 Toshifumi Nomura
Toshifumi Nomura- Department of Dermatology, Faculty of Medicine and Graduate School of Medicine, Hokkaido University, Sapporo, Japan
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition, clinically characterized by boiled cysts, comedones, abscesses, hypertrophic scars, and/or sinus tracts typically in the apocrine-gland-rich areas such as the axillae, groin, and/or buttocks. Although its precise pathogenic mechanisms remain unknown, I herein emphasize the importance of the following three recent discoveries in the pathogenesis of HS: First, heterozygous loss-of-function mutations in the genes encoding γ-secretase, including NCSTN, PSENEN, and PSEN1, have been identified in some patients with HS. Such genetic alterations result in hyperkeratosis, dysregulated hair follicle differentiation, and cyst formation via aberrant Notch signaling. Furthermore, Psen1–/Psen2–, Psen1–, Ncstn+/–, and Notch1–/Notch2– mice share common phenotypes of human HS, suggesting a role of aberrant keratinization in the development of HS. Second, upregulation of interleukin 1β, interleukin-36, caspase-1, and NLRP3 and dysregulation of the Th17:Treg cell axis have been demonstrated in HS samples, suggesting that autoinflammation is a key event in the pathophysiology of the disease. Notably, HS may be complicated with other autoinflammatory diseases such as inflammatory bowel diseases and pyoderma gangrenosum, again highlighting the importance of autoinflammation in HS. Last, biologics such as adalimumab, infliximab, anakinra, ustekinumab, and secukinumab are reportedly effective for moderate-to-severe HS. These findings collectively suggest that HS is closely linked with aberrant keratinization and autoinflammation, raising the question whether it represents an autoinflammatory keratinization disease, a recently proposed disease entity. In this mini review, I introduce the concept of autoinflammatory keratinization disease and attempt to address this clinically important question.
Introduction
Autoinflammatory keratinization diseases (AIKDs) belong to a recently proposed disease concept defined by the following four points (1). First, the primary and main inflammation sites are the epidermis and the upper dermis (1). Second, inflammation in the epidermis and upper dermis leads to hyperkeratosis, which is the main and characteristic phenotype of AIKDs (1). Third, AIKDs have primary genetic causative factors associated with the hyperactivation of innate immunity (autoinflammation), mainly in the epidermis and upper dermis (1). Last, the concept of AIKDs encompasses diseases with mixed pathomechanisms of autoinflammation and autoimmunity (1). In the original report of AIKDs, this novel disease category encompasses several genetic skin diseases caused by mutations in CARD14, IL36RN, or NLRP1 (1). It was recently proposed that hidradenitis suppurativa (HS) and porokeratosis should also be categorized as AIKDs (2–4). This mini review aims to provide a concise overview highlighting the aberrantly keratinizing and autoinflammatory nature of HS and to discuss whether it represents an AIKD.
Clinicopathological Features and Epidemiology of HS
HS, also known as acne inversa, is a chronic inflammatory skin disease of the hair follicle that usually presents after puberty, with a recurrent and progressive disease course (5–8). Clinical features of HS vary in severity and may include inflamed cysts, comedones, papules, pustules, nodules, abscesses, hypertrophic scars, fistulae, and tunneling sinus tracts, most commonly distributed in the apocrine-gland-rich and intertriginous areas such as the axillae, groin, perineum, buttocks, medial thighs, inframammary folds, and postauricular regions (5–8). Patients with HS may experience pain, pruritus, chronic malodorous purulent discharge, scar contracture, and/or sexual dysfunction and distress (5–9). Thus, HS often causes both physical and psychosocial burdens and severely impairs patients' quality of life (9–11).
There is a preponderance of females among HS patients, with an estimated female-to-male ratio of 2–3:1 (12–14). The previously published prevalence estimates of HS vary greatly from 0.05% to 4.1% depending on the types of studies (8, 13, 14); the lower estimates are derived from registry studies and the higher ones from self-reported studies (8). The exact prevalence of HS remains unknown, because, due to the hidden nature of the disease, it is an under-reported condition. Surveys show that the mean delay in the diagnosis of HS is 7.2 years (15), which may result from a lack of awareness of HS or the absence of internationally recognized diagnostic criteria (16). The diagnosis is usually made for a clinical history of recurrent, painful, inflammatory lesions in characteristic apocrine-gland-bearing areas (16).
HS was originally considered a bacterial skin infection in apocrine sweat glands because of the clinical features such as purulent discharge and the common involvement of the apocrine-gland-bearing areas. However, microbiologic screening usually reveals negative cultures or the detection of mixed normal flora and skin commensals as the main bacteria cultured from suppurative discharge (7). Notably, in a histological study of axillary skin excised from 12 patients with HS, the majority of cases (10 out of 12) showed cystic epithelium-lined structures or sinus tracts lined by squamous epithelium, both of which are derived from hair follicles (17). In contrast, only 4 out of 12 cases displayed inflammation in the apocrine glands (17). In another histological study of 60 HS biopsy samples, major findings included follicular occlusion (17/60), folliculitis (17/60), sinus tracts (9/60), epithelial cyst (6/60), and abscess (5/60) (18). Taken together, HS is now regarded as a non-suppurative disease of the hair follicle—rather than a simple bacterial infection—that is characterized by follicular occlusion or cyst formation.
Mutations in NCSTN, PSENEN, and PSEN1 are Responsible for HS
Approximately 34–42% of patients with HS report a family history of the condition, showing an autosomal dominant inheritance pattern (19–21). In 2010, heterozygous loss-of-function mutations in NCSTN, PSENEN, and PSEN1 were identified in six Chinese patients with HS (22). These genes encode components of γ-secretase, an intramembrane protease that cleaves various substrates, including Notch receptors. Subsequent studies in multiple populations such as British, French, African-American, Japanese, and Chinese have robustly confirmed the pathogenic role of these genes in HS patients with a positive family history of the disease (3, 19, 23–29). Interestingly, disease-causing variants have also been identified in four non-familial, sporadic cases. However, the frequency of identifying pathogenic variants in these genes is rare—~5% of overall HS cases (7)—even in familial HS cases. Furthermore, no significant genotype–phenotype correlation has been reported so far (30). Although γ-secretase is composed of presenilin, presenilin enhancer-2, nicastrin, and anterior pharynx defective encoded by PSEN1/PSEN2, PSENEN, NCSTN, and APH1A/APH1B, respectively (31), no disease-causing mutations in PSEN2, APH1A, or APH1B have been identified in HS patients (16). Notably, in the clinical trial of γ-secretase inhibitor nirogacestat in 17 adults, six exhibited follicular and cystic lesions in intertriginous regions (32). Furthermore, mice models such as Psen1–/Psen2–, Psen1–, and Ncstn+/– mice show follicular keratinization, cyst formation, epidermal hyperplasia, follicular atrophy, and/or absent sebaceous glands, which recapitulate the histological features of HS in humans (33, 34). Mice with pharmacologic inhibition of γ-secretase also develop HS-like lesions observed in Ncstn+/– mice (34). These human and mice data indicate that haploinsufficiency of the γ-secretase components, though rare, plays a key role in the development of HS.
Deficient Notch Signaling Plays a Key Role in HS
The Notch pathway regulates cell proliferation, cell differentiation, and cell death in many organs, including the skin (22, 33, 34). Importantly, the discovery of disease-causing mutations in NCSTN, PSENEN, and PSEN1 in HS patients strongly suggests that haploinsufficiency of the γ-secretase components cause HS via reduced Notch signaling (22). To date, more lines of evidence have pointed to the causal role of deficient Notch signaling in HS. First, the phenotypes of Notch1–/Notch2– mice mirror those of Psen1–/Psen2– mice, showing epidermal cysts and hyperkeratosis of the follicular epithelium with the occlusion and dilatation of the follicle by keratin plug formation (33). Second, mutations in POFUT1 and POGLUT1 cause HS and/or Dowling–Degos disease, an autosomal dominant skin disease clinically characterized by flexural hyperpigmentation (35, 36). Given that POFUT1 and POGLUT1 encode GDP-fucose protein O-fucosyltransferase 1 and protein O-glucosyltransferase 1, respectively, both of which are involved in the Notch pathway (35, 36), there should be a causal link between HS and reduced Notch signaling. Furthermore, mutations in PSENEN have also been reported in patients with HS and/or Dowling–Degos disease (37–39), which indicates that haploinsufficiency of presenilin enhancer-2 due to PSENEN mutations and reduced Notch signaling due to POFUT1 and POGLUT1 mutations both result in similar clinical consequences. These studies have placed deficient Notch signaling at the center of HS pathogenesis.
More Genes Associated With HS
Several HS or HS-like cases have been described in association with pachyonychia congenita or steatocystoma multiplex caused by mutations in KRT17 or KRT6A (40, 41). Mutations in FGFR2 also underlie nevus comedonicus and HS-like skin lesions (42). Furthermore, four cases of keratitis–ichthyosis–deafness syndrome, an autosomal-dominant skin disease caused by mutations in GJB2 and occurring in association with the follicular occlusion triad (HS, acne conglobata, and dissecting folliculitis of the scalp) have been reported (43–45). These findings further highlight the importance of aberrant cyst formation and hair follicle occlusion in the pathophysiology of HS.
HS can present as a component of systemic autoinflammatory syndromes like pyoderma gangrenosum, acne, pyogenic arthritis, and HS (PAPASH) and pyoderma gangrenosum, acne, and HS (PASH), which are caused by mutations in PSTPIP1 (46, 47). HS may also be presented by patients with familial Mediterranean fever carrying MEFV mutations (48–50). Notably, the frequency of MEFV mutations in the group of patients with HS was higher than that in healthy controls (49), suggesting that MEFV mutations may contribute to the pathogenesis of HS. Genetic variants in other autoinflammatory genes (e.g., NOD2, LPIN2, NLRP3, NLRP12, PSMB8, MVK, IL1RN) have also been identified in patients with HS or its syndromic forms (51). Thus, autoinflammation is the other key event in the pathogenesis of HS.
These findings collectively suggest that gene mutations leading to aberrant differentiation of the hair follicle epithelium and autoinflammation are associated with HS.
Immunological Features of HS
The precise mechanism leading to inflammation in patients with HS has not been fully elucidated. However, it is thought that dermal seeding of keratin, sebum, bacterial components, and cellular debris can result in a dense infiltration of immune cells consisting of T cells (mainly CD4+, but also CD8+), B cells, macrophages, and neutrophils in HS lesional skin (52). One of the most remarkable immunological features observed in HS lesioned skin is the marked upregulation of interleukin 1β (IL-1β) (53), mainly secreted by macrophages, the most numerous inflammatory cells found in HS infiltrates (54). Highly activated IL-1β pathways induce the strong production of chemokines (e.g., CXCL1, CXCL6), which contributes to the massive infiltration of immune cells, including neutrophils, thereby leading to clinical features of HS such as purulent discharge (53). Furthermore, IL-1β enhances the secretion of matrix metalloproteinases such as MMP3 and MMP10, which appear to be involved in tissue destruction, another major feature of HS (53). Notably, the increased expression of caspase-1, NLRP3, IL-6, IL-18, and IL-36 has also been reported (53). These findings emphasize the role of autoinflammation in the pathophysiology of HS.
Other major immunological characteristics of HS include the upregulation of IL-17 and tumor necrosis factor-α (51–53, 55, 56). The enrichment of Th17 cells and the dysregulation of the Th17:Treg cell axis, likely driven largely by increased IL-1β and IL-6 as a result of inflammasome activation (55), are remarkable features seen in HS lesioned skin (55, 56). Notably, the impaired Th17/Treg balance has also been shown in various autoinflammatory diseases, including inflammatory bowel diseases, Behçet's disease, and spondyloarthritis (56). These findings further indicate that HS has an autoinflammatory nature.
Biologics for HS
Given the immunological features of HS as discussed above, one could envision that immunomodulatory treatments targeting cytokines such as IL-1, IL-17, and tumor necrosis factor-α are beneficial (57). Indeed, the efficacy of biologics such as adalimumab, infliximab, etanercept, anakinra, ustekinumab, and secukinumab have been reported for moderate-to-severe HS (57–60). Particularly, adalimumab is the only biologic approved for this indication worldwide at the time of submission of this manuscript. In two phase 3 randomized controlled trials, adalimumab, 40 mg weekly, achieved significantly higher clinical response rates at week 12 than the placebo in patients with moderate-to-severe HS (58). This positive effect has lasted through week 168 (59). Notably, Moran et al. (55) showed that inhibition of tumor necrosis factor-α led to the reduction of IL-17 and the normalization of a dysregulated Th17:Treg cell axis in the skin, which may explain its therapeutic effects in HS. The significant response to biologics further supports the notion that HS should be considered a disease of aberrant immunity and not simply a bacterial infection.
Conclusion and Future Perspectives
The recent findings discussed above collectively suggest that HS is closely linked with aberrant keratinization and autoinflammation and thereby that, in the broad sense, HS should be regarded as an AIKD (Figure 1). However, caution should be taken in reaching a definitive conclusion as to whether HS should be included as an AIKD in the narrow sense (Figure 1) because it is uncertain whether the inflammatory response precedes the hyperkeratotic events such as hyperkeratosis of the terminal hair follicle epithelium or cyst formation in HS. It is generally believed that follicular occlusion is the primary event in HS. However, given that variants in autoinflammatory genes (e.g., MEFV, NOD2, LPIN2, NLRP3, NLRP12, PSMB8, MVK, IL1RN, PSTPIP1), which should lead to hyperkeratosis preceded by inflammation, have been linked to HS phenotypes (49) and that a loss-of-function mutation in NCSTN was identified in one PASH patient (61), one could predict that at least some HS patients completely fulfill all of the AIKD criteria proposed by Akiyama et al. (1) (Figure 1). Future research on the molecular basis of HS, especially capturing the full picture of the immune milieu and identifying more responsible genes, should address the question at hand and may benefit patients with this intractable disease.
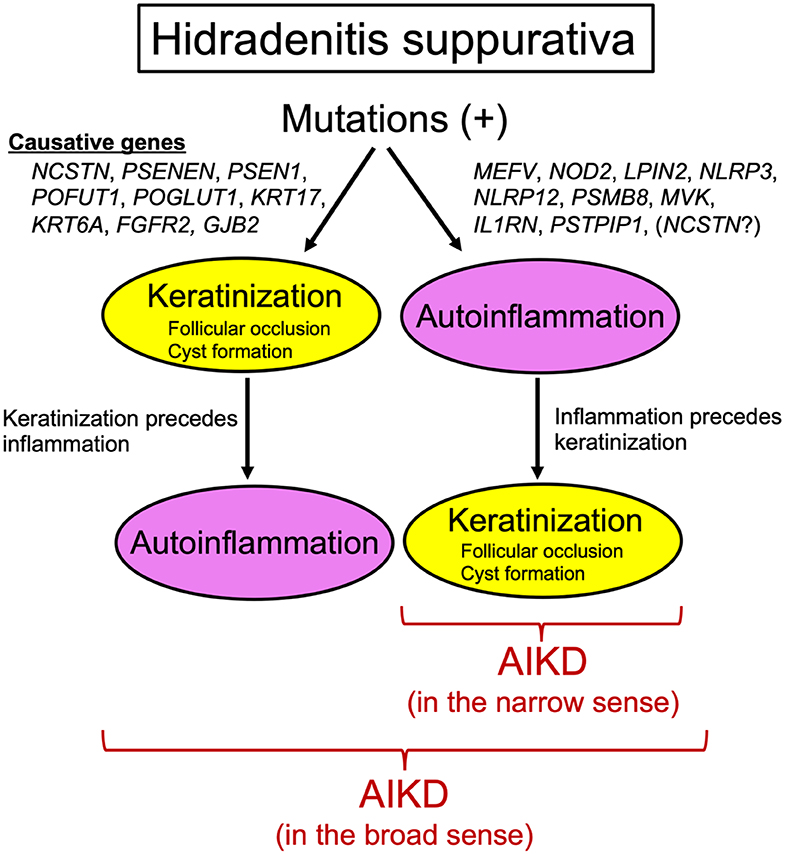
Figure 1. Hidradenitis suppurativa and autoinflammatory keratinization disease. Genes responsible for hidradenitis suppurativa can be divided into two groups. One includes NCSTN, PSENEN, PSEN1, POFUT1, POGLUT1, KRT17, KRT6A, FGFR2, and GJB2, whose mutations result in autoinflammation preceded by keratinization. This hidradenitis suppurativa subtype can be regarded as an autoinflammatory keratinization disease in the broad sense. The other group includes MEFV, NOD2, LPIN2, NLRP3, NLRP12, PSMB8, MVK, IL1RN, PSTPIP1, and possibly NCSTN, whose mutations lead to keratinization preceded by autoinflammation. This hidradenitis suppurativa subtype can be regarded as an autoinflammatory keratinization disease in the narrow sense.
Author Contributions
TN conceived the work, performed the literature review, and wrote the manuscript.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I am indebted to the patients with hidradenitis suppurativa who made this work possible.
Abbreviations
HS, hidradenitis suppurativa; AIKDs, autoinflammatory keratinization diseases; PAPASH, pyoderma gangrenosum, acne, pyogenic arthritis, and HS; PASH, pyoderma gangrenosum, acne, and HS; IL-1β, interleukin 1β.
References
1. Akiyama M, Takeichi T, McGrath JA, Sugiura K. Autoinflammatory keratinization disease. J Allergy Clin Immunol. (2017) 140:1545–7. doi: 10.1016/j.jaci.2017.05.019
2. De Vita V, McGonagle D. Hidradenitis suppurativa as an autoinflammatory keratinization disease. J Allergy Clin Immunol. (2018) 141:1953. doi: 10.1016/j.jaci.2018.01.010
3. Takeichi T, Matsumoto T, Nomura T, Takeda M, Niwa H, Kono M, et al. A novel NCSTN missense mutation in the signal peptide domain causes hideradenitis suppurativa, which has features characteristic of an autoinflammatory keratinization disease. Br J Dermatol. (2019) 182:491–3. doi: 10.1111/bjd.18445
4. Takeichi T, Akiyama M. Familial or sporadic porokeratosis as an autoinflammatory keratinization disease. J Dermatol. (2019)46:e125–6. doi: 10.1111/1346-8138.14666
5. Jemec GB. Clinical practice. Hidradenitis suppurativa. N Engl J Med. (2012) 366:158–64. doi: 10.1056/NEJMcp1014163
6. Revuz J. Hidradenitis suppurativa. J Eur Acad Dermatol Venereol. (2009) 23:985–98. doi: 10.1111/j.1468-3083.2009.03356.x
7. Zouboulis CC, Desai N, Emtestam L, Hunger RE, Ioannides D, Juhasz I, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. (2015) 29:619–44. doi: 10.1111/jdv.12966
8. Saunte DM, Jemec GB. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. (2017) 318:2019–32. doi: 10.1001/jama.2017.16691
9. Matusiak L. Profound consequences of hidradenitis suppurativa: a review. Br J Dermatol. (2018). doi: 10.1111/bjd.16603. [Epub ahead of print].
10. Senthilnathan A, Kolli SS, Cardwell LA, Richardson IM, Feldman SR, Pichardo RO. Even mild hidradenitis suppurativa impairs quality of life. Br J Dermatol. (2019) 181:838–9. doi: 10.1111/bjd.17928
11. Janse IC, Deckers IE, van der Maten AD, Evers AWM, Boer J, van der Zee HH, et al. Sexual health and quality of life are impaired in hidradenitis suppurativa: a multicentre cross-sectional study. Br J Dermatol. (2017) 176:1042–7. doi: 10.1111/bjd.14975
12. Revuz JE, Canoui-Poitrine F, Wolkenstein P, Viallette C, Gabison G, Pouget F, et al. Prevalence and factors associated with hidradenitis suppurativa: results from two case–control studies. J Am Acad Dermatol. (2008) 59:596–601. doi: 10.1016/j.jaad.2008.06.020
13. Jemec GB, Kimball AB. Hidradenitis suppurativa: epidemiology and scope of the problem. J Am Acad Dermatol. (2015) 73:S4–7. doi: 10.1016/j.jaad.2015.07.052
14. Ingram JR, Jenkins-Jones S, Knipe DW, Morgan CLI, Cannings-John R, Piguet V, et al. Population-based clinical practice research datalink study using algorithm modelling to identify the true burden of hidradenitis suppurativa. Br J Dermatol. (2018) 178:917–24. doi: 10.1111/bjd.16101
15. Saunte DM, Boer J, Stratigos A, Szepietowski JC, Hamzavi I, Kim KH, et al. Diagnostic delay in hidradenitis suppurativa is a global problem. Br J Dermatol. (2015) 173:1546–9. doi: 10.1111/bjd.14038
16. Pink AE, Simpson MA, Desai N, Trembath RC, Barker JN. γ-Secretase mutations in hidradenitis suppurativa: new insights into disease pathogenesis. J Invest Dermatol. (2013) 133:601–7. doi: 10.1038/jid.2012.372
17. Yu CC, Cook MG. Hidradenitis suppurativa: a disease of follicular epithelium, rather than apocrine glands. Br J Dermatol. (1990) 122:763–9. doi: 10.1111/j.1365-2133.1990.tb06264.x
18. Jemec GB, Hansen U. Histology of hidradenitis suppurativa. J Am Acad Dermatol. (1996) 34:994–9. doi: 10.1016/S0190-9622(96)90277-7
19. Pink AE, Simpson MA, Desai N, Dafou D, Hills A, Mortimer P, et al. Mutations in the γ-secretase genes NCSTN, PSENEN, and PSEN1 underlie rare forms of Hidradenitis Suppurativa (acne inversa). J Invest Dermatol. (2012) 132:2459–61. doi: 10.1038/jid.2012.162
20. Fitzsimmons JS, Guilbert PR. A family study of hidradenitis suppurativa. J Med Genet. (1985) 22:367–73. doi: 10.1136/jmg.22.5.367
21. Von Der Werth JM, Williams HC, Raeburn JA. The clinical genetics of hidradenitis suppurativa revisited. Br J Dermatol. (2000) 142:947–53. doi: 10.1046/j.1365-2133.2000.03476.x
22. Wang B, Yang W, Wen W, Sun J, Su B, Liu B, et al. Gamma- secretase gene mutations in familial acne inversa. Science. (2010) 330:1065. doi: 10.1126/science.1196284
23. Liu Y, Gao M, Lv YM, Yang X, Ren YQ, Jiang T, et al. Confirmation by exome sequencing of the pathogenic role of NCSTN mutations in acne inversa (hidradenitis suppurativa). J Invest Dermatol. (2011) 131:1570–2. doi: 10.1038/jid.2011.62
24. Nomura Y, Nomura T, Sakai K, Sasaki K, Ohguchi Y, Mizuno O, et al. A novel splice site mutation in NCSTN underlies a Japanese family with Hidradenitis Suppurativa. Br J Dermatol. (2013) 168:206–9. doi: 10.1111/j.1365-2133.2012.11174.x
25. Pink AE, Simpson MA, Brice GW, Smith CH, Desai N, Mortimer PS, et al. PSENEN and NCSTN mutations in familial hidradenitis suppurativa (Acne Inversa). J Invest Dermatol. (2011) 131:1568–70. doi: 10.1038/jid.2011.42
26. Miskinyte S, NassifA, Merabtene F, Ungeheuer MN, Join-Lambert O, Jais JP, et al. Nicastrin mutations in French families with Hidradenitis Suppurativa. J Invest Dermatol. (2012) 132:1728–30. doi: 10.1038/jid.2012.23
27. Chen S, Mattei P, You J, Sobreira NL, Hinds GA. Gamma-secretase mutation in an African American family with Hidradenitis Suppurativa. JAMA Dermatol. (2015) 151:668–70. doi: 10.1001/jamadermatol.2014.5306
28. Nomura Y, Nomura T, Suzuki S, Takeda M, Mizuno O, Ohguchi Y, et al. A novel NCSTN mutation alone may be insufficient for the development of familial Hidradenitis Suppurativa. J Dermatol Sci. (2014) 74:180–2. doi: 10.1016/j.jdermsci.2014.01.013
29. Liu M, Davis JW, Idler KB, Mostafa NM, Okun MM, Waring JF. Genetic analysis of NCSTN for potential association with hidradenitis suppurativa in familial and nonfamilial patients. Br J Dermatol. (2016) 175:414–6. doi: 10.1111/bjd.14482
30. Frew JW, Hawkes JE, Sullivan-Whalen M, Gilleaudeau P, Krueger JG. Inter-rater reliability of phenotypes and exploratory genotype-phenotype analysis in inherited hidradenitis suppurativa. Br J Dermatol. (2019) 181:566–71. doi: 10.1111/bjd.17695
31. Bergmans BA, De Strooper B. Gamma-secretases: from cell biology to therapeutic strategies. Lancet Neurol. (2010) 9:215–26. doi: 10.1016/S1474-4422(09)70332-1
32. O'Sullivan Coyne G, Woodring TS, Lee CR, Chen AP, Kong HH. Hidradenitis Suppurativa-like lesions associated with pharmacologic inhibition of gamma-secretase. J Invest Dermatol. (2018) 138:979–81. doi: 10.1016/j.jid.2017.09.051
33. Pan Y, Lin MH, Tian X, Cheng HT, Gridley T, Shen J, et al. Gamma-secretase functions through Notch signaling to maintain skin appendages but is not required for their patterning or initial morphogenesis. Dev Cell. (2004) 7:731–43. doi: 10.1016/j.devcel.2004.09.014
34. Li T, Wen H, Brayton C, Das P, Smithson LA, Fauq A, et al. Epidermal growth factor receptor and notch pathways participate in the tumor suppressor function of gamma-secretase. J Biol Chem. (2007) 282:32264–73. doi: 10.1074/jbc.M703649200
35. González-Villanueva I, Gutiérrez M, Hispán P, Betlloch I, Pascual JC. Novel POFUT1 mutation associated with hidradenitis suppurativa-Dowling-Degos disease firm up a role for Notch signalling in the pathogenesis of this disorder. Br J Dermatol. (2018) 178:984–6. doi: 10.1111/bjd.16264
36. Li A, Peng Y, Taiclet LM, Tanzi RE. Analysis of hidradenitis suppurativa-linked mutations in four genes and the effects of PSEN1-P242LfsX11 on cytokine and chemokine expression in macrophages. Hum Mol Genet. (2019) 28:1173–82. doi: 10.1093/hmg/ddy414
37. Ralser DJ, Basmanav FB, Tafazzoli A, Wititsuwannakul J, Delker S, Danda S, et al. Mutations in gamma-secretase subunit-encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J Clin Invest. (2017) 127:1485–90. doi: 10.1172/JCI90667
38. Li C, Li W, Xu H, Zhang X, Su B, Zhang W, et al. PSENEN mutation carriers with co-manifestation of acne inversa (AI) and dowling-degos disease (DDD): is AI or DDD the subphenotype? J Invest Dermatol. (2017) 137:2234–6. doi: 10.1016/j.jid.2017.05.021
39. Pavlovsky M, Sarig O, Eskin-Schwartz M, Malchin N, Bochner R, Mohamad J, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. (2018) 178:502–8. doi: 10.1111/bjd.16000
40. Musumeci ML, Fiorentini F, Bianchi L, Cascella R, Giardina E, Caputo V, et al. Follicular occlusion tetrad in a male patient with pachyonychia congenita: clinical and genetic analysis. J Eur Acad Dermatol Venereol. (2019) 33(Suppl. 6):36–9. doi: 10.1111/jdv.15851
41. Pedraz J, Peñas PF, Garcia-Diez A. Pachyonychia congenita and hidradenitis suppurativa: no response to infliximab therapy. J Eur Acad Dermatol Venereol. (2008) 22:1500–1. doi: 10.1111/j.1468-3083.2008.02691.x
42. Higgins R, Pink A, Hunger R, Yawalkar N, Navarini AA. Generalized comedones, acne, and hidradenitis suppurativa in a patient with an FGFR2 missense mutation. Front Med. (2017) 4:16. doi: 10.3389/fmed.2017.00016
43. Montgomery JR, White TW, Martin BL, Turner ML, Holland SM. A novel connexin 26 gene mutation associated with features of the keratitis- ichthyosis-deafness syndrome and the follicular occlusion triad. J Am Acad Dermatol. (2004) 51:377–82. doi: 10.1016/j.jaad.2003.12.042
44. Maintz L, Betz RC, Allam JP, Wenzel J, Jaksche A, Friedrichs N, et al. Keratitis-ichthyosis-deafness syndrome in association with follicular occlusion triad. Eur J Dermatol. (2005) 15:347–52.
45. Lazic T, Li Q, Frank M, Uitto J, Zhou LH. Extending the phenotypic spectrum of keratitis-ichthyosis-deafness syndrome: report of a patient with GJB2 (G12R) Connexin 26 mutation and unusual clinical findings. Pediatr Dermatol. (2012) 29:349–57. doi: 10.1111/j.1525-1470.2011.01425.x
46. Calderon-Castrat X, Bancalari-Diaz D, Roman-Curto C, Romo-Melgar A, Amoros-Cerdan D, Alcaraz-Mas LA, et al. PSTPIP1 gene mutation in a pyoderma gangrenosum, acne and suppurative hidradenitis (PASH) syndrome. Br J Dermatol. (2016) 175:194–8. doi: 10.1111/bjd.14383
47. Marzano AV, Trevisan V, Gattorno M, Ceccherini I, De Simone C, Crosti C. Pyogenic arthritis, pyoderma gangrenosum, acne, and hidradenitis suppurativa (PAPASH): a new autoinflammatory syndrome associated with a novel mutation of the PSTPIP1 gene. JAMA Dermatol. (2013) 149:762–4. doi: 10.1001/jamadermatol.2013.2907
48. Vural S, Gundogdu M, Kundakci N, Ruzicka T. Familial Mediterranean fever patients with hidradenitis suppurativa. Int J Dermatol. (2017) 56:660–3. doi: 10.1111/ijd.13503
49. Vural S, Gündogdu M, Gökpinar Ili E, Durmaz CD, Vural A, Steinmüller-Magin L, et al. Association of pyrin mutations and autoinflammation with complex phenotype hidradenitissuppurativa: a case-control study. Br J Dermatol. (2019) 180:1459–67. doi: 10.1111/bjd.17466
50. Ilgen U, Yayla ME, Eyüpoglu S, Karahan I. Hidradenitis suppurativa and Mediterranean fever gene mutations. JAAD Case Rep. (2019) 5:792–3. doi: 10.1016/j.jdcr.2019.06.035
51. Marzano AV, Damiani G, Ceccherini I, Berti E, Gattorno M, Cugno M. Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis). Br J Dermatol. (2017) 176:1588–98. doi: 10.1111/bjd.15226
52. Tricarico PM, Boniotto M, Genovese G, Zouboulis CC, Marzano AV, Crovella S. An integrated approach to unravel hidradenitis suppurativa etiopathogenesis. Front Immunol. (2019) 10:892. doi: 10.3389/fimmu.2019.00892
53. Witte-Händel E, Wolk K, Tsaousi A, Irmer ML, Mößner R, Shomroni O, et al. The IL-1 pathway is hyperactive in hidradenitis suppurativa and contributes to skin infiltration and destruction. J Invest Dermatol. (2019) 139:1294–305. doi: 10.1016/j.jid.2018.11.018
54. Shah A, Alhusayen R, Amini-Nik S. The critical role of macrophages in the pathogenesis of hidradenitis suppurativa. Inflamm Res. (2017) 66:931–45. doi: 10.1007/s00011-017-1074-y
55. Moran B, Sweeney CM, Hughes R, Malara A, Kirthi S, Tobin AM, et al. Hidradenitis Suppurativa is characterized by dysregulation of the Th17:treg cell axis, which is corrected by anti-TNF therapy. J Invest Dermatol. (2017) 137:2389–95. doi: 10.1016/j.jid.2017.05.033
56. Melnik BC, John SM, Chen W, Plewig G. T helper 17 cell/regulatory T-cell imbalance in hidradenitis suppurativa/acne inversa: the link to hair follicle dissection, obesity, smoking and autoimmune comorbidities. Br J Dermatol. (2018) 179:260–72. doi: 10.1111/bjd.16561
57. Matusiak Ł, Jemec GB, Szepietowski JC. Pharmacological development in hidradenitis suppurativa. Curr Opin Pharmacol. (2019) 46:65–72. doi: 10.1016/j.coph.2019.04.006
58. Kimball AB, Okun MM, Williams DA, Gottlieb AB, Papp KA, Zouboulis CC, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. (2016) 375:422–34. doi: 10.1056/NEJMoa1504370
59. Zouboulis CC, Okun MM, Prens EP, Gniadecki R, Foley PA, Lynde C, et al. Long-term adalimumab efficacy in patients with moderate-to-severe hidradenitis suppurativa/acne inversa: 3-year results of a phase 3 open-label extension study. J Am Acad Dermatol. (2019) 80:60–9.e2. doi: 10.1016/j.jaad.2018.05.040
60. Prussick L, Rothstein B, Joshipura D, Saraiya A, Turkowski Y, Abdat R, et al. Open-label, investigator-initiated, single-site exploratory trial evaluating secukinumab, an anti-interleukin-17A monoclonal antibody, for patients with moderate-to-severe hidradenitissuppurativa. Br J Dermatol. (2019) 181:609–11. doi: 10.1111/bjd.17822
Keywords: hidradenitis suppurativa, autoinflammatory keratinization disease, γ-secretase, Notch signaling, interleukin 1β, interleukin 17, inflammasome, biologics
Citation: Nomura T (2020) Hidradenitis Suppurativa as a Potential Subtype of Autoinflammatory Keratinization Disease. Front. Immunol. 11:847. doi: 10.3389/fimmu.2020.00847
Received: 13 December 2019; Accepted: 14 April 2020;
Published: 20 May 2020.
Edited by:
Kazumitsu Sugiura, Fujita Health University, JapanReviewed by:
Carmelo Carmona-Rivera, National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), United StatesDaisuke Watanabe, Aichi Medical University, Japan
Copyright © 2020 Nomura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toshifumi Nomura, nomura@huhp.hokudai.ac.jp