177.1.511
160. Mohrs M, Shinkai K, Mohrs K, Locksley RM. Analysis of Type 2 Immunity in vivo with a bicistronic IL-4 reporter. Immunity. (2001) 15:303–11. doi: 10.1016/S1074-7613(01)00186-8
161. Steinfelder S, Rausch S, Michael D, Kühl AA, Hartmann S. Intestinal helminth infection induces highly functional resident memory CD4+ T cells in mice. Eur J Immunol. (2017) 47:353–63. doi: 10.1002/eji.201646575
162. Schopf LR, Hoffmann KF, Cheever AW, Urban JF, Wynn TA. IL-10 Is critical for host resistance and survival during gastrointestinal helminth infection. J Immunol. (2002) 168:2383–92. doi: 10.4049/jimmunol.168.5.2383
163. Duque-Correa MA, Karp NA, McCarthy C, Forman S, Goulding D, Sankaranarayanan G, et al. Exclusive dependence of IL-10Rα signalling on intestinal microbiota homeostasis and control of whipworm infection. PLOS Pathog. (2019) 15:e1007265. doi: 10.1371/journal.ppat.1007265
164. Panzer M, Sitte S, Wirth S, Drexler I, Sparwasser T, Voehringer D. Rapid in vivo conversion of effector T cells into Th2 cells during helminth infection. J Immunol. (2012) 188:615–23. doi: 10.4049/jimmunol.1101164
165. Honda K, Littman DR. The Microbiome in infectious disease and inflammation. Annu Rev Immunol. (2012) 30:759–95. doi: 10.1146/annurev-immunol-020711-074937
166. Frank DN, St. Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. (2007) 104:13780–5. doi: 10.1073/pnas.0706625104
167. Peterson DA, Frank DN, Pace NR, Gordon JI. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe. (2008) 3:417–27. doi: 10.1016/j.chom.2008.05.001
168. Lee SC, Tang MS, Lim YAL, Choy SH, Kurtz ZD, Cox LM, et al. Helminth colonization is associated with increased diversity of the gut microbiota. PLoS Negl Trop Dis. (2014) 8:e2880. doi: 10.1371/journal.pntd.0002880
169. Lee SC, Tang MS, Easton AV, Devlin JC, Chua LL, Cho I, et al. Linking the effects of helminth infection, diet and the gut microbiota with human whole-blood signatures. PLoS Pathog. (2019) 15:1–30. doi: 10.1371/journal.ppat.1008066
170. Holm JB, Sorobetea D, Kiilerich P, Ramayo-Caldas Y, Estellé J, Ma T, et al. Chronic Trichuris muris infection decreases diversity of the intestinal microbiota and concomitantly increases the abundance of lactobacilli. PLoS ONE. (2015) 10:1–22. doi: 10.1371/journal.pone.0125495
171. Walk ST, Blum AM, Ewing SAS, Weinstock JV, Young VB. Alteration of the murine gut microbiota during infection with the parasitic helminth Heligmosomoides polygyrus. Inflamm Bowel Dis. (2010) 16:1841–9. doi: 10.1002/ibd.21299
172. Rausch S, Held J, Fischer A, Heimesaat MM, Kühl AA, Bereswill S, et al. Small intestinal nematode infection of mice is associated with increased enterobacterial loads alongside the intestinal tract. PLoS ONE. (2013) 8:e74026. doi: 10.1371/journal.pone.0074026
173. Houlden A, Hayes KS, Bancroft AJ, Worthington JJ, Wang P, Grencis RK, et al. Chronic Trichuris muris infection in C57BL/6 mice causes significant changes in host microbiota and metabolome: effects reversed by pathogen clearance. PLoS ONE. (2015) 10:e0125945. doi: 10.1371/journal.pone.0125945
174. Fricke WF, Song Y, Wang AJ, Smith A, Grinchuk V, Pei C, et al. Type 2 immunity-dependent reduction of segmented filamentous bacteria in mice infected with the helminthic parasite Nippostrongylus brasiliensis. Microbiome. (2015) 3:77. doi: 10.1186/s40168-015-0142-1
175. Reynolds LA, Smith KA, Filbey KJ, Harcus Y, Hewitson JP, Redpath SA, et al. Commensal-pathogen interactions in the intestinal tract: lactobacilli promote infection with, and are promoted by, helminth parasites. Gut Microbes. (2014) 5:522–32. doi: 10.4161/gmic.32155
176. Dea-Ayuela MA, Rama-Iñiguez S, Bolás-Fernandez F. Enhanced susceptibility to Trichuris muris infection of B10Br mice treated with the probiotic Lactobacillus casei. Int Immunopharmacol. (2008) 8:28–35. doi: 10.1016/j.intimp.2007.10.003
177. White EC, Houlden A, Bancroft AJ, Goldrick M, Hayes KS, Roberts IS, et al. Manipulation of host and parasite microbiotas: survival strategies during chronic nematode infection. Sci Adv. (2018) 4:eaap7399. doi: 10.1126/sciadv.aap7399
178. Hayes KS, Bancroft AJ, Goldrick M, Portsmouth C, Roberts IS, Grencis RK. Exploitation of the intestinal microflora by the parasitic nematode Trichuris muris. Science. (2010) 328: 1391–4. doi: 10.1126/science.1187703
179. Mitchell GF. A Note on concomitant immunity in host-parasite relationships: a successfully transplanted concept from tumor immunology. Adv Cancer Res. (1990) 54:319–32. doi: 10.1016/s0065-230x(08)60816-7
Keywords: trichuris muris, trickle infection, Th2 immunity, Heligmosomoides bakeri, mucosal immunology, helminths, parasitism
Citation: Colombo SAP and Grencis RK (2020) Immunity to Soil-Transmitted Helminths: Evidence From the Field and Laboratory Models. Front. Immunol. 11:1286. doi: 10.3389/fimmu.2020.01286
Received: 15 April 2020; Accepted: 21 May 2020;
Published: 23 June 2020.
Edited by:
Mark C. Siracusa, Rutgers Biomedical and Health Sciences, United StatesReviewed by:
Meera G. Nair, University of California, Riverside, United StatesColby Zaph, Monash University, Australia
Copyright © 2020 Colombo and Grencis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard K. Grencis, cmljaGFyZC5ncmVuY2lzQG1hbmNoZXN0ZXIuYWMudWs=