
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 21 May 2020
Sec. Immunological Tolerance and Regulation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00975
This article is part of the Research Topic Retinal Immunobiology and Retinopathy View all 13 articles
 Fauziyya Muhammad1
Fauziyya Muhammad1 Priscilla N. Avalos2
Priscilla N. Avalos2 M. H. Mursalin1Jian-Xing Ma3
M. H. Mursalin1Jian-Xing Ma3 Michelle C. Callegan1,2,4
Michelle C. Callegan1,2,4 Darren J. Lee1,2,4*
Darren J. Lee1,2,4*Experimental autoimmune uveoretinitis (EAU) is a mouse model of human autoimmune uveitis. EAU spontaneously resolves and is marked by ocular autoantigen-specific regulatory immunity in the spleen. Kallikrein binding protein (KBP) or kallistatin is a serine proteinase inhibitor that inhibits angiogenesis and inflammation, but its role in autoimmune uveitis has not been explored. We report that T cells activation is inhibited and EAU is attenuated in human KBP (HKBP) mice with no significant difference in the Treg population that we previously identified both before and after recovery from EAU. Moreover, following EAU immunization HKBP mice have potent ocular autoantigen specific regulatory immunity that is functionally suppressive.
Uveitis is the third leading cause of blindness in Western countries, with an incidence between 25.6–122 cases per 100,000 a year, and a prevalence of 69–623 cases per 100,000 (1–4). While anterior uveitis is the most common form of uveitis, with 33% of these patients becoming chronic (5), posterior uveitis as the second most common form of uveitis is far more devastating. Approximately 17.6% of active uveitis patients experience transient or permanent vision loss (6), so a proper diagnosis and treatment are crucial for maintaining vision. Corticosteroids are the first line of treatment for uveitis patients, but due to the myriad of side-effects they are not a long-term treatment option (7–9). Therefore, immunosuppressive medications are used to control uveitis, with the goal of sustained remission (10–15). However, not all the immunosuppressive medications are effective and some chronic uveitis patients fail multiple treatment regimens. As such, additional immunosuppressive treatment options are necessary for the treatment of autoimmune uveitis.
Kallistatin (or kallikrein-binding protein) is a serine proteinase inhibitor that is encoded by SERPINA4 (16, 17). Kallistatin was first identified as an inhibitor of tissue kallikrein (16, 18). Later, kallistatin has been investigated extensively for its role in diabetes (17, 19, 20). Type 1 diabetic patients have high serum levels of kallistatin (19), but the vitreous of diabetic retinopathy patients have low levels of kallistatin (19, 21). It has also been demonstrated that kallistatin has potent anti-inflammatory properties (22, 23). Therefore, it is of interest to determine if kallistatin could be a therapeutic alternative for autoimmune uveitis.
The most widely used model of human posterior autoimmune uveitis is the mouse model, experimental autoimmune uveitis (EAU). In the B10.RIII background, the disease is more severe and resolves more quickly compared to the more chronic C57BL/6J model. The onset of EAU is 12–14 days after immunization and resolution spontaneously occurs without relapse at 2–3 months after immunization (24–26). At resolution of EAU (post-EAU), regulatory immunity emerges in the spleen (27) and provides resistance to EAU during re-immunization and when adoptively transferred to mice that are immunized for EAU (27–30). Post-EAU regulatory immunity requires a regulatory T cell (Treg) that has been identified as CD4+CD25+Nrp-1−PD-1+PD-L1+Foxp3+ (29, 30).
A transgenic mouse that expresses human kallistatin under a chicken ß-actin promoter (TG-HKBP) has been studied with respect to angiogenesis related to diabetes (31). As such, in this report we asked if kallistatin over-expression affects T cell activation, provides resistance to EAU, and if regulatory immunity that provides resistance to EAU emerges in the spleen of post-EAU TG-HKBP mice. Our observations demonstrate that kallistatin over-expression provides resistance to EAU and does not affect the induction of regulatory immunity in the spleen of post-EAU TG-HKBP mice.
All mouse procedures described in this study were approved by the University of Oklahoma Health Sciences Center Institutional Animal Care and Use Committee (OUHSC IACUC) and all mouse study methods were carried out in accordance with the relevant guidelines approved by the OUHSC IACUC. C57BL/6J mice were purchased from Jackson Laboratories. Transgenic mice expressing human kallikrein binding protein under the chicken β-actin promoter (TG-HKBP or HKBP) were generated in the Jian-Xing Ma Lab on a C57BL/6J background (31, 32). All TG-HKBP mice were genotyped to verify transmission of the transgene before inclusion in this study. Serum was previously collected from TG-HKBP mice to verify elevated expression (31). All mice were housed in the Dean McGee Eye Institute vivarium under specific-pathogen-free conditions in a 12-h light cycle with access to food and water ad libitum. When mice were purchased from Jackson Laboratories they were allowed 2 weeks to acclimate under conventional housing conditions before inclusion in experimental procedures. The methods of euthanasia of all mice were approved by OUHSC IACUC and are consistent with the IACUC policies and the 2013 AVMA Guidelines on Euthanasia. Euthanasia of all mice for this study was done with carbon dioxide or cervical dislocation followed by a secondary method such as decapitation, bilateral thoracotomy, exsanguination, or vital organ removal. Mice were randomly chosen for experimental groups.
EAU was induced in 6 to 10-week-old mice using the previously described immunization procedure (30). Complete Freund's adjuvant (CFA) with 5 mg/mL desiccated M. tuberculosis (Difco Laboratories, Detroit, MI) was emulsified with 2 mg/ml interphotoreceptor retinoid binding protein (peptides 1–20) (IRBP) (Genscript, Piscataway, NJ) to immunize mice for EAU. In order to minimize suffering mice were anesthetized with a ketamine/xylazine (100 mg/10 mg per kg) before immunization. The emulsion was injected subcutaneously at a volume of 200 μL total into two sites in the lower back followed by an intraperitoneal injection of 0.3 μg pertussis toxin. The course of EAU was evaluated every 3–4 days by fundus examination with a slit lamp microscope. Prior to examination of the retina, the iris was dilated with 1% tropicamide, and the cornea was flattened with a glass coverslip. We utilize a clinical scoring system that is validated by at least two members of the lab rather than histological examination because we have an interest in the resolution phase of EAU, so collecting mice over the course of disease for histological evaluation would significantly reduce the power of the study. As previously described, the clinical signs of observable infiltration and vasculitis in the retina were scored on a 5-point scale (33). Because the immunization procedure can elicit a score of 1, when the EAU score is 1 or lower, the disease is considered background or resolved. Both eyes were scored and the higher score was used to represent that mouse for that day, because the more severe score suggests that the systemic immune response is severe enough to elicit the greater score. The average score for the group of mice was then calculated. The highest score for each mouse over the entire course of disease was also determined and plotted for each group. Another masked member of the lab with experience evaluating EAU confirmed the clinical scores.
A single cell suspension that was depleted of red blood cells using RBC lysis buffer (Sigma, St Louis, MO) was made from the spleens collected into 5% FBS in RPMI supplemented with 10 μg/ml Gentamycin (Sigma), 10 mM HEPES (GE Healthcare), 1 mM Sodium Pyruvate (BioWhittaker), Nonessential Amino Acids 0.2% (BioWhittaker). Serum free media (SFM) consisting of RPMI-1640 with 1% ITS+1 solution (Sigma) and 0.1% BSA (Sigma) was used to resuspend the spleen cells with IRBP (50 μg/mL) or α-CD3 (clone 2C11, Biolegend). The spleen cells were then incubated for 48 h at 37°C and 5% CO2 to reactivate antigen specific T cells. After the reactivation cells were collected for flow cytometry analysis or adoptive transfer (1 × 106) into recipient mice.
Mice were anesthetized using isoflurane. CD4+ splenocytes (1 × 106/μL) were injected into the left and right eye of each mouse with a sterile borosilicate glass micropipette (Kimble Glass Inc., Vineland, NJ, USA) beveled to an approximate bore size of 10–20 μm (BV-10 KT Brown Type micropipette beveller, Sutter Instrument Co., Novato, CA, USA). The micropipettes were inserted just posterior to the superior limbus under stereomicroscopic visualization, and 1-μL volume was injected directly into the vitreous. Injection rates and volumes were monitored using a programmable cell microinjector (Microdata Instruments, Plainfield, NJ, USA).
Mouse spleen cells were washed with PBS with 1% BSA (staining buffer), blocked with mouse IgG in staining buffer, then stained with conjugated antibodies. Antibodies used were anti-CD4 (clone RM4-5, Biolegend, San Diego, CA), anti-CD25 (clone PC61, Biolegend), anti-PD-1 (clone 29F.1A12, Biolegend), anti-PD-L1 (clone 10F.9G2 Biolegend), anti-Nrp-1 (Cat FAB5994N, R&D Systems, Minneapolis, MN), anti-FoxP3 (clone FJK-16s, eBiosciences), anti-CD44 (clone IM7, Biolegend), anti-CD62L (clone MEL-14, Biolegend), anti-CD45.1 (clone A20, Biolegend), and anti-CD45.2 (clone 104, Biolegend).
Stained cells were analyzed in the Oklahoma Medical Research Facility (OMRF) Flow Cytometry Core Facility on a BD LSRII (BD Biosciences) data was analyzed using FlowJo Software (Tree Star, Inc., Ashland, OR).
Statistical significance between maximum EAU scores and flow cytometry results was determined using non-parametric Mann-Whitney two-tailed test. Two-way ANOVA was also used to assess significant changes in the tempo of disease between the groups of treated EAU mice with post-test Bonferroni comparison analysis. Statistical significance was designated when P ≤ 0.05 and analyzed using Prism 6 (GraphPad Software, Inc., La Jolla, CA).
We first asked if there is a defect in the T cell response in TG-HKBP mice. Naïve and memory T cells were evaluated in splenic T cells that were non-specifically activated through CD3. As expected, wild-type T cells showed a significant elevation in memory T cells (CD44+) and significant reduction in naïve T cells (CD62L+) (Figures 1A,B,E,F). In contrast TG-HKBP T cells showed no significant change in memory or naïve T cell populations following CD3 activation (Figures 1C–F). This inhibition of T cell activation and the anti-inflammatory properties of kallistatin (kallikrein-binding protein) (23) prompted us to ask if it suppresses experimental autoimmune uveitis (EAU). TG-HKBP mice have been previously characterized in studies related to diabetic retinopathy, and serum kallistatin in these mice is over four-fold higher than the endogenous mouse kallistatin (31, 32). When TG-HKBP mice were immunized for EAU, a significant decrease in the severity and acceleration of resolution was observed in TG-HKBP mice compared to wild-type mice immunized for EAU (Figure 2A). Because the tempo of disease can vary from mouse to mouse over the course of EAU, we also compared the maximum severity of each mouse over the entire course of disease by plotting the maximum score of each mouse (Figure 2B). The maximum EAU score also showed that TG-HKBP mice experienced significantly less severe EAU compared to wild-type mice (Figure 2B). Because the immunization procedure can elicit a score of 1, when the EAU score is 1 or lower, the disease is considered resolved. These observations demonstrate that kallistatin attenuates EAU and accelerates the resolution of EAU. Our clinical scores were also confirmed with fundus photos (Figure 2C). These observations demonstrate that kallistatin inhibits T cell activation resulting in the attenuation of EAU.
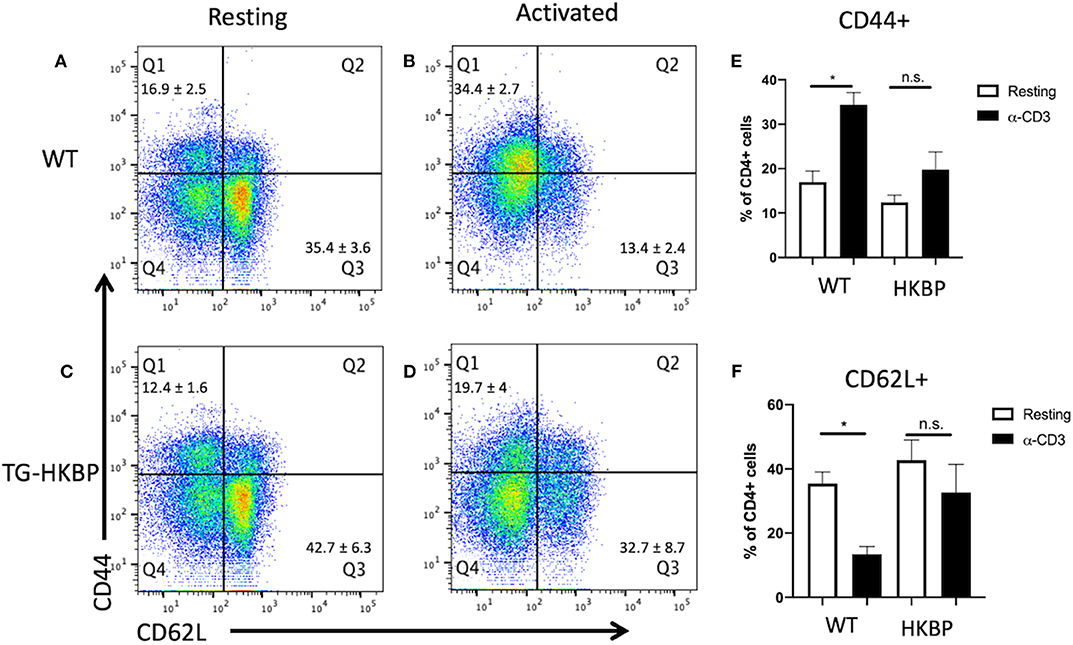
Figure 1. T cell activation in T cells from TG-HKBP mice. Wild-type and TG-HKBP splenocytes were cultured ± α-CD3 for 2 days, stained for CD4, CD44, and CD62L, and analyzed by flow cytometry. Representative dot plots are shown for wild-type (n = 5) (A,B) and TG-HKBP (n = 7) (C,D), and bar graphs show the mean ± SEM for the indicated group (E,F). One to three mice were used for each experiment, and each experiment was repeated three times. Statistical significance (P ≤ 0.05) is designated by *, n.s. indicates no significance.
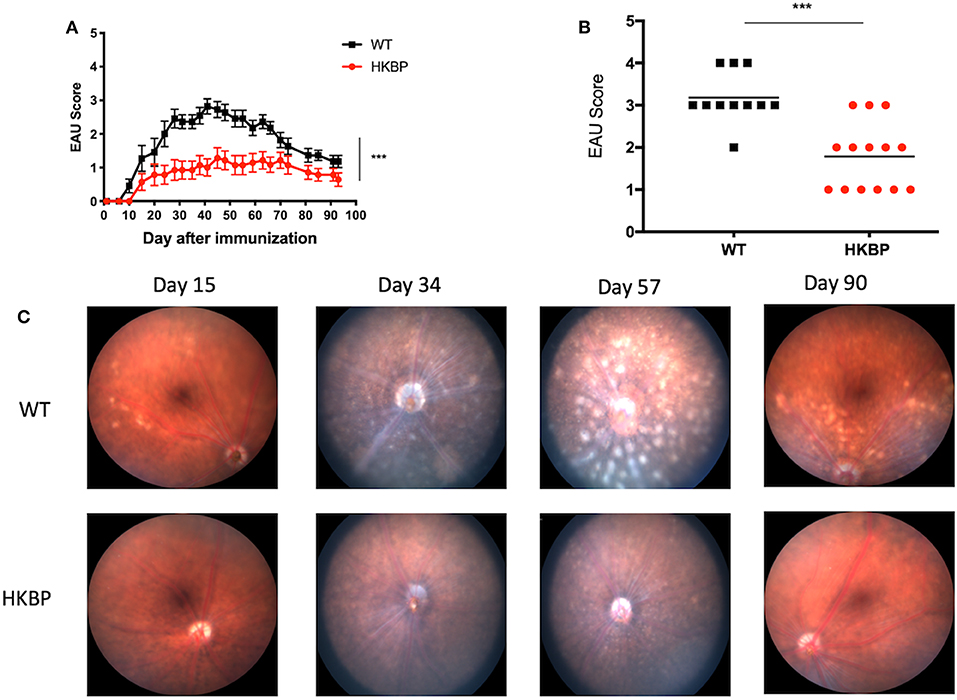
Figure 2. EAU course of disease and maximum scores in TG-HKBP mice immunized for EAU. Wild-type and TG-HKBP mice were immunized for EAU and the clinical EAU score was assessed by slit lamp examination of the retina every 3–4 days. The highest score for each mouse on the indicated day was averaged with all the mice on that day and graphed with the SEM for WT (black squares, n = 11) and HKBP mice (red circle, n = 14) (A). The highest EAU score for each mouse over the course of EAU was also determined (black squares, n = 11) and HKBP mice (red circle, n = 14) (B). Representative images at the indicated time points are shown (C). Statistical significance (P ≤ 0.05) is designated by ***.
We next sought to ask if the TG-HKBP mice have alterations in the Treg compartment. TG-HKBP mice have significantly less circulating B cells and a significantly greater percentage of circulating CD4+ T cells (32). Regulatory T cells (Tregs) are necessary for the resolution of EAU (34) and for providing resistance to relapse (30, 35). Because we observed attenuated EAU in TG-HKBP mice, we asked if there is a change in the Treg compartment. Because the Tregs that provide resistance to relapse have been identified as CD4+CD25+Nrp-1−PD-1+PD-L1+Foxp3+ (30), we asked if TG-HKBP mice had an alteration in the number of these Tregs. The blood and bone marrow showed a trend toward an increase in PD-1+PD-L1+Foxp3+Nrp-1−CD25+CD4+ Tregs, but not a statistically significant increase (Figures 3A–D). These observations suggest suppression of EAU by TG-HKBP mice may be due to suppression of an inflammatory response but not necessarily an increase in the induction of Tregs.
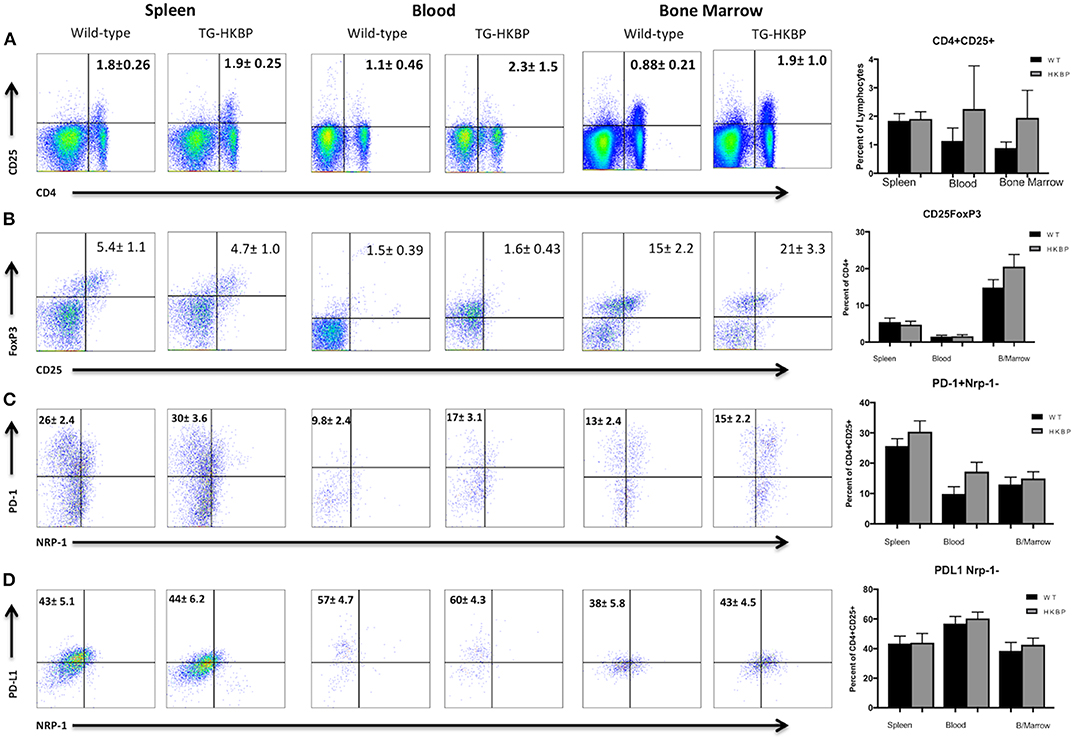
Figure 3. Flow cytometry analysis of TG-HKBP mice for Treg cells. Spleen, blood, and bone marrow T cells from WT and TG-HKBP mice were stained for Treg cell markers. Representative flow plots with the mean and SEM of 3 independent experiments of 3–4 mice per experiment of WT mice (n = 10, black bars) and HKBP mice (n = 11, gray bars). CD4 and CD25 staining on T cells gated on the lymphocyte population based on FSC and SSC (A), CD25 and FoxP3 staining of cells gated on CD4 (B), NRP-1 and PD-1 staining of cells gated on CD4 and CD25 (C), NRP-1 and PD-L1 staining of cells gated on CD4 and CD25 (D).
The post-EAU regulatory immunity that emerges in the spleen of mice at resolution of EAU provides resistance to relapse (29, 30, 34, 35), so that may be a mechanism to provide sustained remission for uveitis patients in the clinical setting. A requirement of post-EAU regulatory immunity is resolution of EAU (27). Because the clinical EAU scores of TG-HKBP mice were significantly lower compared to wild-type mice, we therefore asked if the post-EAU TG-HKBP mice also had CD4+CD25+Nrp-1−PD-1+PD-L1+Foxp3+ Tregs in the spleen at resolution of EAU. The spleens from post-EAU TG-HKBP mice were collected and reactivated in vitro as has been done before (29, 30, 35). There was no significant difference between post-EAU wild-type and post-EAU TG-HKBP Treg cell populations in the spleen (Figures 4A–D). However, the percentage of PD-1 and PD-L1 expressing CD4+CD25+ T cells (Figures 4C,D) was increased compared to the unimmunized unstimulated T cells (Figures 3C,D). Because there is no significant difference between the post-EAU wild-type mice and TG-HKBP mice, these observations demonstrate that this post-EAU Treg population is present in the spleen of TG-HKBP mice.
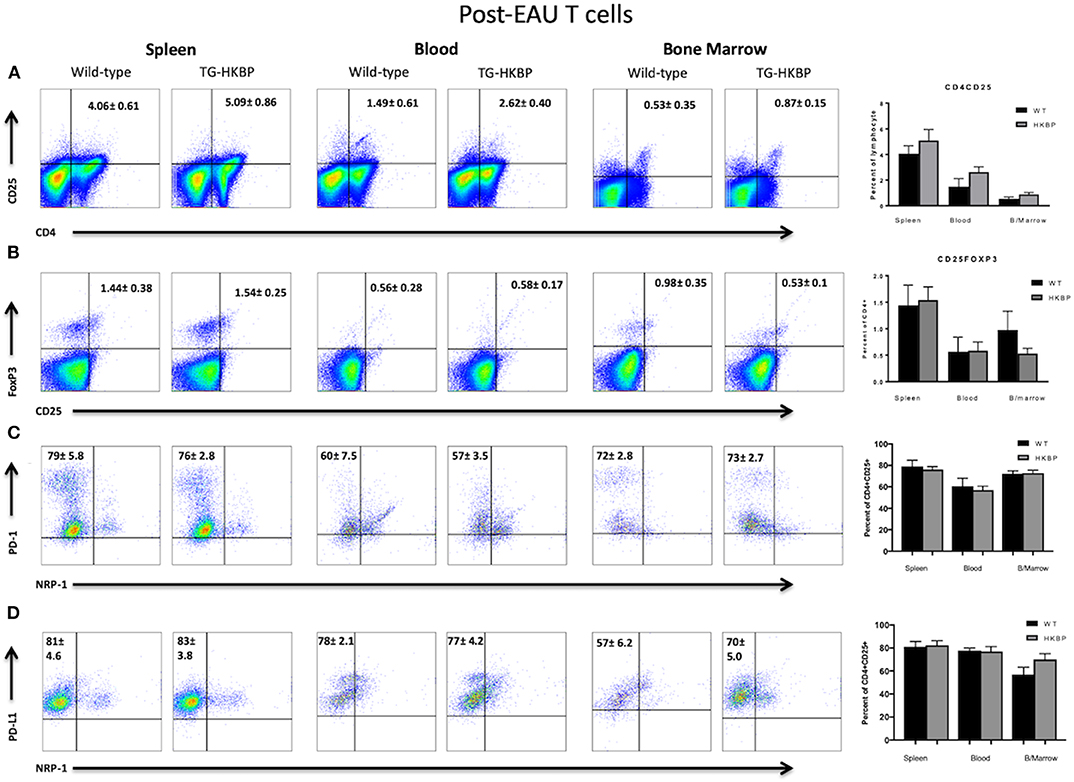
Figure 4. Flow cytometry analysis of post-EAU TG-HKBP mice for Treg cells. Spleen, blood, and bone marrow T cells from WT and TG-HKBP mice were stained for Treg cell markers. Representative flow plots with the mean and SEM of 3 independent experiments of 3–7 mice per experiment of WT mice (n = 6, black bars) and HKBP mice (n = 15, gray bars). CD4 and CD25 staining on T cells gated on the lymphocyte population based on FSC and SSC (A), CD25 and FoxP3 staining of cells gated on CD4 (B), NRP-1 and PD-1 staining of cells gated on CD4 and CD25 (C), NRP-1 and PD-L1 staining of cells gated on CD4 and CD25 (D).
We next asked if the post-EAU regulatory immunity in TG-HKBP mice is functionally suppressive. The spleen of post-EAU TG-HKBP mice was collected and reactivated in vitro as has been done before (29, 30, 35). The reactivated splenocytes were then adoptively transferred to recipient mice immunized for EAU. As expected, mice that received splenocytes from post-EAU wild-type mice showed a significant suppression of EAU (29, 30, 33) compared to EAU mice that did not receive an adoptive transfer (Figures 5A,B). The mice that received post-EAU TG-HKPB splenocytes also showed a significant suppression of EAU (Figure 5D) and significantly lower maximum EAU scores compared to controls (Figure 5E). A representative fundus photo is shown at day 67 to confirm the clinical observations (Figures 5C,F). These observations demonstrate that kallistatin does not affect the emergence of post-EAU regulatory immunity in the spleen.
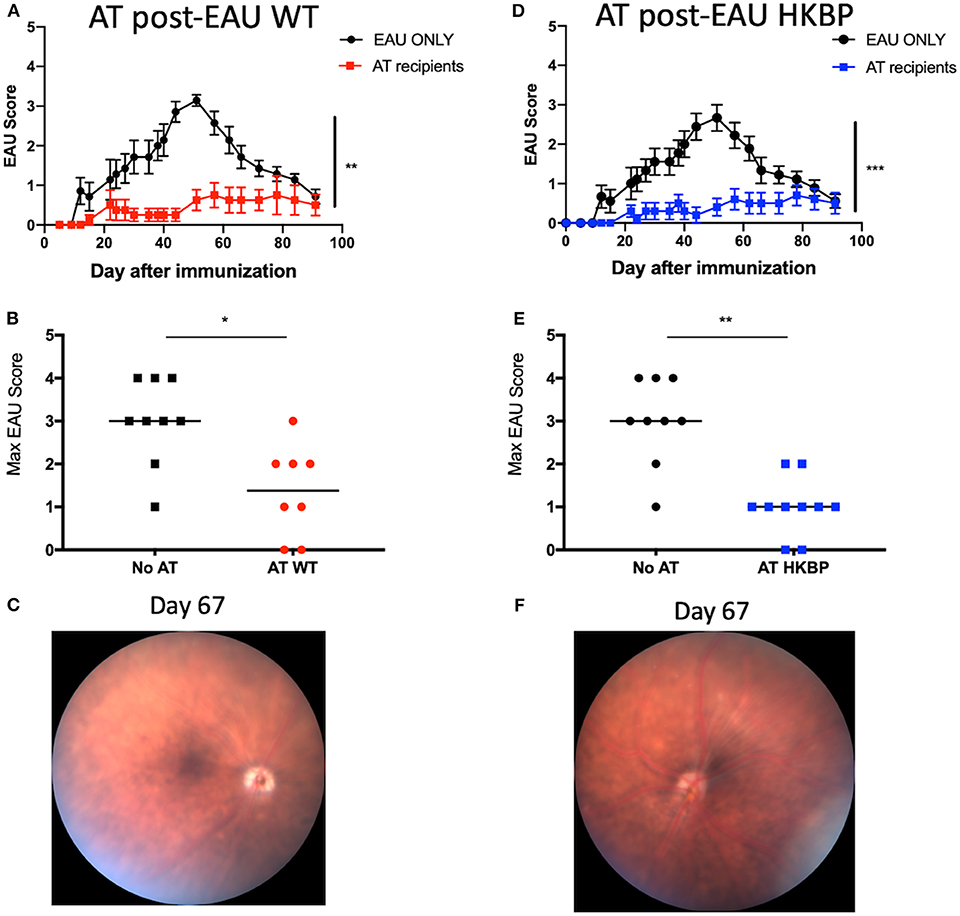
Figure 5. EAU course of disease in recipient mice immunized for EAU that received splenocytes from post-EAU TG-HKBP mice. Post-EAU splenocytes from WT or TG-HKBP mice were reactivated in vitro and transferred to recipient mice that were immunized for EAU. Shown are clinical scores and representative retina images from recipient mice. Recipient scores are compared with EAU mice that did not receive cell transfer (black circles and squares). Mice that received WT post-EAU splenocytes (red squares and circles, n = 8) (A,B) and mice that received HKBP post-EAU splenocytes (blue squares n = 10) (D,E). The same group of control EAU mice that did not receive a transfer are used for comparison (A,B,D,E). A representative fundus image is shown for mice that received an adoptive transfer (C,F). Statistical significance is designated by * (P ≤ 0.05), ** (P ≤ 0.01), and *** (P ≤ 0.005).
In order to determine if kallistatin mediated Treg induction is intrinsic or extrinsic, we transferred CD4+ HKBP cells into wild-type mice to determine if FoxP3 expression is mediated through intrinsic T cell expression of kallistatin. Recipient mice were congenic (CD45.1) to identify the CD45.2 HKBP transferred cells. We found a significant elevation of FoxP3 expression in the transferred cells when HKBP cells were transferred to wild-type mice compared to wild-type cells transferred to the eyes of HKBP mice (Figures 6A,C). These observations show that kallistatin functions intrinsically in T cells. We next asked if this was an ocular specific effect by transferring congenic cells intravenously and assaying the spleen for FoxP3 expression in the transferred T cells. A similar pattern of expression with ocular injected cells was observed with a significant increase in FoxP3 expression in HKBP cells transferred to wild-type mice compared with wild-type cells transferred to HKBP mice (Figures 6B,C). These observations demonstrate that kallistatin functions intrinsically on T cells to maintain or induce Treg cells.
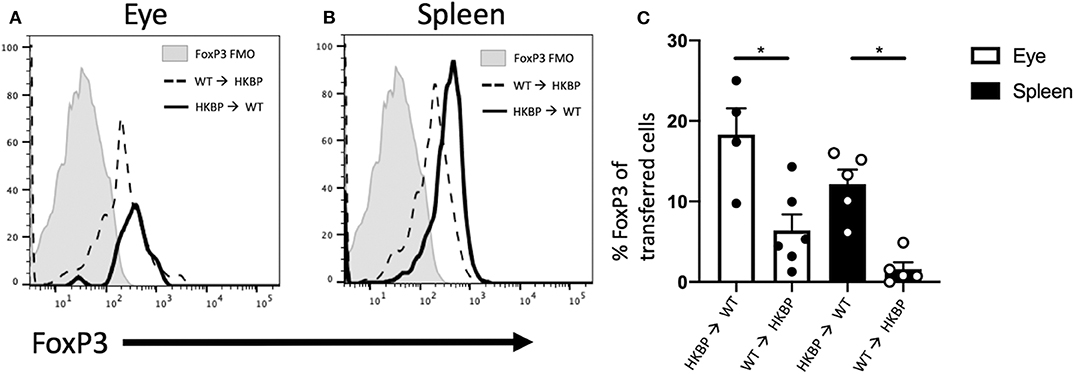
Figure 6. FoxP3 expression in transferred HKBP or WT cells transferred to WT or HKBP mice. CD4+ splenocytes were collected from donor HKBP or WT mice and transferred to recipient congenic mice. Donor cells were injected into the eyes (1 × 105 cells) or intravenously (1 × 106 cells). Forty eight-hours after the transfer the eyes were collected from mice that received an intraocular injection or the spleen was collected from mice that received an intravenous injection. Cells were then analyzed by flow cytometry for CD45.1, CD45.2, and FoxP3. Histograms shown are representative for all experiments, FoxP3 expression in the transferred cells collected from injected eyes (A), or FoxP3 expression in the transferred cells collected from the spleen of intravenously injected mice (B), the same fluorescence minus one (FMO) control is shown in both panels and is from the spleen. The mean ± SEM for all experiments are shown (C). Each experiment included 1–3 mice and the experiment was repeated three times. Statistical significance (P ≤ 0.05) is designated by *.
It has been previously demonstrated that a relationship between kallistatin and inflammation exists. This further illustrates a relationship between angiogenesis and inflammation. Previous reports demonstrate that TG-HKBP mice exhibit a reduction in B cells and a greater percentage of CD4+ T cells (32). It has also been previously shown that TG-HKBP mice also have an impairment of wound healing due to defects in angiogenesis (17). In this report, we asked if kallistatin is capable of suppressing autoimmune uveitis. Our observations demonstrate that EAU is attenuated in TG-HKBP mice and display a reduction in severity and acceleration of resolution. This indicates the anti-inflammatory and anti-angiogenic properties of kallistatin are sufficient to suppress autoimmune uveitis and should be further investigated to develop novel treatments for autoimmune uveitis. However, the anti-angiogenic properties associated with elevated systemic kallistatin will need to be carefully considered if this is translated into a uveitis treatment. Another caveat to consider is that the overexpression of kallistatin is not equivalent to administration of the protein systemically because kallistatin likely has different properties in the eye compared to systemic kallistatin (17, 19, 21). Therefore, it may be the case that kallistatin given locally into the eye may be a suitable alternative to systemic administration.
It has been demonstrated that kallistatin is a Wnt/β-catenin inhibitor. This accounts for the reduction of lymphocytes because of the reduction of hematopoietic stem cells (32, 36–38). A reduction in T cells supports the hypothesis that EAU is suppressed because of a lack of effector T cells that can mediate inflammation. This may still be the case, but because we observe Treg cells that emerge at the resolution of EAU in TG-HKBP mice, it is likely that there are still pathogenic effector T cells being generated. However, given the anti-inflammatory properties and antagonism of TNF-α (23, 39) kallistatin has the capacity to limit the extent to which these effector T cells can mediate disease. Support for a kallistatin-mediated limitation of T cell activation is provided with the significant reduction of CD44+ TG-HKBP T cells following CD3 activation. As such, further studies are necessary to determine the exact mechanism whereby, kallistatin suppresses EAU.
Since mice that recover from EAU have regulatory immunity that provides resistance to EAU, it was of interest to determine if the post-EAU TG-HKBP mice also generate this regulatory immunity (29, 30, 35). Our observations demonstrate that not only is EAU attenuated in TG-HKBP mice, but they also generate regulatory immunity that is capable of providing resistance to relapse. The importance of this discovery is that if a uveitis patient is treated with kallistatin, there is still a generation of regulatory immunity that could provide sustained remission. However, overexpression of kallistatin is not equivalent to the administration of the protein as a therapeutic, but it has been demonstrated to confer greater survival in a sepsis model (40). Therefore, additional study assessing the efficacy of recombinant kallistatin is needed. We further show that the Tregs that emerge in the spleen of TG-HKBP mice share similar markers as the typical Tregs that normally emerge during EAU, so the Tregs likely emerge through similar mechanisms.
Because the TG-HKBP mice express serum levels similar to diabetic patients with vascular complications (17, 19), it may be the case that diabetic patients could be resistant to the development of autoimmune uveitis. However, because there are many mechanisms that promote autoimmune uveitis, this may only represent one such mechanism that provides resistance. As such, it has been observed that patients with diabetic retinopathy experience exacerbated uveitis (41, 42). This discrepancy is likely due to the status of ocular immune privilege, as is observed in diabetic retinopathy patients with lower kallistatin concentration in the vitreous (19, 21), and if systemic regulatory immunity has been established or not. In the case that ocular immune privilege has established regulatory immunity to protect against ocular inflammation, the patient would be protected against uveitis.
These observations demonstrate for the first time to our knowledge that kallistatin is effective in decreasing the severity of EAU and in accelerating the resolution of EAU. This suppression is likely due to multiple mechanisms that may include an inhibition of T cell activation and an intrinsic predisposition to regulatory activity in T cells. Importantly, in mice that overexpress kallistatin and have recovered from EAU, a functionally suppressive regulatory immunity is found in the spleen. This regulatory immunity in the spleen has a similar abundance of post-EAU Tregs that naturally emerge at resolution of EAU. Therefore, kallistatin suppresses autoimmune uveitis while also allowing for the emergence of systemic regulatory immunity that provides resistance to EAU.
The datasets generated for this study are available on request to the corresponding author.
This animal study was reviewed and approved by Institutional Animal Care and Use Committee, University of Oklahoma Health Science Center.
All experiments, analysis, and experimental design of this work were done by DL, PA, FM, MM, MC, and J-XM. The conceptual design of this work and the writing of this manuscript was a collaborative effort between DL and J-XM.
This work was supported by National Institutes of Health (NIH) EY024951 (DL), EY029240 (DL), NIH Core Grant P30 EY021725, EY018659 (J-XM), EY019309 (J-XM), GM104934 (J-XM), EY028810 (MC), EY28066 (MC), Oklahoma Center for the Advancement of Science and Technology HR16-041 (J-XM), and in part by an unrestricted Research to Prevent Blindness grant (New York, NY, USA).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the Oklahoma Medical Research Foundation Flow Cytometry Core facility and the Dean McGee Eye Institute Vivarium staff.
1. Darrell RW, Wagener HP, Kurland LT. Epidemiology of uveitis. Incidence and prevalence in a small urban community. Arch Ophthalmol. (1962) 68:502–14. doi: 10.1001/archopht.1962.00960030506014
2. Reeves SW, Sloan FA, Lee PP, Jaffe GJ. Uveitis in the elderly: epidemiological data from the National Long-term Care Survey Medicare Cohort. Ophthalmology. (2006) 113:307.e1. doi: 10.1016/j.ophtha.2005.10.008
3. Suhler EB, Lloyd MJ, Choi D, Rosenbaum JT, Austin DF. Incidence and prevalence of uveitis in Veterans Affairs Medical Centers of the Pacific Northwest. Am J Ophthalmol. (2008) 146:890–896.e8. doi: 10.1016/j.ajo.2008.09.014
4. Hwang DK, Chou YJ, Pu CY, Chou P. Epidemiology of uveitis among the Chinese population in Taiwan: a population-based study. Ophthalmology. (2012) 119:2371–6. doi: 10.1016/j.ophtha.2012.05.026
5. Natkunarajah M, Kaptoge S, Edelsten C. Risks of relapse in patients with acute anterior uveitis. Br J Ophthalmol. (2007) 91:330–4. doi: 10.1136/bjo.2005.083725
6. Gritz DC, Wong IG. Incidence and prevalence of uveitis in Northern California; the Northern California Epidemiology of Uveitis Study. Ophthalmology. (2004) 111:491–500; discussion: 500. doi: 10.1016/j.ophtha.2003.06.014
7. Lee FF, Foster CS. Pharmacotherapy of uveitis. Expert Opin Pharmacother. (2010) 11:1135–46. doi: 10.1517/14656561003713534
8. Heiligenhaus A, Michels H, Schumacher C, Kopp I, Neudorf U, Niehues T, et al. Evidence-based, interdisciplinary guidelines for anti-inflammatory treatment of uveitis associated with juvenile idiopathic arthritis. Rheumatol Int. (2012) 32:1121–33. doi: 10.1007/s00296-011-2126-1
9. Lerman MA, Rabinovich CE. The future is now: biologics for non-infectious pediatric anterior uveitis. Paediatr Drugs. (2015) 17:283–301. doi: 10.1007/s40272-015-0128-2
10. Jabs DA, Rosenbaum JT, Foster CS, Holland GN, Jaffe GJ, Louie JS, et al. Guidelines for the use of immunosuppressive drugs in patients with ocular inflammatory disorders: recommendations of an expert panel. Am J Ophthalmol. (2000) 130:492–513. doi: 10.1016/S0002-9394(00)00659-0
11. Nguyen QD, Callanan D, Dugel P, Godfrey DG, Goldstein DA, Wilensky JT. Treating chronic noninfectious posterior segment uveitis: the impact of cumulative damage. Proceedings of an expert panel roundtable discussion. Retina. (2006) 26(Suppl.):1–16. doi: 10.1097/01.iae.0000250601.15893.5f
12. Siddique SS, Shah R, Suelves AM, Foster CS. Road to remission: a comprehensive review of therapy in uveitis. Expert Opin Investig Drugs. (2011) 20:1497–515. doi: 10.1517/13543784.2011.617741
13. Kruh J, Foster CS. The philosophy of treatment of uveitis: past, present and future. Dev Ophthalmol. (2012) 51:1–6. doi: 10.1159/000336183
14. Foster CS, Kothari S, Anesi SD, Vitale AT, Chu D, Metzinger JL, et al. The Ocular Immunology and Uveitis Foundation preferred practice patterns of uveitis management. Surv Ophthalmol. (2016) 61:1–17. doi: 10.1016/j.survophthal.2015.07.001
15. Maleki A, Meese H, Sahawneh H, Foster CS. Progress in the understanding and utilization of biologic response modifiers in the treatment of uveitis. Expert Rev Clin Immunol. (2016) 12:775–86. doi: 10.1586/1744666X.2016.1166052
16. Chao J, Chao L. Biochemistry, regulation and potential function of kallistatin. Biol Chem Hoppe Seyler. (1995) 376:705–13.
17. McBride JD, Jenkins AJ, Liu X, Zhang B, Lee K, Berry WL, et al. Elevated circulation levels of an antiangiogenic SERPIN in patients with diabetic microvascular complications impair wound healing through suppression of Wnt signaling. J Invest Dermatol. (2014) 134:1725–34. doi: 10.1038/jid.2014.40
18. Chao J, Li P, Chao L. Kallistatin suppresses cancer development by multi-factorial actions. Crit Rev Oncol Hematol. (2017) 113:71–8. doi: 10.1016/j.critrevonc.2017.03.011
19. Jenkins AJ, McBride JD, Januszewski AS, Karschimkus CS, Zhang B, O'Neal DN, et al. Increased serum kallistatin levels in type 1 diabetes patients with vascular complications. J Angiogenes Res. (2010) 2:19. doi: 10.1186/2040-2384-2-19
20. Yiu WH, Wong DW, Wu HJ, Li RX, Yam I, Chan LY, et al. Kallistatin protects against diabetic nephropathy in db/db mice by suppressing AGE-RAGE-induced oxidative stress. Kidney Int. (2016) 89:386–98. doi: 10.1038/ki.2015.331
21. Ma JX, King LP, Yang Z, Crouch RK, Chao L, Chao J. Kallistatin in human ocular tissues: reduced levels in vitreous fluids from patients with diabetic retinopathy. Curr Eye Res. (1996) 15:1117–23. doi: 10.3109/02713689608995143
22. Shen B, Smith RS Jr, Hsu YT, Chao L, Chao J. Kruppel-like factor 4 is a novel mediator of Kallistatin in inhibiting endothelial inflammation via increased endothelial nitric-oxide synthase expression. J Biol Chem. (2009) 284:35471–8. doi: 10.1074/jbc.M109.046813
23. Lu SL, Tsai CY, Luo YH, Kuo CF, Lin WC, Chang YT, et al. Kallistatin modulates immune cells and confers anti-inflammatory response to protect mice from group A streptococcal infection. Antimicrob Agents Chemother. (2013) 57:5366–72. doi: 10.1128/AAC.00322-13
24. Caspi RR, Roberge FG, Chan CC, Wiggert B, Chader GJ, Rozenszajn LA, et al. A new model of autoimmune disease. Experimental autoimmune uveoretinitis induced in mice with two different retinal antigens. J Immunol. (1988) 140:1490–5.
25. Caspi RR, Silver PB, Luger D, Tang J, Cortes LM, Pennesi G, et al. Mouse models of experimental autoimmune uveitis. Ophthalmic Res. (2008) 40:169–74. doi: 10.1159/000119871
26. Chen J, Qian H, Horai R, Chan CC, Caspi RR. Mouse models of experimental autoimmune uveitis: comparative analysis of adjuvant-induced vs spontaneous models of uveitis. Curr Mol Med. (2015) 15:550–7. doi: 10.2174/1566524015666150731100318
27. Kitaichi N, Namba K, Taylor AW. Inducible immune regulation following autoimmune disease in the immune-privileged eye. J Leukoc Biol. (2005) 77:496–502. doi: 10.1189/jlb.0204114
28. Lee DJ, Taylor AW. Following EAU recovery there is an associated MC5r-dependent APC induction of regulatory immunity in the spleen. Invest Ophthalmol Vis Sci. (2011) 52:8862–7. doi: 10.1167/iovs.11-8153
29. Lee DJ, Taylor AW. Both MC5r and A2Ar Are required for protective regulatory immunity in the spleen of post-experimental autoimmune uveitis in mice. J Immunol. (2013) 191:4103–11. doi: 10.4049/jimmunol.1300182
30. Lee DJ, Taylor AW. Recovery from experimental autoimmune uveitis promotes induction of antiuveitic inducible Tregs. J Leukoc Biol. (2015) 97:1101–9. doi: 10.1189/jlb.3A1014-466RR
31. Liu X, Zhang B, McBride JD, Zhou K, Lee K, Zhou Y, et al. Antiangiogenic and antineuroinflammatory effects of kallistatin through interactions with the canonical Wnt pathway. Diabetes. (2013) 62:4228–38. doi: 10.2337/db12-1710
32. McBride JD, Liu X, Berry WL, Janknecht R, Cheng R, Zhou K, et al. Transgenic expression of a canonical Wnt inhibitor, kallistatin, is associated with decreased circulating CD19+ B lymphocytes in the peripheral blood. Int J Hematol. (2017) 105:748–57. doi: 10.1007/s12185-017-2205-5
33. Namba K, Kitaichi N, Nishida T, Taylor AW. Induction of regulatory T cells by the immunomodulating cytokines alpha-melanocyte-stimulating hormone and transforming growth factor-beta2. J Leukoc Biol. (2002) 72:946–52. doi: 10.1189/jlb.72.5.946
34. Silver PB, Horai R, Chen J, Jittayasothorn Y, Chan CC, Villasmil R, et al. Retina-specific T regulatory cells bring about resolution and maintain remission of autoimmune uveitis. J Immunol. (2015) 194:3011–9. doi: 10.4049/jimmunol.1402650
35. Lee DJ, Preble J, Lee S, Foster CS, Taylor AW. MC5r and A2Ar deficiencies during experimental autoimmune uveitis identifies distinct T cell polarization programs and a biphasic regulatory response. Sci Rep. (2016) 6:37790. doi: 10.1038/srep37790
36. Malhotra S, Baba Y, Garrett KP, Staal FJ, Gerstein R, Kincade PW. Contrasting responses of lymphoid progenitors to canonical and noncanonical Wnt signals. J Immunol. (2008) 181:3955–64. doi: 10.4049/jimmunol.181.6.3955
37. Staal FJ, Langerak AW. Signaling pathways involved in the development of T-cell acute lymphoblastic leukemia. Haematologica. (2008) 93:493–7. doi: 10.3324/haematol.12917
38. Staal FJ, Sen JM. The canonical Wnt signaling pathway plays an important role in lymphopoiesis and hematopoiesis. Eur J Immunol. (2008) 38:1788–94. doi: 10.1002/eji.200738118
39. Yin H, Gao L, Shen B, Chao L, Chao J. Kallistatin inhibits vascular inflammation by antagonizing tumor necrosis factor-alpha-induced nuclear factor kappaB activation. Hypertension. (2010) 56:260–7. doi: 10.1161/HYPERTENSIONAHA.110.152330
40. Li P, Bledsoe G, Yang ZR, Fan H, Chao L, Chao J. Human kallistatin administration reduces organ injury and improves survival in a mouse model of polymicrobial sepsis. Immunology. (2014) 142:216–26. doi: 10.1111/imm.12242
41. Skarbez K, Priestley Y, Hoepf M, Koevary SB. Comprehensive review of the effects of diabetes on ocular health. Expert Rev Ophthalmol. (2010) 5:557–77. doi: 10.1586/eop.10.44
Keywords: autoimmune disease, autoimmune uveitis, kallistatin, experimental autoimmune uveitis, T cells
Citation: Muhammad F, Avalos PN, Mursalin MH, Ma J-X, Callegan MC and Lee DJ (2020) Kallistatin Attenuates Experimental Autoimmune Uveitis by Inhibiting Activation of T Cells. Front. Immunol. 11:975. doi: 10.3389/fimmu.2020.00975
Received: 29 January 2020; Accepted: 24 April 2020;
Published: 21 May 2020.
Edited by:
Anne Fletcher, Monash University, AustraliaReviewed by:
Igal Gery, National Eye Institute (NEI), United StatesCopyright © 2020 Muhammad, Avalos, Mursalin, Ma, Callegan and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Darren J. Lee, ZGFycmVuLWxlZUBvdWhzYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.