 Haiou Li1,2†Yunjiao Zhou1,2†Haizhou Wang1,2†Meng Zhang1,2Peishan Qiu1,2Mengna Zhang1,2Ruike Zhang1,2
Haiou Li1,2†Yunjiao Zhou1,2†Haizhou Wang1,2†Meng Zhang1,2Peishan Qiu1,2Mengna Zhang1,2Ruike Zhang1,2 Qiu Zhao1,2*
Qiu Zhao1,2* Jing Liu1,2*
Jing Liu1,2*- 1Department of Gastroenterology, Zhongnan Hospital of Wuhan University, Wuhan, China
- 2Hubei Clinical Center, Key Lab of Intestinal and Colorectal Diseases, Wuhan, China
Nonalcoholic steatohepatitis (NASH), the advanced stage of nonalcoholic fatty liver disease (NAFLD), is emerging as a leading cause of progressive liver fibrosis and end-stage liver disease. Liver macrophages, mainly composed of Kupffer cells (KCs) and monocyte-derived macrophages (MoMFs), play a vital role in NASH progression and regression. Recent advances suggest that cell–cell communication is a fundamental feature of hepatic microenvironment. The reprogramming of cell–cell signaling between macrophages and surrounding cells contributes to the pathogenesis of NASH. In this review, we summarize the current knowledge of NASH regarding the composition of liver macrophages and their communication with surrounding cells, which are composed of hepatocytes, hepatic stellate cells (HSCs), liver sinusoidal endothelial cells (LSECs) and other immune cells. We also discuss the potential therapeutic strategies based on the level of macrophages.
Introduction
Nonalcoholic fatty liver disease (NAFLD), an increasingly common liver disease worldwide, ranges from relatively benign NAFL to nonalcoholic steatohepatitis (NASH) (1, 2). NASH is strongly associated with progressive liver fibrosis and has further become a major cause of cirrhosis and liver cancer (3). Unlike isolated hepatic steatosis, NASH is characterized as the presence of inflammation, hepatocellular injury, and varying degrees of fibrosis (4). However, the underlying mechanisms involved in pathogenesis of NASH are not fully understood. It was demonstrated that liver macrophages orchestrate both the progression and restoration of NASH (5). Traditionally, liver macrophages mainly comprise liver-resident Kupffer cells (KCs) and circulating monocyte-derived macrophages (MoMFs) (6). The activation of liver macrophages during NASH progression is a dynamic procedure dependent on various stimuli such as cytokines, lipid metabolites, and other signal molecules (7, 8).
Emerging evidence suggests that cellular networks rather than a single cell type modulate NASH progression (9). In conjunction with surrounding cells, liver macrophages can trigger inflammation response, fibrogenesis, vascular remodeling, and so forth. In the development of NASH, hepatocytes contribute to KC activation and MoMF recruitment via multiple signal molecules such as damage-associated molecular patterns (DAMPs), extracellular vesicles (EVs), and harmful lipids (5). In response to those signals, activated macrophages also signal back to modulate hepatocyte fate. Besides, those activated macrophages further mediate the activation of hepatic stellate cells (HSCs) via producing cytokines and chemokines, including transforming growth factor-β (TGFβ), interleukin-1β (IL-1β), platelet—derived growth factor (PDGF) receptor, and CC-chemokine ligand 2 (CCL2) (10). Moreover, liver macrophages influence the biological functions of liver sinusoidal endothelial cells (LSECs) and other immune cells (11, 12). In turn, those surrounding cells can stimulate liver macrophages during NASH progression (13, 14). Understanding the intercellular crosstalk between liver macrophages and their surrounding cells is critical for developing novel therapeutic interventions based on the level of macrophages.
In this review, we summarize the intercellular signaling between liver macrophages and surrounding cells involved in NASH development. The potential macrophage-targeted therapeutic strategies for NASH are also discussed.
The Composition of Liver Macrophages in Nonalcoholic Steatohepatitis
Liver macrophage populations comprise different subsets of cells. In particular, KCs and freshly recruited MoMFs are important mediators of liver inflammation, fibrogenesis, and fibrinolysis in the development of NASH (15, 16). In mice, circulating monocytes were divided into two main subsets: lymphocyte antigen 6C high (Ly-6Chi) and Ly-6C low (Ly-6Clo) expressing monocytes. It was demonstrated that the hepatic infiltration of Ly-6Chi monocytes occurred early in murine NASH models and patients with NASH (16, 17). Those monocytes gave rise to phenotypically distinct populations of MoMFs upon external stimulus. Briefly, KCs and MoMFs could be differentiated toward either a classic proinflammatory phenotype (M1 macrophages) or an alternative anti-inflammatory phenotype (M2 macrophages) in vitro (18). The M1 macrophages produced proinflammatory cytokines such as tumor necrosis factor α (TNFα), IL-1β, CCL2, and CCL5. In contrast, M2 macrophages secreted a distinct set of mediators including IL-13, IL-10, IL-4, and TGFβ (19). It was noted that KCs and MoMFs in NASH liver exhibited a notable shift toward a proinflammatory phenotype on the basis of their gene expression signatures at the single-cell level (20).
In a recent single-cell RNA sequencing (scRNA-seq) study, two distinct subpopulations of liver macrophages are exhibited in western diet (WD)-induced NASH models in mice, including MoMFs with high lysozyme 2 (Lyz2) expression and KCs with high C-type lectin domain family 4 member F (Clec4f) expression (21). Besides, those MoMFs segregated into three subtypes owing to their striking heterogeneity (21). Furthermore, a NASH-specific macrophage population, marked by high expression of triggering receptors expressed on myeloid cells 2 (Trem2), was observed in NASH livers of both mice and humans, termed NASH-associated macrophages (NAMs) (20). Consistently, another scRNA-seq study identified a pathogenic subpopulation of TREM2+CD9+ macrophages in the fibrotic niche of human liver with NASH, named scar-associated macrophages (SAMacs). The expansion of SAMacs was positively correlated with the degree of NASH-induced liver fibrosis (22). More studies are needed to understand the ontology of hepatic macrophage subpopulations in NASH.
Intercellular Crosstalk of Liver Macrophages in Nonalcoholic Steatohepatitis
The growing consensus is that cell–cell communication within liver represents a key aspect that leads to the progression toward NASH (9). The anatomical location of liver macrophages allows them to interact with several liver resident cells and circulating immune cells (23). Histologically, the clusters of KCs were characterized as microgranulomas, and those with lipid droplets were characterized as lipogranulomas in human NAFLD/NASH (24–26). A unique histological structure, where activated macrophages aggregated around hepatocytes with large lipid droplets, was detected in the murine NASH models and patients with NASH, termed hepatic crown-like structures (hCLS) (27). Conversely, activated KCs were not shown to form hCLS in patients and mice with simple steatosis (28). This section focuses on liver macrophage-related crosstalk in NASH (Figure 1).
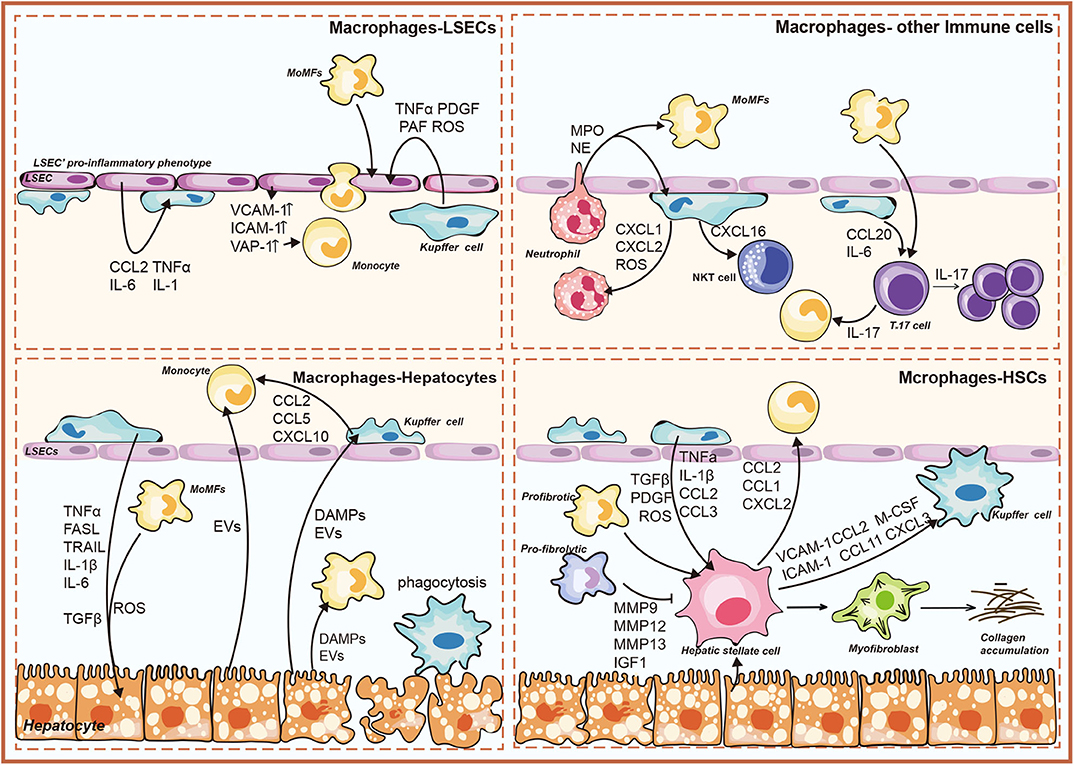
Figure 1. Overview of liver macrophage-related intercellular signaling in nonalcoholic steatohepatitis (NASH). The illustration consists of four groups, as follows: liver macrophages–hepatocytes; liver macrophages–hepatic stellate cells (HSCs); liver macrophages–liver sinusoidal endothelial cells (LSECs); liver macrophages–immune cells. DAMPs, damage-associated molecular patterns; EVs, extracellular vesicles; TNFα, tumor necrosis factor α; TRAIL, TNF-related apoptosis-inducing ligand; FasL, Fas ligand; ROS, reactive oxygen species; CCL, chemokine (C-C) motif ligand; CXCL, chemokine (C-X-C motif) ligand; IL, interleukin; MMP, matrix metalloproteinase; IGF1, insulin-like growth factor 1; TGFβ, transforming growth factor-β; M-CSF, macrophage colony-stimulating factor; PDGF, platelet-derived growth factor; PAF, platelet-activating factor; ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1; VAP-1, vascular adhesion protein-1; MPO, myeloperoxidase; NO, nitric oxide; IFNγ, interferon γ.
Interaction Between Liver Macrophages and Hepatocytes
Lipotoxicity is characterized as a key feature that differentiated NASH from isolated steatosis (29, 30). Various lipotoxic compounds (e.g., free cholesterol, ceramides, and saturated fatty acids) induce metabolic stress, oxidative stress, and endoplasmic reticulum-related stress in hepatocytes, resulting in hepatocyte injury and death (31). Hepatocyte stress and death cause the release of their cellular contents into extracellular space, which contributes to macrophage activation (29).
Kupffer Cells-Hepatocytes
The DAMPs, such as cytosolic proteins, purine nucleotides, and mitochondrial compounds, primarily acted on pattern recognition receptors (PRRs) to promote inflammatory responses of KCs (32). High mobility group box-1 (HMGB1) was a widely studied DAMP that induced cytokine release of macrophages (33). It bound toll-like receptor 4 (TLR4) to induce nuclear factor (NF)-κB translocation and TNFα release in KCs (34). Besides, the mitochondrial DNA (mtDNA) released from damaged hepatocytes activated TLR9 on KCs to promote inflammatory response (35). Recently, it was reported that the mtDNA was recognized by the stimulator of IFN genes (STING) in KCs to induce TNFα and IL-6 production under lipid overload (36). Adenosine triphosphate (ATP) was also released into extracellular space from injured hepatocytes. Being sensed by P2X purinoceptor 7 (P2X7) receptor on KCs, ATP could medicate the induction of NLR family pyrin domain-containing 3 (NLRP3) inflammasome and the consequent production of proinflammatory cytokines (37, 38).
Recent studies implicated lipotoxic hepatocyte-derived EVs (LPC-EVs) in mediating cell–cell communication by transferring various cargos (39). Apoptotic bodies formed by apoptotic hepatocytes fall in the category of EVs. Engulfment of apoptotic bodies by KCs promoted the production of TNFα, TNF-related apoptosis-inducing ligand (TRAIL), and Fas ligand (FasL) (40). These death receptor (DR) ligands further induced hepatocyte apoptosis in a feed-forward loop (Figure 2). Moreover, KCs were shown to aggregate around dead hepatocytes to form hCLSs. Specifically, the cholesterol crystals within remnant lipid droplets of dead hepatocytes were processed by KCs, which then activated the NLRP3 inflammasome in KCs, causing proinflammatory cytokines production (41). In this line, NLRP3 inflammasome blockade improved cholesterol crystal-derived inflammation and fibrosis in experimental NASH (42).
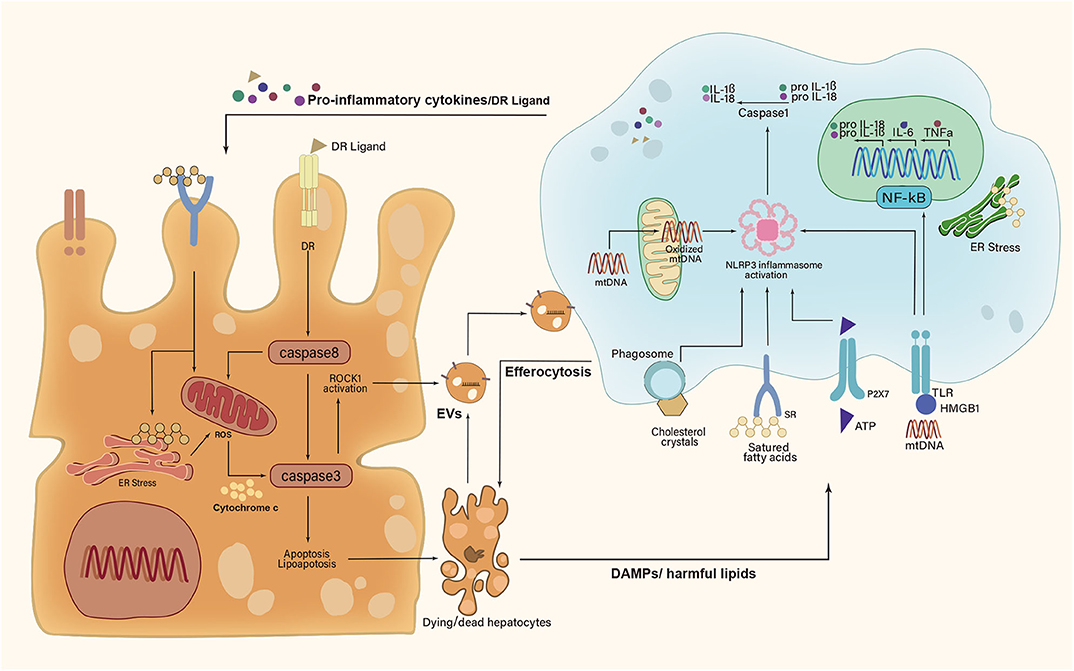
Figure 2. A feed-forward regulatory loop between lipotoxic hepatocytes and Kupffer cells. Upon metabolic stress, dying and dead hepatocytes release damage-associated molecular patterns (DAMPs), extracellular vesicles (EVs), and harmful lipids to activate Kupffer cells (KCs). In turn, activated KCs secrete proinflammatory cytokines and death receptor (DR) ligands to aggravate hepatocyte damage. However, KCs can remove apoptotic hepatocytes via efferocytosis. IL-1β, interleukin-1β; IL-6, interleukin-6; IL-18, interleukin-18; TNFα, tumor necrosis factor α; HMGB1, high mobility group box-1; ATP, adenosine triphosphate; mtDNA, mitochondrial DNA; ROCK1, rho-associated, coiled-coil containing protein kinase 1; NF-κB, nuclear factor-κB; NLRP3, NLR family pyrin domain-containing 3; ER, endoplasmic reticulum; ROS, reactive oxygen species; P2X7, P2X purinoceptor 7; TLR, toll-like receptor; SR, scavenger receptor.
In response to those signals sent by hepatocytes, KCs also signaled back to the hepatocytes and regulated their fate (43–46) (Figure 2). Firstly, activated KCs exerted actions via producing cytokines (e.g., IL-6, TNFα, and IL-1β) (47). TNFα allowed for the activation of caspase-8 in hepatocytes by binding to TNF receptor 1 (TNFR1), which not only triggered apoptotic caspase cascade directly but also induced mitochondrial dysfunction to amplify the signals indirectly (8, 48). In addition, KC-derived IL-1β signaling was associated with de novo lipogenesis in hepatocytes and promoted hepatic lipid deposition (49–51). IL-6 contributed to insulin resistance in hepatocytes by disrupting key steps in the insulin signal transduction (52). Additionally, KCs were shown to remove apoptotic hepatocytes via efferocytosis (9). The efferocytic clearance of dead hepatocytes prevents the release of DAMPs and subsequent DAMP-mediated inflammation. Efferocytosis could be triggered by a series of “eat-me” signals from apoptotic hepatocytes (53). The well-studied “eat-me” signal was the presence of phosphatidylserine (PtdSer) on the outer leaflet of the cell membrane during apoptosis (54). KCs were thought to be the most important hepatic efferocytes with the expression of several different PtdSer receptors, such as T cell immunoglobulin, mucin domain-containing molecule 3 (Tim3), Tim4, macrophage c-mer tyrosine kinase (MerTK), stabilin-1, and stabilin-2 (53). Strikingly, both Tim3 and Tim4 were overexpressed in all detected liver macrophage subsets in methionine- and choline-deficient diet (MCD)-induced NASH mice (55, 56). Their absence led to increased production of reactive oxygen species (ROS), IL-1β, and IL-18 in macrophages, concomitant with the aggravation of steatosis and liver fibrosis (55, 56). Further studies are urgently needed to explore that how those PtdSer receptors participate in efferocytosis mechanisms of macrophages during NASH development.
Monocyte-Derived Macrophages-Hepatocytes
NASH-induced hepatocyte damage recruited MoMFs indirectly by stimulating KCs to release proinflammatory chemokines including CCL2, CCL5, and CXCL10 (57). Lipotoxic hepatocytes also release EVs to induce the hepatic recruitment of MoMFs. TRAIL-enriched LPC-EVs induced the expression of IL-1β and IL-6 via NF-κB activation in mouse bone marrow-derived macrophages (58). Ceramide and chemokine (C-X-C motif) ligand 10 (CXCL10) within EVs contributed to MoMF recruitment to the liver via activating macrophage chemotaxis (59, 60). Besides, integrin β (ITGβ) enriched LPC-EVs mediated monocyte adhesion to LSECs, an essential step for hepatic recruitment of MoMFs in murine NASH (61).
Interaction Between Liver Macrophages and Hepatic Stellate Cells
Studies revealed that macrophages were key regulators in the pathogenesis of NASH-driven fibrosis (9, 62). Similarly, therapeutic inhibition of macrophage infiltration accelerated liver fibrosis regression in murine NASH (16, 63, 64). Besides, macrophages aggregated to form hCLS where they could interact with HSCs (27). The hCLS was located close to fibrogenic lesions and the number of hCLS significantly linked to the extent of liver fibrosis (27, 41). In turn, activated HSCs were shown to regulate macrophage accumulation and proliferation through paracrine effects (65). Moreover, a scRNA-seq analysis showed that activated HSCs were implicated in modulating the functions of macrophages via a series of stellakines (e.g., CCL2, CCL11, and CXCL2) in murine NASH models (20).
Kupffer Cells–Hepatic Stellate Cells
On the molecular level, KCs regulated HSC activation by producing cytokines and chemokines such as TGFβ, PDGF, TNFα, and IL-1β (10). KC-derived TGFβ promoted HSC differentiation into a profibrogenic phenotype, concomitant with increased collagen and α-smooth muscle actin expression (66). Recently, Cai et al. proved that the MerTK signaling in KCs promoted HSC activation and liver fibrosis in NASH mice via TGFβ1 production (67). Moreover, TGFβ induced oxidative DNA damage in HSCs through downregulation of cytoglobin (68). Besides, in murine NASH models, the enhancement of TNFα signaling following KC activation facilitated HSC survival via activating the NF-κB pathway in HSCs (69, 70). Activated KCs caused the HSC migration and recruitment through the secretion of CCL2 and CCL5 (71, 72). On the other hand, the HSC-derived chemokines that included CCL2 and macrophage colony-stimulating factor (M-CSF) further activated KCs, amplifying the inflammatory response (73). In response to lipopolysaccharide (LPS), HSCs secreted intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin to induce KC migration (74). The underlying mechanisms governing this process have not been fully elucidated.
Monocyte-Derived Macrophages–Hepatic Stellate Cells
Infiltrating MoMFs are divided into two major subsets: Ly-6Chi macrophages and Ly-6Clo macrophages. Similar to KCs, proinflammatory Ly-6Chi macrophages activated HSCs by secreting TGFβ, IL-1β, PDGF, and CCL2, enhancing the fibrotic response. Recently, Ramachandran P et al. demonstrated that the TREM2+CD9+ SAMacs, differentiating from circulating monocytes, performed a profibrogenic characteristic with multiple profibrogenic genes expression (22). Of note, during the regression stage, the pro-restorative Ly-6Clo macrophages promoted HSC apoptosis and accelerated extracellular matrix degradation by increasing the expression of matrix metalloproteinase 9 (MMP9), MMP12, MMP13, and insulin-like growth factor 1 (IGF1) (75, 76). This pro-restorative subpopulation also expressed chemokine (C-X3-C motif) receptor 1 (CX3CR1), and its ligand CX3C ligand 1 (CX3CL1) was mainly expressed by HSCs (77). The CX3CL1–CX3CR1 interaction negatively regulated the inflammatory properties in macrophages (78).
Interaction Between Liver Macrophages and Liver Sinusoidal Endothelial Cells
LSECs constituted a unique vascular bed with fenestrae in liver and interacted directly with the immune cells and antigens in the blood flow (79). Monocyte's adhesion to LSECs is a crucial step for inflammation response in NASH, which verified the “gatekeeper” role of LSECs in the progression from simple steatosis to NASH (80).
Kupffer Cells–Liver Sinusoidal Endothelial Cells
At the early stage of NAFLD, LSECs exhibited an anti-inflammatory property by inhibiting KC activation and monocyte migration (81, 82). At the stage of NASH, LSEC capillarization happened, and capillarized LSECs were necessary for activation of KCs (83). LSECs acquired a proinflammatory phenotype to produce proinflammatory mediators, leading to KC activation (14). Activated KCs were shown to be involved in angiogenesis through the secretion of ROS and cytokines including TNFα, PDGF, and platelet-activating factor (PAF) (84).
Monocyte-Derived Macrophages–Liver Sinusoidal Endothelial Cells
In NASH, the proinflammatory phenotype of LSECs increased proinflammatory chemokine CCL2 to facilitate hepatic recruitment of monocytes (14). Moreover, in mice models of NASH, the overexpression of adhesion molecules ICAM-1, VCAM-1, and vascular adhesion protein-1 (VAP-1) in LSECs were critical for the adhesion and transmigration of monocytes to amplify local inflammatory response (14, 85, 86). Little is known about the pathophysiological roles of MoMFs toward LSECs in NASH.
Interaction Between Liver Macrophages and Other Immune Cells
Kupffer Cells–Other Immune Cells
The interactions of immune cells in homeostasis and disease have been reviewed in detail elsewhere (87). Firstly, KCs contribute to the hepatic infiltration of neutrophils in NASH. The inflammatory activation of KCs resulted in the production of chemokines (e.g., CXCL1, CXCL2, and CXCL8) and ROS, which stimulated neutrophil recruitment to expanded inflammation (44, 72). Hepatic neutrophil content and neutrophil elastase (NE) activity were significantly increased in high-fat diet (HFD)-fed mice. NE treatment caused the proinflammatory markers of macrophages to largely increase (88). Neutrophil-derived myeloperoxidase (MPO) was also associated with the formation of hCLS in NASH (89). Besides, activated KCs promote natural killer T (NKT) cell over-activation and subsequent deficiency in the pathogenesis of NAFLD (90). KC-derived IL-12 was associated with the reduced numbers of hepatic NKT cells in hepatosteatosis (91). Conversely, Syn et al. described that NKT cells were associated with NASH-related fibrosis (92). CXCL16 secreted by KCs triggered the hepatic accumulation of CXCR6+ NKT cells, thereby accentuating liver inflammation and fibrosis in murine liver (93).
Monocyte-Derived Macrophages–Other Immune Cells
A proinflammatory phenotype of macrophages showed a close relationship with diverse T-cell subsets by secreting IL-6, TNFα, IL-1β, IL-12, and IL-23 in the pathogenesis of NAFLD (12). Although these cytokines are well-established drivers of T-cell differentiation, their roles in controlling T-cell differentiation in NASH are not fully understood (87). T helper type 17 (Th17) cells and their production of IL-17 facilitated the transition from simple steatosis to steatohepatitis in NAFLD (94). They favored the further activation of monocytes, leading to the release of proinflammatory cytokines that, in turn, amplified liver inflammation (95).
Macrophage-Targeted Therapeutic Interventions in Nonalcoholic Steatohepatitis
Currently, there are still no Food and Drug Administration (FDA)-approved effective drugs for NASH despite its high prevalence. Owing to their critical roles in NASH, liver macrophages are emphasized as attractive targets for NASH treatment. Specifically, there are some options that exert potential therapeutic effects by regulating cell–cell communication in NASH.
Because the recruited MoMFs widely interact with resident cells, interfering with recruiting signals would disrupt intercellular communication at the level of macrophages. Cenicriviroc (CVC), a dual CCR2/5 antagonist, efficiently reduced the hepatic recruitment of MoMFs that ameliorated hepatic inflammation and fibrosis in NASH mice models (64). This drug was evaluated in a phase II clinical trial in NASH patients and was found to be effective in reducing fibrosis after CVC administration (96). The RNA-aptamer molecule mNOX-E36 also relieved steatohepatitis and accelerated regression of liver fibrosis in experimental mouse models via antagonizing CCL2 (63). Maraviroc, a CCL5 inhibitor, ameliorated hepatic steatosis in HFD-induced NAFLD in mice (97). Moreover, monocyte's adhesion to LSECs is an essential step for hepatic recruitment of MoMFs. The VAP-1 inhibitor, also called amine oxidase copper containing three (AOC3) inhibitor, decreased inflammatory cell recruitment and reduced fibrosis (85). This drug was tested in a phase II trial in patients with NASH, but it was discontinued owing to the risk of drug interactions in NASH patients (98).
Another potential NASH treatment is to regulate KC activation. Because hepatocyte-derived DAMPs trigger the sterile inflammatory response of KCs by acting on PRRs, targeting released DAMPs or PRRs can inhibit KC activation, thus ameliorating liver inflammation (99). HMGB1 neutralizing antibodies and PRR antagonists (e.g., TLR2, TLR3, and TLR4 antagonists) were shown to attenuate liver inflammation in murine models (100, 101). Targeting macrophage-derived profibrogenic molecules may be promising to improve NASH fibrosis. Galectin-3 is a profibrogenic protein that is highly expressed in macrophages surrounding lipotoxic hepatocytes. Treatment with galectin-3 inhibitor (GR-MD-02) markedly improved fibrosis in a murine model of NASH (102). A phase IIb trial showed that GR-MD-02 reduced the hepatic-portal vein pressure gradient in patients with NASH cirrhosis (103).
Another potential option is regulating intracellular pathways in macrophages, which has been reviewed elsewhere (5). Notably, non-coding RNAs (ncRNAs) offered new possibilities in developing therapeutic strategies for NASH on the basis of the level of macrophages (104, 105). For instance, in murine fibrotic NASH models, treatment with miR-223-3p mimic ameliorated activation of HSCs and fibrosis development through its NLRP3-targeted effect in KCs (106). In addition, miR-146b acted as a promising approach to attenuate HFD-induced NASH in mice by directly targeting the IL-1 receptor-associated kinase 1 and TNFR-associated factor 6 in macrophages, resulting in suppression of TNFα and IL-6 (107). A cell-specific delivery system with efficiency and safety is essential for the clinical application of those miRNAs.
Conclusion and Future Perspectives
Multiple studies have shown that liver macrophages play a central role in the progression and regression of NASH. They sense various external signals and act as key mediators of hepatic inflammation. Importantly, owing to their strategic location, liver macrophages can interact with different cells such as hepatocytes, HSCs, and LSECs. However, there are several issues that need to be addressed. Firstly, most of the observed interactive effects are in specific cytokine-dependent manner. The core intracellular pathways of macrophages in mediating intercellular signaling in NASH are still unclear, which points out a future research goal. Secondly, owing to their striking heterogeneity, more studies are needed to reveal the complex cell–cell communication network based on the large spectrum of macrophage phenotypes. In addition, most findings from murine models are insufficient to reflect the complex cellular networks during NASH progression in humans. Further exploration of the macrophage function in human NASH liver is warranted.
Moreover, liver macrophages are identified as attractive targets for NASH treatment. As described in this review, some signaling pathways that mediated cellular crosstalk are potentially druggable. Besides, the rapid advancement in nanomedicine allows for targeted delivery of drugs to macrophages, such as miRNA mimic. Taken together, deciphering macrophage function and their role in intercellular signaling network will facilitate the design of novel targeted therapies to treat NASH.
Author Contributions
HL and YZ searched the literature and wrote the manuscript. HW prepared the figures. MengZ, PQ, MengnaZ, and RZ carefully checked the manuscript and helped to improve paragraphs. QZ and JL designed and revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a research grant from the National Natural Science Foundation of China (JL, grant no. 81472735); Wuhan University (JL, 2042019kf0206); and National Basic Research Program of China (973 program, 2015CB932600).
References
1. Di Sessa A, Cirillo G, Guarino S, Marzuillo P, Miraglia Del Giudice E. Pediatric non-alcoholic fatty liver disease: current perspectives on diagnosis and management. Pediatric Health Med Ther. (2019) 10:89–97. doi: 10.2147/PHMT.S188989
2. Diehl AM, Day C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med. (2017) 377:2063–72. doi: 10.1056/NEJMra1503519
3. Doycheva I, Issa D, Watt KD, Lopez R, Rifai G, Alkhouri N. Nonalcoholic steatohepatitis is the most rapidly increasing indication for liver transplantation in young adults in the united states. J Clin Gastroenterol. (2018) 52:339–46. doi: 10.1097/MCG.0000000000000925
4. Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S. Nonalcoholic steatohepatitis: a Review. JAMA. (2020) 323:1175–83. doi: 10.1001/jama.2020.2298
5. Kazankov K, Jorgensen SMD, Thomsen KL, Moller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. (2019) 16:145–59. doi: 10.1038/s41575-018-0082-x
6. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. (2014) 60:1090–6. doi: 10.1016/j.jhep.2013.12.025
7. Hundertmark J, Krenkel O, Tacke F. Adapted immune responses of myeloid-Derived cells in fatty liver disease. Front Immunol. (2018) 9:2418. doi: 10.3389/fimmu.2018.02418
8. Hirsova P, Gores GJ. Death receptor-Mediated cell death and proinflammatory signaling in nonalcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol. (2015) 1:17–27. doi: 10.1016/j.jcmgh.2014.11.005
9. Schwabe RF, Tabas I, Pajvani UB. Mechanisms of fibrosis development in nASH. Gastroenterology. (2020) 158:1913–28. doi: 10.1053/j.gastro.2019.11.311
10. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. (2017) 14:397–411. doi: 10.1038/nrgastro.2017.38
11. Ramirez-Pedraza M, Fernandez M. Interplay between macrophages and angiogenesis: a Double-Edged sword in liver disease. Front Immunol. (2019) 10:2882. doi: 10.3389/fimmu.2019.02882
12. Van Herck MA, Weyler J, Kwanten WJ, Dirinck EL, De Winter BY, Francque S M, et al. The differential roles of t Cells in non-alcoholic fatty liver disease and obesity. Front Immunol. (2019) 10:82. doi: 10.3389/fimmu.2019.00082
13. Cai J, Zhang XJ, Li H. The role of innate immune cells in nonalcoholic steatohepatitis. Hepatology. (2019) 70:1026–37. doi: 10.1002/hep.30506
14. Hammoutene A, Rautou PE. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol. (2019) 70:1278–91. doi: 10.1016/j.jhep.2019.02.012
15. Reid DT, Reyes JL, McDonald BA, Vo T, Reimer RA, Eksteen B. Kupffer cells undergo fundamental changes during the development of experimental nASH and are critical in initiating liver damage and inflammation. PLoS ONE. (2016) 11:e0159524. doi: 10.1371/journal.pone.0159524
16. Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through cCR2. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G1310–21. doi: 10.1152/ajpgi.00365.2011
17. Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, Horsfall L, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology. (2014) 59:1393–405. doi: 10.1002/hep.26937
18. Zhou D, Yang K, Chen L, Wang Y, Zhang W, Xu Z, et al. Macrophage polarization and function: new prospects for fibrotic disease. Immunol Cell Biol. (2017) 95:864–9. doi: 10.1038/icb.2017.64
19. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in nAFLD/NASH. Dig Dis Sci. (2016) 61:1294–303. doi: 10.1007/s10620-016-4049-x
20. Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of intercellular crosstalk in healthy and nASH liver revealed by single-Cell secretome gene analysis. Mol Cell. (2019) 75:644–60 e5. doi: 10.1016/j.molcel.2019.07.028
21. Krenkel O, Hundertmark J, Abdallah AT, Kohlhepp M, Puengel T, Roth T, et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut. (2020) 69:551–63. doi: 10.1136/gutjnl-2019-318382
22. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu N T, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. (2019) 575:512–8. doi: 10.1038/s41586-019-1631-3
23. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. (2017) 17:306–21. doi: 10.1038/nri.2017.11
24. Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol. (2009) 175:1473–82. doi: 10.2353/ajpath.2009.080999
25. Brunt EM. Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. (2010) 7:195–203. doi: 10.1038/nrgastro.2010.21
26. Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. (2010) 5:145–71. doi: 10.1146/annurev-pathol-121808-102132
27. Itoh M, Kato H, Suganami T, Konuma K, Marumoto Y, Terai S, et al. Hepatic crown-like structure: a unique histological feature in non-alcoholic steatohepatitis in mice and humans. PLoS ONE. (2013) 8:e82163. doi: 10.1371/journal.pone.0082163
28. Ioannou GN, Haigh WG, Thorning D, Savard C. Hepatic cholesterol crystals and crown-like structures distinguish nASH from simple steatosis. J Lipid Res. (2013) 54:1326–34. doi: 10.1194/jlr.M034876
29. Caligiuri A, Gentilini A, Marra F. Molecular pathogenesis of nASH. Int J Mol Sci. (2016) 17:1575. doi: 10.3390/ijms17091575
30. Pan X, Wang P, Luo J, Wang Z, Song Y, Ye J, et al. Adipogenic changes of hepatocytes in a high-fat diet-induced fatty liver mice model and non-alcoholic fatty liver disease patients. Endocrine. (2015) 48:834–47. doi: 10.1007/s12020-014-0384-x
31. Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in nASH pathogenesis. J Hepatol. (2018) 68:280–95. doi: 10.1016/j.jhep.2017.11.014
32. Mihm S. Danger-Associated molecular patterns (DAMPs): molecular triggers for sterile inflammation in the liver. Int J Mol Sci. (2018) 19:3104. doi: 10.3390/ijms19103104
33. Yang H, Wang H, Chavan SS, Andersson U. High mobility group box protein 1 (HMGB1): the prototypical endogenous danger molecule. Mol Med. (2015) 21 Suppl 1:S6–S12. doi: 10.2119/molmed.2015.00087
34. Li L, Chen L, Hu L, Liu Y, Sun H Y, Tang J, et al. Nuclear factor high-mobility group box1 mediating the activation of toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology. (2011) 54:1620–30. doi: 10.1002/hep.24552
35. Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, et al. Hepatocyte mitochondrial dNA drives nonalcoholic steatohepatitis by activation of tLR9. J Clin Invest. (2016) 126:859–64. doi: 10.1172/JCI83885
36. Yu Y, Liu Y, An W, Song J, Zhang Y, Zhao X. STING-mediated inflammation in kupffer cells contributes to progression of nonalcoholic steatohepatitis. J Clin Invest. (2019) 129:546–55. doi: 10.1172/JCI121842
37. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. (2009) 461:282–6. doi: 10.1038/nature08296
38. Ishimaru M, Yusuke N, Tsukimoto M, Harada H, Takenouchi T, Kitani H, et al. Purinergic signaling via p2Y receptors up-mediates iL-6 production by liver macrophages/Kupffer cells. J Toxicol Sci. (2014) 39:413–23. doi: 10.2131/jts.39.413
39. Hirsova P, Ibrahim SH, Verma VK, Morton LA, Shah VH, LaRusso NF, et al. Extracellular vesicles in liver pathobiology: small particles with big impact. Hepatology. (2016) 64:2219–33. doi: 10.1002/hep.28814
40. Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. (2003) 38:1188–98. doi: 10.1053/jhep.2003.50472
41. Ioannou GN, Subramanian S, Chait A, Haigh WG, Yeh MM, Farrell GC, et al. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine nASH. J Lipid Res. (2017) 58:1067–79. doi: 10.1194/jlr.M072454
42. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental nASH in mice. J Hepatol. (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
43. Krenkel O, Tacke F. Macrophages in nonalcoholic fatty liver disease: a Role model of pathogenic immunometabolism. Semin Liver Dis. (2017) 37:189–97. doi: 10.1055/s-0037-1604480
44. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in nASH. Nat Rev Gastroenterol Hepatol. (2018) 15:349–64. doi: 10.1038/s41575-018-0009-6
45. Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, et al. Depletion of liver kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes. (2010) 59:347–57. doi: 10.2337/db09-0016
46. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine cCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. (2012) 61:416–26. doi: 10.1136/gutjnl-2011-300304
47. Oates JR, McKell MC, Moreno-Fernandez ME, Damen M, Deepe GS Jr, et al. Macrophage function in the pathogenesis of non-alcoholic fatty liver disease: the mac attack. Front Immunol. (2019) 10:2893. doi: 10.3389/fimmu.2019.02893
48. Liedtke C, Trautwein C. The role of tNF and fas dependent signaling in animal models of inflammatory liver injury and liver cancer. Eur J Cell Biol. (2012) 91:582–9. doi: 10.1016/j.ejcb.2011.10.001
49. Negrin KA, Roth Flach RJ, DiStefano MT, Matevossian A, Friedline RH, Jung D, et al. IL-1 signaling in obesity-induced hepatic lipogenesis and steatosis. PLoS ONE. (2014) 9:e107265. doi: 10.1371/journal.pone.0107265
50. Almog T, Kandel Kfir M, Levkovich H, Shlomai G, Barshack I, Stienstra R, et al. Interleukin-1alpha deficiency reduces adiposity, glucose intolerance and hepatic de-novo lipogenesis in diet-induced obese mice. BMJ Open Diabetes Res Care. (2019) 7:e000650. doi: 10.1136/bmjdrc-2019-000650
51. Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, et al. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology. (2010) 51:511–22. doi: 10.1002/hep.23337
52. Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes. (2002) 51:3391–9. doi: 10.2337/diabetes.51.12.3391
53. Horst AK, Tiegs G, Diehl L. Contribution of macrophage efferocytosis to liver homeostasis and disease. Front Immunol. (2019) 10:2670. doi: 10.3389/fimmu.2019.02670
54. Morioka S, Maueroder C, Ravichandran KS. Living on the edge: efferocytosis at the interface of homeostasis and pathology. Immunity. (2019) 50:1149–62. doi: 10.1016/j.immuni.2019.04.018
55. Du X, Wu Z, Xu Y, Liu Y, Liu W, Wang T, et al. Increased tim-3 expression alleviates liver injury by regulating macrophage activation in mCD-induced nASH mice. Cell Mol Immunol. (2019) 16:878–86. doi: 10.1038/s41423-018-0032-0
56. Liu W, Bai F, Wang H, Liang Y, Du X, Liu C, et al. Tim-4 inhibits nLRP3 inflammasome via the lKB1/AMPKalpha pathway in macrophages. J Immunol. (2019) 203:990–1000. doi: 10.4049/jimmunol.1900117
57. Lanthier N. Targeting kupffer cells in non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: why and how? World J Hepatol. (2015) 7:2184–8. doi: 10.4254/wjh.v7.i19.2184
58. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid-Induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology. (2016) 150:956–67. doi: 10.1053/j.gastro.2015.12.037
59. Kakazu E, Mauer AS, Yin M, Malhi H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an iRE1alpha-dependent manner. J Lipid Res. (2016) 57:233–45. doi: 10.1194/jlr.M063412
60. Ibrahim SH, Hirsova P, Tomita K, Bronk SF, Werneburg NW, Harrison SA, et al. Mixed lineage kinase 3 mediates release of c-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology. (2016) 63:731–44. doi: 10.1002/hep.28252
61. Guo Q, Furuta K, Lucien F, Gutierrez Sanchez LH, Hirsova P, Krishnan A, et al. Integrin beta1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine nASH. J Hepatol. (2019) 71:1193–205. doi: 10.1016/j.jhep.2019.07.019
62. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. (2014) 14:181–94. doi: 10.1038/nri3623
63. Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N, et al. Pharmacological inhibition of the chemokine c-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing ly-6C(+) macrophage infiltration in mice. Hepatology. (2014) 59:1060–72. doi: 10.1002/hep.26783
64. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. (2018) 67:1270–83. doi: 10.1002/hep.29544
65. Cai X, Wang J, Wang J, Zhou Q, Yang B, He Q, et al. Intercellular crosstalk of hepatic stellate cells in liver fibrosis: new insights into therapy. Pharmacol Res. (2020) 155:104720. doi: 10.1016/j.phrs.2020.104720
66. Kiagiadaki F, Kampa M, Voumvouraki A, Castanas E, Kouroumalis E, Notas G. Activin-A causes hepatic stellate cell activation via the induction of tNFalpha and tGFbeta in kupffer cells. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:891–9. doi: 10.1016/j.bbadis.2017.12.031
67. Cai B, Dongiovanni P, Corey KE, Wang X, Shmarakov IO, Zheng Z, et al. Macrophage merTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab. (2020) 31:406–21 e7. doi: 10.1016/j.cmet.2019.11.013
68. Okina Y, Sato-Matsubara M, Matsubara T, Daikoku A, Longato L, Rombouts K, et al. TGF-beta-driven reduction of cytoglobin leads to oxidative dNA damage in stellate cells during non-alcoholic steatohepatitis. J Hepatol. (2020) doi: 10.1016/j.jhep.2020.03.051. [Epub ahead of print].
69. Tomita K, Tamiya G, Ando S, Ohsumi K, Chiyo T, Mizutani A, et al. Tumour necrosis factor alpha signalling through activation of kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut. (2006) 55:415–24. doi: 10.1136/gut.2005.071118
70. Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. (2013) 58:1461–73. doi: 10.1002/hep.26429
71. Seki E, De Minicis S, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, et al. CCR1 and cCR5 promote hepatic fibrosis in mice. J Clin Invest. (2009) 119:1858–70. doi: 10.1172/jci37444
72. Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology. (2014) 147:577–94 e1. doi: 10.1053/j.gastro.2014.06.043
73. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. (2008) 88:125–72. doi: 10.1152/physrev.00013.2007
74. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. (2017) 127:55–64. doi: 10.1172/JCI88881
75. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A. (2012) 109:E3186–95. doi: 10.1073/pnas.1119964109
76. Campana L, Iredale JP. Regression of liver fibrosis. Semin Liver Dis. (2017) 37:1–10. doi: 10.1055/s-0036-1597816
77. Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. (2015) 61:1066–79. doi: 10.1002/hep.27332
78. Karlmark KR, Zimmermann HW, Roderburg C, Gassler N, Wasmuth HE, Luedde T, et al. The fractalkine receptor cX(3)CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology. (2010) 52:1769–82. doi: 10.1002/hep.23894
79. Sorensen KK, Simon-Santamaria J, McCuskey RS, Smedsrod B. Liver sinusoidal endothelial cells. Compr Physiol. (2015) 5:1751–74. doi: 10.1002/cphy.c140078
80. Knolle PA, Wohlleber D. Immunological functions of liver sinusoidal endothelial cells. Cell Mol Immunol. (2016) 13:347–53. doi: 10.1038/cmi.2016.5
81. McMahan RH, Porsche CE, Edwards MG, Rosen HR. Free fatty acids differentially downregulate chemokines in liver sinusoidal endothelial cells: insights into non-Alcoholic fatty liver disease. PLoS ONE. (2016) 11:e0159217. doi: 10.1371/journal.pone.0159217
82. Tateya S, Rizzo NO, Handa P, Cheng AM, Morgan-Stevenson V, Daum G, et al. Endothelial nO/cGMP/VASP signaling attenuates kupffer cell activation and hepatic insulin resistance induced by high-fat feeding. Diabetes. (2011) 60:2792–801. doi: 10.2337/db11-0255
83. Miyao M, Kotani H, Ishida T, Kawai C, Manabe S, Abiru H, et al. Pivotal role of liver sinusoidal endothelial cells in nAFLD/NASH progression. Lab Invest. (2015) 95:1130–44. doi: 10.1038/labinvest.2015.95
84. Coulon S, Heindryckx F, Geerts A, Van Steenkiste C, Colle I, Van Vlierberghe H. Angiogenesis in chronic liver disease and its complications. Liver Int. (2011) 31:146–62. doi: 10.1111/j.1478-3231.2010.02369.x
85. Weston CJ, Shepherd EL, Claridge LC, Rantakari P, Curbishley SM, Tomlinson JW, et al. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J Clin Invest. (2015) 125:501–20. doi: 10.1172/JCI73722
86. Miyachi Y, Tsuchiya K, Komiya C, Shiba K, Shimazu N, Yamaguchi S, et al. Roles for cell-Cell adhesion and contact in obesity-Induced hepatic myeloid cell accumulation and glucose intolerance. Cell Rep. (2017) 18:2766–79. doi: 10.1016/j.celrep.2017.02.039
87. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol. (2016) 13:88–110. doi: 10.1038/nrgastro.2015.200
88. Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. (2012) 18:1407–12. doi: 10.1038/nm.2885
89. Rensen SS, Bieghs V, Xanthoulea S, Arfianti E, Bakker JA, Shiri-Sverdlov R, et al. Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PLoS ONE. (2012) 7:e52411. doi: 10.1371/journal.pone.0052411
90. Tang T, Sui Y, Lian M, Li Z, Hua J. Pro-inflammatory activated kupffer cells by lipids induce hepatic nKT cells deficiency through activation-induced cell death. PLoS ONE. (2013) 8:e81949. doi: 10.1371/journal.pone.0081949
91. Kremer M, Thomas E, Milton R J, Perry AW, van Rooijen N, Wheeler MD, et al. Kupffer cell and interleukin-12-dependent loss of natural killer t cells in hepatosteatosis. Hepatology. (2010) 51:130–41. doi: 10.1002/hep.23292
92. Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, et al. Accumulation of natural killer t cells in progressive nonalcoholic fatty liver disease. Hepatology. (2010) 51:1998–2007. doi: 10.1002/hep.23599
93. Wehr A, Baeck C, Heymann F, Niemietz PM, Hammerich L, Martin C, et al. Chemokine receptor cXCR6-dependent hepatic nK t Cell accumulation promotes inflammation and liver fibrosis. J Immunol. (2013) 190:5226–36. doi: 10.4049/jimmunol.1202909
94. Rau M, Schilling AK, Meertens J, Hering I, Weiss J, Jurowich C, et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of th17 cells in the liver and an increased th17/Resting regulatory t Cell ratio in peripheral blood and in the liver. J Immunol. (2016) 196:97–105. doi: 10.4049/jimmunol.1501175
95. Tang Y, Bian Z, Zhao L, Liu Y, Liang S, Wang Q, et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin Exp Immunol. (2011) 166:281–90. doi: 10.1111/j.1365-2249.2011.04471.x
96. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. (2018) 67:1754–67. doi: 10.1002/hep.29477
97. Perez-Martinez L, Perez-Matute P, Aguilera-Lizarraga J, Rubio-Mediavilla S, Narro J, Recio E, et al. Maraviroc, a cCR5 antagonist, ameliorates the development of hepatic steatosis in a mouse model of non-alcoholic fatty liver disease (NAFLD). J Antimicrob Chemother. (2014) 69:1903–10. doi: 10.1093/jac/dku071
98. Sumida Y, Yoneda M, Ogawa Y, Yoneda M, Okanoue T, Nakajima A. Current and new pharmacotherapy options for non-alcoholic steatohepatitis. Expert Opin Pharmacother. (2020) 2020:1–15. doi: 10.1080/14656566.2020.1744564
99. van der Heide D, Weiskirchen R, Bansal R. Therapeutic targeting of hepatic macrophages for the treatment of liver diseases. Front Immunol. (2019) 10:2852. doi: 10.3389/fimmu.2019.02852
100. Li X, Wang LK, Wang LW, Han XQ, Yang F, Gong ZJ. Blockade of high-mobility group box-1 ameliorates acute on chronic liver failure in rats. Inflamm Res. (2013) 62:703–9. doi: 10.1007/s00011-013-0624-1
101. Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. (2013) 59:583–94. doi: 10.1016/j.jhep.2013.03.033
102. Traber PG, Zomer E. Therapy of experimental nASH and fibrosis with galectin inhibitors. PLoS ONE. (2013) 8:e83481. doi: 10.1371/journal.pone.0083481
103. Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, et al. Effects of belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology. (2019) 12:534–45. doi: 10.1053/j.gastro.2019.11.296
104. Sulaiman SA, Muhsin NIA, Jamal R. Regulatory non-coding rNAs network in non-alcoholic fatty liver disease. Front Physiol. (2019) 10:279. doi: 10.3389/fphys.2019.00279
105. Su Q, Kumar V, Sud N, Mahato RI. MicroRNAs in the pathogenesis and treatment of progressive liver injury in nAFLD and liver fibrosis. Adv Drug Deliv Rev. (2018) 129:54–63. doi: 10.1016/j.addr.2018.01.009
106. Jimenez Calvente C, Del Pilar H, Tameda M, Johnson CD, Feldstein AE. MicroRNA 223 3p negatively regulates the nLRP3 inflammasome in acute and chronic liver injury. Mol Ther. (2020) 28:653–63. doi: 10.1016/j.ymthe.2019.09.013
Keywords: nonalcoholic steatohepatitis, cellular crosstalk, liver macrophages, liver cells, therapeutic strategies
Citation: Li H, Zhou Y, Wang H, Zhang M, Qiu P, Zhang M, Zhang R, Zhao Q and Liu J (2020) Crosstalk Between Liver Macrophages and Surrounding Cells in Nonalcoholic Steatohepatitis. Front. Immunol. 11:1169. doi: 10.3389/fimmu.2020.01169
Received: 24 February 2020; Accepted: 12 May 2020;
Published: 24 June 2020.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Paramananda Saikia, Cleveland Clinic, United StatesPayel Sil, National Institute of Environmental Health Sciences (NIEHS), United States
Copyright © 2020 Li, Zhou, Wang, Zhang, Qiu, Zhang, Zhang, Zhao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiu Zhao, cWl1emhhbyYjeDAwMDQwO3dodS5lZHUuY24=; Jing Liu, bGl1amluZ19HSSYjeDAwMDQwO3dodS5lZHUuY24=
†These authors have contributed equally to this work