
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 June 2020
Sec. Microbial Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01250
This article is part of the Research Topic Emerging Concepts of Innate Immune Responses to Neglected Tropical Diseases View all 15 articles
 Omar A. Alfituri1
Omar A. Alfituri1 Juan F. Quintana2
Juan F. Quintana2 Annette MacLeod2
Annette MacLeod2 Paul Garside2
Paul Garside2 Robert A. Benson2
Robert A. Benson2 James M. Brewer2
James M. Brewer2 Neil A. Mabbott1
Neil A. Mabbott1 Liam J. Morrison1
Liam J. Morrison1 Paul Capewell3*
Paul Capewell3*African trypanosomes are single-celled extracellular protozoan parasites transmitted by tsetse fly vectors across sub-Saharan Africa, causing serious disease in both humans and animals. Mammalian infections begin when the tsetse fly penetrates the skin in order to take a blood meal, depositing trypanosomes into the dermal layer. Similarly, onward transmission occurs when differentiated and insect pre-adapted forms are ingested by the fly during a blood meal. Between these transmission steps, trypanosomes access the systemic circulation of the vertebrate host via the skin-draining lymph nodes, disseminating into multiple tissues and organs, and establishing chronic, and long-lasting infections. However, most studies of the immunobiology of African trypanosomes have been conducted under experimental conditions that bypass the skin as a route for systemic dissemination (typically via intraperitoneal or intravenous routes). Therefore, the importance of these initial interactions between trypanosomes and the skin at the site of initial infection, and the implications for these processes in infection establishment, have largely been overlooked. Recent studies have also demonstrated active and complex interactions between the mammalian host and trypanosomes in the skin during initial infection and revealed the skin as an overlooked anatomical reservoir for transmission. This highlights the importance of this organ when investigating the biology of trypanosome infections and the associated immune responses at the initial site of infection. Here, we review the mechanisms involved in establishing African trypanosome infections and potential of the skin as a reservoir, the role of innate immune cells in the skin during initial infection, and the subsequent immune interactions as the parasites migrate from the skin. We suggest that a thorough identification of the mechanisms involved in establishing African trypanosome infections in the skin and their progression through the host is essential for the development of novel approaches to interrupt disease transmission and control these important diseases.
African trypanosomiasis has historically been the cause of large outbreaks of human disease, likely contributing to the deaths of millions of people across sub-Saharan Africa in the early twentieth century (1, 2) and inflicting substantial economic damage on the African agriculture industry to this day (3, 4). African trypanosomes include an array of vector-borne, single cell hemoflagellate protozoa (order Kinetoplastida), although three species cause major disease: Trypanosoma brucei, T. congolense, and T. vivax. Two subspecies of T. brucei, T. b. rhodesiense, and T. b. gambiense, cause human African trypanosomiasis (HAT), also known as sleeping sickness (5–7), with more than 57 million people at risk of infection (6, 8). T. congolense, T. vivax, and T. brucei are also the most significant contributors to disease in livestock animals (animal African trypanosomiasis or AAT). Classically, the trypanosome lifecycle starts with the tsetse fly (Glossina spp.) depositing an inoculum of metacyclic trypomastigotes in the skin when taking a blood meal (9–11). Following intradermal (i.d.) inoculation of metacyclic forms, the parasites differentiate into long-slender trypomastigotes that are proliferative and able to establish patent infections in the vertebrate host. However, the timing and the mechanisms controlling these events remain unclear (3, 12, 13). From the initial site of infection, the proliferative long-slender form trypanosomes travel to the local draining lymph nodes via afferent lymphatic vessels before disseminating systemically and establishing a patent infection in the bloodstream (14–17). African trypanosomes (and T. brucei in particular) also actively colonize multiple tissues in the vertebrate host, including the skin. Skin-dwelling parasites functionally and behaviorally adapt to their microenvironment, allowing them to thrive and persist (18, 19). These recent studies demonstrate that there is a previously underappreciated heterogeneity in the population of parasites residing within the vertebrate host, with important implications for understanding the biology of trypanosomes and the way in which the host responds to infection. The presence of trypanosomes in the skin has been demonstrated in both animal models of infection and human clinical samples, suggesting that it is a central aspect of transmission. Nonetheless, the mechanisms deployed by trypanosomes to inhabit and migrate from the cutaneous environment, and the interplay between resident skin cells (including immune cells), and trypanosomes during the onset of the infection, remain largely unexplored. In this review, we aim to (i) highlight current knowledge on trypanosome establishment of infection in the skin; (ii) examine the interactions between the host immune system and trypanosomes in the skin; (iii) explore the mechanisms of trypanosome migration from the skin toward systemic infection and further transmission; and finally (iv) discuss the potential of novel therapeutic and intervention strategies being developed as a consequence of these studies.
Upon infection, metacyclic trypomastigotes must circumvent several environmental challenges in order to develop into the proliferative long-slender form trypomastigotes. This series of events ultimately leads to parasite dissemination in the host bloodstream but involves interactions between the developmental stages of the parasite, the host cells in the dermis, and the immune cells recruited to the site of infection. Mammalian skin is a large, highly complex organ that acts as a protective barrier between the internal components of the host and the external environment (Figure 1) (20, 21). The mechanism by which the skin protects the host is not simply through providing a physical barrier, but also the collection of immune cells, biological factors, layers of tissue, and networks of lymphatic and blood vessels (21–23). The three main components of the skin are the epidermis, dermis, and subcutaneous layer, each containing various immune cells involved in innate responses, inflammation, and surveillance (Figure 1A) (21, 24). The dermis is primarily connective tissue produced by dermal fibroblasts. Local immune responses generated within the tissue are initiated by dermal macrophages, dermal dendritic cells, natural killer (NK) cells, mast cells, αβ/γδ T cells, and natural killer T (NKT) cells (25, 26). The skin also contains numerous blood and lymphatic vessels, nerves, and (in humans but not mice) sweat glands (23, 27, 28). Together, these layers create a highly organized body compartment that represents a strong physical and biological barrier to pathogens and systemic infections.
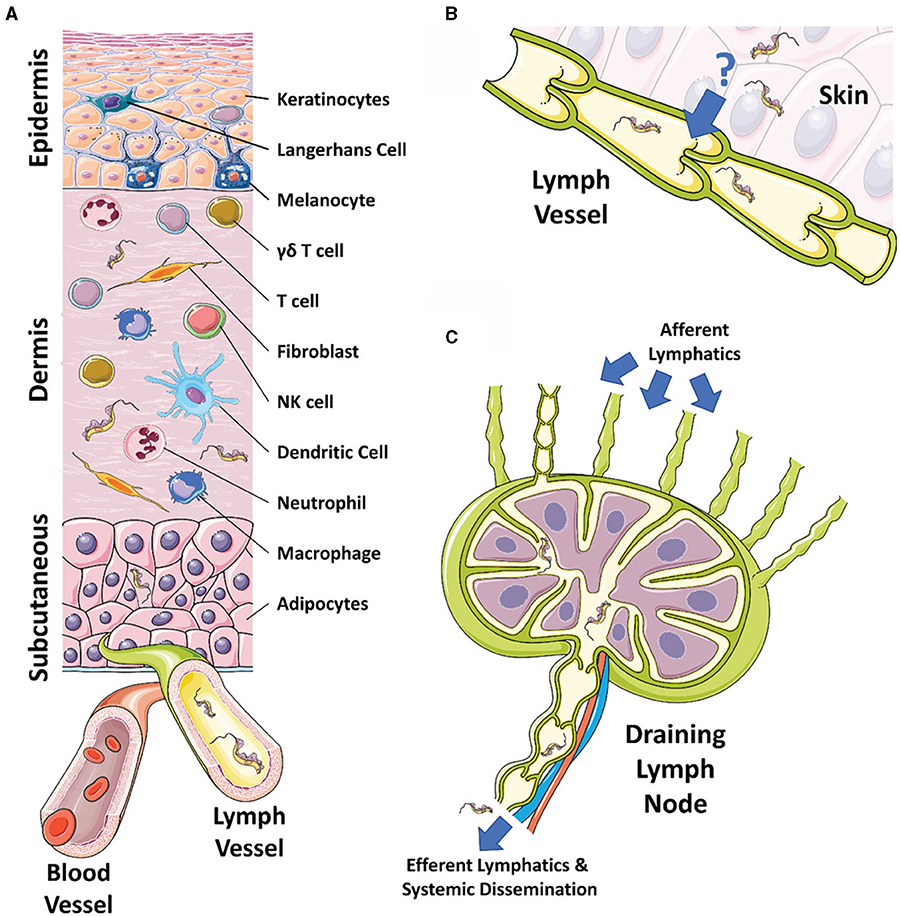
Figure 1. The skin, draining lymphatics, and lymph nodes. (A) Diagram of the cellular composition of the epidermal, dermal, and subcutaneous layers of mammalian skin. The outermost epidermal layer consists of a layer of corneocytes above a layer of keratinocytes. These cells manage the tight junctions and the stratum corneum. Langerhans cells and intraepithelial T cells survey the epidermis for antigen to be presented. The central dermal layer contains fibroblasts that produce extracellular matrix proteins to provide structural support and elasticity. Immune responses are initiated by dermal macrophages, dermal dendritic cells, NK cells, and T cells. The inner subcutaneous layer primarily consists of adipocytes. Local lymphatic and blood vessels allow for the trafficking of cells, proteins, and waste. The initial tsetse fly bite injects trypanosomes into the dermis. From the dermis, the parasites exhibit tropism that leads to migration toward the afferent lymph vessels in the skin disseminating to the blood and other regions of the body. (B) The mechanism behind directional migration of trypanosomes from the skin to the lymphatics is unknown. Parasites may be responding to an unreported chemical cue in a chemotactic manner and they may crawl along lymph vessels, access open junctions, or are drawn into the lymphatics through hydrodynamic flow force and pressure. (C) Afferent lymphatic vessels in the skin allow for the drainage of leukocytes and antigen into the draining lymph node. Lymph, containing activated T and B cells, plasma cells, and antibody, passes into the medullary sinus, before exiting via efferent lymphatic vessels. Trypanosomes enter the draining lymph nodes, causing lymphadenopathy, and exit via the efferent lymphatics. Systemic dissemination of the host is reached via the main lymphatic ducts.
Within an infected tsetse fly, T. brucei undergoes a range of complex developmental stages (29). When first taking a blood-meal from an infected mammalian host, parasites are ingested, and become proliferative procyclic trypomastigote forms in the midgut of the tsetse fly. These procyclic trypomastigotes divide rapidly before transforming into mesocyclic forms in the alimentary tract that then invade the salivary glands and transform into the rapidly proliferating epimastigotes. Tsetse saliva provides an environment that promotes trypanosome adherence to epithelial surfaces, the initiation of binary fission, and also triggers their transformation into mammal-infective metacyclic trypomastigotes (29–31). Without the presence of these salivary components, metacyclic trypomastigotes have reduced infectivity (32). Following an infected tsetse fly feeding on a mammalian host, trypanosomes are deposited into the dermis of the host skin (11). During this process, the tsetse fly proboscis inflicts significant trauma on the skin and associated tissues, while also introducing a concoction of active compounds via the saliva (33). In humans, a trypanosome-filled lesion known as a chancre often develops 5–15 days post-inoculation, which comprises indurated and inflamed patches of skin (34, 35). Similar lesions also occur in goats and cattle (36). The development of a chancre may be related to several components of tsetse saliva that have been shown to affect inflammation at the site of infection, including 5′nucleotidase-related (5′Nuc), tsetse thrombin inhibitor (TTI), and both thrombin serine protease, and esterase inhibitors (30, 37–40). This altered immune state is characterized by elevated interleukin IL-4 and IL-10 production (30, 32, 40–42). Murine models of infection have also shown that tsetse saliva suppresses T and B cell responses systemically, skewing the host toward a Th2 immune environment, leading to increased IL-4 and IL-10, alongside decreased interferon-γ (IFN-γ) titers, in the draining lymph nodes of infected mice. There is also an associated immunoglobulin (Ig) IgG1 and IgE antibody response. In addition, bites from tsetse flies infected with trypanosomes showed significantly reduced thrombin inhibition and less anticoagulation compared to bites by naïve tsetse flies. This results in a more prolonged feeding period that contributes to an increased likelihood of parasite transmission (9). Together, these anti-inflammatory and anti-coagulative processes act to increase the efficiency of parasite transmission from the fly vector to the mammalian host (32), leading to the induction of microenvironmental conditions more suitable for metacyclic trypomastigotes. This facilitates their differentiation into proliferative long-slender trypomastigotes. In this context, it appears likely that metacyclic trypomastigotes are equipped with various molecular tools (e.g., secreted virulence factors) to initiate tissue colonization events and overcome the acute response elicited by skin-resident and recruited immune cells. This leads to a scenario in which the acute immune response at the site of infection may act as a bottleneck that selects for more infectious parasites. Consequently, the route of infection could potentially shape downstream interactions and responses in other body compartments, affecting parasitaemia and longer-term infection dynamics.
Despite the skin being the first point of contact between metacyclic trypomastigotes and the vertebrate host, the importance of the skin-stage of disease in trypanosome infections has largely been overlooked in experimental studies. The majority of prior experiments have examined infections initiated via needle challenge, predominantly by intraperitoneal (i.p.) or intravenous (i.v.) routes (Table 1). However, a small number of comparative experiments have revealed that inoculation routes can substantially affect outcome (43, 44). For example, it has been shown that the percentage of BALB/c mice with detectable parasitemia after infection by T. brucei differed between animals infected i.p. vs. i.d. This study also found that the proportions of mice displaying detectable parasitemia were significantly reduced in the i.d. infected mice compared to i.p. infected mice, and that i.d. infected mice were 100-fold less susceptible to trypanosome infection than i.p. infected mice in a dose-dependent manner (43). Moreover, the impact of infection route on infectivity also differs between trypanosome species (44). For example, T. congolense infected intramuscularly (i.m.) had an earlier onset of parasitemia compared to those infected via an i.d. route. However, animals in which T. brucei parasites were administered i.p. led to an earlier onset of parasitemia than both i.m. and i.d. administration. These observations suggest that parasites face tissue-specific challenges in the skin (e.g., resident immune cells or nutrient availability) that delineate their capacity to disseminate systemically within the vertebrate host (44).
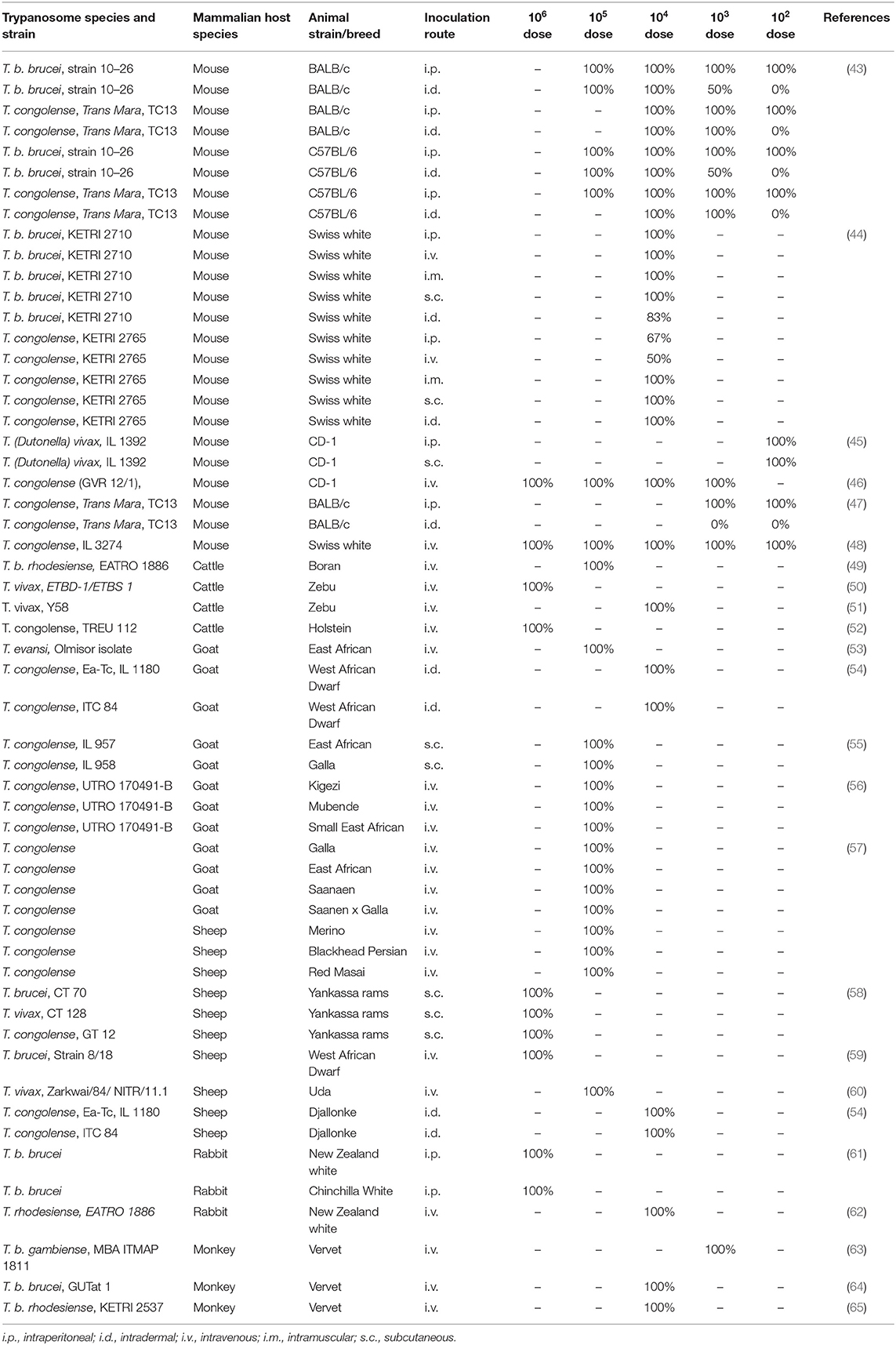
Table 1. Susceptibility of various animal models to different trypanosome strains represented by the proportion of animals displaying a patent infection, depending on route and dose.
In addition to infection route, inoculum dose has also been shown to influence the outcome of trypanosome infection (44). For example, goats infected i.d. with T. congolense showed a delay in the onset of a local skin reaction (chancre formation) as the inoculum dose decreased (66), with a concomitant decrease in the size of the chancre. Consistent with these observations, a similar effect was reported for BALB/c mice infected i.d. with T. brucei, showing reduced infection rates at lower doses compared to higher doses (43). In this study, all animals infected with doses ranging from 1 × 105 to 1 × 104 parasites developed detectable parasitemia. Conversely, doses of 1 × 102 parasites led to no patent infections and 1 × 103 parasites resulted in only 50% of the animals developing a patent infection. In contrast, all the mice infected with 1 × 102 parasites i.p. developed detectable parasitemia at the same time point, indicating that the skin poses a significant barrier for systemic dissemination and infection dynamics. These results suggest that there is a minimal infective parasite dose able to survive the initial challenge mounted by the local immune response in the skin.
The dose and route of infection has been shown to affect the dynamics of several protozoan infections, including the related trypanomastid Leishmania. These parasites are similarly transmitted to the skin of the mammalian host through the bites of female sand fly vectors (67), although they differ from extracellular African trypanosomes in that the inoculated lifecycle stage invades mammalian cells and replicates intracellularly. Upon feeding, the sand fly regurgitates metacyclic promastigote forms into the skin that are then phagocytosed by host macrophages. Promastigotes then develop into amastigote forms and replicate within the host cell. Subcutaneous (s.c.) injections of Leishmania major and L. tropica in mice result in a lower systemic parasite burden and increased protective immunity compared to i.p. and i.d. infections (68, 69). An increased Th1-associated resistance to Leishmania infection was induced in BALB/c mice following a low dose of parasite inoculum administered either i.d. or s.c. (69, 70), whereas a high inoculum induced a more Th2-skewed immune response that led to higher susceptibility and systemic parasite burden. These observations in related parasites highlight the importance of the initial parasite dose and site of infection for disease outcome. More importantly, these initial interactions might favor further dissemination (e.g., by infecting recruited immune cells) or the formation of quiescent parasite foci relevant for diagnostics and infection recrudescence upon treatment. It is likely that there are similar effects occurring during the initial skin stages of African trypanosome infections, although the factors determining parasite survival and migration remain to be fully explored.
African trypanosomes exist entirely extracellularly within the mammalian host and are constantly exposed to the host innate and adaptive immune systems. Previous studies examining systemic immune responses in mice using artificial inoculation routes (primarily i.p. and i.v.) have found numerous components that are vital to controlling the initial stages of trypanosome infection, including macrophages, monocytes, dendritic cells, neutrophils, and NK cells (Figure 2) (71–75). The early immune response (<2 weeks) is characterized by an induction of a strong pro-inflammatory profile, including the expression of IFN-γ, tumor necrosis factor (TNF), IL-6, and the production of nitric oxide (NO) (3, 76–82). An adaptive B cell response is also elicited, leading to the production of antigen-specific antibodies that target the immunodominant variant surface glycoprotein (VSG) at the parasite surface. However, the artificial inoculation routes used in these studies overlook the events that occur in the skin early during infection. Through understanding the processes involved in establishing infection in the mammalian host and establishing how the immune system interacts with the parasites in the initial stages, skin-targeted research could provide important information on how the disease progresses within the host. This additional information may reveal methods to develop novel methods for controlling disease transmission in humans and animals. It is also likely that the immune responses in the skin have wider impacts on the systemic infection, similar to those observed in Leishmania infections (69, 70) and recent data have shown that there is a population of African trypanosomes present in the skin of the host (11, 18, 83). These parasites are integral to transmission, but their persistence suggests that the parasites can avoid or co-opt the immune response in the skin. Understanding how this is achieved could lead to methods to limit this population, reducing transmission. This skin-dwelling population also presents issues for new therapeutics targeting African trypanosomes that have been developed for parasites in the bloodstream and CNS. Although there has been little work to date on skin immune responses during trypanosome challenge, inferences can be made from systemic studies and related parasites, in addition to the small number of studies that have been performed.
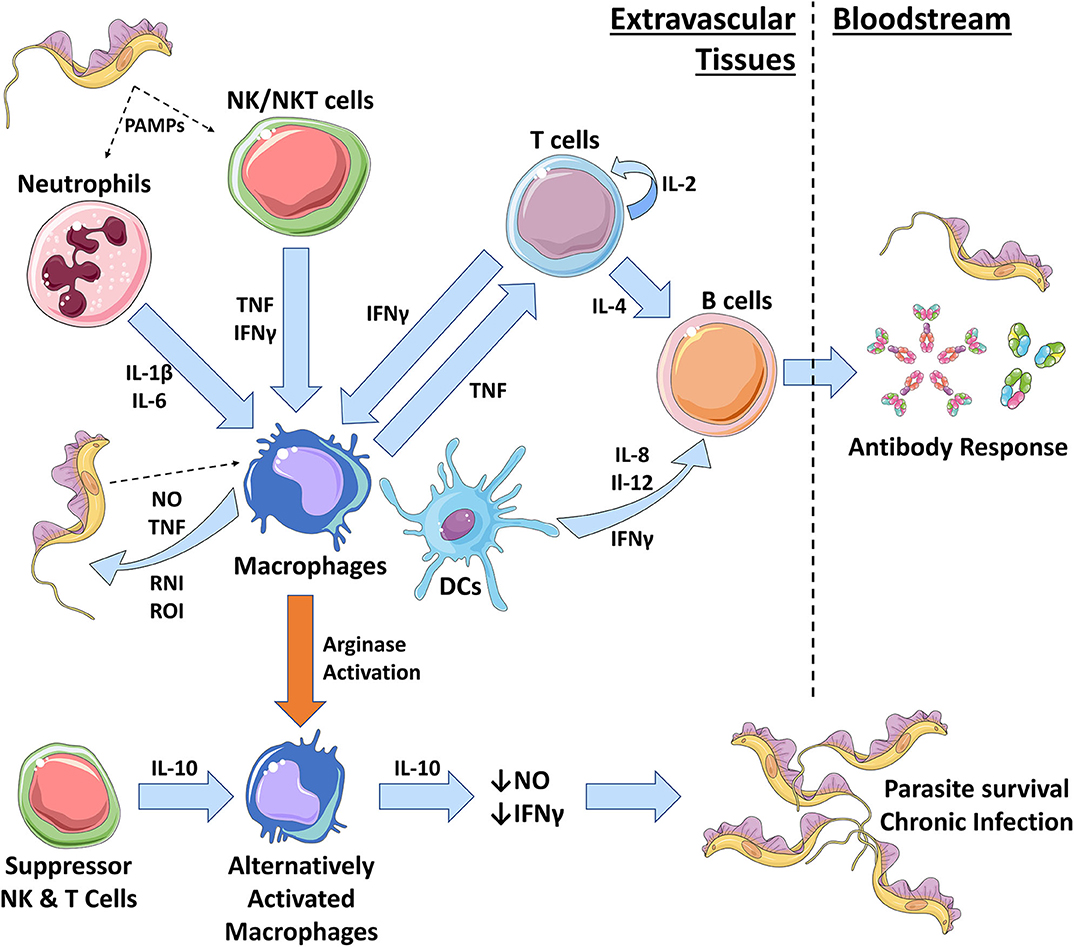
Figure 2. The role of innate immune cells during African trypanosomiasis. During early trypanosome infection, a strong Th1 immune response is initiated by the host. In the skin, neutrophils and NK cells are the first to respond to trypanosomal pathogen-associated molecular patterns (PAMPs), such as VSG and CpG DNA. Neutrophils are heavily involved in repairing the initial wound caused by the tsetse fly bite and also produce pro-inflammatory IL-1β and IL-6. NK cells produce pro-inflammatory TNF and IFN-γ that results in the classical activation of pro-inflammatory macrophages (Mθ) via iNOS activation. Macrophages can also be activated through interactions with trypanosomal PAMPs. Classically activated macrophages produce further pro-inflammatory molecules, including TNF, nitric oxide (NO), and reactive nitrogen intermediates (RNIs) and reactive oxygen intermediates (ROIs). These chemicals can directly kill trypanosomes in extravascular spaces and tissues and allows for parasite control. Macrophage secretion of TNF can also recruit and activate T cells which self-renew via autocrine IL-2 secretion. T cells produce IFN-γ to further activate macrophages and IL-4 to activate B cells. Macrophages and dendritic cells (DCs) will further activate B cells during a Th1 response, via IL-6, IL-12, and IFN-γ, to promote the production of antibodies that can target the VSG trypanosomes, inducing waves of parasite clearance in the bloodstream. However, antigenic variation hinders the effective clearance of trypanosome populations. Macrophages can also become alternatively activated, resulting in a Th2 immunosuppressive response. Cytokines such as IL-10, IL-4, and TGF-β initiate this type of response by promoting arginase activation in macrophages. As a result, alternatively activated macrophages produce immunosuppressive IL-10 and suppress production of trypanostatic NO and IFN-γ. This promotes parasite growth and survival, leading to a chronic infection.
It is likely that some of the earliest cellular responders to trypanosome inoculation in the skin are neutrophils, NK cells, and NKT cells (3, 84, 85). Neutrophils are some of the most ubiquitous leukocytes in the human immune system and are involved in the killing of many pathogens (including protozoa). They act through phagocytosis, the release of reactive oxygen species, and neutrophil extracellular traps (NETs) (86–89). They are also important mediators of tissue repair and wound healing, producing various pro-inflammatory cytokines that include transforming growth factor-β (TGF-β), IL-4, IL-12, and IL-13 (88). They may also potentially release IFN-γ, although it is unclear whether neutrophils are a true source of IFN-γ, especially in humans (90). As such, our understanding of these mechanisms relies heavily on murine data that may not be applicable to human mechanisms of immunity. In these model experiments, neutrophils have been shown to be the primary responders to tsetse fly bites and are recruited to the dermal bite site within 4.5 h (85). At the same time as neutrophil recruitment, there is an induction of pro-inflammatory IL-1β and IL-6, as well as anti-inflammatory IL-10 (85), from unidentified sources. Neutrophils may also produce trypanolytic antimicrobial peptides, such as cathelicidins and defensins (91), although this early neutrophil response does not appear to contribute to trypanosome killing in the skin (85).
Cytotoxic NK and NKT cells are also commonly employed during the earliest periods of pathogen infection. However, the roles of NK cells during trypanosome infection are poorly understood and investigation is limited to systemic studies. NK cell-deficient mice infected with T. congolense are unable to control the levels of parasitemia due to lower levels of IFN-γ and TNF, leading to rapid onset of death due and uncontrollable parasitemia (92). This lethal phenotype was rescued when NK cells were transferred into the NK cell-deficient mice prior to infection. Moreover, mice infected i.p. with T. congolense show a systemic recruitment of NK cells that are thought to provide early production of IFN-γ and TNF in the blood, spleen, lungs, and liver (92). NK cell activity in trypanotolerant strains of mice (strains that display reduced clinical disease when infected with trypanosomes compared to susceptible counterparts) infected with T. congolense has been suggested to be due to an increase in the production of IFN-γ during infection (72). The activation of neutrophils, NK cells, and NK/T cells in the skin following exposure to trypanosomal pathogen-associated molecular patterns (PAMPs) also results in the production of IFN-γ and TNF that can induce the activation of classically activated macrophages (93). In this context, we speculate that recruitment of NK cells in the skin also provide early protective immunity in trypanotolerant hosts, utilizing pro-inflammatory cytokines to promote further immune activation and parasite killing. Studies regarding the spatio-temporal recruitment of immune effectors to the skin during T. brucei infection might shed lights into these processes.
Similar to African trypanosomes, Leishmania spp. are transmitted into the subdermal layer of the skin by female phlebotomine sand flies (67, 94). In this case, it has been shown that neutrophils in the skin are the initial responders to infection and phagocytose Leishmania metacyclic promastigotes (95). Two-photon intravital imaging has shown that 40–60 min post-infection, a rapid early recruitment of neutrophils is induced at the site of infection in the skin following sand fly feeding (96). Here, the neutrophils occupy the epidermis in large numbers where they directly kill promastigotes using NETs (86, 97) and active phagocytosis of parasites (96). However, there is substantial evidence that promastigotes can persist within neutrophils, escaping effective killing, and potentially taking advantage of neutrophils as a means to continue their life cycle within the mammalian host (98, 99). The promastigotes in the infected neutrophils are in turn phagocytosed by macrophages and dendritic cells where they develop into the amastigote form and replicate (94, 95). This neutrophil recruitment response is similar to that observed following an infected tsetse fly bite (85). However, while direct killing by neutrophils has been shown with Leishmania parasites, it has not been shown during African trypanosome infections. One hypothesis is that the rapid recruitment of neutrophils during trypanosome infection occurs in response to the tissue damage inflicted by the tsetse fly and is required for wound repair. In this regard, the initial recruitment of neutrophils upon T. brucei infection in mice (via i.p.) is prolonged after the initial peak of parasitemia. This is thought to contribute to the suppression of NK, NK/T cells, and both T and B lymphocytes in the spleen at later stages of infection (100). This may be through disruption of the splenic microarchitecture due to significant pro-inflammatory responses. How these immunosuppressive mechanisms relate to early infection in the skin, as well as the activation of the skin-resident immune population, remain unknown and should therefore be an important area for further investigation.
The skin contains an abundance of macrophages with the potential to combat infection but again, little is known about the role of skin-resident macrophages during trypanosome infection. During systemic infections, macrophages are considered to play an essential role in combating African trypanosome infections in the mammalian host and are central to mediating the immune response in the extravascular tissues (Figures 2, 3) (3, 74, 101–110). After infection, trypanosome associated PAMPs trigger the activation of these innate mononuclear phagocytes via interactions with pattern recognition receptors (PRRs) on the surfaces of host immune cells. These are triggered by parasite-derived molecules such as CpG DNA (recognized by TLR-9) and soluble glycosylphosphatidyl inositol (GPI)-anchored VSG (recognized by scavenger receptor PRRs) (36, 74, 84, 103, 105, 108, 111–115). The release of soluble VSG further stimulates immune cells in a type 1-dependent manner (112). These trigger signaling pathways that lead to the acquisition of a classically activated phenotype in macrophages (and the release of pro-inflammatory cytokines such as IFN-γ and TNF), which is important for quickly controlling invading trypanosomes (Figure 3) (36, 84, 103, 105, 108, 112–114).
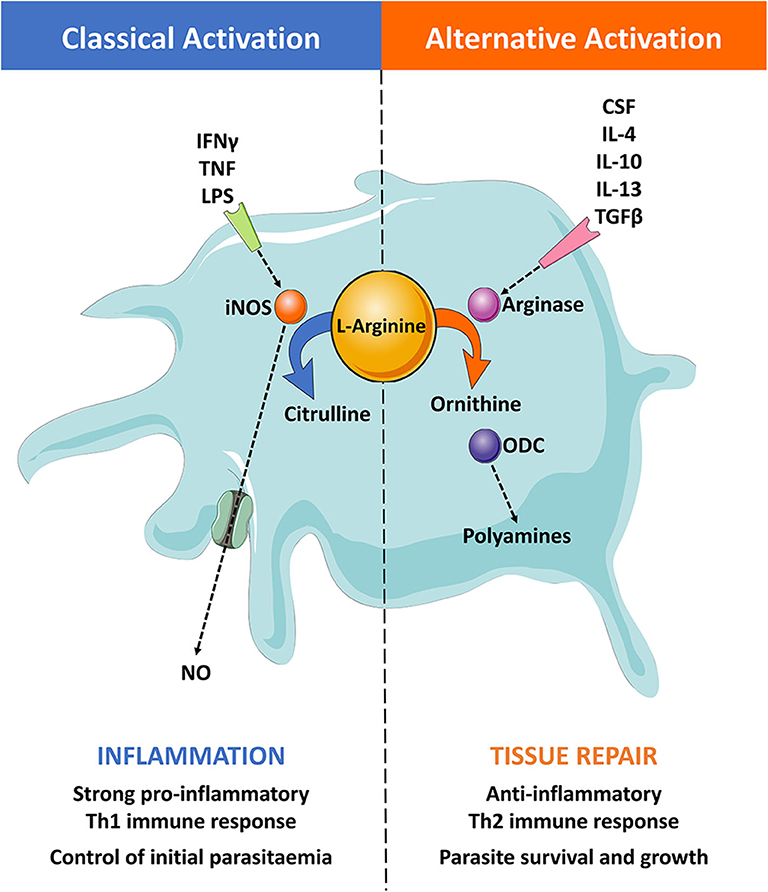
Figure 3. The roles of classically and alternatively activated macrophages. (Left) Pro-inflammatory stimuli, such as IFN-γ, TNF-α, and LPS, classically activates macrophages. Classically activated macrophages induce the expression of the inducible nitric oxide synthase (iNOS) enzyme, which catabolizes the substrate L-arginine to produce NO and citrulline. This results in a pro-inflammatory Th1 immune response that can effectively control the initial parasitaemia peak. (Right) Anti-inflammatory stimuli, such as CSF, IL-4, IL-10, IL-13, and TGF-β, alternatively activate macrophages. These induce the expression of the arginase enzyme that catabolizes the substrate L-arginine to produce ornithine. The enzyme ornithine decarboxylase (ODC) catalyzes the breakdown of ornithine to produce polyamines, resulting in an anti-inflammatory Th2 immune response and tissue repair. This type of response typically leads to an immune environment that promotes trypanosome growth and survival.
The strong pro-inflammatory type 1 (Th1) immune response initiated during the early onset of infection, characterized by elevated levels of IFN-γ and IL-2, leads to an increase in macrophage numbers in the spleen, liver, and bone marrow (106). During clinical disease, macrophage migrating inhibitory factor (MIF) mRNA levels are also increased in the blood of infected patients (116). This could explain the elevated levels of recruited macrophages observed in peripheral immune organs. Macrophages and liver-resident Kupffer cells phagocytose trypanosomes that are opsonized by parasite-specific Ig. These cells are aided by various soluble factors derived from the host complement system (106, 117–119). Experiments using T. brucei-infected mice have shown that Kupffer cells in the liver are involved in most of the parasite clearance that occurs via complement and antibody-mediated phagocytosis (120). Classically activated macrophages can also utilize the inducible nitric oxide synthase (iNOS) enzyme to produce highly reactive and toxic nitric oxide (NO) via the L-arginine metabolic pathway (Figure 3) (121, 122). Additionally, these mononuclear phagocytes can produce further pro-inflammatory cytokines such as TNF, IL-1, IL-6, IL-8, and IL-12 (93). Interestingly, several pro-inflammatory cytokines have been shown to display potent trypanostatic activities, further highlighting the importance of these cytokines during the onset of the infection (3, 72, 84, 123–130). One such trypanostatic effect is believed to be mediated by NO as experimental infections with T. congolense and T. brucei have shown that NO production can inhibit trypanosome growth and specifically control the first wave of parasitemia (131, 132). However, the role of NO is contentious as in vivo studies have reported that NO readily binds to hemoglobin and interacts with red blood cells (76–78). This would mean that NO would be quickly quenched in the bloodstream, removing its inhibitory effect. However, NO may still be effective in the microenvironment of extravascular spaces in which hemoglobin is much reduced or absent, such as the skin (133). Importantly, not all interactions with cytokines appear to be detrimental to trypanosomes. Trypanosomes are known to secrete trypanosome lymphocyte triggering factor (TLTF) that can trigger the production of IFN-γ from CD8+ T cells (134, 135), inducing a potent classically-activated macrophage response. This seemingly contradictory effect may suggest that African trypanosomes can benefit from this specific T cell interaction for survival, or alternatively, that TLTF might play an important role in inducing the development of a more immunotolerant environment for the parasite to sustain survival. This remains speculative but merits further investigation.
The induction of type 2 macrophages is a prominent feature of the immunosuppression that occurs during chronic T. brucei infections. Studies have shown that trypanosomes trigger a switch in macrophage activation from pro-inflammatory (or classically-activated) cells to a more anti-inflammatory (or alternatively activated) state (74, 104, 110, 136, 137). This switch in macrophage character skews the host immune response from Th1 to Th2, resulting in an anti-inflammatory environment that promotes parasite survival in the host (138). The implications for this profound switch in immunological state are a matter of active research but the process may contribute to reducing the deleterious effects on the host of sustained inflammation, as well as favoring tissue repair and regeneration (93, 139–143). Macrophages can become alternatively activated through stimulation by macrophage colony-stimulating factor (CSF-1), IL-4, IL-10, IL-13, and TGF-β (Figure 3) (93, 144–146). One of the main hallmarks of alternatively-activated macrophages (AAM) is the expression of arginases upon stimulation, which in turn compete with iNOS and induce ornithine and urea production via the L-arginine pathway instead of NO and citrulline (147).
A recent study has also shown that T. brucei can actively skew macrophage and glial cell activation by secreting metabolites that suppress their pro-inflammatory functions (148). These macrophage responses varied depending on the strain of the parasite, suggesting it is a parasite-derived factor determining host response, supporting earlier work describing the existence of a parasite-driven virulence factor (149). These trypanosomal factors can trigger macrophages to switch toward a Th2 phenotype and include the T. brucei kinesin heavy chain isoform (TbKHC1) that actively induces IL-10 production and arginase activity, resulting in decreased NO production (150). When wild-type mice were infected with TbKHC1 KO trypanosomes, parasitemia was reduced and the survival of the host enhanced (150). These data suggest that the release of TbKHC1 by T. brucei enables the parasites to manipulate host cell metabolism by biasing the L-arginine pathway toward arginase enzyme activity, although it remains unclear whether TbKHC1 is constitutively released by viable parasites or is a byproduct from decaying or dead parasites. It has also been suggested that immunosuppression in the skin during intradermal trypanosome infection could be mediated by the combination of a mixed classical/alternative macrophage response and suppressor T cell response (151). These findings suggest that a pro-inflammatory macrophage/Th1 response is needed for the effective control of trypanosomes in the skin, although a Th2 response seems necessary to sustain host survival in the face of a chronic infection. As such, these specific macrophage-trypanosome interactions should be investigated thoroughly for opportunities for blocking disease progression.
Dendritic cells (DCs) are a group of antigen-presenting cells (APCs) that recognize and capture antigen for presentation to T cells (152, 153). Studies using i.p. T. b. rhodesiense infections in mice have shown that splenic DCs may be the primary APCs involved in activating VSG-specific T helper (Th) cell responses in trypanotolerant mouse lines through the upregulation of co-stimulatory receptors, the presentation of VSG peptide antigen, and the production of IL-12 (154, 155). However, subsequent VSG antigen processing and presentation was notably reduced during high parasite burdens in the mice, suggesting a trypanosome factor may be interfering with the ability of APCs to process and present antigen to T cells. Alternatively, there may be a potential impairment of the antigen presentation capacity or DC maturation during chronic infection. This remains speculative for African trypanosome infections but has been described for other infections associated with immunotolerant states (156, 157).
Skin-residing DCs include epidermal Langerhans cells and dermal DCs (158–160). Langerhans cells sample and present antigen from the epidermis to promote the adaptive immune response (161, 162). They have been shown to have a suppressive function in Leishmania infections by modifying the behavior of regulatory T cells (Tregs) but their role in Africa trypanosomiasis is completely unknown and remains to be established (163). Langerhans cells and dermal DCs migrate from the epidermis and dermis, respectively, to the local cutaneous draining lymph nodes to present sampled pathogen antigen to T cells (158–160, 162). Dermal DCs have also been shown to act rapidly to dermis invading protozoan parasites such as L. major (164). It has previously been reported in L. major-infected mice that epidermal Langerhans cells localized at the sand-fly bite site become activated and upregulate major histocompatibility complex molecules and co-stimulatory receptors (165). This results in cytokine release (IL-12) and the promotion of a Th1 cellular response. African trypanosomes have recently been shown to form active foci in the skin and it is likely that this would be modulated and controlled by resident DCs and Langerhans cells, similar to related trypanomastids (11, 83). Moreover, DCs might elicit a more immunotolerant state in the skin, ultimately allowing the persistence of skin foci. However, the specific roles of skin resident DCs during trypanosome infection remain to be elucidated.
Skin-resident T cells are another group of immune cells that survey the tissue for pathogens. The epidermal layer is patrolled by αβ effector T cells and more innate-like γδ T cells, in addition to group 2 innate lymphoid cells (ILC2s) (166–168). The γδ T cell population act with innate cells to survey tissue during the early stages of infection before the more conventional adaptive immune cells become involved. Dermal γδ T cells also express many receptors and cytokines that can alter Th1 and Th17 responses and affect extracellular pathogen, including IFN-γ, TNF, and IL-17 (169–171). ILC2s are dependent on IL-7 and constitutively secrete IL-13, thought to be central for interaction with granulocytes (167). Although T cells during skin diseases have been well-studied, particularly for psoriasis and allergies (172–174), the roles of specific γδ T cells, ILC2s, and their associated cytokines during human trypanosome infections are unclear. A study using cattle found that tsetse-transmitted T. congolense infection leads to the increased numbers of γδ T cells and that these cells are activated in the trypanotolerant N'Dama breed but not the more susceptible Boran breed (175). However, cattle and other ruminants possess a substantially higher proportion of γδ T cells in the peripheral blood mononuclear lymphocyte population (15–60%) than both humans and mice (<5%) (176) and their role may differ between these different hosts. Importantly, due to the induction of AAM activity observed during chronic infection, a reduction in IL-2 secretion and expression of the α-chain of the IL-2 cytokine receptor in lymph node T cells leads to inhibition of immune responses during T. brucei and T. congolense infections in mice and cattle (177–181).
While it is unclear what the potential role of skin-resident T cells may be during African trypanosome infection, insights could be gained from the pathogenesis of other parasites. For example, during infection with Plasmodium spp., sporozoites induce rapid immunosuppression in the skin (182–184) that affects both T and B cell functionality. Plasmodium-specific Tregs are also induced in the skin that expand upon re-exposure to Plasmodium antigens and suppress immunity to infection (185). This was found to be linked to parasite specific factors and when the sporozoite surface protein CSP was injected into the skin at low doses with CpG DNA, Plasmodium-specific CD8+ effector T cells were significantly inhibited (182). This is hypothesized to involve regulatory B cells (Bregs) that produce immunosuppressive IL-10 and TGF-β as depletion of B cells rescued effector T cell function during malaria infection (186). Similarly, it is possible that the suppressive phenotypes observed during African trypanosomiasis also occur in the skin, hindering the successful elimination of trypanosomes. Indeed, very little inflammation or other immune responses were observed in the skin of mice with heavy burdens of skin-dwelling T. brucei parasites (83).
Following skin infection, trypanosomes trigger a series of immunological events that activate resident immune cells and promote recruitment to the site of infection. This in turn delineates long-term disease outcome, specifically by determining how the parasite will disseminate systemically and establish infection. In order to achieve this, trypanosomes must overcome the initial immunological challenge mounted in the skin and travel to the afferent lymphatics, entering the local draining lymph nodes (Figure 1) (11, 14–17, 151). Historical studies using dogs found that T. brucei equiperdum leaves the dermis via the afferent lymphatics, spreading into the draining lymph nodes before reaching the bloodstream (15). A similar sequence of events has also been shown in cattle, goats (14, 187), and recently mice (11). In this recent mouse study, trypanosomes were first detected within the local draining lymph nodes prior to their detection in the bloodstream (18 h compared to 42 h post-infection, respectively) (11). Intravital imaging of infected mice following tsetse fly bites of the skin has also been used to elucidate some of these mechanisms (17). In these experiments, a large number of parasites exhibited directional migration following trypanosome injection into the skin, traveling back and forth toward the lymphatic vessels. In addition to parasites found in the skin interstitium, parasites were also found within the afferent lymphatic vessels of the dermis. These parasites were characterized by significantly higher velocities than their extravascular counterparts, suggesting behavioral diversity. This is similar to the diverse ranges in parasite motility previously described for T. carassii in zebrafish (188). This would indicate that there are unknown mechanisms through which African trypanosomes are attracted toward and then invade the afferent lymphatics of the skin, allowing their subsequent systemic dissemination in the host (Figures 1B,C). Similar to African trypanosomes, large numbers of Plasmodium sporozoites have been shown to remain in the dermis while others drain to the local lymph nodes (189). These skin-resident sporozoites “glide” through the dermis and can invade the local dermal blood and lymphatic vessels before reaching the liver (190–192). Imaging studies in rodents have shown that skin sporozoites largely drain via the lymphatics having been phagocytosed by dendritic cells. Conversely, a minority enter directly into the bloodstream (193). Intravital imaging has shown that immunization of mice with attenuated sporozoites and P. berghei circumsporozoite protein inhibits sporozoite motility in the skin, resulting in inhibition of dermal blood vessel invasion (194). In addition, Leishmania parasites are known to form reservoirs in the skin that enhance their onward transmission to the sand fly vector (195). However, several species of Leishmania also disseminate systemically around the host and this has been suggested to involve infected macrophages or dendritic cells carrying the parasites to the local draining lymph nodes (94). Although African trypanosomes do not invade cells in the host, it is possible that they may be chaperoned into the local draining lymph nodes via macrophages or dendritic cells, either through an unknown attachment mechanism or a chemical cue.
It is also plausible that lymphatic accumulation may be driven by hydrostatic pressure, protein and/or chemical gradients, or the sensing of lymph flow (196–201). These environmental cues could direct trypanosomes toward open junctions in the lymphatic epithelium, similar to the systems used by lymphocytes for lymphatic invasion (199, 202, 203). For example, dendritic cells have been shown to respond to gradients of CCL19 and CCL21 chemokines expressed in the lymphatic vessels (204), facilitating entry into the lymphatics in the skin (205), and CXCL12 gradients have been shown to be important for the initiation of dendritic cell responses in the skin (206). However, there was no evidence for the role of several host-derived chemokines in attracting trypanosomes in a recent study (17). As African trypanosomes possess chemosensory capabilities through receptors found on the flagellum and flagellar pocket (207), it is feasible that they could respond to non-chemokine chemical gradients within the host to reach the lymphatics. For example, glucose is crucial for the metabolism of bloodstream form trypanosomes (208) and glucose concentrations in canines have been shown to be higher in the lymph than the blood (209). Glucose, lipids, or other factors essential for trypanosome metabolism, could therefore act as chemical chemoattractants for trypanosomes. The identification of potential these (tissue-specific) parasite chemoattractants merits further investigation as these might be key for understanding tissue colonization and the development of transmission-inhibiting therapeutics. Recent in vivo imaging has also shown that the presence of a hydrodynamic flow impacts substrate binding and swimming in trypanosomes, suggesting that the forces acting on these parasites can directly lead to changes in behavior that promote dissemination (188). Regardless of mechanisms involved, two central questions remain: (i) what are the processes deployed by trypanosomes to circumvent the immunological and physical barriers that they encounter in the skin en route to the lymphatic system? And (ii) are these interactions established by direct cell-cell contact between trypanosomes and host cells, or mediated by secreted factors (e.g., soluble virulence factors or extracellular vesicles)? An intriguing hypothesis is that upon differentiation, the proliferative long-slender trypomastigotes, in addition to immune cells recruited to the site of infection (e.g., neutrophils and macrophages), may actively remodel tissue architecture in a manner that facilitates movement from the site of infection and the establishment of a systemic infection. Understanding these mechanisms could again lead to tools that disrupt these behaviors, affecting transmission and the establishment of systemic infections in humans and animals.
In summary, lymphatic tropism leads to the accumulation of trypanosomes within the lymph nodes, triggering a myriad of adaptive immune responses before systemic dissemination (17, 94, 189–195). Ultimately, entry into the lymphatic system enables direct access to the bloodstream as fluids and cells drain through the thoracic duct and right lymphatic ducts re-joining the systemic circulation via the subclavian veins. Dissemination of the parasite and continued interaction with the host immune system would drive many of the consequent pathologies associated with infection. Recirculation through the dermal capillary beds also presents accessible trypanosomes to infect the tsetse fly vector, facilitating transmission and disease persistence. However, recent evidence has emerged of extravascular, skin-dwelling parasites that are also involved in transmission (11, 18, 83). Understanding this new anatomical niche has therefore become key to ongoing efforts to control the disease, particularly in the context of recently described latent HAT infections that may also be infective (210, 211).
Historically, the presence of African trypanosomes in the host skin was a widely recognized aspect of infection that has been largely supplanted by the repeated description of T. brucei as a bloodstream parasite (212). Re-discovering this overlooked anatomical reservoir, and the potential implications for transmission, treatment, and control, has become a focus of trypanosome research (213). This reassessment includes the description of a metabolically unique population of T. brucei parasites found in the adipose tissues of various organs in the mammalian host (18). These adipose tissue form (ATF) parasites metabolize fatty acids through β-oxidation and utilize the tricarboxylic acid cycle rather than glycolysis, making them more similar to the procyclic forms found in the tsetse fly midgut rather than mammalian bloodstream forms (214). While this study did not directly find ATF trypanosomes resident in the skin, the large deposits of subcutaneous adipose (particularly in non-murine hosts) would make it an ideal environment for the parasites. Further studies have since confirmed that T. brucei parasites are indeed present in the skin, both interacting with adipocytes (11) and throughout the extravascular space between the panniculus carnosus and the dermis (83). However, it is currently unclear whether these skin-dwelling parasites are ATF or transcriptionally distinct.
Nevertheless, video evidence demonstrates that skin-resident African trypanosomes are motile and undergo division (83, 215). There is little overt inflammation associated with trypanosome numbers in the skin (83), although there is a rise in temperature that may serve to attract tsetse flies to sites of infection (11). Within the skin, the number of parasites cycles with the characteristic peaks and troughs associated with trypanosome infections but the cycle does not appear linked to numbers in the blood, suggesting limited transfer between compartments (83). The extent of parasite exchange between the extravascular compartment and the blood is an aspect of infection that requires further study as this has implications for treatment and pathogenesis. Skin-dwelling parasites also proceed through their life cycle and develop into characteristic “stumpy” forms that are pre-adapted to survive in the tsetse fly vector (216). The presence of stumpy trypanosomes was unequivocally demonstrated via the detection of the stumpy-specific marker PAD-1 (217) in parasites in the skin (83). Importantly, these extravascular trypanosomes contribute to tsetse fly transmission, revealing that the skin is not a “dead-end” for this supposed blood parasite. Initially, a study involving dual infection with two fluorescently tagged trypanosome strains used RT-qPCR quantification to establish that tsetse flies were predominantly infected by parasites resident in the skin rather than the blood (11). Subsequent experiments comparing infectivity with tsetse flies fed on patches of mouse skin, either with or without tissue resident trypanosomes, showed that blood and skin parasites contribute to infection, with both required for maximal transmission (83). These experiments also showed that tsetse flies could be infected by skin-resident trypanosomes in apparently aparasitemic hosts.
As HAT approaches elimination in humans, there has been an increased emphasis on understanding how the disease avoided elimination in the past. In addition to animal reservoirs (218, 219), the role of asymptomatic or latent human infections has begun to receive attention (210, 211). Counter to decades of dogma, field studies have now shown that there are individuals able to tolerate T. b. gambiense as latent infections without developing symptoms (211). This latent period can be extremely protracted, with a patient recently described who was infected for at least 29 years without symptoms (220). Latent individuals rarely display detectable blood parasites and are diagnosed via serology (211). However, murine experiments demonstrating that skin-dwelling trypanosomes can infect tsetse flies raise the possibility that these latent infections can contribute to transmission and act as a reservoir of infection. Recent predictive modeling also indicates that aparasitemic but infective hosts are required for a disease focus to be stable in the absence of an animal reservoir (221). This hypothesis is supported by xenodiagnosis experiments showing tsetse flies can be infected by apparently aparasitemic hosts (222, 223) and the identification of trypanosomes in historical skin samples from HAT foci (83). To date, there has been no thorough examination of trypanosomes in the skin of domestic or peri-domestic animals and this large potential reservoir requires further study to fully understand the impact on both human and animal disease.
In summary, the notion that African trypanosomiasis is a disease of just the lymphatic and circulatory systems is being re-assessed. There is strong evidence for skin-resident parasites prior to and long after systemic parasite burden, with the potential for transmission from apparently asymptomatic hosts. This has wider impacts for understanding pathogenesis, developing new therapeutics, and identifying overlooked reservoirs. These field and laboratory data also suggest that overlooked parasites in the skin of human and animal reservoirs threaten the WHO goal of eliminating HAT transmission by 2030 (218). Fully understanding the role of the skin as a biological niche for African trypanosomes will therefore likely continue to shape the field of African trypanosomiasis research.
African trypanosomes have evolved sophisticated mechanisms to swiftly adapt to rapid changes in their microenvironment, like those encountered by metacyclic trypomastigotes delivered by the tsetse fly in the vertebrate skin when taking a blood meal. These changes are not only physical (e.g., changes in temperature and oxygen pressure) but also mechanical (e.g., transitioning from tsetse fly saliva to solid tissues such as the skin), and immunological (e.g., activation and/or recruitment of immune cells upon infection). In this scenario, it seems plausible that a combination of extrinsic factors, such as those encountered in the skin, exerts a selection pressure for trypomastigotes that are able to overcome these barriers when migrating to nearby lymphatics, leading to the establishment of systemic infections. The skin is therefore the natural point of first contact between trypanosomes and hosts, playing an active role in infection establishment and disease outcome. However, several questions remain unanswered. For example, it is unclear whether metacyclic trypomastigotes use different environmental cues in the skin as drivers for differentiation, and if so, what the chemical nature of these differentiation signals may be. It also remains to be determined whether the initial parasite population at the site of infection remains in the skin, forming a skin-resident parasite subpopulation that is distinct from the parasites found in other tissues and organs (e.g., bloodstream forms), or whether the skin is colonized multiple times as a results of parasite migration to and from the bloodstream.
From the perspective of host-pathogen interactions, it is clear that trypanosomes release a myriad of virulence factors, including soluble products and extracellular vesicles, thought to be critical to modulate the immune response against them (224–228). In this case, it would be important to understand whether the secretion of virulence factors differs between metacyclic and long slender trypomastigotes and to what extent these secreted molecules aid in the establishment of chronic infections in the skin. Similarly, it is still unclear whether the local immune response in the skin at the site of infection helps shape the population structure of parasites that systemically disseminate within the host, and the mechanisms involved in the activation of resident skin immune cells (e.g., Langerhans cells and γδ T cells). The application of novel approaches to identify cellular heterogeneity (e.g., single cell transcriptomics and spatial transcriptomics) would help to clarify the series of events that take place in the skin before parasite dissemination; from parasite differentiation and diversity, to the activation of resident and recruited immune cells.
Finally, it is now clear that the parasites residing in the skin are central to disease transmission and present in both murine models and human clinical samples. Screening of the skin in both humans and animals is likely required to fully understand the true extent of African trypanosome infections in the field, especially as latent infections are likely still infective to tsetse flies due to skin-dwelling parasites. However, our understanding of the skin as an active immune organ delineating disease outcome in human African trypanosomiasis is in its infancy, and we anticipate that future studies should address a myriad of key basic questions in this novel field of research. As such, we need to better understand the specific mechanisms involved in establishing infection in the skin, parasitic migration from the skin, and their subsequent invasion of the lymphatics that leads to systemic infection of the host. It is also important that newly developed drugs targeting the parasite can enter the skin and remain functional, otherwise transmission will be maintained alongside increasing treatment failures as skin-dwelling parasites are selected for. In addition, it is also important to understand the host-parasite interactions that are occurring in the skin and whether they can be manipulated to act as a potential therapeutic or transmission limiting tool? How are trypanosomes reaching the lymphatics from the skin and can they be inhibited from doing so? These are all questions that need to be addressed to if we are to better understand the pathogenesis of African trypanosomiasis and develop new methods of controlling and limiting disease transmission in humans and animals.
The figures use art adapted from material provided by Servier Medical Art under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
OA, JQ, AM, PG, RB, JB, NM, LM, and PC drafted and edited the manuscript. All authors approved the final version of the manuscript.
LM, NM, and OA were supported by Institute Strategic Programme Grant funding (BBS/E/D/20231762 and BBS/E/D/20002174) and a Strategic Skills Award and EASTBIO doctoral training programme studentship (BB/J01446X/1) from the Biotechnology and Biological Sciences Research Council. AM and JQ were supported by a Wellcome Trust Senior Fellowship (209511/Z/17/Z) awarded to AM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. de Raadt P. The history of sleeping sickness. In: Fourth International Course on African Trypanosomiasis. (2005)
2. Steverding D. The history of African trypanosomiasis. Parasites Vectors. (2008) 1:3. doi: 10.1186/1756-3305-1-3
3. Baral TN. Immunobiology of African trypanosomes: need of alternative interventions. J Biomed Biotechnol. (2010) 2010:389153. doi: 10.1155/2010/389153
4. Morrison LJ, Vezza L, Rowan T, Hope JC. Animal African trypanosomiasis: time to increase focus on clinically relevant parasite and host species. Trends Parasitol. (2016) 32:599–607. doi: 10.1016/j.pt.2016.04.012
5. Barrett MP, Burchmore RJ, Stich A, Lazzari JO, Frasch AC, Cazzulo JJ, et al. The trypanosomiases. Lancet. (2003) 362:1469–80. doi: 10.1016/s0140-673614694-6
6. Brun R, Blum J, Chappuis F, Burri C. Human African trypanosomiasis. Lancet. (2010) 375:148–59. doi: 10.1016/S0140-673660829-1
7. Aksoy S, Buscher P, Lehane M, Solano P, Van Den Abbeele J. Human African trypanosomiasis control: achievements and challenges. PLoS Neglect Trop Dis. (2017) 11:e0005454. doi: 10.1371/journal.pntd.0005454
8. Organization WH. Control and Surveillance of Human African Trypanosomiasis. Geneva: WHO Press (2013).
9. Van Den Abbeele J, Caljon G, De Ridder K, De Baetselier P, Coosemans M. Trypanosoma brucei modifies the tsetse salivary composition, altering the fly feeding behavior that favors parasite transmission. PLoS Pathogens. (2010) 6:e1000926. doi: 10.1371/journal.ppat.1000926
10. Aksoy S, Weiss BL, Attardo GM. Trypanosome transmission dynamics in tsetse. Curr Opin Insect Sci. (2014) 3:43–9. doi: 10.1016/j.cois.2014.07.003
11. Caljon G, Van Reet N, De Trez C, Vermeersch M, Pérez-Morga D, Van Den Abbeele J. The dermis as a delivery site of Trypanosoma brucei for tsetse flies. PLoS Pathogens. (2016) 12:e1005744. doi: 10.1371/journal.ppat.1005744
12. Matthews KR. The developmental cell biology of Trypanosoma brucei. J Cell Sci. (2005) 118:283–90. doi: 10.1242/jcs.01649
13. Farr H, Gull K. Cytokinesis in trypanosomes. Cytoskeleton. (2012) 69:931–41. doi: 10.1002/cm.21074
14. Emery DL, Barry JD, Moloo SK. The appearance of Trypanosoma (Duttonella) vivax in lymph following challenge of goats with infected Glossina morsitans morsitans. Acta Tropica. (1980) 37:375–9.
15. Theis JH, Bolton V. Trypanosoma equiperdum: movement from the dermis. Exp Parasitol. (1980) 50:317–30.
16. Barry JD, Emery DL. Parasite development and host responses during the establishment of Trypanosoma brucei infection transmitted by tsetse fly. Parasitology. (1984) 88:67–84.
17. Alfituri OA, Ajibola O, Brewer JM, Garside P, Benson RA, Peel T, et al. Effects of host-derived chemokines on the motility and viability of Trypanosoma brucei. Parasite Immunol. (2019) 41:e12609. doi: 10.1111/pim.12609
18. Trindade S, Rijo-Ferreira F, Carvalho T, Pinto-Neves D, Guegan F, Aresta-Branco F, et al. Trypanosoma brucei parasites occupy and functionally adapt to the adipose tissue in mice. Cell Host Microbe. (2016) 19:837–48. doi: 10.1016/j.chom.2016.05.002
19. Smith TK, Bringaud F, Nolan DP, Figueiredo LM. Metabolic reprogramming during the Trypanosoma brucei life cycle. F1000 Res. (2017) 6:2. doi: 10.12688/f1000research.10342.2
20. Salmon JK, Armstrong CA, Ansel JC. The skin as an immune organ. Western J Med. (1994) 160:146–52.
21. Richmond JM, Harris JE. Immunology and skin in health and disease. Cold Spring Harbor Perspect Med. (2014) 4:a015339. doi: 10.1101/cshperspect.a015339
22. Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nat Rev Immunol. (2009) 9:679–91. doi: 10.1038/nri2622
23. Heath WR, Carbone FR. The skin-resident and migratory immune system in steady state and memory: innate lymphocytes, dendritic cells and T cells. Nat Immunol. (2013) 14:978–85. doi: 10.1038/ni.2680
24. Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol. (2014) 14:289–301. doi: 10.1038/nri3646
25. Kupper TS, Fuhlbrigge RC. Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev Immunol. (2004) 4:211. doi: 10.1038/nri1310
26. Summerfield A, Meurens F, Ricklin ME. The immunology of the porcine skin and its value as a model for human skin. Mol Immunol. (2015) 66:14–21. doi: 10.1016/j.molimm.2014.10.023
27. Wong R, Geyer S, Weninger W, Guimberteau J-C, Wong JK. The dynamic anatomy and patterning of skin. Exp Dermatol. (2015) 25:92–8. doi: 10.1111/exd.12832
28. Ono S, Egawa G, Kabashima K. Regulation of blood vascular permeability in the skin. Inflam Regenerat. (2017) 37:11. doi: 10.1186/s41232-017-0042-9
29. Rotureau B, Van Den Abbeele J. Through the dark continent: African trypanosome development in the tsetse fly. Front Cell Infect Microbiol. (2013) 3:53. doi: 10.3389/fcimb.2013.00053
30. Van Den Abbeele J, Caljon G, Dierick JF, Moens L, De Ridder K, Coosemans M. The Glossina morsitans tsetse fly saliva: general characteristics and identification of novel salivary proteins. Insect Biochem Mol Biol. (2007) 37:1075–85. doi: 10.1016/j.ibmb.2007.06.006
31. Sharma R, Gluenz E, Peacock L, Gibson W, Gull K, Carrington M. The heart of darkness: growth and form of Trypanosoma brucei in the tsetse fly. Trends Parasitol. (2009) 25:517–24. doi: 10.1016/j.pt.2009.08.001
32. Caljon G, Van Den Abbeele J, Stijlemans B, Coosemans M, De Baetselier P, Magez S. Tsetse fly saliva accelerates the onset of Trypanosoma brucei infection in a mouse model associated with a reduced host inflammatory response. Infect Immunity. (2006) 74:6324–30. doi: 10.1128/iai.01046-06
33. Somda MB, Bengaly Z, Dama E, Poinsignon A, Dayo G-K, Sidibe I, et al. First insights into the cattle serological response to tsetse salivary antigens: a promising direct biomarker of exposure to tsetse bites. Vet Parasitol. (2013) 197:332–40. doi: 10.1016/j.vetpar.2013.05.018
34. Cochran R, Rosen T. African Trypanosomiasis in the United States. Archiv Dermatol. (1983) 119:670–4. doi: 10.1001/archderm.1983.01650320044014
35. Kennedy PGE. Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol. (2013) 12:186–94. doi: 10.1016/S1474-4422(12)70296-X
36. Akol GW, Murray M. Early events following challenge of cattle with tsetse infected with Trypanosoma congolense: development of the local skin reaction. Vet. Rec. (1982) 110:295–302.
37. Parker KR, Mant MJ. Effects of tsetse (Glossina morsitans morsitans Westw.) (Diptera: Glossinidae) salivary gland homogenate on coagulation and fibrinolysis. Thrombosis Haemostasis. (1979) 42:743–51.
38. Cappello M, Bergum PW, Vlasuk GP, Furmidge BA, Pritchard DI, Aksoy S. Isolation and characterization of the tsetse thrombin inhibitor: a potent antithrombotic peptide from the saliva of Glossina morsitans morsitans. Am J Trop Med Hygiene. (1996) 54:475–80.
39. Cappello M, Li S, Chen X, Li CB, Harrison L, Narashimhan S, et al. Tsetse thrombin inhibitor: bloodmeal-induced expression of an anticoagulant in salivary glands and gut tissue of Glossina morsitans morsitans. PNAS. (1998) 95:14290–5.
40. Caljon G, De Ridder K, De Baetselier P, Coosemans M, Van Den Abbeele J. Identification of a tsetse fly salivary protein with dual inhibitory action on human platelet aggregation. PLoS ONE. (2010) 5:e9671. doi: 10.1371/journal.pone.0009671
41. Caljon G, Van Den Abbeele J, Sternberg JM, Coosemans M, De Baetselier P, Magez S. Tsetse fly saliva biases the immune response to Th2 and induces anti-vector antibodies that are a useful tool for exposure assessment. Int J Parasitol. (2006) 36:1025–35. doi: 10.1016/j.ijpara.2006.05.002
42. Awuoche EO. Tsetse fly saliva: could it be useful in fly infection when feeding in chronically aparasitemic mammalian hosts. Open Vet J. (2012) 2:95–105.
43. Wei G, Bull H, Zhou X, Tabel H. Intradermal infections of mice by low numbers of African trypanosomes are controlled by innate resistance but enhance susceptibility to reinfection. J Infect Dis. (2011) 203:418–29. doi: 10.1093/infdis/jiq051
44. Ndungu K, Thungu D, Wamwiri F, Mireji P, Ngae G, Gitonga P, et al. Route of inoculation influences Trypanosoma congolense and Trypanosoma brucei brucei virulence in Swiss white mice. PLoS ONE. (2019) 14:e0218441. doi: 10.1371/journal.pone.0218441
45. D'Archivio S, Cosson A, Medina M, Lang T, Minoprio P, Goyard S. Non-invasive in vivo study of the Trypanosoma vivax infectious process consolidates the brain commitment in late infections. PLoS Negl Trop Dis. (2013) 7:e1976. doi: 10.1371/journal.pntd.0001976
46. Sones KR, Holmes PH. The influence of the size of the initial inoculum on the efficacy of isometamidium (samorin) on a stock of Trypanosoma congolense. Acta Trop. (1992) 51:213–6. doi: 10.1016/0001-706x(92)90039-z
47. Onyilagha C, Okwor I, Kuriakose S, Singh R, Uzonna J. Low-dose intradermal infection with trypanosoma congolense leads to expansion of regulatory T cells and enhanced susceptibility to reinfection. Infect Immun. (2014) 82:1074–83. doi: 10.1128/IAI.01028-13
48. Mamman M, Gettinby G, Murphy NB, Kemei S, Peregrine AS. Frequency of diminazene-resistant trypanosomes in populations of Trypanosoma congolense arising in infected animals following treatment with diminazene aceturate. Antimicrob Agents Chemother. (1995) 39:1107–13. doi: 10.1128/aac.39.5.1107
49. Wellde BT, Duxbury RE, Sadun EH, Langbehn HR, Lotzsch R, Deindl G, et al. Experimental infections with African trypanosomes. IV immunization of cattle with gamma-irradiated Trypanosoma rhodesiense. Exp Parasitol. (1973) 34:62–8. doi: 10.1016/0014-489490063-5
50. Dagnachew S, Terefe G, Abebe G, Sirak A, Bollo E, Barry D, et al. Comparative clinico-pathological observations in young Zebu (Bos indicus) cattle experimentally infected with Trypanosoma vivax isolates from tsetse infested and non-tsetse areas of Northwest Ethiopia. BMC Vet Res. (2015) 11:307. doi: 10.1186/s12917-015-0625-0
51. Esievo KA, Saror DI. Leukocyte response in experimental Trypanosoma vivax infection in cattle. J Comp Pathol. (1983) 93:165–9. doi: 10.1016/0021-997590002-6
52. Kobayashi A, Tizard IR, Woo PT. Studies on the anemia in experimental African trypanosomiasis. II the pathogenesis of the anemia in calves infected with Trypanosoma congolense. Am J Trop Med Hyg. (1976) 25:401–6. doi: 10.4269/ajtmh.1976.25.401
53. Ngeranwa JJ, Gathumbi PK, Mutiga ER, Agumbah GJ. Pathogenesis of Trypanosoma (brucei) evansi in small east African goats. Res Vet Sci. (1993) 54:283–9. doi: 10.1016/0034-528890124-x
54. Osaer S, Goossens B, Clifford DJ, Kora S, Kassama M. A comparison of the susceptibility of Djallonke sheep and West African Dwarf goats to experimental infection with two different strains of Trypanosoma congolense. Vet Parasitol. (1994) 51:191–204. doi: 10.1016/0304-401790156-2
55. Whitelaw DD, Kaaya GP, Moulton JE, Moloo SK, Murray M. Susceptibility of different breeds of goats in Kenya to experimental infection with Trypanosoma congolense. Trop Anim Health Prod. (1985) 17:155–65. doi: 10.1007/BF02355876
56. Katunguka-Rwakishaya E, Murray M, Holmes PH. Susceptibility of three breeds of Ugandan goats to experimental infection with Trypanosoma congolense. Trop Anim Health Prod. (1997) 29:7–14. doi: 10.1007/BF02632337
57. Griffin L, Allonby E. Trypanotolerance in breeds of sheep and goats with an experimental infection of Trypanosoma congolense. Vet Parasitol. (1979) 5:97–105.
58. Omotainse SO, Anosa VO. Comparative histopathology of the lymph nodes, spleen, liver and kidney in experimental ovine trypanosomosis. Onderstepoort J Vet Res. (2009) 76:377–83. doi: 10.4102/ojvr.v76i4.21
59. Ikede BO, Losos GJ. Pathogenesin of Trypanosoma brucei infection is sheep. I clinical signs. J Comp Pathol. (1975) 85:23–31. doi: 10.1016/0021-997590080-8
60. Maikaje DB, Sannusi A, Kyewalabye EK, Saror DI. The course of experimental Trypanosoma vivax infection in Uda sheep. Vet Parasitol. (1991) 38:267–74. doi: 10.1016/0304-401790139-m
61. Oyewusi J, Saba A, Oridupa O. The course of trypanosomiasis in laboratory rabbits following experimental infection, treatment and re-infection: a haematological study. Euro J Sci Res. (2010) 42:411–9.
62. Nagle RB, Dong S, Guillot JM, McDaniel KM, Lindsley HB. Pathology of experimental African trypanosomiasis in rabbits infected with Trypanosoma rhodesiense. Am J Trop Med Hyg. (1980) 29:1187–95. doi: 10.4269/ajtmh.1980.29.1187
63. Ouwe-Missi-Oukem-Boyer O, Mezui-Me-Ndong J, Boda C, Lamine I, Labrousse F, Bisser S, et al. The vervet monkey (Chlorocebus aethiops) as an experimental model for Trypanosoma brucei gambiense human African trypanosomiasis: a clinical, biological and pathological study. Trans Royal Soc Trop Med Hygiene. (2006) 100:427–36. doi: 10.1016/j.trstmh.2005.07.023
64. Waema M, Maina N, Karanja S, Gachie B, Ngotho M, Kagira J. Development of a safer laboratory vervet monkey model for the study of human African trypanosomiasis. Afr J Lab Med. (2014) 3:100. doi: 10.4102/ajlm.v3i1.100
65. Ngotho M, Maina N, Kagira J, Royo F, Farah IO, Hau J. IL-10 is up regulated in early and transitional stages in vervet monkeys experimentally infected with Trypanosoma brucei rhodesiense. Parasitol Int. (2006) 55:243–8. doi: 10.1016/j.parint.2006.06.004
66. Dwinger RH, Lamb G, Murray M, Hirumi H. Dose and stage dependency for the development of local skin reactions caused by Trypanosoma congolense in goats. Acta Tropica. (1987) 44:303–14.
67. Burza S, Croft SL, Boelaert M. Leishmaniasis. Lancet. (2018) 392:951–70. doi: 10.1016/S0140-673631204-2
68. Mahmoudzadeh-Niknam H, Khalili G, Abrishami F, Najafy A, Khaze V. The route of Leishmania tropica infection determines disease outcome and protection against Leishmania major in BALB/c mice. Korean J Parasitol. (2013) 51:69–74. doi: 10.3347/kjp.2013.51.1.69
69. Loeuillet C, Banuls AL, Hide M. Study of Leishmania pathogenesis in mice: experimental considerations. Parasite Vectors. (2016) 9:144. doi: 10.1186/s13071-016-1413-9
70. Kaur S, Kaur T, Garg N, Mukherjee S, Raina P, Athokpam V. Effect of dose and route of inoculation on the generation of CD4+ Th1/Th2 type of immune response in murine visceral leishmaniasis. Parasitol Res. (2008) 103:1413–9. doi: 10.1007/s00436-008-1150-x
71. Veer M, Kemp J, Meeusen E. The innate defence against nematode parasites. Parasite Immunol. (2007) 29:1–9. doi: 10.1111/j.1365-3024.2006.00910.x
72. Namangala B. Contribution of innate immune responses towards resistance to African trypanosome infections. Scand J Immunol. (2012) 75:5–15. doi: 10.1111/j.1365-3083.2011.02619.x
73. Abbas A, Lichtman AH, Pillai S, Baker DL, Baker A. Cellular and Molecular Immunology. 9th Ed. Amsterdam: Elsevier (2018). p. 57–97.
74. Stijlemans B, De Baetselier P, Magez S, Van Ginderachter JA, De Trez C. African trypanosomiasis-associated anemia: the contribution of the interplay between parasites and the mononuclear phagocyte system. Front Immunol. (2018) 9:218. doi: 10.3389/fimmu.2018.00218
75. McDonald DR, Levy O. Innate immunity−3. In: RR Rich, TA Fleisher, WT Shearer, HW Schroeder, AJ Frew, CM Weyand, editors. Clinical Immunology. 5th Ed. London: Content Repository Only! (2019). p. 39–53.e31.
76. Mabbott NA, Sutherland IA, Sternberg JM. Trypanosoma brucei is protected from the cytostatic effects of nitric oxide under in vivo conditions. Parasitol Res. (1994) 80:687–90.
77. Sternberg JM, Mabbott N, Sutherland I, Liew FY. Inhibition of nitric oxide synthesis leads to reduced parasitaemia in murine Trypanosoma brucei infection. Infect Immunity. (1994) 62:2135–7.
78. Mabbott NA, Sutherland IA, Sternberg JM. Suppressor macrophages in Trypanosoma brucei infection: nitric oxide is related to both suppressive activity and lifespan in vivo. Parasite Immunol. (1995) 17:143–50.
79. Mansfield JM, Paulnock DM. Regulation of innate and acquired immunity in African trypanosomiasis. Parasite Immunol. (2005) 27:361–71. doi: 10.1111/j.1365-3024.2005.00791.x
80. Namangala B, De Baetselier P, Beschin A. Both type-I and type-II responses contribute to murine trypanotolerance. J Vet Med Sci. (2009) 71:313–8. doi: 10.1292/jvms.71.313
81. Ponte-Sucre A. An overview of Trypanosoma brucei infections: an intense host–parasite interaction. Front Microbiol. (2016) 7:2126. doi: 10.3389/fmicb.2016.02126
82. Bakari SM, Ofori JA, Kusi KA, Aning GK, Awandare GA, Carrington M, et al. Serum biochemical parameters and cytokine profiles associated with natural African trypanosome infections in cattle. Parasites Vectors. (2017) 10:312. doi: 10.1186/s13071-017-2255-9
83. Capewell P, Cren-Travaille C, Marchesi F, Johnston P, Clucas C, Benson RA, et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. (2016) 5:e17716. doi: 10.7554/eLife.17716
84. Stijlemans B, Caljon G, Van Den Abbeele J, Van Ginderachter JA, Magez S, De Trez C. Immune evasion strategies of Trypanosoma brucei within the mammalian host: progression to pathogenicity. Front Immunol. (2016) 7:233. doi: 10.3389/fimmu.2016.00233
85. Caljon G, Mabille D, Stijlemans B, De Trez C, Mazzone M, Tacchini-Cottier F, et al. Neutrophils enhance early Trypanosoma brucei infection onset. Sci Rep. (2018) 8:11203. doi: 10.1038/s41598-018-29527-y
86. Guimarães-Costa AB, Nascimento MTC, Froment GS, Soares RPP, Morgado FN, Conceição-Silva F, et al. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc Natl Acad Sci USA. (2009) 106:6748. doi: 10.1073/pnas.0900226106
87. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. (2010) 31:318–24. doi: 10.1016/j.it.2010.05.006
88. Perobelli SM, Galvani RG, Goncalves-Silva T, Xavier CR, Nobrega A, Bonomo A. Plasticity of neutrophils reveals modulatory capacity. Brazil J Med Biol Res. (2015) 48:665–75. doi: 10.1590/1414-431x20154524
89. Silva LM, Munoz-Caro T, Burgos RA, Hidalgo MA, Taubert A, Hermosilla C. Far beyond phagocytosis: phagocyte-derived extracellular traps act efficiently against protozoan parasites in vitro and in vivo. Mediators Inflamm. (2016) 2016:5898074. doi: 10.1155/2016/5898074
90. Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: facts beyond expression. Front Immunol. (2014) 5:508. doi: 10.3389/fimmu.2014.00508
91. Harrington JM. Antimicrobial peptide killing of African trypanosomes. Parasite Immunol. (2011) 33:461–9. doi: 10.1111/j.1365-3024.2011.01294.x
92. Onyilagha C, Kuriakose S, Ikeogu N, Kung SKP, Uzonna JE. NK cells are critical for optimal immunity to experimental Trypanosoma congolense infection. J Immunol. (2019) 203:964–71. doi: 10.4049/jimmunol.1900103
93. Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. (2014) 5:1–12. doi: 10.3389/fimmu.2014.00491
94. Kaye P, Scott P. Leishmaniasis: complexity at the host-pathogen interface. Nat Rev Microbiol. (2011) 9:604–15. doi: 10.1038/nrmicro2608
95. Liu D, Uzonna J. The early interaction of Leishmania with macrophages and dendritic cells and its influence on the host immune response. Front Cell Infect Microbiol. (2012) 2:83. doi: 10.3389/fcimb.2012.00083
96. Peters N, Egen J, Secundino N, Debrabant A, Kimblin N, Kamhawi S, et al. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science. (2008) 321:970–4. doi: 10.1126/science.1159194
97. de Menezes JP, Saraiva EM, da Rocha-Azevedo B. The site of the bite: Leishmania interaction with macrophages, neutrophils and the extracellular matrix in the dermis. Parasites Vectors. (2016) 9:264. doi: 10.1186/s13071-016-1540-3
98. Gueirard P, Laplante A, Rondeau C, Milon G, Desjardins M. Trafficking of Leishmania donovani promastigotes in non-lytic compartments in neutrophils enables the subsequent transfer of parasites to macrophages. Cell Microbiol. (2008) 10:100–11. doi: 10.1111/j.1462-5822.2007.01018.x
99. Carlsen ED, Liang Y, Shelite TR, Walker DH, Melby PC, Soong L. Permissive and protective roles for neutrophils in leishmaniasis. Clin Exp Immunol. (2015) 182:109–18. doi: 10.1111/cei.12674
100. Deleeuw V, Phạm HTT, De Poorter I, Janssens I, De Trez C, Radwanska M, et al. Trypanosoma brucei brucei causes a rapid and persistent influx of neutrophils in the spleen of infected mice. Parasite Immunol. (2019) 41:e12664. doi: 10.1111/pim.12664
101. Grosskinsky CM, Askonas BA. Macrophages as primary target cells and mediators of immune dysfunction in African trypanosomiasis. Infect Immunol. (1981) 33:149–55.
102. Fierer J, Askonas BA. Trypanosoma brucei infection stimulates receptor-mediated phagocytosis by murine peritoneal macrophages. Infect Immunity. (1982) 37:1282–4.
103. Grosskinsky CM, Ezekowitz RA, Berton G, Gordon S, Askonas BA. Macrophage activation in murine African trypanosomiasis. Infect Immunity. (1983) 39:1080–6.
104. Baetselier PD, Namangala B, Noel W, Brys L, Pays E, Beschin A. Alternative versus classical macrophage activation during experimental African trypanosomosis. Int J Parasitol. (2001) 31:575–87. doi: 10.1016/s0020-7519(01)00170-9
105. Paulnock DM, Coller SP. Analysis of macrophage activation in African trypanosomiasis. J Leukocyte Biol. (2001) 69:685–90. doi: 10.1189/jlb.69.5.685
106. Vincendeau P, Bouteille B. Immunology and immunopathology of African trypanosomiasis. Anais da Academia Brasileira de Ciencias. (2006) 78:645–65. doi: 10.1590/s0001-37652006000400004
107. Paulnock DM, Freeman BE, Mansfield JM. Modulation of innate immunity by African trypanosomes. Parasitology. (2010) 137:2051–63. doi: 10.1017/s0031182010001460
108. Stijlemans B, Vankrunkelsven A, Caljon G, Bockstal V, Guilliams M, Bosschaerts T, et al. The central role of macrophages in trypanosomiasis-associated anemia: rationale for therapeutical approaches. Endocrine Metabolic Immune Disord. (2010) 10:71–82. doi: 10.2174/187153010790827966
109. de Sousa KP, Atouguia JM, Silva MS. Induced cytokine network during experimental African trypanosomiasis. Int J Interferon Cytokine Mediator Res. (2011) 2011:71–8. doi: 10.2147/IJICMR.S25914
110. Kuriakose SM, Singh R, Uzonna J. Host intracellular signaling events and pro-inflammatory cytokine production in African trypanosomiasis. Front Immunol. (2016) 7:181. doi: 10.3389/fimmu.2016.00181
111. Mensa-Wilmot K, Englund PT. Glycosyl phosphatidylinositol-specific phospholipase C of Trypanosoma brucei: expression in Escherichia coli. Mol Biochem Parasitol. (1992) 56:311–21. doi: 10.1016/0166-685190180-r
112. Magez S, Stijlemans B, Baral T, De Baetselier P. VSG-GPI anchors of African trypanosomes: their role in macrophage activation and induction of infection-associated immunopathology. Microb Infect. (2002) 4:999–1006. doi: 10.1016/s1286-4579(02)01617-9
113. Drennan MB, Stijlemans B, Van den Abbeele J, Quesniaux VJ, Barkhuizen M, Brombacher F, et al. The induction of a type 1 immune response following a Trypanosoma brucei infection is MyD88 dependent. J Immunol. (2005) 175:2501–9. doi: 10.4049/jimmunol.175.4.2501
114. Leppert BJ, Mansfield JM, Paulnock DM. The soluble variant surface glycoprotein of African trypanosomes is recognized by a macrophage scavenger receptor and induces I kappa B alpha degradation independently of TRAF6-mediated TLR signaling. J Immunol. (2007) 179:548–56. doi: 10.4049/jimmunol.179.1.548
115. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
116. Kaboré JW, Camara O, Ilboudo H, Capewell P, Clucas C, Cooper A, et al. Macrophage migrating inhibitory factor expression is associated with Trypanosoma brucei gambiense infection and is controlled by trans-acting expression quantitative trait loci in the Guinean population. Infect Genet Evol. (2019) 71:108–15. doi: 10.1016/j.meegid.2019.03.021
117. Dempsey WL, Mansfield JM. Lymphocyte function in experimental African trypanosomiasis. V Role of antibody and the mononuclear phagocyte system in variant-specific immunity. J Immunol. (1983) 130:405–11.
118. Shi M, Wei G, Pan W, Tabel H. Trypanosoma congolense infections: antibody-mediated phagocytosis by Kupffer cells. J Leukocyte Biol. (2004) 76:399–405. doi: 10.1189/jlb.1003500
119. Guirnalda P, Murphy NB, Nolan D, Black SJ. Anti-Trypanosoma brucei activity in Cape buffalo serum during the cryptic phase of parasitemia is mediated by antibodies. Int J Parasitol. (2007) 37:1391–9. doi: 10.1016/j.ijpara.2007.04.019
120. Macaskill JA, Holmes PH, Whitelaw DD, McConnell I, Jennings FW, Urquhart GM. Immunological clearance of 75Se-labelled Trypanosoma brucei in mice. II Mechanisms in immune animals. Immunology. (1980) 40:629–35.
121. Satriano J. Arginine pathways and the inflammatory response: interregulation of nitric oxide and polyamines: review article. Amino Acids. (2004) 26:321–9. doi: 10.1007/s00726-004-0078-4
122. Wijnands KA, Castermans TM, Hommen MP, Meesters DM, Poeze M. Arginine and citrulline and the immune response in sepsis. Nutrients. (2015) 7:1426–63. doi: 10.3390/nu7031426
123. Vincendeau P, Daulouede S, Veyret B, Darde ML, Bouteille B, Lemesre JL. Nitric oxide-mediated cytostatic activity on Trypanosoma brucei gambiense and Trypanosoma brucei brucei. Exp Parasitol. (1992) 75:353–60.
124. Magez S, Lucas R, Darji A, Songa EB, Hamers R, De Baetselier P. Murine tumour necrosis factor plays a protective role during the initial phase of the experimental infection with Trypanosoma brucei brucei. Parasite Immunol. (1993) 15:635–41.
125. Magez S, Geuskens M, Beschin A, del Favero H, Verschueren H, Lucas R, et al. Specific uptake of tumor necrosis factor-alpha is involved in growth control of Trypanosoma brucei. J Cell Biol. (1997) 137:715–27.
126. Hertz CJ, Filutowicz H, Mansfield JM. Resistance to the African trypanosomes is IFN-gamma dependent. J Immunol. (1998) 161:6775–83.
127. Sternberg JM. Human African trypanosomiasis: clinical presentation and immune response. Parasite Immunol. (2004) 26:469–76. doi: 10.1111/j.0141-9838.2004.00731.x
128. Magez S, Radwanska M, Drennan M, Fick L, Baral TN, Brombacher F, et al. Interferon-γ and nitric oxide in combination with antibodies are key protective host immune factors during Trypanosoma congolense Tc13 infections. J Infect Dis. (2006) 193:1575–83. doi: 10.1086/503808
129. Barkhuizen M, Magez S, Atkinson RA, Brombacher F. Interleukin-12p70-dependent interferon- gamma production is crucial for resistance in African trypanosomiasis. J Infect Dis. (2007) 196:1253–60. doi: 10.1086/521681
130. Magez S, Radwanska M, Drennan M, Fick L, Baral TN, Allie N, et al. Tumor necrosis factor (TNF) receptor-1 (TNFp55) signal transduction and macrophage-derived soluble TNF are crucial for nitric oxide-mediated Trypanosoma congolense parasite killing. J Infect Dis. (2007) 196:954–62. doi: 10.1086/520815
131. Duleu S, Vincendeau P, Courtois P, Semballa S, Lagroye I, Daulouède S, et al. Mouse strain susceptibility to trypanosome infection: an arginase-dependent effect. J Immunol. (2004) 172:6298. doi: 10.4049/jimmunol.172.10.6298
132. Lu W, Wei G, Pan W, Tabel H. Trypanosoma congolense infections: induced nitric oxide inhibits parasite growth in vivo. J Parasitol Res. (2011) 2011:316067. doi: 10.1155/2011/316067
133. Vincendeau P, Daulouede S. Macrophage cytostatic effect on Trypanosoma musculi involves an L-arginine-dependent mechanism. J Immunol. (1991) 146:4338–43.
134. Hamadien M, Lycke N, Bakhiet M. Induction of the trypanosome lymphocyte-triggering factor (TLTF) and neutralizing antibodies to the TLTF in experimental african trypanosomiasis. Immunology. (1999) 96:606–11.
135. Nishimura K, Hamashita K, Okamoto Y, Kawahara F, Ihara H, Kozaki S, et al. Differential effects of interferon-gamma on production of trypanosome-derived lymphocyte-triggering factor by Trypanosoma brucei gambiense and Trypanosoma brucei brucei. J Parasitol. (2004) 90:740–5. doi: 10.1645/ge-211r1
136. Namangala B, Brys L, Magez S, De Baetselier P, Beschin A. Trypanosoma brucei brucei infection impairs MHC class II antigen presentation capacity of macrophages. Parasite Immunol. (2000) 22:361–70. doi: 10.1046/j.1365-3024.2000.00314.x
137. Noël W, Hassanzadeh G, Raes G, Namangala B, Daems I, Brys L, et al. Infection stage-dependent modulation of macrophage activation in Trypanosoma congolense-resistant and -susceptible mice. Infect Immunity. (2002) 70:6180–7. doi: 10.1128/IAI.70.11.6180-6187.2002
138. Raes G, Beschin A, Ghassabeh GH, De Baetselier P. Alternatively activated macrophages in protozoan infections. Curr Opin Immunol. (2007) 19:454–9. doi: 10.1016/j.coi.2007.05.007
139. Hume DA, MacDonald KPA. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood. (2012) 119:1810. doi: 10.1182/blood-2011-09-379214
140. Gause WC, Wynn TA, Allen JE. Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat Rev Immunol. (2013) 13:607–14. doi: 10.1038/nri3476
141. Hamilton TA, Zhao C, Pavicic PG, Datta S. Myeloid colony-stimulating factors as regulators of macrophage polarization. Front Immunol. (2014) 5:554. doi: 10.3389/fimmu.2014.00554
142. Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity. (2015) 43:29–40. doi: 10.1016/j.immuni.2015.07.007
143. Boulakirba S, Pfeifer A, Mhaidly R, Obba S, Goulard M, Schmitt T, et al. IL-34 and CSF-1 display an equivalent macrophage differentiation ability but a different polarization potential. Sci Rep. (2018) 8:256. doi: 10.1038/s41598-017-18433-4
144. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
145. Hume DA. The many alternative faces of macrophage activation. Front Immunol. (2015) 6:370. doi: 10.3389/fimmu.2015.00370
146. Roszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflam. (2015) 2015:816460. doi: 10.1155/2015/816460
147. Rath M, Müller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. (2014) 5:532. doi: 10.3389/fimmu.2014.00532
148. Campbell NK, Williams DG, Fitzgerald HK, Barry PJ, Cunningham CC, Nolan DP, et al. Trypanosoma brucei secreted aromatic ketoacids activate the Nrf2/HO-1 pathway and suppress pro-inflammatory responses in primary murine glia and macrophages. Front Immunol. (2019) 10:2137. doi: 10.3389/fimmu.2019.02137
149. Morrison LJ, McLellan S, Sweeney L, Chan CN, MacLeod A, Tait A, et al. Role for parasite genetic diversity in differential host responses to Trypanosoma brucei infection. Infect Immun. (2010) 78:1096–108. doi: 10.1128/IAI.00943-09
150. De Muylder G, Daulouède S, Lecordier L, Uzureau P, Morias Y, Van Den Abbeele J, et al. A Trypanosoma brucei Kinesin heavy chain promotes parasite growth by triggering host arginase activity. PLoS Pathogens. (2013) 9:e1003731. doi: 10.1371/journal.ppat.1003731
151. Tabel H, Wei G, Bull HJ. Immunosuppression: Cause for failures of vaccines against African trypanosomiases. PLoS Neglected Tropical Diseases. (2013) 7:e2090. doi: 10.1371/journal.pntd.0002090
152. Klechevsky E. Functional diversity of human dendritic cells. In: SP Schoenberger, PD Katsikis, B Pulendran, editors. Crossroads Between Innate and Adaptive Immunity V. SP Schoenberger, PD Katsikis, B Pulendran: Springer International Publishing (2015). p. 43–54.
153. Waisman A, Lukas D, Clausen BE, Yogev N. Dendritic cells as gatekeepers of tolerance. Semin Immunopathol. (2017) 39:153–63. doi: 10.1007/s00281-016-0583-z
154. Dagenais TR, Freeman BE, Demick KP, Paulnock DM, Mansfield JM. Processing and presentation of variant surface glycoprotein molecules to T cells in African trypanosomiasis. J Immunol. (2009) 183:3344. doi: 10.4049/jimmunol.0802005
155. Geiger A, Bossard G, Sereno D, Pissarra J, Lemesre J-L, Vincendeau P. Escaping deleterious immune response in their hosts: lessons from trypanosomatids. Front. Immunol. (2016) 7:212. doi: 10.3389/fimmu.2016.00212
156. Auffermann-Gretzinger S, Keeffe EB, Levy S. Impaired dendritic cell maturation in patients with chronic, but not resolved, hepatitis C virus infection. Blood. (2001) 97:3171–6. doi: 10.1182/blood.v97.10.3171
157. Sukriti S, Pati NT, Bose S, Hissar SS, Sarin SK. Impaired antigen processing and presentation machinery is associated with immunotolerant state in chronic hepatitis B virus infection. J Clin Immunol. (2010) 30:419–25. doi: 10.1007/s10875-010-9379-4
158. Malissen B, Tamoutounour S, Henri S. The origins and functions of dendritic cells and macrophages in the skin. Nat Rev Immunol. (2014) 14:417–28. doi: 10.1038/nri3683
159. Haniffa M, Gunawan M, Jardine L. Human skin dendritic cells in health and disease. J Dermatol Sci. (2015) 77:85–92. doi: 10.1016/j.jdermsci.2014.08.012
160. Nirschl CJ, Anandasabapathy N. Duality at the gate: skin dendritic cells as mediators of vaccine immunity and tolerance. Hum Vaccines Immunother. (2016) 12:104–16. doi: 10.1080/21645515.2015.1066050
161. Kaplan DH. Ontogeny and function of murine epidermal Langerhans cells. Nat Immunol. (2017) 18:1068–75. doi: 10.1038/ni.3815
162. Deckers J, Hammad H, Hoste E. Langerhans cells: sensing the environment in health and disease. Front Immunol. (2018) 9:93. doi: 10.3389/fimmu.2018.00093
163. Kautz-Neu K, Noordegraaf M, Dinges S, Bennett CL, John D, Clausen BE, et al. Langerhans cells are negative regulators of the anti-Leishmania response. J Exp Med. (2011) 208:885–91. doi: 10.1084/jem.20102318
164. Ng LG, Hsu A, Mandell MA, Roediger B, Hoeller C, Mrass P, et al. Migratory dermal dendritic cells act as rapid sensors of protozoan parasites. PLoS Pathogens. (2008) 4:e1000222. doi: 10.1371/journal.ppat.1000222
165. von Stebut E, Belkaid Y, Jakob T, Sacks DL, Udey MC. Uptake of Leishmania major amastigotes results in activation and interleukin 12 release from murine skin-derived dendritic cells: implications for the initiation of anti-Leishmania immunity. J Exp Med. (1998) 188:1547–52. doi: 10.1084/jem.188.8.1547
166. Strid J, Tigelaar RE, Hayday AC. Skin immune surveillance by T cells–a new order? Semin Immunol. (2009) 21:110–20. doi: 10.1016/j.smim.2009.03.002
167. Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat Immunol. (2013) 14:564–73. doi: 10.1038/ni.2584
168. Paul S, Shilpi Lal G. Role of gamma-delta (gammadelta) T cells in autoimmunity. J Leukocyte Biol. (2015) 97:259–71. doi: 10.1189/jlb.3RU0914-443R
169. Mueller SN, Zaid A, Carbone FR. Tissue-resident T cells: dynamic players in skin immunity. Front Immunol. (2014) 5:332. doi: 10.3389/fimmu.2014.00332
170. O'Brien RL, Born WK. Dermal gammadelta T cells–what have we learned? Cell Immunol. (2015) 296:62–9. doi: 10.1016/j.cellimm.2015.01.011
171. Lawand M, Déchanet-Merville J, Dieu-Nosjean M-C. Key features of gamma-delta T-cell subsets in human diseases and their immunotherapeutic implications. Front Immunol. (2017) 8:761. doi: 10.3389/fimmu.2017.00761
172. Cruz MS, Diamond A, Russell A, Jameson JM. Human alphabeta and gammadelta T cells in skin immunity and disease. Front Immunol. (2018) 9:1304. doi: 10.3389/fimmu.2018.01304
173. Ho AW, Kupper TS. T cells and the skin: from protective immunity to inflammatory skin disorders. Nat Rev Immunol. (2019) 19:490–502. doi: 10.1038/s41577-019-0162-3
174. Sabat R, Wolk K, Loyal L, Docke WD, Ghoreschi K. T cell pathology in skin inflammation. Semin Immunopathol. (2019) 41:359–77. doi: 10.1007/s00281-019-00742-7
175. Flynn JN, Sileghem M. Involvement of gamma delta T cells in immunity to trypanosomiasis. Immunology. (1994) 83:86–92.
176. Guzman E, Hope J, Taylor G, Smith AL, Cubillos-Zapata C, Charleston B. Bovine γδ T cells are a major regulatory T cell subset. J Immunol. (2014) 193:208–22. doi: 10.4049/jimmunol.1303398
177. Sileghem M, Darji A, Hamers R, Van de Winkel M, De Baetselier P. Dual role of macrophages in the suppression of interleukin 2 production and interleukin 2 receptor expression in trypanosome-infected mice. Eur J Immunol. (1989) 19:829–35. doi: 10.1002/eji.1830190508
178. Sileghem M, Flynn JN. Suppression of interleukin 2 secretion and interleukin 2 receptor expression during tsetse-transmitted trypanosomiasis in cattle. Eur J Immunol. (1992) 22:767–73. doi: 10.1002/eji.1830220321
179. Darji A, Beschin A, Sileghem M, Heremans H, Brys L, De Baetselier P. In vitro simulation of immunosuppression caused by Trypanosoma brucei: active involvement of gamma interferon and tumor necrosis factor in the pathway of suppression. Infect Immunity. (1996) 64:1937–43.
180. Uzonna JE, Kaushik RS, Gordon JR, Tabel H. Immunoregulation in experimental murine Trypanosoma congolense infection: anti-IL-10 antibodies reverse trypanosome-mediated suppression of lymphocyte proliferation in vitro and moderately prolong the lifespan of genetically susceptible BALB/c mice. Parasite Immunol. (1998) 20:293–302.
181. Stijlemans B, Radwanska M, De Trez C, Magez S. African Trypanosomes undermine humoral responses and vaccine development: link with inflammatory responses? Front Immunol. (2017) 8:582. doi: 10.3389/fimmu.2017.00582
182. Guilbride DL, Guilbride PDL, Gawlinski P. Malaria's deadly secret: a skin stage. Trends Parasitol. (2012) 28:142–50. doi: 10.1016/j.pt.2012.01.002
183. Sinnis P, Zavala F. The skin: where malaria infection and the host immune response begin. Semin Immunopathol. (2012) 34:787–92. doi: 10.1007/s00281-012-0345-5
184. Menard R, Tavares J, Cockburn I, Markus M, Zavala F, Amino R. Looking under the skin: the first steps in malarial infection and immunity. Nat Rev Microbiol. (2013) 11:701–12. doi: 10.1038/nrmicro3111
185. Guilbride DL, Gawlinski P, Guilbride PD. Why functional pre-erythrocytic and bloodstage malaria vaccines fail: a meta-analysis of fully protective immunizations and novel immunological model. PLoS ONE. (2010) 5:e10685. doi: 10.1371/journal.pone.0010685
186. Overstreet MG, Freyberger H, Cockburn IA, Chen Y-C, Tse S-W, Zavala F. CpG-enhanced CD8+ T-cell responses to peptide immunization are severely inhibited by B cells. Eur J Immunol. (2010) 40:124–33. doi: 10.1002/eji.200939493
187. Adams ARD. Trypanosomiasis of stock in Mauritius. Ann Trop Med Parasitol. (1936) 30:521–31. doi: 10.1080/00034983.1936.11684957
188. Doro E, Jacobs SH, Hammond FR, Schipper H, Pieters RP, Carrington M, et al. Visualizing trypanosomes in a vertebrate host reveals novel swimming behaviours, adaptations, and attachment mechanisms. Elife. (2019) 8:e48388. doi: 10.7554/eLife.48388
189. Hafalla JC, Silvie O, Matuschewski K. Cell biology and immunology of malaria. Immunol Rev. (2011) 240:297–316. doi: 10.1111/j.1600-065X.2010.00988.x
190. Frischknecht F, Baldacci P, Martin B, Zimmer C, Thiberge S, Olivo-Marin JC, et al. Imaging movement of malaria parasites during transmission by Anopheles mosquitoes. Cell Microbiol. (2004) 6:687–94. doi: 10.1111/j.1462-5822.2004.00395.x
191. Amino R, Giovannini D, Thiberge S, Gueirard P, Boisson B, Dubremetz J-F, et al. Host cell traversal is important for progression of the malaria parasite through the dermis to the liver. Cell Host Microbe. (2008) 3:88–96. doi: 10.1016/j.chom.2007.12.007
192. Vaughan AM, Aly ASI, Kappe SHI. Malaria parasite pre-erythrocytic stage infection: gliding and hiding. Cell Host Microbe. (2008) 4:209–18. doi: 10.1016/j.chom.2008.08.010
193. Amino R, Thiberge S, Martin B, Celli S, Shorte S, Frischknecht F, et al. Quantitative imaging of Plasmodium transmission from mosquito to mammal. Nat Med. (2006) 12:220–4. doi: 10.1038/nm1350
194. Vanderberg JP, Frevert U. Intravital microscopy demonstrating antibody-mediated immobilisation of Plasmodium berghei sporozoites injected into skin by mosquitoes. Int J Parasitol. (2004) 34:991–6. doi: 10.1016/j.ijpara.2004.05.005
195. Doehl JSP, Bright Z, Dey S, Davies H, Magson J, Brown N, et al. Skin parasite landscape determines host infectiousness in visceral leishmaniasis. Nat Commun. (2017) 8:57–57. doi: 10.1038/s41467-017-00103-8
196. Casley-Smith JR. The influence of tissue hydrostatic pressure and protein concentration on fluid and protein uptake by diaphragmatic initial lymphatics; effect of calcium dobesilate. Microcirc Endothelium Lymphatics. (1985) 2:385–415.
198. Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med. (2007) 204:2349–62. doi: 10.1084/jem.20062596
199. Tal O, Lim HY, Gurevich I, Milo I, Shipony Z, Ng LG, et al. DC mobilization from the skin requires docking to immobilized CCL21 on lymphatic endothelium and intralymphatic crawling. J Exp Med. (2011) 208:2141–53. doi: 10.1084/jem.20102392
200. Yao L-C, Baluk P, Srinivasan RS, Oliver G, McDonald DM. Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am J Pathol. (2012) 180:2561–75. doi: 10.1016/j.ajpath.2012.02.019
201. Wang Y, Simons M. Flow-regulated lymphatic vasculature development and signaling. Vascular Cell. (2014) 6:14–14. doi: 10.1186/2045-824X-6-14
202. Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. (2005) 5:617–28. doi: 10.1038/nri1670
203. Dadiani M, Kalchenko V, Yosepovich A, Margalit R, Hassid Y, Degani H, et al. Real-time imaging of lymphogenic metastasis in orthotopic human breast cancer. Cancer Res. (2006) 66:8037–41. doi: 10.1158/0008-5472.can-06-0728
204. Russo E, Teijeira A, Vaahtomeri K, Willrodt AH, Bloch JS, Nitschké M, et al. Intralymphatic CCL21 promotes tissue egress of dendritic cells through afferent lymphatic vessels. Cell Rep. (2016) 14:1723–34. doi: 10.1016/j.celrep.2016.01.048
205. Weber M, Hauschild R, Schwarz J, Moussion C, de Vries I, Legler DF, et al. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science. (2013) 339:328–32. doi: 10.1126/science.1228456
206. Kabashima K, Shiraishi N, Sugita K, Mori T, Onoue A, Kobayashi M, et al. CXCL12-CXCR4 engagement is required for migration of cutaneous dendritic cells. Am J Pathol. (2007) 171:1249–57. doi: 10.2353/ajpath.2007.070225
207. Ralston KS, Kabututu ZP, Melehani JH, Oberholzer M, Hill KL. The Trypanosoma brucei flagellum: moving parasites in new directions. Ann Rev Microbiol. (2009) 63:335–62. doi: 10.1146/annurev.micro.091208.073353
208. Creek DJ, Mazet M, Achcar F, Anderson J, Kim DH, Kamour R, et al. Probing the metabolic network in bloodstream-form Trypanosoma brucei using untargeted metabolomics with stable isotope labelled glucose. PLoS Pathogens. (2015) 11:e1004689. doi: 10.1371/journal.ppat.1004689
209. Hendrix BM, Sweet JE. A study of amino nitrogen and glucose in lymph and blood before and after the injection of nutrient solutions in the intestine. J Biol Chem. (1917) 32:299–307.
210. Koffi M, Solano P, Denizot M, Courtin D, Garcia A, Lejon V, et al. Aparasitemic serological suspects in Trypanosoma brucei gambiense human African trypanosomiasis: a potential human reservoir of parasites? Acta Tropica. (2006) 98:183–8. doi: 10.1016/j.actatropica.2006.04.001
211. Jamonneau V, Ilboudo H, Kabore J, Kaba D, Koffi M, Solano P, et al. Untreated human infections by Trypanosoma brucei gambiense are not 100% fatal. PLoS Neglect Trop Dis. (2012) 6:e1691. doi: 10.1371/journal.pntd.0001691
212. Goodwin LG. Pathological effects of Trypanosoma brucei on small blood vessels in rabbit ear-chambers. Trans Royal Soc Trop Med Hygiene. (1971) 65:82–8. doi: 10.1016/0035-920390189-1
214. Mantilla BS, Marchese L, Casas-Sánchez A, Dyer NA, Ejeh N, Biran M, et al. Proline metabolism is essential for Trypanosoma brucei brucei survival in the tsetse vector. PLoS Pathogens. (2017) 13:e1006158. doi: 10.1371/journal.ppat.1006158
215. Calvo-Alvarez E, Cren-Travaillé C, Crouzols A, Rotureau B. A new chimeric triple reporter fusion protein as a tool for in vitro and in vivo multimodal imaging to monitor the development of African trypanosomes and Leishmania parasites. Infect Genet Evol. (2018) 63:391–403. doi: 10.1016/j.meegid.2018.01.011
216. Cayla M, Rojas F, Silvester E, Venter F, Matthews KR. African trypanosomes. Parasit Vectors. (2019) 12:190. doi: 10.1186/s13071-019-3355-5
217. Dean S, Marchetti R, Kirk K, Matthews KR. A surface transporter family conveys the trypanosome differentiation signal. Nature. (2009) 459:213–7. doi: 10.1038/nature07997
218. Buscher P, Bart JM, Boelaert M, Bucheton B, Cecchi G, Chitnis N, et al. Do cryptic reservoirs threaten gambiense-sleeping sickness elimination? Trends Parasitol. (2018) 34:197–207. doi: 10.1016/j.pt.2017.11.008
219. Mehlitz D, Molyneux DH. The elimination of Trypanosoma brucei gambiense? Challenges of reservoir hosts and transmission cycles: expect the unexpected. Parasite Epidemiol Control. (2019) 6:e00113. doi: 10.1016/j.parepi.2019.e00113
220. Sudarshi D, Lawrence S, Pickrell WO, Eligar V, Walters R, Quaderi S, et al. Human African trypanosomiasis presenting at least 29 years after infection—what can this teach us about the pathogenesis and control of this neglected tropical disease? PLoS Neglect Trop Dis. (2014) 8:e3349. doi: 10.1371/journal.pntd.0003349
221. Capewell P, Atkins K, Weir W, Jamonneau V, Camara M, Clucas C, et al. Resolving the apparent transmission paradox of African sleeping sickness. PLoS Biol. (2019) 17:e3000105. doi: 10.1371/journal.pbio.3000105
222. Frezil JL. Application of xenodiagnosis in the detection of T. gambiense trypanosomiasis in immunologically suspect patients. Bull Soc Pathol Exot Filiales. (1971) 64:871–8.
223. Wombou Toukam CM, Solano P, Bengaly Z, Jamonneau V, Bucheton B. Experimental evaluation of xenodiagnosis to detect trypanosomes at low parasitaemia levels in infected hosts. Parasite. (2011) 18:295–302. doi: 10.1051/parasite/2011184295
224. Holzmuller P, Biron DG, Courtois P, Koffi M, Bras-Gonçalves R, Daulouede S, et al. Virulence and pathogenicity patterns of Trypanosoma brucei gambiense field isolates in experimentally infected mouse: differences in host immune response modulation by secretome and proteomics. Microb Infect. (2008) 10:79–86. doi: 10.1016/j.micinf.2007.10.008
225. Torrecilhas AC, Schumacher RI, Alves MJM, Colli W. Vesicles as carriers of virulence factors in parasitic protozoan diseases. Microb Infect. (2012) 14:1465–74. doi: 10.1016/j.micinf.2012.07.008
226. Szempruch AJ, Dennison L, Kieft R, Harrington JM, Hajduk SL. Sending a message: extracellular vesicles of pathogenic protozoan parasites. Nat Rev Microbiol. (2016) 14:669. doi: 10.1038/nrmicro.2016.110
227. Szempruch AJ, Sykes SE, Kieft R, Dennison L, Becker AC, Gartrell A, et al. Extracellular vesicles from Trypanosoma brucei mediate virulence factor transfer and cause host anemia. Cell. (2016) 164:246–57. doi: 10.1016/j.cell.2015.11.051
228. Dauchy FA, Contin-Bordes C, Nzoumbou-Boko R, Bonhivers M, Landrein N, Robinson DR, et al. Trypanosoma brucei gambiense excreted/secreted factors impair lipopolysaccharide-induced maturation and activation of human monocyte-derived dendritic cells. Parasite Immunol. (2019) 41:e12632. doi: 10.1111/pim.12632
Keywords: African trypanosomiasis, Trypanosoma brucei, skin, transmission, innate immunity, neglected tropical disease
Citation: Alfituri OA, Quintana JF, MacLeod A, Garside P, Benson RA, Brewer JM, Mabbott NA, Morrison LJ and Capewell P (2020) To the Skin and Beyond: The Immune Response to African Trypanosomes as They Enter and Exit the Vertebrate Host. Front. Immunol. 11:1250. doi: 10.3389/fimmu.2020.01250
Received: 14 January 2020; Accepted: 18 May 2020;
Published: 12 June 2020.
Edited by:
Yasuyuki Goto, The University of Tokyo, JapanReviewed by:
Guy Caljon, University of Antwerp, BelgiumCopyright © 2020 Alfituri, Quintana, MacLeod, Garside, Benson, Brewer, Mabbott, Morrison and Capewell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul Capewell, cGF1bC5jYXBld2VsbEBnbGFzZ293LmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.