 Man Sup Kwak1,2†
Man Sup Kwak1,2† Hee Sue Kim
Hee Sue Kim Myoungsun Son
Myoungsun Son Jeon-Soo Shin
Jeon-Soo Shin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 June 2020
Sec. Molecular Innate Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01189
This article is part of the Research Topic The Role of HMGB1 in Immunity View all 10 articles
Most extracellular proteins are secreted via the classical endoplasmic reticulum (ER)/Golgi-dependent secretion pathway; however, some proteins, including a few danger-associated molecular patterns (DAMPs), are secreted via non-classical ER/Golgi-independent secretion pathways. The evolutionarily conserved high mobility group box1 (HMGB1) is a ubiquitous nuclear protein that can be released by almost all cell types. HMGB1 lacks signal peptide and utilizes diverse non-canonical secretion mechanisms for its extracellular export. Although the post-translational modifications of HMGB1 were demonstrated, the oxidation of HMGB1 and secretion mechanisms are not highlighted yet. We currently investigated that peroxiredoxins I and II (PrxI/II) induce the intramolecular disulfide bond formation of HMGB1 in the nucleus. Disulfide HMGB1 is preferentially transported out of the nucleus by binding to the nuclear exportin chromosome-region maintenance 1 (CRM1). We determined the kinetics of HMGB1 oxidation in bone marrow-derived macrophage as early as a few minutes after lipopolysaccharide treatment, peaking at 4 h while disulfide HMGB1 accumulation was observed within the cells, starting to secrete in the late time point. We have shown that HMGB1 oxidation status, which is known to determine the biological activity in extracellular HMGB1, is crucial for the secretion of HMGB1 from the nucleus. This review summarizes selected aspects of HMGB1 redox biology relevant to the induction and propagation of inflammatory diseases. We implicate the immunological significance and the need for novel HMGB1 inhibitors through mechanism-based studies.
High mobility group box 1 (HMGB1) is an abundant non-histone nuclear protein that was discovered over four decades ago. The protein was isolated from calf thymus chromatin by 0.35 M NaCl extraction (1) and was then biochemically characterized (2). Based on its mobility during polyacrylamide gel electrophoresis, Goodwin et al. termed the proteins “high mobility group,” or HMG proteins; however, the group of proteins that migrated more slowly during polyacrylamide gel electrophoresis were termed “low-mobility group” proteins. HMG proteins were therefore divided into two groups based on their molecular weight: higher—HMG-1 and HMG-2 (now HMGB1 and HMGB2), lower—HMG-14 and HMG-17 (now HMGN1 and HMGN2), and HMG-I, -Y (now HMGA1a and HMGA1b) (3–9).
HMG proteins are categorized into three superfamilies based on the specific functional domains or motifs via which they recognize individual DNA structures on chromatin: HMGA, HMGB, and HMGN. Proteins in the HMGA family contain an AT-hook, which is a DNA-binding motif with a preference for A/T rich regions. In contrast, those in the HMGB family contain A-box and B-box functional motifs and those in the HMGN family contain a nucleosomal binding domain (NBD). HMGB proteins are ubiquitous and abundant in most cells and can bind to DNA without sequence specificity (10–12).
The human HMGB1 protein has 215 amino acid (aa) residues (MW: 25–30 kDa) that form two homologous DNA-binding domains (A-box, 1–79 aa; B-box, 89–162 aa) and a negatively charged C-terminal acidic tail (186–215 aa; Figure 1A) (9, 13). HMGB1 is located in the nucleus as a result of bipartite nuclear localization signals (NLS; NLS1, 28–44 aa; NLS2, 179–185 aa) mediated by the nuclear importin karyopherin (KAP)-α1; however, the affinity between the two molecules is decreased by HMGB1 phosphorylation (14, 15). Conversely, the DNA-binding domain of HMGB1 contains a nuclear-export signal (NES), and its cytoplasmic localization is mediated by the nuclear exportin chromosome-region maintenance 1 (CRM1) (16). The acidic C-terminal of HMGB1 regulates DNA binding and bending by interacting with its DNA-binding domains (8, 17) or histones H1/H3 (18); thus, HMGB1 lacking the C-terminal domain displays improved DNA looping and binding abilities (19).
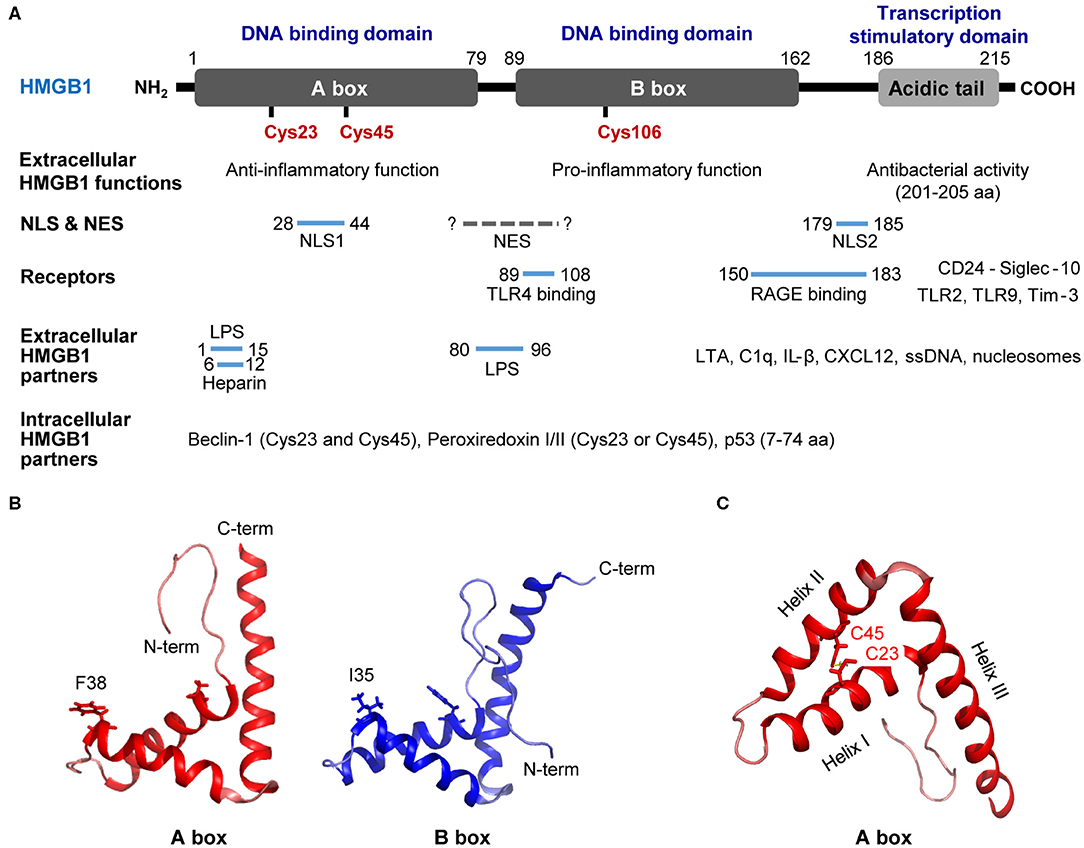
Figure 1. Structure of HMGB1. (A) Detailed summary of various domains and motifs in HMGB1. (B) Signature L-loop structure of the A-box and B-box structures of HMGB1. (C) Overall structure of HMGB1 showing the functional A-box cysteines (C23 and C45) and their geometric distribution in HMGB1. A-box and B-box structures modified based on 2YRQ [Protein Data Bank (PDB) IP: 2yrq] using PyMol. NLS, nuclear localization sequence; NES, nuclear export signal; TLR4, toll-like receptor 4; RAGE, receptor for advanced glycation endproducts; Tim-3, T-cell immunoglobulin and mucin domain-containing protein-3; LPS, lipopolysaccharide; LTA, lipoteichoic acid; C1q, complement component 1q; IL-1β, interleukin 1β; CXCL12, C-X-C motif chemokine 12; F, phenylalanine; I, isoleucine; C, cysteine; C-term, C-terminal; N-term, N-terminal. Solid line; known binding domain or sequence of HMGB1, Dotted grey line; unknown sequence of HMGB1.
The C-terminal acidic domain of HMGB1 also functions as a transcriptional activator (20, 21), while HMGB1 B-box has been reported to induce pro-inflammatory signals upon extracellular stimulation, and the A-box induces antagonistic effects (22). In particular, the 201–205 aa residues in the C-terminal acidic tail play a crucial role in the antibacterial activity of HMGB1 (23). Moreover, interactions with diverse receptors, extracellular partners, and intracellular partners play important roles in the activity and biological functions of HMGB1. HMGB1 residues 89–108 bind to Toll-like receptor (TLR) 4 and increase pro-inflammatory signaling (22), whereas residues 150–183 interact with the receptor for advanced glycation end products (RAGE) to regulate cell migration (24) and stimulate inflammation (25). HMGB1 has also been shown to bind to dendritic cell (DC)-derived TIM-3 and suppress nucleic acid-mediated innate immune responses (26). In addition, residues 1–15 and 80–96 have been found to inhibit lipopolysaccharide (LPS)-induced cytokine production in a subclinical endotoxemia mouse model (27). HMGB1 binds to lipoteichoic acid (LTA) and enhances proinflammatory responses by mediating the transfer of LTA to CD14 and TLR2 (28). Furthermore, HMGB1 residues 6–12 are responsible for binding heparin and compete with binding between RAGE and HMGB1 (29). A- and B-box of HMGB1 bind to C1q, but only B-box of HMGB1 can induce the complement activation leading to sterile inflammation (30). In addition, complex formation between HMGB1 and IL-1β enhances inflammation and destruction mechanisms in arthritic joints (31), whereas the HMGB1 and C-X-C motif chemokine ligand 12 (CXCL12) complex binds to C-X-C chemokine receptor 4 (CXCR4) and promotes the recruitment of inflammatory cells (32). Extracellular HMGB1 binds to single-stranded oligonucleotides, forming HMGB1-5'-C-phosphate-G (CpG)-DNA complex, interacting with TLR9 to augments cytokine production (33, 34). Also, HMGB1 released from apoptosis binds to the nucleosomes and induces cytokine production or dendritic cells (DCs) activation through interaction with TLR2 (35). In contrary, HMGB1-CD24 complex selectively represses the tissue damage-induced inflammation via interaction with Siglec-10 protein (36). Cytoplasmic HMGB1 binds to Beclin 1 using intramolecular disulfide bridge (Cys23 and Cys45) to affect autophagosome formation (37). Cys23 or Cys45 in HMGB1 can also bind to reactive cysteine residues in peroxiredoxins I and II (PrxI/II) to form intramolecular disulfide bonds that promote its secretion in response to inflammatory stimuli (38). Residues 7–74 are responsible for binding the p53 transactivation domain and thus increasing gene transcription (39) (Figure 1A).
Studies have modified HMGB1 A-box and B-box structures using the PyMol program based on 2YRQ [Protein Data Bank (PBD) ID: 2yrq]. These two DNA-binding domains consist of three alpha helices (helix-I, -II, and -III) and two loops (loop-I and -II) that form an L-shaped structure (40). HMGB1 binds to the minor groove of pre-bent or linear DNA with little sequence specificity (41, 42); however, both A- and B-box have the remarkable ability to unwind and bend DNA with different properties. For instance, the A-box domain recognizes pre-bent or linear DNA, whereas the B-box domain binds to mini-circles and bends linear DNA (43–45). In the crystal structure of HMGB1 showing A-box domains and an AT-rich DNA fragment, the two HMGB1 A-box domains were found to collaborate in order to interact with pre-bent or kinked DNA. The Phe37 (Phe38 in HMGB1 described here) residues from both domains were shown to play important roles in initiating intercalation with CG base pairs and thus generating highly kinked DNA (Figure 1B) (46). The B-box domain is structurally similar to the A-box in its DNA-binding characteristics and its Ile34 (Ile35 in B-box or Ile122 in the full HMGB1 sequence described here) residue is sterically comparable to the Phe37 residue in the A-box domain (Figure 1B).
HMGB1 senses and coordinates the cellular stress response. As mentioned earlier, HMGB1 contains three conserved cysteines: Cys23, Cys45, and Cys106 (Figure 1A). Cys23 and Cys45 can form an intramolecular disulfide bond depending on the reactive oxygen species (ROS) concentration and environmental conditions under which HMGB1 binds to its ligands (Figure 1C) (47). Indeed, the half-life of all-thiol-HMGB1 ranges from ~17 min in human serum and saliva to 3 h in cell culture medium (47). The oxidation state of HMGB1 determines its interactions with diverse receptors (32, 48) and its DNA-binding affinity (49). Depending on its redox status, extracellular HMGB1 can trigger numerous effects: (1) all-thiol-HMGB1 can exert chemoattractive effects by binding to CXCR4; (2) all-thiol-HMGB1 can prompt autophagy by binding to RAGE (50); (3) disulfide-HMGB1 can exert pro-inflammatory effects by binding to TLR4; and (4) fully oxidized-HMGB1 is inert. Cys106 plays a crucial role in the translocation of HMGB1 from the nucleus to the cytosol (51). Moreover, C23-C45 oxidation induces a shift between helix I and helix II in the A-box domain that reduces DNA binding affinity by altering the orientation of Phe37 (52), resulting in cytoplasmic translocation. HMGB1 can also affect transcription in the nucleus, requiring rapid transition between the all-thiol-and disulfide forms of HMGB1 (53).
Purification of HMGB1 under its native conditions yields both homodimers and oligomeric forms of the protein; however, these forms are dissociated when acid-extracted (54). Our group has also described the Cys106-mediated formation of HMGB1 dimers under conditions of excessive ROS generation at the cellular level (unpublished data). As previously discussed, HMGB1 is a versatile molecule because of intra- and inter-molecular interactions in its different domains; moreover, the protein can be either actively secreted by activated immune cells or passively released due to necrotic cell death where it acts as a damage-associated molecular pattern (DAMP). Because of its versatile and variable nature, it is important to understand the mechanism underlying the secretion of HMGB1 to fully appreciate its therapeutic and pathological potential. In this review, we briefly summarize the conventional and non-conventional mechanisms of cytokine secretion, and describe in detail the mechanisms of HMGB1 oxidation and secretion that have been determined so far, with a focus on immunological function.
Most soluble secretory proteins utilize a well-known conventional secretion system involving the endoplasmic reticulum (ER) and Golgi network, and contain a signal peptide to target them to the ER (55). When such proteins are synthesized in the ribosome, the signal peptide is recognized by a signal recognition particle (SRP) complex. The protein-ribosome-SRP complex then moves to the Sec61 translocon complex in the ER outer membrane and proteins translocate into the ER lumen via the translocon complex (56, 57). Within the ER lumen, the protein meets chaperone proteins such as Bip and undergoes modification (glycosylation) followed by protein folding (58, 59). These proteins are then translocated to the Golgi and plasma membrane via several processes, concluding the conventional protein secretion pathway. A recent study found that when the ER/Golgi pathway is blocked, some proteins are secreted via an independent mechanism. These proteins lack a signal peptide targeting them to the ER and are secreted under specific conditions, such as ROS accumulation, inflammation, and cell growth factors. These forms of protein secretion are considered unconventional, and the secreted proteins are generally involved in immune surveillance, cell survival, and cellular stress (60).
Cytokines regulate immunological functions via their secretion in immune environments; therefore, controlling cytokine secretion is crucial for regulating immune function. Many cytokines such as tumor necrosis factor-α (TNF-α), interleukin (IL)-2, and IL-12 are secreted via the conventional secretion mechanism; however, some, including HMGB1, IL-1β, fibroblast growth factor (FGF)-1/2, and galectins, utilize non-conventional secretion mechanisms. Here, we briefly explore different cytokines that use various secretion mechanisms, and summarize them in Table 1.

Table 1. Conventional and non-conventional secretion of cytokines.
TNF-α was first identified for its anti-tumor activity but is now also known to act as a multifunctional cytokine in host defense mechanisms during inflammatory responses (77). Since it contains an ER signal peptide, TNF-α is translocated across the ER and through the Golgi apparatus to the plasma membrane (61). Newly synthesized TNF-α is localized in the trans-Golgi network (TGN) with golgin and p230/golgin-245 for intracellular trafficking and cell surface delivery (78). TNF-α is then released from the granule via fusion with the plasma membrane (62) and is cleaved at the cell surface by the tumor necrosis factor-α converting enzyme (TACE) between Ala76 and Val77 (79).
IL-1β plays important roles in the cytokine response to inflammation and immunity during bacterial or viral infection and is mainly secreted by monocytes, macrophages, and CD in response to inflammasome activation under conditions such as LPS and adenosine triphosphate (ATP) stimulation (80). Although IL-1β is passively released during pyroptotic cell death (63), secretion of cleaved-IL-1β requires cytosolic compartments such as the secretory lysosome (64), microvesicles shed from the plasma membrane (81) or exosomal release (65). Previous studies have reported that caspase-1/11 cleave gasdermin D (GSDMD), whose N-terminal then forms pores in the plasma membrane that are crucial for the passive release of IL-1β during pyroptosis (66, 67). Although the pathway is unknown, IL-1β secretion is known to require ABC transporters whose knockdown or inhibition is reported to ameliorate IL-1β secretion (82, 83).
FGF-1 and -2 belong to a family of heparin-binding growth factors (84) and control mitogenic activity (85) and tumor-induced angiogenesis (86). FGF-1/2 are not only secreted via the ER/Golgi-dependent secretion pathway, but also the non-conventional secretion system (68–70); however, the secretion pathways of FGF-1 and FGF-2 are distinct (87). Unlike FGF-1, FGF-2 secretion is sensitive to Na+/K+-ATPase inhibition and is dependent on forming higher-order complexes with Na+/K+-ATPase ion transporters, with its export occurring in a membrane potential-independent manner (71). Conversely, FGF-1 secretion is increased by cellular stresses such as heat shock (88), hypoxia (72), and serum starvation (73), while copper also can induce FGF-1 secretion by forming multiprotein aggregates in response to stress (89); however, FGF1 folding does not prevent its export (90).
Galectins are a family of abundant β-galactoside-specific lectins that reside in the extracellular matrix and are implicated in many cellular processes, such as proliferation, differentiation, and apoptosis (91, 92). Since galectins lack the signal peptides found in IL-1β and FGF-1/2 for ER/Golgi-mediated secretion, their secretion is not blocked by the ER/Golgi-dependent inhibitors brefeldin A and monensin (74, 93). Moreover, galectin-1/3 are not packaged into vesicles during extracellular export (74, 75, 94, 95) but accumulate at the plasma membrane and induce the formation of exosomes that are pinched off and released into extracellular space (74, 75, 94, 95). Secreted galectins bind to the extracellular surface of the plasma membrane or extracellular matrix (75, 76) via the N- and O-glycosylated β-galactose-terminated oligosaccharide side chains of glycoproteins (74, 92).
Generally, cytokines containing a leader sequence undergo secretion via the ER/Golgi secretion pathway; however, the non-histone nuclear protein HMGB1 lacks this signal peptide, and studies have suggested that HMGB1 secretion involves diverse unconventional secretion pathways. For instance, infection or cellular stress has been shown to increase the cytoplasmic accumulation of HMGB1, which is then passively released into the extracellular space or actively secreted via secretory lysosomes (9) (Figure 2). Here, we describe in detail the mechanisms that participate in HMGB1 secretion.
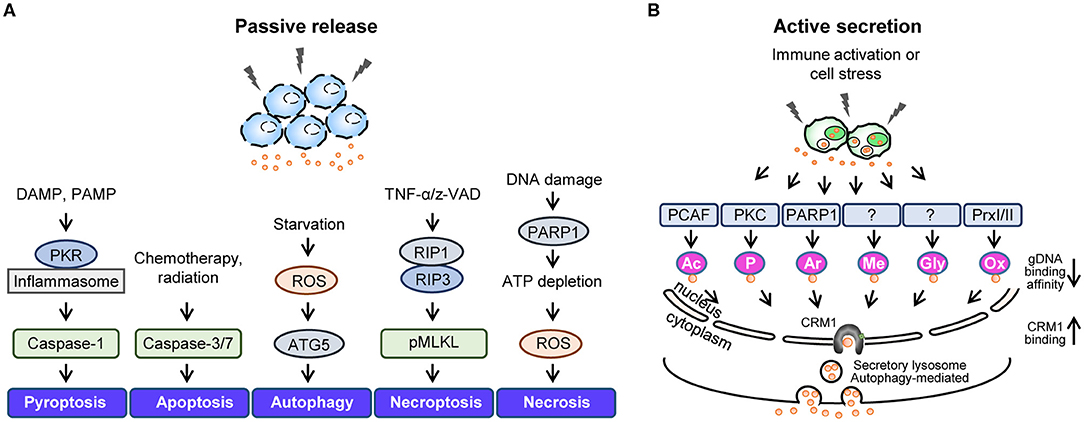
Figure 2. Active secretion and passive release of HMGB1. Summary of stimuli leading to passive HMGB1 release (A) and active HMGB1 secretion (B). (A) Passive release mechanisms involve the disruption of the plasma membrane via various cell death mechanisms. (B) Active secretion involves various HMGB1 post-translational modifications that reduce its genomic DNA binding activity and increase its CRM1 binding affinity. DAMP, danger-associated molecular pattern; PAMP, pathogen-associated molecular pattern; PKR, double-stranded RNA-dependent protein kinase; ROS, reactive oxygen species; ATG5, autophagy related 5; z-VAD, carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]; RIP1, receptor-interacting serine/threonine-protein kinase 1; RIP3, receptor-interacting protein kinase 3; pMLKL, Phosphorylated mixed linage kinase domain like; PARP1, poly [ADP-ribose] polymerase 1; PCAF, P300/CBP-associated factor.
HMGB1 can be passively released during various forms of cell death, including pyroptosis, apoptosis, autophagy, necroptosis, and necrosis (Figure 2A). Pyroptosis refers to inflammatory programmed cell death that occurs after inflammasome formation caused by bacterial or viral infection. During pyroptosis, double-stranded RNA-dependent protein kinase (PKR) induces inflammasome formation, caspase-1 activation, and HMGB1 release upon exposure to diverse inflammasome-activating agents (96). Apoptosis is another form of programmed cell death that occurs when cells die due to injury and involves caspase-3/7, which belong to a family of protease enzymes. Apoptotic cells induce HMGB1 release, and it has been reported that Z-VAD, a pan-caspase inhibitor, can reduce the levels of HMGB1 released (97). Autophagy is an intracellular degradation system that balances energy sources in response to nutrient stress by regulating the degradation of cellular material using lysosomes or vacuoles; however, excessive autophagy can lead to cell death. Indeed, studies have shown that epithelial and glioblastoma tumor cells release HMGB1 when treated with the autophagy-inducing agent epidermal growth factor receptor-targeted diphtheria toxin (DT-EGF) (98), also ATG5 knock-out bone marrow-derived macrophages (BMDMs) reduced HMGB1 secretion under EBSS starvation conditions (99). In addition, autophagosome-mediated HMGB1 secretion has been identified, with ATG5 deficient cells or those treated with an early autophagy inhibitor displaying all-thiol-HMGB1 secretion (data not published). Necrosis is a form of premature cell death caused by the loss of membrane integrity, intracellular organelle swelling, and ATP depletion, and it has been shown that HMGB1 is passively released by necrotic or damaged cells (100). Necroptosis is a form of programmed necrosis mediated by death signals that cause the phosphorylated mixed lineage kinase domain-like protein (MLKL) to be inserted into and permeabilize the plasma membrane (101). Moreover, TNF-α/Z-VAD-induced necroptosis has been shown to phosphorylate MLKL proteins and increase HMGB1 secretion levels (102).
HMGB1 can undergo several extensive PTMs that increase its cytoplasmic accumulation and extracellular secretion during infection or cell stress, including acetylation (16), phosphorylation (14, 15), ADP-ribosylation (103), methylation (104), glycosylation (105), and oxidation (38, 51) (Figure 2B). Various PTMs increase the interaction between HMGB1 and the nuclear transport receptor CRM1, thus favoring its translocation from the nucleus to the cytoplasm. PTM-mediated HMGB1 secretion is caused by lysosomal exocytosis wherein cytoplasmic HMGB1 co-localizes with the lysosomal marker LAMP1 for secretion (106). The PTMs and subsequent events that HMGB1 undergoes are summarized below and visualized in Figure 2B.
1) Acetylation is a major PTM that can affect protein function by altering properties such as hydrophobicity, solubility, and surface properties. Protein acetylation refers to the reaction during which the acetyl group of acetyl coenzyme A (Ac-CoA) is transferred to the lysine (Lys) residue of the target protein. HMGB1 has two acetylation clusters at Lys27–29 and Lys181–183, and it has been shown that nuclear localization is unaffected by mutating either Lys cluster (16). The poly(ADP-ribose) polymerase-1 (PARP1) induces the cytoplasmic translocation and extracellular secretion of HMGB1 by catalyzing its acetylation (107). Compare to HMGB1, mimicking acetylated HMGB1 (six lysine residues for glutamines) increases the TNF-α production in RAW264.7 cells and reduces DC maturation (108). Various triggers which induces HMGB1 acetylation includes inflammatory signal such as LPS or TNF-α (109), and cell stress triggered by chemotherapeutic reagent such as cisplatin (110). Such conditions can be experimentally mimicked using trichostatin A (TSA), an inhibitor of histone deacetylase complex (HDAC) (111).
2) Phosphorylation is a molecular mechanism via which amino acid residues are phosphorylated by a protein kinase to regulate the functional response of proteins to various extra- or intracellular stimuli. HMGB1 phosphorylation is mediated by classical protein kinase C (cPKC) in a calcium-dependent manner via the PI3K-PKC signaling pathway (14). In HMGB1, Ser35, 39, 42, and 46 in NLS1, 181 in NLS2, and 53 close to NLS1 have been shown to be phosphorylated in macrophages after TNF-α and okadaic acid treatment (15), while Ser39, 53, and 182 of HMGB1 are phosphorylated by PKC-ζ in colon cancer cells (112). Moreover, HMGB1 phosphorylation has been found to reduce its binding affinity with the nuclear import protein KAP-α1 and promote its cytoplasmic translocation and extracellular secretion (15).
3) ADP-ribosylation refers to the process wherein one or more ADP-ribose moieties are added to target proteins, and includes mono-ADP-ribosylation, poly-ADP-ribosylation, ADP-ribose cyclization, and O-acetyl-ADP-ribose formation. PARP activation regulates the translocation of HMGB1 from the nucleus to the cytoplasm during DNA-alkylating damage (103), while hyper poly(ADP)-ribosylated HMGB1 has been shown to inhibit efferocytosis by binding to phosphatidylserine (PS) on apoptotic cells and RAGE on macrophages (113). Such activation was reported in cell death related stimuli such as activation of tumor necrosis factor [ligand] superfamily member 10 (TNFSF10)—TNF-related apoptosis-inducing ligand (TRAIL) pathway (114) or daunorubicin treatment (115), and inflammatory assault with LPS (103).
4) Methylation is a PTM in which a methyl group is added to proteins, usually on the side-chain nitrogens of arginine and lysine or carboxyl groups of glutamate and leucine. During the process of neutrophilic differentiation, Lys42 in HMGB1 can be mono-methylated which significantly reduces its DNA binding activity, causing its translocation from the nucleus to the cytoplasm (104). Lys112 has also been found to be mono-methylated in HMGB1 and contribute toward its cytoplasmic localization (116). However, it is still unclear which stimuli exclusively leads to the methylation of HMGB1 during active secretion.
5) Glycosylation is a common PTM characterized by the attachment of sugar moieties to proteins. HMGB1 derived from calf thymus and chicken erythrocytes undergoes O-linked GlcNac glycosylation with sugars such as Fuc, Man, GalNH2, GlcNH2, and Gal monosaccharides (117), while it has recently been reported that HMGB1 can also undergo N-linked glycosylation at two consensus (Asn37 and Asn137) residues and one non-consensus (Asn135) residue. N-glycosylation of HMGB1, induced by PMA, TSA and LPS, can persuade its secretion into the extracellular space by reducing its DNA binding affinity and increasing the association with CRM1 (105).
6) Oxidation is a covalent modification that proteins undergo during redox reactions involving the transfer of oxygen, hydrogen, and electrons. HMGB1 contains three redox-sensitive cysteines: Cys23, Cys45, and Cys106. Under mild oxidative stress, Cys23 and Cys45 rapidly form an intramolecular disulfide bond that increases the cytoplasmic localization and extracellular secretion of HMGB1 (51). In response to inflammatory stimuli, PrxI and PrxII induce HMGB1 oxidation to its disulfide form and lead to its nucleocytoplasmic translocation and secretion (38). Such oxidative stresses may come from external sources such as H2O2 or glucose oxidase, or by internal activation of molecules by LPS, TNF-α, or interferon-γ which in turn causes HMGB1 release through TNF-dependent manner (118). Since the oxidation status and immunological properties of HMGB1 crucially influence its biological function, we dedicated a separate section to discussing HMGB1 redox biology in detail.
The function and secretion of HMGB1 are dependent on its redox status, which is controlled by three redox-sensitive cysteines: Cys23 and Cys45 in A-box and Cys106 in B-box. Thus, HMGB1 can take three different oxidation forms: an all-thiol form, disulfide form (Cys23-Cys45 intramolecular disulfide bond with Cys106 thiol form), and a fully oxidized form. During the active secretion of HMGB1, the Cys23-Cys45 intramolecular disulfide bond induces HMGB1 cytoplasmic translocation. This process also requires Cys106, as demonstrated by the nuclear localization of Cys106-mutated even with Cys23 and Cys45 mutations (51). Hydrogen peroxide is a ROS that induces the release of HMGB1 from macrophages and monocytes, reportedly by increasing its interaction with CRM1 and thus increasing HMGB1 secretion (119). Under elevated ROS conditions, PrxI and PrxII cause disulfide-HMGB1 formation (38) (Figure 3). The diversity of HMGB1 redox status also affects its passive release from necrotic and apoptotic cells, with the majority of HMGB1 released from necrotic cells being in an all-thiol state but that released from apoptotic cells being in a fully oxidized form. Moreover, HMGB1 oxidation status plays an important role in receptor binding and subsequent cytokine-like activities.
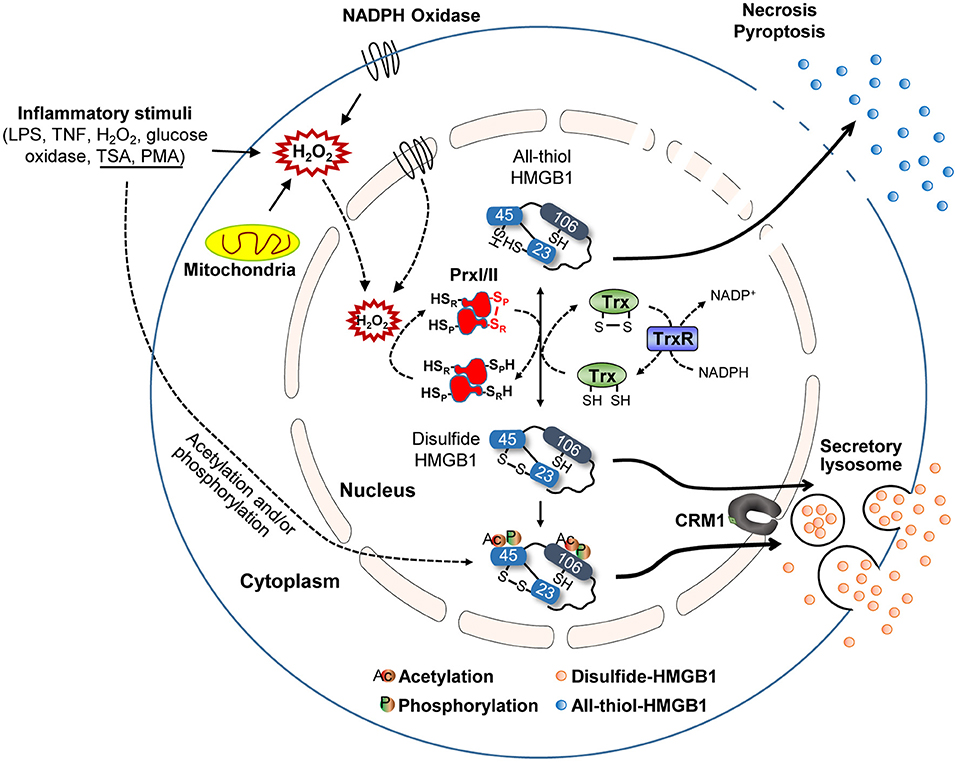
Figure 3. HMGB1 redox biology. Summary of HMGB1 redox biology and the crucial role of peroxiredoxin and thioredoxin. Various stimuli cause oxidative stress that promotes HMGB1 oxidation via the peroxiredoxin-dependent pathway. NADPH, nicotinamide adenine dinucleotide phosphate; SP, peroxidatic cysteine; SR, resolving cysteine; H, hydrogen; Trx, thioredoxin; TrxR, thioredoxin reductases; NADP, nicotinamide adenine dinucleodebtide phosphate.
Hoppe et al. previously described the mechanism of HMGB1 oxidation (51), identifying that HMGB1 interacts with the de-glutathionylation enzyme glutaredoxin (Grx) during the nuclear extraction of Chinese hamster ovary (CHO) cells after diamide treatment. Electrophoretic mobility assays revealed that HMGB1 oxidation increases in a diamide concentration-dependent manner, while disulfide-HMGB1 could be reversed by incubating diamide-treated retinal pigment epithelium (RPE) cells with thioredoxin (Trx) or Grx/glutathione (Figure 3). Conversely, we found that HMGB1 can be oxidized by PrxI/II in the nucleus after exposure to inflammatory stimuli (Figure 3) (38). PrxI/II can interact with all-thiol-HMGB1 generated by mutagenesis (Cys23-to-Ser or Cys45-to-Ser) after hydrogen peroxide stimulation; however, such HMGB1 oxidation is suppressed in PrxI/II-deficient mouse embryonic fibroblast (MEF) cells, even when exposed to inflammatory stimuli. All-thiol-HMGB1 cannot translocate into the cytoplasm or extracellular space, while PrxI/II-deficient BMDMs lack the ability to secrete HMGB1 despite treatment with diverse inflammatory stimuli, such as LPS, phorbol-12-myristate-13-acetate (PMA), trichostatin A (TSA), or TNF-α (38). HMGB1 phosphorylation by PMA and/or acetylation by TSA promotes its nuclear transport and extracellular secretion (15, 16). Although cells treated with PMA or TSA display increased HMGB1 secretion, this secretion is inhibited by treatment with the antioxidant N-acetylcysteine (NAC). Moreover, HMGB1 secretion induced by PMA can be inhibited by the NADPH oxidase inhibitor diphenyleneiodonium (DPI), indicating that PMA-induced HMGB1 secretion requires H2O2 production by the mitochondria and/or Nox. Intracellular disulfide bond formation in HMGB1 (Cys23-Cys45) has been shown to be important for its nucleocytoplasmic translocation and extracellular secretion. Indeed, HMGB1 mutants with defective phosphorylation or acetylation sites undergo less translocation into the extracellular space than WT HMGB1 (38). In summary, these findings indicate that the mechanisms of HMGB1 oxidation and reduction are induced by PrxI/II and Trx or Grx/glutathione, respectively (Figure 3). It is possible that, like acetylated-HMGB1, oxidized-HMGB1 may be less favored for nuclear import and thus accumulates in the cytosol. Oxidized-HMGB1 in the cytosol is packed into lysosomes through an as yet unknown mechanism and then secreted. Nevertheless, extracellular HMGB1 can induce an immune response and HMGB1 oxidation decides its immune function. Also, as mentioned previously, HMGB1 oxidation has a more substantial influence on its secretion compared to acetylation and phosphorylation. Thus, control of HMGB1 oxidation both in intracellular and extracellular is important for the therapeutic approach based on blockade of HMGB1 secretion and immune response.
The PTMs of HMGB1 in the nucleus occurs rapidly after exposure to diverse stimuli; however, after binding to CRM1 in the nucleus, the export of modified HMGB1 into the cytoplasm is known to take around 6–8 h, whereas it can take up to 18 h for HMGB1 secretion into the extracellular space to peak. The degree of HMGB1 oxidation has been reported to be crucial for its immune function (9, 51, 120) and is also very important for its secretion. For instance, HMGB1 secretion induced by PTMs such as phosphorylation or acetylation also requires oxidation, with anti-oxidant treatment reducing HMGB1 secretion even when treated with PMA or TSA (38). Therefore, we examined instances when HMGB1 PTMs and extracellular secretion occur under oxidative conditions by treating mouse BMDMs with 100 ng/mL of LPS and separating their nuclei at a series of time points to determine the HMGB1 oxidation ratio. HMGB1 oxidation increased with time, with oxidation first detectable after just 30 min, and disulfide-HMGB1 was maintained for up to 4 h and then gradually decreased after 8 h. Despite rapid HMGB1 oxidation in the nucleus, its secretion began only after 4 h and increased up to 16 h (Figure 4). Further studies investigating the mechanism underlying the delay between oxidation and secretion would improve our understanding of HMGB1 secretion kinetics. It is possible that secretion-ready cytosolic HMGB1 is packed into a secretory lysosome or autophagosome and secreted via non-conventional secretion mechanisms, requiring a very complex, as yet unknown, packaging mechanism. Conversely, a recent article by Wang et al. observed HMGB1 localization in mitochondria and peroxisome in neuron cells via electron microscopy and immunofluorescence, but not in lysosomes (121). In macrophages or macrophage lineages, the release of HMGB1 occurred through a lysosomal pathway after acetylation of the HMGB1 (16). It has also been demonstrated that LPS-induced HMGB1 secretion by monocytes is mediated by lysosomal exocytosis (106). Various explanations may be available for such discrepancy; however, the diverse origin of cells (macrophage, epithelial, neuronal, etc.) may have different mechanisms that translocated HMGB1 takes to be secreted to the extracellular milieu. A complete understanding of the oxidation kinetics mechanism of HMGB1 is necessary to predict the fate of HMGB1-mediated inflammation.
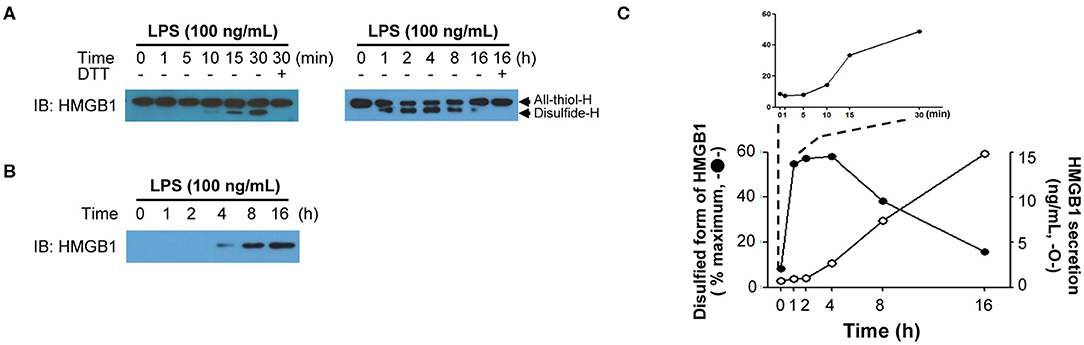
Figure 4. HMGB1 redox kinetics. (A) Short-term (left) and long-term (right) changes of HMGB1 redox status upon LPS stimulation. Western blot showing the All-thiol-H (all-thiol-HMGB1) or Disulfide-H (disulfide-HMGB1) expression in BMDM whole-cell lysates, which was treated with LPS (100 ng/mL) for indicated times. Methods were used as our previous study (38). (B) HMGB1 secretion timeframe. Disulfide-H from culture supernatant was measured by Western blot. (C) Graphical representation of the relationship between HMGB1 oxidation (left y-axis, closed circle) quantified as % maximum from (A) and HMGB1 secretion (right y-axis, opened circle). The level of secreted HMGB1 was determined by ELISA (38).
As mentioned in the Introduction, extracellular HMGB1 via active secretion or passive release binds to diverse partners (Figure 1A). HMGB1 associates and shows interactions with several molecules, such as Heparin, LPS, LTA, IL-1β, RNA, and DNA, CXCL12, nucleosomes and C1q (9, 27–30). HMGB1-partner complex (or HMGB1 alone) interacts with immune receptors or surface molecules such as TLRs and RAGE, which then activate the downstream signaling pathways such as MyD88, IFN regulatory factors (IRFs), nuclear factor κB (NF-κB), MAPKs and phosphatidylinositol 3-kinase (PI3K) to enhance the inflammation and immune response (9, 22, 25, 26, 28, 34, 36). Besides, HMGB1 oxidation status alters its receptor bindings and subsequent cytokine-like activities (9, 51). As mentioned in section Post-Translational Modifications (PTMs) and Active Secretion of HMGB1, multiple PTMs are involved in extracellular secretion of HMGB1. Effect of PTM in receptor-ligand interaction kinetics between extracellular HMGB1 and its receptors, however, are understood to a lesser degree. One example of PTMs positively affecting the HMGB1-receptor interaction is portrayed using acetylation-mimicking mutant HMGB1 (six lysine residues for glutamines), increasing TNF-α production in RAW264.7 cells (108). Thus, although specific effects for each PTM are yet to be reported, diversely modified HMGB1 may have an important role in the receptor binding and downstream signaling pathway. Of all PTMs associated with HMGB1, only the effects of oxidation are studied in detail, hence we will focus our scope to the redox status of extracellular HMGB1 in regards to its activity. HMGB1 binds to different receptors depending on the redox state of its three cysteines (C23, C45, C106), which subsequently determines its functions (120). HMGB1 containing three thiol-form cysteines exerts chemoattractive effects by forming a heterocomplex with CXCL12, which then binds to CXCR4 and induces cell migration. The formation of a complex between HMGB1 and CXCR4 induces conformational changes in aa residues 3–12 of CXCL12 alongside specific conformational changes in the CXCR4 homodimer, which promotes better chemotactic abilities than CXCL12 alone (32). All-thiol-HMGB1 can also bind to RAGE and promote autophagy (122) by inhibiting mTOR and promoting Beclin 1-Ptdlns3KC3 complex formation (123). ROS induces HMGB1 oxidation and cytosolic translocation from the nucleus. Cytoplasmic HMGB1 binds to Beclin 1 using an intramolecular disulfide bridge (Cys23 and Cys45) to enhance autophagic flux (37, 124, 125). Moreover, the interaction between HMGB1 and RAGE activates NF-κB, the MAP kinase pathways and affects cell migration by inducing the expression of adhesion molecules (126, 127). Extracellular HMGB1 can also stimulate RAGE expression (128). Conversely, disulfide-HMGB1 stimulates cytokine production and inflammation by forming a complex with CD14 and MD-2 via TLR4. The disulfide bond between C23-C45 and the thiol form of C106 residues are not only required for binding TLR4 but also inducing the translocation of NF-κB and release of TNF-α (48). These findings were confirmed in apoptotic cells where fully oxidized-HMGB1 produced by excessive ROS contributed toward immunological inertness and apoptotic cell death (32). Although its specific functions remain unclear, fully oxidized-HMGB1 is known to prevent the cytokine or chemokine activities of other HMGB1 forms and ultimately induce immune tolerance (32) (Figure 5). In contrast, CD24-Siglec-10 and TIM-3 are negative receptors that inhibit HMGB1 immune activity in macrophages, DCs and tumor cells. HMGB1 can bind to CD24, first identified as a B cell differentiation marker, and selectively represses the tissue damage-induce inflammation through induction of CD24-Siglec-10 complex formation, negatively regulating NF-κB (36). Also, HMGB1 can bind to TIM-3, a member of the T-cell immunoglobulin domain and mucin domain family, and its binding suppresses the nucleic acid-mediated antitumor immunity via A-box competing with nucleic acid (26). The aforementioned reports indicate extracellular HMGB1 have not only pro-inflammatory effects but also anti-inflammatory effects according to the microenvironment.
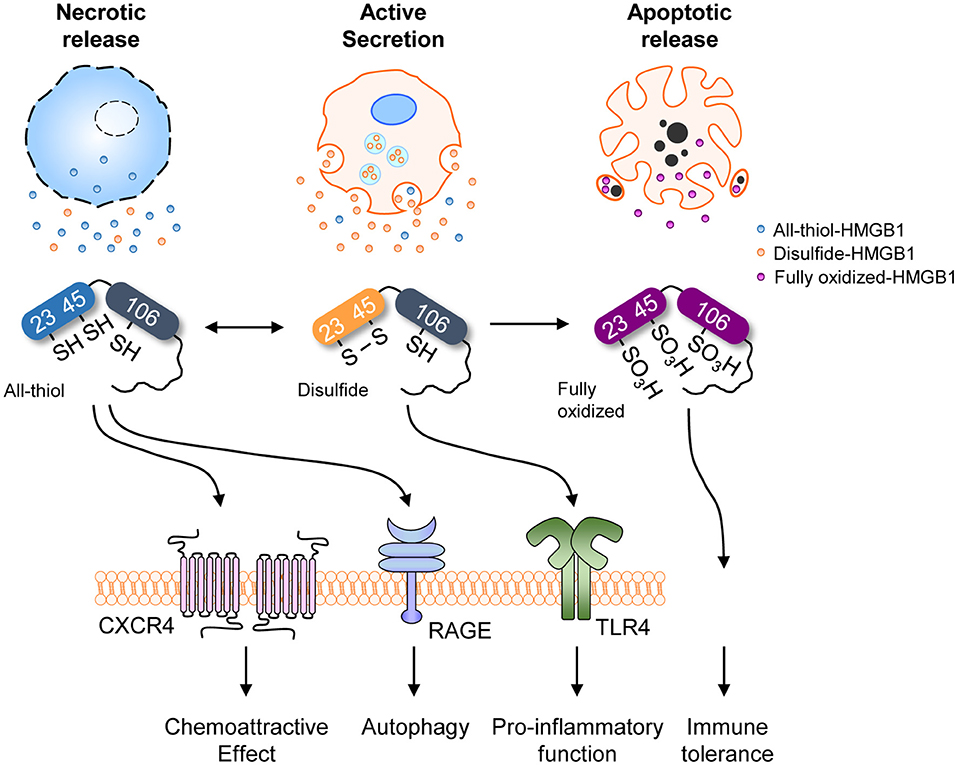
Figure 5. HMGB1 redox status and receptors. Redox status of HMGB1 from various sources with different receptors and their representative functions. Different redox states are associated with different release mechanisms, each linked to various immunological and cell biological functions.
Extracellular HMGB1 not only differ in functions by its PTM derivations, but also by the types of cells responding to HMGB1. Monocytes exposed to HMGB1 polarized toward pro-inflammatory (M1) macrophages, upregulating the production of inflammatory cytokines both in vitro and in vivo (129, 130). Silencing of HMGB1, on the other hand, prevents macrophage polarization to the M1 phenotype following LPS stimulation (131). Similarly, neutrophils react to extracellular HMGB1 by promoting its neutrophil extracellular trap formation (132) and heighten its immune reactions (133). DCs consider HMGB1 as an endogenous adjuvant to boost its effectiveness in antigen-presenting to its adaptive counterparts (134). Extracellular HMGB1, as discussed above, has exhibited significance in various immunological and physiological contexts, sparking an interest in suppressing its functions. Controlling HMGB1 as a potential therapeutic target in the immune diseases must be exquisitely controlled depending on its purpose.
The involvement of HMGB1 in various pathologies ranging from inflammatory diseases to cancer has been discussed thoroughly and has resulted in the development of HMGB1 secretion inhibitors. Currently, several companies and research centers sought to control the effects of HMGB1 by modulating its expression, translocation, secretion, and receptor binding ability using diverse chemicals as an approach to develop therapeutic agents. These strategies for suppressing HMGB1 secretion can be divided into three categories: (1) small molecules inhibiting HMGB1 release; (2) neutralizing HMGB1 itself; and (3) blocking HMGB1 receptors (Figure 6).
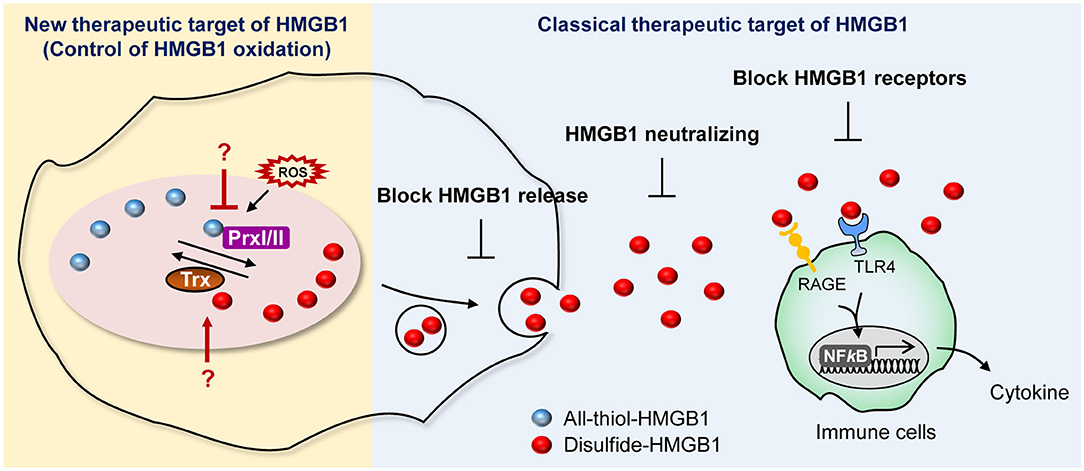
Figure 6. Current and potential strategies for HMGB1 inhibition. Summary of newly proposed inhibition targets (left), targets utilized by conventional inhibitors (right).
Numerous studies have reported small molecules capable of inhibiting HMGB1 secretion, from newly synthesized molecules to those isolated from natural sources. For instance, naturally isolated small molecules such as glycyrrhizin have been reported to be effective in treating numerous pathological conditions, such as septic shock, neuroinflammation, atopic dermatitis, and Pseudomonas aeruginosa keratitis (135–138). Synthetic molecules such as ethyl pyruvate, atorvastatin, and simvastatin have also demonstrated promising therapeutic activity by targeting HMGB1. In addition, the natural flavonoid kaempferol was found to alleviate neuroinflammation by suppressing HMGB1 release and down regulating the TLR4/MyD88 pathway (139), and the rare ginsenosides Rk1 and Rg5 have shown promise by reducing HMGB1 release and thereby improving survival in cecum ligation- and puncture-induced murine sepsis models (140). Inflachromene, a novel small molecule developed as a potential anti-inflammatory drug, was also found to inhibit HMGB1 secretion via directly binding to HMGB1 and inhibiting autophagy (141, 142). Despite most of the candidates are yet to be approved by the Food and Drug Administration (FDA), Metformin, clinically approved drug for metabolic disease and type 2 diabetes, has investigated as an inhibitor for HMGB1 through direct binding each other, inhibiting the cytosolic translocation within the cells and receptor binding in the extracellular space (143, 144). Other candidates of HMGB1 secretion inhibitors are also being discovered through drug repositioning efforts, such as salicylic acid, methotrexate, and (-)-epigallocatechin-3-gallate (145–147).
Neutralizing antibodies against HMGB1 have been used to confirm its involvement in mouse models of various pathologies, such as arthritis, suggesting that HMGB1-neutralizing antibodies could be used therapeutically (148, 149). Indeed, neutralizing the effects of HMGB1 by competitively inhibiting its activity with soluble receptors or neutralizing antibodies could be a straightforward and approach. A soluble form of RAGE was reported to effectively reduce neutrophilic asthma attacks and angiotensin II-induced cardiomyocyte hypertrophy by inhibiting the HMGB1-RAGE axis (150, 151). Similarly, studies have reported the inhibition of the HMGB1-receptor signaling pathway using neutralizing antibodies against its receptors. For instance, neutralizing monoclonal antibodies recognizing TLR4 were used to reduce IL-8 secretion upon LPS stimulation in human primary monocytes, and efforts to use HMGB1 neutralizing antibodies in stroke patients are being continually made (152, 153). Moreover, the continual administration of neutralizing antibodies against RAGE in murine models of neurological pain were reported to reverse mechanical hyperalgesia (154, 155) Further information about HMGB1 inhibitors could be found in this Frontiers Research Topics of “The Role of HMGB1 in Immunity” by Yang et al. (156).
We suggest expanding on the importance of modulating the HMGB1 oxidation mechanism. HMGB1 oxidation is the major PTM that drives secretion and its oxidized form of extracellular HMGB1 induce inflammatory signaling, which leads to many diseases, including neuroinflammation, hyperalgesia, drug-induced liver injury, and sepsis (157–159). Of particular note is the fact that it is important to develop specific inhibitors targeting the enzymes involved in altering HMGB1 redox status. Prx, which is a major direct modulator of HMGB1 redox status, could possibly be a plausible candidate for inhibition (38), whereas well-established redox enzymes with a strong connection to Prx, such as Trx and sulforedoxins, could also be potential targets for inhibition (160) (Figure 6).
Although several strategies have been shown successfully in inhibiting HMGB1-dependent inflammatory processes (156), there is still a lack of specificity originating from HMGB1's involvement in pathologies. This review aims to overcome the aforementioned weak points by suggesting various plausible aspects of inhibition, increasing the specificity of inhibition therapies. We provide an overview of the protein secretion mechanisms and discuss the HMGB1 secretion mechanisms and pathways in depth. Besides, we highlight the importance of multiple PTMs and the redox biology of HMGB1, with a particular focus on the important role of HMGB1 oxidation in its secretion. Finally, we discuss multiple immunological and non-immunological diseases involving HMGB1, as well as attempts to inhibit its secretion, extracellular activity, or the receptors that bind to HMGB1. The next step should to unveil fine-tuned process of HMGB1 PTMs in physiological and pathological conditions. Future researches would benefit from extensive quantitative analysis of extracellular HMGB1 and its PTM patterns in various cell types and different pathological conditions to further develop disease-specific inhibition strategies.
MK and HK wrote the manuscript, contributed to the conception, design, and analysis of the study. BL and YK contributed to interpretation of the study and drafted the manuscript. MK assisted in manuscript editing and figure preparation. MS and J-SS contributed to the conception and design of the study and the discussion, writing, supervision, and critical revision of the manuscript.
This work was supported by grants from the National Research Foundation (NRF) of Korea, which was funded by the Korean Government (2017R1A2B3006704, 2019R1A6A1A03032869, and 2019R1I1A1A01058308), the Research Center Program of Institute for Basic Science (IBS) in Korea (IBS-R026-D1), the Brain Korea 21 PLUS Project for Medical Science, and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health in USA (R01AI135063 [MS]).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Songhee H. Kim for kindly providing the graphics for the A-box and B-box structures of HMGB1.
1. Goodwin GH, Johns EW. Isolation and characterisation of two calf-thymus chromatin non-histone proteins with high contents of acidic and basic amino acids. Eur J Biochem. (1973) 40:215–9. doi: 10.1111/j.1432-1033.1973.tb03188.x
2. Goodwin GH, Sanders C, Johns EW. A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur J Biochem. (1973) 38:14–9. doi: 10.1111/j.1432-1033.1973.tb03026.x
3. Brown E, Goodwin GH, Mayes EL, Hastings JR, Johns EW. Heterogeneity of proteins resembling high-mobility-group protein HMG-T in trout testes nuclei. Biochem J. (1980) 191:661–4. doi: 10.1042/bj1910661
4. Cockerill PN, Goodwin GH, Cary PD, Turner C, Johns EW. Comparisons of the structures of the chromosomal high mobility group proteins HMG1 and HMG2 prepared under conditions of neutral and acidic pH. Biochim Biophys Acta. (1983) 745:70–81. doi: 10.1016/0167-4838(83)90171-1
5. Goodwin GH, Brown E, Walker JM, Johns EW. The isolation of three new high mobility group nuclear proteins. Biochim Biophys Acta. (1980) 623:329–38. doi: 10.1016/0005-2795(80)90260-3
6. Goodwin GH, Rabbani A, Nicolas PH, Johns EW. The isolation of the high mobility group non-histone chromosomal protein HMG 14. FEBS Lett. (1977) 80:413–6. doi: 10.1016/0014-5793(77)80488-2
7. Walker JM, Hastings JR, Johns EW. The primary structure of a non-histone chromosomal protein. Eur J Biochem. (1977) 76:461–8. doi: 10.1111/j.1432-1033.1977.tb11616.x
8. Lund T, Holtlund J, Skalhegg B, Laland SG. Method for complete separation of the high mobility group (HMG) proteins HMG I and HMG Y from HMG 14 and HMG 17 and a procedure for purification of HMG I and HMG Y. J Chromatogr. (1986) 369:341–8. doi: 10.1016/S0021-9673(00)90140-7
9. Lee SA, Kwak MS, Kim S, Shin JS. The role of high mobility group box 1 in innate immunity. Yonsei Med J. (2014) 55:1165–76. doi: 10.3349/ymj.2014.55.5.1165
10. Murphy FVT, Sweet RM, Churchill ME. The structure of a chromosomal high mobility group protein-DNA complex reveals sequence-neutral mechanisms important for non-sequence-specific DNA recognition. EMBO J. (1999) 18:6610–8. doi: 10.1093/emboj/18.23.6610
11. Murphy FVT, Churchill ME. Nonsequence-specific DNA recognition: a structural perspective. Structure. (2000) 8:R83–9. doi: 10.1016/S0969-2126(00)00126-X
12. Allain FH, Yen YM, Masse JE, Schultze P, Dieckmann T, Johnson RC, et al. Solution structure of the HMG protein NHP6A and its interaction with DNA reveals the structural determinants for non-sequence-specific binding. EMBO J. (1999) 18:2563–79. doi: 10.1093/emboj/18.9.2563
13. Bianchi ME, Falciola L, Ferrari S, Lilley DM. The DNA binding site of HMG1 protein is composed of two similar segments (HMG boxes), both of which have counterparts in other eukaryotic regulatory proteins. EMBO J. (1992) 11:1055–63. doi: 10.1002/j.1460-2075.1992.tb05144.x
14. Oh YJ, Youn JH, Ji Y, Lee SE, Lim KJ, Choi JE, et al. HMGB1 is phosphorylated by classical protein kinase C and is secreted by a calcium-dependent mechanism. J Immunol. (2009) 182:5800–9. doi: 10.4049/jimmunol.0801873
15. Youn JH, Shin JS. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol. (2006) 177:7889–97. doi: 10.4049/jimmunol.177.11.7889
16. Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. (2003) 22:5551–60. doi: 10.1093/emboj/cdg516
17. Stros M. DNA bending by the chromosomal protein HMG1 and its high mobility group box domains. Effect of flanking sequences. J Biol Chem. (1998) 273:10355–61.
18. Cato L, Stott K, Watson M, Thomas JO. The interaction of HMGB1 and linker histones occurs through their acidic and basic tails. J Mol Biol. (2008) 384:1262–72. doi: 10.1016/j.jmb.2008.10.001
19. Stros M, Stokrova J, Thomas JO. DNA looping by the HMG-box domains of HMG1 and modulation of DNA binding by the acidic C-terminal domain. Nucleic Acids Res. (1994) 22:1044–51. doi: 10.1093/nar/22.6.1044
20. Landsman D, Bustin M. Assessment of the transcriptional activation potential of the HMG chromosomal proteins. Mol Cell Biol. (1991) 11:4483–9. doi: 10.1128/MCB.11.9.4483
21. Banerjee S, Kundu TK. The acidic C-terminal domain and A-box of HMGB-1 regulates p53-mediated transcription. Nucleic Acids Res. (2003) 31:3236–47. doi: 10.1093/nar/gkg412
22. Li J, Kokkola R, Tabibzadeh S, Yang R, Ochani M, Qiang X, et al. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med. (2003) 9:37–45. doi: 10.1007/BF03402105
23. Gong W, Li Y, Chao F, Huang G, He F. Amino acid residues 201–205 in C-terminal acidic tail region plays a crucial role in antibacterial activity of HMGB1. J Biomed Sci. (2009) 16:83. doi: 10.1186/1423-0127-16-83
24. Huttunen HJ, Fages C, Kuja-Panula J, Ridley AJ, Rauvala H. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. (2002) 62:4805–11.
25. He Q, You H, Li XM, Liu TH, Wang P, Wang BE. HMGB1 promotes the synthesis of pro-IL-1beta and pro-IL-18 by activation of p38 MAPK and NF-kappaB through receptors for advanced glycation end-products in macrophages. Asian Pac J Cancer Prev. (2012) 13:1365–70. doi: 10.7314/APJCP.2012.13.4.1365
26. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. (2012) 13:832–42. doi: 10.1038/ni.2376
27. Youn JH, Kwak MS, Wu J, Kim ES, Ji Y, Min HJ, et al. Identification of lipopolysaccharide-binding peptide regions within HMGB1 and their effects on subclinical endotoxemia in a mouse model. Eur J Immunol. (2011) 41:2753–62. doi: 10.1002/eji.201141391
28. Kwak MS, Lim M, Lee YJ, Lee HS, Kim YH, Youn JH, et al. HMGB1 Binds to lipoteichoic acid and enhances TNF-alpha and IL-6 production through HMGB1-mediated transfer of lipoteichoic acid to CD14 and TLR2. J Innate Immun. (2015) 7:405–16. doi: 10.1159/000369972
29. Ling Y, Yang ZY, Yin T, Li L, Yuan WW, Wu HS, et al. Heparin changes the conformation of high-mobility group protein 1 and decreases its affinity toward receptor for advanced glycation endproducts in vitro. Int Immunopharmacol. (2011) 11:187–93. doi: 10.1016/j.intimp.2010.11.014
30. Kim SY, Son M, Lee SE, Park IH, Kwak MS, Han M, et al. High-mobility group box 1-induced complement activation causes sterile inflammation. Front Immunol. (2018) 9:705. doi: 10.3389/fimmu.2018.00705
31. Wahamaa H, Schierbeck H, Hreggvidsdottir HS, Palmblad K, Aveberger AC, Andersson U, et al. High mobility group box protein 1 in complex with lipopolysaccharide or IL-1 promotes an increased inflammatory phenotype in synovial fibroblasts. Arthritis Res Ther. (2011) 13:R136. doi: 10.1186/ar3450
32. Schiraldi M, Raucci A, Munoz LM, Livoti E, Celona B, Venereau E, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med. (2012) 209:551–63. doi: 10.1084/jem.20111739
33. Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. (2007) 8:487–96. doi: 10.1038/ni1457
34. Ivanov S, Dragoi AM, Wang X, Dallacosta C, Louten J, Musco G, et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood. (2007) 110:1970–81. doi: 10.1182/blood-2006-09-044776
35. Urbonaviciute V, Fürnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. (2008) 205:3007–18. doi: 10.1084/jem.20081165
36. Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science. (2009) 323:1722–5. doi: 10.1126/science.1168988
37. Kang R, Livesey KM, Zeh HJ, Loze MT, Tang D. HMGB1: a novel Beclin 1-binding protein active in autophagy. Autophagy. (2010) 6:1209–11. doi: 10.4161/auto.6.8.13651
38. Kwak MS, Kim HS, Lkhamsuren K, Kim YH, Han MG, Shin JM, et al. Peroxiredoxin-mediated disulfide bond formation is required for nucleocytoplasmic translocation and secretion of HMGB1 in response to inflammatory stimuli. Redox Biol. (2019) 24:101203. doi: 10.1016/j.redox.2019.101203
39. Rowell JP, Simpson KL, Stott K, Watson M, Thomas JO. HMGB1-facilitated p53 DNA binding occurs via HMG-Box/p53 transactivation domain interaction, regulated by the acidic tail. Structure. (2012) 20:2014–24. doi: 10.1016/j.str.2012.09.004
40. Hardman CH, Broadhurst RW, Raine AR, Grasser KD, Thomas JO, Laue ED. Structure of the A-domain of HMG1 and its interaction with DNA as studied by heteronuclear three- and four-dimensional NMR spectroscopy. Biochemistry. (1995) 34:16596–607. doi: 10.1021/bi00051a007
41. Werner MH, Huth JR, Gronenborn AM, Clore GM. Molecular basis of human 46X,Y sex reversal revealed from the three-dimensional solution structure of the human SRY-DNA complex. Cell. (1995) 81:705–14. doi: 10.1016/0092-8674(95)90532-4
42. Dragan AI, Liggins JR, Crane-Robinson C, Privalov PL. The energetics of specific binding of AT-hooks from HMGA1 to target DNA. J Mol Biol. (2003) 327:393–411. doi: 10.1016/s0022-2836(03)00050-0
43. Teo SH, Grasser KD, Thomas JO. Differences in the DNA-binding properties of the HMG-box domains of HMG1 and the sex-determining factor SRY. Eur J Biochem. (1995) 230:943–50. doi: 10.1111/j.1432-1033.1995.tb20640.x
44. Müller S, Bianchi ME, Knapp S. Thermodynamics of HMGB1 interaction with duplex DNA. Biochemistry (2001) 40:10254–61. doi: 10.1021/bi0100900
45. Paull TT, Haykinson MJ, Johnson RC. The nonspecific DNA-binding and -bending proteins HMG1 and HMG2 promote the assembly of complex nucleoprotein structures. Genes Dev. (1993) 7:1521–34. doi: 10.1101/gad.7.8.1521
46. Sanchez-Giraldo R, Acosta-Reyes FJ, Malarkey CS, Saperas N, Churchill ME, Campos JL. Two high-mobility group box domains act together to underwind and kink DNA. Acta Crystallogr D Biol Crystallogr. (2015) 71(Pt. 7):1423–32. doi: 10.1107/S1399004715007452
47. Zandarashvili L, Sahu D, Lee K, Lee YS, Singh P, Rajarathnam K, et al. Real-time kinetics of high-mobility group box 1 (HMGB1) oxidation in extracellular fluids studied by in situ protein NMR spectroscopy. J Biol Chem. (2013) 288:11621–7. doi: 10.1074/jbc.M113.449942
48. Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA. (2010) 107:11942–7. doi: 10.1073/pnas.1003893107
49. Polanska E, Pospisilova S, Stros M. Binding of histone H1 to DNA is differentially modulated by redox state of HMGB1. PLoS ONE. (2014) 9:e89070. doi: 10.1371/journal.pone.0089070
50. Kang R, Tang D, Loze MT, Zeh HJ. Apoptosis to autophagy switch triggered by the MHC class III-encoded receptor for advanced glycation endproducts (RAGE). Autophagy. (2011) 7:91–3. doi: 10.4161/auto.7.1.13852
51. Hoppe G, Talcott KE, Bhattacharya SK, Crabb JW, Sears JE. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp Cell Res. (2006) 312:3526–38. doi: 10.1016/j.yexcr.2006.07.020
52. Wang J, Tochio N, Takeuchi A, Uewaki J, Kobayashi N, Tate S. Redox-sensitive structural change in the A-domain of HMGB1 and its implication for the binding to cisplatin modified DNA. Biochem Biophys Res Commun. (2013) 441:701–6. doi: 10.1016/j.bbrc.2013.10.085
53. Stros M, Polanska E, Kucirek M, Pospisilova S. Histone H1 Differentially inhibits DNA bending by reduced and oxidized HMGB1 protein. PLoS ONE. (2015) 10:e0138774. doi: 10.1371/journal.pone.0138774
54. Marekov LN, Demirov DG, Beltchev BG. Isolation of high-mobility-group proteins HMG1 and HMG2 in non denaturing conditions and comparison of their properties with those of acid-extracted proteins. Biochim Biophys Acta. (1984) 789:63–8. doi: 10.1016/0167-4838(84)90061-X
55. Palade G. Intracellular aspects of the process of protein synthesis. Science. (1975) 189:347–58. doi: 10.1126/science.1096303
56. Deshaies RJ, Sanders SL, Feldheim DA, Schekman R. Assembly of yeast Sec proteins involved in translocation into the endoplasmic reticulum into a membrane-bound multisubunit complex. Nature. (1991) 349:806–8. doi: 10.1038/349806a0
57. Gorlich D, Rapoport TA. Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell. (1993) 75:615–30. doi: 10.1016/0092-8674(93)90483-7
58. Dierks T, Volkmer J, Schlenstedt G, Jung C, Sandholzer U, Zachmann K, et al. A microsomal ATP-binding protein involved in efficient protein transport into the mammalian endoplasmic reticulum. EMBO J. (1996) 15:6931–42. doi: 10.1002/j.1460-2075.1996.tb01085.x
59. Tyedmers J, Lerner M, Wiedmann M, Volkmer J, Zimmermann R. Polypeptide-binding proteins mediate completion of co-translational protein translocation into the mammalian endoplasmic reticulum. EMBO Rep. (2003) 4:505–10. doi: 10.1038/sj.embor.embor826
60. Murray RZ, Stow JL. Cytokine secretion in macrophages: SNAREs, Rabs, and membrane trafficking. Front Immunol. (2014) 5:538. doi: 10.3389/fimmu.2014.00538
61. Stanley AC, Lacy P. Pathways for cytokine secretion. Physiology. (2010) 25:218–29. doi: 10.1152/physiol.00017.2010
62. Mócsai A, Ligeti E, Lowell CA, Berton G. Adhesion-dependent degranulation of neutrophils requires the Src family kinases Fgr and Hck. J Immunol. (1999) 162:1120–6.
63. Martin-Sanchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, Bagnall J, et al. Inflammasome-dependent IL-1beta release depends upon membrane permeabilisation. Cell Death Differ. (2016) 23:1219–31. doi: 10.1038/cdd.2015.176
64. Andrei C, Dazzi C, Lotti L, Torrisi MR, Chimini G, Rubartelli A. The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol Biol Cell. (1999) 10:1463–75. doi: 10.1091/mbc.10.5.1463
65. Qu Y, Franchi L, Nunez G, Dubyak GR. Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol. (2007) 179:1913–25. doi: 10.4049/jimmunol.179.3.1913
66. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
67. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
68. Jackson A, Friedman S, Zhan X, Engleka KA, Forough R, Maciag T. Heat shock induces the release of fibroblast growth factor 1 from NIH 3T3 cells. Proc Natl Acad Sci USA. (1992) 89:10691–5. doi: 10.1073/pnas.89.22.10691
69. Trudel C, Faure-Desire V, Florkiewicz RZ, Baird A. Translocation of FGF2 to the cell surface without release into conditioned media. J Cell Physiol. (2000) 185:260–8. doi: 10.1002/1097-4652(200011)185:2<260::AID-JCP11>3.0.CO;2-X
70. Mignatti P, Morimoto T, Rifkin DB. Basic fibroblast growth factor, a protein devoid of secretory signal sequence, is released by cells via a pathway independent of the endoplasmic reticulum-Golgi complex. J Cell Physiol. (1992) 151:81–93. doi: 10.1002/jcp.1041510113
71. Florkiewicz RZ, Anchin J, Baird A. The inhibition of fibroblast growth factor-2 export by cardenolides implies a novel function for the catalytic subunit of Na+, K+-ATPase. J Biol Chem. (1998) 273:544–51. doi: 10.1074/jbc.273.1.544
72. Mouta Carreira C, Landriscina M, Bellum S, Prudovsky I, Maciag T. The comparative release of FGF1 by hypoxia and temperature stress. Growth Factors. (2001) 18:277–85. doi: 10.3109/08977190109029116
73. Shin JT, Opalenik SR, Wehby JN, Mahesh VK, Jackson A, Tarantini F, et al. Serum-starvation induces the extracellular appearance of FGF-1. Biochim Biophys Acta. (1996) 1312:27–38. doi: 10.1016/0167-4889(96)00013-4
74. Hughes RC. Secretion of the galectin family of mammalian carbohydrate-binding proteins. Biochim Biophys Acta. (1999) 1473:172–85. doi: 10.1016/S0304-4165(99)00177-4
75. Cooper DN, Barondes SH. Evidence for export of a muscle lectin from cytosol to extracellular matrix and for a novel secretory mechanism. J Cell Biol. (1990) 110:1681–91. doi: 10.1083/jcb.110.5.1681
76. Lutomski D, Fouillit M, Bourin P, Mellottee D, Denize N, Pontet M, et al. Externalization and binding of galectin-1 on cell surface of K562 cells upon erythroid differentiation. Glycobiology. (1997) 7:1193–9. doi: 10.1093/glycob/7.8.1193
77. Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. (2012) 119:651–65. doi: 10.1182/blood-2011-04-325225
78. Lieu ZZ, Lock JG, Hammond LA, La Gruta NL, Stow JL, Gleeson PA. A trans-Golgi network golgin is required for the regulated secretion of TNF in activated macrophages in vivo. Proc Natl Acad Sci USA. (2008) 105:3351–6. doi: 10.1073/pnas.0800137105
79. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. (1997) 385:729–33. doi: 10.1038/385729a0
80. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. (2004) 430:213–8. doi: 10.1038/nature02664
81. MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A. Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity. (2001) 15:825–35. doi: 10.1016/S1074-7613(01)00229-1
82. Hamon Y, Luciani MF, Becq F, Verrier B, Rubartelli A, Chimini G. Interleukin-1beta secretion is impaired by inhibitors of the Atp binding cassette transporter, ABC1. Blood. (1997) 90:2911–5. doi: 10.1182/blood.V90.8.2911
83. Marty V, Medina C, Combe C, Parnet P, Amedee T. ATP binding cassette transporter ABC1 is required for the release of interleukin-1beta by P2X7-stimulated and lipopolysaccharide-primed mouse Schwann cells. Glia. (2005) 49:511–9. doi: 10.1002/glia.20138
84. Burgess WH, Maciag T. The heparin-binding (fibroblast) growth factor family of proteins. Annu Rev Biochem. (1989) 58:575–606. doi: 10.1146/annurev.bi.58.070189.003043
85. Schweigerer L, Neufeld G, Friedman J, Abraham JA, Fiddes JC, Gospodarowicz D. Capillary endothelial cells express basic fibroblast growth factor, a mitogen that promotes their own growth. Nature. (1987) 325:257–9. doi: 10.1038/325257a0
86. Stan AC, Nemati MN, Pietsch T, Walter GF, Dietz H. In vivo inhibition of angiogenesis and growth of the human U-87 malignant glial tumor by treatment with an antibody against basic fibroblast growth factor. J Neurosurg. (1995) 82:1044–52. doi: 10.3171/jns.1995.82.6.1044
87. Prudovsky I, Mandinova A, Soldi R, Bagala C, Graziani I, Landriscina M, et al. The non-classical export routes: FGF1 and IL-1alpha point the way. J Cell Sci. (2003) 116(Pt 24):4871–81. doi: 10.1242/jcs.00872
88. Mignatti P, Rifkin DB. Release of basic fibroblast growth factor, an angiogenic factor devoid of secretory signal sequence: a trivial phenomenon or a novel secretion mechanism? J Cell Biochem. (1991) 47:201–7. doi: 10.1002/jcb.240470303
89. Landriscina M, Bagala C, Mandinova A, Soldi R, Micucci I, Bellum S, et al. Copper induces the assembly of a multiprotein aggregate implicated in the release of fibroblast growth factor 1 in response to stress. J Biol Chem. (2001) 276:25549–57. doi: 10.1074/jbc.M102925200
90. Graziani I, Doyle A, Sterling S, Kirov A, Tarantini F, Landriscina M, et al. Protein folding does not prevent the nonclassical export of FGF1 and S100A13. Biochem Biophys Res Commun. (2009) 381:350–4. doi: 10.1016/j.bbrc.2009.02.061
91. Perillo NL, Pace KE, Seilhamer JJ, Baum LG. Apoptosis of T cells mediated by galectin-1. Nature. (1995) 378:736–9. doi: 10.1038/378736a0
92. Perillo NL, Marcus ME, Baum LG. Galectins: versatile modulators of cell adhesion, cell proliferation, and cell death. J Mol Med. (1998) 76:402–12. doi: 10.1007/s001090050232
93. Lindstedt R, Apodaca G, Barondes SH, Mostov KE, Leffler H. Apical secretion of a cytosolic protein by Madin-Darby canine kidney cells. Evidence for polarized release of an endogenous lectin by a nonclassical secretory pathway. J Biol Chem. (1993) 268:11750–7.
94. Sato S, Burdett I, Hughes RC. Secretion of the baby hamster kidney 30-kDa galactose-binding lectin from polarized and nonpolarized cells: a pathway independent of the endoplasmic reticulum-Golgi complex. Exp Cell Res. (1993) 207:8–18. doi: 10.1006/excr.1993.1157
95. Mehul B, Hughes RC. Plasma membrane targeting, vesicular budding and release of galectin 3 from the cytoplasm of mammalian cells during secretion. J Cell Sci. (1997) 110(Pt. 10):1169–78.
96. Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. (2012) 488:670–4. doi: 10.1038/nature11290
97. Qin S, Wang H, Yuan R, Li H, Ochani M, Ochani K, et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med. (2006) 203:1637–42. doi: 10.1084/jem.20052203
98. Thorburn J, Horita H, Redzic J, Hansen K, Frankel AE, Thorburn A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. (2009) 16:175–83. doi: 10.1038/cdd.2008.143
99. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. (2011) 30:4701–11. doi: 10.1038/emboj.2011.398
100. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. (2002) 418:191–5. doi: 10.1038/nature00858
101. Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, et al. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol Cell. (2016) 61:589–601. doi: 10.1016/j.molcel.2016.01.011
102. Negroni A, Colantoni E, Pierdomenico M, Palone F, Costanzo M, Oliva S, et al. RIP3 AND pMLKL promote necroptosis-induced inflammation and alter membrane permeability in intestinal epithelial cells. Dig Liver Dis. (2017) 49:1201–10. doi: 10.1016/j.dld.2017.08.017
103. Ditsworth D, Zong WX, Thompson CB. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J Biol Chem. (2007) 282:17845–54. doi: 10.1074/jbc.M701465200
104. Ito I, Fukazawa J, Yoshida M. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J Biol Chem. (2007) 282:16336–44. doi: 10.1074/jbc.M608467200
105. Kim YH, Kwak MS, Park JB, Lee SA, Choi JE, Cho HS, et al. N-linked glycosylation plays a crucial role in the secretion of HMGB1. J Cell Sci. (2016) 129:29–38. doi: 10.1242/jcs.176412
106. Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. (2002) 3:995–1001. doi: 10.1093/embo-reports/kvf198
107. Yang Z, Li L, Chen L, Yuan W, Dong L, Zhang Y, et al. PARP-1 mediates LPS-induced HMGB1 release by macrophages through regulation of HMGB1 acetylation. J Immunol. (2014) 193:6114–23. doi: 10.4049/jimmunol.1400359
108. Chen X, Xu Y, Xiong P, Tan Z, Gong F, Hou X, et al. Effects of mimicked acetylated HMGB1 on macrophages and dendritic cells. Mol Med Rep. (2018) 18:5527–35. doi: 10.3892/mmr.2018.9584
109. Hwang JS, Choi HS, Ham SA, Yoo T, Lee WJ, Paek KS, et al. Deacetylation-mediated interaction of SIRT1-HMGB1 improves survival in a mouse model of endotoxemia. Sci Rep. (2015) 5:15971. doi: 10.1038/srep15971
110. He Y, Ding Y, Wang D, Zhang W, Chen W, Liu X, et al. HMGB1 bound to cisplatin-DNA adducts undergoes extensive acetylation and phosphorylation in vivo. Chem Sci. (2015) 6:2074–8. doi: 10.1039/C4SC03650F
111. Zou JY, Crews FT. Release of neuronal HMGB1 by ethanol through decreased HDAC activity activates brain neuroimmune signaling. PLoS ONE. (2014) 9:e87915. doi: 10.1371/journal.pone.0087915
112. Lee H, Park M, Shin N, Kim G, Kim YG, Shin JS, et al. High mobility group box-1 is phosphorylated by protein kinase C zeta and secreted in colon cancer cells. Biochem Biophys Res Commun. (2012) 424:321–6. doi: 10.1016/j.bbrc.2012.06.116
113. Davis K, Banerjee S, Friggeri A, Bell C, Abraham E, Zerfaoui M. Poly(ADP-ribosyl)ation of high mobility group box 1 (HMGB1) protein enhances inhibition of efferocytosis. Mol Med. (2012) 18:359–69. doi: 10.2119/molmed.2011.00203
114. Yang M, Liu L, Xie M, Sun X, Yu Y, Kang R, et al. Poly-ADP-ribosylation of HMGB1 regulates TNFSF10/TRAIL resistance through autophagy. Autophagy. (2015) 11:214–24. doi: 10.4161/15548627.2014.994400
115. Li Y, Xie J, Li X, Fang J. Poly (ADP-ribosylation) of HMGB1 facilitates its acetylation and promotes HMGB1 translocation-associated chemotherapy-induced autophagy in leukaemia cells. Oncol Lett. (2020) 19:368–78. doi: 10.3892/ol.2019.11116
116. Wu F, Zhao ZH, Ding ST, Wu HH, Lu JJ. High mobility group box 1 protein is methylated and transported to cytoplasm in clear cell renal cell carcinoma. Asian Pac J Cancer Prev. (2013) 14:5789–95. doi: 10.7314/APJCP.2013.14.10.5789
117. Chao YB, Scovell WM, Yan SB. High mobility group protein, HMG-1, contains insignificant glycosyl modification. Protein Sci. (1994) 3:2452–4. doi: 10.1002/pro.5560031230
118. Rendon-Mitchell B, Ochani M, Li J, Han J, Wang H, Yang H, et al. IFN-gamma induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J Immunol. (2003) 170:3890–7. doi: 10.4049/jimmunol.170.7.3890
119. Tang D, Shi Y, Kang R, Li T, Xiao W, Wang H, et al. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J Leukoc Biol. (2007) 81:741–7. doi: 10.1189/jlb.0806540
120. Tang D, Billiar TR, Lotze MT. A Janus tale of two active high mobility group box 1 (HMGB1) redox states. Mol Med. (2012) 18:1360–2. doi: 10.2119/molmed.2012.00314
121. Wang D, Liu K, Fukuyasu Y, Teshigawara K, Fu L, Wake H, et al. HMGB1 translocation in neurons after ischemic insult: subcellular localization in mitochondria and peroxisomes. Cells. (2020) 9:643. doi: 10.3390/cells9030643
122. Tang D, Kang R, Zeh HJ III, Lotze MT. High-mobility group box 1 and cancer. Biochim Biophys Acta. (2010) 1799:131–40. doi: 10.1016/j.bbagrm.2009.11.014
123. Kang R, Livesey KM, Zeh HJ III, Lotze MT, Tang D. Metabolic regulation by HMGB1-mediated autophagy and mitophagy. Autophagy. (2011) 7:1256–8. doi: 10.4161/auto.7.10.16753
124. Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. (2010) 190:881–92. doi: 10.1083/jcb.200911078
125. Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. (2010) 29:5299–310. doi: 10.1038/onc.2010.261
126. Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger CJ, Arnold B, et al. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. (2005) 61:1–9. doi: 10.1111/j.0300-9475.2005.01534.x
127. Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. (2003) 101:2652–60. doi: 10.1182/blood-2002-05-1300
128. Li J, Qu X, Schmidt AM. Sp1-binding elements in the promoter of RAGE are essential for amphoterin-mediated gene expression in cultured neuroblastoma cells. J Biol Chem. (1998) 273:30870–8. doi: 10.1074/jbc.273.47.30870
129. Son M, Porat A, He M, Suurmond J, Santiago-Schwarz F, Andersson U, et al. C1q and HMGB1 reciprocally regulate human macrophage polarization. Blood. (2016) 128:2218–28. doi: 10.1182/blood-2016-05-719757
130. Tian S, Zhang L, Tang J, Guo X, Dong K, Chen SY. HMGB1 exacerbates renal tubulointerstitial fibrosis through facilitating M1 macrophage phenotype at the early stage of obstructive injury. Am J Physiol Renal Physiol. (2015) 308:F69–75. doi: 10.1152/ajprenal.00484.2014
131. Jiang Y, Chen R, Shao X, Ji X, Lu H, Zhou S, et al. HMGB1 silencing in macrophages prevented their functional skewing and ameliorated EAM development: nuclear HMGB1 may be a checkpoint molecule of macrophage reprogramming. Int Immunopharmacol. (2018) 56:277–84. doi: 10.1016/j.intimp.2018.01.013
132. Tadie JM, Bae HB, Jiang S, Park DW, Bell CP, Yang H, et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol. (2013) 304:L342–9. doi: 10.1152/ajplung.00151.2012
133. Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. (2007) 26:1129–39. doi: 10.1038/sj.emboj.7601552
134. Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. (2004) 5:825–30. doi: 10.1038/sj.embor.7400205
135. Zhao F, Fang Y, Deng S, Li X, Zhou Y, Gong Y, et al. Glycyrrhizin protects rats from sepsis by blocking HMGB1 signaling. Biomed Res Int. (2017) 2017:9719647. doi: 10.1155/2017/9719647
136. Kong ZH, Chen X, Hua HP, Liang L, Liu LJ. The oral pretreatment of glycyrrhizin prevents surgery-induced cognitive impairment in aged mice by reducing neuroinflammation and Alzheimer's-related pathology via HMGB1 inhibition. J Mol Neurosci. (2017) 63:385–95. doi: 10.1007/s12031-017-0989-7
137. Wang Y, Zhang Y, Peng G, Han X. Glycyrrhizin ameliorates atopic dermatitis-like symptoms through inhibition of HMGB1. Int Immunopharmacol. (2018) 60:9–17. doi: 10.1016/j.intimp.2018.04.029
138. Ekanayaka SA, McClellan SA, Barrett RP, Kharotia S, Hazlett LD. Glycyrrhizin reduces HMGB1 and bacterial load in Pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci. (2016) 57:5799–809. doi: 10.1167/iovs.16-20103
139. Cheng X, Yang Y-L, Yang H, Wang Y-H, Du G-H. Kaempferol alleviates LPS-induced neuroinflammation and BBB dysfunction in mice via inhibiting HMGB1 release and down-regulating TLR4/MyD88 pathway. Int Immunopharmacol. (2018) 56:29–35. doi: 10.1016/j.intimp.2018.01.002
140. Kim J-E, Lee W, Yang S, Cho S-H, Baek M-C, Song G-Y, et al. Suppressive effects of rare ginsenosides, Rk1 and Rg5, on HMGB1-mediated septic responses. Food Chem Toxicol. (2019) 124:45–53. doi: 10.1016/j.fct.2018.11.057
141. Cho W, Koo JY, Park Y, Oh K, Lee S, Song JS, et al. Treatment of sepsis pathogenesis with high mobility group box protein 1-regulating anti-inflammatory agents. J Med Chem. (2017) 60:170–9. doi: 10.1021/acs.jmedchem.6b00954
142. Kim YH, Kwak MS, Shin JM, Hayuningtyas RA, Choi JE, Shin JS. Inflachromene inhibits autophagy through modulation of Beclin 1 activity. J Cell Sci. (2018) 131:jcs211201. doi: 10.1242/jcs.211201
143. Horiuchi T, Sakata N, Narumi Y, Kimura T, Hayashi T, Nagano K, et al. Metformin directly binds the alarmin HMGB1 and inhibits its proinflammatory activity. J Biol Chem. (2017) 292:8436–46. doi: 10.1074/jbc.M116.769380
144. Han Y, Yuan F, Deng C, He F, Zhang Y, Shen H, et al. Metformin decreases LPS-induced inflammatory response in rabbit annulus fibrosus stem/progenitor cells by blocking HMGB1 release. Aging. (2019) 11:10252–65. doi: 10.18632/aging.102453
145. VanPatten S, Al-Abed Y. High Mobility Group box-1 (HMGb1): current wisdom and advancement as a potential drug target. J Med Chem. (2018) 61:5093–107. doi: 10.1021/acs.jmedchem.7b01136
146. Kuroiwa Y, Takakusagi Y, Kusayanagi T, Kuramochi K, Imai T, Hirayama T, et al. Identification and characterization of the direct interaction between methotrexate (MTX) and high-mobility group box 1 (HMGB1) protein. PLoS ONE. (2013) 8:e63073. doi: 10.1371/journal.pone.0063073
147. Meng XY, Li B, Liu S, Kang H, Zhao L, Zhou R. EGCG in green tea induces aggregation of HMGB1 protein through large conformational changes with polarized charge redistribution. Sci Rep. (2016) 6:22128. doi: 10.1038/srep22128
148. Kokkola R, Li J, Sundberg E, Aveberger AC, Palmblad K, Yang H, et al. Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum. (2003) 48:2052–8. doi: 10.1002/art.11161
149. Schierbeck H, Lundback P, Palmblad K, Klevenvall L, Erlandsson-Harris H, Andersson U, et al. Monoclonal anti-HMGB1 (high mobility group box chromosomal protein 1) antibody protection in two experimental arthritis models. Mol Med. (2011) 17:1039–44. doi: 10.2119/molmed.2010.00264
150. Zhang F, Su X, Huang G, Xin XF, Cao EH, Shi Y, et al. sRAGE alleviates neutrophilic asthma by blocking HMGB1/RAGE signalling in airway dendritic cells. Sci Rep. (2017) 7:14268. doi: 10.1038/s41598-017-14667-4
151. Lim S, Lee ME, Jeong J, Lee J, Cho S, Seo M, et al. sRAGE attenuates angiotensin II-induced cardiomyocyte hypertrophy by inhibiting RAGE-NFkappaB-NLRP3 activation. Inflamm Res. (2018) 67:691–701. doi: 10.1007/s00011-018-1160-9
152. Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. (2006) 26:174–9. doi: 10.1097/01.shk.0000225404.51320.82
153. Tian X, Liu C, Shu Z, Chen G. Review: therapeutic targeting of HMGB1 in stroke. Curr Drug Deliv. (2017) 14:785–90. doi: 10.2174/1567201813666160808111933
154. Allette YM, Due MR, Wilson SM, Feldman P, Ripsch MS, Khanna R, et al. Identification of a functional interaction of HMGB1 with Receptor for Advanced Glycation End-products in a model of neuropathic pain. Brai Behav Immun. (2014) 42:169–77. doi: 10.1016/j.bbi.2014.06.199
155. Nakamura Y, Morioka N, Abe H, Zhang FF, Hisaoka-Nakashima K, Liu K, et al. Neuropathic pain in rats with a partial sciatic nerve ligation is alleviated by intravenous injection of monoclonal antibody to high mobility group box-1. PLoS ONE. (2013) 8:e73640. doi: 10.1371/journal.pone.0073640
156. Yang H, Wang H, Andersson U. Targeting inflammation driven by HMGB1. Front Immunol. (2020) 11:484. doi: 10.3389/fimmu.2020.00484
157. Frank MG, Weber MD, Fonken LK, Hershman SA, Watkins LR, Maier SF. The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: a role for the NLRP3 inflammasome. Brain Behav Immun. (2016) 55:215–24. doi: 10.1016/j.bbi.2015.10.009
158. Lian YJ, Gong H, Wu TY, Su WJ, Zhang Y, Yang YY, et al. Ds-HMGB1 and fr-HMGB induce depressive behavior through neuroinflammation in contrast to nonoxid-HMGB1. Brain Behav Immun. (2017) 59:322–32. doi: 10.1016/j.bbi.2016.09.017
159. Yamasoba D, Tsubota M, Domoto R, Sekiguchi F, Nishikawa H, Liu K, et al. Peripheral HMGB1-induced hyperalgesia in mice: redox state-dependent distinct roles of RAGE and TLR4. J Pharmacol Sci. (2016) 130:139–42. doi: 10.1016/j.jphs.2016.01.005
Keywords: high mobility group box1 (HMGB1), oxidation, inflammation, therapeutic target, danger-associated molecular pattern (DAMP)
Citation: Kwak MS, Kim HS, Lee B, Kim YH, Son M and Shin J-S (2020) Immunological Significance of HMGB1 Post-Translational Modification and Redox Biology. Front. Immunol. 11:1189. doi: 10.3389/fimmu.2020.01189
Received: 16 March 2020; Accepted: 13 May 2020;
Published: 10 June 2020.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Masahiro Nishibori, Okayama University Graduate School of Medicine Dentistry and Pharmaceutical Sciences, JapanCopyright © 2020 Kwak, Kim, Lee, Kim, Son and Shin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeon-Soo Shin, anNzaGluNjIwM0B5dWhzLmFj
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.