 Omar A. Alfituri
Omar A. Alfituri Barry M. Bradford
Barry M. Bradford Edith Paxton
Edith Paxton Liam J. Morrison
Liam J. Morrison Neil A. Mabbott
Neil A. Mabbott
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 03 June 2020
Sec. Microbial Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01118
This article is part of the Research Topic Emerging Concepts of Innate Immune Responses to Neglected Tropical Diseases View all 15 articles
Infection of the mammalian host with African trypanosomes begins when the tsetse fly vector injects the parasites into the skin dermis during blood feeding. After injection into the skin, trypanosomes first accumulate in the draining lymph node before disseminating systemically. Whether this early accumulation within the draining lymph node is important for the trypanosomes to establish infection was not known. Lymphotoxin-β-deficient mice (LTβ−/− mice) lack most secondary lymphoid tissues, but retain the spleen and mesenteric lymph nodes. These mice were used to test the hypothesis that the establishment of infection after intradermal (ID) T. brucei infection would be impeded in the absence of the skin draining lymph nodes. However, LTβ−/− mice revealed greater susceptibility to ID T. brucei infection than wild-type mice, indicating that the early accumulation of the trypanosomes in the draining lymph nodes was not essential to establish systemic infection. Although LTβ−/− mice were able to control the first parasitemia wave as effectively as wild-type mice, they were unable to control subsequent parasitemia waves. LTβ−/− mice also lack organized B cell follicles and germinal centers within their remaining secondary lymphoid tissues. As a consequence, LTβ−/− mice have impaired immunoglobulin (Ig) isotype class-switching responses. When the disturbed microarchitecture of the B cell follicles in the spleens of LTβ−/− mice was restored by reconstitution with wild-type bone marrow, their susceptibility to ID T. brucei infection was similar to that of wild-type control mice. This effect coincided with the ability to produce significant serum levels of Ig isotype class-switched parasite-specific antibodies. Thus, our data suggest that organized splenic microarchitecture and the production of parasite-specific Ig isotype class-switched antibodies are essential for the control of ID African trypanosome infections.
African trypanosomes are single-celled protozoan parasites that are transmitted between mammalian host species via blood-feeding tsetse flies of the genus Glossina. The Trypanosoma brucei rhodesiense and T. b. gambiense subspecies cause human African trypanosomiasis in endemic regions of sub-Saharan Africa. Animal African trypanosomiasis is caused by Trypanosoma congolense, Trypanosoma vivax and T. brucei, and inflicts substantial economic strains on the African livestock industry. These parasites exist entirely extracellularly within their hosts, and the life cycle in the mammalian host is initiated by the intradermal (ID) injection of metacyclic trypomastigotes into the skin dermis by the tsetse fly vector. After ID injection the parasites can directly infect the lymphatics (1) before disseminating to the draining lymph nodes and then systemically via the bloodstream (2–5). Soon after ID injection the parasites also undergo morphological change into the long-slender bloodstream form stages that are specifically adapted for survival within the hostile environment of the mammalian bloodstream.
Although much attention has been given to the study of experimental trypanosome infections initiated by intraperitoneal (IP) or intravenous injection, comparatively little is known of the host-specific factors that are important for the effective control of ID transmitted trypanosome infections. However, Wei and colleagues have shown that infection of mice by the IP route establishes a detectable parasitemia earlier than after ID injection, with a higher parasite burden (6). Furthermore, the same study suggested mice were ~100X less susceptible to ID injected T. brucei infection when compared to mice infected by the IP route.
Lymphotoxin-α (LTα) and lymphotoxin-β (LTβ) are members of the TNF superfamily and form a heterotrimer (LTα1β2) to allow signaling through the LTβ receptor (LTβR) (7). Signaling via LTβR during embryonic development is essential for the development of the peripheral lymph nodes (8). Subsequently, post-natal lymphocyte-derived LTα1β2-mediated stimulation is also essential for the maintenance of organized tissue microarchitecture within the secondary lymphoid organs. As a consequence, mice deficient in LTα, LTβ, or LTβR lack most peripheral lymph nodes and have grossly disorganized microarchitecture in their remaining secondary lymphoid organs such as the spleen (8–15). For example, these mice have disturbed B cell follicles, lack germinal centers (GC) and as a consequence have a significantly reduced ability to produce high affinity antigen-specific class-switched immunoglobulins (Ig) upon immunization. Therefore, in this study we used LTβ−/− mice that lack most peripheral lymph nodes to determine the requirement of the draining lymph nodes for the effective establishment of infection after intradermal (ID) inject with T. brucei. Using these mice we show that the early accumulation of the trypanosomes in the draining lymph nodes was not essential to establish systemic infection. Although LTβ−/− mice were able to control the first parasitemia wave as effectively as wild-type mice, they were unable to control subsequent parasitemia waves. Subsequent experiments suggested that the inability of LTβ−/− mice to control the subsequent parasitemia waves coincided with the lack of organized B cell follicles in their spleens, and their impaired ability to mount parasite-specific Ig isotype class-switched antibody responses. This study provides important insight into the important host factors that are essential for the efficient control of ID trypanosome infections.
Six to 8 weeks old female C57BL/6J mice (Charles Rivers, Harlow, England) and lymphotoxin-β-deficient (LTβ−/−) mice (15) were used throughout this study. Mice were maintained in individually ventilated cages and provided food and water ad libitum. All procedures using experimental mice were carried out under the authority of the appropriate project and personal licenses, in accordance with the United Kingdom Home Office regulations and the Animals (Scientific Procedures) Act 1986. Ethical approvals were obtained from The Roslin Institute's and University of Edinburgh's ethics committees.
Where indicated, recipient female LTβ−/− mice (~20 g each, 6–8 weeks old) were γ-irradiated twice (5 Grays each) at a 4 h interval. Bone marrow from the long bones of donor mice was aseptically prepared as a single cell suspension at ~1 × 107 cells/ml in HBSS (Life Technologies, Paisley, UK). Approximately 18 h after the irradiation the recipient mice each received 100 μl of fresh donor bone marrow by intravenous injection into the tail vein. Recipient mice were allowed to recover for 10 weeks prior to their use in subsequent experiments.
The pleomorphic wild-type T. b. brucei strain STIB247 was used throughout this study. These trypanosomes were originally isolated from a hartebeeste (Alcelaphus buselaphus) in Tanzania's Serengeti National Park in 1971 (16). The trypanosomes were axenically cultivated in vitro as described previously (1). Prior to their use in the in vivo studies described, ~1x105 axenically cultivated trypanosomes were first injected IP into groups of C57BL/6J mice. Blood was collected at the peak of the first parasitemia and used as a fresh source of in vivo-adapted parasites for each experiment. In the experiments described the mice were infected ID with ~1 × 102 or 1 × 105 T. b. brucei STIB247 parasites, where indicated.
Serum IgM and IgG isotype levels were measured by ELISA. For total Ig measurements a capture ELISA was used whereby 96 well plates (Immulon 4HBX 96-Well Micro Plate, SLS, UK) were first coated with 50 μl of either purified rat anti-mouse IgM coating antibody (BD 553435 (II/41), BD Biosciences, USA) or purified polyclonal goat anti-mouse coating Ig antibody (BD 553998, BD Biosciences, USA), each prepared at 5 μg/ml in p-nitrophenyl phosphate substrate buffer. The plates were sealed and incubated overnight at 4°C. The plates were then blocked using 100 μl of 1% bovine serum albumin (BSA)/PBS (Sigma-Aldrich) to each well and incubated at 37°C for 1 h, and then washed 5 times in 0.05% Tween/PBS. Sera (50 μl/well) was then added in 0.1% BSA/PBS and incubated for 1 h at 37°C at the following dilutions: IgM, 1:400; IgG1 1:800; IgG2c, 1:50; IgG3, 1:800. The serum dilutions used were based on previously established titrations. Serial dilutions of normal mouse IgM (clone RMM-1), normal mouse IgG1 (clone RMG1-1), normal mouse IgG2c (clone RMG2a62) or normal mouse IgG3 (clone R40-82) (all from Biolegend, USA) were used to establish standard curves. After washing, 50 μl of Ig-subclass-specific biotinylated secondary antibodies (anti-IgM, clone G155-228, BD Biosciences; anti-IgG1, clone MG1-45, BioLegend; anti-IgG2c, clone MG2a-53, BioLegend; anti-IgG3, clone MG3-25, clone MG3-25) were applied in 1% skimmed milk/0.1% BSA/PBS (1/500 dilution), and incubated for 1 h at 37°C. Plates were subsequently washed and 50 μl streptavidin-conjugated horseradish peroxidase was added (1/1,000 dilution) and incubated at 37°C for 1 h. After a final wash, bound peroxidase activity was revealed by adding 50 μl of SureBlue TMB microwell Peroxidase Substrate to each well (KPL, SeraCare 5120-0075, Massachusetts, USA), and the reaction stopped using HCl (1M). Optical density (OD) was then determined using a Perkin Elmer Wallac 1420 Victor2 Microplate Reader (GMI, USA) at 450 nm with 620 nm used as the reference OD value.
To estimate trypanosome-specific Ig levels an indirect ELISA was used whereby the plates were first coated with 50 μl of trypanosome lysate (0.7 μg protein/ml) in 0.1 M bicarbonate buffer, in place of the purified rat anti-mouse IgM purified polyclonal goat anti-mouse coating Ig antibodies. The remainder of the assay was repeated as above. A potential limitation of this approach being that the binding characteristics of the coating Ig antibodies used in the capture ELISA may differ from that of the trypanosome lysate used in the indirect assay.
Spleens were snap frozen at the temperature of liquid nitrogen, and 10 μm thick sections cut using a cryostat. Sections were then fixed in acetone and follicular dendritic cells detected using mAb 8C12 to detect CD35 (BD Biosciences, Oxford, UK) and B cells detected using rat anti-mouse B220 mAb (clone RA3-6B2, Life Technologies). Sections were subsequently stained with species-specific secondary antibodies conjugated to Alexa Fluor 488 (green) or Alexa Fluor 594 (red) dyes (Life Technologies). Sections were then imaged using a Zeiss LSM710 confocal microscope (Zeiss, Welwyn Garden City, UK).
ELISA data were tested for normal distribution using Shapiro-Wilk and Kolmogorov Smirnov tests. Data that passed these normality tests were subsequently compared using a Student's t-test. Data that did not pass these tests were compared using a Mann-Whitney U test. These analyses were performed using GraphPad Prism v.8.0 (GraphPad Software Inc. San Diego, USA). To compare the parasitemias between different groups, linear mixed effects models were performed using RStudio. These were used to statistically compare the quadratic (squared) and cubic curve effect of the infected mouse parasitemia across the observation period. Mean peak parasitemias were compared using a Student's t-test or Tukey's multiple comparisons test. P < 0.05 were accepted as significant.
First, LTβ−/− mice and C57BL/6J wild-type (WT) control mice were injected IP with Chicago Sky Blue 6B ink and the macroscopic presence of their secondary lymphoid tissues determined at post-mortem 7 d later. Throughout this study the ear pinna was used as the site of ID parasite injection. The mandibular lymph nodes, sub-mandibular lymph nodes and superficial parotid lymph nodes are considered to drain this anatomical region. As anticipated, these lymph nodes were absent in LTβ−/− mice (Table 1) as stimulation through the LTβR during embryonic development is essential for their formation (12, 17, 18). However, the LTβ−/− mice retained the spleen (Table 1).
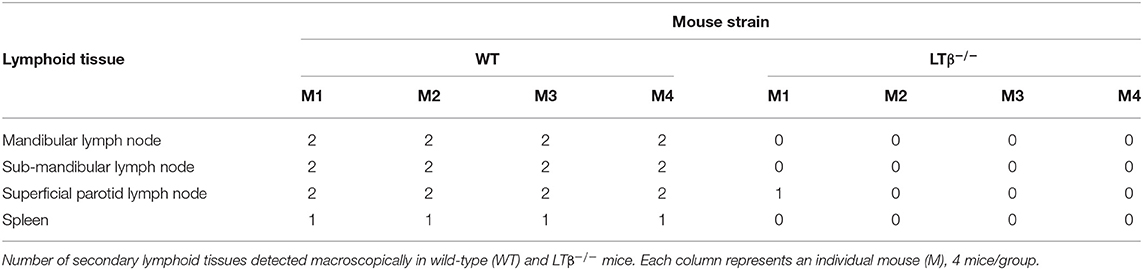
Table 1. Incidence of lymphoid tissues in WT and LTβ−/− mice.
Independent studies using the T. brucei strain 10–26 have estimated that the ID route of infection is ~100X less efficient than the IP route (6). Although infected tsetse flies are likely to transmit low numbers of trypanosomes during blood feeding, ID injection of WT mice with a 1 × 102 dose of T. brucei strain 10–26 parasites was insufficient to establish infection (6). Here, WT control mice (n = 8/group) were also injected ID into the ear pinna with 1 × 102 T. brucei STIB 247 parasites, and blood parasitemias assessed daily for 30 d using the rapid matching method (19). As this assay has a detection threshold of ~4 × 105 parasites/ml blood, parasitemias below this level were classified as below the limit of detection. Consistent with the above study, our data show that when WT mice were injected ID with 1 × 102 trypanosomes, only two of eight mice had a detectable parasitemia for short duration during the 30 d observation period (Figure 1A). Therefore, since mice are considered to be at least 100X less susceptible to ID T. brucei infection, in the subsequent experiments below the mice were injected ID with 1 × 105 trypanosomes to ensure that all the recipient WT mice developed a detectable parasitemia.
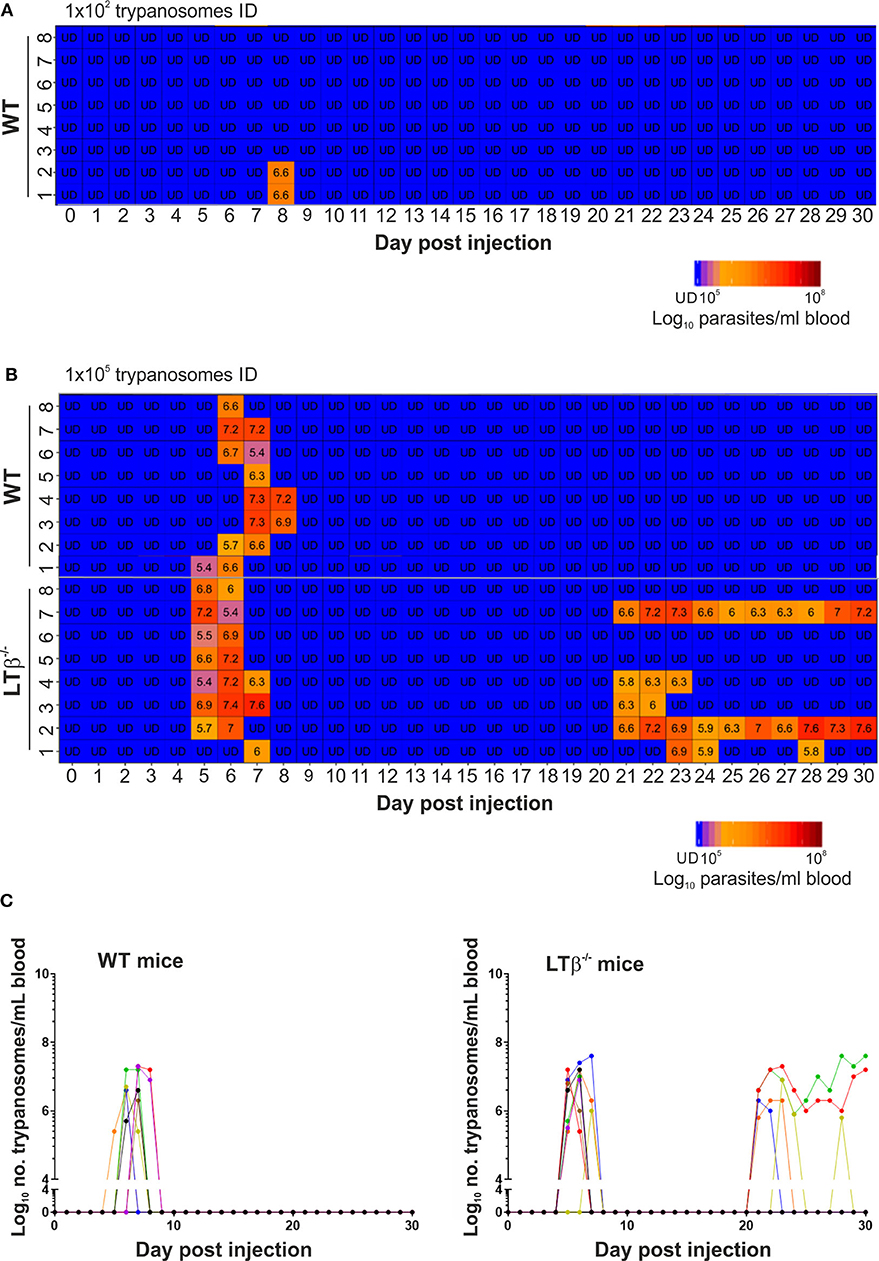
Figure 1. Enhanced susceptibility of LTβ−/− mice to ID infection with T. brucei. (A) Wild-type (WT) mice (n = 8) were injected ID with a 1 × 102 dose of T. brucei STIB 247 parasites and blood parasitemia levels determined at daily intervals. Heatmap shows the blood parasitemia (log10 number of trypanosomes/ml of blood) in each mouse. Each row represents data from an individual mouse. UD = below the limit of detection, ~5.4 log10 parasites/ml. (B) Groups of WT and LTβ−/− mice (n = 8 mice/group) were injected ID with a 1 × 105 dose of T. brucei STIB 247 parasites and blood parasitemia levels determined at daily intervals. Heatmap shows the blood parasitemia (log10 number of trypanosomes/ml of blood) in each mouse. Each row represents data from an individual mouse. UD, below the limit of detection, ~5.4 log10 parasites/ml. (C) Charts show parasitemia profiles (log10 number of trypanosomes/ml of blood) in each mouse from each group following ID injection with 1 × 105 dose of T. brucei STIB 247 parasites. Each line represents data from an individual mouse.
Next, groups of LTβ−/− mice and WT control mice (n = 8/group) were injected ID with 1 × 105 T. brucei STIB 247 parasites, and blood parasitemias assessed daily for 30 d. After infection with this dose of parasites all the mice developed a detectable parasitemia. Furthermore, the onset and duration of the first detectable parasitemia wave in the bloodstream of each mouse group was similar (Figures 1B,C): WT mice, 6–8 d post-infection (dpi); LTβ−/− mice, 5–7 dpi. Furthermore, the mean parasite burdens at the peak of the first parasitemia wave were also similar in each mouse group (Figures 1B,C): WT mice, 8 × 106/ml parasites/ml; LTβ−/− mice, 1 × 107 parasites/ml; P = 0.460, Student's t-test, n = 8/group.
None of the ID-injected WT mice displayed any relapse in their parasitemias during the remainder of the 30 d observation period. In contrast, five of eight of the LTβ−/− mice relapsed between 21 and 30 dpi, displaying subsequent parasitemia waves (Figures 1B,C). These data clearly show that ID injected T. brucei can successfully establish infection in the bloodstream of LTβ−/− mice despite the absence of the draining lymph nodes. However, the LTβ−/− mice have a reduced ability to control an ID T. brucei infection when compared to WT mice.
Constitutive post-natal LTβR-stimulation is also important for the maintenance of organized B cell follicles and the stromal follicular dendritic cells (FDC) within them (20). As a consequence, the spleens of LTβ−/− mice lacked CD35-expressing FDC and had disturbed B cell follicles that presented as ring-like structures (Figure 2A).
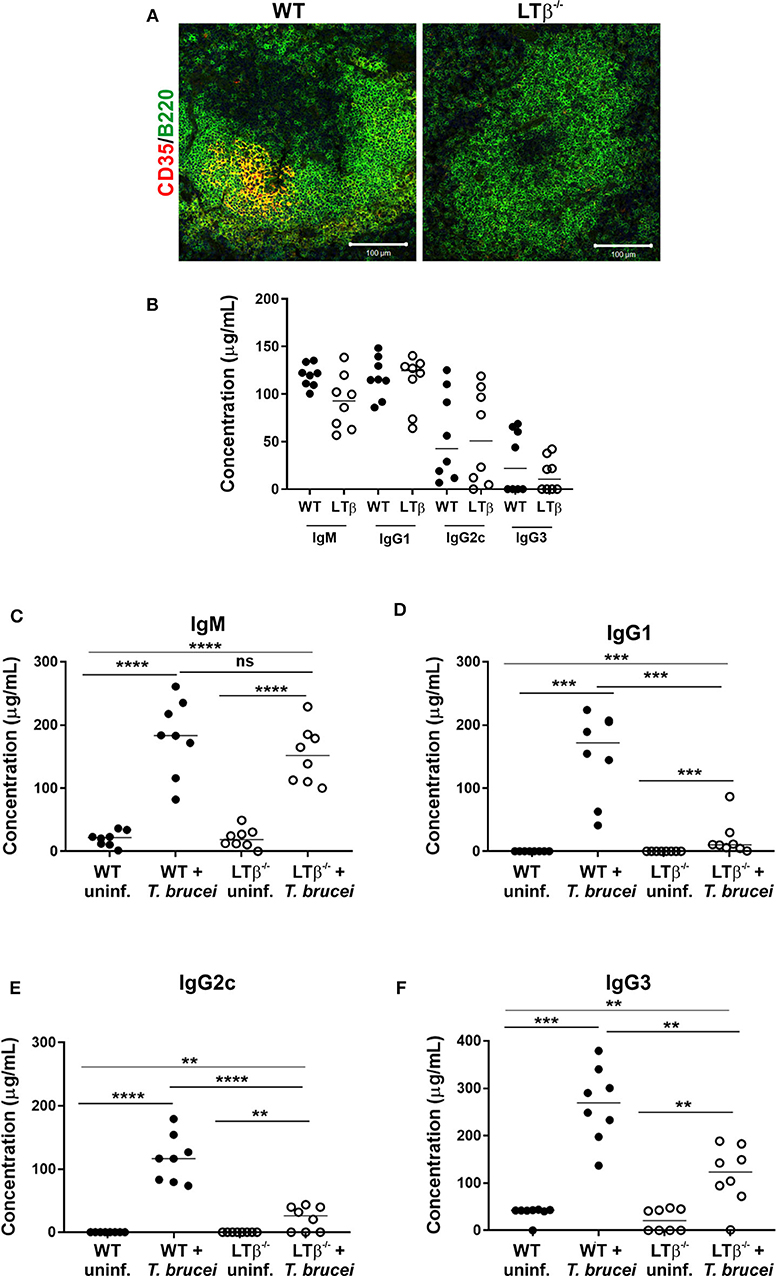
Figure 2. LTβ−/− mice have disturbed splenic microarchitecture and reduced serum class-switched immunoglobulin levels after trypanosome infection. (A) Spleen sections from wild-type (WT) and LTβ−/− mice were immunostained to detect follicular dendritic cells (FDC, CD35+ cells, red) and B cells (B220+ cells, green). The spleens of LTβ−/− mice lacked FDC networks and had disturbed B cell follicles. (B) Total serum IgM, IgG1, IgG2c, and IgG3 concentrations in uninfected naïve WT and LTβ−/− mice determined by ELISA (n = 8 mice/group). (C–F) Groups of WT and LTβ−/− mice (n = 8 mice/group) were injected ID with a 1 × 105 dose of T. brucei STIB 247 parasites and serum concentrations of trypanosome-specific (C) IgM, (D) IgG1, (E) IgG2c and (F) IgG3 determined by ELISA. Uninf., uninfected. Closed circles, WT mice; open circles, LTβ−/− mice. Each point represents data from an individual mouse. Horizontal bar, median. Statistical analyses: (B) Student's t-test; (C) Student's t-test; (D) Mann-Whitney U-test; (E) Student's t-test; (F) Mann-Whitney U-test; **P < 0.01; ***P < 0.001; ****P < 0.0001.
The retention of antigens on the surface of FDC is essential for GC formation and the production of high-affinity antigen-specific immunoglobulin (Ig) isotype class-switched antibodies by B cells (21). The levels of total IgM, IgG1, IgG2c, and IgG3 antibodies in the serum of uninfected naïve WT and LTβ−/− mice were similar (Figure 2B), consistent with previous data (10). By 30 d following ID infection with T. brucei, the sera of infected mice from each group contained significantly elevated levels of trypanosome-specific IgM (Figure 2C). However, whereas elevated levels of trypanosome-specific class-switched IgG1, IgG2c and IgG3 antibodies were detected in the sera of WT mice, significantly lower levels, if any, were detected in the sera of LTβ−/− mice (Figures 2D–F).
We next determined whether the increased susceptibility of LTβ−/− mice to ID T. brucei infection was a consequence of their impaired ability to produce trypanosome-specific class-switched Ig isotypes. Constitutive LTβR-mediated stimulation via lymphocyte-derived LTα1β2 is essential for the maintenance of FDC networks and lymphoid tissue microarchitecture (18, 20, 22). As mature FDC are important for the efficient production of antigen-specific class-switched antibodies by B cells (21, 23), FDC differentiation was induced in the remaining lymphoid tissues of LTβ−/− mice. Thus, the reconstitution of LTβ−/− mice with LT-expressing donor bone marrow from WT mice (termed WT → LTβ−/− mice, hereafter) stimulated the differentiation of FDC from stromal-derived precursor cells in the spleens of the recipient LTβ−/− mice (Figure 3). However, this treatment does not induce the development of the missing secondary lymphoid tissues (24). Conversely, when LTβ−/− mice were reconstituted with LTβ−/− donor bone marrow as a negative control (termed LTβ−/− → LTβ−/− mice, here after), no induction of FDC network formation was observed (Figure 3).
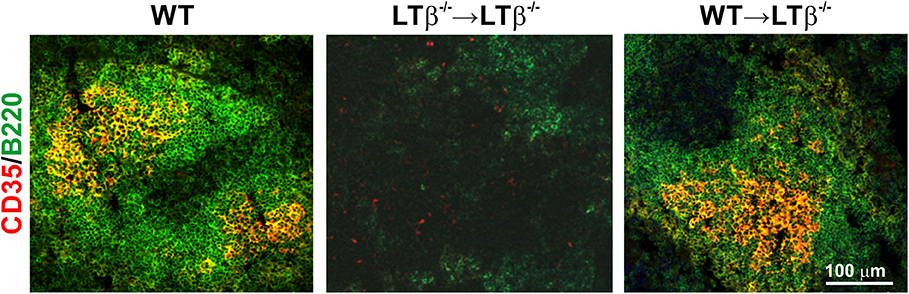
Figure 3. Reconstitution of LTβ−/− mice with wild-type (WT) bone marrow restores the disturbed splenic microarchitecture. LTβ−/− mice were γ-irradiated and reconstituted with donor bone marrow from WT mice (WT → LTβ−/− mice) or LTβ−/− mice (LTβ−/− → LTβ−/− mice). Ten weeks later spleens were collected and sections immunostained to detect follicular dendritic cells (FDC, CD35+ cells, red) and B cells (B220+ cells, green). FDC differentiation was restored in the spleens of WT → LTβ−/− mice.
Next, groups of WT → LTβ−/− mice, LTβ−/− → LTβ−/− mice and un-irradiated WT control mice were ID injected with 1x105 T. brucei STIB 247 parasites and blood parasitemias assessed daily for 30 d. The onset and duration of the first detectable parasitemia waves in the bloodstream of each mouse group were similar (Figures 4A,B). However, noticeable differences in the parasitemia kinetics were evident during the later stages of the infections. The trypanosome infection relapsed in most of the LTβ−/− → LTβ−/− mice from ~21 dpi with the detection of subsequent parasitemia waves (Figures 4A,B). These data show that LTβ−/− → LTβ−/− mice, like LTβ−/− mice (Figure 1), have increased susceptibility to ID T. brucei infection. In contrast, none of the WT → LTβ−/− mice, and only one of eight of the ID injected un-irradiated WT mice, had any detectable relapses in their parasitemias during the remainder of the experiment (Figures 4A,B).
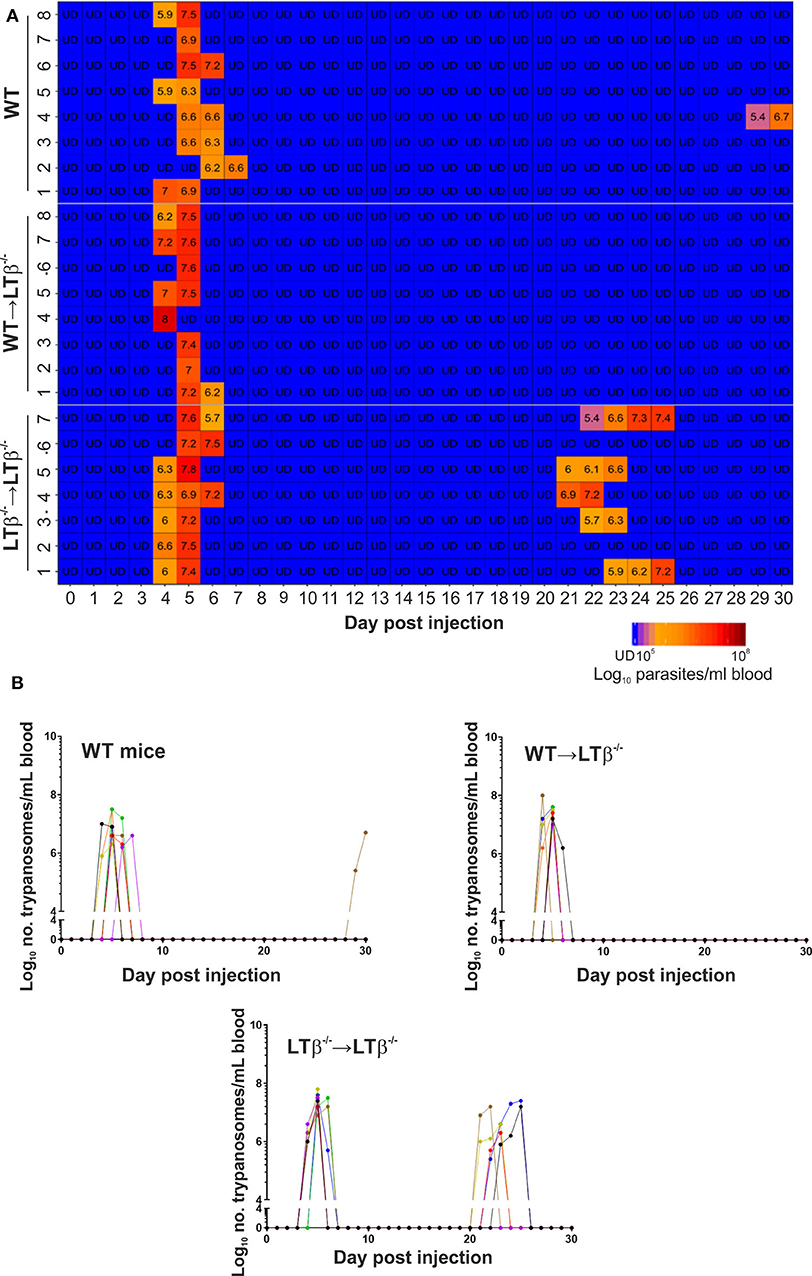
Figure 4. Enhanced susceptibility of LTβ−/− mice to ID infection with T. brucei. Groups of LTβ−/− mice were γ-irradiated and reconstituted with donor bone marrow from wild-type (WT) mice (WT → LTβ−/− mice) or LTβ−/− mice (LTβ−/− → LTβ−/− mice). Ten weeks later groups of WT mice, WT → LTβ−/− mice and LTβ−/− → LTβ−/− mice (n = 7–8 mice/group) were injected ID with a 1 × 105 dose of T. brucei STIB 247 parasites and blood parasitemia levels determined at daily intervals. (A) Heatmap shows the blood parasitemia (log10 number of trypanosomes/ml of blood) in each mouse. Each row represents data from an individual mouse. UD, below the limit of detection, ~5.4 log10 parasites/ml. (B) Charts show parasitemia profiles (log10 number of trypanosomes/ml of blood) in each mouse from each group following ID injection with 1 × 105 dose of T. brucei STIB 247 parasites. Each line represents data from an individual mouse.
We next determined whether the reconstitution of LTβ−/− mice with WT bone marrow induced the ability to produce class-switched Ig isotypes. By 30 d following ID injection with T. brucei, similarly elevated levels of total IgM were detected in the sera of WT mice, WT → LTβ−/− mice and LTβ−/− → LTβ−/− mice (Figure 5A). However, the sera of WT → LTβ−/− mice contained significantly elevated levels of total class-switched IgG isotypes when compared to uninfected WT controls (Figures 5B–D). These data suggest that the restoration of splenic microarchitecture in WT → LTβ−/− mice coincided with the ability to produce class-switched Ig isotypes.
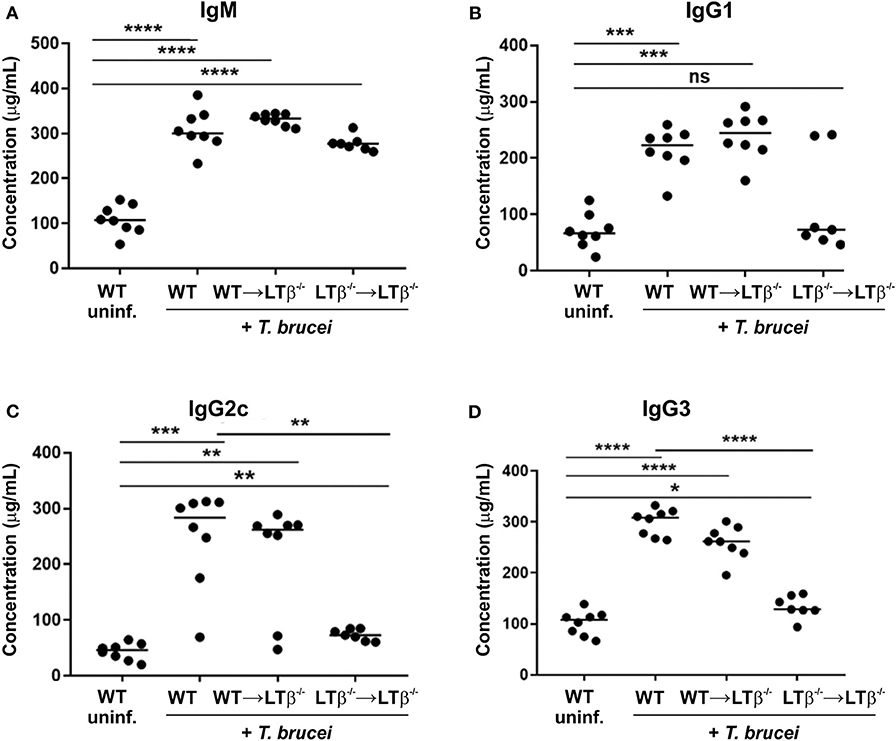
Figure 5. Reconstitution of LTβ−/− mice with wild-type (WT) bone marrow restores their ability to produce Ig isotype class-switched antibodies. Groups of LTβ−/− mice were γ-irradiated and reconstituted with donor bone marrow from WT mice (WT → LTβ−/− mice) or LTβ−/− mice (LTβ−/− → LTβ−/− mice). Ten weeks later groups of WT mice, WT → LTβ−/− mice and LTβ−/− → LTβ−/− mice (n = 7–8 mice/group) were injected ID with 1x105 dose of T. brucei STIB 247 parasites and 30 d later concentrations of total serum (A) IgM, (B) IgG1, (C) IgG2c, and (D) IgG3 determined by ELISA. Uninf., uninfected. Each point represents data from an individual mouse. Horizontal bar, median. Statistical analyses: (A) Student's t-test; (B) Mann-Whitney U-test; (C) Mann-Whitney U test; (D) Student's t-test; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Finally, we compared the levels of trypanosome-specific antibodies in the sera of infected WT mice, WT → LTβ−/− mice and LTβ−/− → LTβ−/− mice. Sera from infected mice from each group contained significantly elevated levels of trypanosome-specific IgM (Figure 6A). However, whereas WT mice and WT → LTβ−/− mice produced similarly elevated levels of trypanosome-specific class-switched IgG1, IgG2c, and IgG3 antibodies, much lower levels if any were detected in the sera of LTβ−/− → LTβ−/− mice (Figures 6B,C). Thus, these data suggest that the restoration of the microarchitecture in the spleens of WT → LTβ−/− mice enabled the production of trypanosome-specific class-switched antibodies.
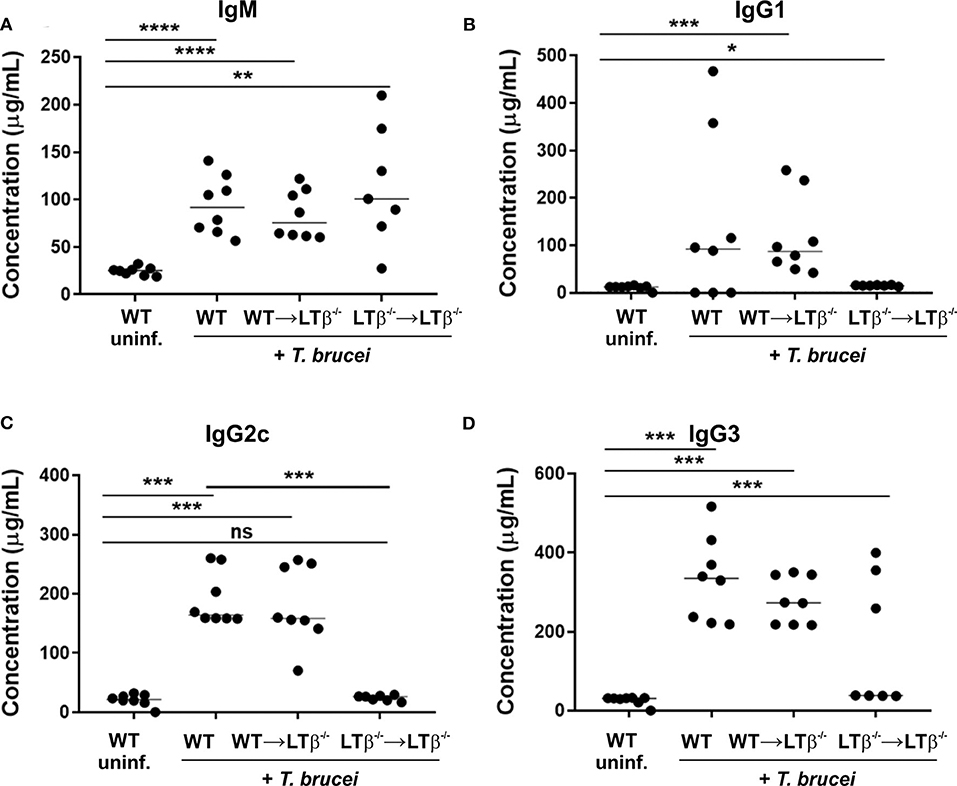
Figure 6. Reconstitution of LTβ−/− mice with wild-type (WT) bone marrow restores their ability to produce trypanosome-specific Ig isotype class-switched antibodies. Groups of LTβ−/− mice were γ-irradiated and reconstituted with donor bone marrow from WT mice (WT → LTβ−/− mice) or LTβ−/− mice (LTβ−/− → LTβ−/− mice). Ten weeks later groups of WT mice, WT → LTβ−/− mice and LTβ−/− → LTβ−/− mice (n = 7–8 mice/group) were injected ID with a 1 × 105 dose of T. brucei STIB 247 parasites and 30 d later concentrations of total serum (A) IgM, (B) IgG1, (C) IgG2c, and (D) IgG3 determined by ELISA. Uninf., uninfected. Each point represents data from an individual mouse. Horizontal bar, median. Statistical analyses: (A) Student's t-test; (B) Mann-Whitney U test; (C) Mann-Whitney U test; (D) Student's t-test; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Following ID injection into the host's skin the extracellular trypanosomes invade the afferent lymphatics (1) from where they infect the draining lymph nodes and begin to disseminate systemically (4, 5). For example, during the initial period following ID infection of cattle and goats with T. vivax by tsetse fly bite, the parasites were first encountered in the draining pre-scapular lymph nodes before the bloodstream (2, 3). Similar disease kinetics have also been reported in mice infected with T. brucei by tsetse fly bite (5). Circulating lymphocytes/leukocytes, antigens and other components within the lymph fluid can enter the bloodstream via the right lymphatic duct on the right side of the body, or the thoracic duct on the left side of the body. From these lymphatic ducts, the passage of lymph into the blood circulatory system is achieved either via the right or left subclavian vein. We show that the ability of the ID injected trypanosomes to establish systemic infection was not compromised in mice lacking the draining lymph nodes. Our data therefore suggest that following their initial invasion into the host's lymphatics, the trypanosomes can directly enter the bloodstream via the lymphatic ducts and disseminate further without the requirement for prior adaptation or amplification within the draining lymph nodes.
Although the LTβ−/− mice lacked most peripheral lymph nodes, had disorganized microarchitecture within their spleens and lacked the ability to mount trypanosome-specific class-switched IgG responses, the magnitude and duration of the initial parasitemia wave was similar when compared to WT mice. The increased ability of C57BL/6 mice to control the first parasitemia wave when compared to C3H/He mice, has been suggested to be associated with their relative ability to produce trypanosome-specific Ig (25). During the first few days after trypanosome infection the initial and predominant parasite-specific antibodies that are produced by the host are of the IgM isotype that recognize parasite surface antigens (26–29). The production of parasite-specific IgM was shown to be important for control of T. b. evansi infection (30). In contrast, a study using IgM-deficient mice has suggested only a limited role for IgM in controlling infection with clonal pleomorphic T. brucei AnTat 1.1E parasites (31). However, the compensatory effects of other Ig isotypes in host protection could not be entirely excluded. Macrophages have also been suggested to play an important role in the innate immune system's ability to control the infection by phagocytosing and destroying the parasites in tissues (32). For example, the Kupffer cells in the liver can aid the elimination of T. congolense from the bloodstream through IgM- and IgG-dependent phagocytosis of the parasites (33). In the current study the levels of parasite-specific IgM detected in the sera of LTβ−/− mice were similar to those in WT controls. This implies that a combination of parasite-specific IgM and the responses and actions of innate phagocytes are likely to play an important role in controlling the initial phase of the infection to the infecting parasite clones via antibody-dependent phagocytosis. It remains to be determined whether B1 B-cell subsets or B2 B-cell subsets, including GC B-cell, follicular B-cell and marginal zone (MZ) B cells, are the sources of the parasite-specific IgM in the current study. However, since mice that lack LTβR-signaling also lack GC, B cell follicles and MZ in their spleens (34, 35), B2 B cells are unlikely to be a major source of trypanosome-specific IgM in the ID infected LTβ−/− mice. An independent study has also suggested that MZ B cells are depleted following infection with trypanosomes by the IP route (36). The requirement for GC B cells is less clear. Whereas, data suggest that GC are not induced in the spleen during trypanosome infections (37), GC B cells have been reported to accumulate in this tissue during the later stages of IP infection with pleomorphic T. brucei AnTat 1.1E parasites (36). In contrast, GC formation has been described in the spleens of mice after IP infection with T. congolense (38). Whether GC are also induced in the skin draining lymph nodes during T. brucei infection is not known.
Closer analysis suggested that the onset of the first parasitemia wave in the ID infected LTβ−/− mice typically occurred a day earlier than ID infected WT mice. The precise reason for this earlier onset is uncertain, but it is plausible that after ID infection in WT mice some of the parasites are initially retained in, or migration is slowed by, the local draining lymph node. Alternatively, some parasites may also be phagocytosed and destroyed by resident macrophages as they travel through the draining lymph node. Since LTβ−/− mice lacked the draining lymph nodes it is possible that these effects were avoided, enabling a higher burden of parasites to initially enter the blood-stream, leading to the slightly earlier development of the first parasitemia peak.
As the infection progressed the LTβ−/− mice were unable to successfully control the parasitemia when compared to WT mice. Following the initial parasitemia wave the production of trypanosome-specific Ig isotype class-switched IgG antibodies is essential for the control of the subsequent relapses in the parasitemia (29, 31, 39, 40). The parasite class-switched IgG antibodies that are induced during this phase of the disease target the trypanosome's variable surface glycoprotein coat with high affinity (40, 41). Furthermore, differences in the magnitude of the host's parasite-specific class-switched IgG response can have a direct influence on the pathogenesis of T. congolense infection in mice (42) and cattle (43). For example, relatively resistant C57BL/6 mice produce high levels of trypanosome-specific IgG isotypes when compared to relatively susceptible A/J mice (42).
Here, the increased susceptibility of LTβ−/− mice to ID T. brucei infection coincided with significantly reduced serum levels of parasite-specific class-switched Ig isotypes. Since stimulation through the LTβR is essential for the organization of the B cell follicles within the secondary lymphoid tissues, LTβ−/− mice have disorganized B cell follicles (9, 12) and a significantly reduced ability to elicit high affinity antigen-specific class-switched IgG responses (35). Consistent with these studies the ID infected LTβ−/− mice also had significantly reduced serum levels of trypanosome-specific IgG1, IgG2c, and IgG3. This implies that the reduced ability of the LTβ−/− mice to effectively control the parasitemia during the later stages of the infection was at least in part a consequence of their reduced ability to produce parasite-specific class-switched IgG antibodies. To test this hypothesis the splenic microarchitecture and the ability of LTβ−/− mice to produce antigen-specific Ig class-switched antibodies was restored by reconstitution with bone marrow from WT donor mice. Our data clearly show that the WT → LTβ−/− mice produced significant levels of trypanosome-specific class-switched IgG1, IgG2c and IgG3, and this coincided with the ability of the WT → LTβ−/− mice to control further relapses in the parasitemia to a similar extent as WT mice. Future studies using transgenic mice with specific deficiencies in IgG1 (44) or IgG3 (45) will help resolve the individual roles of parasite-specific class-switched antibody isotypes in the control of ID T. brucei infections.
Our studies using LTβ−/− mice suggest that the presence of the draining lymph nodes has little, if any, impact on susceptibility to ID T. brucei infection. However, our data indicate that the status of the microarchitecture of the spleen has an important role in controlling the trypanosome infection. In the current study the disorganized microarchitecture in the spleens of LTβ−/− mice and LTβ−/− → LTβ−/− mice coincided with increased susceptibility to ID T. brucei infections. Conversely, the organized splenic microarchitecture in WT mice and WT → LTβ−/− mice coincided with their increased ability to control subsequent parasitemia waves. These data are consistent with data in an independent study, which showed that in the absence of the spleen, parasite-specific IgG2a/c and IgG3 responses were impaired and this coincided with increased susceptibility to T. congolense infection (40). Our data are also consistent with those in a study using mice lacking the B cell adaptor molecule Bam32 (38). When compared to WT controls, these mice also had increased disease severity, which correlated with impaired splenic GC formation and a diminished ability to produce parasite-specific IgG after IP T. congolense infection. Others, however, have suggested that the induction of anti-trypanosome antibody responses can occur independently of the lymph nodes and spleen, during experimental IP infection with T. brucei AnTat 1.1E parasites (46). Furthermore, another study suggested that LTβ−/− mice had reduced susceptibility to IP infection with T. congolense (47). The reason for the discrepancy between these studies and our own is uncertain, but may be related to differences in route of infection, parasite burden and parasite strains. When LTα−/− mice (as used by Magez and colleagues, 2002, (46) are immunized with low doses of T-cell-dependent antigens their ability to produce high affinity antigen-specific IgG1 is reduced. Conversely, when these mice are immunized with high doses of antigens they produce similar levels of high affinity antigen-specific IgG1 to WT mice (13, 34). Studies in mice also show that the route of trypanosome injection can have a significant influence on disease susceptibility and pathogenesis, with ID infections with T. brucei or T. congolense being 100X less efficient than IP infections (6). Using the T. brucei strain 10-26, Wei and colleagues showed that a detectable parasitemia was established in all mice injected with as few as 102 parasites by the IP route. However, the same dose was insufficient to establish a detectable infection by the ID route. Indeed, a detectable parasitemia was only observed in all of the recipient mice when injected ID with 104 parasites. Wei and colleagues also showed that when the mice were infected by the IP route the trypanosomes established a detectable parasitemia earlier than after ID injection, with a higher parasite burden (6). In the studies by Magez et al. (46) and Okwor et al. (47) the mice were similarly injected with trypanosomes by the IP route. Thus, it is plausible that the IP infections led to a significantly higher and earlier antigen burden in the LTα−/− mice enabling high affinity parasite-specific IgG responses to be induced despite the absence of organized splenic microarchitecture (13, 34). To resolve these issues additional studies are now required to definitively address the contributions of the skin draining lymph nodes and the spleen for the induction and maintenance of parasite-specific antibody responses after ID trypanosome infection. This could include comparison of parasitemia kinetics, disease susceptibility and parasite-specific B cell responses in intact and splenectomised WT mice and LTβ−/− mice.
When considered in the context of similar studies, our data imply that the first wave of the parasitemia after ID infection with T. brucei is predominantly controlled by innate mechanisms, most likely including non-class switched IgM and the engulfment of the parasites by phagocytes. However, control of the subsequent parasitemia waves requires organized B cell follicles in the spleen and the production of trypanosome-specific class-switched IgG. Drug resistance, as well as adverse side-effects, continue to significantly impact on the ability to successfully treat and control trypanosome infections. A more thorough understanding of the host factors that are essential for the efficient control of ID trypanosome infections could identify novel therapeutics and aid the development of protective vaccines.
All datasets generated for this study are included in the article/supplementary material.
The animal study was reviewed and approved by The Roslin Institute's and University of Edinburgh's ethics committees.
LM and NM conceived the study and obtained funding. OA, LM, and NM designed the study. OA, EP, and BB performed the experiments and acquired these data. All authors interpreted these data and contributed to the final version of the manuscript.
This study was supported by project Institute Strategic Programme Grant funding (BBS/E/D/20231762 and BBS/E/D/20002174) and a Strategic Skills Award and EASTBIO doctoral training partnership studentship (BB/J01446X/1) from the Biotechnology and Biological Sciences Research Council.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Alfituri OA, Ajibola O, Brewer JM, Garside P, Benson RA, Peel T, et al. Effects of host-derived chemokines on the motility and viability of Trypanosoma brucei. Parasite Immunol. (2019) 41:e12609. doi: 10.1111/pim.12609
2. Adams ARD. Trypanosiomiasis of stock in Mauritius III. The diagnosis and course of untreated T vivax infections in domestic animals. Ann Tropical Med Parasitol. (1936) 30:521–31. doi: 10.1080/00034983.1936.11684957
3. Emery DL, Barry JD, Moloo SK. The appearance of Trypanosoma (Duttonella) vivax in lymph following challenge of goats with infected Glossina moritans moritans. Acta Trop. (1980) 37:375–9.
4. Tabel H, Wei G, Bull HJ. Immunosuppression: cause for failures of vaccines against African trypanosomiases. PLoS Negl Trop Dis. (2013) 7:e2090. doi: 10.1371/journal.pntd.0002090
5. Caljon G, Van Reet N, De Trez C, Vermeersch M, Perez-Morga D, Van Den Abbeele J. The dermis as a delivery site of Trypanosoma brucei for tsetse flies. PLoS Pathog. (2016) 12:e1005744. doi: 10.1371/journal.ppat.1005744
6. Wei G, Bull H, Zhou X, Tabel H. Intradermal infections of mice by low numbers of African trypanosomes are controlled by innate rsistance but enhance susceptibility to reinfection. Journal of Infectious Diseases. (2011) 203:418–29. doi: 10.1093/infdis/jiq051
7. Chaplin DD, Fu X. Cytokine regulation of secondary lymphoid organ development. Curr Opin Immunol. (1998) 10:289–97. doi: 10.1016/S0952-7915(98)80167-2
8. Rennert PD, Browning JL, Hochman PS. Selective disruption of lymphotoxin ligands reveals a novel set of mucosal lymph nodes and unique effects on lmyph node cellular organization. Int Immunol. (1997) 9:1627–39. doi: 10.1093/intimm/9.11.1627
9. de Togni P, Goellner J, Ruddle NH, Streeter PR, Fick A, Mariathasan S, et al. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. (1994) 264:703–7. doi: 10.1126/science.8171322
10. Banks TA, Rouse BT, Kerley MK, Blair PJ, Godfrey VL, Kuklin NA, et al. Lymphotoxin-a-deficient mice. J Immunol. (1995) 155:1685–93.
11. Ettinger R, Browning JL, Michie SA, van Ewijk W, McDevitt HO. Disrupted splenic architecture, but normal lymph node development in mice expressing a soluble lymphotoxin-b receptor-IgG1 fusion protein. Proc Natl Acad Sci USA. (1996) 93:13102–7. doi: 10.1073/pnas.93.23.13102
12. Alimzhanov MB, Kuprash DV, Kosco-Vilbois MH, Luz A, Turetskaya RL, Tarakhovsky A, et al. Abnormal development of secondary lymphoid tissues in lymphotoxin b-deficient mice. Proc Natl Acad Sci USA. (1997) 94:9302–7. doi: 10.1073/pnas.94.17.9302
13. Fu Y-X, Molina H, Matsumoto M, Huang G, Min J, Chaplin DD. Lymphotoxin-a (LTa) supports development of splenic follicular structure that is required for IgG response. J Exp Med. (1997) 185:2111–20. doi: 10.1084/jem.185.12.2111
14. Fütterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K. The lymphotoxin b receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. (1998) 9:59–70. doi: 10.1016/S1074-7613(00)80588-9
15. Ngo VN, Korner H, Gunn MD, Schmidt KN, Riminton DS, Cooper MD, et al. Lymphotoxin a/b and tumour necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J Exp Med. (1999) 189:403–12. doi: 10.1084/jem.189.2.403
16. Jenni L. Comparisons of antigenic types of Trypanosoma (T) brucei strains transmitted by Glossina m. morsitans Acta Trop. (1977) 34:35–41.
17. Koni PA, Flavell RA. A role for tumour necrosis factor receptor type 1 in gut-associated lymphoid tissue development: genetic evidence of synergism with lymphotoxin b. J Exp Med. (1998) 187:1977–83. doi: 10.1084/jem.187.12.1977
18. Tumanov AV, Kuprash DV, Lagarkova MA, Grivennikov SI, Abe K, Shakhov A, et al. Distinct role of surface lymphotoxin epxressed by B cells in the organization of secondary lymphoid tissues. Immunity. (2002) 239:239–50. doi: 10.1016/S1074-7613(02)00397-7
19. Herbert WJ, Lumsden WH. Trypanosoma brucei: a rapid “matching” method for estimating the host's parasitemia. Exp Parasitol. (1976) 40:427–31. doi: 10.1016/0014-4894(76)90110-7
20. Mackay F, Browning JL. Turning off follicular dendritic cells. Nature. (1998) 395:26–7. doi: 10.1038/25630
21. Heesters BA, Myers RC, Carroll MC. Follicular dendritic cells: dynamic antigen libraries. Nat Rev Immunol. (2014) 14:495–504. doi: 10.1038/nri3689
22. Krautler NJ, Kana V, Kranich J, Tian Y, Perera D, Lemm D, et al. Follicular dendritic cells emerge from ubiquitous perivascular precursors. Cell. (2012) 150:194–206. doi: 10.1016/j.cell.2012.05.032
23. Victoratos P, Lagnel J, Tzima S, Alimzhanov MB, Rajewsky K, Pasparakis M, et al. FDC-specific functions of p55TNFR and IKK2 in the development of FDC networks and of antibody responses. Immunity. (2006) 24:65–77. doi: 10.1016/j.immuni.2005.11.013
24. Glaysher BR, Mabbott NA. Role of the GALT in scrapie agent neuroinvasion from the intestine. J Immunol. (2007) 178:3757–66. doi: 10.4049/jimmunol.178.6.3757
25. Black SJ, Sendashonga CN, Webster P, Koch GLE, Shapiro SZ. (1986) Regulation of parasite-specific antibody responses in resistant (C57BL/6) and susceptible (C3H/HE) mice infected with Trpanosoma (trypanozoon) brucei brucei. Parasite Immunol 8, 425–442. doi: 10.1111/j.1365-3024.1986.tb00859.x
26. Reinitz DM, Mansfield JM. T-cell-independent and T-cell-dependent B-cell responses to exposed variant surface glycoprotein epitopes in trypanosome-infected mice. Infect Immun. (1990) 58:2337–42. doi: 10.1128/IAI.58.7.2337-2342.1990
27. Pan W, Ogunremi O, Wei G, Shi M, Tabel H. CR3 (CD11b/CD18) is the major macrophage receptor for IgM antibody-mediated phagocytosis of African trypanosomes: diverse effect on subsequent synthesis of tumor necrosis factor alpha and nitric oxide. Microbes Infect. (2006) 8:1209–18. doi: 10.1016/j.micinf.2005.11.009
28. Vincendeau P, Bouteille B. Immunology and immunopathology of African trypanosomiasis. An Acad Bras Cienc. (2006) 78:645–65. doi: 10.1590/S0001-37652006000400004
29. Guirnalda P, Murphy NB, Nolan D, Black SJ. Anti-Trypanosoma brucei activity in Cape buffalo serum during the cryptic phase of parasitemia is mediated by antibodies. Int J Parasitol. (2007) 37:1391–9. doi: 10.1016/j.ijpara.2007.04.019
30. Baral TN, De Baetselier P, Brombacher F, Magez S. Control of trypanosoma evansi infection is igm mediated and does not require a type i inflammatory response. J Infect Dis. (2007) 195:1513–20. doi: 10.1086/515577
31. Magez S, Schwegmann A, Atkinson R, Claes F, Drennan M, De Baetselier P, et al. The role of B-cells and IgM antibodies in parasitemia, anemia, and VSG switching in Trypanosoma brucei-infected mice. PLoS Pathog. (2008) 4:e1000122. doi: 10.1371/journal.ppat.1000122
32. Dempsey WL, Mansfield JM. Lymphocyte function in experimental African trypanosomiasis. V Role of antibody and the mononuclear phagocyte system in variant-specific immunity. J Immunol. (1983) 130:405–11.
33. Shi M, Wei G, Pan W, Tabel H. Trypanosoma congolense infections: antibody-mediated phagocytosis by Kupffer cells. J Leukoc Biol. (2004) 76:399–405. doi: 10.1189/jlb.1003500
34. Matsumoto M, Lo SF, Carruthers CJL, Min J, Mariathasan S, et al. Affinity maturation without germinal centres in lymphotoxin-a-deficient mice. Nature. (1996) 382:462–6. doi: 10.1038/382462a0
35. Koni PA, Sacca R, Lawton P, Browning JL, Ruddle NH, Flavell RA. Distinct roles in lymphoid organogenesis for lymphotoxins a and b revealed in lymphotoxin b-deficient mice. Immunity. (1997) 6:491–500. doi: 10.1016/S1074-7613(00)80292-7
36. Radwanska M, Guirnalda P, De Trez C, Ryffel B, Black S, Magez S. Trypanosomiasis-induced B cell apoptosis results in loss of protective anti-parasite antibody responses and abolishment of vaccine-induced memory responses. PLoS Pathog. (2008) 4:e1000078. doi: 10.1371/journal.ppat.1000078
37. Morrison WI, Murray M, Bovell DL. Response of the murine lymphoid system to a chronic infection with Trypanosoma congolense. I The spleen. Lab Invest. (1981) 45:547–57.
38. Onyilagha C, Jia P, Jayachandran N, Hou S, Okwor I, Kuriakose S, et al. The B cell adaptor molecule Bam32 Is critically important for optimal antibody response and resistance to Trypanosoma congolense infection in mice. PLoS Negl Trop Dis. (2015) 9:e0003716. doi: 10.1371/journal.pntd.0003716
39. Campbell GH, Esser KM, Weinbaum FI. Trypanosoma rhodesiense infection in B-cell-deficient mice. Infect Immun. (1977) 18:434–8. doi: 10.1128/IAI.18.2.434-438.1977
40. Magez S, Radwanska M, Drennan M, Fick L, Baral TN, Brombacher F, et al. Interferon-g and nitric oxide in combination with antibodies are key protective host immune factors during Trypanosoma congolense TC13 infections. J Infect Dis. (2006) 193:1575–83. doi: 10.1086/503808
41. Stijlemans B, Radwanska M, De Trez C, Magez S. (2017). African trypanosomes undermine humoral responses and vaccine development: link with inflammatory responses? Front Immunol 8:582. doi: 10.3389/fimmu.2017.00582
42. Morrison WI, Murray M. The role of humoral immune responses in determining susceptibility of A/J and C57BL/6 mice to infection with Trypanosoma congolense. Parasite Immunol. (1985) 7:63–79. doi: 10.1111/j.1365-3024.1985.tb00479.x
43. Taylor KA, Lutje V, Kennedy D, Authie E, Boulange A, Logan-Henfrey L, et al. Trypanosoma congolense: B-lymphocyte responses differ between trypanotolerant and trypanosusceptible cattle. Exp Parasitol. (1996) 83:106–16. doi: 10.1006/expr.1996.0054
44. Lorenz M, Jung S, Radbruch A. Switch transcripts in immunoglobulin class switching. Science. (1995) 267:1825–8. doi: 10.1126/science.7892607
45. Shapiro DA, Threadgill DS, Copfer MJ, Corey DA, McCool TL, McCormick L, et al. γ3 gene-disrupted mice selectively deficient in the dominant IgG subclass made to bacterial polysaccharides undergo normal isotype switching after immunization with polysaccharide-protein conjugate vaccines. J Immunol. (1998) 161:3393–9.
46. Magez S, Stijlemans B, Caljon G, Eugster H-P, De Baetselier P. (2002). Control of experimental Trypanosoma brucei infection occurs independently of lymphotoxin-a induction. Infect. Immun. 70, 1342–1351. doi: 10.1128/IAI.70.3.1342-1351.2002
Keywords: Trypanosoma brucei, trypanosomes, spleen, lymph nodes, B cells, lymphotoxin
Citation: Alfituri OA, Bradford BM, Paxton E, Morrison LJ and Mabbott NA (2020) Influence of the Draining Lymph Nodes and Organized Lymphoid Tissue Microarchitecture on Susceptibility to Intradermal Trypanosoma brucei Infection. Front. Immunol. 11:1118. doi: 10.3389/fimmu.2020.01118
Received: 09 January 2020; Accepted: 07 May 2020;
Published: 03 June 2020.
Edited by:
Malcolm Scott Duthie, HDT Biotech Corporation, United StatesReviewed by:
Guy Caljon, University of Antwerp, BelgiumCopyright © 2020 Alfituri, Bradford, Paxton, Morrison and Mabbott. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neil A. Mabbott, bmVpbC5tYWJib3R0QHJvc2xpbi5lZC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.