 Lumin Zhang
Lumin Zhang Meena B. Bansal
Meena B. Bansal
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 16 June 2020
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01086
This article is part of the Research Topic Advances in Liver Inflammation and Fibrosis due to Infectious Diseases View all 10 articles
While the interactions between HIV and various liver cell populations have been explored, the relevance of these interactions when patients are well-controlled on ART is less clear. Therefore, we focus this perspective on HIV-related alterations that may drive hepatic inflammation and fibrosis in aviremic patients, with a focus on Kupffer cells and Hepatic Stellate Cells. Persistent CD4+ T cell depletion in the gut resulting in increased gut permeability has been postulated to play a role in systemic immune activation in HIV patients. The liver, with its unique location, remains the gatekeeper between the gut and the systemic circulation. The resident liver macrophage, Kupffer cell, is responsible for clearing and responding to these products. We propose that changes in Kupffer cell biology, in the context of HIV infection, creates a mileu that drives hepatic inflammation and fibrosis in response to microbial translocation. Targeting these pathways may be helpful in improving liver-related outcomes in HIV patients.
End-stage liver disease is a major cause of non-AIDS related mortality in HIV+ patients even with effective anti-retroviral therapy, accounting for almost 15% of deaths (1–7). As a result of shared routes of transmission, HCV and HBV are the most common liver diseases in HIV-infected patients, although other chronic liver diseases are emerging (8, 9). Most data, therefore, regarding fibrosis progression rates is derived from those with coinfection. These patients have a higher relative risk (RR) of cirrhosis, increased development of decompensated cirrhosis and accelerated fibrosis progression rates compared with those who are only infected with HCV or HBV (10, 11). Furthermore, rapid fibrosis correlates with reduced CD4+ T cell counts and detectable plasma HIV levels. HIV patients are also more susceptible to other liver diseases, which synergize to accelerative liver fibrosis. Alcohol consumption is associated with increased relative risk of fibrosis progression in HIV mono-infected patients (12) while NASH is emerging as a major cause of liver disease, with half of mono-infected patients with unexplained liver enzyme elevations having NASH (13). While many may have an unrecognized chronic liver injury, a higher frequency of liver fibrosis was demonstrated in HIV-1–monoinfected patients (range 11–40.9%) compared with uninfected patients even without coinfection of hepatitis viruses and alcohol abuse, suggesting a correlation between HIV-1 infection and advanced liver fibrosis (14–19). Therefore, persistent HIV-1 infection and viral associated liver immune dysfunction may independently contribute to the progression of liver diseases (20). Lastly, in those on ART, drug-induced liver injury and increased rates of NASH due to both medications and metabolic derangements common in HIV are being observed. While hepatic stellate cells are the downstream effector of liver fibrosis, this perspective focuses on the role Kupffer cells play in promoting a mileu conducive to fibrosis progression in patients with HIV infection, particularly in aviremic patients.
Shortly after HIV infection, a severe CD4+ T cell depletion in the gut-associated lymphoid tissues leads to a disruption of the intestinal barrier, consequently promoting translocation of microbial products into the portal circulation. The liver, which derives the majority of its blood flow from the portal circulation, is uniquely positioned to protect the systemic circulation from gut-derived products. In particular, the resident hepatic macrophage, the Kupffer cell, located within the hepatic sinusoid is charged with clearing translocated bacterial products in an immunotolerant manner. However, when products provoke a pro-inflammatory response by Kuppffer cells, a cascade of intrahepatic inflammatory respones is initiated with numerous secreted cytokines, such as IL-1β, TNF-α, and IL-6, serving as major drivers in the progression of liver injury and fibrosis.
Kupffer cells (KCs) are the largest population of resident tissue macrophages in the liver. They reside within the hepatic sinusoid in close proximity to hepatic stellate cells, liver sinusoidal endothelia cells, and intrahepatic lymphocytes. Both the low flow state of the portal circulation and the uniquely fenestrated endothelium create a conducive environment for interaction of KCs with neighboring cells and circulating cells of the immune system. Physiologically, KCs are the first line of defense to eliminate macromolecules, immune complexes, senescent cells, virally-infected cells, and translocated microbial products from the gut to avoid liver injury and systemic immune responses (21). Given the dynamic nature of cell surface receptor expression on macrophage populations and some controversy regarding their origins, CD163 or CD68, CD14 and CD16 are often used to identify human KCs. However, murine KCs display phenotypic patterns characterized by F4/80+, MHCII, and CD11bInt expression. A detailed discussion of markers for various macrophage subpopulations within the liver is beyond the scope of this perspective and discussed elsewhere (22). The focus of this perspective is on the role of CD68+human KCs in promoting liver inflammation and fibrosis in patients with HIV.
The importance of KCs in liver injury and inflammation have been established with depletion studies wherein GdCl3 was associated with AST reduction and inflammation in an alcohol model of liver injury (23). Crosstalk between KCs and HSCs is also evidenced by KC depletion as mRNA levels of TGF-β, α-SMA and collagen I are significantly decreased (24). Although GdCl3 is not specific to KCs, and thus interpretation is complex, GdCl3 treatment dramatically decreased cytokines predominantly produced by KCs, TNF-α, IL-6, and IL-1β, in response to LPS stimulation in murine livers (25, 26). Similarly, liposome/clodronate can suppress pro-inflammatory responses through the depletion of KCs (27).
In homeostasis, KCs are central to intrahepatic immune tolerance through an antigen-mediated induction of functional arrest of CD4 cells and regulatory T cells. However, in an inflamed microenvironment this delicate equilibrium is disrupted resulting in immune dysfunction and tolerance break (28). Indeed, knockout of TREM-1 (Triggering receptor expressed on myeloid cells), which is highly expressed on KCs in liver fibrosis, reduced liver fibrosis through the inhibition of TNF-α and IL-6 responses in a number of chronic injury models (29). Similarly, knock down of Jun N-terminal kinase (JNK-1/2) from KCs reversed liver fibrosis in a choline-deficient L-aminoacid-defined (CDAA) model, with a decline in inflammatory responses, including TNF-α, IL6, IL-1β, and TGF-β (30).
While KCs display M1-like features in acute liver injury, with protracted chronic inflammation, due to exhaustion of M1-like macrophages and immune cells, M2-like macrophages emerge and secrete protective cytokines upon chronic cytotoxic stimulation such as IL-4, IL-10, and TGF-β (31, 32). IL-10, an anti-inflammatory cytokine, down-regulates macrophage effector functions and differentiation of neighboring cells to maintain immune microenvironment homeostasis. For example, administration of IL-10 decreased TNF-α produced from LPS-treated KCs (33). While very complex, the manipulation of KC mediated immune responses or approaches to limit their stimulation may be exploited therapeutically.
The impact of translocated microbial products on KCs is well-established. pretreatment with 2.5% dextran sulphate sodium (DSS) causes increased intestinal permeability and promotes translocation of microbial products into the portal blood in mice. The resulting amplified TLR4 mediated inflammatory responses in KCs resulted in significant livery injury (34). Using a liver slice model, LPS stimulation increased IL-1β and TNF-α production compared to the control (35). Consistently, in mouse models, LPS administration rapidly induces the release of inflammatory cytokines in the liver with a higher IL-6 production obtained from LPS stimulated KCs than splenic and alveolar macrophages (36).
TLR4, as one member of Toll-like receptors, belongs to the pattern recognition receptor (PRR) family. After stimulation by TLR4 ligands, for example lipopolysaccharides, TLR4 is activated through conformational changes and interaction with TIR-domain-containing adapter proteins via hydrophilic interactions. Intracellular TLR4 signaling is mediated by two classical pathways: the TIRAP–MyD88-NF-κB pathway and the TRIF–TRAM-interferon regulatory factor-3 (IRF3)-NF-κB pathway. TLR4 signaling participates in the initiation of pro-inflammatory response, especially TRIF mediated TNF-α and synthesis of chemokines and have been reveiwed in detail elswhere (37).
In addition to TNF-α, TLR4 signaling also contributes to the transmission of two priming signals for the IL-1β pathway through the NLRP3 inflammasome. IL-1β is a crucial proinflammatory cytokine in response to microbial infection. IL-1β from LPS-treated KCs can produce a deleterious effect on hepatocytes and promote the secretion of VLDL apo B and lipid (38). IL-1β was also found to inhibit IFN-α induced STAT1 activation in hepatocytes, attenuating the innate immune response to viral infection in hepatocytes (39). In general, NLRP3 mediated-cleavage of caspase 1 is the critical step to promote the maturation of IL-1β. The formation of the NLRP3 inflammasome is initiated by ATP or microbial stimulation (40). Blockage of NLRP3 activation in KCs decreased IL-1β response to Ischemia/Reperfusion induced liver injury and improved survival (41, 42). Administration of MCC950, a small molecule selective inhibitor of NLRP3, suppressed LPS primed IL-1β response in NPC cells, subsequently, decreasing liver injury (43). Given the important role of TLR4 signaling in KCs, the modulation of this pathway in the context of HIV infection and persistent microbial translocation is critical.
In addition to CD4, both CCR5 and CXCR4, HIV-1 co-receptors, are detected on human KCs isolated from non-HIV-1 individuals, suggesting that KCs are permissive for HIV-1 infection. HIV-1 infection of KCs in viremic patients has been shown by in situ hybridization for HIV-1 RNA and PCR for proviral DNA on FACS-purifed KCs from livers of patients with Acquired Immunodeficiency Sydnrome (AIDs) (44–46). Moreover, retrieval of HIV-1 from KCs derived from patients either not on ART (47) or on ART for short durations has been shown and supported by studies in SIVDH12R-infected macaques (48, 49). Recently it has been shown that KCs derived from patients on long term ART, while containing evidence of HIV-1 transcripts, do not secrete replication competent virus (50). While macrophages are known to be able to transmit infectious virus to susceptible CD4+ cells via cell-cell contact (51, 52), the ability of KCs in patients on long-term ART to do so has not yet been explored though warrants investigation.
We have shown that Kupffer cells are highly permissive for HIV-1 infection in vitro with robust and sustained viral replication (53). HIV-1BaL, a laboratory adapted CCR5-tropic HIV, infection rendered KCs more sensitive to LPS treatment through an increase in CD14 and TLR4 expression on the cell surface, resulting in increased secretion of TNF-α and IL-6, which was blocked by a small molecule TLR4 inhibitor. Interestingly, despite AZT and ritonavir abrogated viral replication, KCs maintained their sensitivity to the pro-inflammatory response to LPS. These findings suggest that even in patients on ART, KC biology may be impacted and promote a mileu supporting hepatic inflammation and fibrosis in response to microbial translocation. While no change in IFNα or IFNβ expression in HIV-1 infected KCs was observed, IL-1β mRNA and both intracellular and secreted IL-1β was increased by HIV-1BaL infection. Similar to the IL-6 and TNF-α response, this HIV-related sensitization was found to be TLR4-dependent and further determined to be via the NLRP3-caspase 1 pathway. Immunostaining on liver tissue derived from aviremic HIV+ patients demonstrated an increased expression of IL-1β compared to normal liver with a high degree of colocalization in CD68+ macrophages (54). These studies show that TLR4 mediated NLRP3 activation is critical for the inflammatory responses to microbial products in KCs. Importantly, liver injury and resulting damage-associated molecular patterns (DAMPS) also activate TLR4 signals in KCs and thus may play a role in other forms of liver injury in HIV patients such as drug-induced liver injury. Interestingly, it has also been shown that CCR5 and TLR may co-cluster on monocyte-derived macrophages (MDMs) as secretion of CCL2 and CXCL8 in response to either R5 gp120, recombinant envelope protein from CCR5-tropic HIV-1, or LPS can be blocked by either a CCR5 inhibitor or TLR4 blocking. These results suggest another mechanism for synergistic effects of HIV and LPS on macrophage biology and should be specifically examined in human KCs (55).
While beyond the scope of this perspective, HIV-1 infection impacts a number of other cells critical to the inflammatory response in the liver. In line with circulating CD4+ T cells, HIV infection leads to a depletion of CD4+ T cell in the liver with relative reversal of CD4/CD8 ratio typically seen. Viral infection also makes IL2+ CD4+ T cells dysfunctional and attenuates hepatic immune response to microbial infection (56–58). CD4+ T cells from HIV mono-infected patients exhibit a low regulatory effect on Natural killer (NK). Co-cultured with NK cells, CD4+ T cells from HIV-1+ individuals greatly reduced anti-fibrotic effect of NK cells on HSCs (59). Therefore, reduction in CD4+ T cells influences progression of liver fibrosis in HIV+ patients (10, 60) while increased relative CD8+T cells correlates with a higher fibrosis scores in HIV-1 infected patients (61).
NK cells, which account for up to 30–50% human liver lymphocytes, play an important role in clearing virally infected cells trough NK cell antibody dependent cell cytotoxicity (ADCC). The activation and NK cellular numbers are spontaneously increased early in response to HIV-1 infection but with chronic infection exhaustion results in NK dysfunction with persistent viremia (62).
While the role of DCs in HIV-1 infection and progression and ability to transmit infectious virus to CD4 cells by cell-cell contact has been shown, HIV interaction with DCs in the liver is less studied. TLR7 is constitutively expressed by human pDCs. The delivery of HIV-1 viral nucleic acids in early endosome of pDCs can be blocked by TLR7 inhibitor (63), suggesting that TLR7 is involved in the antigen presentation by pDCs. In addition, mDC express TLR4 and the frequency of mDCs, especially CXCL16-producing mDCs has been shown to be associated with the level of microbial products in the liver of HIV+ patients (64).
In vitro studies have shown that the envelope protein, HIV gp 120, which binds either CXCR4 (X4) or CCR5 (R5) on its target cell can promote hepatocyte apoptosis (65) and along with the HCV glycoprotein E2 promote the secretion of the pro-inflammatory cytokine IL-8 (66, 67). It has been known that Kupffer cells play a primary role in the clearance of apoptotic hepatocytes/cellular debris within the liver and thus play a key role in sterile inflammation and repair (68). More recently, Ganesan et. al demonstrated that ethanol exposure promotes HIV accumulation within hepatocytes, ultimately leading to increased oxidative stress and apoptosis. These apoptotic hepatocytes then stimulate inflammasome activation in KCs and pro-fibrogenic genes in hepatic stellate cells (69). Moreover, hepatic stellate cells can also engulf apoptotic hepatocytes resulting in NAPDH oxidation, stellate cell activation, and fibrogenesis (70). Therefore, effects on HIV on hepatocytes can promote both KC and HSC activation, synergistically driving hepatic inflammation and fibrosis.
HSCs express both HIV CCR5 and CXCR4 co-receptors. We have shown that HIV and its envelope protein gp120 promote HSC activation, collagen I production, and CCL2 secretion through interactions with CXCR4 (71) and others have shown that the envelope protein on HIV that preferentially uses CCR5 for cellular entry (R5 gp120) promotes HSC chemotaxis and CCL2 secretion (72). While HIV can infect HSCs in vitro, infection in vivo has not been established. Similar to what has recently been shown for KCs, in vitro infected HSCs do not secrete replication competent virus though, like DCs, may be able to transmit virus by cell-cell contact (71, 73, 74). Similar to what has been shown on MDMs, CCR5 and TLR4 seem to co-cluster on HSCs and result in increased CCL2 and CXCL8 in response to gp120 and LPS, with effects of ligands blocked by inhibiting either receptor alone (55). As CCL2 is an important chemokine for attracting circulating monocytes into the liver, this may be important for propagating hepatic inflammation. R5 gp120 also promotes IL-6 secretion from HSCs through Jun-NF-kB activation (75). These studies suggest that HIV promotes inflammation and fibrosis by interacting with CXCR4 and CCR5 via gp120 and synergizes with TLR4 signals. While the latter is important in viremic patients, relevance for those on ART are not clear. For those on ART, the impact of HIV on microbial translocation and KC biology may be more important.
While the ultimate effector cell in liver fibrosis is the hepatic stellate cell, the signals generated by KCs are critically important in promoting the activation of HSCs and then perpetuating the activated state. TLR4 activation on HSCs results in downregulation of the TGFβ1 pseudoreceptor, BAMBI, which sensitizes HSCs to the pro-fibrogenic effects of TGFβ1 (76), much of which is derived by KCs. Therefore, in the context of HIV-1 and associated microbial translocation, the effects on both KCs and HSCs are compounded and drive hepatic inflammation and fibrosis (Figure 1). Overall association between microbial translocation and liver fibrosis progression has been shown in a variety of liver diseases and thus HIV simply compounds this effect.
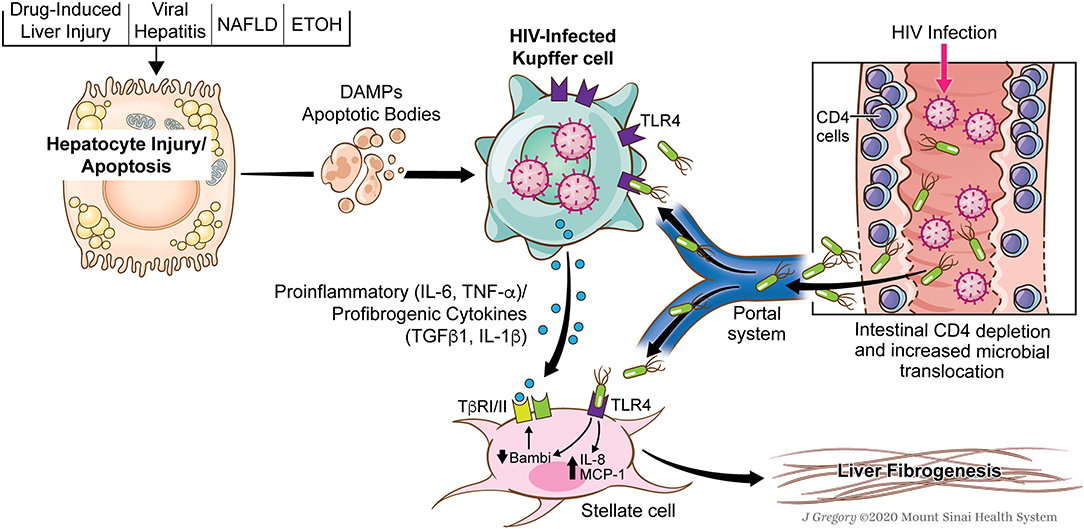
Figure 1. Role of HIV and associated microbial translocation in driving hepatic inflammation and fibrosis through kupffer cell and stellate cell interactions. HIV infection causes early CD4 gut depletion which promotes increased microbial translocation and the delivery of pathogen-associated molecular patterns to the liver (PAMPs). At the same time, patients with HIV have multiple secondary chronic liver injuries (NAFLD, DILI, HCV, HBV, Alcohol) which promote direct injury to hepatocytes and give rise to damage-associated molecular patterns (DAMPs) or endogenous TLR ligands. HIV infected KCs are sensitized to effects of both the translocated microbial products as well as DAMPs. These signals converge on the HIV-infected KCs resulting in the secretion of a number of pro-inflammatory (IL-6, TNF-α) and pro-fibrogenic cytokines (IL-1β and TGFβ1). Additionally, TLR ligands have pro-inflammatory effects on HSCs and sensitize them to KC-derived TGFβ1. Used with permission from @Mount Sinai Health System.
As patients with HIV live longer, liver disease will continue to emerge as a leading cause of morbidity and mortality. Understanding how HIV may set the stage for hepatic injury, inflammation and fibrosis may lead to novel therapeutic strategies. While treatment of underlying diseases, ranging from viral hepatitis to NASH or alcohol, remains the most imporant strategy, the alterations unique to this population need to be kept in mind. With this perspective, HIV related alterations in KC biology and microbial translocation may be at the nexus of creating a milue conducive to hepatic fibrosis. Targeting either the KC response to TLR4 ligands, PAMPs or DAMPS, decreasing the burden of microbial products from reaching the portal circulation, or blocking downstream pro-inflammatory or pro-fibrogenic effects on stellate cells are important to consider. Much will be learned from current treatments undergoing investigation for non-HIV related liver fibrosis that may be additionally leveraged for this special population.
LZ and MB wrote and reviewed this manuscript.
This work was supported by research grants NIH R01DK108364 to MB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Lifson AR, Group ICODW, Belloso WH, Carey C, Davey RT, Duprez D, et al. Determination of the underlying cause of death in three multicenter international HIV clinical trials. HIV Clin Trials. (2008) 9:177–85. doi: 10.1310/hct0903-177
2. Data Collection on Adverse Events of Anti-HIV drugs (D:A:D) Study Group, Smith C, Sabin CA, Lundgren JD, Thiebaut R, Weber R, et al. Factors associated with specific causes of death amongst HIV-positive individuals in the D:A:D Study. AIDS. (2010) 24:1537–48. doi: 10.1097/QAD.0b013e32833a0918
3. Price JC, Thio CL. Liver disease in the HIV-infected individual. Clin Gastroenterol Hepatol. (2010) 8:1002–12. doi: 10.1016/j.cgh.2010.08.024
4. Leone S, Gregis G, Quinzan G, Velenti D, Cologni G, Soavi L, et al. Causes of death and risk factors among HIV-infected persons in the HAART era: analysis of a large urban cohort. Infection. (2011) 39:13–20. doi: 10.1007/s15010-010-0079-z
5. Puoti M, Moioli MC, Travi G, Rossotti R. The burden of liver disease in human immunodeficiency virus-infected patients. Semin Liver Dis. (2012) 32:103–13. doi: 10.1055/s-0032-1316473
6. Weber R, Ruppik M, Rickenbach M, Spoerri A, Furrer H, Battegay M, et al. Decreasing mortality and changing patterns of causes of death in the Swiss HIV Cohort Study. HIV Med. (2013) 14:195–207. doi: 10.1111/j.1468-1293.2012.01051.x
7. Salmon-Ceron D, Rosenthal E, Lewden C, Bouteloup V, May T, Burty C, et al. Emerging role of hepatocellular carcinoma among liver-related causes of deaths in HIV-infected patients: the French national Mortalite 2005 study. J Hepatol. (2009) 50:736–45. doi: 10.1016/j.jhep.2008.11.018
8. Vallet-Pichard A, Mallet V, Pol S. Nonalcoholic fatty liver disease and HIV infection. Semin Liver Dis. (2012). 32:158–66. doi: 10.1055/s-0032-1316471
9. Stabinski L, Reynolds SJ, Ocama P, Laeyendecker O, Ndyanabo A, Kiggundu V. et al. High prevalence of liver fibrosis associated with HIV infection: a study in rural Rakai, Uganda. Antivir Ther. (2011) 16:405–11. doi: 10.3851/IMP1783
10. Benhamou Y, Bochet M, Di Martino V, Charlotte F, Azria F, Coutellier A, et al. Liver fibrosis progression in human immunodeficiency virus and hepatitis C virus coinfected patients. The Multivirc Group Hepatol. (1999) 30:1054–8. doi: 10.1002/hep.510300409
11. Macías J, Berenguer J, Japón MA, Girón JA, Rivero A, López-Cortés LF, et al., Fast fibrosis progression between repeated liver biopsies in patients coinfected with human immunodeficiency virus/hepatitis C virus. Hepatology. (2009) 50:1056–63. doi: 10.1002/hep.23136
12. Chaudhry AA, Sulkowski MS, Chander G, Moore RD. Hazardous drinking is associated with an elevated aspartate aminotransferase to platelet ratio index in an urban HIV-infected clinical cohort. HIV Med. (2009) 10:133–42. doi: 10.1111/j.1468-1293.2008.00662.x
13. Joshi D, O'grady J, Dieterich D, Gazzard B, Agarwal K. Increasing burden of liver disease in patients with HIV infection. Lancet. (2011) 377:1198–209. doi: 10.1016/S0140-6736(10)62001-6
14. Dallapiazza M, Amorosa VK, Localio R, Kostman JR, Lo Re V III. Prevalence and risk factors for significant liver fibrosis among HIV-monoinfected patients. BMC Infect Dis. (2010). 10:116. doi: 10.1186/1471-2334-10-116
15. Merchante N, Perez-Camacho I, Mira JA, Rivero A, Macias J, Camacho A, et al. Prevalence and risk factors for abnormal liver stiffness in HIV-infected patients without viral hepatitis coinfection: role of didanosine. Antivir Ther. (2010) 15:753–63. doi: 10.3851/IMP1612
16. Han SH, Kim SU, Kim CO, Jeong SJ, Park JY, Choi JY, et al. Abnormal liver stiffness assessed using transient elastography (Fibroscan(R)) in HIV-infected patients without HBV/HCV coinfection receiving combined antiretroviral treatment. PLoS ONE. (2013) 8:e52720. doi: 10.1371/journal.pone.0052720
17. Tahiri M, Sodqi M, Lahdami FE, Marih L, Lamdini H, Hliwa W, et al. Risk factors for liver fibrosis among human immunodeficiency virus monoinfected patients using the FIB4 index in Morocco. World J Hepatol. (2013) 5:584–8. doi: 10.4254/wjh.v5.i10.584
18. Rockstroh JK, Mohr R, Behrens G, Spengler U. Liver fibrosis in HIV: which role does HIV itself, long-term drug toxicities and metabolic changes play? Curr Opin HIV AIDS. (2014) 9:365–70. doi: 10.1097/COH.0000000000000064
19. Anadol E, Lust K, Boesecke C, Schwarze-Zander C, Mohr R, Wasmuth JC, et al. Exposure to previous cART is associated with significant liver fibrosis and cirrhosis in human immunodeficiency virus-infected patients. PLoS ONE. (2018) 13:e0191118. doi: 10.1371/journal.pone.0191118
20. Lamers SL, Rose R, Maidji E, Agsalda-Garcia M, Nolan DJ, Fogel GB, et al. HIV DNA is frequently present within pathologic tissues evaluated at autopsy from combined antiretroviral therapy-treated patients with undetectable viral loads. J Virol. (2016) 90:8968–83. doi: 10.1128/JVI.00674-16
21. Shi J, Fujieda H, Kokubo Y, Wake K. Apoptosis of neutrophils and their elimination by Kupffer cells in rat liver. Hepatology. (1996) 24:1256–63. doi: 10.1002/hep.510240545
22. Guillot A, Tacke F. Liver macrophages: old dogmas and new insights. Hepatol Commun. (2019) 3:730–43. doi: 10.1002/hep4.1356
23. Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology. (1994) 20:453–60. doi: 10.1002/hep.1840200227
24. Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. (2003) 38:1188–98. doi: 10.1053/jhep.2003.50472
25. Rizzardini M, Zappone M, Villa P, Gnocchi P, Sironi M, Diomede L, et al. Kupffer cell depletion partially prevents hepatic heme oxygenase 1 messenger RNA accumulation in systemic inflammation in mice: role of interleukin 1beta. Hepatology. (1998) 27:703–10. doi: 10.1002/hep.510270311
26. Tomiyama K, Ikeda A, Ueki S, Nakao A, Stolz DB, Koike Y, et al. Inhibition of Kupffer cell-mediated early proinflammatory response with carbon monoxide in transplant-induced hepatic ischemia/reperfusion injury in rats. Hepatology. (2008) 48:1608–20. doi: 10.1002/hep.22482
27. Ju C, Reilly TP, Bourdi M, Radonovich MF, Brady JN, George JW, et al. Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem Res Toxicol. (2002) 15:1504–13. doi: 10.1021/tx0255976
28. Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology. (2015) 62:279–91. doi: 10.1002/hep.27793
29. Nguyen-Lefebvre AT, Ajith A, Portik-Dobos V, Horuzsko DD, Arbab AS, Dzutsev A, et al. The innate immune receptor TREM-1 promotes liver injury and fibrosis. J Clin Invest. (2018) 128:4870–83. doi: 10.1172/JCI98156
30. Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, De Minicis S, et al. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology. (2009) 137:1467–77 e1465. doi: 10.1053/j.gastro.2009.06.045
31. Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. (2010) 22:231–7. doi: 10.1016/j.coi.2010.01.009
32. Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. (2013) 339:286–91. doi: 10.1126/science.1232227
33. Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. (2014) 59:130–42. doi: 10.1002/hep.26607
34. El Kasmi KC, Anderson AL, Devereaux MW, Fillon SA, Harris JK, Lovell MA, et al. Toll-like receptor 4-dependent Kupffer cell activation and liver injury in a novel mouse model of parenteral nutrition and intestinal injury. Hepatology. (2012) 55:1518–28. doi: 10.1002/hep.25500
35. Olinga P, Merema MT, De Jager MH, Derks F, Melgert BN, Moshage H, et al. Rat liver slices as a tool to study LPS-induced inflammatory response in the liver. J Hepatol. (2001) 35:187–94. doi: 10.1016/S0168-8278(01)00103-9
36. Ogle CK, Wu JZ, Mao X, Szczur K, Alexander JW, Ogle JD. Heterogeneity of Kupffer cells and splenic, alveolar, and peritoneal macrophages for the production of TNF, IL-1, and IL-6. Inflammation. (1994) 18:511–23. doi: 10.1007/BF01560698
37. Vaure C, Liu Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol. (2014) 5:316. doi: 10.3389/fimmu.2014.00316
38. Bartolome N, Arteta B, Martinez MJ, Chico Y, Ochoa B. Kupffer cell products and interleukin 1beta directly promote VLDL secretion and apoB mRNA up-regulation in rodent hepatocytes. Innate Immun. (2008) 14:255–66. doi: 10.1177/1753425908094718
39. Tian Z, Shen X, Feng H, Gao B. IL-1 beta attenuates IFN-alpha beta-induced antiviral activity and STAT1 activation in the liver: involvement of proteasome-dependent pathway. J Immunol. (2000) 165:3959–65. doi: 10.4049/jimmunol.165.7.3959
40. He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
41. Huang H, Chen HW, Evankovich J, Yan W, Rosborough BR, Nace GW, et al. Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J Immunol. (2013) 191:2665–79. doi: 10.4049/jimmunol.1202733
42. Xu Y, Yao J, Zou C, Zhang H, Zhang S, Liu J, et al. Asiatic acid protects against hepatic ischemia/reperfusion injury by inactivation of Kupffer cells via PPARgamma/NLRP3 inflammasome signaling pathway. Oncotarget. (2017) 8:86339–55. doi: 10.18632/oncotarget.21151
43. Mridha AR, Wree A, Robertson A, a,B, Yeh MM, Johnson CD, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
44. Housset C, Lamas E, Brechot C. Detection of HIV1 RNA and p24 antigen in HIV1-infected human liver. Res Virol. (1990) 141:153–9. doi: 10.1016/0923-2516(90)90017-D
45. Hoda SA, White JE, Gerber MA. Immunohistochemical studies of human immunodeficiency virus-1 in liver tissues of patients with AIDS. Mod Pathol. (1991) 4:578–81.
46. Schmitt MP, Steffan AM, Gendrault JL, Jaeck D, Royer C, Schweitzer C, et al. Multiplication of human immunodeficiency virus in primary cultures of human Kupffer cells–possible role of liver macrophage infection in the physiopathology of AIDS. Res Virol. (1990) 141:143–52. doi: 10.1016/0923-2516(90)90016-C
47. Schweitzer C, Keller F, Schmitt MP, Jaeck D, Adloff M, Schmitt C, et al. Morphine stimulates HIV replication in primary cultures of human Kupffer cells. Res Virol. (1991) 142:189–95. doi: 10.1016/0923-2516(91)90056-9
48. Persidsky Y, Steffan AM, Gendrault JL, Hurtrel B, Berger S, Royer C, et al. Permissiveness of Kupffer cells for simian immunodeficiency virus (SIV) and morphological changes in the liver of rhesus monkeys at different periods of SIV infection. Hepatology. (1995) 21:1215–25. doi: 10.1002/hep.1840210502
49. Igarashi T, Brown CR, Endo Y, Buckler-White A, Plishka R, Bischofberger N, et al. Macrophage are the principal reservoir and sustain high virus loads in rhesus macaques after the depletion of CD4+ T cells by a highly pathogenic simian immunodeficiency virus/HIV type 1 chimera (SHIV): implications for HIV-1 infections of humans. Proc Natl Acad Sci USA. (2001) 98:658–63. doi: 10.1073/pnas.98.2.658
50. Kandathil AJ, Sugawara S, Goyal A, Durand CM, Quinn J, Sachithanandham J, et al. No recovery of replication-competent HIV-1 from human liver macrophages. J Clin Invest. (2018) 128:4501–9. doi: 10.1172/JCI121678
51. Poli G. Cell-to-cell vs. cell-free HIV-1 transmission from macrophages to CD4+ T lymphocytes: lessons from the virology textbook. AIDS. (2013) 27:2307–8. doi: 10.1097/QAD.0b013e328363619a
52. Bracq L, Xie M, Benichou S, Bouchet J. Mechanisms for Cell-to-Cell Transmission of HIV-1. Front Immunol. (2018) 9:260. doi: 10.3389/fimmu.2018.00260
53. Mosoian A, Zhang L, Hong F, Cunyat F, Rahman A, Bhalla R, et al. Frontline Science: HIV infection of Kupffer cells results in an amplified proinflammatory response to LPS. J Leukoc Biol. (2017) 101:1083–90. doi: 10.1189/jlb.3HI0516-242R
54. Zhang L, Mosoian A, Schwartz ME, Florman SS, Gunasekaran G, Schiano T, et al. HIV infection modulates IL-1beta response to LPS stimulation through a TLR4-NLRP3 pathway in human liver macrophages. J Leukoc Biol. (2019) 105:783–95. doi: 10.1002/JLB.4A1018-381R
55. Del Corno M, Cappon A, Donninelli G, Varano B, Marra F, Gessani S. HIV-1 gp120 signaling through TLR4 modulates innate immune activation in human macrophages and the biology of hepatic stellate cells. J Leukoc Biol. (2016) 100:599–606. doi: 10.1189/jlb.4A1215-534R
56. Ostrowski SR, Gerstoft J, Pedersen BK, Ullum H. Impaired production of cytokines is an independent predictor of mortality in HIV-1-infected patients. AIDS. (2003) 17:521–30. doi: 10.1097/00002030-200303070-00007
57. Sun Z, Denton PW, Estes JD, Othieno FA, Wei BL, Wege AK, et al. Intrarectal transmission, systemic infection, and CD4+ T cell depletion in humanized mice infected with HIV-1. J Exp Med. (2007) 204:705–14. doi: 10.1084/jem.20062411
58. Erikstrup C, Kronborg G, Lohse N, Ostrowski SR, Gerstoft J, Ullum H. T-cell dysfunction in HIV-1-infected patients with impaired recovery of CD4 cells despite suppression of viral replication. J Acquir Immune Defic Syndr. (2010) 53:303–10. doi: 10.1097/QAI.0b013e3181ca3f7c
59. Glassner A, Eisenhardt M, Kokordelis P, Kramer B, Wolter F, Nischalke HD, et al. Impaired CD4(+) T cell stimulation of NK cell anti-fibrotic activity may contribute to accelerated liver fibrosis progression in HIV/HCV patients. J Hepatol. (2013) 59:427–33. doi: 10.1016/j.jhep.2013.04.029
60. Brau N, Salvatore M, Rios-Bedoya CF, Fernandez-Carbia A, Paronetto F, Rodriguez-Orengo JF, et al. Slower fibrosis progression in HIV/HCV-coinfected patients with successful HIV suppression using antiretroviral therapy. J Hepatol. (2006) 44:47–55. doi: 10.1016/j.jhep.2005.07.006
61. Kooij KW, Wit FW, Van Zoest RA, Schouten J, Kootstra NA, Van Vugt M, et al. Liver fibrosis in HIV-infected individuals on long-term antiretroviral therapy: associated with immune activation, immunodeficiency and prior use of didanosine. AIDS. (2016) 30:1771–80. doi: 10.1097/QAD.0000000000001119
62. Lucar O, Reeves RK, Jost S. A Natural Impact: NK Cells at the Intersection of Cancer and HIV Disease. Front Immunol. (2019) 10:1850. doi: 10.3389/fimmu.2019.01850
63. Beignon AS, Mckenna K, Skoberne M, Manches O, Dasilva I, Kavanagh DG, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest. (2005) 115:3265–75. doi: 10.1172/JCI26032
64. Evans TI, Li H, Schafer JL, Klatt NR, Hao XP, Traslavina RP, et al. SIV-induced translocation of bacterial products in the liver mobilizes myeloid dendritic and natural killer cells associated with liver damage. J Infect Dis. (2016) 213:361–9. doi: 10.1093/infdis/jiv404
65. Vlahakis S, Villasis-Keever A, Gomez T, Al E. Human immunodeficiency virus-induced apoptosis of human hepatocytes via CXCR4. J Infect Dis. (2003) 188:1455–60. doi: 10.1086/379738
66. Balasubramanian S, Ganju R, Groopman J. Hepatitis C virus and HIV envelope proteins collaboratively mediate interleukin-8 secretion through activation of p38 MAP kinase and SHP2 in hepatocytes. J Biol Chem. (2003) 278:35755–66. doi: 10.1074/jbc.M302889200
67. Munshi N, Balasbramanian A, Koziel M, Ganju R, Groopman J. Hepatitis C and human immunodeficiency virus envelope proteins cooperatively induce hepatocytic apoptosis via an innocent bystander mechanism. J Infect Dis. (2003) 188:1192–204. doi: 10.1086/378643
68. Yang L, Seki E. Toll-like receptors in liver fibrosis: cellular crosstalk and mechanisms. Front Physiol. (2012) 3:138. doi: 10.3389/fphys.2012.00138
69. Ganesan M, New-Aaron M, Dagur RS, Makarov E, Wang W, Kharbanda KK, et al. Alcohol metabolism potentiates HIV-induced hepatotoxicity: contribution to end-stage liver disease. Biomolecules. (2019) 9:20851. doi: 10.3390/biom9120851
70. Zhan SS, Jiang JX, Wu J, Halsted C, Friedman SL, Zern MA, et al. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. (2006) 43:435–43. doi: 10.1002/hep.21093
71. Hong F, Saiman Y, Si C, Mosoian A, Bansal MB. X4 Human immunodeficiency virus type 1 gp120 promotes human hepatic stellate cell activation and collagen I expression through interactions with CXCR4. PLoS ONE. (2012) 7:e33659. doi: 10.1371/journal.pone.0033659
72. Bruno R, Galastri S, Sacchi P, Cima S, Caligiuri A, Defranco R, et al. gp120 modulates the biology of human hepatic stellate cells: a link between HIV infection and liver fibrogenesis. Gut. (2010) 59:513–20. doi: 10.1136/gut.2008.163287
73. Tuyama AC, Hong F, Saiman Y, Wang C, Ozkok D, Mosoian A, et al. Human immunodeficiency virus (HIV)-1 infects human hepatic stellate cells and promotes collagen I and monocyte chemoattractant protein-1 expression: implications for the pathogenesis of HIV/hepatitis C virus-induced liver fibrosis. Hepatology. (2010) 52:612–22. doi: 10.1002/hep.23679
74. Salloum S, Holmes JA, Jindal R, Bale SS, Brisac C, Alatrakchi N, et al. Exposure to human immunodeficiency virus/hepatitis C virus in hepatic and stellate cell lines reveals cooperative profibrotic transcriptional activation between viruses and cell types. Hepatology. (2016) 64:1951–68. doi: 10.1002/hep.28766
75. Gupta D, Rani M, Khan N, Jameel S. HIV-1 infected peripheral blood mononuclear cells modulate the fibrogenic activity of hepatic stellate cells through secreted TGF-beta and JNK signaling. PLoS ONE. (2014) 9:e91569. doi: 10.1371/journal.pone.0091569
Keywords: liver fibrosis, HIV - human immunodeficiency virus, hepatic stellate cell (HSCs), Kupffer cells, microbial translocation
Citation: Zhang L and Bansal MB (2020) Role of Kupffer Cells in Driving Hepatic Inflammation and Fibrosis in HIV Infection. Front. Immunol. 11:1086. doi: 10.3389/fimmu.2020.01086
Received: 10 February 2020; Accepted: 05 May 2020;
Published: 16 June 2020.
Edited by:
M. Victoria Delpino, CONICET Institute of Immunology, Genetics and Metabolism (INIGEM), ArgentinaReviewed by:
Larisa Y. Poluektova, University of Nebraska Medical Center, United StatesCopyright © 2020 Zhang and Bansal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meena B. Bansal, bWVlbmEuYmFuc2FsQG1zc20uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.