 Estefanía García-Guerrero
Estefanía García-Guerrero Belén Sierro-Martínez
Belén Sierro-Martínez Jose Antonio Pérez-Simón*
Jose Antonio Pérez-Simón*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 05 June 2020
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01128
This article is part of the Research TopicEngineered Immune Cells in Cancer Immunotherapy (EICCI)View all 47 articles
Multiple myeloma (MM) remains an incurable disease regardless of recent advances in the field. Therefore, a substantial unmet need exists to treat patients with relapsed/refractory myeloma. The use of novel agents such as daratumumab, elotuzumab, carfilzomib, or pomalidomide, among others, usually cannot completely eradicate myeloma cells. Although these new drugs have had a significant impact on the prognosis of MM patients, the vast majority ultimately become refractory or can no longer be treated due to toxicity of prior treatment, and thus succumb to the disease. Cellular therapies represent a novel approach with a unique mechanism of action against myeloma with the potential to defeat drug resistance and achieve long-term remissions. Genetic modification of cells to express a novel receptor with tumor antigen specificity is currently being explored in myeloma. Chimeric antigen receptor gene-modified T-cells (CAR T-cells) have shown to be the most promising approach so far. CAR T-cells have shown to induce durable complete remissions in other advanced hematologic malignancies like acute lymphocytic leukemia (ALL) and diffuse large B-cell lymphoma (DLBCL). With this background, significant efforts are underway to develop CAR-based therapies for MM. Currently, several antigen targets, including CD138, CD19, immunoglobulin kappa (Ig-Kappa) and B-cell maturation antigen (BCMA), are being used in clinical trials to treat myeloma patients. Some of these trials have shown promising results, especially in terms of response rates. However, the absence of a plateau is observed in most studies which correlates with the absence of durable remissions. Therefore, several potential limitations such as lack of effectiveness, off-tumor toxicities, and antigen loss or interference with soluble proteins could hamper the efficacy of CAR T-cells in myeloma. In this review, we will focus on clinical outcomes reported with CAR T-cells in myeloma, as well as on CAR T-cell limitations and how to overcome them with next generation of CAR T-cells.
Multiple myeloma (MM) is an hematological malignancy characterized by the clonal proliferation of malignant plasma cells (1). Myeloma develops from a pre-malignant monoclonal proliferation of plasma cells (monoclonal gammopathy of undetermined significance) which progresses to smoldering myeloma and finally to symptomatic disease (1, 2). With an incidence of 5.6 cases per 100.000 people/year in Western countries it accounts for 1% of all cancers and around 10% of hematological malignancies (3). Diagnosis of MM is based on the presence of clonal plasma cells plus monoclonal protein in serum or urine and clinical manifestations including hypercalcemia, renal impairment, anemia and/or bone lesions (acronym: CRAB) (4, 5). Levels of albumin, β2microglobulin and LDH together with the presence or not of high risk cytogenetic abnormalities, including del(17p), and/or t(4;14) and/or t(14;16), allows to identify subgroups of patients with very different outcomes varying from 82% overall survival (OS) at 5 years for the low risk, 62% for the intermediate risk and 40% for the high risk subgroups (6).
Great advances have been achieved in the last decade in the treatment of MM with the discovery of new therapeutic agents such as immunomodulatory drugs (thalidomide, lenalidomide, pomalidomide), proteasome inhibitors (bortezomib, carfilzomib), monoclonal antibodies (daratumumab and elotuzumab), and the use of hematopoietic stem cell transplantation (1, 4, 7). However, MM remains an incurable disease as patients almost invariably relapse upon treatment and the probabilities to obtain disease response decrease after each relapse and the time to progression does shorten in every relapse. It is therefore necessary to develop more efficient MM therapies (8).
Engineered T-cells expressing chimeric antigen receptors (CARs) have demonstrated encouraging results in the treatment of relapsed/refractory hematological malignancies (9–12). CARs are synthetic receptor proteins integrated by an extracellular antigen-binding domain derived from a single-chain variable fragment (scFv) of a monoclonal antibody linked to a T cell receptor (TCR)-derived CD3ζ chain, subsequently redirecting cytolytic T-lymphocytes to cells expressing this specific antigen in a human leukocyte antigen (HLA)-independent manner (3, 13). Second- and third-generation CARs present further costimulatory domains such as CD28, 4-1BB, or OX40 to potentiate T-cell activation. Fourth-generation CAR T-cells may include controllable on-off switch proteins, a suicide gene or molecules to potentiate T-cell function, expansion and reduce exhaustion (3, 14).
Two anti-CD19 CAR T-cell products, Tisagenlecleucel (Kymriah) and Axicabtagene Ciloleucel (Yescarta), have been approved by US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of acute lymphoblastic leukemia (ALL) and diffuse large B cell lymphoma (DLBCL). Sustained durable complete remissions have been accomplished with CD19 CAR T-cell products in relapsed/refractory ALL patients which have prompted attention to a possible alternative to overcome actual treatment limitations in MM (10, 12).
As far as MM is concerned, numerous CAR T-cell products are under development. B-cell maturation antigen (BCMA) is the predominantly used target against MM based on its high expression in the surface of malignant plasma cells and restricted expression in normal tissues/cells except for a low-level expression in mature B-cells. BCMA is vital for the survival and proliferation of MM cells, it is expressed in most MM patient samples (60–100%) and its efficacy as a MM antigen for targeted immunotherapy has been tested in several clinical trials. Other targets under development include CD38, CD138, CD19, or immunoglobulin kappa light chain (Ig-Kappa) (3, 15, 16). Despite the promising results achieved by CAR T-cell administration in MM in terms of response rates, the absence of a plateau corresponding with the absence of durable remissions is common to all studies. Clinical experience with CAR T-cell therapy has pointed out several limitations of this technology such as lack of effectiveness, toxicities, antigen loss, interference with soluble proteins or manufacturing issues (15, 17).
In this review, we will report clinical outcomes achieved so far with CAR T-cells for the treatment of MM, as well as focus on their limitations and how to overcome these restrictions with next generation CAR T-cells.
Selection of a suitable antigen is essential for the development of an optimal CAR T-cell product. As the recognition of an antigen by the CAR is HLA-independent, the target must be expressed in the cell surface. Besides, the antigen must be homogeneously expressed in tumor cells and have an essential role in their proliferation and survival to avoid escape from CAR T-cells recognition. It is also essential that the chosen antigen is not expressed in vital healthy tissues to avoid undesired on-target, off-tumor toxicities (15). Although BCMA is the predominantly used target for CAR T-cell products for the treatment of MM, several other antigens have been studied and some are being evaluated in clinical trials.
CD38 is a transmembrane glycoprotein implicated in calcium regulation, signal transduction and cell adhesion. Among the hematological cell lineages, CD38 is highly expressed on precursor B-cells, plasma cells, NK cells and myeloid precursors. CD38 is also expressed on gut, prostate cells, pancreas, nervous system, muscle cells and osteoclasts (18). Since the FDA approval of several anti-CD38 monoclonal antibodies for the treatment of MM in 2015 (daratumumab, istuximab) (19, 20) the generation of CD38 CAR T-cells has been extensively studied preclinically (21). The wide expression of CD38 among hematopoietic cells might be a critical inconvenient for its clinical application due to possible on-target, off-tumor toxicities. To date CD38 CAR T-cells are under clinical investigation in several trials. In the study NCT03464916, a CD38 CAR T-cell product is being used as monotherapy for relapsed/refractory myeloma to evaluate its efficacy and safety although no outcomes have been posted yet. Moreover, CD38 is also being evaluated within clinical trials in combination with other target antigens including a dual specificity CD38 and BCMA CAR T-cell product (NCT03767751), and a combination CAR T therapy with CD19 (NCT03125577). Other approaches targeting several MM antigens including CD38, BCMA, CD138 and CD56 are being explored (NCT03271632, NCT03473496). A fourth generation CAR T-cell product targeting multiple antigens, including CD38, and expressing simultaneously interleukin-7 (IL-7) and chemokine (C-C motif) ligand 19 (CCL19), is also under clinical investigation for the treatment of relapsed/refractory MM patients (NCT03778346). Nevertheless, results from these CD38-targeted CAR T therapies have not been published to date.
CD138 belongs to the syndecan family type I transmembrane proteoglycans and it is implicated in wound healing, cell adhesion and endocytosis. CD138 is expressed on the surface of mature epithelial cells, nonetheless its expression is restricted within the hematopoietic system to normal and tumor plasma cells (22). It has been correlated with survival and disease progression of MM, and its inhibition promotes apoptosis of myeloma cells (23, 24). CD138 has been proven to be an effective target antigen for the treatment of MM in preclinical studies (25). To date, there is only one published clinical trial for the study of autologous CD138 CAR T-cells in relapsed/refractory MM patients pretreated with chemotherapy and stem cell transplantation. Five patients were treated with a single average dose of 0.756 × 107 cells/kg of CD138 CAR T-cells. The CAR gene was detectable in peripheral blood in all patients and high levels were persistent for at least 4 weeks after infusion. No severe toxicities were observed apart from infusion-related fever (grade 3) and nausea and vomiting (grade 2). Four patients experienced myeloma regression after CD138 CAR T-cells infusion for 3–7 months, while the other patient progressed despite the presence of CAR in bone marrow until day 90 post-infusion. No complete responses (CR) were achieved in this clinical trial (26) (Tables 1, 2).
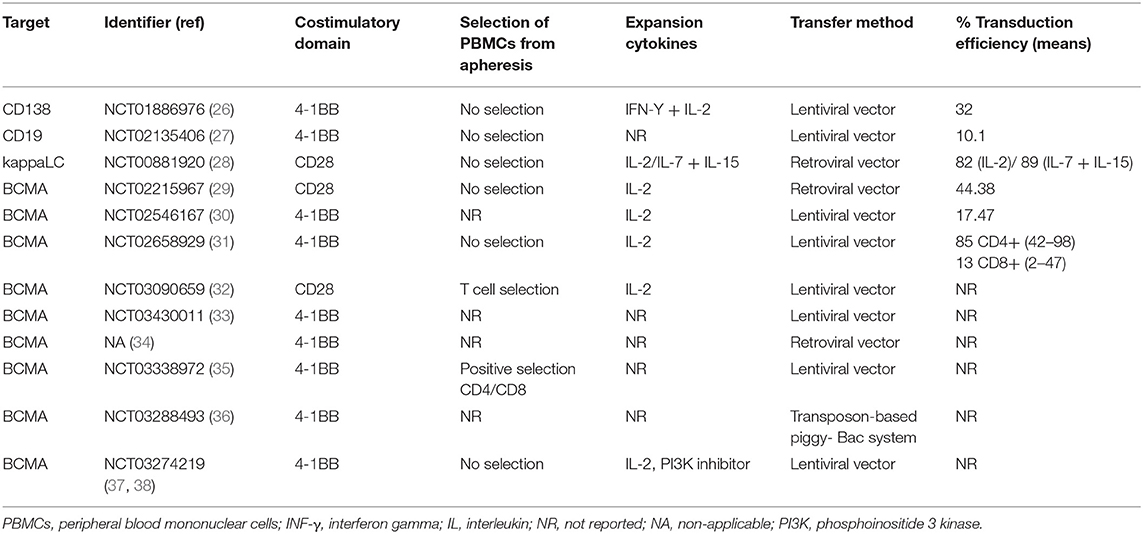
Table 1. Characteristics of T-cell products generated in each clinical trial.
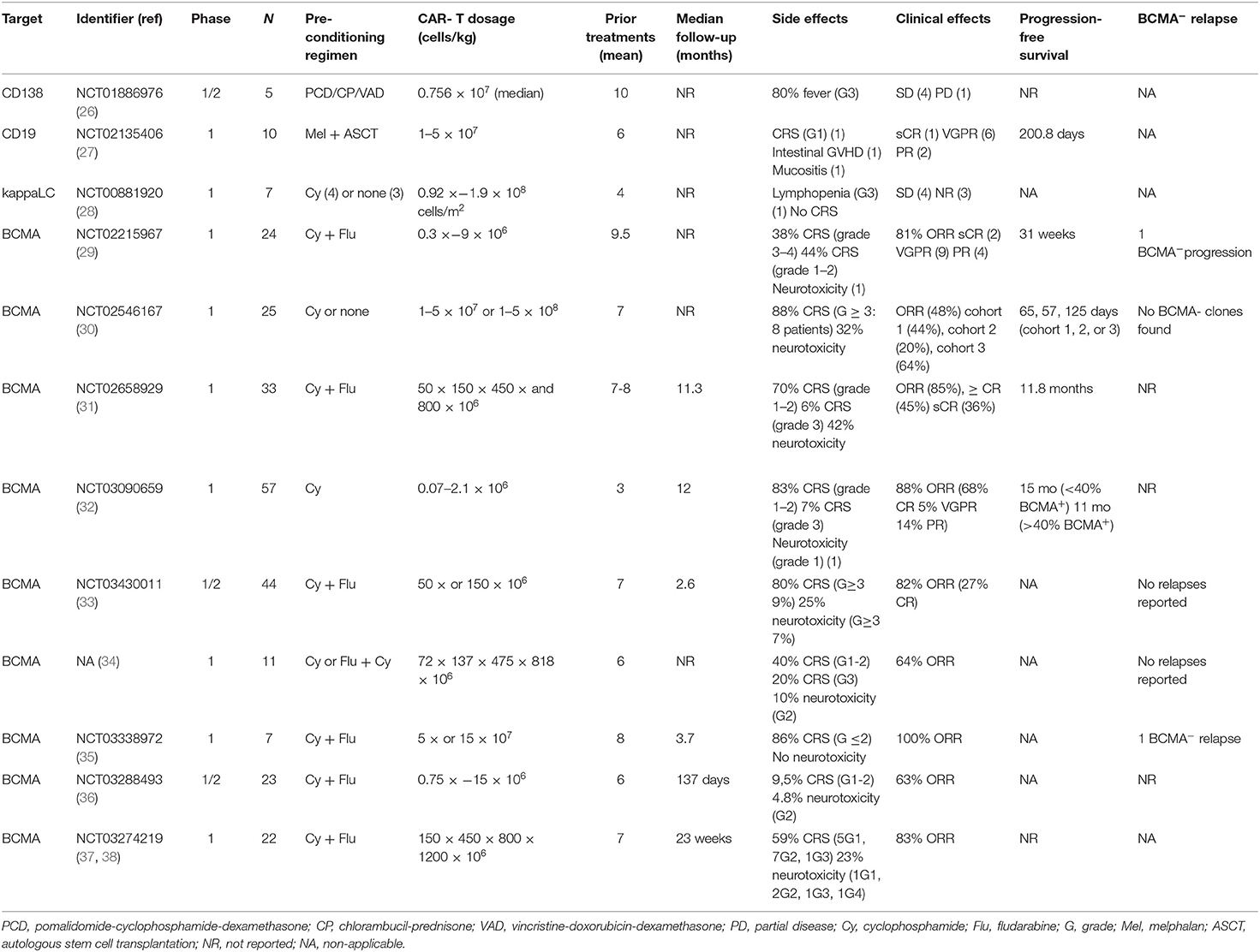
Table 2. Published clinical trials of CAR T-cell therapy in multiple myeloma.
CD19 is a B lymphocyte-specific surface protein which constitutes a component of the B-cell co-receptor complex and belongs to the immunoglobulin superfamily. It is expressed throughout B-cell differentiation, from pre- to mature B-cells (39, 40). Although expression of CD19 is rare in plasma cells, there is a small population of CD19positive myeloma cells which has been discovered to be more pre-mature and might constitute the myeloma-initiating or myeloma-stem cells. They have been associated with high-risk disease, poor prognosis, relapses and reduced survival (41, 42). CD19 is the most widely studied target antigen for the development of CAR T therapies with two products (Kymriah and Yescarta) approved for the treatment of ALL and DLBCL, and multiple published and ongoing clinical trials (43). Therefore, targeting CD19 in MM represents an interesting strategy to focus on this CD19positive myeloma cell subset. In the study NCT02135406, 10 refractory MM patients were infused with autologous CD19 CAR T-cells after autologous stem cell transplantation (ASCT). All patients included received previously a first ASCT resulting in poor response with progression-free survival (PFS) of <1 year. CD19 expression in myeloma cells was assessed by flow cytometry and, as expected, the predominant myeloma population was CD19negative in all patients. However, seven out of nine evaluable patients presented a small CD19positive subset (from 0.04 to 1.6%) (27). Patients were infused with 1–5 x 107 cells/kg CD19 CAR T-cells (CTL019) 2 weeks after high-dose melphalan and a second ASCT. In 2015, the clinical outcome of the first treated patient was reported with a sustained complete remission lasting for at least 12 months in spite of CD19 expression-absence in most of the myeloma cells (44). Six out of 10 patients infused obtained a very good partial response (VGPR) at day 100 post-transplantation. To find out whether CTL019 infusion did increase PFS after ASCT, they compared PFS from each subject after prior ASCT alone vs. ASCT+CTL019 treatment. Two patients significantly increased PFS after CTL019 treatment (479 vs. 181 days; 249 vs. 127 days). These results highlight the recognition of target antigen by the CAR even when it is present in very low intensity or non-detectable by flow cytometry (45) (Tables 1, 2). The same group conducted a phase II clinical trial (NCT02794246) to study the efficacy of CD19 CAR T-cells infusion 60 days post-ASCT in 5 MM patients. No results have been published yet. The combination of autologous/allogenic CD19 CAR T-cells and BCMA CAR T-cells has also been explored.
Despite the success achieved with CD19 CAR T-cells in hematological malignancies, sustained clinical responses need long-term in vivo CAR persistence which is linked to B-cell aplasia and therefore impaired humoral immunity. This toxicity occurs due to the expression of CD19 in normal B-lymphocytes as it is a pan-B-cell expression marker. New antigens with more restricted distribution need to be explored to reduce cytotoxicity and allow normal humoral immunity recovery even with in vivo CAR persistence (10, 28). Expression of surface immunoglobulin, with either kappa (κ) or lambda (λ) light chain, is limited to mature B-cells and mature B-cell malignancies. Although normal plasma cells do not maintain immunoglobulin expression, a clonogenic MM-initiating population has been described which expresses surface immunoglobulin (46). Directing CAR T-cells to a certain type of immunoglobulin light chain (κ or λ) would eliminate the MM-monoclonal cells expressing the target light chain while avoiding cytotoxicity against normal mature B-cells expressing the remaining one. Therefore, targeting immunoglobulin kappa light chain (IgkLC) might be a feasible strategy to direct CAR T therapy to MM while being more restrained within the whole B-cell subset. In Ramos et al. (28) a clinical trial (NCT00881920) is described to evaluate safety and efficacy of κCAR T-cells in chronic lymphocytic leukemia (CLL), non-Hodgkin lymphoma (NHL) and MM patients. Seven relapsed/refractory MM patients were infused with 0.92–1.9 × 108 cells/m2 after cyclophosphamide preconditioning. No serious CAR-related adverse events were reported excluding a patient with grade 3 lymphopenia. According to clinical responses, four out of seven patients reached stable disease (SD) from 6 to 24 months. The other three patients did not respond to the therapy (28) (Tables 1, 2).
B-cell maturation antigen is the ultimate target studied for the development of CAR T therapies for MM with up to 53 clinical trials worldwide. BCMA is a transmembrane glycoprotein which constitutes part of the tumor necrosis factor receptor (TNFR) superfamily. It participates in the B-cell differentiation into plasma cells and in its long-term survival and proliferation (16, 47). Besides, expression of BCMA was confirmed by Friedman et al. (48) in malignant MM cells in 100% of the patients analyzed, though levels were variable. Several clinical trials have assessed the benefits of targeting BCMA for the treatment of MM either with anti-BCMA bispecific T-cell engagers (BiTE) or anti-BCMA antibody-drug conjugates (49, 50). Indeed, multiple clinical trials have explored the effect of BCMA CAR T-cells in the treatment of MM (51).
The first clinical trial designed with anti-BCMA CAR T-cells was carried out in the National Cancer Institute (NCT02215967) and results were presented by Brudno et al. (29) in 2018. They enrolled 24 patients, 10 in a dose escalation phase (0.3 × −3 x 106 cells/kg) and 16 were infused with the highest dose (9 × 106 cells/kg). They reported an overall response rate (ORR) of 81% among the 16 patients treated with the highest dose with 2 patients achieving a stringent complete response (sCR), 8 a VGPR, 3 a partial response (PR) and 3 non-responding to treatment. Peak CAR+ cell levels in peripheral blood, occurring 7 days-post-infusion, were associated with anti-myeloma responses. Cytokine release syndrome (CRS), resulting from T-cell activation after CAR T engagement, and neurotoxicity are the major CAR T-related adverse events described to date (52). CRS grade 3–4 was reported in 5 out of 16 patients infused with the highest dose (38%) and mild CRS was present in 7 patients (44%). Neurotoxicity was not reported or limited to delirium or confusion except for patient 15 who presented encephalopathy. Higher levels of bone marrow plasma cells were also associated with a more severe CRS. BCMA expression was also assessed in myeloma cells pre- and post-treatment and patient 11 was found to have BCMAnegative myeloma cells at week 56 post-infusion followed by myeloma progression at week 68 with mixed BCMA expression (29) (Tables 1, 2).
Cohen et al. (30) reported a phase I clinical trial (NCT02546167) to evaluate safety and efficacy of BCMA CAR T-cells in relapsed/refractory MM patients. Three different cohorts were studied, cohort 1: 1–5 × 108 BCMA CAR T-cells/kg infused (9 patients), cohort 2: 1–5 × 107 cells/kg + Cyclophosphamide as preconditioning (5 patients), and cohort 3: 1–5 × 108 cells/kg + Cyclophosphamide as preconditioning (11 patients). In this trial, all 25 patients were infused in a 3-dose-split protocol over 3 days (30). CRS was reported in 22 out of 25 patients (88%) (grade 3–4) and neurotoxicity was observed in 8 out of 25 patients (32%). Three patients presented severe neurotoxicity (grade 3–4) which correlated with high tumor burden, a dose of 5 × 108 cells/kg and grade 3-4 CRS. The objective responses within the cohorts were: 44% in cohort 1, 20% in cohort 2 and 64% in cohort 3 (1 CR, 5 VGPR and 1sCR). The ORR was 48% (12 out of 25), with 1-5 x 108 cells/kg being the most effective dose (11 responding-patients) (30) (Tables 1, 2).
In the Bluebird study NCT02658929, 33 patients with relapsed/refractory MM were treated with anti-BCMA CAR T-cells (bb2121) in a dose/escalation study (50–800 × 106 cells/kg) (31). In this study, 25 patients presented CRS, among them 23 (70%) had grade 1–2 and 2 (6%) had grade 3–4. Neurologic toxicities were also remarkable occurring in 14 patients (42%). The ORR was 85% with 15 complete responses (45%), and all responding-patients had negative minimal residual disease (MRD). However, six out of the 15 patients with CR finally relapsed (31) (Tables 1, 2).
In Zhao et al. (32) the authors developed a BCMA-directed CAR T-cell containing two heavy chain-only antibodies (VHH) which targets two different BCMA epitopes (LCAR-B38M). The safety and efficacy of this CAR T-cell product were studied in a clinical trial with 57 patients enrolled (NCT03090659) (32). They reported 51 (90%) patients who developed CRS on variable grades (Table 2). One patient presented neurotoxicity grade 1 with seizure-like activity, agitation and aphasia. The authors described an ORR of 88% with 68% CR, 5% VGPR and 14% PR. They analyzed BCMA expression on all patients but no correlation among BCMA expression, progression-free survival, overall survival or clinical response was found. Correlation between CAR T-cell dose and clinical response was not found either (32) (Tables 1, 2).
The Memorial Sloan Kettering Cancer Center (MSKCC) has developed different CAR T-cell products containing human-derived or fully human scFv antibodies against BCMA named JCARH125, MCARH171, and FCARH143. All CAR T-cell products have been included in phase 1 clinical trials. JCARH125 is an anti-BCMA CAR T-cell product studied in a multicenter clinical trial in the United States (EVOLVE, NCT03430011). In this study, 44 patients were infused with two different dose levels (50 × 106 or 150 × 106 cells/kg). With a 2.6 months median follow-up, 82% overall responses were reported with 27% CR. CRS was present in 80% of patients and neurotoxicity in 25% (33, 51) (Tables 1, 2). MCARH171 expresses a different human-derived scFv antibody than JCARH125 and T-cells were transduced with γ-retrovirus instead of using a lentiviral vector. Safety and efficacy were evaluated in a cohort of 11 patients with an ORR of 64%. CRS grade 1–2 occurred in 40% of patients and 20% had CRS grade 3. Neurotoxicity was only experienced by one patient with grade 1 encephalopathy (34) (Tables 1, 2). It has been described that defined ratios of CD4+:CD8+ in the T-cell product might benefit expansion and function (53). FCARH143 employs the same construction as JCARH125 but CD4+ and CD8+ T-cells were cultured separately ex vivo and infused in a defined 1:1 ratio. Seven patients have been reported to date with an ORR of 100% at 28 days post-infusion. CAR+ T-cells were detectable 90 days after infusion. One patient relapsed at day 60 and tumor biopsy demonstrated the presence of a BCMAnegative plasma cell population (35) (Tables 1, 2).
A novel CAR T-cell product designed with a non scFv antibody but a different BCMA-specific antibody-mimetic binding domain has been evaluated in a clinical trial. Besides, a different non-viral transfection method was used, named transposon piggy-bac system, which allows more cargo capacity and preferential transfection of stem cell memory T-cells with a lower cost (Table 1). To date, 23 patients have been treated in 5 different dose groups (Table 2). Two patients experienced CRS (grade 1–2) (9.5%) and neurotoxicity was reported in one patient (grade 3) (4.8%). ORR goes from 50% to 100% depending on dose group with a median of 63% in all patients (36, 51) (Table 2).
In Shah et al. (37) the authors designed a clinical trial with a next-generation CAR T-cell (bb21217) using the same construct as bb2121 (31) but with a novel approach by employing phosphoinositide 3 kinase (PI3K) inhibitor bb007 during ex vivo expansion to enrich the product in memory-like T-cells (Table 1). In the update presented at the American Society of Hematology Annual Meeting 2019, they reported 22 infused patients with an ORR of 83% (15/18 evaluable patients). CRS occurred in 59% of patients and neurotoxicity in 23% (38) (Table 2).
With an increasing number of CAR T-cell-treated patients, observations of therapy-related toxicity and disease relapse are showing the current limitations of this therapeutic modality (17, 54, 55). Key challenges related to CAR T-cell therapy include toxicities, antigen escape, suboptimal activation and persistence of CAR T-cells.
Immunotherapy with adoptive T-cells targeting myeloma-associated antigens are at various stages of development and have brought a new hope for cure (31, 38, 56, 57). Nevertheless, severe toxicities accompany this promising technology as it has been reported with the increasing clinical experience with CAR T-cell therapy (58–63). CAR T-cell related toxicities can be divided into two categories: (1) general toxicities due to T-cell recognition and activation against tumor cells and followed by uncontrolled release of high levels of cytokines (on-target, on-tumor toxicities); and (2) toxicities appearing from specific binding between CAR T-cell and its target antigen expressed in normal cells (on-target, off-tumor toxicities) (17, 64).
Severe and sometimes lethal increases in systemic cytokine levels have been observed in patients treated with CAR T-cells in many clinical trials. Robust interactions between CAR modified T-cells either with tumor or host immune cells may result in CAR T-cell activation and expansion. In some cases, this immune cell activation and uncontrolled cytokine release can be toxic for patients (64). Typically observed CAR T-cell related toxicities are cytokine release syndrome (CRS) and neurotoxicity. CRS is the most-common toxicity of cellular immunotherapy and it appears as a consequence of accelerated expansion and activation of CAR T-cells. This pronounced activation provokes an extreme release of serum levels of interferon gamma, interleukin 6, among other inflammatory cytokines. CRS commonly appears within the first 2 weeks post-infusion of CAR T-cells. Clinical symptoms of this on-target, on-tumor toxicity goes from mild fevers, malaise and flu-like symptoms to severe sepsis-like indications such as high-grade fevers, hypotension, hypoxemia, organ dysfunction, coagulopathy, and pancytopenia which may require intensive care admission (64–66). Treatment with corticosteroids could reduce CRS but to the detriment of effectiveness of CAR T-cells due to suppression of T-cell function and/or induction of T-cell apoptosis (67, 68). Interleukin 6 blockade by anti-interleukin 6 receptor antibodies, such as tocilizumab, is the most commonly used treatment of severe CRS. Besides, tocilizumab does not seem to have a major impact on CAR T-cell efficacy (64–66). Another common adverse event of CAR T-cells is neurotoxicity. This event is observed in up to 42% of patients receiving anti-BCMA CAR T-cells and usually overlaps or appears shortly after CRS (Table 2). Neurotoxicity is commonly restricted to low grade symptoms such as few days of mild confusion, somnolence, and/or word-finding difficulties. Eventually, neurotoxicity can evolve to more severe immune effector cell-associated neurotoxicity syndrome (ICANS) (66) which includes focal deficits, seizures, and fatal cerebral edema (69). Pathophysiology of neurotoxicity is not well-described, but high levels of inflammatory cytokines within central nervous system associated with an increase in endothelial cell activation and vascular permeability have been observed (65). Some factors associated with a higher risk of neurotoxicity include high tumor burden, and more rapid and severe CRS; however, more studies are needed to better understand this toxicity (69).
CAR T-cell infusion can also be associated to on-target, off-tumor toxicities due to their uncontrolled growth and excess cytokine release after recognition of target antigen expressed on non-malignant cells (58, 70, 71). The challenge to find tumor-restricted antigens that are not expressed on normal cells, is the major concern in CAR T-cell development (51, 72). Current target antigens of CAR T-cells are usually present on the surface of non-malignant cells, even though at low level. However, this low level could eventually lead to severe off-tumor toxicities (73). B-cell aplasia after CD19 CAR T-cells infusion, is an excellent example of on-target, off-tumor toxicity. The functionality of CD19 CAR T-cells is always followed by B-cell aplasia as a result of the depletion of CD19positive B-cell progenitors. As a consequence, reduced immunoglobulin level is observed in serum of these patients (74). However, while this side effect can be addressed by immunoglobulin replacement (27, 75, 76), damage to more essential organs or tissues can be fatal (61, 73). Among MM target antigens, CD38 has been reported to have medium-expression level on normal hematopoietic cells and non-hematopoietic tissues including prostate epithelial cells. The potency of CAR T-cells does not guarantee safety when targeting this widely expressed protein despite the currently use of anti-CD38 monoclonal antibodies clinically approved (77). Moreover, expression of CD38 on activated T-cells may suppose a detrimental effect because of fratricide cytotoxicity among CD38 CAR T-cells (78). Therefore, the affinity of the scFv antibody used to develop CD38 CAR T-cell product, needs to be accurately optimized to avoid on-target, off-tumor side effects (21, 71). The same happens with CD138 target antigen. Its widely expression leads to concerns with regard to off-tumor toxicities using CD138 CAR T-cells in myeloma patients. Although no severe epithelial toxicities have been noted at present (25), more studies evaluating CD138 CAR T-cells should include strategies to avoid off-tumor toxicities while maintaining anti-tumor effect. BCMA is the most commonly targeted antigen in CAR T-cell therapies for MM. BCMA plays a fundamental role in long-term plasma cell survival and B-cell differentiation into plasma cells (47) with an increasing expression during B-cell differentiation. It is found only in late memory B cells and normal and malignant plasma cells at varying intensities (79–81). Therefore, B-cell depletion is not anticipated when targeting BCMA, as the majority of normal B-cells are BCMAnegative. Although the toxicity profile of BCMA CAR T-cells seems to be manageable (80–82), alternative approaches have been developed to overcome this CAR T-cell therapy limitation.
Antigen escape post-infusion of modified CAR T-cells is an emerging issue (17, 83, 84). Relapses have been observed in long term follow-up studies, but the mechanisms for these relapses need to be better described. The most common reason for relapse after CAR T-cell therapy is the emergence of tumor cells with loss or downregulation of target antigen expression. The use of CD19 CAR T-cells in ALL patients has highlighted the loss of CD19 antigen expression due to therapeutic pressure. Consequently, CD19negative tumor cells, have been reported in relapses. This can be caused by lymphoid to myeloid trans-differentiation, alternative splicing leading to target epitope loss, or selection of pre-existing antigen-negative leukemia cell clones (85–87). In the myeloma setting, BCMA loss or downregulation on residual MM cells after BCMA CAR T therapy has been reported in several clinical trials to date (29, 30, 88). Antigen escape in MM is more likely to occur due to the coexistence of numerous tumor subclones in treated patients resulting in a potential advantage to emerging or preexisting BCMAlow or BCMAnegative subclones during treatment (51). In the NCI trial, one patient presented persisting BCMAnegative myeloma cells in the bone marrow 56 weeks after BCMA CAR T-cell infusion and at the time of myeloma progression (29). In the UPENN trial, 12 out of 18 (67%) evaluable patients had significantly diminished levels of BCMA expression intensity commonly 1 month after CAR T-cell administration (30). This reduction in BCMA intensity was more frequent on residual myeloma cells from patients responding to therapy than non-responder patients. Regarding to FCARH143 trial, the authors reported a patient who relapsed presenting a BCMAnegative myeloma subclone and an overall reduction of 70% BCMA expression level in the BCMApositive myeloma cell population (35). The observed loss and downregulation of BCMA expression on the surface of myeloma cells after CAR T-cell infusion in several clinical trials, highlights an imperative need to investigate the mechanism of resistance to BCMA CAR T-cell therapy.
A potential limitation for the clinical application of CAR engineered T-cells would be the presence of soluble protein antigens in the serum of patients. Target antigens can be cleaved from the cell membrane and released into blood circulation, therefore CAR T-cell therapy could be limited due to binding to soluble target antigens. Thus, the functionality of CAR T-cells could be abrogated by soluble antigens (89). In the myeloma setting, CD138 extracellular domain has been reported to be cleaved from cell surface, which could abrogate CAR T-cells function by blocking their antigen-binding domain and, hence, resulting in immune escape (90). Cleavage of the target antigen is not exclusive to CD138 protein, but it has been well-described that BCMA can be released from myeloma cells into the serum of patients. BCMA expression on the surface of myeloma cells can be modified by a protease called γ-secretase (GS), which mediates BCMA shedding within the transmembrane domain, leading to the release of a soluble fragment of BCMA (sBCMA) (91). Consequently, recognition of tumor cells by CAR T-cells could be hampered by soluble BCMA as a result of reducing BCMA density on the tumor cell surface and/or blocking the antigen-binding domain of the CAR. Thus, the presence of sBCMA in the serum of MM patients is being actively discussed as an obstacle for BCMA CAR T-cell therapy. In this sense, controversial data have been described. Several studies have reported that BCMA CAR T-cells were not abrogated by soluble BCMA protein (48, 80). However, alternative BCMA CAR T-cells have recently shown that sBCMA in concentrations of 10 ng/mL decreases the frequency of cytokine-producing BCMA CAR T-cells, at the same time that did not compromise CD19 CAR T-cells function. Moreover, high concentrations (333–1,000 ng/ml) of soluble BCMA protein affected cytotoxic capacity of CAR T-cells against 1 out of 3 BCMApositive cell lines (92). These contradicting observations might be due to the use of different BCMA CAR T-cells directed to distinct epitopes, therefore it could happen that the BCMA-CAR target-epitope is not accessible in the soluble BCMA conformation.
Response rates of 64–85% have been achieved in myeloma patients treated with BCMA CAR T-cells (29, 30). However, only 8-39% of patients had a sustained VGPR or CR/sCR and this, together with the fact that most of the current clinical trials to date have reported BCMApositive relapses, highlights a loss of efficacy of BCMA CAR T-cells against malignant plasma cells. This loss of efficacy might be a consequence of limited persistence of the CAR T-cells in vivo. Accordingly, long-term responses (>2 years) were unusually seen after BCMA CAR T-cell infusion (29–31). Nowadays, most of the CAR T-cell products are generated from autologous T-cells (17). Although this personalized cellular therapy has reported notable success in clinical trials (30, 31), the generation of CAR T-cells from patients could limit their application for multiple reasons. The first limiting factor is the harvesting of an adequate T-cell number from cancer patients who commonly present lymphopenia due to disease or previous treatments (17). Besides, the generation of autologous CAR T-cells might be a long procedure and progression of the disease could happen during manufacturing in advance-stage cancer patients (63, 93). Finally, intrinsic characteristics of apheresis products may be another limitation factor for the generation of autologous CAR T-cells (94). In the myeloma setting, harvesting the sufficient number of T-cells from patients seems to be feasible to produce CAR T-cells [e.g., the bb2121 clinical trial has shown that in 100% of patients who underwent leukapheresis, BCMA CAR T-cells were successfully generated (31)]. However, the quality of harvested T-cells in myeloma patients is likely a significant limiting factor due to deterioration of the immune system in these patients. Thus, the most solid indicator associated with response in the myeloma arena is the level of in vivo expansion and persistence of infused CAR T-cells (95). On the other hand, patient age (96, 97), number of prior lines of treatment (98), and the disease itself (99, 100) may limit the number and quality of patient-derived T-cells, potentially influencing the potency and variability of the CAR T products. Furthermore, the logistics of clinical manufacturing using patient-derived T-cells limits the accessibility of these therapies.
To overcome CAR T therapy limitations in MM, new strategies have been developed in order to create next generation CAR T-cells to treat myeloma patients (Figure 1).
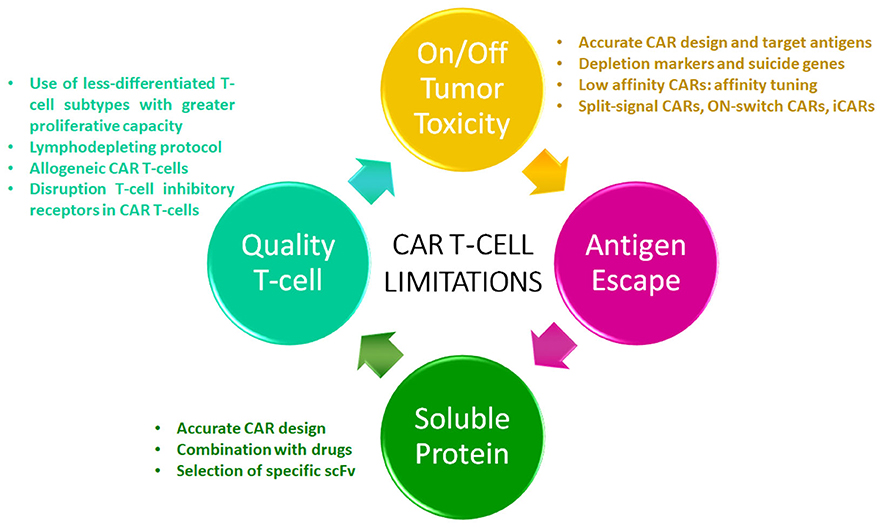
Figure 1. CAR T-cell limitations. The diagram shows the current limitations of this therapeutic modality in multiple myeloma. Strategies to overcome these limitations are listed.
To achieve clinical efficacy while avoiding on-target, on-tumor toxicities, CAR T-cells activation and cytokine release must remain under a controlled level. The affinity of the antigen-binding domain for the tumor epitope, tumor burden, antigen density on the surface of cancer cells, and costimulatory domains present in the CAR, along with other factors, are implicated in the kinetics of CAR T-cell activation (101, 102). Dose-escalation schedules in phase I clinical trials are needed to determine the therapeutic window of CAR T-cell activation for each CAR due to differences in CAR design. In order to optimize this therapeutic window, diverse regions of the CAR gene can be accurately modified (17). In this sense, the costimulatory domain is considered one of the key points to optimize CAR design to reduce on-target, on-tumor toxicities. The most common costimulatory domains used in CAR T-cells are CD28 and 4-1BB. The use of CD28 costimulatory region has been correlated with a high and pronounced CAR T-cell activation and, hence, immune exhaustion phenotype. On the other hand, CAR T-cells with 4-1BB costimulatory region present lower peak of expansion, resulting in prolonged persistence and a lower risk of cytokine-mediated toxicities (103). Therefore, the costimulatory domain chosen in the CAR design may explain, at least in part, the toxicity-pattern differences observed in patients treated with CD28 containing CAR T-cells in which earlier onset CRS is more commonly observed as compared to patients treated with 4-1BB containing CAR T-cells (66). Some studies have also reported significant neurotoxicity and death from cerebral edema when CD28 containing CAR T-cell products were used (62). However, other clinical trials described no difference in the rate or grade of CRS and/or ICANS between CAR T-cells containing CD28 vs. 4-1BB costimulatory domains (63, 104, 105). Further studies are needed to better elucidate the link between costimulatory domain and toxicity events in patients receiving CAR T-cells. Others aspects of the CAR design such as the extracellular hinge region and/or transmembrane domain can also be optimized to reduce on-target, on-tumor toxicities. In fact, Ying et al. (86) generated a new anti-CD19 CAR variant [CD19-BBz (86)], which released lower levels of cytokines, expressed higher levels of anti-apoptotic molecules and proliferated more slowly than the prototype CD19-BBz CAR T-cells, while retained robust cytolytic activity. A phase I clinical trial was developed to evaluate this new variant and no significant CRS or ICANS events were reported while achieving a CR rate of 54.5% (106). In addition, novel strategies to control CAR T-cells toxicity include the engineering of depletion markers [e.g., truncated epidermal growth factor receptor [EGFRt]] and suicide genes (e.g., iCasp9) into the CAR design, providing a way to delete CAR T-cells if on-target, on-tumor (and/or on-target, off-tumor) toxicities appear. Accordingly, co-expression of the CAR gene together with the epitope recognized by clinically approved-monoclonal antibodies has been explored. For example, CD20 and EGFRt, which are targetable with rituximab and cetuximab, respectively (107–109). Furthermore, both depletion markers can be used to monitor T-cell transduction. Likewise, apoptosis of CAR T-cells can be induced by caspase pathway activation in iCasp9 next-generation CAR T-cells after the addition of the dimerization drug (109, 110).
Multiple strategies to reduce off-tumor toxicities are now under development and are likely to offer novel clinically effective CAR T-cell products (111). Some of these approaches include the restriction of the recognition of normal cells by the CAR optimizing the specific interaction with tumor cells, either by (1) including the recognition of more restricted tumor-antigens or recognition of multiple antigens; and/or (2) limiting the spatial and temporal activity of CAR T-cells. In this sense, a novel target antigen has been reported for the immunotherapy of MM, GPRC5D. GPRC5D is a human orphan family C G protein-coupled receptor recently described to be expressed on 98% of CD138positive cells (112, 113). The expression pattern of GPRC5D has proven to be very restricted in non-plasma tissues with the only exception of hair follicle cells. Consequently, GPRC5D CAR T-cells were generated by Smith et al. (113) showing anti-tumor efficacy against myeloma cells both in vitro and in a mouse model. Of note, GPRC5D CAR T-cells were also effective in eradication of myeloma after BCMA CAR T-cell treatment in a murine model which might be an option to overcome BCMA antigen escape. These data suggest that GPRC5D CAR T-cells could be an attractive alternative treatment in MM.
A distinct approach to limit off-tumor toxicities when a tumor-restricted antigen is not identified, could be the “affinity-tuning,” which is based on differences in density of antigen level among tumor and normal cells. Low-affinity CAR T-cells (low affinity scFv antibodies) could be generated to target antigens expressed at higher density on tumor cells than on normal cells. This is based upon published observations that T-cell activation initiated through the endogenous αβ TCR may result from reaching an activation threshold level, which can be triggered by binding a few high-affinity TCRs or greater number of low-affinity TCRs (114, 115). Several studies have demonstrated that a CAR with reduced affinity rendered T-cells preferentially activated by high, but not low, density of target antigen (116–119). However, immune escape has also been observed if downregulation of target antigen occurs (116, 120). In the myeloma setting, off-tumor toxicity profile of low affinity CD38 CAR T-cells has been evaluated. Drent et al. (71) showed that CD38 CAR T-cells with up to 1,000-fold decreased affinity were effective against CD38positive myeloma cells, whereas CD38positive healthy hematopoietic cells resulted unaffected.
Novel approaches to prevent off-tumor toxicities are the generation of CAR T-cells which require the recognition of two tumor antigens for their activation. In this regard, “split-CARs” co-expressing two different modules, one containing a scFv antibody along with CD3ζ (signal 1) and the second containing a different scFv antibody along with costimulatory domain (CD28, 4-1BB) have been explored (121, 122). This strategy may result in suboptimal CAR T-cell activation and limited off-tumor toxicity where only one antigen recognition occurs, which may be the case in normal cells. On the other hand, when both antigens are present, as in tumor cells, robust CAR T-cell activation is reached after double recognition (122). An alternative strategy consists of “ON-switch CARs” in which the antigen binding domain is dissociated from the signaling domain (CD3ζ), and CAR T-cell activation is controlled by a small molecule that induces dimerization (123). Subsequently, the intensity of CAR T-cells responses relies on the dose of dimerization molecule. Recently, “AND-gate CARs” has arisen as a promising technology. In this approach, the recognition of the first antigen by the synthetic Notch receptor induces the excision of the transcription factor which allows the expression of the CAR gene targeted against the second antigen. The expression of the CAR on the cell surface allows T-cell activation after antigen recognition (124). Therefore, CAR expression and CAR T-cell activation and hence, tumor elimination, can only happen when both target antigens are present. Nevertheless, the loss of the first antigen targeted by the synthetic Notch receptor which may result in immune escape, as well as slow activation kinetics, are the major limitations of this approach.
Another next-generation technology is the “inhibitory CAR” (iCAR) which incorporates the signaling region of an immunoinhibitory receptor (i.e., PD-1 or CTLA-4) to limit on-target, off-tumor toxicity. This novel strategy consists on the expression of a conventional CAR together with an iCAR in the same T-cell (125). The recognition of the target antigen by the iCAR restricts T-cell activation while the absence of this antigen allows CAR T-cell activation. Again, the challenge of this technology is finding antigens with an optimal expression pattern. First, iCAR should recognize antigens that are strictly expressed on normal cells whereas conventional CAR should recognize antigens that are specifically express on tumor cells.
CAR engineered T-cells targeting multiple antigens can be generated to address the antigen loss and therefore, reduce relapse rates (55). Next generation CAR T-cells can be developed by (1) combining different CAR T-cell products directed against single target antigens pre-infusion (co-administration of two or more different CAR T-cell products) (Figure 2A) or (2) transducing the same T-cell with two different complete CAR constructs (“bicistronic CAR T-cells”) (Figure 2B). Alternatively, bi-specific CAR T-cells, called also “tandem CAR T-cells,” can be generated by designing a single CAR construct with two (or more) different binding domains against two different antigens (126, 127) (Figure 2C). One important factor in the CAR design is the length of the transgene. In this sense, “tandem CAR T-cells,” which have smaller transgene length, show an advantage compared to “bicistronic CAR T-cells.” On the other hand, “tandem CAR T-cells” need design optimization including linker sequence, spacer size and VH-VL orientation between both scFv antibodies to accomplish desired antigens recognition and CAR T-cell activation (128). In 2019, Yan et al. (129) evaluated the efficacy and safety of the co-infusion of CD19 and BCMA CAR T-cells in relapse/refractory MM patients (NCT 03455972). CAR T-cell products were co-administrated on day 14–20 after autologous transplantation. The authors reported an ORR of 92.6% with 11/28 (40.7%) CRs or sCR, 8/28 (29.6%) VGPR and 5/28 (18.5%) PR. The median OS was 16 months. CRS was grade 1–2 in 19 (67.9%) patients (129). Although the exact mechanism of co-infusion remains unclear, expanding the coverage of MM cell targets might lead to better depletion of MM cell clonogenicity improving the duration of responses avoiding antigen escape. Co-infusion of two CAR T-cell products could likely reduce antigen escape, at the expense of a possible increase in toxicities as a result of simultaneous targeting of two or more antigens. Further studies with longer follow up and larger cohort of patients are needed in order to make conclusions regarding the use of combinatory CAR T-cell products. Another possible combination to overcome antigen escape limitation in MM is targeting both BCMA and TACI on myeloma cells. However, it has not been described whether BCMA-directed therapy affects TACI expression on the surface of residual myeloma cells. APRIL, a proliferation-inducing ligand, is the natural ligand of BCMA and TACI, and it is secreted in a trimeric form (130–132). In the preclinical setting, APRIL CAR T-cells have been successfully developed targeting both BCMA and TACI (133). Consequently, a clinical trial phase I/II is ongoing to evaluate the safety and efficacy of APRIL CAR T-cells in myeloma patients (NCT03287804) (134). Relapsed/refractory MM patients were infused with 15 × 106 (1/11 pts), 75 × 106 (3/11 pts), 225 × 106 (3/11 pts), 600 × 106 (3/11 pts), and 900 × 106 (1/11 pts) APRIL CAR T-cells. The ORR was 43% (28 PRs and 14% VGPRs) in patients receiving ≥ 225 × 106 CAR T-cells (134). Long-term follow up is needed to assess the efficacy of this dual-targeting CAR. Recently, Maus et al. (135) designed a novel human APRIL CAR T-cell product preserving its trimeric conformation (TriPRIL) showing enhance killing of both BCMA myeloma cell lines and primary myeloma cells. More recently, bicistronic BCMA/GPRC5D CAR T-cells are being explored and, preclinical data demonstrated that BCMA/GPRC5D CAR T-cells can limit BCMA antigen escape-mediated relapse in a murine model (136).
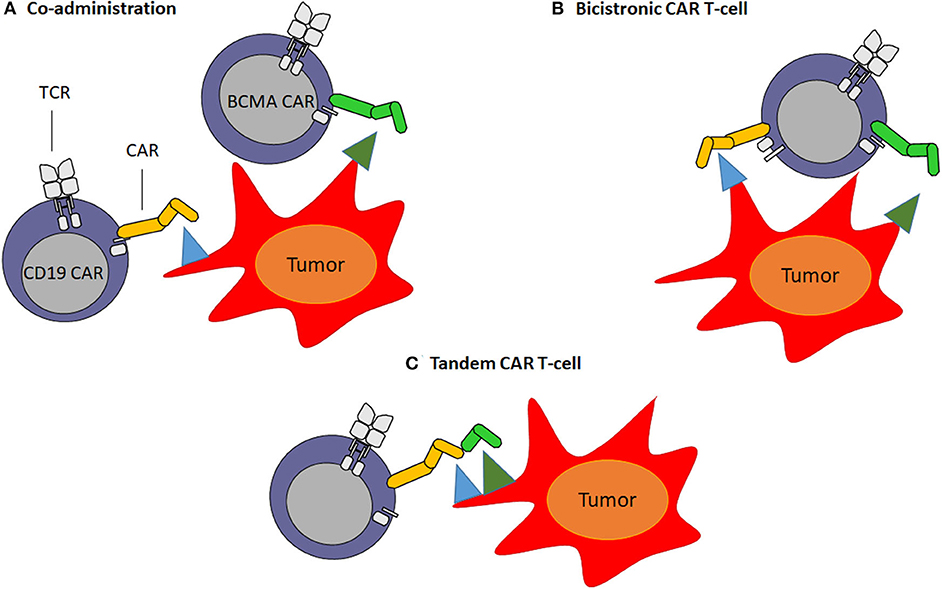
Figure 2. Multi-targeted CAR T-cell approaches. (A) Co-administration: involves production of two separate CAR T-cell products infused together or sequentially. (B) Bicistronic CAR T-cells: consist of the expression of two different CARs on the same T-cell. (C) Tandem CAR T-cells: consist of encoding two different scFv antibodies on same chimeric antigen receptor protein using a single vector.
Blocking the cleavage of target antigens from myeloma cells could be an interesting strategy to avoid the release of these target antigens as soluble proteins. In this sense, Pont et al. (92) have recently published that exposure to γ-secretase inhibitors (GSIs) efficiently blocks the release of BCMA from myeloma cells. Consequently, BCMA surface expression on MM cells is increased leading to a higher anti-tumor capacity of BCMA CAR T-cells, as well as improved cytokine production and proliferation in preclinical models (92). Relapsed myeloma patients with downregulated target antigen expression after BCMA-directed treatments could benefit from this novel GSI strategy. However, an optimal dose of GSI should be defined in order to avoid adverse effects on CAR T-cell function since the inhibition of GS did not impair viability or cytolytic activity but reduced IL2 production and proliferation of BCMA CAR T-cells (92). An alternative strategy might be the use of scFv antibodies to generate CAR T-cells directed against epitopes which are not accessible in the soluble protein conformation or belong to the extracellular region of the target protein that remains after cleavage. Thus, it has been reported that sBCMA folds and participates in the formation of high-molecular-weight complexes under physiological conditions (130, 137). Furthermore, data from three trials uncover the absence of correlation between sBCMA concentrations and extent of response (29, 56, 57). Therefore, the selection of the antigen-binding domain is a key factor in CAR design to overcome abrogation of CAR T-cells by soluble protein.
A promising approach to increase CAR T-cell persistence is the use of T-cell products containing a higher frequency of less-differentiated T-cell subtypes such as naïve T-cells (TN), stem cell memory T-cells (TSCM) and central memory T-cells (TCM), which have a superior proliferation capacity showing a delayed exhaustion or senescence immunophenotype (138–140). Compared with conventional CAR T-cell products, less-differentiated CAR T-cells have shown a greater proliferation and killing capacity in preclinical studies. To generate less-differentiated CAR T-cells, several strategies have been developed such as previous pre-selection of TN/TSCM subtypes or manufacturing in the presence of kinase inhibitors (138, 141, 142). In this sense, Shah et al. (37) designed a clinical trial with a next-generation CAR T-cell (bb21217) using the same construct as bb2121 but with a novel approach by employing phosphoinositide 3 kinase (PI3K) inhibitor bb007 during ex vivo expansion to enrich the CAR T-cell product in memory-like T-cells. To date, similar ORR and toxicity profile were observed in eight myeloma patients treated with the lowest dose of bb21217 (150 × 106 CAR T-cells). However, longer follow-up is required. Clinical trials are currently ongoing using CAR T-cell products selectively generated from CD8+ TCM cells to evaluate safety and efficacy (NCT01087294) (142). Another strategy could be to define the CD4:CD8 ratio into the CAR T-cell product. In this sense, increased CAR T-cell expansion has been observed in patients after the infusion of a defined 1:1 (CD4:CD8) CAR T-cell ratio (35, 105, 143).
In most clinical trials conventional CAR T-cells derive from autologous T-cells. Generation of autologous CAR T-cells is a lengthy and elaborated process, time-consuming and logistically challenging, which comprises multiple steps including T-cell isolation and selection, transduction, expansion and infusion into patients. Along with that, the majority of patients are in relapsed/refractory stage and have already received numerous lines of toxic treatments, which further weaken T-cells quality and hence, reduce their immune response against tumor cells. Therefore, the quality/fitness of T-cells from which CAR T-cells are generated might also have an important role in CAR T-cell expansion and persistence, and anti-tumor capacity. An allogeneic CAR T-cell approach might have the potential to circumvent all these challenges by using healthy donor-derived T-cells to produce CAR T-cells which can be available as an off-the-shelf product (144–146). However, the use of allogeneic CAR T-cells could induce graft-versus-host disease (GVHD). In this context, host allo-antigens can be recognized via TCR by allogeneic CAR T-cells. On the other hand, the infused allogeneic CAR T-cells could be attacked by the host immune system owing to the HLA disparity resulting in CAR T-cell elimination. In consequence, further genetic modifications are needed in order to create next generation off-the-shelf allogeneic CAR T-cells. These genetic modifications consist of disrupting the endogenous TCR using gene-editing technology such as CRISPR/Cas9 and TALEN to limit the risk of GVHD, and the addition of a safety element to limit toxicities (14, 147, 148). Moreover, suppression of HLA class I expression by disrupting the HLA-A or b2-microglobulin genes is a novel strategy that is being evaluated to avoid allogeneic CAR T-cell elimination by the host immune system (149). Alternative approaches to prevent early allogeneic CAR T-cell rejection have been explored for instance, the use of more intensive lymphodepletion protocols. Nevertheless, the later immune system reconstitution of the lymphodepleted patient may be an obstacle for the allogenic CAR T-cell persistence (51). Advances in gene-editing techniques are leading to a new scenario in CAR T-cell therapy resulting in the development of universal CAR T-cells from allogeneic healthy donors. In this regard, preclinical results of allogenic second-generation BCMA CAR T-cells, called ALLO-715, were reported (150). In this study the authors, after CAR-transduction, transfected CAR T-cells with messenger RNA of TALEN. In particular, both CD52 and T-cell receptor α-chain genes were specifically disrupted resulting in CAR T-cell resistance to anti-CD52 lymphodepletion treatment, as well as preventing GvHD, respectively, without compromising CAR-mediated cytotoxicity. Moreover, this ALLO-715 BCMA-CAR incorporates a rituximab-sensitive safety switch. Currently, a phase I trial is ongoing to evaluate the safety and efficacy of the ALLO-715 product.
CRISPR/Cas9 gene-editing tool constitutes a promising technology to create next-generation CAR T-cells. This technology offers a wide range of CAR T-cell products, such as more potent CAR T-cells by disrupting inhibitory genes, allogeneic CAR T-cells by endogenous TCR and HLA elimination and novel CAR T-cells by knock-out of the targeted antigens to avoid fratricide effect. However, off-target toxicity is the major barrier of CRISPR/Cas9 gene-editing technology. These off-target toxicities can provoke non-desired deleterious consequences such as activating oncogenes or disrupting tumor-suppressor genes (151). To overcome this limitation, several approaches have been explored such as optimized sgRNA design and Cas9 activity, prior off-target detection assays, and careful selection of the target site to reduce off-target effects (152, 153). Therefore, a deeply understanding of the potential toxic effects of gene-editing in CAR T-cells is required.
Finally, to improve T-cell persistence and, hence T-cell response, we might target several T-cell inhibitory receptors such as PD-1, TIM-3, LAG-3, and CTLA-4 which send inhibitory signals into the T-cell. The expression of these inhibitory receptors on CAR T-cells leads to T-cell exhaustion. Recent studies show that tumor cells can take advantage of these T-cell inhibitory receptors in order to evade the immune response. For example, tumor cells upregulate PD-1 ligand (PD-L1) which causes reduced immune responses through PD1/PD-L1 pathway (154). Therapeutic strategies specifically designed to inhibit these inhibitory signals by immune checkpoint inhibitors, such as anti-PD-1 and anti-CTLA-4, have been described showing promising results in the treatment of solid tumors in addition to hematological malignancies (155, 156). This background has led to design directed-CRISPR/Cas9 technology in order to disrupt immune checkpoints. Some studies suggest an improvement in the anti-tumor efficacy and clinical outcome using these next generation modified CAR T-cells (157–159).
CAR T-cell therapy is an outbreaking technology to treat hematologic cancers, however, several limitations need to be overcome in order to reach optimal patient response. Promising strategies have been proposed to optimize conventional CAR T-cells increasing safety and efficacy and improving manufacturing feasibility. Fundamental modifications in CAR design can be a promising strategy to reduce CAR T-cell toxicities. Although it is still not clear the influence of soluble antigens in CAR T-cell therapy, the existence of soluble proteins in serum of myeloma patients is being actively discussed as an obstacle for this immunotherapy. Therefore, the selection of the antigen-binding domain is a key factor in CAR design to overcome abrogation of CAR T-cells by soluble protein. In the myeloma setting, CAR T-cell treatment will probably become soon a key strategy for the treatment of relapsed/refractory patients. Likewise, resistance mechanisms to CAR T-cell therapy, such as antigen loss, need to be explored and strategies to overcome these limitations will be essential to ensure optimal efficacy in myeloma treatment. Development of novel strategies to increase long-term responses by combining CAR T-cell therapy with different drugs which increase antigen density specifically in myeloma cells avoiding antigen escape and toxicities is mandatory. Nowadays, CAR T-cells in myeloma are usually administered in patients who are refractory after numerous previous lines of treatment. Consequently, finding an adequate bridging therapy for these patients while CAR T-cell products are generated can be difficult. Can we obtain a better efficacy of CAR T-cells in earlier lines of treatment? Ongoing trials are looking at this question. Moreover, a new era is coming in CAR T-cell therapy in myeloma: allogeneic CAR T-cells derived from healthy donors can overcome some limitations as compared to conventional autologous CARs like manufacturing labor, production time and costs.
BCMA CAR T-cells have already proven safety and efficacy in relapsed/refractory patients and CAR T-cell therapy in myeloma is in great development with major efforts to optimize this approach all over the world. Even though there are more unsolved issues regarding the use of CAR T-cell therapy in MM, the field is still arising, and its full potential is about to discover.
EG-G wrote and revised the manuscript, figures and references, and supervised the tables. BS-M wrote the manuscript and tables, assisted in the elaboration of the references list. JP-S supervised the manuscript, figures and references.
The authors would like to thank the CIBERONC (CB16/12/00480) and Red TerCel, and ISCIII (RD16/0011/0015, RD16/0011/0035).
EG-G is an inventor on a patent related to chimeric antigen receptor technologies in myeloma that have been filed by the University of Wurzburg (Wurzburg, Germany).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Palumbo A, Bringhen S, Ludwig H, Dimopoulos MA, Bladé J, Mateos MV, et al. Personalized therapy in multiple myeloma according to patient age and vulnerability: a report of the European Myeloma Network (EMN). Blood. (2011) 118:4519–29. doi: 10.1182/blood-2011-06-358812
3. Timmers M, Roex G, Wang Y, Campillo-Davo D, Van Tendeloo VFI, Chu Y, et al. Chimeric antigen receptor-modified T cell therapy in multiple myeloma: beyond B cell maturation antigen. Front Immunol. (2019) 10:1–12. doi: 10.3389/fimmu.2019.01613
4. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. (2011) 364:1046–60. doi: 10.1056/NEJMra1011442
5. Rajkumar SV, Kumar S. Multiple myeloma: diagnosis and treatment. Mayo Clin Proc. (2016) 91:101–19. doi: 10.1016/j.mayocp.2015.11.007
6. Palumbo A, Avet-Loiseau H, Oliva S, Lokhorst HM, Goldschmidt H, Rosinol L, et al. Revised international staging system for multiple myeloma: a report from international myeloma working group. J Clin Oncol. (2015) 33:2863–9. doi: 10.1200/JCO.2015.61.2267
7. Rajkumar SV. Treatment of multiple myeloma. Nat Rev Clin Oncol. (2011) 8:479–91. doi: 10.1038/nrclinonc.2011.63
8. Mikkilineni L, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood. (2017) 130:2594–602. doi: 10.1182/blood-2017-06-793869
9. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RPT, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. (2015) 33:540–9. doi: 10.1200/JCO.2014.56.2025
10. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
11. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
12. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. (2015) 125:4017–23. doi: 10.1182/blood-2014-12-580068
13. Stoiber S, Cadilha BL, Benmebarek M-R, Lesch S, Endres S, Kobold S. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells. (2019) 8:472. doi: 10.3390/cells8050472
14. Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol. (2018) 11:25–9. doi: 10.1186/s13045-018-0677-2
15. Danhof S, Hudecek M, Smith EL. CARs and other T cell therapies for MM: the clinical experience. Best Pract Res Clin Haematol. (2018) 31:147–57. doi: 10.1016/j.beha.2018.03.002
16. Cho SF, Anderson KC, Tai YT. Targeting B cell maturation antigen (BCMA) in multiple myeloma: potential uses of BCMA-based immunotherapy. Front Immunol. (2018) 9:1821. doi: 10.3389/fimmu.2018.01821
17. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. (2019) 17:147–67 doi: 10.1038/s41571-019-0297-y
18. Deaglio S, Mehta K, Malavasi F. Human CD38: A (r)evolutionary story of enzymes and receptors. Leuk Res. (2001) 25:1–12. doi: 10.1016/S0145-212600093-X
19. Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. (2016) 375:754–66. doi: 10.1056/NEJMoa1606038
20. Morandi F, Horenstein AL, Costa F, Giuliani N, Pistoia V, Malavasi F. CD38: a target for immunotherapeutic approaches in multiple myeloma. Front Immunol. (2018) 9:2722. doi: 10.3389/fimmu.2018.02722
21. Drent E, Groen RWJ, Noort WA, Themeli M, van Bueren JJL, Parren PWHI, et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica. (2016) 101:616–25. doi: 10.3324/haematol.2015.137620
22. O'Connell FP, Pinkus JL, Pinkus GS. CD138 (Syndecan-1), a plasma cell marker immunohistochemical profile in hematopoietic and nonhematopoietic neoplasms. Am J Clin Pathol. (2004) 121:254–63. doi: 10.1309/617DWB5GNFWXHW4L
23. Mccarron MJ, Park PW, Fooksman DR. CD138 mediates selection of mature plasma cells by regulating their survival. Blood. (2017) 129:2749–59. doi: 10.1182/blood-2017-01-761643
24. Kawano Y, Fujiwara S, Wada N, Izaki M, Yuki H, Okuno Y, et al. Multiple myeloma cells expressing low levels of CD138 have an immature phenotype and reduced sensitivity to lenalidomide. Int J Oncol. (2012) 41:876–84. doi: 10.3892/ijo.2012.1545
25. Sun C, Mahendravada A, Ballard B, Kale B, Ramos C, West J, et al. Safety and efficacy of targeting CD138 with a chimeric antigen receptor for the treatment of multiple myeloma. Oncotarget. (2019) 10:2369–83. doi: 10.18632/oncotarget.26792
26. Guo B, Chen M, Han Q, Hui F, Dai H, Zhang W, et al. CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma. J Cell Immunother. (2016) 2:28–35. doi: 10.1016/j.jocit.2014.11.001
27. Garfall AL, Stadtmauer EA, Hwang WT, Lacey SF, Melenhorst JJ, Krevvata M, et al. Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI insight. (2018) 3:e120505. doi: 10.1172/jci.insight.120505
28. Ramos CA, Savoldo B, Torrano V, Ballard B, Zhang H, Dakhova O, et al. Clinical responses with T lymphocytes targeting malignancy-associated κ light chains. J Clin Invest. (2016) 126:2588–96. doi: 10.1172/JCI86000
29. Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, et al. T cells genetically modified to express an anti-B-Cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. (2018) 36:2267–80. doi: 10.1200/JCO.2018.77.8084
30. Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest. (2019) 129:2210–21. doi: 10.1172/JCI126397
31. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, Turka A, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. (2019) 380:1726–37. doi: 10.1056/NEJMoa1817226
32. Zhao W-H, Liu J, Wang B-Y, Chen Y-X, Cao X-M, Yang Y, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. (2018) 11:141. doi: 10.1186/s13045-018-0681-6
33. Mailankody S, Htut M, Lee KP, Bensinger W, Devries T, Piasecki J, et al. JCARH125, anti-BCMA CAR T-cell Therapy for Relapsed/Refractory Multiple Myeloma: Initial Proof of concept results from a phase 1/2 multicenter study (EVOLVE). Blood. (2018) 132:957. doi: 10.1182/blood-2018-99-113548
34. Mailankody S, Ghosh A, Staehr M, Purdon TJ, Roshal M, Halton E, et al. Clinical Responses and pharmacokinetics of MCARH171, a human-derived bcma targeted CAR T cell therapy in relapsed/refractory multiple myeloma: final results of a phase I clinical trial. Blood. (2018) 132:959. doi: 10.1182/blood-2018-99-119717
35. Green DJ, Pont M, Sather BD, Cowan AJ, Turtle CJ, Till BG, et al. Fully human bcma targeted chimeric antigen receptor T cells administered in a defined composition demonstrate potency at low doses in advanced stage high risk multiple myeloma. Blood. (2018) 132:1011. doi: 10.1182/blood-2018-99-117729
36. Costello CL, Gregory TK, Ali SA, Berdeja JG, Patel KK, Shah ND, et al. Phase 2 study of the response and safety of P-Bcma-101 CAR-T cells in patients with relapsed/refractory (r/r) multiple myeloma (MM) (PRIME). Blood. (2019) 134:3184. doi: 10.1182/blood-2019-129562
37. Shah N, Alsina M, Siegel DS, Jagannath S, Madduri D, Kaufman JL, et al. Initial results from a phase 1 clinical study of bb21217, a next-generation anti bcma CAR T therapy. Blood. (2018) 132:488. doi: 10.1182/blood-2018-99-116953
38. Berdeja JG, Alsina M, Shah ND, Siegel DS, Jagannath S, Madduri D, et al. Updated results from an ongoing phase 1 clinical study of bb21217 anti-bcma CAR T Cell therapy. Blood. (2019) 134:927. doi: 10.1182/blood-2019-126660
39. Ishikawa H, Tsuyama N, Mahmoud MS, Fujii R, Abroun S, Liu S, et al. CD19 expression and growth inhibition of tumours in human multiple myeloma. Leuk Lymphoma. (2002) 43:613–6. doi: 10.1080/10428190290012146
40. Carter RH, Fearon DT. CD19: Lowering the threshold for antigen receptor stimulation of B lymphocytes. Science. (1992) 256:105–7. doi: 10.1126/science.1373518
41. Mateo G, Montalbán MA, Vidriales MB, Lahuerta JJ, Mateos MV, Gutiérrez N, et al. Prognostic value of immunophenotyping in multiple myeloma: a study by the PETHEMA/GEM cooperative study groups on patients uniformly treated with high-dose therapy. J Clin Oncol. (2008) 26:2737–44. doi: 10.1200/JCO.2007.15.4120
42. Matsui W, Wang Q, Barber JP, Brennan S, Smith BD, Borrello I, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. (2008) 68:190–7. doi: 10.1158/0008-5472.CAN-07-3096
44. Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med. (2015) 373:1040–7. doi: 10.1056/NEJMoa1504542
45. Nerreter T, Letschert S, Götz R, Doose S, Danhof S, Einsele H, et al. Super-resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimination by CD19 CAR-T. Nat Commun. (2019) 10:3137. doi: 10.1016/j.clml.2019.09.277
46. Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, Tanhehco Y, et al. Characterization of clonogenic multiple myeloma cells. Blood. (2004) 103:2332–6. doi: 10.1182/blood-2003-09-3064
47. O'connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med J Exp Med. (2004) 199:91–7. doi: 10.1084/jem.20031330
48. Friedman KM, Garrett TE, Evans JW, Horton HM, Latimer HJ, Seidel SL, et al. Effective Targeting of multiple B-cell maturation antigen-expressing hematological malignances by anti-B-cell maturation antigen chimeric antigen receptor t cells. Hum Gene Ther. (2018) 29:585–601. doi: 10.1089/hum.2018.001
49. Topp MS, Duell J, Zugmaier G, Attal M, Moreau P, Langer C, et al. Evaluation of AMG 420, an anti-BCMA bispecific T-cell engager (BiTE) immunotherapy, in R/R multiple myeloma (MM) patients: updated results of a first-in-human (FIH) phase I dose escalation study. J Clin Oncol. (2019) 37:8007. doi: 10.1200/JCO.2019.37.15_suppl.8007
50. Trudel S, Lendvai N, Popat R, Voorhees PM, Reeves B, Libby EN, et al. Targeting B-cell maturation antigen with GSK2857916 antibody-drug conjugate in relapsed or refractory multiple myeloma: a dose-escalation and expansion phase 1 trial (BMA117159) HHS public access. Lancet Oncol. (2018) 19:1641–53. doi: 10.1016/S1470-204530576-X
51. Agostino MD, Raje N. Anti-BCMA CAR T-cell therapy in multiple myeloma : can we do better? Leukemia. (2020) 34:21–34. doi: 10.1038/s41375-019-0669-4
52. Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity. CA Cancer J Clin. (2020) 70:86–104. doi: 10.3322/caac.21596
53. Lee DH, Cervantes-Contreras F, Lee SY, Green DJ, Till BG. Improved expansion and function of CAR T Cell products from cultures initiated at defined CD4:CD8 ratios. Blood. (2018) 132:3334. doi: 10.1182/blood-2018-99-111576
54. Lesch S, Benmebarek MR, Cadilha BL, Stoiber S, Subklewe M, Endres S, et al. Determinants of response and resistance to CAR T cell therapy. Semin Cancer Biol. (2019) 24:563–71. doi: 10.1016/j.semcancer.2019.11.004
55. Song MK, Park BB, Uhm JE. Resistance mechanisms to CAR T-cell therapy and overcoming strategy in B-cell hematologic malignancies. Int J Mol Sci. (2019) 20:5010. doi: 10.3390/ijms20205010
56. Berdeja JG, Lin Y, Raje N, Munshi NC, Siegel D, Liedtke M, et al. Durable clinical responses in heavily pretreated patients with relapsed/refractory multiple myeloma: updated results from a multicenter study of bb2121 anti-bcma car t cell therapy. Blood. (2017) 130:740. doi: 10.1182/blood.V130.Suppl_1.740.740
57. Cohen AD, Garfall AL, Stadtmauer EA, Lacey SF, Lancaster E, Vogl DT, et al. Safety and efficacy of B-Cell Maturation Antigen (BCMA)-Specific chimeric antigen receptor T Cells (CART-BCMA) with cyclophosphamide conditioning for refractory multiple myeloma (MM). Blood. (2017) 130:505. doi: 10.1182/blood.V130.Suppl_1.505.505
58. Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. (2014) 20:119–22. doi: 10.1097/PPO.0000000000000035
59. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. (2014) 24:188–95. doi: 10.1182/blood-2014-05-552729
60. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood. (2016) 127:3321–30. doi: 10.1182/blood-2016-04-703751
61. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of t cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
62. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-Cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
63. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
64. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
65. Cohen AD. CAR T Cells and Other Cellular Therapies for Multiple Myeloma:2018 Update. Am Soc Clin Oncol Educ B. (2018) 38:e6–15. doi: 10.1200/EDBK_200889
66. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, Lin Y, Jain N, Daver N, et al. Chimeric antigen receptor T-cell therapy-assessment and management of toxicities. Nat Rev Clin Oncol. (2018) 15:47–62. doi: 10.1038/nrclinonc.2017.148
67. Lanza L, Scudeletti M, Puppo F, Bosco O, Peirano L, Filaci G, et al. Prednisone increases apoptosis in in vitro activated human peripheral blood T lymphocytes. Clin Exp Immunol. (2007) 103:482–90. doi: 10.1111/j.1365-2249.1996.tb08306.x
68. Paliogianni F, Ahuja SS, Balow JP, Balow JE, Boumpas DT. Novel mechanism for inhibition of human T cells by glucocorticoids. Glucocorticoids inhibit signal transduction through IL-2 receptor. J Immunol. (1993) 151:4081–9.
69. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. (2017) 7:1404–19. doi: 10.1158/2159-8290.CD-17-0698
70. Drent E, Poels R, Mulders MJ, van de Donk NWCJ, Themeli M, Lokhorst HM, et al. Feasibility of controlling CD38-CAR T cell activity with a Tet-on inducible CAR design. PLoS ONE. (2018) 13:1–16. doi: 10.1371/journal.pone.0197349
71. Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J, et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol Ther. (2017) 25:1946–58. doi: 10.1016/j.ymthe.2017.04.024
72. Sadelain M. CAR therapy: The CD19 paradigm. J Clin Invest. (2015) 125:3392–400. doi: 10.1172/JCI80010
73. Lamers CHJ, Klaver Y, Gratama JW, Sleijfer S, Debets R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells - A completed study overview. Biochem Soc Trans. (2016) 44:951–9. doi: 10.1042/BST20160037
74. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. (2011) 3:95ra73. doi: 10.1126/scitranslmed.3002842
75. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. (2012) 119:2709–20. doi: 10.1182/blood-2011-10-384388
76. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. (2010) 116:4099–102. doi: 10.1182/blood-2010-04-281931
77. Kramer G, Steiner G, Fodinger D, Fiebiger E, Rappersberger C, Binder S, et al. High Expression of a CD38-Like Molecule in Normal Prostatic Epithelium and its Differential Loss in Benign and Malignant Disease. J Urol. (1995) 154:1636–41. doi: 10.1097/00005392-199511000-00004
78. Muñoz P, Navarro MDC, Pavón EJ, Salmerón J, Malavasi F, Sancho J, et al. CD38 Signaling in T Cells Is Initiated within a Subset of Membrane Rafts Containing Lck and the CD3-ζ Subunit of the T Cell Antigen Receptor. J Biol Chem. (2003) 278:50791–802. doi: 10.1074/jbc.M308034200
79. Novak AJ, Darce JR, Arendt BK, Harder B, Henderson K, Kindsvogel W, et al. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism for growth and survival. Blood. (2004) 103:689–94. doi: 10.1182/blood-2003-06-2043
80. Carpenter RO, Evbuomwan MO, Pittaluga S, Rose JJ, Raffeld M, Yang S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. (2013) 19:2048–60. doi: 10.1158/1078-0432.CCR-12-2422
81. Bu D xiu, Singh R, Choi EE, Ruella M, Cruz SN, Mansfield KG, et al. Pre-clinical validation of B cell maturation antigen (BCMA) as a target for T cell immunotherapy of multiple myeloma. Oncotarget. (2018) 9:25764–80. doi: 10.18632/oncotarget.25359
82. Köhler M, Greil C, Hudecek M, Lonial S, Raje N, Wäsch R, et al. Current developments in immunotherapy in the treatment of multiple myeloma. Cancer. (2018) 124:2075–85. doi: 10.1002/cncr.31243
83. Rafiq S, Brentjens RJ. Tumors evading CARs-the chase is on. Nat Med. (2018) 24:1492–3. doi: 10.1038/s41591-018-0212-6
84. Majzner RG, Mackall CL. Tumor antigen escape from car t-cell therapy. Cancer Discov. (2018) 8:1219–26. doi: 10.1158/2159-8290.CD-18-0442
85. Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. (2016) 127:2406–10. doi: 10.1182/blood-2015-08-665547
86. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. (2015) 5:1282–95. doi: 10.1158/2159-8290.CD-15-1020
87. Ruella M, Kenderian SS, Shestova O, Fraietta JA, Qayyum S, Zhang Q, et al. The addition of the btk inhibitor ibrutinib to anti-cd19 chimeric antigen receptor T Cells (CART19) improves responses against mantle cell lymphoma. Clin Cancer Res. (2016) 22:2684–96. doi: 10.1158/1078-0432.CCR-15-1527
88. Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. (2016) 128:1688–700. doi: 10.1182/blood-2016-04-711903
89. Bachmann M, Cartellieri M, Feldmann A, Bippes C, Stamova S, Wehner R, et al. Chimeric antigen receptor-engineered T cells for immunotherapy of cancer. J Biomed Biotechnol. (2010) 2010:956304. doi: 10.1155/2010/956304
90. Grigoriadis G, Whitehead S. CD138 shedding in plasma cell myeloma. Br J Haematol. (2010) 150:249. doi: 10.1111/j.1365-2141.2010.08203.x
91. Laurent SA, Hoffmann FS, Kuhn PH, Cheng Q, Chu Y, Schmidt-Supprian M, et al. γ-secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun. (2015) 6:7333. doi: 10.1038/ncomms8333
92. Pont MJ, Hill T, Cole GO, Abbott JJ, Kelliher J, Salter AI, et al. G-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood. (2019) 134:1585–97. doi: 10.1182/blood.2019000050
93. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric antigen receptor T Cells in refractory B-Cell lymphomas. N Engl J Med. (2017) 377:2545–54. doi: 10.1056/NEJMoa1708566
94. Elavia N, Panch SR, McManus A, Bikkani T, Szymanski J, Highfill SL, et al. Effects of starting cellular material composition on chimeric antigen receptor T-cell expansion and characteristics. Transfusion. (2019) 59: 1755–64. doi: 10.1111/trf.15287
95. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. (2019) 16:372–85. doi: 10.1038/s41571-019-0184-6
96. Palmer S, Albergante L, Blackburn CC, Newman TJ. Thymic involution and rising disease incidence with age. Proc Natl Acad Sci USA. (2018) 115:1883–8. doi: 10.1073/pnas.1714478115
97. Salam N, Rane S, Das R, Faulkner M, Gund R, Kandpal U, et al. T cell ageing: Effects of age on development, survival & function. Indian J Med Res. (2013) 138:595–608.
98. Singh N, Perazzelli J, Grupp SA, Barrett DM. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci Transl Med. (2016) 8:320ra3. doi: 10.1126/scitranslmed.aad5222
99. Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. (2016) 127:1117–27. doi: 10.1182/blood-2015-11-679134
100. Hoffmann JM, Schubert ML, Wang L, Hückelhoven A, Sellner L, Stock S, et al. Differences in expansion potential of naive chimeric antigen receptor T cells from healthy donors and untreated chronic lymphocytic leukemia patients. Front Immunol. (2018) 8:1956. doi: 10.3389/fimmu.2017.01956
101. Van Der Stegen SJC, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. (2015) 14:499–509. doi: 10.1038/nrd4597
102. Milone MC, Bhoj VG. The Pharmacology of T Cell Therapies. Mol Ther - Methods Clin Dev. (2018) 8: 210–21. doi: 10.1016/j.omtm.2018.01.010
103. Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal. (2018) 11:eaat6753. doi: 10.1126/scisignal.aat6753
104. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
105. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
106. Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. (2019) 25:947–53. doi: 10.1038/s41591-019-0421-7
107. Griffioen M, Van Egmond EHM, Kester MGD, Willemze R, Falkenburg JHF, Heemskerk MHM. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica. (2009) 94:1316–20. doi: 10.3324/haematol.2008.001677
108. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. (2014) 124:1277–87. doi: 10.1182/blood-2014-01-545020
109. Wang X, Chang WC, Wong CLW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. (2011) 118:1255–63. doi: 10.1182/blood-2011-02-337360
110. Di Stasi A, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N Engl J Med. (2011) 365:1673–83. doi: 10.1056/NEJMoa1106152
111. Caruso HG, Heimberger AB, Cooper LJN. Steering CAR T cells to distinguish friend from foe. Oncoimmunology. (2019) 8:e1271857. doi: 10.1080/2162402X.2016.1271857
112. Bräuner-Osborne H, Jensen AA, Sheppard PO, Brodin B, Krogsgaard-Larsen P, O'Hara P. Cloning and characterization of a human orphan family C G-protein coupled receptor GPRC5D. Biochim Biophys Acta. (2001) 1518:237–48. doi: 10.1016/S0167-4781(01)00197-X
113. Smith EL, Harrington K, Staehr M, Masakayan R, Jones J, Long TJ, et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci Transl Med. (2019) 11:eaau7746. doi: 10.1126/scitranslmed.aau7746
114. Corse E, Gottschalk RA, Allison JP. Strength of TCR-Peptide/MHC interactions and in vivo T cell responses. J Immunol. (2011) 186:5039–45. doi: 10.4049/jimmunol.1003650
115. Tan MP, Gerry AB, Brewer JE, Melchiori L, Bridgeman JS, Bennett AD, et al. T cell receptor binding affinity governs the functional profile of cancer-specific CD8+ T cells. Clin Exp Immunol. (2015) 180:255–70. doi: 10.1111/cei.12570
116. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. (2015) 75:3505–18. doi: 10.1158/0008-5472.CAN-15-0139
117. Arcangeli S, Rotiroti MC, Bardelli M, Simonelli L, Magnani CF, Biondi A, et al. Balance of Anti-CD123 Chimeric Antigen Receptor Binding Affinity and Density for the Targeting of Acute Myeloid Leukemia. Mol Ther. (2017) 25:1933–45. doi: 10.1016/j.ymthe.2017.04.017
118. Han C, Sim SJ, Kim SH, Singh R, Hwang S, Kim YI, et al. Desensitized chimeric antigen receptor T cells selectively recognize target cells with enhanced antigen expression. Nat Commun. (2018) 9:468. doi: 10.1038/s41467-018-02912-x
119. Hughes-Parry HE, Cross RS, Jenkins MR. The evolving protein engineering in the design of chimeric antigen receptor T cells. Int J Mol Sci. (2020) 21:204. doi: 10.3390/ijms21010204
120. Song DG, Ye Q, Poussin M, Liu L, Figini M, Powell DJ. A fully human chimeric antigen receptor with potent activity against cancer cells but reduced risk for off-tumor toxicity. Oncotarget. (2015) 6:21533–46. doi: 10.18632/oncotarget.4071
121. Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, et al. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. (2013) 1:43–53. doi: 10.1158/2326-6066.CIR-13-0008
122. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. (2013) 31:71–5. doi: 10.1038/nbt.2459
123. Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule–gated chimeric receptor. Science. (2015) 350:aab4077. doi: 10.1126/science.aab4077
124. Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision Tumor Recognition by T Cells with Combinatorial Antigen-Sensing Circuits. Cell. (2016) 164:770–9. doi: 10.1016/j.cell.2016.01.011
125. Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. (2013) 5:215ra172. doi: 10.1126/scitranslmed.3006597
126. Wu C, Zhang L, Brockman QR, Zhan F, Chen L. Chimeric antigen receptor T cell therapies for multiple myeloma. J Hematol Oncol. (2019) 12:120. doi: 10.1186/s13045-019-0823-5
127. Lin Q, Zhao J, Song Y, Liu D. Recent updates on CAR T clinical trials for multiple myeloma. Mol Cancer. (2019) 18:154. doi: 10.1186/s12943-019-1092-1
128. Zah E, Lin MY, Anne SB, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. (2016) 4:498–508. doi: 10.1158/2326-6066.CIR-15-0231
129. Yan L, Yan Z, Shang J, Shi X, Jin S, Kang L, et al. Sequential CD19- and Bcma-Specific Chimeric Antigen Receptor T Cell Treatment for RRMM: Report from a Single Center Study. Blood. (2019) 134:578. doi: 10.1182/blood-2019-129740
130. Hymowitz SG, Patel DR, Wallweber HJA, Runyon S, Yan M, Yin JP, et al. Structures of APRIL-receptor complexes: Like BCMA, TACI employs only a single cysteine-rich domain for high affinity ligand binding. J Biol Chem. (2005) 280:7218–722. doi: 10.1074/jbc.M411714200
131. Patel DR, Wallweber HJA, Yin JP, Shriver SK, Marsters SA, Gordon NC, et al. Engineering an APRIL-specific B Cell Maturation Antigen. J Biol Chem. (2004) 279:16727–35. doi: 10.1074/jbc.M312316200
132. Vincent FB, Morand EF, Schneider P, MacKay F. The BAFF/APRIL system in SLE pathogenesis. Nat Rev Rheumatol. (2014) 10:365–73. doi: 10.1038/nrrheum.2014.33
133. Lee L, Draper B, Chaplin N, Philip B, Chin M, Galas-Filipowicz D, et al. An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood. (2018) 131:746–58. doi: 10.1182/blood-2017-05-781351
134. Popat R, Zweegman S, Cavet J, Yong K, Lee L, Faulkner J, et al. Phase 1 first-in-human study of AUTO2, the first chimeric antigen receptor (CAR) T cell targeting APRIL for patients with relapsed/refractory multiple myeloma (RRMM). Blood. (2019) 134:3112. doi: 10.1182/blood-2019-126689
135. Schmidts A, Ormhøj M, Choi BD, Taylor AO, Bouffard AA, Scarfò I, et al. Rational design of a trimeric April-based CAR-binding domain enables efficient targeting of multiple myeloma. Blood Adv. (2019) 3:3248–60. doi: 10.1182/bloodadvances.2019000703
136. Fernandez de Larrea C, Staehr M, Lopez A, Chen Y, Purdon TJ, Ng KY, et al. Optimal dual-targeted CAR construct simultaneously targeting bcma and GPRC5D prevents Bcma-escape driven relapse in multiple myeloma. Blood. (2019) 134:136. doi: 10.1182/blood-2019-126145
137. Liu Y, Hong X, Kappler J, Jiang L, Zhang R, Xu L, et al. Ligand-receptor binding revealed by the TNF family member TALL-1. Nature. (2003) 423:49–56. doi: 10.1038/nature01543
138. Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature. (2017) 545:423–31. doi: 10.1038/nature22395
139. Busch DH, Fräßle SP, Sommermeyer D, Buchholz VR, Riddell SR. Role of memory T cell subsets for adoptive immunotherapy. Semin Immunol. (2016) 28:28–34. doi: 10.1016/j.smim.2016.02.001
140. McLellan AD, Ali Hosseini Rad SM. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol Cell Biol. (2019) 97:664–74. doi: 10.1111/imcb.12254
141. Petersen CT, Hassan M, Morris AB, Jeffery J, Lee K, Jagirdar N, et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kd inhibitors and VIP antagonists. Blood Adv. (2018) 2:210–23. doi: 10.1182/bloodadvances.2017011254
142. Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of clinical-grade CD19-specific CAR-modified CD81 memory stem cells for the treatment of human B-cell malignancies. Blood. (2016) 128:519–28. doi: 10.1182/blood-2015-11-683847
143. Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. (2016) 8:355ra116. doi: 10.1126/scitranslmed.aaf8621
144. Philip LPB, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, Potrel P, et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. (2015) 75:3853–64. doi: 10.1158/0008-5472.CAN-14-3321
145. Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. (2018) 32:1970–83. doi: 10.1038/s41375-018-0065-5
146. Georgiadis C, Preece R, Nickolay L, Etuk A, Petrova A, Ladon D, et al. Long Terminal Repeat CRISPR-CAR-Coupled “Universal” T Cells Mediate Potent Anti-leukemic Effects. Mol Ther. (2018) 26:1215–27. doi: 10.1016/j.ymthe.2018.02.025
147. Valton J, Guyot V, Marechal A, Filhol JM, Juillerat A, Duclert A, et al. A multidrug-resistant engineered CAR T cell for allogeneic combination immunotherapy. Mol Ther. (2015) 23:1507–18. doi: 10.1038/mt.2015.104
148. Bailey SR, Maus MV. Gene editing for immune cell therapies. Nat Biotechnol. (2019) 37:1425–34. doi: 10.1038/s41587-019-0137-8
149. Guedan S, Calderon H, Posey AD, Maus MV. Engineering and Design of Chimeric Antigen Receptors. Mol Ther Methods Clin Dev. (2019) 12:145–56. doi: 10.1016/j.omtm.2018.12.009
150. Sommer C, Boldajipour B, Kuo TC, Bentley T, Sutton J, Chen A, et al. Preclinical Evaluation of Allogeneic CAR T Cells Targeting BCMA for the Treatment of Multiple Myeloma. Mol Ther. (2019) 27:1126–38. doi: 10.1016/j.ymthe.2019.04.001
151. Peng R, Lin G, Li J. Potential pitfalls of CRISPR/Cas9-mediated genome editing. FEBS J. (2016) 283:1218–31. doi: 10.1111/febs.13586
152. Liu J, Zhou G, Zhang L, Zhao Q. Building potent chimeric antigen receptor T cells with CRISPR genome editing. Front Immunol. (2019) 10:456. doi: 10.3389/fimmu.2019.00456
153. Li H, Yang Y, Hong W, Huang M, Wu M, Zhao X. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct Target Ther. (2020) 5:1–23. doi: 10.1038/s41392-019-0089-y
154. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
155. Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-Line Nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med. (2017) 376:2415–26. doi: 10.1056/NEJMoa1613493
156. Eggermont AMM, Chiarion-Sileni V, Grob J-J, Dummer R, Wolchok JD, Schmidt H, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. (2016) 375:1845–55. doi: 10.1056/NEJMoa1611299
157. Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, et al. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. (2017) 27:154–7. doi: 10.1038/cr.2016.142
158. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-Tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. (2017) 7:737. doi: 10.1038/s41598-017-00462-8
Keywords: CAR T-cell, myeloma, toxicities, antigen escape, soluble protein, allogeneic CAR T-cell
Citation: García-Guerrero E, Sierro-Martínez B and Pérez-Simón JA (2020) Overcoming Chimeric Antigen Receptor (CAR) Modified T-Cell Therapy Limitations in Multiple Myeloma. Front. Immunol. 11:1128. doi: 10.3389/fimmu.2020.01128
Received: 02 March 2020; Accepted: 07 May 2020;
Published: 05 June 2020.
Edited by:
Francisco Martin, Andalusian Autonomous Government of Genomics and Oncological Research (GENYO), SpainReviewed by:
Bryan M. Burt, Baylor College of Medicine, United StatesCopyright © 2020 García-Guerrero, Sierro-Martínez and Pérez-Simón. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jose Antonio Pérez-Simón, am9zZWEucGVyZXouc2ltb24uc3NwYUBqdW50YWRlYW5kYWx1Y2lhLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.