
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 10 June 2020
Sec. Primary Immunodeficiencies
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00900
This article is part of the Research Topic Advances in Primary Immunodeficiency in Central-Eastern Europe View all 12 articles
 Svetlana O. Sharapova1*
Svetlana O. Sharapova1* Małgorzata Skomska-Pawliszak2
Małgorzata Skomska-Pawliszak2 Yulia A. Rodina3Beata Wolska-Kuśnierz2Nel Dabrowska-Leonik2
Yulia A. Rodina3Beata Wolska-Kuśnierz2Nel Dabrowska-Leonik2 Bozena Mikołuć4
Bozena Mikołuć4 Olga E. Pashchenko5
Olga E. Pashchenko5 Srdjan Pasic6
Srdjan Pasic6 Tomáš Freiberger7,8
Tomáš Freiberger7,8 Tomáš Milota9,10Renata Formánková11,12Anna Szaflarska13
Tomáš Milota9,10Renata Formánková11,12Anna Szaflarska13 Maciej Siedlar13,14
Maciej Siedlar13,14 Tadej Avčin15,16
Tadej Avčin15,16 Gašper Markelj15,16Peter Ciznar17
Gašper Markelj15,16Peter Ciznar17 Krzysztof Kalwak18
Krzysztof Kalwak18 Sylwia Kołtan19,20
Sylwia Kołtan19,20 Teresa Jackowska21
Teresa Jackowska21 Katarzyna Drabko22
Katarzyna Drabko22 Alenka Gagro23,24
Alenka Gagro23,24 Małgorzata Pac2Elissaveta Naumova25
Małgorzata Pac2Elissaveta Naumova25 Snezhina Kandilarova25
Snezhina Kandilarova25 Katarzyna Babol-Pokora26
Katarzyna Babol-Pokora26 Dzmitry S. Varabyou27
Dzmitry S. Varabyou27 Barbara H. Barendregt28Elena V. Raykina29Tatiana V. Varlamova29Anna V. Pavlova29
Barbara H. Barendregt28Elena V. Raykina29Tatiana V. Varlamova29Anna V. Pavlova29 Hana Grombirikova7,8
Hana Grombirikova7,8 Maruša Debeljak15,16
Maruša Debeljak15,16 Irina V. Mersiyanova29
Irina V. Mersiyanova29 Anastasiia V. Bondarenko30Liudmyla I. Chernyshova30
Anastasiia V. Bondarenko30Liudmyla I. Chernyshova30 Larysa V. Kostyuchenko31Marina N. Guseva32
Larysa V. Kostyuchenko31Marina N. Guseva32 Jelena Rascon33Audrone Muleviciene33Egle Preiksaitiene34
Jelena Rascon33Audrone Muleviciene33Egle Preiksaitiene34 Christoph B. Geier35Alexander Leiss-Piller35
Christoph B. Geier35Alexander Leiss-Piller35 Yasuhiro Yamazaki36Tomoki Kawai36
Yasuhiro Yamazaki36Tomoki Kawai36 Jolan E. Walter37,38†
Jolan E. Walter37,38† Irina V. Kondratenko39†
Irina V. Kondratenko39† Anna Šedivá9,10†
Anna Šedivá9,10† Mirjam van der Burg40,41†
Mirjam van der Burg40,41† Natalia B. Kuzmenko42†
Natalia B. Kuzmenko42† Luigi D. Notarangelo36†
Luigi D. Notarangelo36† Ewa Bernatowska2†
Ewa Bernatowska2† Olga V. Aleinikova1†
Olga V. Aleinikova1†Background: Variants in recombination-activating genes (RAG) are common genetic causes of autosomal recessive forms of combined immunodeficiencies (CID) ranging from severe combined immunodeficiency (SCID), Omenn syndrome (OS), leaky SCID, and CID with granulomas and/or autoimmunity (CID-G/AI), and even milder presentation with antibody deficiency.
Objective: We aim to estimate the incidence, clinical presentation, genetic variability, and treatment outcome with geographic distribution of patients with the RAG defects in populations inhabiting South, West, and East Slavic countries.
Methods: Demographic, clinical, and laboratory data were collected from RAG-deficient patients of Slavic origin via chart review, retrospectively. Recombinase activity was determined in vitro by flow cytometry-based assay.
Results: Based on the clinical and immunologic phenotype, our cohort of 82 patients from 68 families represented a wide spectrum of RAG deficiencies, including SCID (n = 20), OS (n = 37), and LS/CID (n = 25) phenotypes. Sixty-seven (81.7%) patients carried RAG1 and 15 patients (18.3%) carried RAG2 biallelic variants. We estimate that the minimal annual incidence of RAG deficiency in Slavic countries varies between 1 in 180,000 and 1 in 300,000 live births, and it may vary secondary to health care disparities in these regions. In our cohort, 70% (n = 47) of patients with RAG1 variants carried p.K86Vfs*33 (c.256_257delAA) allele, either in homozygous (n = 18, 27%) or in compound heterozygous (n = 29, 43%) form. The majority (77%) of patients with homozygous RAG1 p.K86Vfs*33 variant originated from Vistula watershed area in Central and Eastern Poland, and compound heterozygote cases were distributed among all Slavic countries except Bulgaria. Clinical and immunological presentation of homozygous RAG1 p.K86Vfs*33 cases was highly diverse (SCID, OS, and AS/CID) suggestive of strong influence of additional genetic and/or epigenetic factors in shaping the final phenotype.
Conclusion: We propose that RAG1 p.K86Vfs*33 is a founder variant originating from the Vistula watershed region in Poland, which may explain a high proportion of homozygous cases from Central and Eastern Poland and the presence of the variant in all Slavs. Our studies in this cohort of RAG1 founder variants confirm that clinical and immunological phenotypes only partially depend on the underlying genetic defect. As access to HSCT is improving among RAG-deficient patients in Eastern Europe, we anticipate improvements in survival.
Recombination-activating gene 1 (RAG1) and 2 (RAG2) encode lymphoid-specific proteins that are expressed during the early stages of T-cell and B-cell development and initiate the process of V(D)J recombination by introducing DNA double-strand breaks (DSBs) for recognizing millions of possible antigens (1). Genotype–phenotype correlation is strong, as null variants of RAG1 and RAG2 genes result in the T-B- severe combined immune deficiency (SCID) phenotype, whereas hypomorphic RAG variants have been associated with distinct clinical entities including Omenn syndrome (OS) and combined immunodeficiency with granuloma and/or autoimmunity (CID/G-AI) with herpesvirus infections and lymphoproliferation (2–5) and with selective polysaccharides antibody deficiency (6). Furthermore, in the era of widespread next-generation sequencing, RAG deficiency is being identified among adults with variants of antibody deficiencies with a frequency of 1 in 500 patients (7).
The RAG1 and RAG2 genes are polymorphic. Described clinical phenotypes are associated with a variety of variants including non-sense, frameshift, in-frame deletion or insertion, and missense variants of the RAG1 and RAG2 genes that affect various domains of the proteins (1). Among numerous RAG variants, some of them were observed in a Jewish population with a high rate of consanguineous marriages (8). In our previous report of 11 OS patients from the East Slavic regions, we described the high rate of c.256_257delAA (p.K86Vfs*33) in the RAG1 gene (n = 4, 50%) (9). This variant was also observed in Polish and Serbian patients with OS and SCID phenotypes (10–13), which suggests a founder effect.
Currently, there is no published systematic evaluation of Slavic patients with RAG deficiency. In this report, we aim to describe the genetic landscape of RAG deficiency by estimating the incidence, genetic diversity, clinical and immunological presentation, and survival rate of a large cohort of Slavic patients. In addition, we focus on RAG1 p.K86Vfs*33 as a candidate founder variant among Slavic patients by studying the geographic distribution of allele and genotype frequencies of RAG1 p.K86Vfs*33 mutation among patients in major Slavic populations.
Patients with pathogenic RAG variants were recruited retrospectively for this study through extensive collaboration with clinical immunologists who collected the data of national primary immunodeficiency (PID) registries from East Slavs (Russia, Belarus, and Ukraine), West Slavs (Poland, Czech Republic, and Slovakia), and South Slavs (Serbia, Slovenia, Montenegro, Croatia, and Bulgaria). The patients were divided into ethnic groups (East, West, and South Slavs) according to their country of origin.
Informed consent forms were signed by the parents as requested and approved by the institutional review boards of various institutions involved. The protocol of study was approved by the institutional review board of Belarusian Research Center for Pediatric Oncology, Hematology, and Immunology (IRB0012-2015).
A detailed questionnaire was completed by the physicians including demographic data (gender, country, place of birth, and year of birth), variants, and clinical data (age at disease manifestation, age at diagnosis, clinical and immunologic phenotype, and outcome).
Corresponding clinicians from each country assigned patients to one of the following subgroups—SCID, OS, atypical SCID (AS), or combined immunodeficiency (CID)—on the basis of clinical and immunologic phenotype, and age at manifestation.
The Primary Immunodeficiency Treatment Consortium (PIDTC) diagnostic subdivides SCID into three categories: typical SCID, atypical SCID, and OS, based on total T-cell count, lymphocyte proliferation, presence of maternal T cells, characteristic phenotypic features, and gene defects. CID was classified by late and mild clinical presentation according to published criteria (4, 14). Roifman et al. distinguished CID from SCID based on a total CD3+ T-cell count of >500/μl (15). The 2019 ESID criteria for diagnosis of CID requires a symptomatic patient (infections, immune dysregulation) or history of affected family members with immune phenotype of two of the four parameters (low CD3 or CD4 or CD8 T cells, low naïve CD4 and/or CD8 T cells, elevated γδ T cells, and reduced proliferation to mitogen or TCR stimulation). This ESID 2019 criterion does not discuss underlying genetic defects for CID patients (16). The IUIS 2020 classification does list genetic defects for CID but fails to include RAG deficiency in this category (17).
To estimate the number of newborns with RAG variant in each country, the information about affected siblings in families with RAG variants was collected through hospital or personal records of affected families.
Mapping was performed by the ArcGIS 10.5 program on the map of Central–Eastern Europe and Russia.
Gene sequencing was performed using standard techniques (panel-based, Sanger) in local laboratories in Europe. The reference DNA and protein sequences for RAG1 are from NIH RefSeq NM_000448.2 and NP_000439.1 and those for RAG2 are from NM_001243785.1 and NP_001230714.1, respectively.
For single-allele mutation, assay is based on a v-Abl Rag1/Rag2−/− pro-B cell line containing a single pMX-INV integrated cassette (18).
TRECs and KRECs quantity was measured by RQ-PCR using plasmid standards, and ALB was taken as an internal control according to manufacturer instructions.
Allele frequencies were compared by chi-square test or Fisher's exact tests. Kaplan–Meier curve was computed for survival post-hematopoietic stem cell transplant (HSCT). p < 0.05 was considered significant. Statistical analysis was performed using GraphPad Prism version 6.0 (GraphPad Software Inc., San Diego, CA, USA).
We retrospectively collected and studied 82 patients with RAG deficiency from 67 families in 12 countries, born in the period of 1992–2018. Thirty-five of 82 (43%) patients were reported previously (9–13, 19–23). Distribution was even between males (n = 40, 49%) and females (n = 42, 51%). The median age at diagnosis of RAG was 1.3 years (range, 1 day to 24 years).
At the time of manuscript submission (March 2020), 43 of the 82 patients died (52%, 8 out of 43 after HSCT). All patients alive received HSCT (ranging from 1 to 18 years) except three females with LS/CID presentation from Poland (42_f), Bulgaria (54_f), and Czech Republic (55_f) who are alive without transplantation at the ages of 3, 6, and 25 (Supplementary Tables E1 and E2 in this article's Online Repository).
The largest group of patients (n = 40, 48.8%) was West Slavs from Poland (n = 29, n = 1 from Lithuania, Polish origin), Czech Republic (n = 8) and Slovakia (n = 2). The second largest group was East Slavs (n = 30, 36.5%) from Russia (n = 24), Ukraine (n = 4), and Belarus (n = 2). The smallest group was South Slavs (n = 12, 14.6%) from Serbia (n = 6), Slovenia (n = 3), Croatia (n = 1), Montenegro (n = 1), and Bulgaria (n = 1).
All patients have genetically confirmed pathogenic RAG1 or RAG2 variants. RAG1 variants were detected in 67 patients in 54 families (82%) and RAG2 variants were detected in 15 patients from 13 families (18%). Nineteen children (23%) out of 16 families and 8 children (10%) out of 7 families were homozygotes while 47 patients (59%) out of 39 families and 8 patients (10%) out of 6 families were compound heterozygotes for RAG1 and RAG2 variants, respectively (Supplementary Tables E1 and E3).
Clinical phenotypes, as determined by attending clinician following immunological and clinical guidelines, included SCID (n = 20), OS (n = 37), AS (n = 21), and CID (n = 4) with highly mixed immune phenotypes (different numbers of T cells in all groups and domination of B+ phenotype in AS group); however, in vitro relative recombinase activity showed good correlation between genotype and clinical cathegories. (Figure 1C, and Supplementary Tables E1 and E3 in this article's Online Repository). Four (10%) out of 41 vaccinated SCID patients with RAG1/2 variants developed BCGosis infection, and 12 (3%) BCGitis were noted. As expected, the AS group had increase frequency of autoimmune cytopenias and history of herpesvirus infections (Figure 1A,B and Supplementary Table E2).
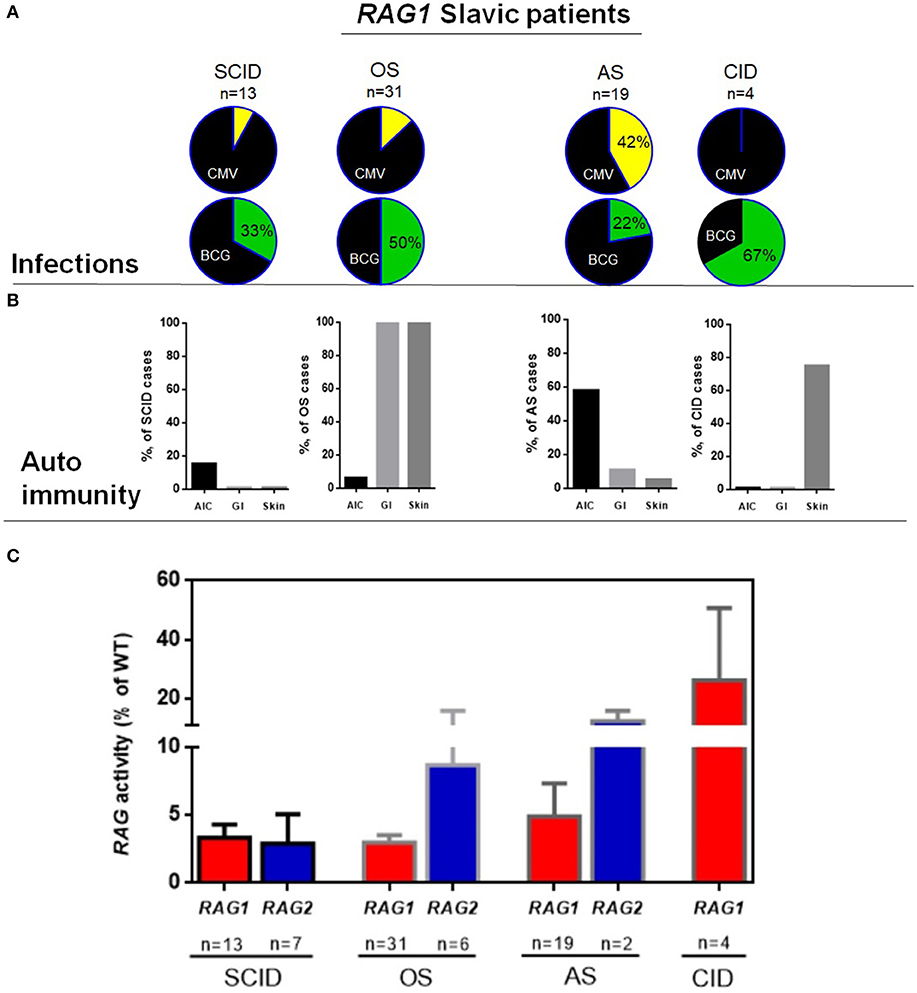
Figure 1. Characteristics of RAG Slavic cohort. (A) Infectious complications in RAG1 patients, upper row CMV in four groups, lower row BCG complications among patients, who received BCG vaccination. (B) Frequency of autoimmunity in 4 groups of RAG1 deficiency. (C) Recombinase activity of RAG1/RAG2 variants for the 82 patients divided into four major clinical presentations for RAG1 and three clinical presentations for RAG2.
Patients with RAG1 variants are described in Supplementary Tables E1 and E2. Most patients presented with OS (46%) (31 children from 26 families) followed by AS (28%) (19 patients from 16 families), SCID (19%) (13 patients from 11 families), and CID (6%) (4 patients from 4 families).
Patients diagnosed as having OS and SCID were characterized by classical clinical and immunologic phenotype (Supplementary Table E1). Their clinical phenotype allowed for diagnosis before the age of 1 year. On the other hand, the median age at the time of diagnosis in AS and CID was 4 years (± 6.02 years) and ranged from 5 months to 24 years (Supplementary Tables E1 and E2).
In the AS group, infectious complications were notable for high prevalence of CMV infection (n = 9, 42%) in both localized (n = 2, retinitis) and systemic forms (n = 7). Among patients who received BCG vaccination, only a fraction (4 of 18, 22%) developed BCG infections (five with BCGitis and one with BCGosis) (Supplementary Table E2 and Figure 1A).
Autoimmune findings in the AS and CID group were observed in 12/19 and 4/4 of patients (Supplementary Table E2), with autoimmune cytopenia being most prevalent in AS (n = 11/19, 58%) including autoimmune hemolytic anemia (n = 8), isolated immune thrombocytopenia (ITP) (n = 2), and multiple lineage cytopenia (n = 2). Autoimmune colitis was diagnosed in 9% (2/19) of AS patients with autoimmunity. In the CID group, one patient had skin granuloma, one developed vasculitis, and one had vitiligo (Supplementary Table E2 and Figure 1B).
Immunological phenotype of the AS group is summarized in Supplementary Table E1. Curiously, high fraction of the patients had preserved fraction of B cells (>65% in 13 of 19 patients) at the time of diagnosis. However, 2 of 7 (28%) patients had complete absence of KRECs. This suggests abnormal B cell maturation even in the setting of preserved fraction of total B cells. Four patients presented with normal IgG level, and one had IgA deficiency at the age of 4 years that progressed to agammaglobulinemia with age.
In this study, we additionally determined the recombination activity of patient's RAG1/RAG2 mutant proteins. Of the RAG variants, 38 variants were unique, including frameshift (n = 7), non-sense (n = 4), and missense (n = 27), and 23 RAG1 mutations were new in our cohort, and for all of them, recombination activity was determined (Supplementary Tables E1 and E2). We had the possibility to demonstrate that mutations associated with SCID and OS had lower residual activity than mutations detected in patients with less severe clinical presentations (AS and CID) (Figure 1C). Nevertheless, genotype–phenotype correlation was less evident in our cohort; we have not found significant differences between OS/SCID and AI/CID in clinical and laboratory markers. Among the patients with homozygous RAG1 p.K86Vfs*33 (c.256_257delAA) variant (n = 18), the clinical and immunological presentation was highly diverse, 4 patients presented as OS, 1 as SCID, and 13 as AS (Supplementary Tables E1 and E2); these findings were supported by Supplementary Table E1 Pasic et al. (12). This is suggestive of the strong influence of other genetic and/or epigenetic factors in shaping the final clinical and immunological phenotype.
Homozygous RAG1 variants were detected in 18 families, but consanguinity was identified only in 4 (22%) [3 families from Poland (2a_m, 2b_m; 39_f; 41a_m, 41b_m) and 1 from Serbia (44a_m, 44b_f, 44c_m)].
The most frequent genetic RAG1 anomaly among patients of all groups and in 10 out of 11 countries was RAG1 p.K86Vfs*33 (c.256_257delAA) (Supplementary Table E1). This deletion occurred in one or both alleles in 22 out of 30 patients with OS, in 7 out of 13 SCID patients, and in 16 out of 19 patients with AI and 2/4 CID phenotype. Because of the enrichment of the p.K86Vfs*33 variant among all cohorts, we calculated the frequency of this allele among East, West, and South Slavic patients (Figures 2A–F) and their families (data not shown). Nine families had two or three children with immunodeficiency, and therefore, we conducted a separate analysis of families and analyzed frequency between countries counting one case per family.
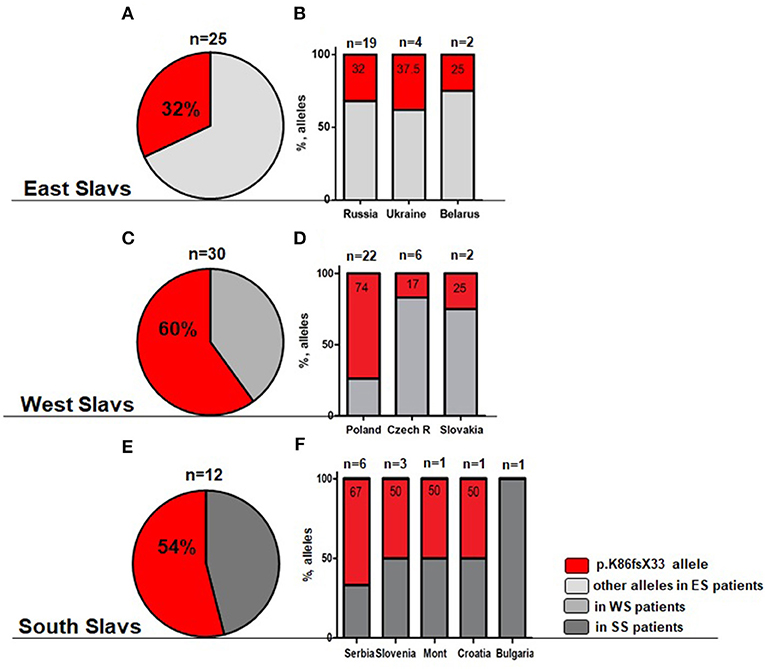
Figure 2. Overall frequency of c.256_257delAA allele (p.K86Vfs*33) among all cohort of RAG1 patients. (A,B) Light gray—East Slavs (ES); (C,D) Gray—West Slavs (WS); (E,F) Dark gray—South Slavs (SS); (B,D,F) Percentage of p.K86Vfs*33 allele in studied countries. In Montenegro and Croatia, only one patient was studied.
Patients of South and West Slavic origin demonstrated the highest frequency of c.256_257delAA allele (Figures 2C,E). We analyzed the relative frequency of this predominant allele among Slavic patients and families. The differences in the allele frequency of c.256_257delAA frequency among the patients (χ2 = 8.4; p = 0.015) and the families (χ2 = 22.8; p < 0.0001) of the East/West/South Slavs proved to be statistically significant. In pairwise comparisons, the West Slavic population had a higher relative frequency of this allele than the East Slavic (families p = 0.0006; patients p = 0.006), but it did not differ from the South Slavic population (families p = 0.14; patients p = 0.81).
Besides the analysis of different Slavic cohorts, we calculated the contribution of each country to the overall frequency of RAG1 c.256_257delAA allele and showed the highest occurrence in Poland (Figure 2D). RAG1 c.256_257delAA allele (p.K86Vfs*33) was present in over 50% of RAG1-deficient patients from Serbia, Slovenia, Montenegro, and Croatia, but extrapolating this finding to the whole South Slavic population is premature because only one patient was from Croatia and Montenegro (Figure 2F).
Among East and South Slavs, we observed only two families (one from Ukraine and one from Serbia) where p.K86Vfs*33 was detected in a homozygous constitution, but consanguinity was proved only in the Serbian patients. The Polish cohort of families showed 12 homozygous genotypes among 18 RAG1 positive families, with only three cases of proven consanguinity.
Due to the high frequency of the RAG1 p.K86Vfs*33 variant in Poland and homozygous genotypes, we studied its geographical distribution in West, East, and South Slavic populations. Our findings are illustrated on the map of East Europe and Russian Federation based on the patient's place of origin (one patient from each family) (Figure 3).
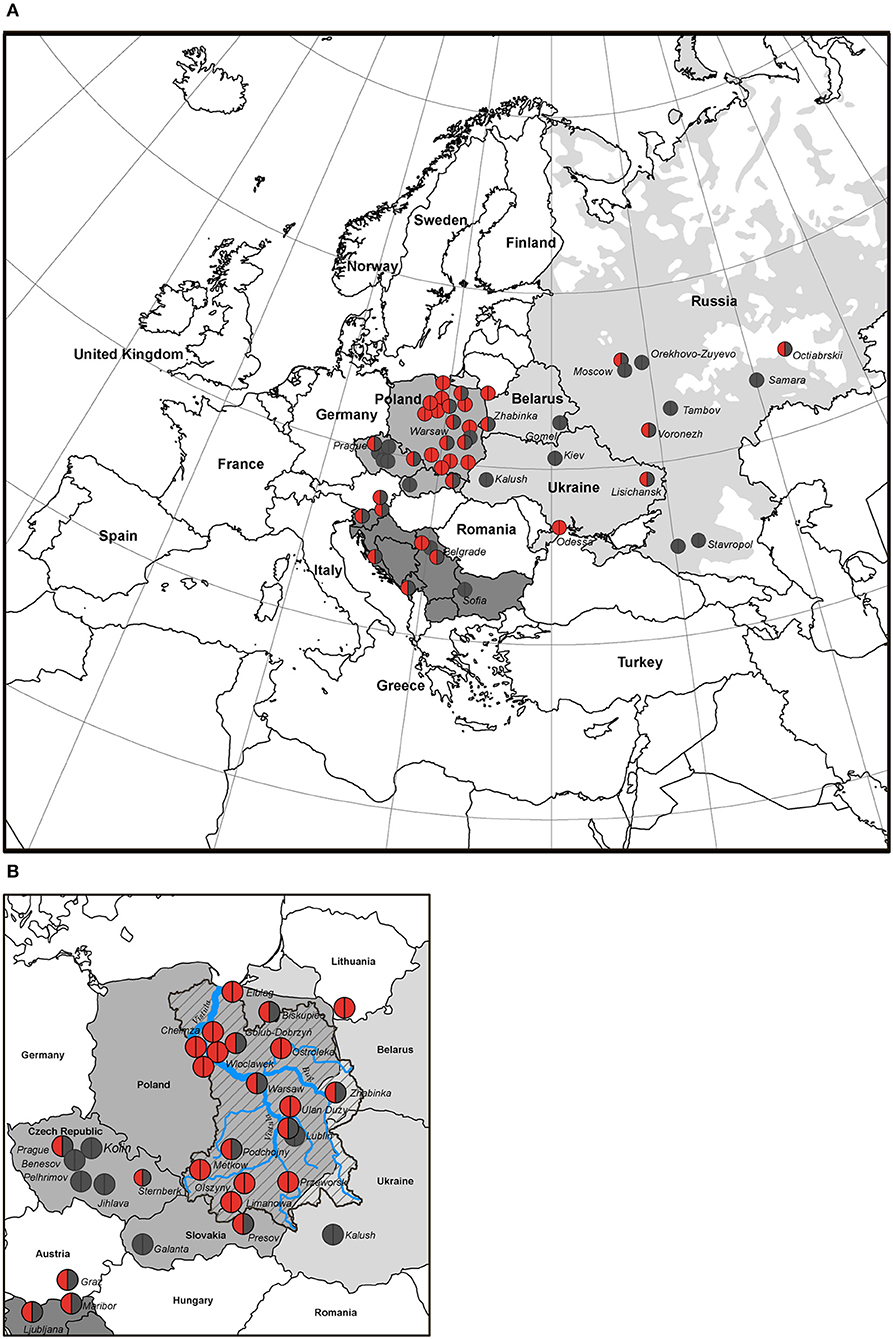
Figure 3. Distribution map of families with RAG1 p.K86Vfs*33 variant in Slavic countries (50 of 55 families). (A) The birthplace of the patients is indicated by the location of the circles; homozygous p.K86Vfs*33 variant is represented by red circles; heterozygous p.K86Vfs*33 variant is half red/half gray, and other variants are gray. (B) Map of the Western Slavs; the Vistula River basin is overlaid on the map of Poland. The blue line is the Vistula River, a zone marked by an oblique black line—the Vistula River basin, the geographic area coincides with the region of the largest concentration of families where patients with p.K86Vfs*33 homozygous variants were born.
We studied the distribution of RAG1 p.K86Vfs*33 variant in 11 out of 13 Slavic ethnic groups and it was widespread and found in 10 out of 11 countries except Bulgaria (Figure 3). The occurrence of p.K86Vfs*33 was observed from Ljubljana (Slovenia) in the West to Vladivostok in the East and from Moscow in the North to Cetinje (Montenegro) in the South (Figures 3, 4). One patient who originated from Slovenia was born in Austria (Graz), but after delivery, he was treated in the homeland of Slovenia (Figure 3).
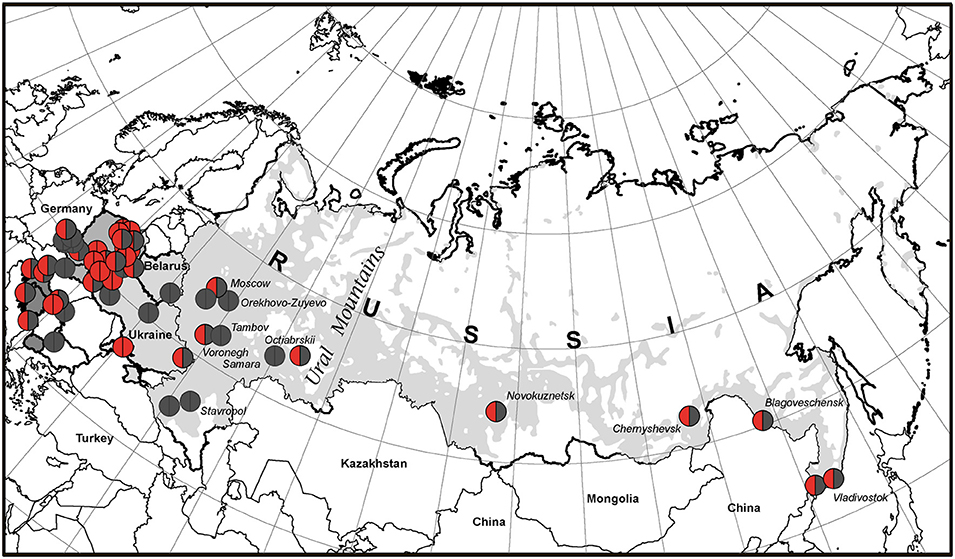
Figure 4. The geographic picture of the p.K86Vfs*33 distribution among East Slavs. Light gray zone on the map of Russia is the territory of predominant Russian settlement (24).
The geographic center of the presumed origin of the RAG1 p.K86Vfs*33 variant corresponds to the Vistula watershed area of Central and Eastern region in Poland in which the variant was the most frequent (Figure 3B) and suggestive of shared origin for the p.K86Vfs*33 alleles in all Slavic populations.
Due to a small number of cases, gradual decrease in the prevalence of p.K86Vfs*33 among East and South Slavs could not be determined, but it is clearly shown that heterozygous families are spread among other Slavic countries from the Polish area of homozygosity (Figure 3).
The broadest area of East Slavs in Russia is located mainly in the European part of the Russian Federation (25). In our cohort of patients from Russia, the majority (11/19) is concentrated on the territory west of the Ural Mountains, but 8 patients from 5 families were born in Siberia and the Russian Far East, and all inherited our studied variant (Figure 4). This finding does not contradict our assumption that p.K86Vfs*33 has a Slavic origin. According to the map of the resettlement of peoples in Russia, most Russians live in the central part or in the south and northwest of Russia as well as in the Ural region. To the east of the Ural Mountains, Russians live predominantly near the southern border of Russia, which is shown in Figure 3 and in Figure 4 as a light gray area on the white map of the Russian Federation.
Patients with RAG2 variants were detected only in three countries among West (Poland and Czech Republic) and East Slavs (Russia) (Supplementary Table E3). Among 15 patients from 13 families, 7/15 presented as SCID, 6/15 presented as OS, and 2/15 presented as AS. Two Polish females manifested as atypical SCID/CID and one had a sister with SCID (8a_ f). In one Russian family, one male presented as OS (3b_ m) and one presented as SCID (3a_ m).
Females with late manifestation of RAG2 variant presented with encephalitis, HSV, bronchitis, pneumonia, neutropenia and pneumonia (8b_f), local BCGitis, and failure to thrive (12_f).
Interestingly, the obtained data showed that 53% (8/15) of patients had homozygous variants in the RAG2 gene, but consanguinity was established only in one family history (1_m). Despite the marked diversity of the genotypes, there are some repeated variants, p.Y434H in Russian, p.R229Q in Polish and Czech children (in homo and in heterozygous state), and p.W453R only among Polish patients. However, an obvious dominating region among variants was not established due to a small number of the studied cases.
The RAG2 variants in Slavic cohort showed a wide range of recombination activity (Supplementary Table E3). Mutations associated with severe combined immune deficiency and OS had lower activity than those detected in patients with atypical SCID presentations (Supplementary Table E3 and Figure 1D). Our data support genotype–phenotype correlation in RAG2 deficiency in Slavic children (18).
We examined the incidence of all types of RAG deficiency together (RAG1/RAG2 genotype; OS/SCID/AS/CID phenotype) in each country during different periods. The average incidence of RAG deficiency was calculated based on the number of all diagnosed patients and their confirmed siblings and the number of live births in studied countries during the period from the year of the first registered patient in the country to the last one (Table 1). The average 10-year incidence was estimated by the number of all patients of Slavic origin diagnosed since 2008 to 2017 in each country (Table 1, last column).
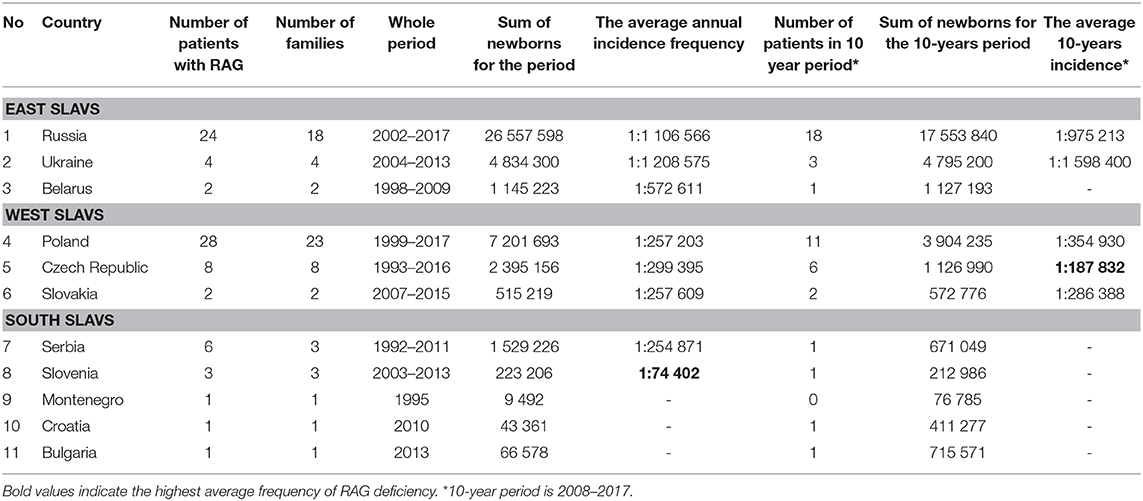
Table 1. Average annual incidence of RAG deficiency in Slavic countries.
The obtained data showed that frequency of diagnosed RAG deficiency ranged from 1 per 75,000 in Slovenia to one per 1,100,000–1,600,000 live births in Russia and Ukraine.
A wide range in frequency among Slavic countries suggest underdiagnosis of atypical late-onset cases. We compared the two largest RAG cohorts from Russia (n = 24) and Poland (n = 30). In further analysis, we graphically illustrated the number of diagnosed patients in the two countries (the age at diagnosis was added to the year of birth) and showed that Russia achieved great success in identification of RAG patients in the last 8 years (Figure 5A, lower). Exponential growth (Figure 5A, upper) was observed; 19 out of 24 RAG patients were diagnosed from 2011 to 2017, and their geography covered the entire country (Figure 4). Poland, as the largest West Slav country, uses advanced immunologic diagnostics services and one to three patients have been confirmed approximately each year since 1999 (Figure 5B).
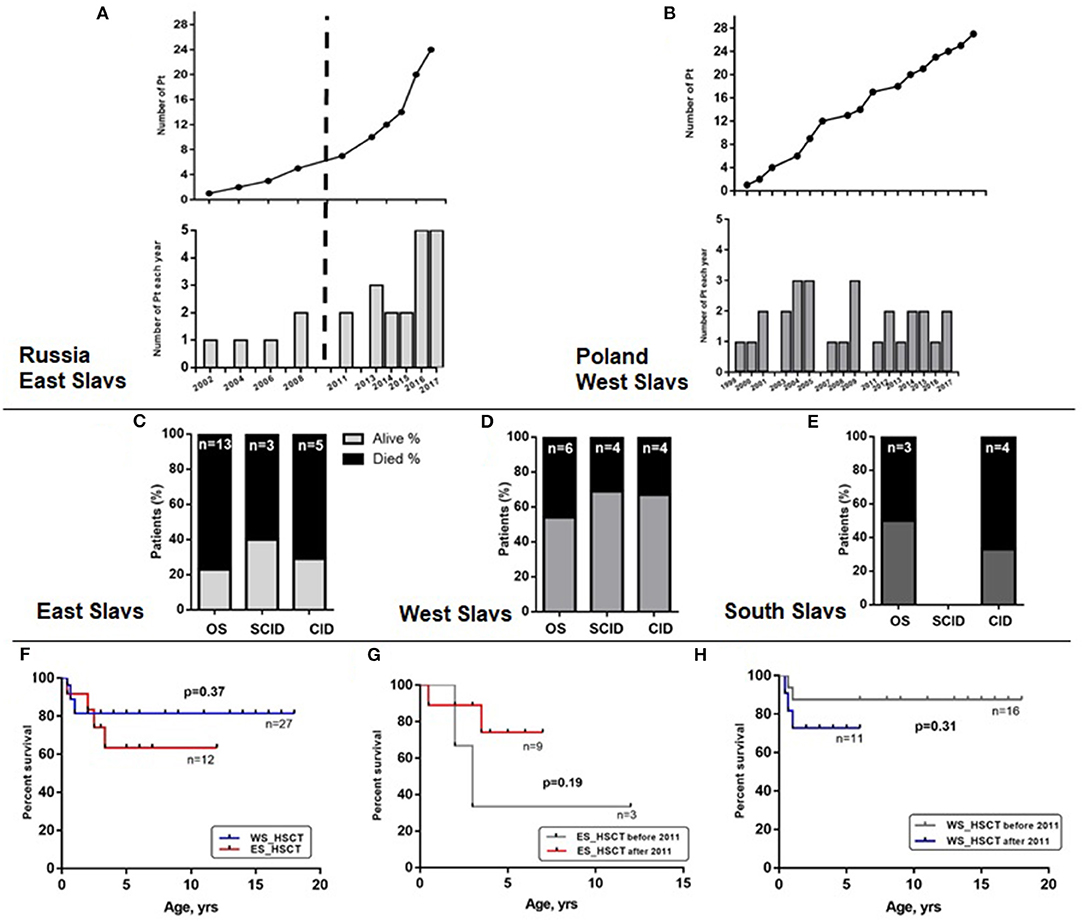
Figure 5. Identification and survival of Slavic patients. Number of patients diagnosed in Russia (A) and Poland (B). Percentage of dead patients (black region) among clinical groups of patients with RAG variants and East (C), West (D), or South Slavic (E) origin. Overall survival displayed as Kaplan–Meier survival curve of WS and ES patient groups with HSCT (F). Overall survival curve of ES (G) and WS (H) patient transplanted before and after 2011.
Despite an increased awareness for SCID and OS variants and improved access for HSCT in most Slavic countries, the mortality rate of RAG patients is still high in Eastern Europe. We studied relative mortality rate among OS/SCID/CID groups of patients with RAG deficiency (Figure 5C—ES, Figure 5D—WS, and Figure 5E—SS). Neither comparison of transplanted patients in two groups (WS and ES) as biggest cohorts revealed any statistically significant difference in survival (Figure 5F). Additionally, we compared the results of HSCT before and after 2011, due to changing the protocols in last years and we were able to show that survival of East Slavic patients became much better after 2011 and reached the same percentage as in West Slavs (Figures 5G,H). We suggest that poor outcome in HSCT before 2011 (three out of five died after HSCT) in East Slavs was due to delayed diagnosis in AS/CID patients and associated complications (chronic infections and immune dysregulation) (Supplementary Figure E1C). Patients with OS were transplanted with a high rate of survival in ES/WS/SS (Supplementary Figure E1B) and with SCID in ES/WS countries (Supplementary Figure E1D).
Several cohort studies with international collaborators are published, and a recent review of literature enumerate the number of published cases with RAG deficiency over 400 (2, 4, 7–9, 13, 14, 23). Among the population with high rate of consanguity, founder variants for RAG genes were proposed among the Amish and in the Middle East (26–28).
In this study, we present the largest geographically defined cohort of patients with RAG deficiency. Our patients are predominantly of Slavic origin from 11 European countries retrospectively collected in a 27-year period (1992–2018). In our current and previous study of patients with RAG1 p.K86Vfs*33 (c.256_257delAA), we confirmed that different clinical phenotypes can been found in patients with identical variants even within the same family (9, 13). Family studies for homozygous RAG1 p.S480G and RAG2 p.M459L were also reported in consanguineous marriages of Arabic descent (29, 30). Furthermore, similar non-familiar findings were reported with patients with RAG1 c.519delT (p.E174SfsX27) (13). Among all these reports, our study with 18 homozygous patients RAG1 p.K86Vfs*33 (c.256_257delAA) is the largest in the literature within a geographically linked population.
The data from California's SCID newborn screening identified RAG1/2 variants in 28.6% of SCID/OS cases, with an incidence of about 1:250,000 (31). In populations with domination of consanguineous marriages, RAG deficiency was diagnosed in 48% (21/44) of Turkish (32) and 51% (19/37) of Iranian SCID patients (33). The incidence of RAG deficiency is difficult to establish, due to the highly variable phenotype, late age of manifestation of hypomorphic variants, and different level of diagnostics capacities in different countries (5). Recently, it was identified that RAG deficiency among adults with a variety of antibody abnormalities occurs with a frequency of 1 in 500 patients with any variant of antibody deficiency (7). That is much higher than the expected number of RAG1/2 SCID or OS cases. Based on our data reported in this study, we determined that the minimal incidence of RAG deficiency varied from 1:189,000 in the Czech Republic to 1:1,200,000 in Ukraine; the average was 1 per 190,000–300,000 live newborns.
It is still unclear if all RAG patients with AS/CID phenotype (with residual recombination activity) can be detected by newborn screening (NBS) (14). Retrospective testing of NBS card showed absence of TRECs in case reports (34). Current observation showed that KREC level can support and underlying RAG deficiency is relevant and further justifies the consideration of KREC in universal NBS for SCID and AS/CID. Our retrospective data suggest that most of the patients with late-onset forms of RAG deficiency (CID) are likely missed, but the level of discovery of SCID/OS patients is reasonable in most Slavic countries. There is a great need for early identification of these patients before infectious and autoimmune complications occur. The availability of next-generation sequencing and the access to HSCT service make it possible to improve the outcome for this group.
In the present study, 27% of patients were classified by attending physicians as atypical SCID. This phenotype was first described by Schuetz C. et al. in 2008, which they named “atypical/leaky SCID.” During the last 10 years, there is growing awareness of these patients (2, 4, 7, 9, 13, 19, 20, 22, 30). Distinct clinical features include inflammatory and/or autoimmune complications. Autoimmune cytopenias (AIC) were the most frequent in our cohort, found in 58% (11/19 RAG1 patients with AS phenotype). In the general population, ITP is the most common AIC. This is comparable to published data (23, 35). Regarding immunological features, although one third of the RAG1 patients had absence of very low B cell count, over two thirds of the cases had presence of low to normal B cells and immunoglobulins that progressive decline with age. Of note, many of our patients did not have full immune evaluation.
Slavs are the largest ethno-linguistic group in Europe (25, 36). In 10 out of 11 countries, the majority of patients carried p.K86Vfs*33 in RAG1 in homo or heterozygous state. Based on our data reported in this study, we established the geographic center of the origin of this specific RAG1 allele; it corresponds to the Vistula watershed area in Central and Eastern Poland (Figure 3B). Since SCID (Bernatowska E. unpublished data) and RAG2 patients are evenly diagnosed in Poland, the lower frequency of absence RAG1 variant p.K86Vfs*33 in the Western part of Poland may be likely related to distance from the geographic center of the founder variant or due to lower medical care in this region. Together, these data suggest that it is most likely that the founder RAG1 variant p.K86Vfs*33 is of a Slavic origin.
Location of the Slavic homeland prior to their great expansion in the fifth to sixth centuries is one of the key questions of European history. Although it is assumed that prehistorically the original habitat of Slavs was Asia, from which they migrated in the third or second millennium BC to populate parts of Eastern Europe, a debate concerning the European homeland of Slavs seems to remain unsolved. Different theories concerning the Slavs' geographic origin based on archaeological, anthropological, and/or linguistic data have been formulated. One places the cradle of Slavs in the watershed of the Vistula and Oder rivers (present-day Poland), and the other locates it in the watershed of the middle Dnieper (present-day Ukraine) (32). The obtained data strengthen the hypothesis that the Slavs could originate from the watershed of the Vistula due to the high number of families with homozygous variant from that region (Figure 3B). However, at present, it is impossible to deny that the variant could be found in the watershed of the middle Dnieper due to low number of cases and low incidence of RAG diagnostics in the Ukraine. Additionally, the only homozygous variant among East Slavs was found in the Ukraine (Odessa), where the parents denied their consanguinity. Collectively, these data indicate that p.K86Vfs*33 could be established as a “Slavic” RAG1 variant or a founder variant.
Population migrations in Europe have led to the distribution of ethnic groups and cultures, and consequently to genetic mixing. Migrations, together with other factors, have also determined the prevalence of genetic variants among other populations (25, 36). In the GnomAD database, this deletion was found in heterozygous state in six people of European population (non-Finnish) of 282,486 individuals [https://gnomad.broadinstitute.org/variant/11-36595109-TAA-T]. Reviewing published cases, we established that patients with homo- and heterozygous p.K86Vfs*33 RAG1 variants were detected in European cohorts (2, 13, 37–40). A recent Turkey study reported one patient with homozygous p.K86Vfs*33 in RAG1 among the cohort of 44 SCID patients (32) and a SCID infant with EBV-positive B-cell lymphoma of the liver with homozygous p.K86Vfs*33 was diagnosed in Austria (40). It should be noted that in all these previous studies, the ethnic origin of the patients was not documented.
Our study highlight that a founder RAG1 variant is frequently present in Eastern Europe, especially in the watershed area of the Vistula. As cases accumulate in a specific region in Poland, screening of carriers of pathogenic RAG variants may be of high importance and could be offered in case of parental interest to reduce the incidence of RAG deficiency in this population. Our study underscores the importance of region of birth and ethnic background in genetic diagnosis. Further studies are needed to explore the carrier frequency of p.K86Vfs*33 in Central and Eastern Poland, as well in otherSlavic countries.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Informed consent forms were signed by the parents as requested and approved by the institutional review boards of various institutions involved. The protocol of study was approved by the institutional review board of Belarusian Research Center for Pediatric Oncology, Hematology and Immunology (IRB0012-2015). Written, informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
SS designed the research, collected data, interpreted, analyzed the results and wrote the manuscript. DV prepared the maps. YY and TK studied RAG activity. MS-P, YR, BW-K, ND-L, BM, OP, PS, TF, TM, RF, AS, MS, TA, GM, PC, KK, SKo, TJ, KD, AG, MP, EN, SKa, KB-P, BB, ER, TV, AP, HG, MD, IM, AB, LC, LK, MG, JR, AM, EP, CG, and AL-P provided patient's information. JW, AS, TF, LN, OA, NK, and MB guided the writing of the manuscript. All authors have read and approved the contents of the manuscript.
LN was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health. The Jeffrey Modell Foundation partly funded the SCID project through grant CHILDREN (SS). This work was supported by the National Institutes of Health (sub-R01AI100887-05 to JW), Robert A. Good Endowment, and University of South Florida (to JW). The genetic studies in Slovenia were partially supported by the Slovenian Research Agency Grant L7-8274.
MS-P, BW-K, ND-L, MB, AS, MS, KK, SK, TJ, KD, MP, KB-P, EB have served as a consultants, and/or speakers, and/or principal investigator/investigators, and/or have provided expert testimony for Baxter/Baxalta/Shire, CSL Behring, Grifols, Biotest, Octapharma and have received one or more payments for above activities. TA served as a speaker for Octapharma and Shire/Takeda.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer YL declared a past co-authorship with one of the authors to the handling editor.
We sincerely thank all our patients and their family members who made this study possible and the referring doctors for their help. We thank Professor Anna Shcherbina for critical input in the data analysis and discussion, Dr. Vasily Pankratov for recommendations on population genetics, Dr. Joseph Dasso for assisting in editing the manuscript, and Ms. Irina Guryanova for technical assistance. We also thank Professor Krystyna Chrzanowska for valuable comments concerning genetic issues of PID patients in Poland.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00900/full#supplementary-material
1. Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol. (2016) 16:234–46. doi: 10.1038/nri.2016.28
2. Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol. (2014) 133:1099–108. doi: 10.1016/j.jaci.2013.10.007
3. Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med. (2008) 358:2030–8. doi: 10.1056/NEJMoa073966
4. Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest. (2015) 125:4135–48. doi: 10.1172/JCI80477
5. Kumanovics A, Lee YN, Close DW, Coonrod EM, Ujhazi B, Chen K, et al. Estimated disease incidence of RAG1/2 mutations: a case report and querying the exome aggregation consortium. J Allergy Clin Immunol. (2017) 139:690–2.e3. doi: 10.1016/j.jaci.2016.07.027
6. Geier CB, Piller A, Linder A, Sauerwein KM, Eibl MM, Wolf HM. Leaky RAG deficiency in adult patients with impaired antibody production against bacterial polysaccharide antigens. PLoS ONE. (2015) 10:e0133220. doi: 10.1371/journal.pone.0133220
7. Lawless D GC, Farmer JR, Lango Allen H, Thwaites D, Atschekzei F, Brown M, et al. Prevalence and clinical challenges among adults with primary immunodeficiency and recombination-activating gene deficiency. J Allergy Clin Immunol. (2018) 141:2303–6. doi: 10.1016/j.jaci.2018.02.007
8. Dalal I, Tasher D, Somech R, Etzioni A, Garti BZ, Lev D, et al. Novel mutations in RAG1/2 and ADA genes in israeli patients presenting with T-B-SCID or omenn syndrome. Clin Immunol. (2011) 140:284–90. doi: 10.1016/j.clim.2011.04.011
9. Sharapova SO, Guryanova IE, Pashchenko OE, Kondratenko IV, Kostyuchenko LV, Rodina YA, et al. Molecular characteristics, clinical and immunologic manifestations of 11 children with omenn syndrome in East slavs (Russia, Belarus, Ukraine). J Clin Immunol. (2016) 36:46–55. doi: 10.1007/s10875-015-0216-7
10. Szaflarska A, Noworolska APD, Kowalczyk D, Zembala M, van der Burg M, van Dongen JJM. Omenn's syndrome in cousins: different clinical course and identical RAG1 mutation. Pediatria Polska. (2009). 84:367–72. doi: 10.1016/S0031-3939(09)70126-X
11. Szaflarska A, Rutkowska-Zapala M, Kotula M, Gruca A, Grabowska A, Lenart M, et al. Mutation c.256_257delAA in RAG1 gene in polish children with severe combined immunodeficiency: diversity of clinical manifestations. Arch Immunol Ther Exp. (2016) 64(Suppl. 1):177–83. doi: 10.1007/s00005-016-0447-1
12. Pasic S, Djuricic S, Ristic G, Slavkovic B. Recombinase-activating gene 1 immunodeficiency: different immunological phenotypes in three siblings. Acta Paediatr. (2009) 98:1062–4. doi: 10.1111/j.1651-2227.2009.01250.x
13. IJspeert H, Driessen GJ, Moorhouse MJ, Hartwig NG, Wolska-Kusnierz B, Kalwak K, et al. Similar recombination-activating gene (RAG) mutations result in similar immunobiological effects but in different clinical phenotypes. J Allergy Clin Immunol. (2014) 133:1124–33. doi: 10.1016/j.jaci.2013.11.028
14. Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the primary immune deficiency treatment consortium experience. J Allergy Clin Immunol. (2014) 133:1092–8. doi: 10.1016/j.jaci.2013.09.044
15. Roifman CM, Somech R, Kavadas F, Pires L, Nahum A, Dalal I, et al. Defining combined immunodeficiency. J Allergy Clin Immunol. (2012) 130:177–83. doi: 10.1016/j.jaci.2012.04.029
16. Seidel MG, Kindle G, Gathmann B, Quinti I, Buckland M, van Montfrans J, et al. The European society for immunodeficiencies (ESID) registry working definitions for the clinical diagnosis of inborn errors of immunity. J Allergy Clin Immunol Pract. (2019) 7:1763–70. doi: 10.1016/j.jaip.2019.02.004
17. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2020) 40:66–81. doi: 10.1007/s10875-020-00763-0
18. Tirosh I, Yamazaki Y, Frugoni F, Ververs FA, Allenspach EJ, Zhang Y, et al. Recombination activity of human recombination-activating gene 2 (RAG2) mutations and correlation with clinical phenotype. J Allergy Clin Immunol. (2018) 143:726–35. doi: 10.1016/j.jaci.2018.04.027
19. Pasic S, Vujic D, Veljkovic D, Slavkovic B, Mostarica-Stojkovic M, Minic P, et al. Severe combined immunodeficiency in Serbia and montenegro between years 1986 and 2010: a single-center experience. J Clin Immunol. (2014) 34:304–8. doi: 10.1007/s10875-014-9991-9
20. Felgentreff K, Perez-Becker R, Speckmann C, Schwarz K, Kalwak K, Markelj G, et al. Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clin Immunol. (2011) 141:73–82. doi: 10.1016/j.clim.2011.05.007
21. Sharapova SO, Migas A, Guryanova I, Aleshkevich S, Kletski S, Durandy A, et al. Late-onset combined immune deficiency associated to skin granuloma due to heterozygous compound mutations in RAG1 gene in a 14 years old male. Hum Immunol. (2013) 74:18–22. doi: 10.1016/j.humimm.2012.10.010
22. Wu KY, Purswani P, Ujhazi B, Csomos K, Snezhina M, Elissaveta N, et al. Arthritis in two patients with partial recombination activating gene deficiency. Front Pediatr. (2019) 7:235. doi: 10.3389/fped.2019.00235
23. Farmer JR, Foldvari Z, Ujhazi B, de Ravin SS, Chen K, Bleesing JJH, et al. Outcomes and treatment Strategies for autoimmunity and hyperinflammation in patients with RAG deficiency. J Allergy Clin Immunol Pract. (2019) 7:1970–85.e4. doi: 10.1016/j.jaip.2019.02.038
24. National Atlas of Russia [Maps]. The Population & Economy, Cartography. Pozdnyak GV, Editor. Vol. 3. Moscow: Roskartografiya (2008). p. 97–101
25. Malyarchuk BA, Derenko MV. Mitochondrial DNA variability in Russians and Ukrainians: implication to the origin of the Eastern Slavs. Ann Hum Genet. (2001) 65:63–78. doi: 10.1046/j.1469-1809.2001.6510063.x
26. Strauss KA, Puffenberger EG, Bunin N, Rider NL, Morton MC, Eastman JT, et al. Clinical application of DNA microarrays: molecular diagnosis and HLA matching of an Amish child with severe combined immune deficiency. Clin Immunol. (2008) 128:31–8. doi: 10.1016/j.clim.2008.02.016
27. Al-Herz W, Massaad MJ, Chou J, Notarangelo LD, Geha RS. DNA recombination defects in Kuwait: clinical, immunologic and genetic profile. Clin Immunol. (2018) 187:68–75. doi: 10.1016/j.clim.2017.10.006
28. Greenberg-Kushnir N, Lee YN, Simon AJ, Lev A, Marcus N, Abuzaitoun O, et al. A large cohort of RAG1/2-deficient SCID patients-clinical, immunological, and prognostic analysis. J Clin Immunol. (2019) 40:211–22. doi: 10.1007/s10875-019-00717-1
29. Schuetz C, Pannicke U, Jacobsen EM, Burggraf S, Albert MH, Honig M, et al. Lesson from hypomorphic recombination-activating gene (RAG) mutations: why asymptomatic siblings should also be tested. J Allergy Clin Immunol. (2014) 133:1211–5. doi: 10.1016/j.jaci.2013.10.021
30. Chou J, Hanna-Wakim R, Tirosh I, Kane J, Fraulino D, Lee YN, et al. A novel homozygous mutation in recombination activating gene 2 in 2 relatives with different clinical phenotypes: Omenn syndrome and hyper-IgM syndrome. J Allergy Clin Immunol. (2012) 130:1414–6. doi: 10.1016/j.jaci.2012.06.012
31. Kwan A, Church JA, Cowan MJ, Agarwal R, Kapoor N, Kohn DB, et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California: results of the first 2 years. J Allergy Clin Immunol. (2013) 132:140–50. doi: 10.1016/j.jaci.2013.04.024
32. Ulusoy E, Karaca NE, Azarsiz E, Berdeli A, Aksu G, Kutukculer N. Recombinase activating gene 1 deficiencies without omenn syndrome may also present with Eosinophilia and bone marrow fibrosis. J Clin Med Res. (2016) 8:379–84. doi: 10.14740/jocmr2316w
33. Abolhassani H, Chou J, Bainter W, Platt CD, Tavassoli M, Momen T, et al. Clinical, immunologic, and genetic spectrum of 696 patients with combined immunodeficiency. J Allergy Clin Immunol. (2018) 141:1450–8. doi: 10.1016/j.jaci.2017.06.049
34. Goda V, Malik A, Kalmar T, Maroti Z, Patel B, Ujhazi B, et al. Partial RAG deficiency in a patient with varicella infection, autoimmune cytopenia, and anticytokine antibodies. J Allergy Clin Immunol Pract. (2018) 6:1769–71.e2. doi: 10.1016/j.jaip.2018.01.015
35. Delmonte OM, Schuetz C, Notarangelo LD. RAG Deficiency: Two Genes, Many Diseases. J Clin Immunol. (2018) 38:646–55. doi: 10.1007/s10875-018-0537-4
36. Rebala K, Mikulich AI, Tsybovsky IS, Sivakova D, Dzupinkova Z, Szczerkowska-Dobosz A, et al. Y-STR variation among slavs: evidence for the slavic homeland in the middle dnieper basin. J Hum Genet. (2007) 52:406–14. doi: 10.1007/s10038-007-0125-6
37. Abraham RS, Recher M, Giliani S, Walter JE, Lee YN, Frugoni F, et al. Adult-onset manifestation of idiopathic T-cell lymphopenia due to a heterozygous RAG1 mutation. J Allergy Clin Immunol. (2013) 131:1421–3. doi: 10.1016/j.jaci.2012.09.016
38. de Villartay JP, Lim A, Al-Mousa H, Dupont S, Dechanet-Merville J, Coumau-Gatbois E, et al. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Invest. (2005) 115:3291–9. doi: 10.1172/JCI25178
39. Villa A, Sobacchi C, Notarangelo LD, Bozzi F, Abinun M, Abrahamsen TG, et al. V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood. (2001) 97:81–8. doi: 10.1182/blood.V97.1.81
Keywords: RAG1, RAG2, primary immunodeficiency, geographic distribution, incidence, Slavic children
Citation: Sharapova SO, Skomska-Pawliszak M, Rodina YA, Wolska-Kuśnierz B, Dabrowska-Leonik N, Mikołuć B, Pashchenko OE, Pasic S, Freiberger T, Milota T, Formánková R, Szaflarska A, Siedlar M, Avčin T, Markelj G, Ciznar P, Kalwak K, Kołtan S, Jackowska T, Drabko K, Gagro A, Pac M, Naumova E, Kandilarova S, Babol-Pokora K, Varabyou DS, Barendregt BH, Raykina EV, Varlamova TV, Pavlova AV, Grombirikova H, Debeljak M, Mersiyanova IV, Bondarenko AV, Chernyshova LI, Kostyuchenko LV, Guseva MN, Rascon J, Muleviciene A, Preiksaitiene E, Geier CB, Leiss-Piller A, Yamazaki Y, Kawai T, Walter JE, Kondratenko IV, Šedivá A, van der Burg M, Kuzmenko NB, Notarangelo LD, Bernatowska E and Aleinikova OV (2020) The Clinical and Genetic Spectrum of 82 Patients With RAG Deficiency Including a c.256_257delAA Founder Variant in Slavic Countries. Front. Immunol. 11:900. doi: 10.3389/fimmu.2020.00900
Received: 04 March 2020; Accepted: 20 April 2020;
Published: 10 June 2020.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Sinisa Savic, University of Leeds, United KingdomCopyright © 2020 Sharapova, Skomska-Pawliszak, Rodina, Wolska-Kuśnierz, Dabrowska-Leonik, Mikołuć, Pashchenko, Pasic, Freiberger, Milota, Formánková, Szaflarska, Siedlar, Avčin, Markelj, Ciznar, Kalwak, Kołtan, Jackowska, Drabko, Gagro, Pac, Naumova, Kandilarova, Babol-Pokora, Varabyou, Barendregt, Raykina, Varlamova, Pavlova, Grombirikova, Debeljak, Mersiyanova, Bondarenko, Chernyshova, Kostyuchenko, Guseva, Rascon, Muleviciene, Preiksaitiene, Geier, Leiss-Piller, Yamazaki, Kawai, Walter, Kondratenko, Šedivá, van der Burg, Kuzmenko, Notarangelo, Bernatowska and Aleinikova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Svetlana O. Sharapova, c2hhcmFwb3Zhc3ZAZ21haWwuY29t; c2hhcmFwb3ZhQG9uY29sb2d5LmJ5
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.