 Livia Sophie Hofer1
Livia Sophie Hofer1 Melanie Ramberger1,2
Melanie Ramberger1,2 Viktoria Gredler1
Viktoria Gredler1 Anna Sophie Pescoller1
Anna Sophie Pescoller1 Kevin Rostásy3
Kevin Rostásy3 Mireia Sospedra4
Mireia Sospedra4 Harald Hegen1
Harald Hegen1 Thomas Berger5
Thomas Berger5 Andreas Lutterotti4
Andreas Lutterotti4 Markus Reindl1*
Markus Reindl1*- 1Clinical Department of Neurology, Medical University of Innsbruck, Innsbruck, Austria
- 2Oxford Autoimmune Neurology Group, Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, United Kingdom
- 3Paediatric Neurology, Children's Hospital Datteln, Witten/Herdecke University, Datteln, Germany
- 4Department of Neuroimmunology, University of Zurich, Zurich, Switzerland
- 5Department of Neurology, Medical University of Vienna, Vienna, Austria
Autoantibodies against aquaporin-4 (AQP4-Ab) and myelin oligodendrocyte glycoprotein (MOG-Ab) are associated with rare central nervous system inflammatory demyelinating diseases like neuromyelitis optica spectrum disorders (NMOSD). Previous studies have shown that not only antibodies, but also autoreactive T-cell responses against AQP4 are present in NMOSD. However, no study has yet analyzed the presence of MOG reactive T-cells in patients with MOG antibodies. Therefore, we compared AQP4 and MOG specific peripheral T-cell response in individuals with AQP4-Ab (n = 8), MOG-Ab (n = 10), multiple sclerosis (MS, n = 8), and healthy controls (HC, n = 14). Peripheral blood mononuclear cell cultures were stimulated with eight AQP4 and nine MOG peptides selected from previous studies and a tetanus toxoid peptide mix as a positive control. Antigen-specific T-cell responses were assessed using the carboxyfluorescein diacetate succinimidyl ester proliferation assay and the detection of granulocyte macrophage colony-stimulating factor (GM-CSF), interferon (IFN)-ɤ and interleukin (IL)-4, IL-6, and IL-17A in cell culture supernatants. Additionally, human leukocyte antigen (HLA)-DQ and HLA-DR genotyping of all participants was performed. We classified a T-cell response as positive if proliferation (measured by a cell division index ≥3) was confirmed by the secretion of at least one cytokine. Reactivity against AQP4 peptides was observed in many groups, but the T-cell response against AQP4 p156-170 was present only in patients with AQP4-Ab (4/8, 50%) and absent in patients with MOG-Ab, MS and HC (corrected p = 0.02). This AQP4 p156-170 peptide specific T-cell response was significantly increased in participants with AQP4-Ab compared to those without [Odds ratio (OR) = 59.00, 95% confidence interval-CI 2.70–1,290.86]. Moreover, T-cell responses against at least one AQP4 peptide were also more frequent in participants with AQP4-Ab (OR = 11.45, 95% CI 1.24–106.05). We did not observe any significant differences for the other AQP4 peptides or any MOG peptide. AQP4-Ab were associated with HLA DQB1*02 (OR = 5.71, 95% CI 1.09–30.07), DRB1*01 (OR = 9.33, 95% CI 1.50–58.02) and DRB1*03 (OR = 6.75, 95% CI = 1.19–38.41). Furthermore, HLA DRB1*01 was also associated with the presence of AQP4 p156-170 reactive T-cells (OR = 31.67, 95% CI 1.30–772.98). To summarize, our findings suggest a role of AQP4-specific, but not MOG-specific T-cells, in NMOSD.
Introduction
Autoantibodies targeting the aquaporin-4 (AQP4) water channel protein and the myelin oligodendrocyte glycoprotein (MOG) are associated with a broad spectrum of human central nervous system (CNS) demyelinating diseases (1, 2). While AQP4-specific antibodies target the AQP4 water channel protein expressed on astrocyte end-feet processes causing a severe astrocytopathy called neuromyelitis optica (NMO) (3, 4), MOG-specific antibodies target the extracellular N-terminal immunoglobulin variable (IgV)-domain of MOG expressed on myelin-forming oligodendrocytes (2, 5, 6). Autoantibodies against AQP4 (AQP4-Ab) have emerged as highly sensitive and specific biomarker for the diagnosis of NMO (3). However, not all patients presenting with clinical features suggestive of an NMO-disease phenotype are positive for AQP4-Ab (7), and a significant proportion of those seronegative patients harbor antibodies to MOG. This created a diagnostic uncertainty reflected in the pathogenetically undefined category of NMO spectrum disorders (NMOSD) proposed in 2015 (1, 2, 8).
Several lines of evidence suggest that autoreactive CD4+ T lymphocytes are key players in the pathogenesis of Ab-associated demyelinating CNS diseases. First, passive transfer models using AQP4-specific human IgG are not considered pathogenic without T-cell induced disruption of the blood brain barrier (BBB) (9–11). The exact relevance of T-cell independent pathogenicity of a high affinity rodent monoclonal AQP4-Ab (12) remains to be determined, as serum AQP4-Ab in human NMOSD patients are polyclonal, with a wide range of affinities and often much lower antibody titers (9, 10, 13–15). The serum concentration of AQP4-Ab is many times higher than in the cerebrospinal fluid (CSF) (13, 16–18), and peripheral B cells have the capacity to produce AQP4-Ab in vitro (19, 20). Thus, it is supposed that these antibodies are produced outside the CNS and that T effector cells might initiate CNS inflammation leading to BBB disruption and entry of antibodies (6, 9–11, 21–23). Local activation of CD4+ T-cells in the CNS is indispensable for providing an inflammatory microenvironment that also enables the initiation of CNS inflammation orchestrating BBB breakdown, lesion location and formation and thus facilitates Ab-mediated disease propagation (6, 11, 23). Second, AQP4-Ab and MOG-Ab are class-switched complement-fixing antibodies depending on T-cell help to be generated emphasizing the pivotal role of antigen-specific T-cell responses. Finally, there is ample evidence that activated T-cells are enriched at lesion sites (11, 24, 25) and that the pathogenic effectors are CD4+ T-cells of either T helper (Th)-1 lineage producing pro-inflammatory interferon (IFN)-ɤ or of Th17 lineage producing pro-inflammatory interleukin (IL)-17A (26, 27). Moreover, NMOSD patients also display a higher proportion of Th17 cells or cytokines like IL-6 (28–34).
While the high diagnostic value of AQP4-Ab as hallmark serologic marker in NMOSD has been shown and AQP4-specific T-cells have been examined in NMOSD patients (31, 35–38), the role of MOG-Ab or MOG-specific T-cells is less clear. Since MOG-Ab can be found in up to 50% of AQP4-Ab seronegative NMOSD patients, it is possible that MOG-specific T-cells could play a role in NMOSD development. So far there is no published information about MOG-specific T-cells in NMOSD and related conditions. Until now all studies focused on MOG-specific T-cell responses from MS patients (39–41) or in experimental autoimmune encephalomyelitis (42–44).
Here, we aimed to analyze the T-cell reactivity in response to selected eight AQP4 and nine MOG peptides and their possible restriction to a particular human leukocyte antigen (HLA)-DQ and HLA-DR genotype, and to examine the functional phenotype of autoreactive CD4+ T-cells in patients with AQP4-Ab or MOG-Ab.
Materials and Methods
Patients and Control Subjects
Eight NMOSD patients with AQP4-Ab, 10 patients with MOG-Ab, 8 patients with MS and 14 healthy controls (HC) were included in this study. NMOSD and MS was diagnosed according to recently published criteria (1, 45, 46). Within the MOG-Ab positive group, one patient also fulfilled the 2015 diagnostic criteria for NMOSD (1), 8 of the other 9 patients had related clinical presentations (three bilateral and one unilateral monophasic optic neuritis, one recurrent optic neuritis, one monophasic and one recurrent myelitis, one acute demyelinating encephalomyelitis with recurrent optic neuritis and one recurrent demyelinating disease) and one patient fulfilled the diagnostic criteria for MS.
Demographic and clinical data of all participants are shown in Table 1.
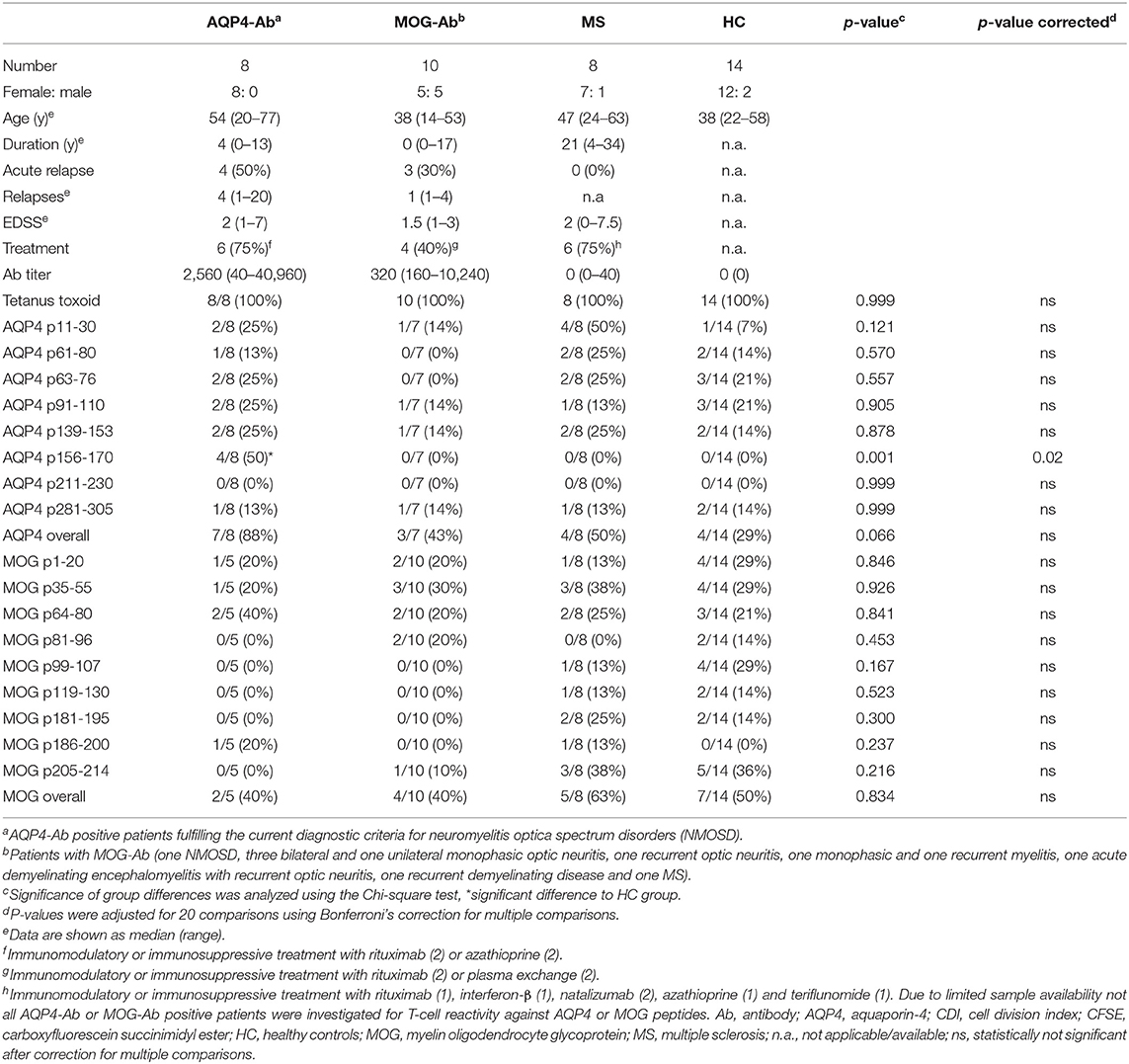
Table 1. CD4+ T-cell proliferation with a cell division index ≥ 3 of T-cell cultures after stimulation with AQP4 or MOG peptides.
All samples were collected between 2008 and 2018 at the Clinical Department of Neurology Innsbruck and at the Section for Neuroimmunology and MS Research (NIMS), Department of Neurology, University Hospital Zurich and stored at the Neurological Research Laboratory Innsbruck.
The study was approved by the local Ethics Committee of Medical University of Innsbruck, Austria (study number AN3041) and University of Zürich, Switzerland (KEK ZH 2013-0001) and all patients or their caregivers and controls gave written informed consent.
AQP4-Ab and MOG-Ab Detection Assays
Serum AQP4-Ab were analyzed using live cell-based immunofluorescence assays as described previously (47). Serum MOG-Ab were analyzed using recombinant live cell-based immunofluorescence assays with HEK293A cells transfected with full-length MOG (human MOG α-1 EGFP fusion protein) as described previously (47). Sera were tested at dilutions of 1:20 and 1:40 and MOG-Ab positivity was titrated with serial dilutions with a threshold of 1:160 to define MOG-Ab positivity. Isolated IgM reactivity was excluded using IgG constant chain (Fc)-specific secondary antibodies (48, 49).
T-Cell Epitope Mapping Using the CFSE Proliferation Assay
Peripheral blood mononuclear cells (PBMC) were isolated by density gradient centrifugation over Histopaque 1077 (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer's instructions and aliquots at a concentration of 1–2 × 107 cells/ml freezing medium (50% Roswell Park Memorial Institute (RPMI) 1,640 medium, 40% fetal calf serum (FCS), 10% dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO, USA) were cryopreserved in liquid nitrogen until use. After thawing adopting a warm and slow processing method as recommended previously (50) to ensure high viability, isolated PBMC of patients and controls at a concentration of 2 × 107 cells/ml were stained with 0.4 μM carboxyfluorescein diacetate succinimidyl ester (CFSE; Life Technologies, Carlsbad, CA, USA) following the manufacturer's instructions and cells were cultivated in X-Vivo 15 growth medium (Lonza, Basel, Switzerland). For the expansion of antigen-specific T-cells, PBMC were exposed to 20 μg/ml of selected AQP4 and MOG peptides. Eight AQP4 and nine MOG peptides were selected based on their encephalitogenicity in animals and/or their immunodominance in humans, in particular AQP4-specific T-cell responses of PBMC from NMOSD patients and MOG-specific T-cell responses of PBMC from MS patients (Figure 1, Table 2; (21, 22, 31, 35–41, 43, 51–56). Peptide lengths varied from 9 to 25 amino acids (aa) and were synthesized by Peptides & Elephants (Potsdam, Germany). As positive control, 5 μg/ml tetanus toxoid pool (TTX; Peptides&Elephants, Potsdam, Germany) and as vehicle control, DMSO (Sigma-Aldrich, St. Louis, MO, USA) was used. Since DMSO was used to dissolve the peptides at a maximum of 35% (v/v) in Dulbecco's phosphate-buffered saline (DPBS; Sigma-Aldrich, St. Louis, MO, USA), a 35% DMSO/65% DPBS mix was added at an equal volume to match the volume of added peptide solution. Moreover, a second positive control with the strong mitogen phytohaemagglutinin (PHA; Sigma-Aldrich, St. Louis, MO, USA) was included. Wells from PHA-stimulated cells were evaluated on day 4 for color change of the medium (from red to yellow) indicative of a high metabolic and proliferative activity and by dilution of the CFSE staining using flow cytometry. Cells were seeded at a density of 2 x 105 cells/200 μl in tissue culture test plates 96 U (TPP, Trasadingen, Switzerland), each six wells per condition. After eight days, cells were re-stimulated with half the amount of respective peptides (10 μg/ml per peptide) or vehicle and positive control and 100 μl of the supernatant were replaced with fresh medium containing 20 U/ml IL-2 (Peprotech, Hamburg, Germany), and supernatants were stored at −80°C for later cytokine analyses. After a further 3 days, PBMC were harvested and the proliferation of CD4+ T-cells in response to single peptides was analyzed via the dilution of the CFSE staining using flow cytometry. For flow cytometry analysis, PBMC were stained with Peridinin-Chlorophyll-Protein (PerCp)-anti-CD3 (SK7), phycoerythrin (PE)-anti-CD8 (SK1) and allophycocyanin (APC)-anti-CD4 (SK3) antibodies (all BD Bioscience, Franklin Lakes, NJ, USA) and analyzed on an Accuri C6 flow cytometer (BD Bioscience, Franklin Lakes, NJ, USA). The gating strategy is shown in Figure 2. For analysis of a positive T-cell proliferative response, the cell division index (CDI) was calculated as follows, whereby a CDI ≥ 3 was considered as significant proliferation:
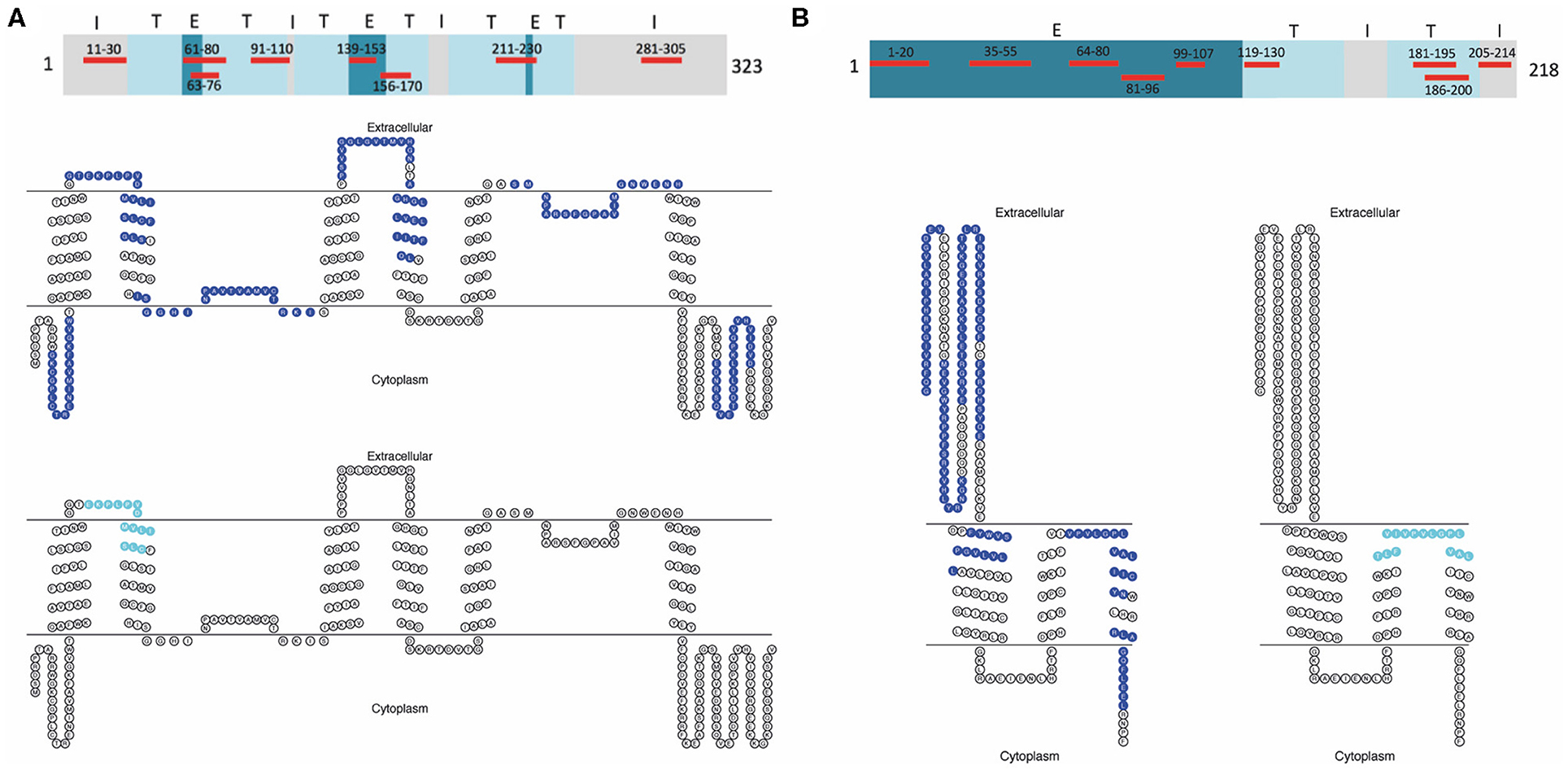
Figure 1. AQP4 and MOG peptides used for CD4+ T-cell stimulation. (A) Topological map of the human AQP4 protein (323 aa). Eight selected AQP4 peptides corresponding to intracellular (I), extracellular (E), and transmembrane (T) sequences of human AQP4 used for CD4+ T-cell stimulation are highlighted in red. Below, seven AQP4 determinants (blue) and further down one overlapping AQP4 determinant (aqua) are represented within a human AQP4 topological diagram using TOPO2 transmembrane protein display software (http://www.sacs.ucsf.edu/TOPO2/). (B) Topological map of the human MOG protein (218 aa). Nine selected MOG peptides corresponding to intracellular (I), extracellular (E), and transmembrane (T) sequences of human MOG used for CD4+ T-cell stimulation are highlighted in red. Below on the left, eight MOG determinants (blue) and on the right, one overlapping MOG determinant (aqua) are represented within a human MOG topological diagram using TOPO2 transmembrane protein display software (http://www.sacs.ucsf.edu/TOPO2/). AQP4, aquaporin-4; MOG, myelin oligodendrocyte glycoprotein.
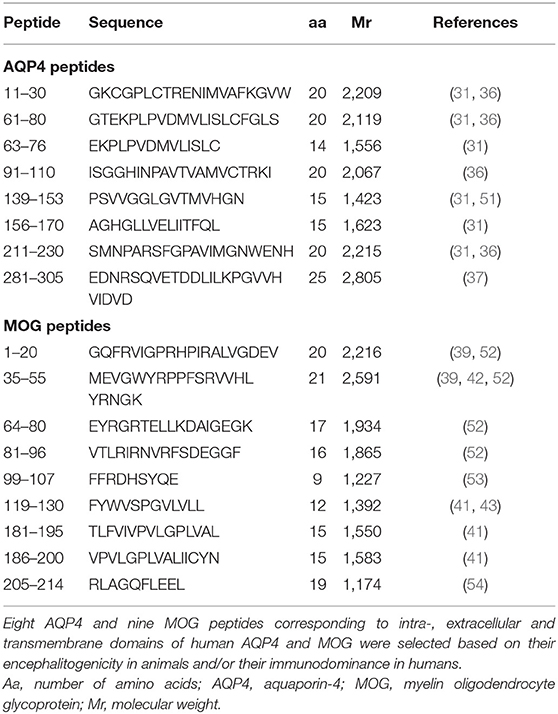
Table 2. AQP4 and MOG peptides used for CD4+ T-cell stimulation.
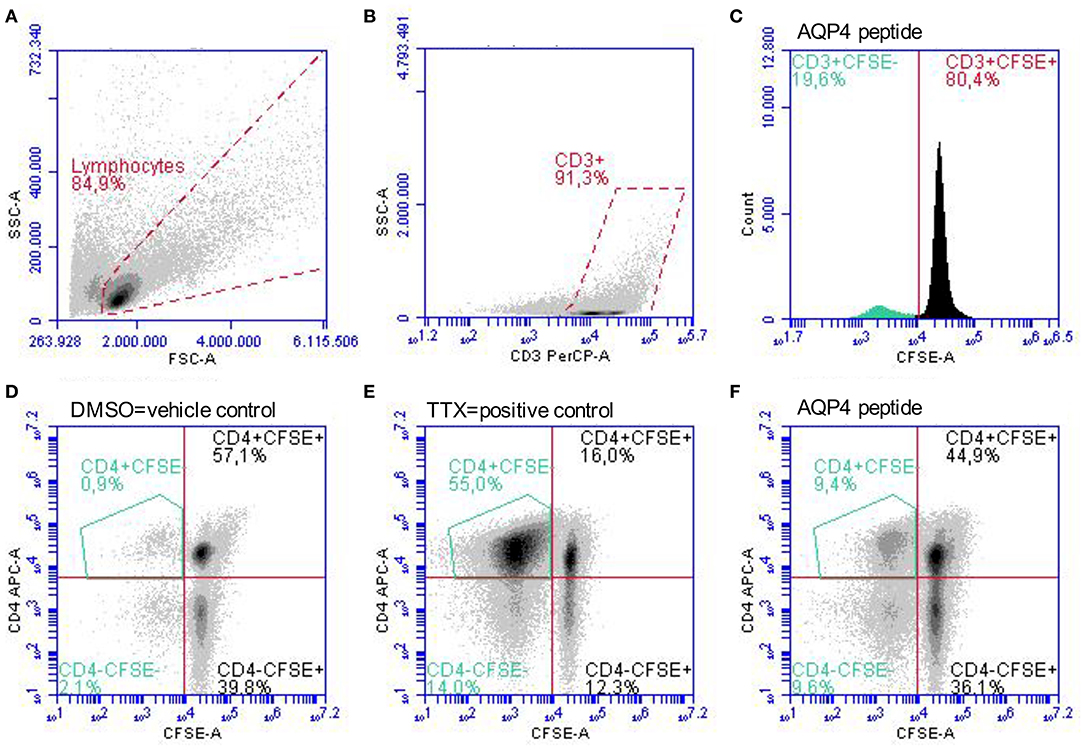
Figure 2. Gating strategy for the identification of proliferated CD4+CFSE− T-cells. PBMC stimulated with single AQP4 and MOG peptides or the vehicle control DMSO and the positive control TTX were analyzed after 11 days in culture. (A) Gating of lymphocytes according to empirical values of size (FSC) and granularity (SSC) followed by (B) gating of CD3+ T-cells is shown. (C) Dilution of the CFSE staining due to proliferating of CD4+ T-cells in response to an AQP4 peptide. (D–F) Gating of proliferated CD4+CFSE− T-cells. Representative scatter plots of proliferated CD4+CFSE− T-cells in response to the vehicle control DMSO (D), to the positive control TTX (E), and to an AQP4 peptide (F) are depicted. APC, allophycocyanin; AQP4, aquaporin-4; CFSE, carboxyfluorescein succinimidyl ester; DMSO, dimethyl sulfoxide; FSC, forward scatter; MOG, myelin oligodendrocyte glycoprotein; PBMC, peripheral blood mononuclear cells; PerCP, peridinin-chlorophyll-protein; SSC, side scatter; TTX, tetanus toxoid.
Evaluation of Cytokine Secretion Using ELISA
For the evaluation of cytokine secretion of autoreactive T-cells in response to either AQP4 or MOG peptides, commercial ELISA kits specific for human granulocyte-macrophage-colony-stimulating factor (GM-CSF) and IFN-ɤ (BioLegend; San Diego, USA) and for human IL-4, IL-6, and IL-17A (Thermo Fischer Scientific, Waltham, MA, USA) were purchased and cell culture supernatants collected after 11 days (72 h after re-stimulation) were analyzed following the manufacturer's instructions. The stimulation index (SI) was calculated as follows, whereby a SI ≥ 3 was considered as significant secretion:
HLA Typing by Sequence-Specific Primers (PCR-SSP-HLA Typing)
Since binding of peptides to major histocompatibility complex (MHC) molecules of antigen presenting cells (APC) is an important prerequisite for T-cell responsiveness, an HLA-DQ und -DR type determination was performed using polymerase chain reaction with sequence-specific primer (PCR-SSP) technique according to the manufacturer's instructions (Olerup SSP, Stockholm, Sweden).
Statistical Analysis
The primary hypothesis of this study was that T-cell responses are associated with auto-antibody responses, i.e., AQP4-specific T-cells are increased in participants with AQP4-Ab and MOG-specific T-cells are increased in participants with MOG-Ab. This hypothesis was tested for nominal data (i.e., proliferation with a CDI ≥ 3) using the Chi-square test (with Fisher's exact test and Bonferroni's correction for multiple comparison for subgroups). Statistical significance was defined as two-sided p < 0.05 after Bonferroni's correction for multiple comparisons (i.e., the number of different peptides used). According to recently published recommendations to avoid the overuse and misinterpretation of p-values, the analysis of all secondary and other endpoints focused on estimates (common odds ratio, OR) and 95% confidence intervals (CI) (57). Statistical analyses were performed using IBM SPSS software (IBM SPSS Statistics; Version 24.0. Armonk, NY: IBM Corp.), GraphPad Prism 8 (GraphPad Software, La Jolla, CA) and OpenMetaAnalyst (http://www.cebm.brown.edu/openmeta/).
Results
AQP4-Ab Are Associated With AQP4-Specific CD4+ T-Cell Reactivity
We adopted a cell division analysis procedure based on the quantitative dilution of the fluorescent dye CFSE to investigate the CD4+ T-cell autoreactivity of individuals with AQP4-Ab, MOG-Ab, MS and HC against selected AQP4 peptides. All participants showed a positive CD4+ T-cell proliferative response with a CDI ≥ 3 to the positive control TTX (Figures 3, 4A and Table 1). T-cell proliferation with a CDI ≥ 3 for at least one AQP4 peptide was observed in the majority of patients with AQP4-Ab (88%), 43% of patients with MOG-Ab, 50% of MS patients and 29% of HC. These proliferative T-cell responses against at least one AQP4 peptide were more frequent in participants with AQP4-Ab as compared to HC (OR = 17.50, 95% CI 1.60–191.89) or all AQP4-Ab negative participants (OR = 11.45, 95% CI 1.24–106.05; Figure 5). Amongst the different AQP4 peptides, a statistically significant response was only seen for AQP4 p156-170: T-cell proliferation with a CDI ≥ 3 was observed in 4/8 (50%) of patients with AQP4-Ab but in none of the other groups (corrected p-value 0.02). Proliferative T-cell responses against AQP4 p156-170 were significantly more frequent in participants with AQP4-Ab as compared to HC (OR = 29.00, 95% CI 1.30–648.44) or all AQP4-Ab negative participants (OR = 59.00, 95% CI 2.70–1,290.86; Figure 5).
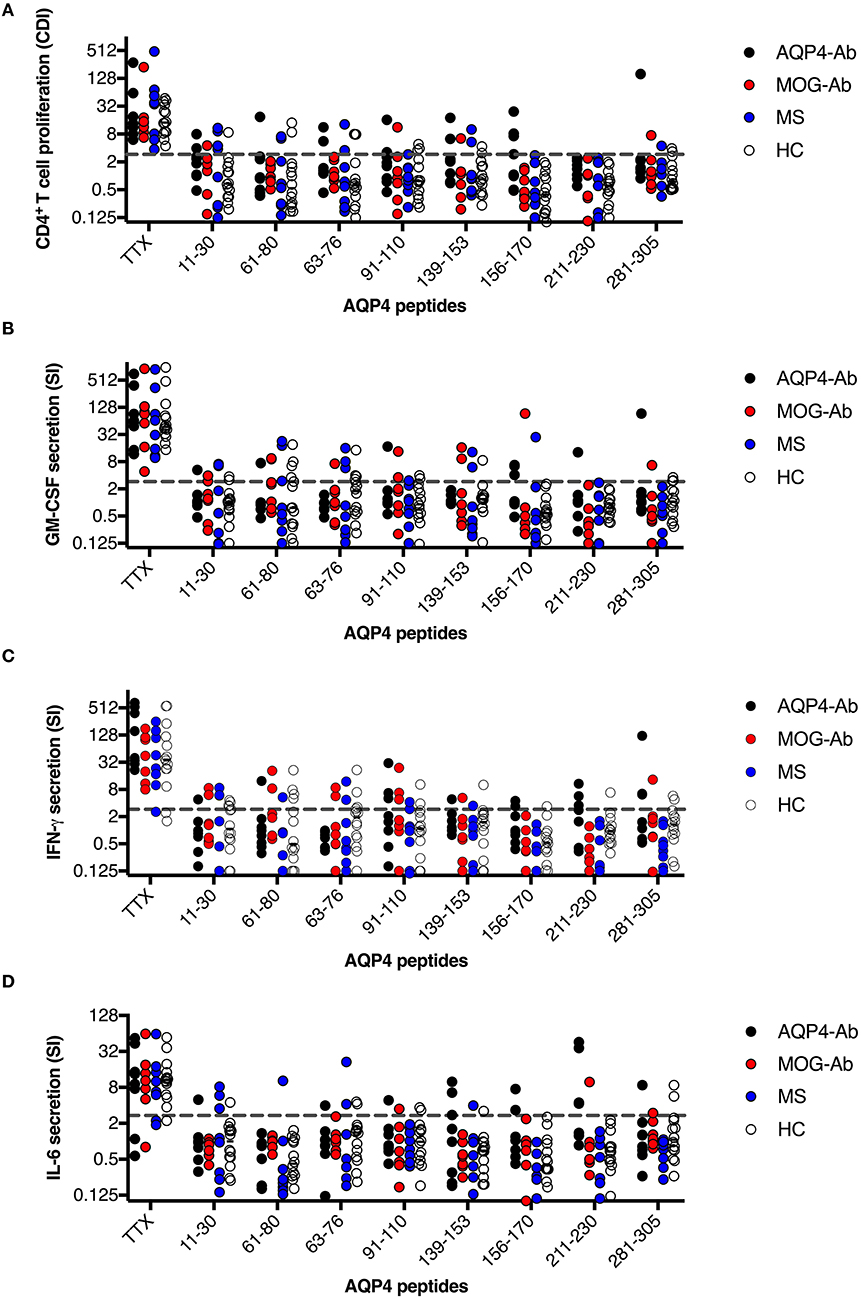
Figure 3. CD4+ T-cell reactivity to AQP4 peptides in participants with AQP4-Ab (n = 8), MOG-Ab (n = 7), MS (n = 8) and HC (n = 14). (A) CD4+ T-cell proliferation after challenging with respective AQP4 peptides and the positive control TTX. The cut-off value of a CDI ≥ 3 is indicated by a gray dashed line. Secretion of GM-CSF (B), IFN-γ (C) and IL-6 (D) after challenging with respective AQP4 peptides and the positive control TTX. The cut-off SI ≥ 3 is indicated by a gray dashed line. AQP4-Ab, aquaporin-4 antibody positive; CDI, cell division index; GM-CSF, granulocyte-macrophage-colony-stimulating factor; HC, healthy controls; IFN, interferon; IL, interleukin; MOG-Ab, myelin oligodendrocyte glycoprotein antibody positive; MS, multiple sclerosis; SI, stimulation index; TTX, tetanus toxoid.
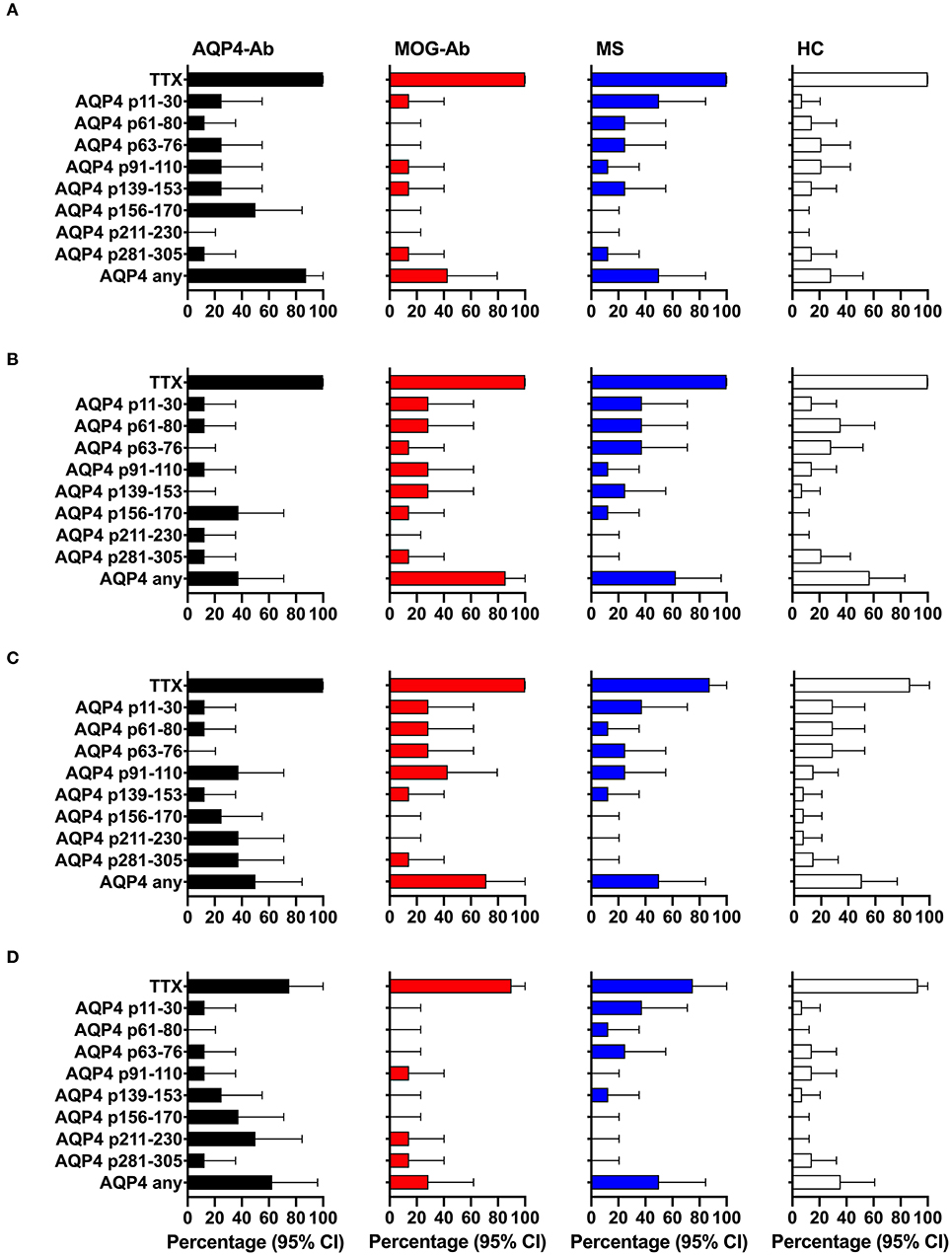
Figure 4. CD4+ T-cell reactivity to AQP4 peptides in participants with AQP4-Ab (n = 8), MOG-Ab (n = 7), MS (n = 8) and HC (n = 14). (A) percentage of participants with positive CD4+ T-cell proliferation (CDI ≥ 3) after challenging with respective AQP4 peptides and the positive control TTX. Percentage of participants with positive secretion (SI ≥ 3) of GM-CSF (B), IFN-γ (C) and IL-6 (D) after challenging with respective AQP4 peptides and the positive control TTX. The 95% confidence intervals are indicated by the error bars. AQP4-Ab, aquaporin-4 antibody positive; CDI, cell division index; GM-CSF, granulocyte-macrophage-colony-stimulating factor; HC, healthy controls; IFN, interferon; IL, interleukin; MOG-Ab, myelin oligodendrocyte glycoprotein antibody positive; MS, multiple sclerosis; SI, stimulation index; TTX, tetanus toxoid.
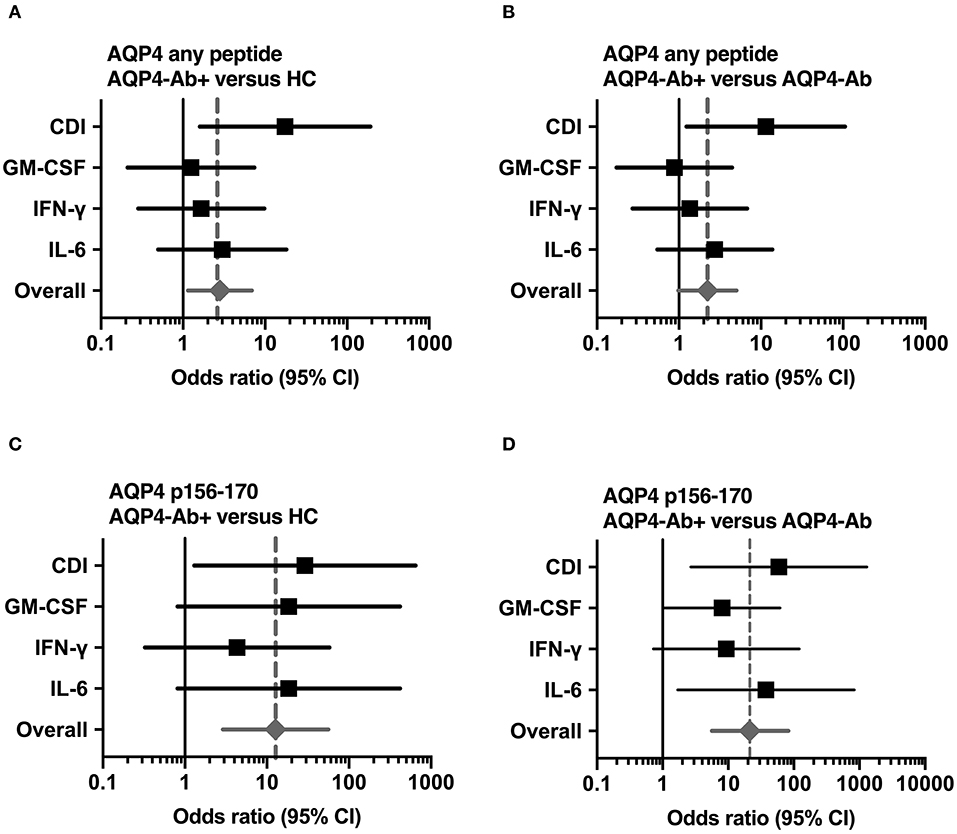
Figure 5. Significant CD4+ T-cell reactivity to AQP4 peptides. Forest plot displaying Mantel-Haenszel common odds ratio estimates (symbols) with asymptomatic 95% CI (horizontal lines) for response to AQP4 peptides. (A) Reactivity against at least one AQP4 peptide, comparison of patients with AQP4-Ab (n = 8) vs. HC (n = 14). (B) Reactivity against at least one AQP4 peptide, comparison of patients with AQP4-Ab (n = 8) vs. all AQP4-Ab negative participants (n = 29). (C) Reactivity against AQP4 p156-170, comparison of patients with AQP4-Ab (n = 8) vs. HC (n = 14). (D) Reactivity against AQP4 p156-170, comparison of patients with AQP4-Ab (n = 8) vs. all AQP4-Ab negative participants (n = 29). AQP4-Ab+, aquaporin-4 antibody positive; AQP4-Ab-, aquaporin-4 antibody negative, CDI, cell division index; CI, confidence interval; GM-CSF, granulocyte-macrophage-colony-stimulating factor; HC, healthy controls; IFN, interferon; IL, interleukin.
The clinical presentation of the 4 patients who had increased T-cell reactivity to AQP4 p156-170 were not substantially different from the other AQP4-Ab NMOSD patients. All four cases (all female, age 20–53 years, disease duration 0.4–13.2 years) had a relapsing NMOSD disease course (2–8 relapses), three of them were treated with rituximab and the fourth patient was under high-dose corticosteroids before the initiation of rituximab treatment. Two of the four patients had a relapse at the time of blood sampling, one of them before the initiation of rituximab treatment.
The functional phenotype of proliferating T-cells was characterized by investigating the secretion of the cytokines IL-4, IL-6, IL-17A, GM-CSF, and IFN-ɤ into cell culture supernatants of the CFSE proliferation assay using ELISA. Cytokine concentrations of IL-4 and IL-17A after stimulation with AQP4 peptides, but not after stimulation with TTX, were below the detection limit of ELISA. Quantitative and qualitative values for GM-CSF, IFN-ɤ, or IL-6 levels are shown in Figures 3, 4B–D. A comprehensive analysis of all proliferative and cytokine responses to any AQP4 peptide and p156-170 is shown in Figure 5. From this figure it is evident that overall these responses are increased in AQP4-Ab positive patients as compared to HC or all AQP4-Ab negative participants.
No Association of MOG-Ab With MOG-Specific CD4+ T-Cell Reactivity
In a next step, we analyzed the CD4+ T-cell autoreactivity of patients with AQP4-Ab, MOG-Ab, MS and HC against selected MOG peptides. All participants showed a positive CD4+ T-cell proliferative response with a CDI ≥ 3 to the positive control TTX. We observed no statistically significant differences after challenge with the different MOG peptides between groups (Figures 6, 7A and Table 1). T-cell proliferation with a CDI ≥ 3 for at least one MOG peptide was observed in 40% of AQP4-Ab positive patients, 40% of MOG-Ab positive patients, 63% of MS patients and 50% of HC.
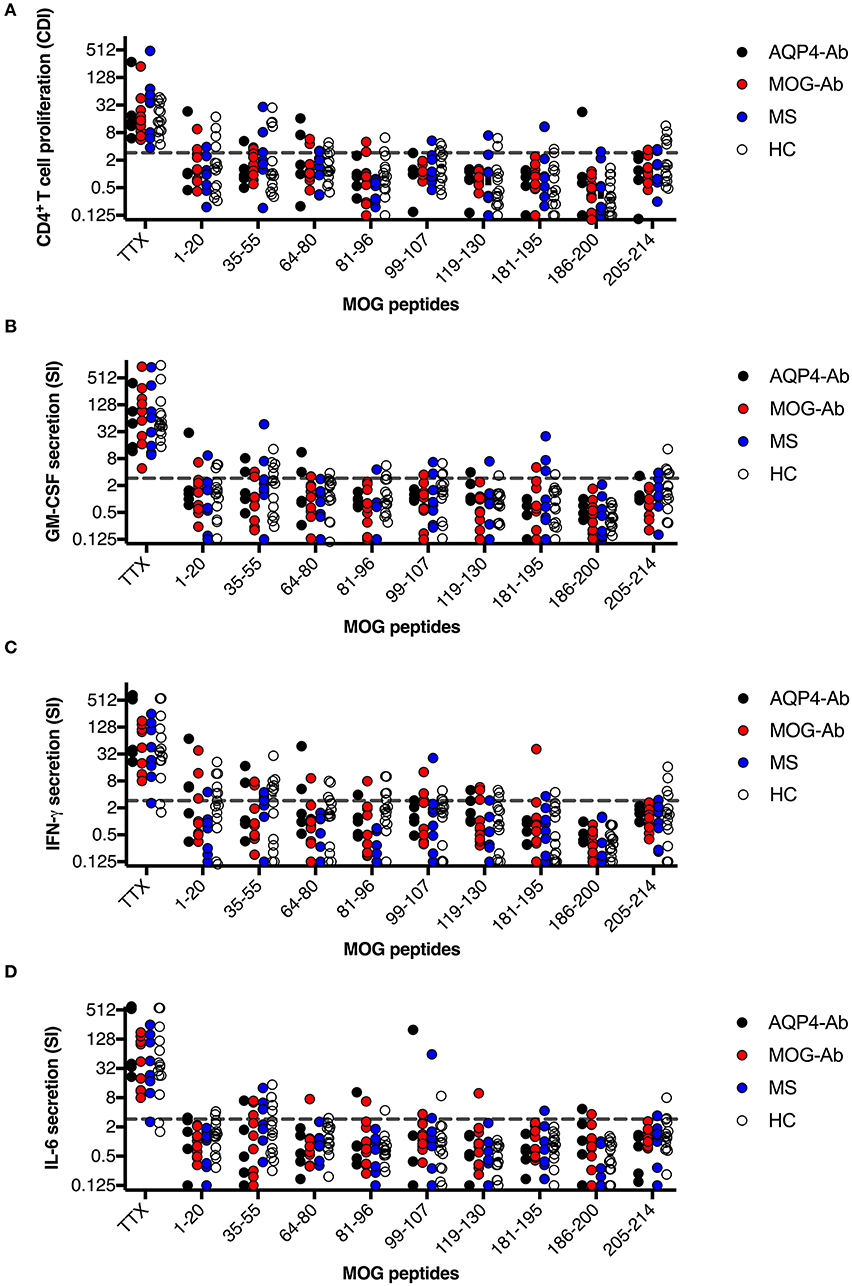
Figure 6. CD4+ T-cell reactivity to MOG peptides in participants with AQP4-Ab (n = 5), MOG-Ab (n = 10), MS (n = 8) and HC (n = 14). (A) CD4+ T-cell proliferation after challenging with respective MOG peptides and the positive control TTX. The cut-off value of a CDI ≥ 3 is indicated by a gray dashed line. Secretion of GM-CSF (B), IFN-γ (C) and IL-6 (D) after challenging with respective MOG peptides and the positive control TTX. The cut-off value of a SI ≥ 3 is indicated by a gray dashed line. AQP4-Ab, aquaporin-4 antibody positive; CDI, cell division index; GM-CSF, granulocyte-macrophage-colony-stimulating factor; HC, healthy controls; IFN, interferon; IL, interleukin; MOG-Ab, myelin oligodendrocyte glycoprotein antibody positive; MS, multiple sclerosis; SI, stimulation index; TTX, tetanus toxoid.
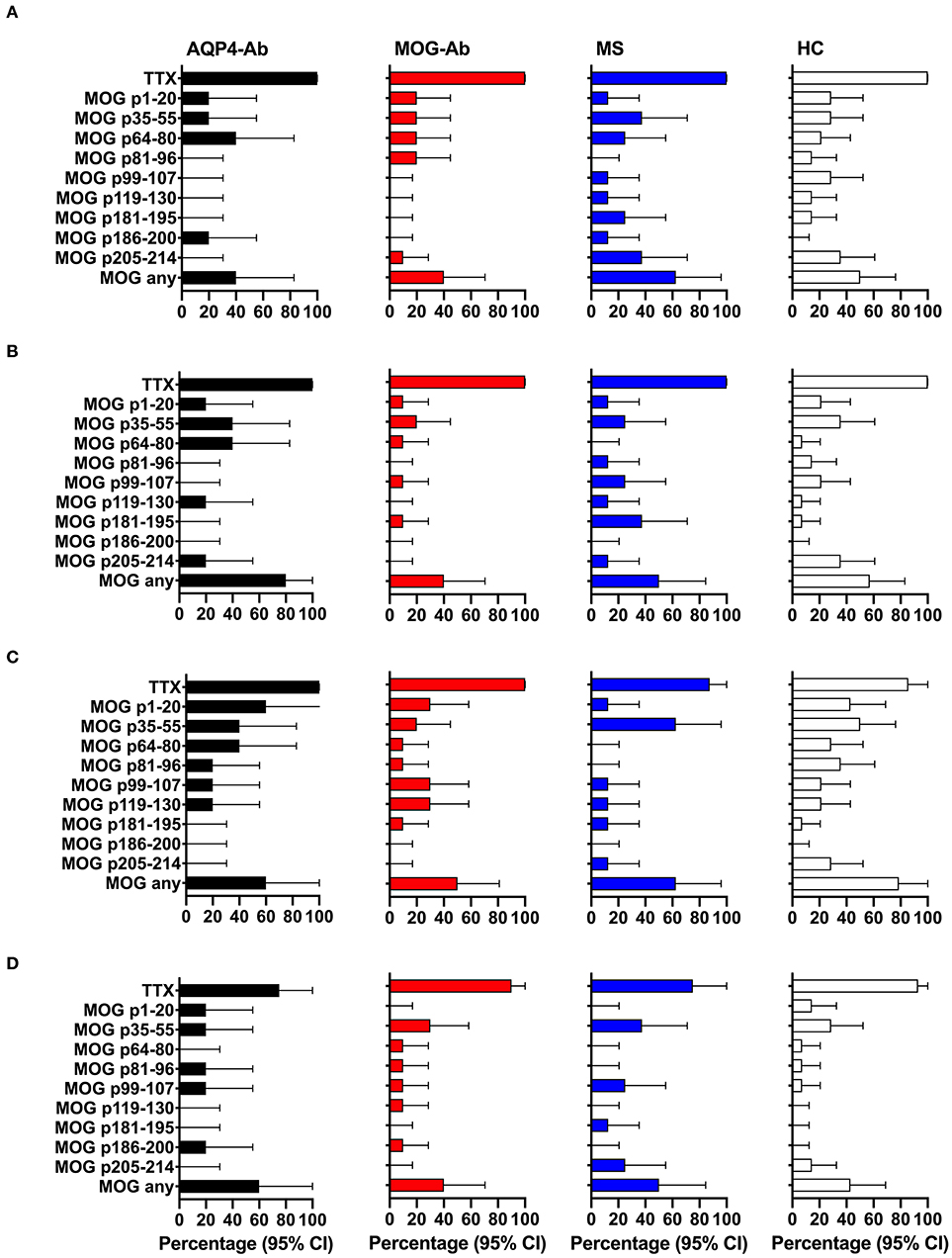
Figure 7. CD4+ T-cell reactivity to MOG peptides in participants with AQP4-Ab (n = 5), MOG-Ab (n = 10), MS (n = 8) and HC (n = 14). (A) Percentage of participants with positive CD4+ T-cell proliferation (CDI ≥ 3) after challenging with respective MOG peptides and the positive control TTX. Percentage of participants with positive secretion (SI ≥ 3) of GM-CSF (B), IFN-γ (C) and IL-6 (D) after challenging with respective MOG peptides and the positive control TTX. The 95% confidence intervals are indicated by the error bars. AQP4-Ab, aquaporin-4 antibody positive; CDI, cell division index; GM-CSF, granulocyte-macrophage-colony-stimulating factor; HC, healthy controls; IFN, interferon; IL, interleukin; MOG-Ab, myelin oligodendrocyte glycoprotein antibody positive; MS, multiple sclerosis; SI, stimulation index; TTX, tetanus toxoid.
The functional phenotype of proliferating T-cells was characterized by investigating the secretion of the cytokines IL-4, IL-6, IL-17A, GM-CSF, and IFN-ɤ into cell culture supernatants of the CFSE proliferation assay using ELISA. Cytokine concentrations of IL-4 and IL-17A after stimulation with AQP4 peptides, but not after stimulation with TTX, were below the detection limit of ELISA. Quantitative and qualitative values for GM-CSF, IFN-ɤ, or IL-6 levels are shown in Figures 6, 7B–D.
HLA Association of AQP4-Specific T-Cell Reactivity
Figure 8 shows the overall frequencies of HLA-DQB1, HLA-DRB1 and HLA-DRB3 alleles in participants with AQP4-Ab, MOG-Ab, MS, and healthy controls. The following HLA genotypes were overrepresented in participants with AQP4-Ab (n = 8) compared to those without AQP4-Ab (n = 31): DQB1*02 (OR = 5.71, 95% CI 1.09–30.07), DRB1*01 (OR = 9.33, 95% CI 1.50–58.02) and DRB1*03 (OR = 6.75, 95% CI 1.19–38.41).
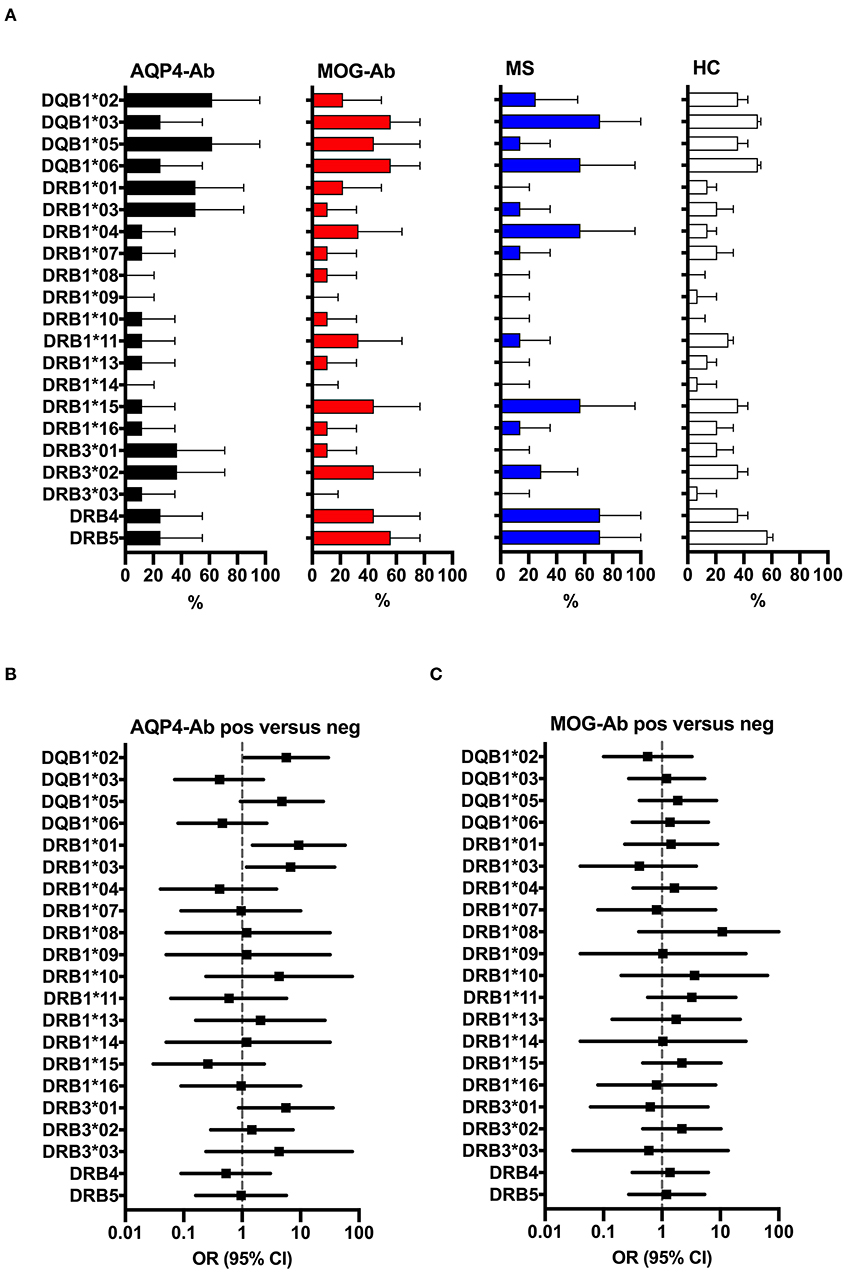
Figure 8. (A) HLA allele frequency (in% with 95% confidence intervals indicated by the error bars) of participants with AQP4-Ab (n = 8), MOG-Ab (n = 9), MS (n = 8) and HC (n = 14). (B) Forest plot displaying Mantel-Haenszel common odds ratio estimates (symbols) with asymptomatic 95% CI (horizontal lines) for the different HLA alleles in participants with AQP4-Ab (n = 8) vs. all AQP4-Ab negative participants (n = 31). HLA alleles DQB1*02, DRB1*01 and DRB1*03 were overrepresented in participants with AQP4-Ab. (C) Forest plot displaying Mantel-Haenszel common odds ratio estimates (symbols) with asymptomatic 95% CI (horizontal lines) for the different HLA alleles in participants with MOG-Ab (n = 9) vs. all MOG-Ab negative participants (n = 30). AQP4-Ab, aquaporin-4 antibody positive; HC, healthy controls; HLA, human leucocyte antigen; MOG-Ab, myelin oligodendrocyte glycoprotein antibody positive; MS, multiple sclerosis.
The four study participants reactive with AQP4 p156-170 peptide 156–170 had the following HLA genotypes: Nr. 1 (DQB1*02, DQB1*05, DRB1*01, DRB1*03, DRB3*01), Nr. 2 (DQB1*03, DQB1*05, DRB1*10, DRB1*11, DRB3*02), Nr. 3 (DQB1*02, DQB1*06, DRB1*07, DRB1*15, DRB4, DRB5), and Nr. 4 (DQB1*03, DQB1*05, DRB1*01, DRB1*04, DRB4). However, only HLA DRB1*01 was associated with the presence of AQP4 p156-170 reactive T-cells (OR = 31.67, 95% CI 1.30–772.98).
Discussion
In this study we analyzed peripheral blood T-cell responses to AQP4 and MOG peptides in individuals with AQP4-Ab, MOG-Ab, MS, and HC. We identified significantly increased AQP4-specific CD4+ T-cell reactivity to AQP4 peptide 156–170 in 4 of 8 AQP4-Ab positive NMOSD patients, but in none of the other groups. In contrast, we could not detect any significant disease-specific T-cell response to other AQP4 or MOG peptides. AQP4 peptide 156–170 has already been described as a T-cell epitope in NMOSD patients (31) and is also one of the most important B-cell epitopes recognized by AQP4-Ab (14, 15, 58, 59). Three other immunodominant T-cell epitopes/peptides of the AQP4 protein have been described by Varrin-Doyer et al. (31), which were also included in our study. However, we and other authors could not confirm immunodominance for these particular determinants (38, 60). The possible reasons for this discrepancy could be explained by the different methods used (CFSE, 3H-thymidine incorporation proliferation assays, cytokine secretion) and the different genetic background, i.e., HLA associations of the study populations. Several studies have identified over-representation of HLA-DPB1*0501, HLA-DRB1*0301, or HLA-DRB3 in NMO patients (31, 61–63). However, only DRB1*0301 but not any of the other HLA alleles was overrepresented in our study population, indicating differences in the genetic background. In contrast, we found an overrepresentation of HLA-DQB1*02 and HLA-DRB1*01 in our study.
Some AQP4-specific T-cells (particularly against AQP4 peptides 11–30, 61–80, 63–76, 91–110, 139–153, and 281–305) were also present in participants with MOG-Ab, MS, and HC, consistent with previous studies demonstrating antibody response against linear AQP4 peptides (64). The presence of these AQP4-specific T-cell responses may reflect unspecific bystander activation, i.e. T-cell receptor (TCR)-independent activation of autoreactive T-cells by pro-inflammatory cytokines during inflammation, and/or epitope spreading (65).
We found no differences in MOG-specific T-cells between the four different groups with our experimental approach. The reason for the observed results could be explained by ignorance of the immune system of the MOG protein (66). In contrast to the AQP4 protein, which is also highly expressed in the periphery (67, 68) and hence underlies highly regulated mechanisms of self-tolerance (69, 70), MOG is only expressed in the CNS at very low levels (71, 72) and therefore not subject to intense immune surveillance. This might explain why HC showed lower response to AQP4 determinants, but in vitro stimulation with MOG peptides also caused profound T cell response in some healthy subjects.
Synthetic MOG peptides used in this study may not accurately represent naturally processed antigen and MHC-presented peptides in an in vivo setting (73). One of the major pathogenic mechanisms of MOG-Ab is considered the enhanced presentation of native MOG protein to T cells via Fc receptor mediated internalization of the antigen-Ab complex (74–76). Therefore, it is possible that MOG-reactive T cells can only be detected using intact MOG protein as the antigen. Indeed, Bronge et al. were able to identify increased frequencies of IFN-γ, IL-22 and IL-17A producing MOG-specific T-cells in patients with MS using bead-bound MOG as the antigen (77).
A major limitation of our study is the small number of included participants. This number reflects the expected number for our clinical centers, since NMOSD and MOG-related disorders are rare with a worldwide prevalence of 1–4/100,000 comparatively similar in most populations. Other limitations are that most patients received immunosuppressive therapy during sample collection related to their severe clinical presentations. Most AQP4-Ab positive patients (6/8) and 4/10 MOG-Ab positive patients received immunomodulatory or immunosuppressive treatment at the time of sample collection. Although B-cell depleting therapy is known to affect T-cell responses in patients with MS (78), 3/4 AQP4-Ab patients who had increased T-cell reactivity to AQP4 peptide 156-170 were treated with rituximab and the fourth patient was under high-dose corticosteroids before the initiation of rituximab treatment.
Importantly, the detailed characterization of single peptides, i.e. known “candidate antigens” based on their encephalitogenicity in animal models and/or their immunodominance in humans is crucial for a potential use in antigen-specific tolerance induction therapies (60, 79). For future studies, the implementation of new unbiased approaches may provide additional perspectives. These new strategies differ from previous studies by using combinatorial peptide libraries, which e.g., cover the entire protein or which allow the discovery of novel “unknown” antigens (80). Discrepancies to other studies might be explained by the use of different assays. We and all other investigators in this field face the issue of very low precursor frequencies of CNS antigen-specific T cells in PBMC preparations. However, the CFSE dilution assay used here is a powerful and sensitive method for directly detecting proliferation of rare autoantigen-specific human T-cells (31, 81). Moreover, the late addition of IL-2 during a re-stimulation step (82–84) offers high sensitivity and specificity. This strategy effectively increases the sensitivity for rare antigen-specific T-cells by selectively facilitating the proliferation of T lymphocytes that express the IL-2 receptor alpha-chain CD25 following antigen recognition (82, 85). However, even though our culture conditions promote the survival of mostly proliferating T-cells, other cells might skew the cytokine response. Indeed, it is well acknowledged that the key Th17-polarizing cytokine IL-6 is produced by myeloid cells (monocytes) rather than T-cells. Furthermore, the timing of sample collection might influence the relative abundance of the different cytokines and therefore explain differences to previous studies. Finally, additional factors such as the TCR avidity or the peptide concentration in different assays may play a role for various specificities. The application of (i) a lower peptide concentration (here used 20 μg/ml per peptide is higher compared to other studies), (ii) the use of native protein antigens instead of peptides, (iii) purified memory T-cells instead of PBMC, and/or (iv) the co-culture of T-cells with autologous APC, e.g., monocytes or EBV-transformed B cells, may be critical improvements for future experiments.
To conclude, this report investigates AQP4- and MOG-specific T-cell reactivities in human individuals presenting with AQP4-Ab and MOG-Ab positive demyelinating diseases. Our in vitro data corroborates previous findings showing the involvement of AQP4-specific T-cells in AQP4-Ab positive NMOSD and confirms the AQP4 peptide 156-170 as specific T-cell epitope. In contrast, no disease-relevant MOG peptide was identified. Future confirmatory studies using an unbiased approach for epitope discovery in larger cohorts may overcome main limitations of small sample size and the use of a limited collection of synthetic peptides in this study.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.
Ethics Statement
The studies involving human participants were reviewed and approved by the local Ethics Committee of Medical University of Innsbruck, Austria (study number AN3041) and University of Zurich, Switzerland (KEK ZH 2013-0001) and all patients and controls gave written informed consent. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
LH and MRe performed experiments, analyzed and interpreted data, and drafted the manuscript. MRa performed experiments and participated in the analysis and interpretation of the data. VG and MRe designed the study and MRe and AL analyzed and interpreted data and revisited the article critically for important intellectual content. AP performed experiments and analyzed the data. KR, MS, HH, TB, and AL provided patient material and revisited the article critically for important intellectual content. All authors approved the final version of the manuscript.
Funding
This study was supported by research grants from the Austrian Multiple Sclerosis Research Society (LH, MRe), the Tyrolean Science Fund (TWF, GZ: UNI-0404/1235, VG and MRe) and the Austrian Science Fund (FWF P326990, MRe). MRa is supported by the Austrian Science Fund (FWF J4157-B30). AL is supported by the Clinical Research Priority Program of the University of Zurich (PrecisionMS).
Conflict of Interest
The Medical University of Innsbruck and the University Hospital Innsbruck (MRe) receive payments for antibody assays (AQP4 and other anti-neuronal and anti-glial antibodies) and for AQP4 antibody validation assays organized by Euroimmun (Luebeck, Germany). HH has participated in meetings sponsored by, received speaker honoraria or travel funding from Bayer, Biogen, Merck, Novartis, Sanofi-Genzyme, Siemens, Teva, and received honoraria for acting as consultant for Biogen and Teva. KR received speaker honoraria from Merck, Novartis and served as a consultant for PARADIGM-Study, Novartis with no compensation. TB has participated in meetings sponsored by and received honoraria (lectures, advisory boards, consultations) from pharmaceutical companies marketing treatments for multiple sclerosis: Almirall, Bayer, Biogen, Biologix, Bionorica, Genzyme, MedDay, Merck, Novartis, Octapharma, Roche, Sanofi/Genzyme, TG Pharmaceuticals, TEVA-ratiopharm and UCB. His institution has received financial support in the last 12 months by unrestricted research grants (Biogen, Merck, Novartis, Roche, Sanofi/Genzyme) and for participation in clinical trials in multiple sclerosis sponsored by Biogen, Merck, Novartis, Roche, Sanofi/Genzyme, and TEVA. AL received financial compensation and/or travel support for lectures and advice from Biogen, Merck, Novartis, Teva, Genzyme, Bayer, Celgene and he is a co-founder of Cellerys and co-inventor on a patent held by the University of Zurich on the use of peptide-coupled cells for treatment of MS.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Kathrin Schanda and Katharina Steiner (Innsbruck, Austria) for expert technical assistance.
References
1. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
2. Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol. (2018) 15:89–102. doi: 10.1038/s41582-018-0112-x
3. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. (2004) 364:2106–12. doi: 10.1016/S0140-6736(04)17551-X
4. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. (2005) 202:473–7. doi: 10.1084/jem.20050304
5. Iglesias A, Bauer J, Litzenburger T, Schubart A, Linington C. T- and B-cell responses to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis and multiple sclerosis. Glia. (2001) 36:220–34. doi: 10.1002/glia.1111
6. Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder? Neurol Neuroimmunol Neuroinflamm. (2015) 2:e62. doi: 10.1212/NXI.0000000000000062
7. Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. (2012) 9:14. doi: 10.1186/1742-2094-9-14
8. Weber MS, Derfuss T, Bruck W. Anti-myelin oligodendrocyte glycoprotein antibody-associated central nervous system demyelination-a novel disease entity? JAMA Neurol. (2018) 75:903–1028. doi: 10.1001/jamaneurol.2018.1055
9. Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. (2009) 66:617–29. doi: 10.1002/ana.21802
10. Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol. (2009) 66:630–43. doi: 10.1002/ana.21837
11. Pohl M, Kawakami N, Kitic M, Bauer J, Martins R, Fischer MT, et al. T cell-activation in neuromyelitis optica lesions plays a role in their formation. Acta Neuropathol Commun. (2013) 1:85. doi: 10.1186/2051-5960-1-85
12. Hillebrand S, Schanda K, Nigritinou M, Tsymala I, Bohm D, Peschl P, et al. Circulating AQP4-specific auto-antibodies alone can induce neuromyelitis optica spectrum disorder in the rat. Acta Neuropathol. (2019) 137:467–85. doi: 10.1007/s00401-018-1950-8
13. Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M, et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain. (2007) 130:1224–34. doi: 10.1093/brain/awm062
14. Owens GP, Ritchie A, Rossi A, Schaller K, Wemlinger S, Schumann H, et al. Mutagenesis of the aquaporin 4 extracellular domains defines restricted binding patterns of pathogenic neuromyelitis optica IgG. J Biol Chem. (2015) 290:12123–34. doi: 10.1074/jbc.M115.647149
15. Tuller F, Holzer H, Schanda K, Aboulenein-Djamshidian F, Hoftberger R, Khalil M, et al. Characterization of the binding pattern of human aquaporin-4 autoantibodies in patients with neuromyelitis optica spectrum disorders. J Neuroinflammation. (2016) 13:176. doi: 10.1186/s12974-016-0642-3
16. Jarius S, Franciotta D, Paul F, Ruprecht K, Bergamaschi R, Rommer PS, et al. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflammation. (2010) 7:52. doi: 10.1186/1742-2094-7-52
17. Di Pauli F, Mader S, Rostasy K, Schanda K, Bajer-Kornek B, Ehling R, et al. Temporal dynamics of anti-MOG antibodies in CNS demyelinating diseases. Clin Immunol. (2011) 138:247–54. doi: 10.1016/j.clim.2010.11.013
18. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflammation. (2016) 13:279. doi: 10.1186/s12974-016-0717-1
19. Chihara N, Aranami T, Sato W, Miyazaki Y, Miyake S, Okamoto T, et al. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. PNAS. (2011) 108:3701–6. doi: 10.1073/pnas.1017385108
20. Wilson R, Makuch M, Kienzler AK, Varley J, Taylor J, Woodhall M, et al. Condition-dependent generation of aquaporin-4 antibodies from circulating B cells in neuromyelitis optica. Brain. (2018) 141:1063–74. doi: 10.1093/brain/awy010
21. Pohl M, Fischer MT, Mader S, Schanda K, Kitic M, Sharma R, et al. Pathogenic T cell responses against aquaporin 4. Acta Neuropathol. (2011) 122:21–34. doi: 10.1007/s00401-011-0824-0
22. Zeka B, Hastermann M, Hochmeister S, Kogl N, Kaufmann N, Schanda K, et al. Highly encephalitogenic aquaporin 4-specific T cells and NMO-IgG jointly orchestrate lesion location and tissue damage in the CNS. Acta Neuropathol. (2015) 130:783–98. doi: 10.1007/s00401-015-1501-5
23. Bradl M, Reindl M, Lassmann H. Mechanisms for lesion localization in neuromyelitis optica spectrum disorders. Curr Opin Neurol. (2018) 31:325. doi: 10.1097/WCO.0000000000000551
24. Misu T, Hoftberger R, Fujihara K, Wimmer I, Takai Y, Nishiyama S, et al. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. (2013) 125:815–27. doi: 10.1007/s00401-013-1116-7
25. Lucchinetti CF, Guo Y, Popescu BF, Fujihara K, Itoyama Y, Misu T. The pathology of an autoimmune astrocytopathy: lessons learned from neuromyelitis optica. Brain Pathol. (2014) 24:83–97. doi: 10.1111/bpa.12099
26. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
27. Jones MV, Huang H, Calabresi PA, Levy M. Pathogenic aquaporin-4 reactive T cells are sufficient to induce mouse model of neuromyelitis optica. Acta Neuropathol Commun. (2015) 3:28. doi: 10.1186/s40478-015-0207-1
28. Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. (2007) 8:942–9. doi: 10.1038/ni1496
29. Uzawa A, Mori M, Ito M, Uchida T, Hayakawa S, Masuda S, et al. Markedly increased CSF interleukin-6 levels in neuromyelitis optica, but not in multiple sclerosis. J Neurol. (2009) 256:2082–4. doi: 10.1007/s00415-009-5274-4
30. Wang HH, Dai YQ, Qiu W, Lu ZQ, Peng FH, Wang YG, et al. Interleukin-17-secreting T cells in neuromyelitis optica and multiple sclerosis during relapse. J Clin Neurosci. (2011) 18:1313–7. doi: 10.1016/j.jocn.2011.01.031
31. Varrin-Doyer M, Spencer CM, Schulze-Topphoff U, Nelson PA, Stroud RM, Cree BA, et al. Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann Neurol. (2012) 72:53–64. doi: 10.1002/ana.23651
32. Linhares UC, Schiavoni PB, Barros PO, Kasahara TM, Teixeira B, Ferreira TB, et al. The ex vivo production of IL-6 and IL-21 by CD4+ T cells is directly associated with neurological disability in neuromyelitis optica patients. J Clin Immunol. (2013) 33:179–89. doi: 10.1007/s10875-012-9780-2
33. Barros PO, Cassano T, Hygino J, Ferreira TB, Centuriao N, Kasahara TM, et al. Prediction of disease severity in neuromyelitis optica by the levels of interleukin (IL)-6 produced during remission phase. Clin Exp Immunol. (2016) 183:480–9. doi: 10.1111/cei.12733
34. Hofer LS, Mariotto S, Wurth S, Ferrari S, Mancinelli CR, Delogu R, et al. Distinct serum and cerebrospinal fluid cytokine and chemokine profiles in autoantibody-associated demyelinating diseases. Mult Scler J Exp Transl Clin. (2019) 5:2055217319848463. doi: 10.1177/2055217319848463
35. Kampylafka EI, Routsias JG, Alexopoulos H, Dalakas MC, Moutsopoulos HM, Tzioufas AG. Fine specificity of antibodies against AQP4: epitope mapping reveals intracellular epitopes. J Autoimmun. (2011) 36:221–7. doi: 10.1016/j.jaut.2011.01.004
36. Matsuya N, Komori M, Nomura K, Nakane S, Fukudome T, Goto H, et al. Increased T-cell immunity against aquaporin-4 and proteolipid protein in neuromyelitis optica. Int Immunol. (2011) 23:565–73. doi: 10.1093/intimm/dxr056
37. Arellano B, Hussain R, Zacharias T, Yoon J, David C, Zein S, et al. Human aquaporin 4281-300 is the immunodominant linear determinant in the context of HLA-DRB1*03:01: relevance for diagnosing and monitoring patients with neuromyelitis optica. Arch Neurol. (2012) 69:1125–31. doi: 10.1001/archneurol.2012.1300
38. Vaknin-Dembinsky A, Brill L, Kassis I, Petrou P, Ovadia H, Ben-Hur T, et al. T-cell responses to distinct AQP4 peptides in patients with neuromyelitis optica (NMO). Mult Scler Relat Disord. (2016) 6:28–36. doi: 10.1016/j.msard.2015.12.004
39. Bielekova B, Sung MH, Kadom N, Simon R, McFarland H, Martin R. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J Immunol. (2004) 172:3893–904. doi: 10.4049/jimmunol.172.6.3893
40. Vargas-Lowy D, Kivisakk P, Gandhi R, Raddassi K, Soltany P, Gorman MP, et al. Increased Th17 response to myelin peptides in pediatric MS. Clin Immunol. (2013) 146:176–84. doi: 10.1016/j.clim.2012.12.008
41. Varrin-Doyer M, Shetty A, Spencer CM, Schulze-Topphoff U, Weber MS, Bernard CC, et al. MOG transmembrane and cytoplasmic domains contain highly stimulatory T-cell epitopes in MS. Neurol Neuroimmunol Neuroinflamm. (2014) 1:e20. doi: 10.1212/NXI.0000000000000020
42. Kerlero de Rosbo N, Mendel I, Ben-Nun A. Chronic relapsing experimental autoimmune encephalomyelitis with a delayed onset and an atypical clinical course, induced in PL/J mice by myelin oligodendrocyte glycoprotein (MOG)-derived peptide: preliminary analysis of MOG T cell epitopes. Eur J Immunol. (1995) 25, 985–93. doi: 10.1002/eji.1830250419
43. Shetty A, Gupta SG, Varrin-Doyer M, Weber MS, Prod'homme T, Molnarfi N, et al. Immunodominant T-cell epitopes of MOG reside in its transmembrane and cytoplasmic domains in EAE. Neurol Neuroimmunol Neuroinflamm. (2014) 1:e22. doi: 10.1212/NXI.0000000000000022
44. Peschl P, Bradl M, Hoftberger R, Berger T, Reindl M. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. (2017) 8:529. doi: 10.3389/fimmu.2017.00529
45. Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. (2011) 69:292–302. doi: 10.1002/ana.22366
46. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. (2017) 17:162–73. doi: 10.1016/S1474-4422(17)30470-2
47. Mader S, Lutterotti A, Di Pauli F, Kuenz B, Schanda K, Aboul-Enein F, et al. Patterns of antibody binding to aquaporin-4 isoforms in neuromyelitis optica. PLoS ONE. (2010) 5:e10455. doi: 10.1371/journal.pone.0010455
48. Hennes EM, Baumann M, Schanda K, Anlar B, Bajer-Kornek B, Blaschek A, et al. Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology. (2017) 89:900–8. doi: 10.1212/WNL.0000000000004312
49. Reindl M, Schanda K, Woodhall M, Tea F, Ramanathan S, Sagen J, et al. International multicenter examination of MOG antibody assays. Neurol Neuroimmunol Neuroinflamm. (2020) 7:e674. doi: 10.1212/NXI.0000000000000674
50. Ramachandran H, Laux J, Moldovan I, Caspell R, Lehmann PV, Subbramanian RA. Optimal thawing of cryopreserved peripheral blood mononuclear cells for use in high-throughput human immune monitoring studies. Cells. (2012) 1:313–24. doi: 10.3390/cells1030313
51. Kalluri SR, Rothhammer V, Staszewski O, Srivastava R, Petermann F, Prinz M, et al. Functional characterization of aquaporin-4 specific T cells: towards a model for neuromyelitis optica. PLoS ONE. (2011) 6:e16083. doi: 10.1371/journal.pone.0016083
52. Kerlero de Rosbo N, Hoffman M, Mendel I, Yust I, Kaye J, Bakimer R, et al. Predominance of the autoimmune response to myelin oligodendrocyte glycoprotein (MOG) in multiple sclerosis: reactivity to the extracellular domain of MOG is directed against three main regions. Eur J Immunol. (1997) 27:3059–69. doi: 10.1002/eji.1830271144
53. Forsthuber TG, Shive CL, Wienhold W, de Graaf K, Spack EG, Sublett R, et al. T cell epitopes of human myelin oligodendrocyte glycoprotein identified in HLA-DR4 (DRB1*0401) transgenic mice are encephalitogenic and are presented by human B cells. J Immunol. (2001) 167:7119–25. doi: 10.4049/jimmunol.167.12.7119
54. Mars LT, Bauer J, Gross DA, Bucciarelli F, Firat H, Hudrisier D, et al. CD8 T cell responses to myelin oligodendrocyte glycoprotein-derived peptides in humanized HLA-A*0201-transgenic mice. J Immunol. (2007) 179:5090–8. doi: 10.4049/jimmunol.179.8.5090
55. Weissert R, Kuhle J, de Graaf KL, Wienhold W, Herrmann MM, Muller C, et al. High immunogenicity of intracellular myelin oligodendrocyte glycoprotein epitopes. J Immunol. (2002) 169:548–56. doi: 10.4049/jimmunol.169.1.548
56. Nelson PA, Khodadoust M, Prodhomme T, Spencer C, Patarroyo JC, Varrin-Doyer M, et al. Immunodominant T cell determinants of aquaporin-4, the autoantigen associated with neuromyelitis optica. PLoS ONE. (2010) 5:e15050. doi: 10.1371/journal.pone.0015050
57. Harrington D, D'Agostino RBSr, Gatsonis C, Hogan JW, Hunter DJ, Normand ST, et al. New guidelines for statistical reporting in the journal. N Engl J Med. (2019) 381:285–6. doi: 10.1056/NEJMe1906559
58. Pisani F, Mastrototaro M, Rossi A, Nicchia GP, Tortorella C, Ruggieri M, et al. Identification of two major conformational aquaporin-4 epitopes for neuromyelitis optica autoantibody binding. J Biol Chem. (2011) 286:9216–24. doi: 10.1074/jbc.M110.123000
59. Pisani F, Mola MG, Simone L, Rosito S, Alberga D, Mangiatordi GF, et al. Identification of a point mutation impairing the binding between aquaporin-4 and neuromyelitis optica autoantibodies. J Biol Chem. (2014) 289:30578–89. doi: 10.1074/jbc.M114.582221
60. Zubizarreta I, Florez-Grau G, Vila G, Cabezon R, Espana C, Andorra M, et al. Immune tolerance in multiple sclerosis and neuromyelitis optica with peptide-loaded tolerogenic dendritic cells in a phase 1b trial. Proc Natl Acad Sci USA. (2019) 116:8460–70. doi: 10.1073/pnas.1820039116
61. Matsushita T, Matsuoka T, Isobe N, Kawano Y, Minohara M, Shi N, et al. Association of the HLA-DPB1*0501 allele with anti-aquaporin-4 antibody positivity in Japanese patients with idiopathic central nervous system demyelinating disorders. Tissue Antigens. (2009) 73:171–6. doi: 10.1111/j.1399-0039.2008.01172.x
62. Brum DG, Barreira AA, dos Santos AC, Kaimen-Maciel DR, Matiello M, Costa RM, et al. HLA-DRB association in neuromyelitis optica is different from that observed in multiple sclerosis. Mult Scler. (2010) 16:21–9. doi: 10.1177/1352458509350741
63. Deschamps R, Paturel L, Jeannin S, Chausson N, Olindo S, Bera O, et al. Different HLA class II (DRB1 and DQB1) alleles determine either susceptibility or resistance to NMO and multiple sclerosis among the French Afro-Caribbean population. Mult Scler. (2011) 17:24–31. doi: 10.1177/1352458510382810
64. Metz I, Beissbarth T, Ellenberger D, Pache F, Stork L, Ringelstein M, et al. Serum peptide reactivities may distinguish neuromyelitis optica subgroups and multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e204. doi: 10.1212/NXI.0000000000000204
65. Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. (2005) 23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707
66. Delarasse C, Daubas P, Mars LT, Vizler C, Litzenburger T, Iglesias A, et al. Myelin/oligodendrocyte glycoprotein-deficient (MOG-deficient) mice reveal lack of immune tolerance to MOG in wild-type mice. J Clin Invest. (2003) 112:544–53. doi: 10.1172/JCI15861
67. Mobasheri A, Marples D, Young IS, Floyd RV, Moskaluk CA, Frigeri A. Distribution of the AQP4 water channel in normal human tissues: protein and tissue microarrays reveal expression in several new anatomical locations, including the prostate gland and seminal vesicles. Channels. (2007) 1:29–38. doi: 10.4161/chan.3735
68. Verkman AS, Phuan PW, Asavapanumas N, Tradtrantip L. Biology of AQP4 and anti-AQP4 antibody: therapeutic implications for NMO. Brain Pathol. (2013) 23:684–95. doi: 10.1111/bpa.12085
69. Sagan SA, Winger RC, Cruz-Herranz A, Nelson PA, Hagberg S, Miller CN, et al. Tolerance checkpoint bypass permits emergence of pathogenic T cells to neuromyelitis optica autoantigen aquaporin-4. Proc Natl Acad Sci USA. (2016) 113:14781–6. doi: 10.1073/pnas.1617859114
70. Vogel AL, Knier B, Lammens K, Kalluri SR, Kuhlmann T, Bennett JL, et al. Deletional tolerance prevents AQP4-directed autoimmunity in mice. Eur J Immunol. (2017) 47:458–69. doi: 10.1002/eji.201646855
71. Johns TG, Bernard CC. The structure and function of myelin oligodendrocyte glycoprotein. J Neurochem. (1999) 72:1–9. doi: 10.1046/j.1471-4159.1999.0720001.x
72. Greer JM. Autoimmune T-cell reactivity to myelin proteolipids and glycolipids in multiple sclerosis. Mult Scler Int. (2013) 2013:151427. doi: 10.1155/2013/151427
73. Godkin AJ, Smith KJ, Willis A, Tejada-Simon MV, Zhang J, Elliott T, et al. Naturally processed HLA class II peptides reveal highly conserved immunogenic flanking region sequence preferences that reflect antigen processing rather than peptide-MHC interactions. J Immunol. (2001) 166:6720–7. doi: 10.4049/jimmunol.166.11.6720
74. Flach AC, Litke T, Strauss J, Haberl M, Gomez CC, Reindl M, et al. Autoantibody-boosted T-cell reactivation in the target organ triggers manifestation of autoimmune CNS disease. Proc Natl Acad Sci USA. (2016) 113:3323–8. doi: 10.1073/pnas.1519608113
75. Kinzel S, Lehmann-Horn K, Torke S, Hausler D, Winkler A, Stadelmann C, et al. Myelin-reactive antibodies initiate T cell-mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol. (2016) 132:43–58. doi: 10.1007/s00401-016-1559-8
76. Spadaro M, Winklmeier S, Beltran E, Macrini C, Hoftberger R, Schuh E, et al. Pathogenicity of human antibodies against myelin oligodendrocyte glycoprotein. Ann Neurol. (2018) 84:315–28. doi: 10.1002/ana.25291
77. Bronge M, Ruhrmann S, Carvalho-Queiroz C, Nilsson OB, Kaiser A, Holmgren E, et al. Myelin oligodendrocyte glycoprotein revisited-sensitive detection of MOG-specific T-cells in multiple sclerosis. J Autoimmun. (2019) 102:38–49. doi: 10.1016/j.jaut.2019.04.013
78. Bar-Or A, Fawaz L, Fan B, Darlington PJ, Rieger A, Ghorayeb C, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol. (2010) 67:452–61. doi: 10.1002/ana.21939
79. Lutterotti A, Yousef S, Sputtek A, Stürner KH, Stellmann JP, Breiden P, et al. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med. (2013) 5:188ra175. doi: 10.1126/scitranslmed.3006168
80. Sospedra M, Pinilla C, Martin R. Use of combinatorial peptide libraries for T-cell epitope mapping. Methods. (2003) 29:236–47. doi: 10.1016/S1046-2023(02)00346-8
81. Mannering SI, Morris JS, Jensen KP, Purcell AW, Honeyman MC, van Endert PM, et al. A sensitive method for detecting proliferation of rare autoantigen-specific human T cells. J Immunol Methods. (2003) 283:173–83. doi: 10.1016/j.jim.2003.09.004
82. Chain B, McCafferty I, Wallace G, Askenase PW. Improvement of the in vitro T cell proliferation assay by a modified method that separates the antigen recognition and IL-2-dependent steps. J Immunol Methods. (1987) 99:221–8. doi: 10.1016/0022-1759(87)90131-1
83. Kennell AS, Gould KG, Salaman MR. Proliferation assay amplification by IL-2 in model primary and recall antigen systems. BMC Res Notes. (2014) 7:662. doi: 10.1186/1756-0500-7-662
84. Ramberger M, Hogl B, Stefani A, Mitterling T, Reindl M, Lutterotti A. CD4+ T-cell reactivity to orexin/hypocretin in patients with narcolepsy type 1. Sleep. (2017) 40:zsw070. doi: 10.1093/sleep/zsw070
Keywords: neuromyelitis optica spectrum disorder, aquaporin-4, myelin oligodendrocyte glycoprotein, antibody, T-cell
Citation: Hofer LS, Ramberger M, Gredler V, Pescoller AS, Rostásy K, Sospedra M, Hegen H, Berger T, Lutterotti A and Reindl M (2020) Comparative Analysis of T-Cell Responses to Aquaporin-4 and Myelin Oligodendrocyte Glycoprotein in Inflammatory Demyelinating Central Nervous System Diseases. Front. Immunol. 11:1188. doi: 10.3389/fimmu.2020.01188
Received: 21 January 2020; Accepted: 13 May 2020;
Published: 17 June 2020.
Edited by:
Valentina Tomassini, University of Studies G. d'Annunzio Chieti and Pescara, ItalyReviewed by:
Tatsuro Misu, Tohoku University, JapanAnne-Katrin Pröbstel, University Hospital of Basel, Switzerland
Copyright © 2020 Hofer, Ramberger, Gredler, Pescoller, Rostásy, Sospedra, Hegen, Berger, Lutterotti and Reindl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Reindl, bWFya3VzLnJlaW5kbCYjeDAwMDQwO2ktbWVkLmFjLmF0