 Zhihua Ren
Zhihua Ren Ting Ding
Ting Ding Zhicai Zuo1
Zhicai Zuo1 Zhanyong Wei
Zhanyong Wei- 1Key Laboratory of Animal Disease and Human Health of Sichuan Province, College of Veterinary Medicine, Sichuan Agricultural University, Chengdu, China
- 2The College of Animal Science and Veterinary Medicine, Henan Agricultural University, Zhengzhou, China
Viral infection is controlled by host innate immune cells that express specialized receptors for viral components. Engagement of these pattern recognition receptors triggers a series of signaling pathways that culminate in the production of antiviral mediators such as type I interferons. Mitochondrial antiviral-signaling protein (MAVS) acts as a central hub for signal transduction initiated by RIG-I-like receptors, which predominantly recognize viral RNA. MAVS expression and function are regulated by both post-transcriptional and post-translational mechanisms, of which ubiquitination and phosphorylation play the most important roles in modulating MAVS function. Increasing evidence indicates that viruses can escape the host antiviral response by interfering at multiple points in the MAVS signaling pathways, thereby maintaining viral survival and replication. This review summarizes recent studies on the mechanisms by which MAVS expression and signaling are normally regulated and on the various strategies employed by viruses to antagonize MAVS activity, which may provide new insights into the design of novel antiviral agents.
Introduction
The innate immune system is the first line of host defense against pathogens. Innate immune cells express specific pattern-recognition receptors that recognize microbial components known as pathogen-associated molecular patterns (e.g., viral nucleic acids and proteins) and activate intracellular signaling pathways leading to the antiviral response (1). The major pattern recognition receptors include Toll-like receptors (TLRs), which are located both intracellularly and on the cell membrane; RIG-I-like receptors (RLRs) and Nod-like receptors (NLRs), both located in the cytoplasm; and some cytoplasmic DNA receptors such as DAI, IFI16, DDX41, cGAS, and AIM2 (2–4). The cellular localization of invading viral components provides the possibility that the host utilizes cytoplasmic pattern-recognition receptors to respond to these stimuli. Among viral components, double-stranded RNA (dsRNA) and single-stranded RNA (ssRNA) are recognized by TLR3 and TLR7/8 in endolysosomes, respectively (5–7). RLRs bind dsDNA and trigger antiviral response (8). Viral ssRNA and dsRNA are recognized by NLRP3 (9), while NOD2 interacts with ssRNA and induces interferon (IFN) production (10). Intracellular dsDNA of viral origin is also recognized by cytoplasmic DAI, IFI16, DDX41, cGAS, and AIM2 (11–15).
RLRs have been extensively studied as sensors of cytoplasmic viral dsRNA and their importance in controlling RNA virus infection is now clear. The RLR family has three members: retinoic acid-inducible gene I (RIG-I), melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology protein 2 (LGP2). RIG-I and MDA5 are typical pattern recognition receptors, whereas LGP2 is considered to be a regulator of RIG-I- and MDA5-mediated signal transduction (16, 17). Upon viral recognition, RIG-I and MDA5 interact with the mitochondrial antiviral-signaling protein (MAVS, also known as IPS1, VISA, and CARDIF), which induces activation of the transcription factors interferon regulatory factors 3 and 7 (IRF3/7) and nuclear transcription factor-κB (NF-κB). This process ultimately leads to the expression of multiple proinflammatory factors and antiviral genes, such as IFN and IFN-stimulated genes (ISGs), which inhibit viral replication and transmission (18–21). Given that MAVS is the key adapter protein known to be required for defense against RLR-recognized RNA viruses, this review will specifically focus on the regulation of MAVS expression and signaling function and their manipulation by viruses.
Structure and Function of MAVS
MAVS is a 540-amino acid protein encoded by the nuclear genome (19). MAVS is mainly localized on the mitochondrial outer membrane, although it has also been detected on peroxisome and mitochondrial-associated endoplasmic reticulum membranes (22–24). MAVS contains three domains: an N-terminal caspase recruitment domain (CARD), a middle proline-rich region (PRR), and a C-terminal transmembrane (TM) domain (Figure 1). The MAVS CARD binds to similar CARDs present in RIG-I and MDA5, which induces MAVS activation (Figure 2). The MAVS PRR domain binds to the tumor necrosis factor receptor-related factor (TRAF) family members TRAF2, TRAF3, TRAF5, and TRAF6 to promote downstream signal transduction (25), and the TM domain ensures localization of MAVS to the mitochondrial outer membrane (20).
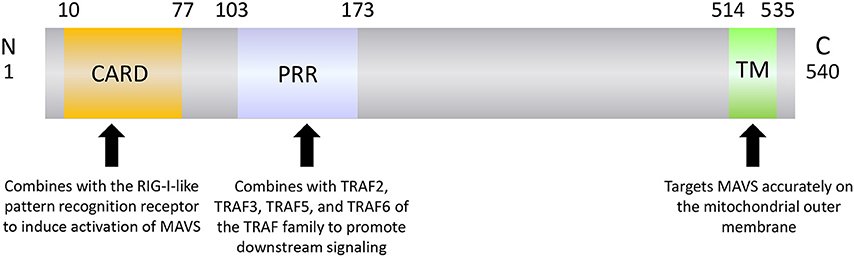
Figure 1. The structure of MAVS.
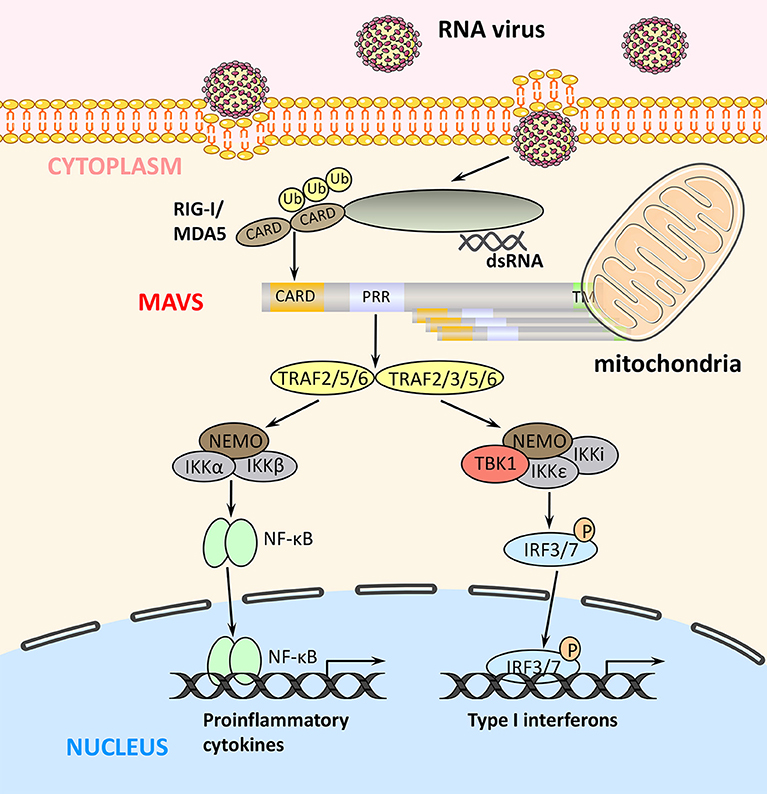
Figure 2. RLR signaling pathways. Binding of viral components to RIG-I and MDA5 induces their interaction with MAVS via their common CARDs. MAVS activates two signaling cascades leading to the production of immune factors. The TRAF2/5/6–IKK complex–NF-κB pathway induces transcription of proinflammatory cytokines, whereas the TRAF2/3/5/6–TBK1 complex–IRF3/7 pathway induces the expression of type I IFN genes.
Upon binding of viral components, RIG-I and MDA5 undergo a conformational change that exposes their N-terminal tandem CARDs and forms the tetramer via CARDs (26), which are modified with K63-polyubiquitin chains by the E3 ubiquitin ligases TRIM25, RNF125, and RIPLET (27). In turn, K63-linked ubiquitination promotes binding of RIG-I and MDA5 to MAVS via their CARDs. The MAVS CARD rapidly forms prion-like aggregates, which convert other MAVS on the mitochondrial outer membrane into prion-like aggregates. This aggregation step is essential for the biological functions of MAVS (28). Next, MAVS binds through its PRR domain to TRAF2, TRAF3, TRAF5, or TRAF6, which promotes activation of TBK1 complex (containing TBK1, IκB kinase [IKK]i/ε, and NEMO) in the case of TRAF2/3/5/6 and of IKK complex (containing IKKα/β and NEMO) in the case of TRAF2/5/6 (Figure 2) (25, 29). The TBK1 complex promotes phosphorylation and homodimerization of IRF3 and/or IRF7, which then translocate to the nucleus where they bind to IFN-stimulated response elements and induce the transcription of target genes such as type I IFNs. Similarly, the TRAF2/5/6-activated IKK complex activates NF-κB to promote the transcription of proinflammatory cytokines (4, 8). Thus, the two MAVS-mediated signaling pathways play distinct but crucial roles in antiviral innate immunity (Figure 2).
Regulation of MAVS Activity Under Physiological Conditions and During Viral Infection
The expression and function of MAVS are tightly regulated at the post-transcriptional and post-translational levels, which ensures that RLR signaling pathways are not only rapidly activated upon viral recognition but also curtailed in a timely manner upon viral clearance to avoid potentially harmful tissue damage.
Post-transcriptional Regulation of MAVS
MAVS mRNA is polycistronic, and the key feature of polycistron is that protein translation can start not only from the N-terminal methionine, but also from the internal methionine, thus encoding for isoforms by alternative translation of distinct start sites (30, 31). MAVS mRNA was identified to produce a full-length and five CARD-deleted proteins (FL MAVS and MAVS-M142/303/358/367/449, respectively) from six distinct translational initiation sites based on methionine distribution. Under physiological conditions, binding of MAVS truncated isoforms to FL MAVS inhibits its spontaneous aggregation, while the formed aggregates are degraded by mitochondrial autophagy, thereby maintaining autoimmune homeostasis (31). In addition, MAVS-M142 cannot prevent the aggregation of FL MAVS during viral infection, but can block the production of IFN after MAVS-M142 binding to TRAF. However, the regulation of MAVS and MAVS-M142 can be achieved by an upstream open reading frame (uORF) in the 5′untranslated region (UTR). The translation of uORF might skip the start site of FL MAVS and initiate the translation of MAVS-M142 (32). Thus, the outcome of MAVS signaling is dictated to a certain extent by the net effects of FL MAVS and MAVS truncated isoforms. At present, only a few genetic studies have investigated the regulation of FL MAVS and MAVS truncated isoforms, and the mechanism underlying their functional competition is still unclear.
It is well-known that mRNA contains a number of regulatory elements in the 3′ and 5′ UTRs. Binding of regulatory factors such as proteins and complementary small RNA molecules to these elements plays an important role in controlling protein translation (33). For example, three AU-rich elements in the 3'UTR of MAVS mRNA mediates binding of human antigen R (HuR), which destabilizes the mRNA and maintains MAVS protein at low levels under normal physiological conditions (34). MicroRNAs (miRNA) are small non-coding RNAs that regulate gene expression by promoting the degradation and/or inhibiting the translation of target mRNAs (35). Four miR-27a-binding sites have been identified in the 3'UTR of MAVS mRNA, and up-regulation of miR-27a can inhibit MAVS expression (34).
The DEAD-box (DDX) family of highly conserved RNA helicases play important roles in RNA synthesis, processing, transport, and degradation (36). A recent study showed that, in virus-infected cells, DDX46 located in the nucleus binds to several conserved CCGGUU elements in MAVS, TRAF3, and TRAF6 mRNAs, which inhibits their translocation to the cytoplasm for translation and thus reduces MAVS, TRAF3, and TRAF6 protein levels (37).
Post-translational Regulation of MAVS
MAVS can undergo a number of post-translational modifications (Table 1) that influence its function in promoting innate immune responses (Figure 3).
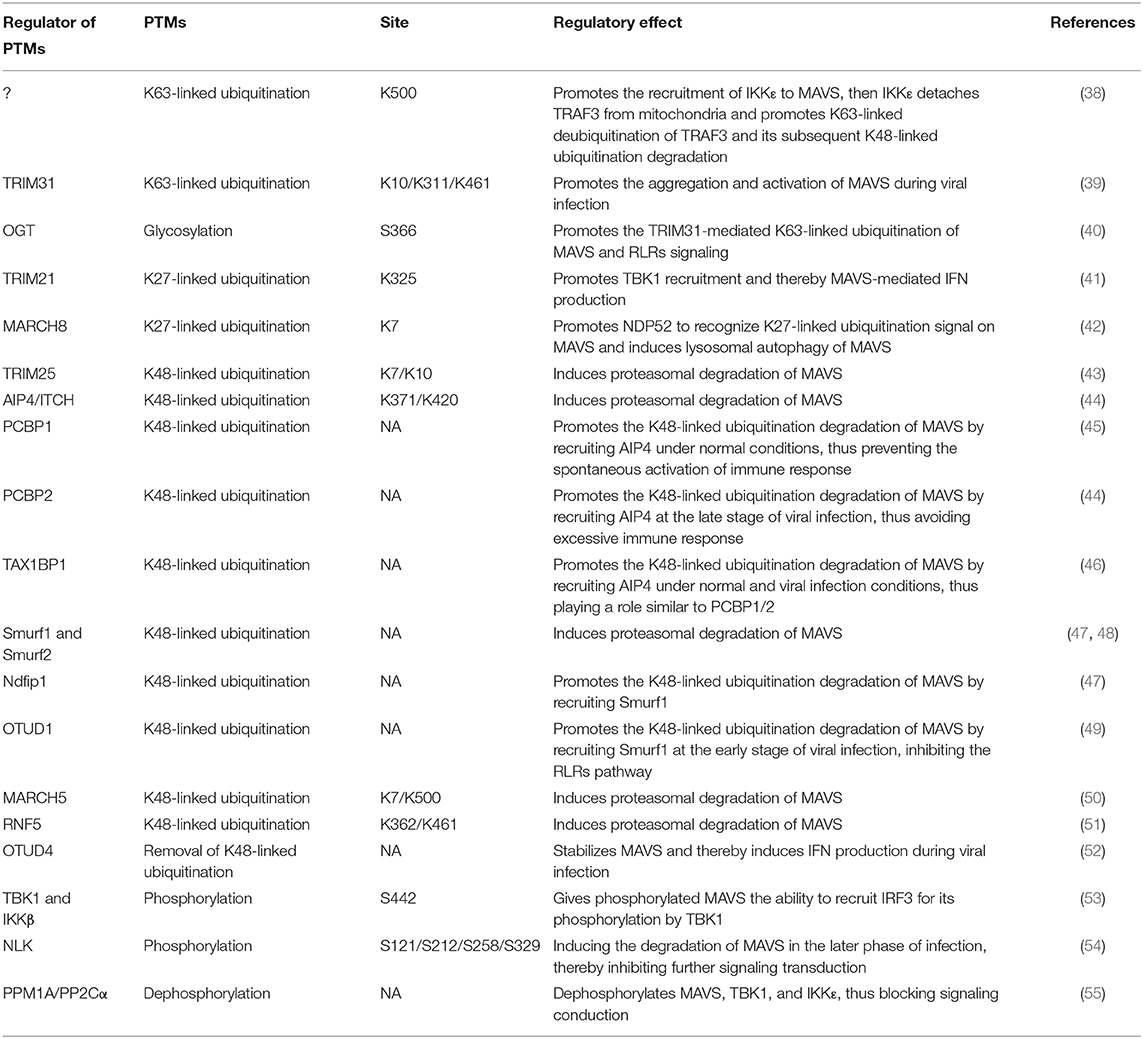
Table 1. The post-translational of modifications (PTMs) of MAVS.
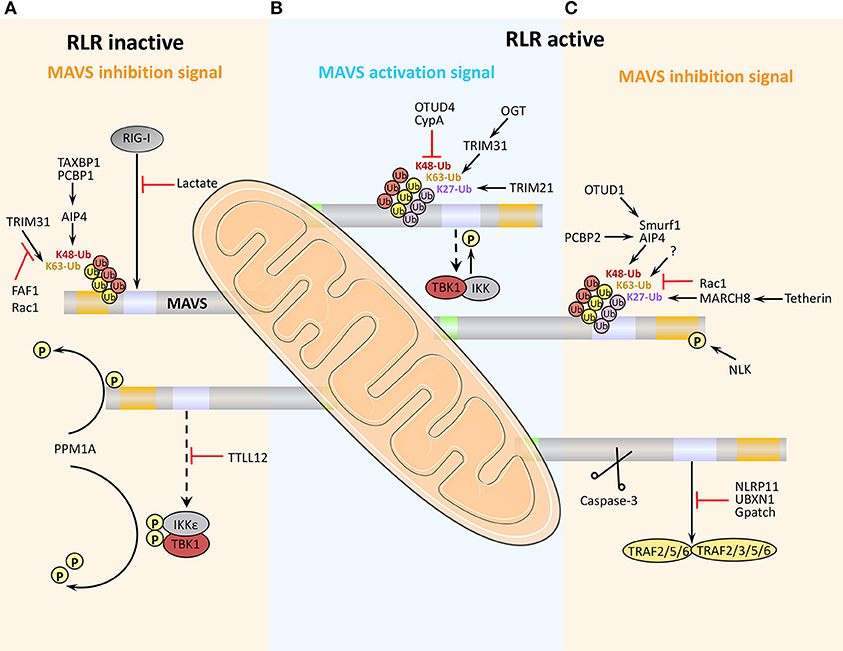
Figure 3. Positive and negative regulation of MAVS activity antiviral immunity. (A) Under physiological conditions, MAVS activity is inhibited to prevent aberrant activation of the immune response. (B) Early after viral infection, MAVS is activated by TRIM31-mediated K63-linked ubiquitination, and the inhibition of MAVS is relieved, thereby initiating the RLR signaling pathway. (C) At later stages of viral infection, MAVS is cleaved and degraded, which effectively arrests RLR-stimulated signaling.
Post-translational Positive Regulation of MAVS
Viral infection leads to K63-linked ubiquitination of MAVS at the mitochondrial outer membrane, which in turn induces MAVS aggregation, a marker of its activation. At present, ubiquitination by the E3 ubiquitin ligase TRIM31 is the only known initiator of MAVS aggregation and activation (Table 1). Notably, TRIM31-mediated MAVS aggregation does not occur under RIG-I deficient or non-viral infection conditions, thus it can be considered that RIG-I engagement is required for TRIM31-mediated MAVS aggregation after viral infection (39). MAVS glycosylation by O-linked N-acetylglucosamine (O-GlcNAc) transferase (OGT) enhances TRIM31-mediated K63-linked ubiquitination of MAVS (Table 1). Viral infection increases glucose metabolism in the host cell, including activation of the hexosamine biosynthesis pathway and elevation of OGT levels (40). Although only 2–5% of intracellular glucose enters the hexosamine biosynthesis pathway under physiological conditions, the ability of OGT to promote RLR signaling highlights the importance of glucose metabolism in antiviral innate immunity. Studies have shown that persistent MAVS aggregation may be associated with the occurrence of autoimmune diseases such as systemic lupus erythematosus. Further, the sustained expression of IFN and its regulatory genes and unsuppressed signals, will eventually result in tissue damages (56).
K27-linked ubiquitination by the E3 ubiquitin ligase TRIM21 also has a positive regulatory effect on MAVS. Viral infection upregulates the expression of TRIM21, thereby promoting the recruitment of TBK1 to MAVS and enhancing downstream signaling (Table 1) (41).
Phosphorylation is another post-translational modification that plays a key role in regulating MAVS signaling. After activation of TBK1 complexes by MAVS, the complex components TBK1 and IKK kinases phosphorylate MAVS, which then gains the ability to recruit IRF3. Recruited IRF3 is then phosphorylated by TBK1, which induces IRF3 homodimerization, translocation to the nucleus, and promotion of IFN transcription (Table 1) (53).
It is well-known that K48-linked ubiquitination of MAVS promotes its proteasomal degradation (44), and proteins that inhibit MAVS K48-linked ubiquitination can thus positively regulate RLR-mediated signaling. For example, the endogenous protein cyclophilin A, which normally competes with TRIM25 for MAVS binding, is upregulated by viral infection, thereby effectively inhibiting TRIM25-mediated MAVS ubiquitination and degradation (43, 57). Ubiquitination is a reversible process, and viral infection also upregulates the expression of ovarian tumor family deubiquitinase 4 (OTUD4), which removes K48-linked ubiquitin from MAVS and thus reduces its degradation (Table 1) (52). These findings suggest that the host cell has evolved mechanisms to inhibit K48-linked ubiquitination and degradation of MAVS during viral infection, thus ensuring that at least some level of signaling for the antiviral response is maintained.
Collectively, these studies have revealed that MAVS signaling is regulated via three main post-translational modifications that act in concert: (1) TRIM31-mediated K63-linked ubiquitination activates MAVS; (2) TRIM21-mediated K27-linked ubiquitination promotes the recruitment of TBK1 to MAVS; and (3) TBK1- and IKK-mediated MAVS phosphorylation promotes the recruitment of IRF3 to MAVS. In addition, inhibition of K48-linked ubiquitination and degradation is necessary to maintain an adequate cellular level of MAVS to ensure antiviral immunity.
Post-translational Negative Regulation of MAVS
At present, the negative post-translational regulation of MAVS is mainly manifested in K48-linked ubiquitination modification, signal blocking, autophagy, and apoptosis (Figure 3).
MAVS regulation by ubiquitination
As noted above, K48-linked ubiquitination of MAVS triggers its proteasomal degradation and thus limits RLR signaling. A number of E3 ubiquitin ligases have been shown to mediate MAVS K48-linked ubiquitination, including TRIM25, AIP4, Smurf1, Smurf2, MARCH5, and RNF5 (see also Table 1 for details). In addition, increasing evidence suggests that binding of various adaptor proteins to MAVS modulates MAVS ubiquitination-mediated degradation. For example, K48-linked ubiquitination of MAVS mediated by the E3 ubiquitin ligase AIP4 (also known as Itch) is promoted by the poly (C)-binding protein 2 (PCBP2), which is upregulated after viral infection. PCBP2 binds to the TM domain of MAVS and recruits AIP4, which promotes MAVS degradation (Table 1) (44). Recent work has established a similar role for the highly homologous PCBP1; however, PCBP1 expression level does not appear to be altered during viral infection (Table 1). Thus, it is possible that MAVS expression is maintained at a low level under physiological conditions in part by PCBP1-mediated promotion of degradation, whereas upregulation of PCBP2 during viral infection promotes MAVS degradation and acts as a negative feedback mechanism to re-establish homeostasis and avoid excessive immune signaling (45). AIP4 is also regulated by the adapter protein Tax1-binding protein 1 (TAX1BP1), which binds to the MAVS CARD and recruits AIP4. TAX1BP1 regulates MAVS degradation under physiological conditions and during viral infection in a similar manner to PCBP1/2 (Table 1) (46). The E3 ubiquitin ligase Smurf1 that mediates the K48-linked degradation of MAVS is known to be positively regulated by two adaptor proteins: OTUD1 and Nedd4 family-interacting protein 1 (Ndfip1). OTUD1 interacts with Smurf1 and functions as a deubiquitinase to remove the K48-linked ubiquitination of Smurf1, prevent its self-ubiquitination degradation, and upregulate its protein level; whereas the binding of Ndfip1 to MAVS recruits Smurf1 to MAVS, and the interaction between Ndfip1 and Smurf1 enhances the self-ubiquitination (possibly k63-linked) and enzyme activity of Smurf1 (Table 1) (47, 49). The diversity of E3 ubiquitin ligase involved in MAVS degradation suggests that they could provide more targets for the treatment of viral infections.
Several proteins negatively regulate antiviral innate immunity by inhibiting TRIM31-mediated K63-linked ubiquitination and activation of MAVS. One is Fas-associated factor 1 (FAF1), a protein that enhances Fas-induced apoptosis and was recently shown to be involved in the MAVS response. Under physiological conditions, the aggregates of FAF1 formed by its UBL domain, bind and antagonize MAVS by competing with TRIM31 for MAVS interaction, thereby inhibiting the MAVS aggregation. However, after viral infection, FAF1 is phosphorylated by activated IKKε, which promotes its depolymerization and lysosomal degradation and thus relieves MAVS inhibition (58). Rac1 is known to promote the growth of tumor cells. However, viral infection leads to geranylgeranylation and palmitoylation of Rac1, which inhibits the TRIM31–MAVS interaction and suppresses innate immunity; interestingly, under physiological conditions, Rac1 can also inhibit MAVS aggregation (59). It is known that TRAF3, an E3 ligase and located in the downstream signaling protein of MAVS, its self-ubiquitination of K63-linked promotes TRAF3-TBK1 interaction (60). However, K63-linked ubiquitination on Lys500 of MAVS (by a currently unidentified enzyme) can promote detachment of TRAF3 from MAVS, and trigger K63-linked deubiquitination on TRAF3 and its subsequent K48-linked ubiquitination degradation, thereby inhibiting TRAF3-mediated signaling (Table 1) (38).
Collectively, these studies suggest that MAVS K48-linked ubiquitination under physiological conditions serves to promote MAVS degradation and prevent spontaneous activation of the immune response. The same event occurring in the late stages of viral infection serves to restore cellular basal activity and prevent excessive production of potentially harmful immune mediators. Similarly, inhibition of TRIM31-mediated K63-linked ubiquitination of MAVS prevents its aggregation and activation, and also serves to maintain MAVS homeostasis.
MAVS blockade via direct protein interaction
Another mechanism by which MAVS signaling is held in check under physiological conditions is via direct binding of tubulin-tyrosine ligase-like protein 12 (TTLL12) to MAVS, TBK1, and IKKε (Figure 3). During viral infection, TTLL12 expression is reduced, thereby releasing the block in MAVS-mediated activation of the immune response (61). Protein phosphatase magnesium-dependent 1A (PPM1A, also known as PP2Cα) is a component of the TBK1/IKKε complex and normally maintains MAVS, TBK1, and IKKε in a dephosphorylated state, preventing MAVS downstream signaling (Table 1). However, upregulation of MAVS expression during viral infection overcomes the PPM1A-mediated block in signaling and serves as a threshold for activation of antiviral signaling (55).
Lactate, the end product of anaerobic glycolysis, has long been considered a metabolic waste product, but recent studies have shown that it has an important role in immunomodulation. Indeed, lactate acts as a key negative regulator of RLR signaling via direct binding to the MAVS TM domain, which prevents MAVS mitochondrial localization, aggregate formation, and signaling function. It is known that lactate level is directly related to glycolysis. Under physiological conditions, the interaction between hexokinase II (HK2), a key rate-limiting enzyme for glycolysis and located on the mitochondrial outer membrane, and MAVS maintains HK2 activity in its basal state, which ensures the operation of glycolysis metabolism and the suppression of MAVS mediated by lactate. However, during viral infection, binding of activated RIG-I to MAVS causes dissociation of HK2, leading to a reduction in glycolysis and lactate production, attenuation of lactate-mediated MAVS inhibition, and upregulation of downstream signaling for IFN production (62, 63). It reveals that the interaction of MAVS and HK2 is associated with its activation and function. Interestingly, downregulation of glucose metabolism and lactate-mediated repression of MAVS occurs only in the early stages of viral infection. This is consistent with the observation that viral infection is accompanied by an overall upregulation of glucose metabolism to ensure adequate energy supplies for replication and proliferation.
During the later stages of viral infection, MAVS activity is negatively regulated by UBX-domain-containing protein 1 (UBXN1), a member of the ubiquitin-binding protein family. The expression of UBXN1 is strongly upregulated late during viral infection and it competes with TRAF3/TRAF6 for binding to MAVS (amino acids 455–460), thus blocking MAVS signaling (64, 65). Gpatch domain-containing protein 3 (GPATCH3), a widely expressed protein, acts similarly to UBXN1. GPATCH3 binding to MAVS prevents the assembly of the MAVS/TRAF6/TBK1 complex during viral infection (66).
MAVS Regulation related to apoptosis and autophagy
When other mechanisms fail, apoptosis can be used as a strategy to inhibit viral replication. However, the cell has also evolved various feedback mechanisms to ensure timely inhibition of immune signaling to avoid excessive tissue damage. For example, NLRP11, a type I IFN-induced Nod-like receptor, is upregulated after viral infection and can inhibit type I IFN production. Recent studies demonstrated that part of NLRP11 translocates from cytoplasm to mitochondria and interacts with MAVS, but NLRP11 neither disrupts the interaction between MAVS and RIG-I nor the ubiquitination of MAVS. However, NLRP11 strongly interacts with TRAF6 and promotes K48-linked ubiquitination degradation of TRAF6, thereby inhibiting the RLR signaling. Interestingly, overexpression of NLRP11 inhibited the cleavage of poly-ADP-ribose polymerase (a molecule involved in apoptosis related to DNA damage) in cells while knockout of TRAF6 abrogated NLRP11-mediated inhibition (67). Because TRAF6 is involved in apoptosis after viral infection (68), NLRP11 can potentially inhibit apoptosis, which also ensures the survival of the host cells. In addition, the knockout of MAVS eliminated the interaction between NLRP11 and TRAF6 (67). These findings suggest that MAVS serves as a platform on which NLRP11 degrades TRAF6 and inhibits TRAF6-dependent apoptosis. The dual blocking role of NLRP11 in the antiviral immune response serves as a negative feedback mechanism for suppression of RLR signaling, IFN production, and apoptosis. Although cysteinyl aspartate specific proteinase-3 (caspase-3) is best known as a key executioner of apoptosis, its non-apoptotic functions are attracting increasing attention. During viral infection, activated caspase-3 cleaves MAVS at Asp429 and Asp490 and inhibits excessive signaling (69). This observation expands our understanding of the functions of caspase-3 and provides new insights for the development of drugs to prevent aberrant immune responses. In addition, the hemagglutinating virus of Japan envelope (HVJ-E) derived from inactivated replication-defective Sendai virus was found to have antitumor activity dependent on RIG-I/MAVS signaling. HVJ-E activates the RIG-I/MAVS signaling to upregulate the expression of its downstream pro-apoptotic genes TRAIL and Noxa, and thus inducing selective apoptosis of tumor cells (including prostate cancer cells, lung cancer cells, and breast cancer cells). Remarkably, this process does not occur in normal immune cells (70, 71).
Autophagy, a cellular degradation process, also acts as an antiviral defense mechanism by clearing intracellular microorganisms and interacting with the innate immune response. For example, Tetherin (also known as BST2 or CD317) is an IFN-induced membrane protein that is upregulated following viral infection. Tetherin induces K27-linked ubiquitination of MAVS by recruiting the E3 ubiquitin ligase MARCH8. The autophagy protein NDP52 then binds to K27-linked ubiquitinated MAVS, leading to lysosomal degradation. Tetherin-mediated autophagic degradation of MAVS can thus be considered another negative feedback mechanism that suppresses excessive signaling (42).
Additional negative regulatory mechanisms in MAVS-mediated antiviral immunity include phosphorylation of MAVS by Nemo-like kinase, which occurs in the later stage of viral infection and induces MAVS degradation (54). The mitochondrial fusion proteins Mfn1 and Mfn2, located on the mitochondrial outer membrane, play opposing roles in targeting MAVS during RLR-induced signaling. Mfn2 interacts with the C-terminal (particularly TM domain) of MAVS through a HR1 region and thus blocking the activation of IRF3 and NF-κB, and the process is based on an Mfn2-dependent complex (~600 kDa). The knockdown of endogenous Mnf2 reduced the relative molecular mass of MAVS complex from high molecular mass to a lower molecular mass and enhanced the antiviral effect of MAVS (72). However, there is evidence that the MAVS aggregate is larger than 700 kDa (28), so speculating that the MAVS complex may be composed of MAVS and its downstream signaling molecules, and this Mfn2-dependent form is not conducive to signaling transmission. In contrast to Mfn2, Mfn1 plays a positive role by regulating the mitochondrial redistribution of MAVS to form its speckled staining pattern in cells, which might relate to the formation of MAVS prion-like structures (73). Collectively, homologous proteins Mfn1 and Mfn2 fine-tune the MAVS-mediated signaling, in addition to ensuring the regulation of mitochondrial fusion. Furthermore, Mfn1/Mfn2-dependent mitochondrial fusion can enhance RLR signaling (74), the specific mechanism remains unclear.
Regulation of MAVS by viral proteins
Viruses can escape the host antiviral immune response by promoting the cleavage or degradation of MAVS and by directly interfering with RLR-activated signaling pathway components. Notably, there is little evidence that viral proteins play a role in enhancing the host innate immune response.
Cleavage of MAVS
Many virus-encoded proteins are proteases and can cleave MAVS independently of proteasomal degradation or apoptosis to inhibit RLR signaling (Figure 4). The first viral protein reported to colocalize with and cleave MAVS at the mitochondria was hepatitis C virus (HCV) serine protease NS3/4A. NS3/4A cleaves MAVS at Cys508, which dislodges the N-terminal fragment of MAVS from the mitochondria, reduces downstream signaling, and enables persistent viral infection (75). The small RNA viruses Seneca Valley virus, human rhinovirus C, and coxsackievirus B3 (CVB3) all encode a cysteine protease, 3Cpro, which cleaves MAVS at Gln148 and inhibits its activity (76–78). CVB3 also produces a second MAVS-cleaving protease, 2Apro, although the specific cleavage site is unclear (79). In addition, a 3C-like serine protease (3CLSP), which is similar to 3Cpro, is produced by porcine reproductive and respiratory syndrome virus and cleaves MAVS at Glu268 (80). The viral-encoded proteases 3ABC (produced by hepatitis A virus, HAV) and 2Apro (enterovirus 71, EV71) are particularly noteworthy. 3ABC produced by bat and human HAV cleave human MAVS at Glu463 and Gln427, respectively, highlighting the possibility that cross-species interference with MAVS signaling may promote the transfer of HAV between species (81). The first viral protein reported to cleave MAVS at multiple amino acid residues was the cysteine protease 2Apro, which is produced by EV71, the main cause of hand, foot and mouth disease. 2Apro cleaves MAVS at Gly209, Gly251, and Gly265 (82).
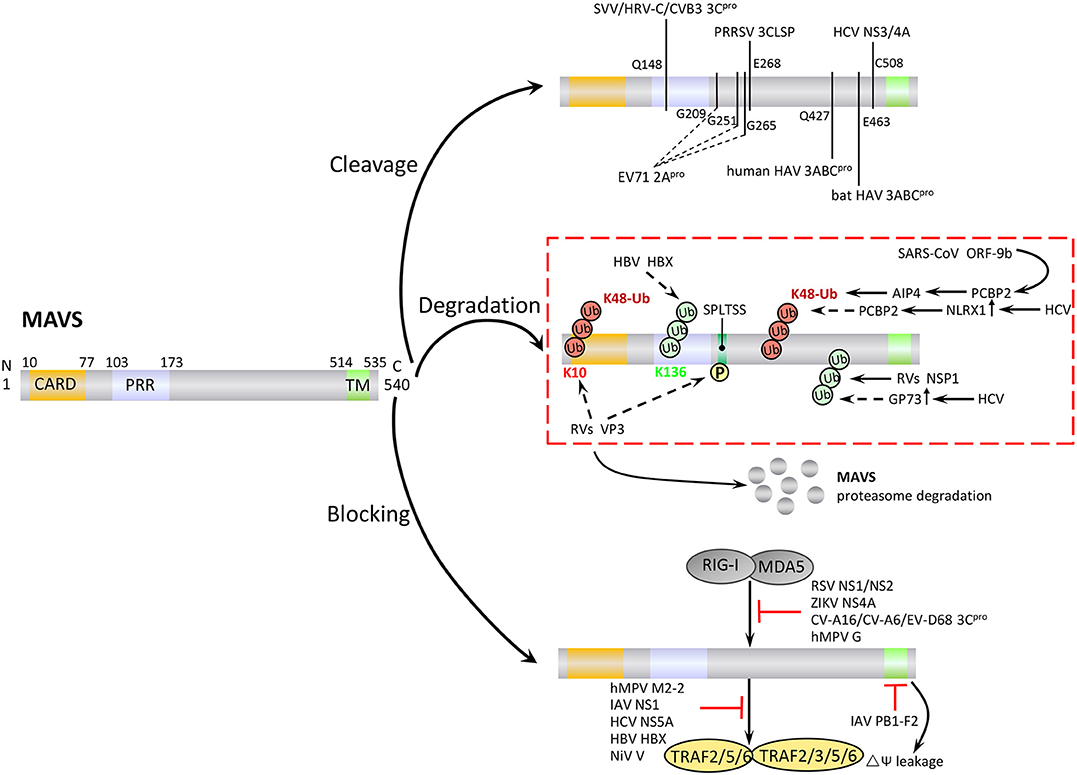
Figure 4. Negative regulation of MAVS signaling by viral proteins. Viruses employ various strategies to escape the host antiviral immune response, including cleavage or proteasomal degradation of MAVS, and direct binding to signaling molecules to block the RLR pathways.
Degradation of MAVS
In addition to the direct cleavage, some viral proteins induce proteasomal degradation of MAVS (Figure 4). For example, the hepatitis B virus (HBV) protein X (HBX) binds to MAVS and promotes its ubiquitination at Lys136, leading to proteasomal degradation. However, it is unclear which E3 ligase is involved in this process (83). Open reading frame 9b (ORF-9b), a protein encoded by coronavirus SARS, catalyzes the K48-linked ubiquitination and degradation of the MAVS signalosome (MAVS, TRAF3, and TRAF6) via the PCBP2–AIP4 axis. The degradation of mitochondrial fission protein Drp1 triggered by ORF-9b can reduce IFN signaling, suggesting that ORF-9b may also contributes to viral escape by manipulating mitochondrial function (84). Rotaviruses (RVs) use multiple mechanisms to degrade RLR pathway signaling components. VP3, an RV structural protein, induces phosphorylation of the newly discovered MAVS motif SPLTSS (residues 188–193), leading to K48-linked ubiquitination and proteasomal degradation of MAVS (85). Interestingly, the NSP1 protein of RVs has E3 ubiquitin ligase-like activity and can target proteins for ubiquitination (86). For example, NSP1 can bind to MVAS CARD or TM domain to promote ubiquitin-dependent proteasomal degradation (87). NSP1 also induces ubiquitin-proteasomal degradation of IRF3/5/7 and the NF-κB activating factor β-TrCP, and induces degradation of RIG-I in a proteasome-independent manner (88–90).
HCV infection upregulates the expression of Golgi protein 73 (GP73), a serum marker of liver disease and hepatocellular carcinoma, and promotes the coiled-coil domain of TRAF6 to recruit GP73 to MAVS. GP73 binding leads to the degradation of MAVS and TRAF6 through a proteasome-dependent pathway, thereby supporting HCV infection (91). HCV infection also upregulates NLRX1 (also known as NOD5/NOD9/NOD26) and induces MAVS K48-linked ubiquitination and degradation via PCBP2. Neither NLRX1 nor PCBP2 has E3 ubiquitin ligase activity, and it seems likely that PCBP2 may recruit AIP4 to mediate the ubiquitination step (92).
Modulation of MAVS Signaling
Viral proteins can block MAVS-mediated signaling by direct or indirect inhibition of individual pathway components (Figure 4). For example, a number of viral proteins bind to and block the interaction between MDA5, RIG-I, and MAVS. These include NS1 and NS2 of respiratory syncytial virus, NS4A of Zika virus, and 3Cpro proteins of coxsackievirus (CV)-A16, CV-A6, and EV-D68 (93–96). Human cytomegalovirus-encoded glycoprotein US9 disrupts mitochondrial integrity and induces loss of membrane potential, which leads to detachment of MAVS (97). The virulence factors M2-2 and glycoprotein G of human metapneumovirus (hMPV) both block RLR signaling. M2-2 binds to MAVS, blocking formation of MAVS/TRAF signalosomes, while glycoprotein G binds to the CARD of RIG-I, thus blocking MAVS binding (98, 99). The capsid protein VP16 of herpes simplex virus 1 blocks the production of early ISGs mediated by peroxisome MAVS, inhibiting the early peroxisome-mediated response to viral infection (100).
Influenza A virus (IAV), the most common type of influenza virus, has caused many worldwide pandemics because of its variability and high pathogenicity. The H5N1 strain of IAV encodes PB1-F2 protein and PB2Δ, a defective but functional polypeptide, which play opposing roles in MAVS-mediated antiviral innate immunity. PB1-F2 binding to the MAVS TM domain reduces the mitochondrial membrane potential and disrupts MAVS signaling, whereas PB2Δ directly interacts with MAVS to promote signaling for type I IFN production, thereby inhibiting virus replication (Figure 4) (101, 102). IAV NS1 inhibits TRAF3–MAVS interactions by removing K63-linked ubiquitination on TRAF3, negatively regulating the antiviral response (103). IAV also inhibits MAVS expression at the post-transcriptional level. For example, IAV upregulates the expression of miRNA-125a and miRNA-125b, which bind to the MAVS mRNA 3'UTR and inhibit its translation (104). In addition, the M2 protein of IAV induces Ca2+-dependent reactive oxygen species production, which enhances MAVS aggregation and activity (105).
Viral proteins can also promote intracellular proteins to inhibit MAVS activity (Figure 4). For example, HCV NS5A promotes the mitochondrial protein leucine-rich PRR motif-containing protein (LRPPRC) binding of MAVS to block the association of MAVS and TRAF3/6 (106). HBX can promote recruitment of the E3 ubiquitin ligase linear ubiquitin chain assembly complex (LUBAC) to the mitochondria, and the subsequent linear ubiquitination of MAVS blocks MAVS–TRAF3 interactions (107). The V protein of Nipah virus inhibits degradation of the negative regulatory factor UBXN1, thereby enhancing blockade of MAVS–TRAF interactions by UBXN1 (65). Several viruses employ multiple strategies to antagonize antiviral immunity. For example, HCV not only cleaves MAVS via N3/4A but also induces the proteasomal degradation of MAVS via GP73 or NLRX1.
Although most current studies focus on hepatitis viruses (HAV, HBV, and HCV) and respiratory viruses (IAV, human rhinovirus C, SRAS coronavirus, respiratory syncytial virus, EV-D68, and hMPV), increasing attention is being paid to viruses such as EV71, CV-A16, CV-A6, Nipah virus, Seneca Valley virus, and Zika virus. This work will undoubtedly add to our understanding of the mechanisms by which viruses evade the host antiviral response.
Conclusion
Recognition that MAVS plays a pivotal role in antiviral immunity has led to a surge in research on this molecule. It is now clear that cells and viruses regulate MAVS at both the post-transcriptional and post-translational levels using multiple mechanisms. Among them, ubiquitination plays a particularly crucial role in initiating, maintaining, and curtailing RLR-mediated signaling pathways, with K27-, K48-, and K63-linked ubiquitination acting in concert to regulate MAVS signaling. Whether ubiquitination of other types (e.g., K6-, K11-, K29-, K33-, and M1-linked ubiquitin chains) can affect the biological functions of MAVS is unclear. The manner in which MAVS is affected by other modifications, such as phosphorylation and glycosylation as well as products of anaerobic glycolysis (lactate) and components of the apoptosis and autophagy pathways are attracting increasing interest. Viral replication requires a considerable uptake of nutrients from the host organism, and whether other components of carbohydrate and protein metabolism can regulate the antiviral immune response is another new field of study. As the key adaptor protein of RLR-mediated signaling, MAVS clearly plays a crucial role in the innate immune response. Despite significant advances in recent years, as illustrated in this review, the mechanisms regulating MAVS-mediated signaling in other species remains unclear. Further work in this area is expected to provide a clearer understanding of MAVS activity in antiviral immunity and to fuel the discovery of new drugs for the treatment of viral infection.
Author Contributions
ZR carried out literature search and designed idea. TD sorted out important background information and drafted the manuscript. ZZ and ZX critically revises the content of the manuscript. JD and ZW performed the manuscript review. All authors read and approved the final manuscript.
Funding
The manuscript was supported by The National Natural Science Foundation of China (U1704231 and 3177131339) and visiting scholar program sponsored by China Scholarship Council (201906915019).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Anne M. O'Rourke, Ph.D., from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
References
1. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
2. Gallucci S, Maffei ME. DNA sensing across the tree of life. Trends Immunol. (2017) 38:719–32. doi: 10.1016/j.it.2017.07.012
3. Holm CK, Paludan SR, Fitzgerald KA. DNA recognition in immunity and disease. Curr Opin Immunol. (2013) 25:13–8. doi: 10.1016/j.coi.2012.12.006
4. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
5. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. (2011) 34:637–50. doi: 10.1016/j.immuni.2011.05.006
6. Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, et al. Structural basis of Toll-like receptor 3 signaling with double-stranded RNA. Science. (2008) 320:379–81. doi: 10.1126/science.1155406
7. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA. (2004) 101:5598–603. doi: 10.1073/pnas.0400937101
8. Loo YM, Gale M. Immune signaling by RIG-I-like receptors. Immunity. (2011) 34:680–92. doi: 10.1016/j.immuni.2011.05.003
9. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The 0NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. (2009) 30:556–65. doi: 10.1016/j.immuni.2009.02.005
10. Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. (2009) 10:1073–80. doi: 10.1038/ni.1782
11. Morrone SR, Matyszewski M, Yu X, Delannoy M, Egelman EH, Sohn J. Assembly-driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat Commun. (2015) 6:7827. doi: 10.1038/ncomms8827
12. Sun LJ, Wu JX, Du FH, Chen X, Chen ZJJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. (2013) 339:786–91. doi: 10.1126/science.1232458
13. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. (2007) 448:501–U14. doi: 10.1038/nature06013
14. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. (2010) 11:997–1042. doi: 10.1038/ni.1932
15. Zhang ZQ, Yuan B, Bao MS, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol. (2011) 12:959–62. doi: 10.1038/ni.2091
16. Brun AM, Leser GP, Lamb RA, Horvath CM. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol Cell. (2014) 55:771–81. doi: 10.1016/j.molcel.2014.07.003
17. Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, et al. Shared and unique functions of the DExD/H-Box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. (2005) 175:2851–8. doi: 10.4049/jimmunol.175.5.2851
18. Kumar H, Kawai T, Kato H, Sato S, Takahashi K, Coban C, et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J Exp Med. (2006) 203:1795–803. doi: 10.1084/jem.20060792
19. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. (2005) 437:1167–72. doi: 10.1038/nature04193
20. Seth RB, Sun LJ, Ea CK, Chen ZJJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell. (2005) 122:669–82. doi: 10.1016/j.cell.2005.08.012
21. Xu LG, Wang YY, Han KJ, Li LY, Zhai ZH, Shu HB. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol Cell. (2005) 19:727–40. doi: 10.1016/j.molcel.2005.08.014
22. Bender S, Reuter A, Eberle F, Einhorn E, Binder M, Bartenschlager R. Activation of type I and III interferon response by mitochondrial and peroxisomal MAVS and inhibition by hepatitis C virus. PLoS Pathog. (2015) 11:e1005264. doi: 10.1371/journal.ppat.1005264
23. Dixit E, Boulant S, Zhang YJ, Lee ASY, Odendall C, Shum B, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. (2010) 141:668–81. doi: 10.1016/j.cell.2010.04.018
24. Horner SM, Liu HM, Park HS, Briley J, Gale M. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci USA. (2011) 108:14590–5. doi: 10.1073/pnas.1110133108
25. Liu SQ, Chen JQ, Cai X, Wu JX, Chen X, Wu YT, et al. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. eLife. (2013) 2:e00785. doi: 10.7554/eLife.00785
26. Peisley A, Wu B, Xu H, Chen ZJJ, Hur S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature. (2014) 509:110–4. doi: 10.1038/nature13140
27. Oshiumi H, Miyashita M, Inoue N, Okabe M, Matsumoto M, Seya T. The ubiquitin ligase riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe. (2010) 8:496–509. doi: 10.1016/j.chom.2010.11.008
28. Hou FJ, Sun LJ, Zheng H, Skaug B, Jiang QX, Chen ZJJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. (2011) 146:448–61. doi: 10.1016/j.cell.2011.06.041
29. Fang R, Jiang Q, Zhou X, Wang C, Guan Y, Tao J, et al. MAVS activates TBK1 and IKKε through TRAFs in NEMO dependent and independent manner. PLoS Pathog. (2017) 13:e1006720. doi: 10.1371/journal.ppat.1006720
30. Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. (1999) 234:187–208. doi: 10.1016/S0378-1119(99)00210-3
31. Qi N, Shi YH, Zhang R, Zhu WT, Yuan BF, Li XY, et al. Multiple truncated isoforms of MAVS prevent its spontaneous aggregation in antiviral innate immune signalling. Nat Commun. (2017) 8:15676. doi: 10.1038/ncomms15676
32. Brubaker SW, Gauthier AE, Mills EW, Ingolia NT, Kagan JC. A bicistronic MAVS transcript highlights a class of truncated variants in antiviral immunity. Cell. (2014) 156:800–11. doi: 10.1016/j.cell.2014.01.021
33. Lytle JR, Yario TA, Steitz JA. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′UTR as in the 3′UTR. Proc Natl Acad Sci USA. (2007) 104:9667–72. doi: 10.1073/pnas.0703820104
34. Xu L, Peng L, Gu TL, Yu DD, Yao YG. The 3′UTR of human MAVS mRNA contains multiple regulatory elements for the control of protein expression and subcellular localization. Biochim Biophys Acta Gene Regul Mech. (2019) 1862:47–57. doi: 10.1016/j.bbagrm.2018.10.017
35. Guo HL, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. (2010) 466:835–40. doi: 10.1038/nature09267
36. Song H, Ji XH. The mechanism of RNA duplex recognition and unwinding by DEAD-box helicase DDX3X. Nat Commun. (2019) 10:3085. doi: 10.1038/s41467-019-11083-2
37. Zheng QL, Hou J, Zhou Y, Li ZY, Cao XT. The RNA helicase DDX46 inhibits innate immunity by entrapping m6A-demethylated antiviral transcripts in the nucleus. Nat Immunol. (2017) 18:1094–103. doi: 10.1038/ni.3830
38. Paz S, Vilasco M, Werden SJ, Arguello M, Joseph-Pillai D, Zhao TJ, et al. A functional C-terminal TRAF3-binding site in MAVS participates in positive and negative regulation of the IFN antiviral response. Cell Res. (2011) 21:895–910. doi: 10.1038/cr.2011.2
39. Liu BY, Zhang M, Chu HL, Zhang HH, Wu HF, Song GH, et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat Immunol. (2016) 18:214–24. doi: 10.1038/ni.3641
40. Li TL, Li XH, Attri KS, Liu CH, Li LP, Herring LE, et al. O-GlcNAc transferase links glucose metabolism to MAVS-mediated antiviral innate immunity. Cell Host Microbe. (2018) 24:791–803. doi: 10.1016/j.chom.2018.11.001
41. Xue BB, Li HY, Guo MM, Wang JJ, Xu Y, Zou X, et al. TRIM21 promotes innate immune response to RNA viral infection through Lys27-linked polyubiquitination of MAVS. J Virol. (2018) 92:e00321–18. doi: 10.1128/JVI.00321-18
42. Jin SH, Tian S, Luo M, Xie WH, Liu T, Duan TH, et al. Tetherin Suppresses type I interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol Cell. (2017) 68:308–22. doi: 10.1016/j.molcel.2017.09.005
43. Castanier C, Zemirli N, Portier A, Garcin D, Bidere N, Vazquez A, et al. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. (2012) 10:44. doi: 10.1186/1741-7007-10-44
44. You FP, Sun H, Zhou X, Sun WX, Liang SM, Zhai ZH, et al. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat Immunol. (2009) 10:1300–8. doi: 10.1038/ni.1815
45. Zhou X, You FP, Chen HH, Jiang ZF. Poly(C)-binding protein 1 (PCBP1) mediates housekeeping degradation of mitochondrial antiviral signaling (MAVS). Cell Res. (2012) 22:717–27. doi: 10.1038/cr.2011.184
46. Choi YB, Shembade N, Parvatiyar K, Balachandran S, Harhaj EW. TAX1BP1 restrains virus-induced apoptosis by facilitating Itch-mediated degradation of the mitochondrial adaptor MAVS. Mol Cell Biol. (2016) 37:e00422. doi: 10.1128/MCB.00422-16
47. Wang YT, Tong XM, Ye X. Ndfip1 negatively regulates RIG-I-dependent immune signaling by enhancing E3 ligase Smurf1-mediated MAVS degradation. J Immunol. (2012) 189:5304–13. doi: 10.4049/jimmunol.1201445
48. Pan Y, Li R, Meng JL, Mao HT, Zhang Y, Zhang J. Smurf2 negatively modulates RIG-I-dependent antiviral response by targeting VISA/MAVS for ubiquitination and degradation. J Immunol. (2014) 192:4758–64. doi: 10.4049/jimmunol.1302632
49. Zhang LT, Liu J, Qian LP, Feng Q, Wang XF, Yuan YK, et al. Induction of OTUD1 by RNA viruses potently inhibits innate immune responses by promoting degradation of the MAVS/TRAF3/TRAF6 signalosome. PLoS Pathog. (2018) 14:e1007067. doi: 10.1371/journal.ppat.1007067
50. Yoo YS, Park YY, Kim JH, Cho H, Kim SH, Lee HS, et al. The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signaling. Nat Commun. (2015) 6:7910. doi: 10.1038/ncomms8910
51. Zhong B, Zhang Y, Tan B, Liu TT, Wang YY, Shu HB. The E3 ubiquitin ligase RNF5 targets virus-induced signaling adaptor for ubiquitination and degradation. J Immunol. (2010) 184:6249–55. doi: 10.4049/jimmunol.0903748
52. Liuyu TZ, Yu KY, Ye LY, Zhang ZD, Zhang M, Ren YJ, et al. Induction of OTUD4 by viral infection promotes antiviral responses through deubiquitinating and stabilizing MAVS. Cell Res. (2019) 29:67–79. doi: 10.1038/s41422-018-0107-6
53. Liu SQ, Cai X, Wu JX, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. (2015) 347:aaa2630. doi: 10.1126/science.aaa2630
54. Li SZ, Shu QP, Song Y, Zhang HH, Liu Y, Jin BX, et al. Phosphorylation of MAVS/VISA by Nemo-like kinase (NLK) for degradation regulates the antiviral innate immune response. Nat Commun. (2019) 10:3233. doi: 10.1038/s41467-019-11258-x
55. Xiang WW, Zhang Q, Lin X, Wu SY, Zhou Y, Meng FS, et al. PPM1A silences cytosolic RNA sensing and antiviral defense through direct dephosphorylation of MAVS and TBK1. Sci Adv. (2016) 2:e1501889. doi: 10.1126/sciadv.1501889
56. Shao WH, Shu DH, Zhen YX, Hilliard B, Priest SO, Cesaroni M, et al. Prion-like aggregation of mitochondrial antiviral signaling protein in lupus patients is associated with increased levels of type I interferon. Arthritis Rheumatol. (2016) 68:2697–707. doi: 10.1002/art.39733
57. Liu W, Li J, Zheng WN, Shang YL, Zhao ZD, Wang SS, et al. Cyclophilin A-regulated ubiquitination is critical for RIG-I-mediated antiviral immune responses. elife. (2017) 6:e24425. doi: 10.7554/eLife.24425
58. Dai T, Wu LM, Wang S, Wang J, Xie F, Zhang ZK, et al. FAF1 regulates antiviral immunity by inhibiting MAVS but is antagonized by phosphorylation upon viral infection. Cell Host Microbe. (2018) 24:776–90. doi: 10.1016/j.chom.2018.10.006
59. Yang SG, Harding AT, Sweeney C, Mlao D, Swan G, Zhou C, et al. Control of antiviral innate immune response by protein geranylgeranylation. Sci Adv. (2019) 5:eaav7999. doi: 10.1126/sciadv.aav7999
60. Kayagaki N, Phung Q, Chan S, Chaudhari R, Quan C, O'Rourke KM, et al. DUBA: a deubiquitinase that regulates type I interferon production. Science. (2007) 318:1628–32. doi: 10.1126/science.1145918
61. Ju LG, Zhu Y, Lei PJ, Yan D, Zhu K, Wang X, et al. TTLL12 inhibits the activation of cellular antiviral signaling through interaction with VISA/MAVS. J Immunol. (2017) 198:1274–84. doi: 10.4049/jimmunol.1601194
62. Roberts DJ, Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Diff . (2015) 22:248–57. doi: 10.1038/cdd.2014.173
63. Zhang WN, Wang GH, Xu ZG, Tu HQ, Hu FQ, Dai J, et al. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell. (2019) 178:176–89. doi: 10.1016/j.cell.2019.05.003
64. Wang PH, Yang L, Cheng G, Yang G, Xu ZY, You FP, et al. UBXN1 interferes with Rig-I-like receptor-mediated antiviral immune response by targeting MAVS. Cell Rep. (2013) 3:1057–70. doi: 10.1016/j.celrep.2013.02.027
65. Uchida S, Horie R, Sato H, Kai C, Yoneda M. Possible role of the Nipah virus V protein in the regulation of the interferon beta induction by interacting with UBX domain-containing protein1. Sci Rep. (2018) 8:7682. doi: 10.1038/s41598-018-25815-9
66. Nie Y, Ran Y, Zhang HY, Huang ZF, Pan ZY, Wang SY, et al. GPATCH3 negatively regulates RLR-mediated innate antiviral responses by disrupting the assembly of VISA signalosome. PLoS Pathog. (2017) 13:e1006328. doi: 10.1371/journal.ppat.1006328
67. Qin YF, Su ZX, Wu YX, Wu CL, Jin SH, Xie WH, et al. NLRP11 disrupts MAVS signalosome to inhibit type I interferon signaling and virus-induced apoptosis. EMBO Rep. (2017) 18:2160–71. doi: 10.15252/embr.201744480
68. Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, Desai A, et al. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. (2010) 29:1762–73. doi: 10.1038/emboj.2010.50
69. Ning XH, Wang YT, Jing M, Sha MY, Lv MZ, Gao PF, et al. Apoptotic caspases suppress type I interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol Cell. (2019) 74:19–31. doi: 10.1016/j.molcel.2019.02.013
70. Kawaguchi Y, Miyamoto Y, Inoue T, Kaneda Y. Efficient eradication of hormone-resistant human prostate cancers by inactivated Sendai virus particle. Int J Cancer. (2009) 124:2478–87. doi: 10.1002/ijc.24234
71. Matsushima-Miyagi T, Hatano K, Nomura M, Liu LW, Nishikawa T, Saga K, et al. TRAIL and Noxa are selectively upregulated in prostate cancer cells downstream of the RIG-I/MAVS signaling pathway by nonreplicating Sendai virus particles. Clin Cancer Res. (2012) 18:6271–83. doi: 10.1158/1078-0432.CCR-12-1595
72. Yasukawa K, Oshiumi H, Takeda M, Ishihara N, Yanagi Y, Seya T, et al. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci Signal. (2009) 2:ra47. doi: 10.1126/scisignal.2000287
73. Onoguchi K, Onomoto K, Takamatsu S, Jogi M, Takemura A, Morimoto S, et al. Virus-infection or 5′ppp-RNA activates antiviral signal through redistribution of IPS-1 mediated by MFN1. PLoS Pathog. (2010) 6:e1001012. doi: 10.1371/journal.ppat.1001012
74. Castanier C, Garcin D, Vazquez A, Arnoult D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. (2010) 11:133–8. doi: 10.1038/embor.2009.258
75. Li XD, Sun LJ, Seth RB, Pineda G, Chen ZJJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA. (2005) 102:17717–22. doi: 10.1073/pnas.0508531102
76. Mukherjee A, Morosky SA, Delorme-Axford E, Dybdahl-Sissoko N, Oberste MS, Wang TY, et al. The Coxsackievirus B 3Cpro protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. (2011) 7:e1001311. doi: 10.1371/journal.ppat.1001311
77. Pang LL, Yuan XH, Shao CS, Li MZ, Wang Y, Wang HM, et al. The suppression of innate immune response by human rhinovirus C. Biochem Biophys Res Commun. (2017) 490:22–8. doi: 10.1016/j.bbrc.2017.05.169
78. Qian SH, Fan WC, Liu TT, Wu MG, Zhang HW, Cui XF, et al. Seneca Valley Virus suppresses host type I interferon production by targeting adaptor proteins MAVS, TRIF, and TANK for cleavage. J Virol. (2017) 91:e00823–e00817. doi: 10.1128/JVI.00823-17
79. Feng Q, Langereis MA, Lork M, Nguyen M, Hato SV, Lanke K, et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J Virol. (2014) 88:3369–78. doi: 10.1128/JVI.02712-13
80. Dong JM, Xu SG, Wang J, Luo R, Wang D, Xiao SB, et al. Porcine reproductive and respiratory syndrome virus 3C protease cleaves the mitochondrial antiviral signalling complex to antagonize IFN-β expression. J Gen Virol. (2015) 96:3049–58. doi: 10.1099/jgv.0.000257
81. Feng H, Sander AL, Moreira-Soto A, Yamane D, Drexler JF, Lemon SM. Hepatovirus 3ABC proteases and evolution of mitochondrial antiviral signaling protein (MAVS). J Hepatol. (2019) 71:25–34. doi: 10.1016/j.jhep.2019.02.020
82. Wang B, Xi XY, Lei XB, Zhang XY, Cui S, Wang JW, et al. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. (2013) 9:e1003231. doi: 10.1371/journal.ppat.1003231
83. Wei CW, Ni CF, Song T, Liu Y, Yang XL, Zheng ZR, et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J Immunol. (2010) 185:1158–68. doi: 10.4049/jimmunol.0903874
84. Shi CS, Qi HY, Boularan C, Huang NN, Abu-Asab M, Shelhamer JH, et al. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol. (2014) 193:3080–9. doi: 10.4049/jimmunol.1303196
85. Ding SY, Zhu S, Ren LL, Feng NG, Song YH, Ge XM, et al. Rotavirus VP3 targets MAVS for degradation to inhibit type III interferon expression in intestinal epithelial cells. eLife. (2018) 7:e39494. doi: 10.7554/eLife.39494
86. Graff JW, Ewen J, Ettayebi K, Hardy ME. Zinc-binding domain of rotavirus NSP1 is required for proteasome-dependent degradation of IRF3 and autoregulatory NSP1 stability. J Gen Virol. (2007) 88:613–20. doi: 10.1099/vir.0.82255-0
87. Nandi S, Chanda S, Bagchi P, Nayak MK, Bhowmick R, Chawla-Sarkar M. MAVS protein is attenuated by rotavirus nonstructural protein 1. PLoS ONE. (2014) 9:e92126. doi: 10.1371/journal.pone.0092126
88. Barro M, Patton JT. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J Virol. (2007) 81:4473–81. doi: 10.1128/JVI.02498-06
89. Graff JW, Ettayebi K, Hardy ME. Rotavirus NSP1 inhibits NFκB activation by inducing proteasome-dependent degradation of β-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog. (2009) 5:e1000280. doi: 10.1371/journal.ppat.1000280
90. Qin L, Ren LL, Zhou Z, Lei XB, Chen L, Xue QH, et al. Rotavirus nonstructural protein 1 antagonizes innate immune response by interacting with retinoic acid inducible gene I. Virol J. (2011) 8:526. doi: 10.1186/1743-422X-8-526
91. Zhang XW, Zhu CL, Wang TC, Jiang H, Ren YH, Zhang Q, et al. GP73 represses host innate immune response to promote virus replication by facilitating MAVS and TRAF6 degradation. PLoS Pathog. (2017) 13:e1006321. doi: 10.1371/journal.ppat.1006321
92. Qin YW, Xue BB, Liu CY, Wang XH, Tian RY, Xie QY, et al. NLRX1 mediates MAVS degradation to attenuate hepatitis C virus-induced innate immune response through PCBP2. J Virol. (2017) 91:e01264–17. doi: 10.1128/JVI.01264-17
93. Boyapalle S, Wong T, Garay J, Teng M, Juan-Vergara HS, Mohapatra S, et al. Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection. PLoS ONE. (2012) 7:e29386. doi: 10.1371/journal.pone.0029386
94. Ling ZH, Tran KC, Teng MN. Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of beta interferon transcription by interacting with RIG-I. J Virol. (2009) 83:3734–42. doi: 10.1128/JVI.02434-08
95. Ma JZ, Ketkar H, Geng TT, Lo E, Wang LL, Xi JM, et al. Zika virus non-structural protein 4A blocks the RLR-MAVS signaling. Front Immunol. (2018) 9:1350. doi: 10.3389/fmicb.2018.01350
96. Rui YJ, Su JM, Wang H, Chang JL, Wang SH, Zheng WW, et al. Disruption of MDA5-mediated innate immune responses by the 3C proteins of Coxsackievirus A16, Coxsackievirus A6, and Enterovirus D68. J Virol. (2017) 91:e00546–17. doi: 10.1128/JVI.00546-17
97. Choi HJ, Park A, Kang SJ, Lee E, Lee TA, Ra EA, et al. Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type I interferon immune responses. Nat Commun. (2018) 9:125. doi: 10.1038/s41467-017-02624-8
98. Bao XY, Kolli D, Ren JP, Liu TS, Garofalo RP, Casola A. Human metapneumovirus glycoprotein G disrupts mitochondrial signaling in airway epithelial cells. PLoS ONE. (2013) 8:e62568. doi: 10.1371/journal.pone.0062568
99. Chen Y, Deng XL, Deng JF, Zhou JH, Ren YP, Liu SX, et al. Functional motifs responsible for human metapneumovirus M2-2-mediated innate immune evasion. Virology. (2016) 499:361–8. doi: 10.1016/j.virol.2016.09.026
100. Zheng CF, Su CH. Herpes simplex virus 1 infection dampens the immediate early antiviral innate immunity signaling from peroxisomes by tegument protein VP16. Virol J. (2017) 14:35. doi: 10.1186/s12985-017-0709-5
101. Boergeling Y, Rozhdestvensky TS, Schmolke M, Resa-Infante P, Robeck T, Randau G, et al. Evidence for a novel mechanism of influenza virus-induced type I interferon expression by a defective RNA-encoded protein. PLoS Pathog. (2015) 11:e1004924. doi: 10.1371/journal.ppat.1004924
102. Varga ZT, Grant A, Manicassamy B, Palese P. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J Virol. (2012) 86:8359–66. doi: 10.1128/JVI.01122-12
103. Qian W, Wei XQ, Guo KL, Li YT, Lin X, Zou Z, et al. The c-terminal effector domain of non-Structural protein 1 of Influenza A virus blocks IFN-β production by targeting TNF receptor-associated factor 3. Front Immunol. (2017) 8:779. doi: 10.3389/fimmu.2017.00779
104. Hsu ACY, Dua K, Starkey MR, Haw TJ, Nair PM, Nichol K, et al. MicroRNA-125a and -b inhibit A20 and MAVS to promote inflammation and impair antiviral response in COPD. JCI Insight. (2017) 2:e90443. doi: 10.1172/jci.insight.90443
105. Wang RF, Zhu YX, Lin X, Ren CW, Zhao JC, Wang FF, et al. Influenza M2 protein regulates MAVS-mediated signaling pathway through interacting with MAVS and increasing ROS production. Autophagy. (2019) 15:1163–81. doi: 10.1080/15548627.2019.1580089
106. Refolo G, Ciccosanti F, Rienzo MD, Perdomo AB, Romani M, Alonzi T, et al. Negative regulation of mitochondrial antiviral signaling protein–mediated antiviral signaling by the mitochondrial protein LRPPRC during Hepatitis C virus infection. Hepatology. (2019) 69:34–50. doi: 10.1002/hep.30149
Keywords: innate immunity, MAVS, molecular regulation, viral replication, immune evasion
Citation: Ren Z, Ding T, Zuo Z, Xu Z, Deng J and Wei Z (2020) Regulation of MAVS Expression and Signaling Function in the Antiviral Innate Immune Response. Front. Immunol. 11:1030. doi: 10.3389/fimmu.2020.01030
Received: 26 November 2019; Accepted: 29 April 2020;
Published: 27 May 2020.
Edited by:
Lucia Lopalco, San Raffaele Hospital (IRCCS), ItalyReviewed by:
Shaoli Lin, University of Maryland, College Park, United StatesJan Rehwinkel, University of Oxford, United Kingdom
Jianzhong Zhu, Yangzhou University, China
Copyright © 2020 Ren, Ding, Zuo, Xu, Deng and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhanyong Wei, emhhbnlvbmdfd2VpJiN4MDAwNDA7MTI2LmNvbQ==
†These authors have contributed equally to this work