
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 06 May 2020
Sec. Vaccines and Molecular Therapeutics
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00823
This article is part of the Research Topic Immunotherapies Towards HIV Cure View all 14 articles
 Beatriz Mothe1,2,3*†
Beatriz Mothe1,2,3*† Miriam Rosás-Umbert1,4†
Miriam Rosás-Umbert1,4† Pep Coll1
Pep Coll1 Christian Manzardo5
Christian Manzardo5 Maria C. Puertas1
Maria C. Puertas1 Sara Morón-López1Anuska Llano1Cristina Miranda2Samandhy Cedeño1Miriam López2
Sara Morón-López1Anuska Llano1Cristina Miranda2Samandhy Cedeño1Miriam López2 Yovaninna Alarcón-Soto6Guadalupe Gómez Melis6
Yovaninna Alarcón-Soto6Guadalupe Gómez Melis6 Klaus Langohr6Ana M. Barriocanal4,7Jessica Toro2
Klaus Langohr6Ana M. Barriocanal4,7Jessica Toro2 Irene Ruiz5Cristina Rovira5
Irene Ruiz5Cristina Rovira5 Antonio Carrillo7Michael Meulbroek8Alison Crook9Edmund G. Wee9Jose M. Miró5Bonaventura Clotet1,2,3,4
Antonio Carrillo7Michael Meulbroek8Alison Crook9Edmund G. Wee9Jose M. Miró5Bonaventura Clotet1,2,3,4 Marta Valle4,10
Marta Valle4,10 Javier Martinez-Picado1,3,11
Javier Martinez-Picado1,3,11 Tomáš Hanke9,12
Tomáš Hanke9,12 Christian Brander1,3,11†José Moltó2,7† and the BCN02 Study Investigators
Christian Brander1,3,11†José Moltó2,7† and the BCN02 Study InvestigatorsKick&kill strategies combining drugs aiming to reactivate the viral reservoir with therapeutic vaccines to induce effective cytotoxic immune responses hold potential to achieve a functional cure for HIV-1 infection. Here, we report on an open-label, single-arm, phase I clinical trial, enrolling 15 early-treated HIV-1-infected individuals, testing the combination of the histone deacetylase inhibitor romidepsin as a latency-reversing agent and the MVA.HIVconsv vaccine. Romidepsin treatment resulted in increased histone acetylation, cell-associated HIV-1 RNA, and T-cell activation, which were associated with a marginally significant reduction of the viral reservoir. Vaccinations boosted robust and broad HIVconsv-specific T cells, which were strongly refocused toward conserved regions of the HIV-1 proteome. During a monitored ART interruption phase using plasma viral load over 2,000 copies/ml as a criterium for ART resumption, 23% of individuals showed sustained suppression of viremia up to 32 weeks without evidence for reseeding the viral reservoir. Results from this pilot study show that the combined kick&kill intervention was safe and suggest a role for this strategy in achieving an immune-driven durable viremic control.
Current antiretroviral therapy (ART) effectively suppresses HIV-1 replication, thus preventing disease progression. However, the infection remains chronic given that a latent HIV-1 reservoir, established early after infection, persists despite suppressive ART (1). Upon ART discontinuation, integrated replication-competent proviruses in the reservoir drive a rapid viral rebound (2). Therapeutic vaccination has been proposed as a possible approach to induce an effective immune control able to contain rebounding virus (3).
Most therapeutic vaccines tested in the past expressed one or several HIV-1 proteins, which expanded HIV-1-specific CD8+ cytotoxic T lymphocyte (CTL) responses to varying levels. However, the responses were ineffective in controlling viremia after ART interruption, likely because of their suboptimal magnitude, breadth, width, specificity, and/or polyfunctionality (4–8), raising the need for novel immunogens and delivery methods to tackle HIV-1 diversity and the virus' ability to escape. New vaccines strategies are being developed to maximize the vaccine coverage of circulating viruses using multivalent mosaic immunogens designed in silico (9, 10). Alternatively, vaccines designs are tested that aim to focus the CTL responses toward more conserved and protective regions of the virus, which are less likely to mutate and escape the T-cell response (11–15). Among the latter, the HIVconsv immunogen is one of the most advanced vaccine candidates in clinical development. HIVconsv immunogen consists of a chimeric protein assembled from 14 highly conserved domains derived from HIV-1 genes Gag, Pol, Vif, and Env alternating, for each domain, the consensus sequence of the four major HIV-1 clades A, B, C, and D (12). Upon delivery to both HIV-1-negative and positive individuals by heterologous prime/boost regimens as DNA or in simian adenovirus of chimpanzee (ChAdV) and poxvirus MVA vectors, HIVconsv vaccines were safe and induced CD8+ T cells with broad inhibitory capacity of HIV-1 in vitro, but showed no effect on the viral reservoir (16–23).
To overcome the limitations of therapeutic vaccines -administered alone- in targeting the viral reservoir, vaccines are combined with latency reversing agents (LRA) in the so called kick&kill strategies (24). This approach intends to activate transcription of HIV-1 using small molecules able to disrupt the viral latency and facilitate effective sensing and clearance of infected cells by vaccine-elicited HIV-1-specific CTL (25, 26). Histone deacetylase inhibitors (HDACi) have been proposed as potential HIV-1 LRA (27–31). Romidepsin (RMD; Istodax®, Celgene Ltd.) is a potent HDACi approved for the treatment of cutaneous and peripheral T-cell lymphomas, which has been shown to induce HIV-1 transcription both in vitro and in vivo (32, 33). The REDUC trial combined RMD with Vacc-4x and rhuGM-CSF in chronically suppressed HIV-1-positive individuals, resulting in a mean reduction of 39.7% in total HIV-1 DNA (34). However, this intervention failed to delay viral rebound after ART interruption, suggesting that the reservoir-purge effect was not sufficient and/or the vaccine-induced response was unable to eliminate cells actively replicating HIV-1. In fact, the increase in cell-associated HIV-1 RNA inversely correlated with time to rebound, supporting that, in the absence of an enhanced HIV-1-specific CTL response, viral reactivation might facilitate viral rebound once ART is interrupted (35).
Here, in this single-arm, open-label, phase I, proof-of-concept study, referred to as BCN02 trial (NCT02616874), we assessed the safety, tolerability, immunogenicity and effect on the viral reservoir of a kick&kill strategy consisting of the combination of HIVconsv vaccines with RMD in suppressed early-treated HIV-1-infected individuals. Participants were rolled-over from the therapeutic vaccine trial BCN01 (NCT01712425), in which individuals who started ART during acute/recent HIV-1 infection had received a prime/boost regimen of the ChAdV63.HIVconsv and MVA.HIVconsv vaccines (CM) (20). Three years after, BCN01 participants who had shown sustained viral suppression and who accepted to participate in BCN02 study were immunized with two doses of MVA.HIVconsv, before and after three weekly-doses of RMD, followed by a monitored antiretroviral pause (MAP) for a period of 32 weeks to assess the ability of the intervention to control viral rebound.
The BCN02 clinical trial was an investigator initiated phase I, open-label, single-arm, multicenter, single-country study to assess the safety, tolerability and efficacy of a combined kick&kill strategy in suppressed HIV-1-infected patients that had initiated ART during acute/recent HIV-infection. Individuals were rolled over from vaccine trial BCN01 (20) and invited to participate after 3 years on suppressive ART. A complete list of inclusion/exclusion criteria is available in the Study Protocol (Appendix). The study took place between February 2016 and October 2017 at two HIV-1 units from universitary hospitals (Hospital Universitari Germans Trias i Pujol -HUGTIP, Badalona and Hospital Clínic, Barcelona) and a community center (BCN-Checkpoint, Barcelona). Before inclusion in the study, all participants signed an informed consent previously discussed, reviewed and approved by the Community Advisory Board of the Barcelona-based vaccine program (HIVACAT). The study was approved by the institutional ethical review board of the participating institutions (Reference Nr AC-15-108-R) and by the Spanish Regulatory Authorities; and was conducted in accordance to the principles of the Helsinki Declaration and local personal data protection law (LOPD 15/1999). The MVA.HIVconsv vaccine was GMP manufactured at IDT Biologika GmbH, Germany, and supplied for the study under an investigator initiated clinical trial contract agreement. Risk of Genetically Modified Organism release to the environment was evaluated by the Spanish Ministry of Environment (B/ES/12/09). RMD was supplied for the study by Celgene Ltd. (Couvet, Switzerland) under an investigator initiated clinical trial contract agreement.
The BCN02 trial design is summarized in Figure 1A. After their inclusion in the study (week 0), all participants received a first dose of 2 × 108 plaque-forming units (pfu) of MVA.HIVconsv (MVA1) administered intramuscularly, followed by three weekly doses of RMD of 5 mg/m2 BSA infused intravenously over 4 hours (RMD1−2−3), and by a second dose of 2 × 108 pfu of MVA.HIVconsv (MVA2) 4 weeks after RMD3 to compensate for any potential impairment in the previous vaccine-induced response caused by RMD. Following RMD prescription information, participants received prophylactic antiemetic treatment with ondasetron before and during 3 days after each RMD dose.
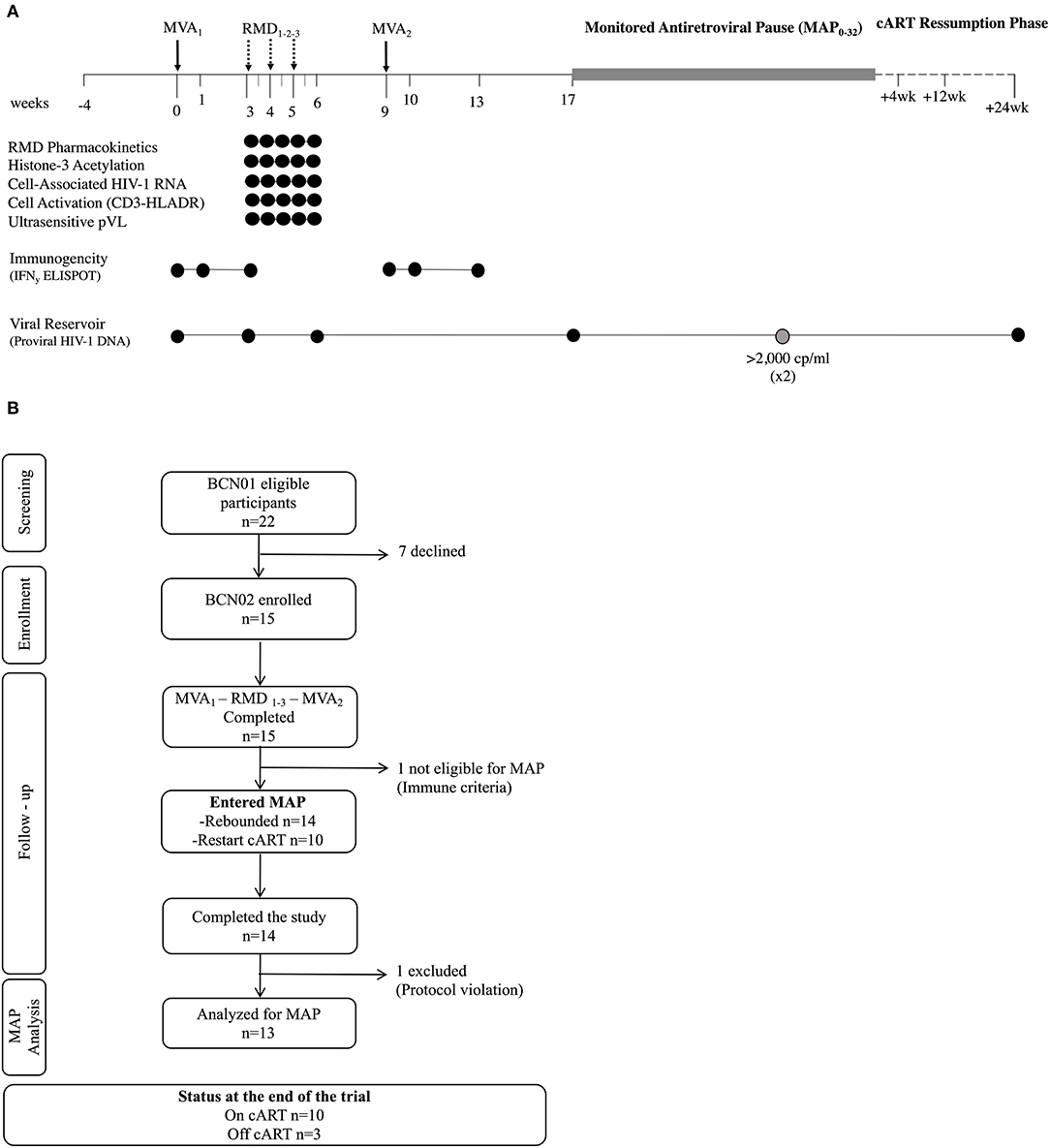
Figure 1. Trial design. (A) Schematic study design. (B) Consolidated Standards of Reporting Trials (CONSORT) flow diagram for the trial. *Viral rebound during MAP was defined as pVL >20 copies/ml and #criteria for ART resumption included pVL over 2,000 copies/ml in two consecutive determinations, CD4+ cell counts decrease over 50% and/or below 500 cells/mm3 and/or development of clinical symptoms suggestive of an acute retroviral syndrome. MVA, MVA.HIVconsv vaccine; RMD, romidepsin; MAP, monitored antiretroviral pause; ART, antiretroviral therapy; pVL, plasma HIV-1 viral load.
Eight weeks after MVA2 (week 17), eligible participants initiated a MAP for a maximum of 32 weeks (MAP0−32) or until any ART resumption criteria were met. To be eligible for the MAP, participants had to maintain undetectable HIV-1 pVL and meet the immune futility criteria, defined as showing a net increase in HIVconsv-specific immune response with MVA2 boost measured in an ex vivo IFN-γ ELISPOT assay. During MAP, participants were allowed to choose either the hospital units or BCN-Checkpoint community center for the follow up visits. Symptoms suggestive of acute retroviral syndrome and sexually transmitted diseases were solicited and viral load was tested using the finger-tip Xpert HIV-1 Qual kit (Cepheid, Sunnyvale, CA, US) in all visits. When a positive result was obtained in the Xpert HIV-1 Qual, participants were called in for a confirmatory quantitative pVL within the next 24 h. If pVL was confirmed to be over 20 copies/ml, a visit was scheduled 3 days after to closely monitor viral rebound and be able to offer prompt ART resumption if required. Details on viral load management during MAP are described in the Study Protocol (Appendix). After ART ressumption, participants were followed at 4, 12, and 24 weeks to assure that they re-attained viral suppression.
BCN02 participants were adult (≥18 years) HIV-1-infected individuals, who had initiated ART >6 months after estimated date of HIV-1 acquisition and who had received a prime/boost heterologous vaccination regimen using ChAdV63.HIVconsv-MVA.HIVconsv in the parental BCN01 study (20). To be eligible for BCN02, participants had to maintain optimal HIV-1 suppression during at least 3 years and CD4+ cell counts ≥500 cells/mm3 at BCN02 baseline visit. Main exclusion criteria included active hepatitis B or C, history of AIDS-defining disease, treatment for cancer or lymphoproliferative disease within 1 year before study entry or use of immunosuppressants within the 3 months prior to the screening visit. Concomitant treatment with strong CYP3A4 inhibitors was not permitted, but switching ART to a non-boosted integrase-inhibitor raltegravir- or dolutegravir-based regimen at least 4 weeks before baseline visit was allowed for those patients receiving ART containing ritonavir or cobicistat at screening.
The primary endpoint of this study was to assess the safety, tolerability and the effect on the viral reservoir size of the combined treatment with HIVconsv vaccines and RMD given as a latency reversing agent. Secondary endpoints included the extent and specificity of the CTL response and the effect of the intervention in controlling viral rebound after ART interruption. Other secondary endpoints included RMD pharmacokinetics and the effects of RMD on histone acetylation in lymphocytes, induction of viral transcription, changes in T-cell activation surface markers, and quantification of plasma viremia.
Safety and tolerability were evaluated by the development of grade ≥3 and serious adverse events (AE). Local and systemic AE were solicited prospectively for a minimum of 7 days following each immunization and RMD administration. Both local and systemic AE were graded according to the Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, version 2.0, November 2014, accessible online at https://rsc.niaid.nih.gov/sites/default/files/daids-ae-grading-table-v2-nov2014.pdf. AE were specified as unrelated, unlikely, probably or definitely related to the investigational products by the investigator.
The concentration of RMD in plasma was determined, for RMD1, at the end of the infusion (4 h) and 4.5, 5, 6, 8, 12, and 24 h after and, for RMD2 and RMD3, at the end of the infusion and 12 h after. RMD concentrations were measured by liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS), according to a validated method. A population pharmacokinetic model for RMD was developed using non-linear mixed-effects modeling with the computer program NONMEM version 7.3 (Icon Development Solution, Ellicot City, MD) (36). Bayesian estimates of the individual parameters of RMD were used to simulate individual drug concentrations, and RMD area under the concentration-time curve (AUC0-inf) was calculated for each individual on each occasion using a non-compartmental approach (Winnonlin software; Phoenix, version 7.0).
The levels of histone H3 acetylation in lymphocytes (based on FSC/SCC scatter) were determined by flow cytometry from samples taken before (0 h) and at the end of each RMD infusion (RMD1−2−3) (4 h), at 8 and 24 h (+1 day) RMD1, and at 72 h (+3 days) and 7 days after (RMD1−2−3). Cryopreserverd PBMC were thawed 4 h before use, and 500,000 cells were blocked with 600 ul of PBS/10% FBS for 20 min and stained with polyclonal rabbit anti-acetyl histoneH3 (10 μg/ml, MerckMillipore #06–599) or normal rabbit serum (control stain, LifeTechnologies #10510) for 30 min. Cells were washed and subsequently incubated with donkey anti-rabbit IgG(H+L) (6 μg/mL, LifeTechnologies #A21206) for 30 min at room temperature in the dark. Cells were washed, re-suspended in 150 μl PBS and analyzed. ~50,000 events were acquired per sample. The median fluorescence intensity (MFI) for each sample was calculated by substracting the background MFI (isotype control stain) from the anti-acetyl histoneH3 stain.
Activation of T cells was determined based upon HLA-DR expression on CD3+ T cells. Cryopreserved PBMC were thawed, and 1,000,000 cells were stained with CD3 APC-Cy7, CD4 FITC, CD8 BV510 and HLA-DR PECy7 (BioLegend #344818, 300538, 301048, and 307616, respectively). Cells were collected on an LSRII instrument (BD), and data analyzed according to the gating criteria shown in Supplementary Figure 1 using FlowJo 10 software.
Cell-associated HIV-1 RNA was quantified in peripheral CD4+ T cells by ddPCR (One-Step RT-ddPCR Advanced Kit for Probes, BioRad) from samples taken before (0 h) and at the end of each RMD infusion (4 h), at 8 and 24 h (+1 day) after RMD1, and 72 h (+3 days) and 7 days after RMD1−2−3. CA HIV-1 RNA was quantified using two different primers/probe sets annealing to the 5′LTR and GAG conserved regions of HIV-1, to circumvent potential primer mismatch in individuals' viral sequence as previously described (37). HIV-1 transcription levels were normalized to the housekeeping gene TATA-binding protein (TBP) and shown as relative to levels before RMD1.
To evaluate HIV-1 RNA below 20 copies/ml, 4–8 ml of plasma samples taken before (0 h) and at the end of each RMD infusion (4 h), at 8 and 24 h (+1 day) after RMD1, and 72 h (+3 days) and 7 days after RMD1−2−3 were ultracentrifugated at 170,000 g at 4°C for 30 min and viral RNA was extracted automatically using the m2000sp Abbot device. HIV-1 RNA copies were quantified using the Abbott Real-Time HIV-1 assay (Abbott Molecular Inc.) and in-house calibration curve sets as described (38). The limit of detection (2 HIV-1 RNA copies/mL) was calculated relative to the plasma volume.
Total HIV-1 and HIVconsv-specific T cells were assessed ex vivo using cryopreserved PBMC obtained the day of vaccination and 1 week afterwards, 3 weeks after MVA1, and 4 weeks after MVA2 using an IFN-γ-detecting enzyme-linked immunoabsorbent spot assay (ELISPOT IFN-γ Mabtech kit). 15-mer peptides overlapping by 11 amino acid were combined into 6 pools of 32–33 peptides per pool corresponding to the HIVconsv vaccine insert (P1-P6, total n = 166 peptides, IN pools) (Figure 3A) and 12 pools of 39–67 peptides per pool spanning the rest of the HIV-1 viral protein sequences (OUT pools for “outside the immunogen,” obtained through the NIH AIDS Reagent Program). All peptides pools were tested in duplicates. The final concentration of individual peptides in the ELISPOT assay was 1.57 μg/ml. Medium only was used as no-peptide negative control in quadruplicate wells, and PHA (50 μg/ml) and a CEF peptide pool (2 μg/ml) consisting of 23 previously defined human CD8+ T-cell epitopes from cytomegalovirus, Epstein-Barr virus and influenza virus (C.T.L. OH, USA) were added as positive controls.
To address the breadth of the vaccine-induced response at the peak immunogenicity time point, an IFN-γ ELISPOT assay with in vitro expanded T cells was performed on stored samples from week 10 and 13 to test individual overlapping peptides covering the HIVconsv immunogen sequence (n = 166 OLP). Briefly, cryopreserved PBMC were thawed and incubated for 3 h at 37°C in R10 before stimulation with an anti-CD3 monoclonal antibody during 2–4 weeks in RPMI 1640 supplemented with FBS and penicillin/streptomycin with 50 U/ml of recombinant IL-2 (39). Before their use in ELISPOT assays, the expanded cells were washed twice with R10 and incubated overnight at 37 °C in the absence of IL-2.
Spots were counted using an automated Cellular Technology Limited (C.T.L., OH, USA) ELISPOT Reader Unit. The threshold for positive responses was set at ≥50 SFC/106 PBMC (5 spots per well), > the mean number of SFC in negative control wells plus 3 SD of the negative control wells, or > 3 × the mean of negative control wells, whichever was higher. To avoid overestimating the breadth of responses, positive responses to two consecutive 15-mer overlapping peptide were counted as one response. The highest magnitude of the sequential responses was taken as the magnitude for each identified response.
To quantify the size of the peripheral blood proviral reservoir, lysed extracts from CD4+ T cells were used to measure total CA HIV-1 DNA by ddPCR. Primers and probes for the RPP30 cellular gene were used for input normalization.
Qualitative variables were represented as mean absolute and relative frequencies, whereas quantitative variables were represented as mean or median and range. Safety endpoints are summarized by the number and percentage of participants reporting local and systemic AE and their grading. The Wilcoxon signed rank test was used to test whether the viral reservoir and the immune parameter changed as an effect of the intervention, without correction for multiple comparisons. The maximum breadth of the T-cell response per individual was estimated as the number of P1-P6 pools eliciting a positive response throughout the study and the number of individual OLP eliciting a response at peak immunogenicity time point from the mapping assay. Reservoir size and immunogenicity were analyzed using GraphPad Prism (v5.01) for Mac OS X (San Diego, CA).
To evaluate the effect of the intervention on viral rebound, a positive pre-defined efficacy signal was established if at least over 20% of patients remained with pVL below 2,000 copies/ml at week 12 of MAP, considering previous data suggesting that early treatment initiation could favor delayed viral rebound/spontaneous viral control in up to 15% of individuals (40). However, BCN02 was an exploratory pilot trial and, due to the absence of a control arm and its final small sample size, the nature of this study only allowed to detect trends in virological effects, which collectively, could be useful to design future studies. To detect possible factors associated to the viremic control observed during the MAP phase, univariate log-binomial regression models were used (41). This model uses the logarithm as a link function, and is a generalized lineal model for a binary outcome where the error terms follows a binomial distribution. The effect size measure of the model is the relative risk. Because of the low number of MAP-C (n = 3), multivariate log-binomial regression models were not fitted. The significance threshold for all univariate analyses was set at a two-sided α = 0.05. The analyses were performed with R Core Team (42) (v3.0.2).
Between February 29th and September 15th 2016, 15 out of the 22 eligible BCN01 participants were enrolled in BCN02. Seven declined to participate due to their inability to attend all the scheduled visits. Baseline characteristics of trial participants are summarized in Table 1. All 15 participants received two doses MVA.HIVconsv (MVA1−2) and three doses of romidepsin (RMD1−2−3) as shown in the study chronogram (Figure 1A), and were included in the safety, immunogenicity and reservoir analyses. One participant was not eligible for MAP due to immune futility pre-defined criteria and 14 participants underwent a MAP for a maximum of 32 weeks. Retrospective analyses of stored plasma samples obtained during MAP revealed the presence of antiretroviral drugs in some samples from one participant, whose MAP data were censored for the viral rebound kinetics analyses (Figure 1B).
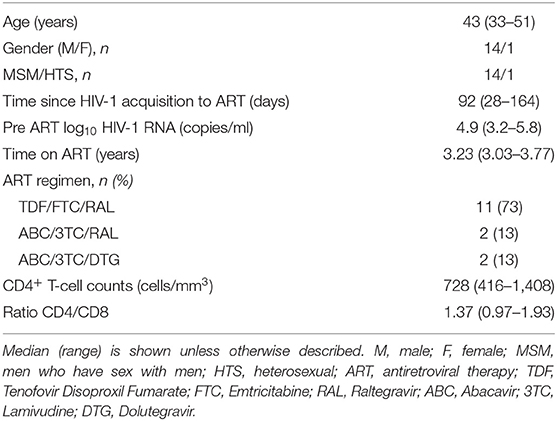
Table 1. Demographic, clinical, and treatment characteristics of study patients at study entry (n = 15).
All participants reported adverse events (AE) related to both study investigational medicinal products. A total of 333 AE were recorded during the study intervention phase, 129 after MVA1−2 and 204 after RMD1−2−3, which were mostly mild or moderate (grade 1-2) (n = 318, 95%). The most frequent AE related to MVA.HIVconsv, summarized in Table 2, were local pain at the injection site and a flu-like syndrome consisting of fatigue, headache, myalgia, and/or low-grade temperature (<38°C). Regarding AE related to RMD (Table 3), the most common grade 1–2 events were headache, fatigue, and gastro-intestinal symptoms. Despite prophylactic ondansetron treatment, 4 (27%) individuals vomited the days of RMD administration. One participant experienced a grade 4 AE consisting in a sepsis by Shigella sonnei that required hospital admission for 24 h, thus fulfilling the criteria of serious adverse event (SAE). The symptoms started within 4 h after RMD3 and therefore, the SAE was considered as possibly related to RMD.
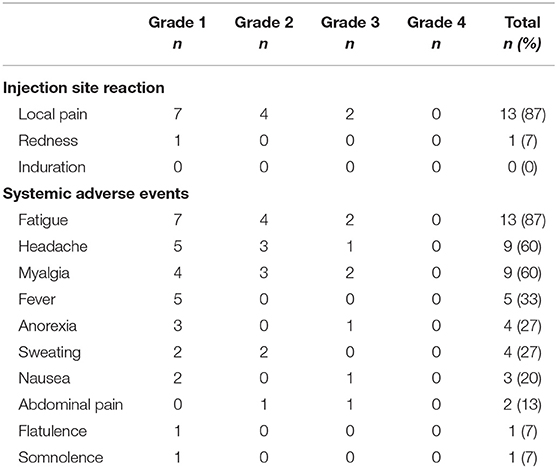
Table 2. Summary of adverse events related to MVA.HIVconsv vaccinations (n = 15).
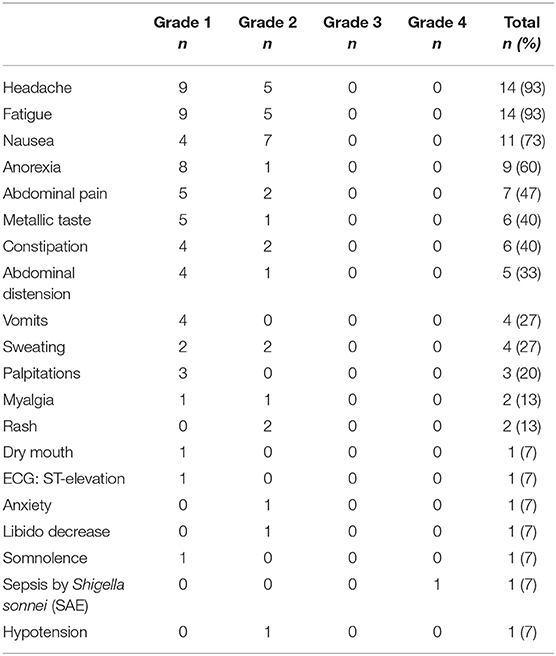
Table 3. Summary of adverse events related to RMD1−2−3 treatment (n = 15).
No laboratory abnormalities related to MVA1−2 were reported. All laboratory abnormalities related to RMD were grade 1-2 (n = 22), the most frequent being hypophosphatemia (8 events) and thrombocytopenia (5 events), except from one case of grade 4 creatinine kinase elevation with normal eGFR which resolved within 7 days without sequelae. Noteworthy, CD4+ T-cell counts showed a transient decrease by a median of 248 cells/mm3 3 days after each RMD administration which was not fully recovered by day 7 after RMD3 (Supplementary Figure 2). Overall, both MVA.HIVconsv and RMD at the regimen and dose administered in this study were well-tolerated and safety profiles were consistent with data previously reported (20, 34).
During the MAP, 12 (86%) participants reported a total of 58 AE, which were all grade 1-2 and not suggestive of acute retroviral syndrome (not shown). Grade 1 anxiety was observed in one participant who repeatedly declined psychological support (same participant with protocol violation during the MAP).
Pharmacokinetics of RMD was comparable to previously described data (43). A population pharmacokinetic model adequately describing RMD concentrations in plasma was developed (36). According to the individual profiles simulated using the model, each infusion was followed by a rapid and polyexponential decline in RMD concentrations in plasma, reaching nearly undetectable levels by 24 h after dosing (Figure 2A).
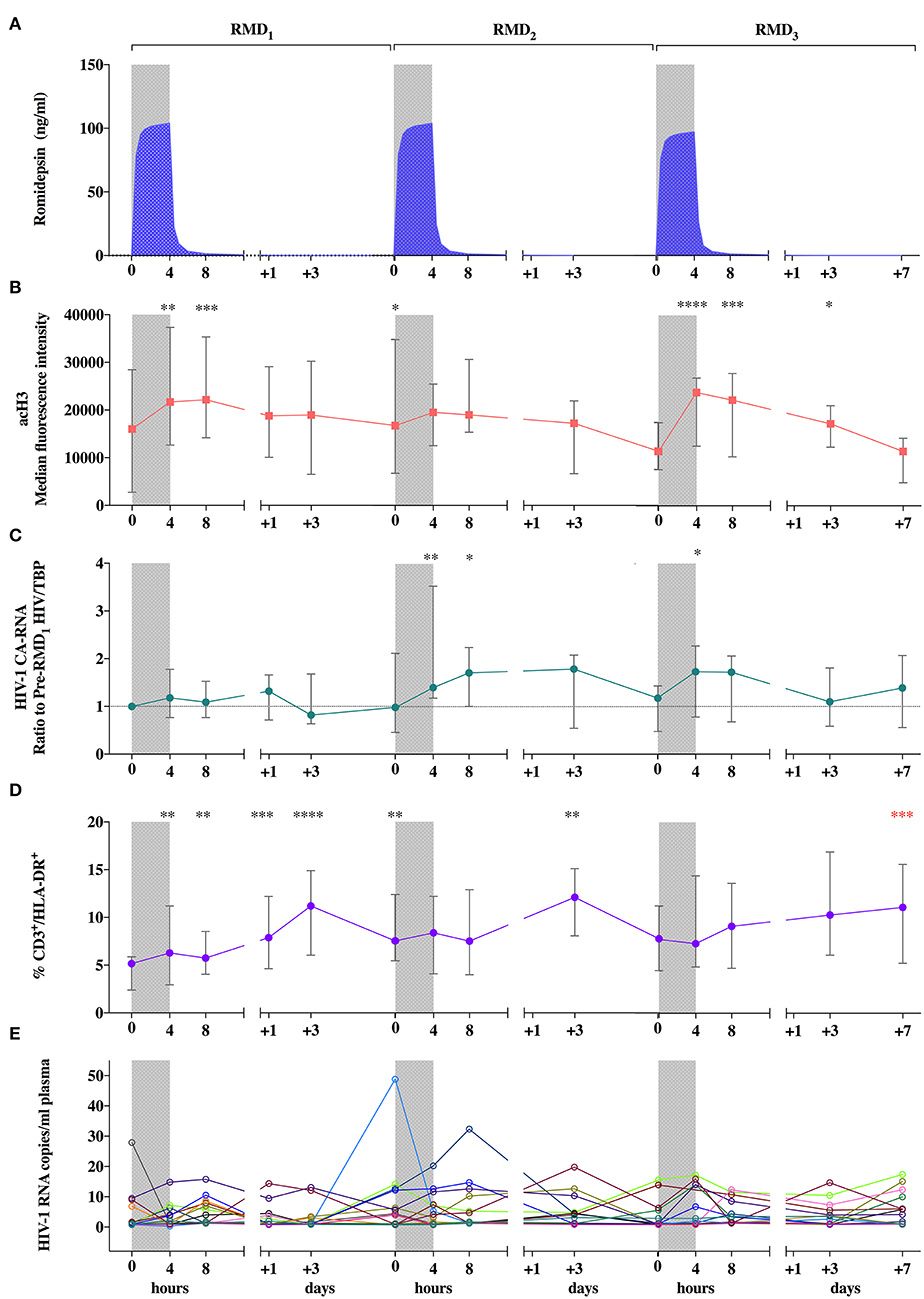
Figure 2. Pharmacokinetic and pharmacodynamic effects of RMD. (A) Mean individual predictions of RMD plasma concentrations. (B) Levels of histone H3 acetylation in peripheral lymphocytes. (C) Viral transcription levels expressed as changes from pre-RMD1 levels of cell-associated HIV-1 RNA in peripheral CD4+ T-cells. (D) Levels of T-cell activation (CD3+/HLA-DR+ cells). (E) individual determinations of pVL. Median of frequencies and IQR (error bars) are represented. Wilcoxon signed-rank p-values compare each represented time point with the corresponding values preceding each RMD administration. *p < 0.05. **p < 0.01. ***p < 0.001 and ****p < 0.0001. The p value resulting from the comparison between the value at day 0 of RMD1 and 7 days after RMD3 is shown in red.
Regarding the direct effect of RMD on chromatin and induction of viral transcription, histone H3 acetylation (acH3) increased rapidly during each RMD infusion, remained high during 4 h, and returned to baseline values 3 days after each dose (Figure 2B), which is consistent with previous reports (33, 34). HIV-1 transcription transiently increased in parallel, with changes more pronouned after RMD2 and RMD3 (Figure 2C). These changes were more evident without normalization for house-keeping genes (Supplementary Figure 3) possibly reflecting the general increase in histone acetylation levels (44) induced by RMD. Increases in T-cell activation, measured by the proportion of CD3+/HLA-DR+ cells, were observed 3 days after each RMD dose. Over the course of the three RMD doses, T-cell activation increased in a progressive manner and was maintained up to 1 week after RMD3 (Figure 2D), suggesting a cumulative effect of RMD.
To evaluate changes in levels of quantifiable plasma HIV-1 RNA, an ultrasensitive single copy assay was used. Kinetics of plasma HIV-1 RNA levels did not follow a clear pattern (Figure 2E), despite the increasing percentage of participants with detectable low-level viremia at the end of each RMD dose (Supplementary Figure 4). Collectively, we reproduced effects on acH3, HIV-1 transcription and T cell activation previously reported in chronically infected individuals (34), suggesting that a lower viral reservoir level achieved by early-treatment initiation does not preclude the reactivation potential of RMD.
Total HIV-1 and HIVconsv-specific T cells were assessed ex vivo by an IFN-γ-detecting enzyme-linked immunoabsorbent spot (ELISPOT) assay using 6 peptide pools covering the HIVconsv immunogen sequence (P1-P6) on week 0 (day of MVA1), 1, 3 (day of RMD1), 9 (day of MVA2), 10, and 13. A total of 90 samples were obtained, of which 3 (3%) were censored due to low positive controls and/or high background. All 15 participants showed an absolute increase in HIVconsv-specific IFN-γ-producing T cells during the study, either after MVA1 (Wilcoxon signed-rank, p = 0.0007) or after MVA2 (Wilcoxon signed-rank, p = 0.0017) (Figure 3B). Median (range) total frequencies of HIVconsv-specific T cells reached 1,965 (530-6,901) spot-forming cells (SFC)/106 PBMC at the peak immunogenicity time point, which represented an absolute median increase of 1,600 (300–6,621) SFC/106 PBMC from baseline (Wilcoxon signed-rank, p < 0.0001).
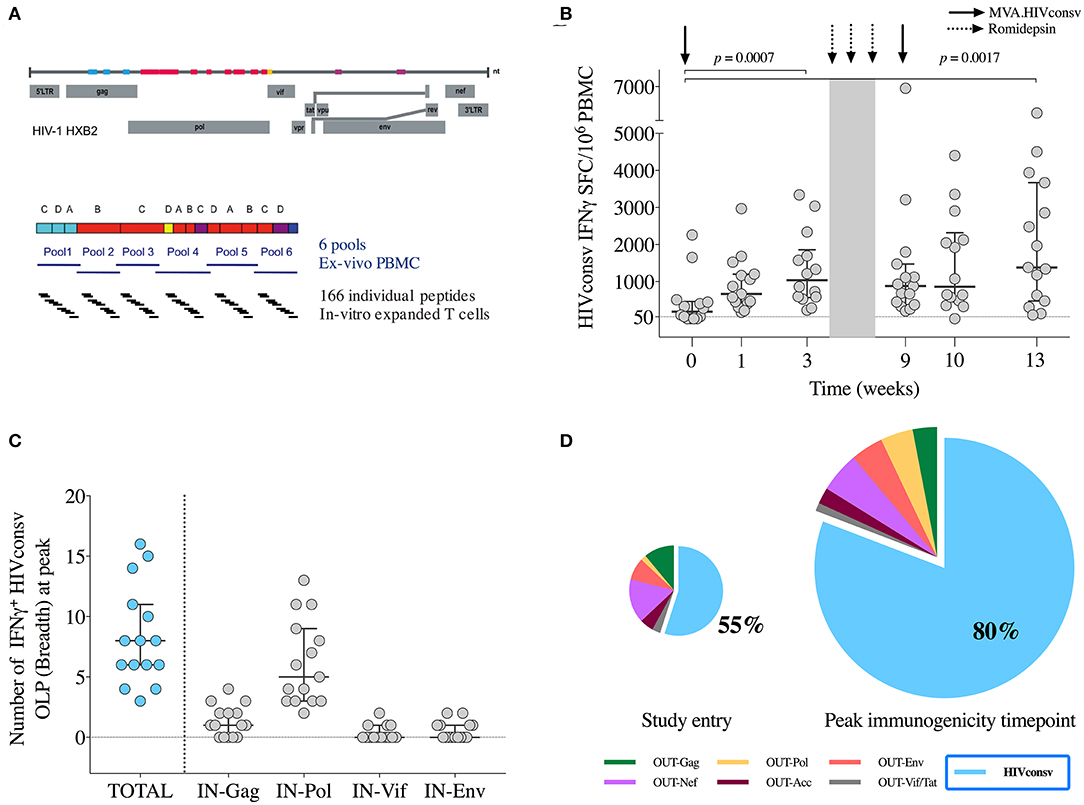
Figure 3. Vaccine immunogenicity. (A) Schematic representation of the selected regions in the HIV-1 proteome from different clades included in the HIVconsv immunogen and distribution of 6 peptide pools (P1-P6) and individual overlapping 15-mer peptides (OLP) used in the IFN-γ ELISPOT assays. (B) Magnitude (sum of SFU/106 PBMC to pools P1-P6) of vaccine-induced responses over the BCN02 study. Horizontal and error bars represent median and IQR, respectively, and p-values correspond to comparisons between the indicated time points using the Wilcoxon signed-rank test. (C) Breadth of vaccine-elicited responses toward individual OLP included in the indicated HIVconsv regions. Horizontal and error bars represent median and IQR, respectively. (D) Average distribution of total HIV-1 T-cells according to their specificity at the indicated time points. HIVconsv-specific responses are shown in blue. Pie charts are scaled according to the total frequencies of responses. IN are peptide pools corresponding to the HIVconsv vaccine insert and OUT peptide pools spanning the rest of HIV-1 proteome “outside the immunogen”.
Over the intervention phase, participants responded to median (range) of 5 (2–6) peptide pools (Supplementary Figure 5). To map the maximum vaccine-induced breadth at peak immunogenicity time point (weeks 10–13), in vitro expanded T cells responding toward individual OLPs covering the HIVconsv immunogen were assessed. Median (range) of 8 (3–16) IFN-γ-producing responses to individual OLPs were found, with a dominance in Pol-specific T cells, consistent with the immunogen composition (Figure 3C).
The dominance of HIVconsv-specific responses was calculated at each time point as the percentage of HIVconsv-specific T-cell frequencies divided by the total HIV-1 proteome-specific T-cell frequencies. At the moment of HIV diagnoses, HIVconsv responses were subdominant (<10% being HIVconsv-specfic) and peaked after the CM vaccination reaching a median (range) of 58% (7%−100%) of the total anti-HIV-1 T-cell responses (BCN01 parental study) (20). In BCN02, 2 years from the last HIVconsv vaccination, the increase in the frequency of HIVconsv-specific T-cell responses after MVA1 or MVA2 further shifted the patterns of T-cell immuno-dominance toward HIVconsv with median (range) of 85% (54%–100%) of the total anti-HIV-1 T-cell responses at peak immunogenicity time point being HIVconsv specific (Figure 3D).
All participants had detectable viral reservoirs, as measured by total CD4+ T cell-associated HIV-1 DNA, throughout the study. Results from 2 samples out of a total of 60 were considered invalid and were censored. At BCN02 study entry, median (range) reservoir size was of 140 (17–752) HIV-1 DNA copies/106 CD4+ T-cells (Figure 4). Proviral DNA showed a tendency to further decrease from baseline to week 17 (Wilcoxon signed-rank, p = 0.0599, Figure 4) to median (range) levels of 120 (11-680) copies/106 CD4+ Tcells.
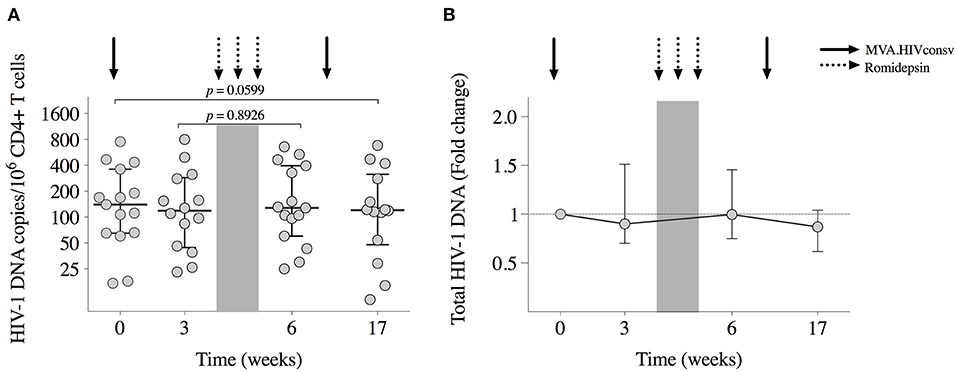
Figure 4. Viral reservoir. (A) Total HIV-1 DNA copies/106 CD4+ T cells for each participant are shown at study entry (week 0), week 3 (day of RMD1), and week 6 (1 week after RMD3) and at week 17 (8 weeks after MVA2). Horizontal and error bars represent median and IQR, respectively, and p-values correspond to comparisons between the indicated time points using the Wilcoxon signed-rank test. (B) Changes in proviral DNA throughout the study with respect to baseline (week 0), which is considered to be 1.
Participants undergoing MAP were monitored weekly for the first 12 weeks and every 2 weeks thereafter for a maximum of 32 weeks (MAP0−32). Criteria for ART resumption included pVL over 2,000 copies/ml in two consecutive determinations, CD4+ cell counts decrease over 50% and/or below 500 cells/mm3 and/or development of clinical symptoms suggestive of an acute retroviral syndrome.
All participants rebounded (detectable pVL over 20 copies/ml) during MAP (Figure 5A). Median (range) time to first detectable pVL was 13 (7–35) days with median (range) of first detectable pVL of 122 (28–3,410) copies/ml. Ten participants resumed ART before MAP12 (MAP-NC for MAP-Non-controllers) with median (range) time to resume ART of 28 (16–59) days. All MAP-NC resumed ART due to the viral load criteria, with median (range) pVL of 19,250 (2,900–179,000) copies/ml at the moment of ART resumption (Figure 5B). None of the participants resumed ART due to immune or clinical criteria. A “late-rebounder” presented the first detectable pVL 5 weeks after ART interruption (MAP5, pVL of 59 copies/ml) and was able to maintain viral load below 2,000 for 3 more weeks, resuming ART at MAP8. In addition to the “late-rebounder,” 3 (23%) other participants remained off ART with sustained pVL <2,000 copies/ml for a total of 32 weeks (MAP-C, from MAP-Controllers). Highest peak pVL determination in the MAP-C was of 3,110 copies/ml at week 12 of MAP in the absence of symptoms followed by 2,460 and 1,100 three and six days later (participant decision to stay off). At week 32, two out of the three MAP-C accepted to stay off cART out of the protocol and were followed under standard of care. One MAP-C showed a late rebound after 48 weeks off cART and the other one, voluntary resumed cART after 1.5 years off cART despite sustained low-level viremia. All participants who restarted ART reached viral re-suppression within 6 months. No evidence of emergence of drug resistance was detected.
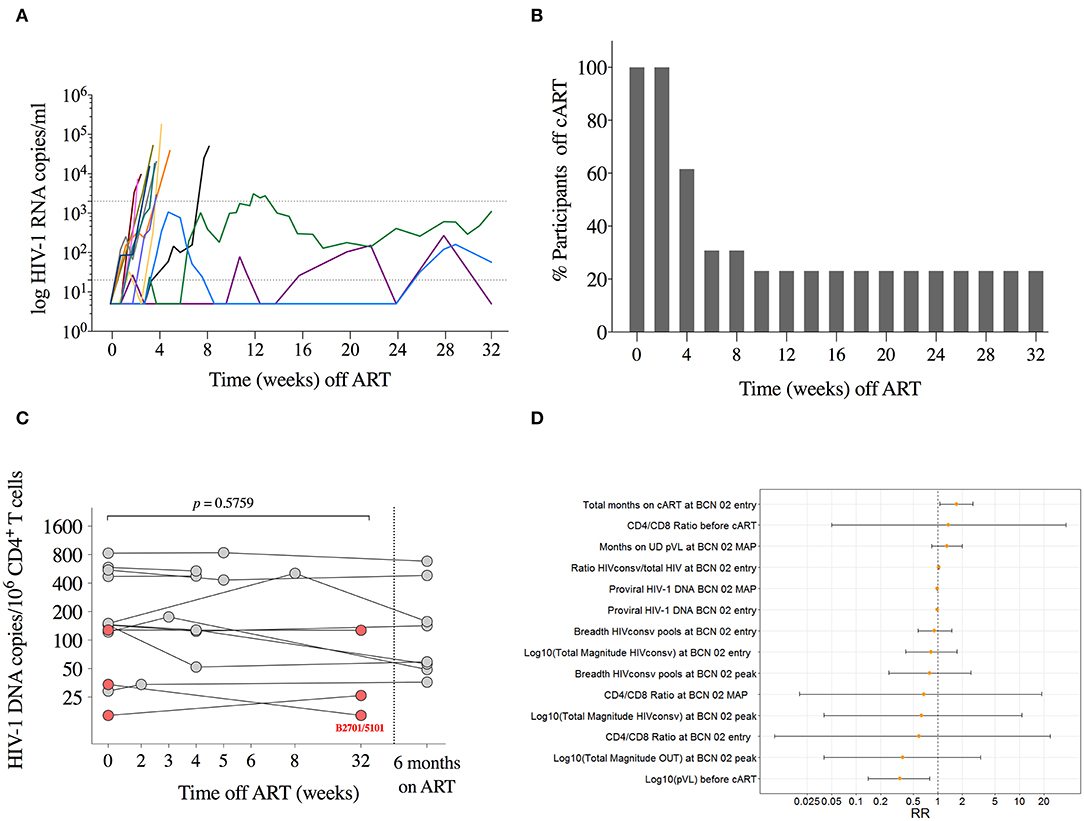
Figure 5. Monitored antiretroviral pause. (A) Individual pVL during MAP is shown for each participant in different colors (n = 13). Lines are interrupted on day of ART resumption. Dotted lines represent detection limit and threshold for ART resumption (20 and 2,000 copies/ml, respectively). (B) Proportion of individuals remaining off ART during MAP. (C) Total HIV-1 DNA copies/106 CD4+ T cells are shown for each participant at MAP initiation, at day of ART ressumption, and 24 weeks after ART. P-value corresponds to comparison between MAP initiation and the day of ART resumption for each individual using the Wilcoxon signed-rank test. Individuals with sustained low-level viremia over 32 weeks are shown in red. (D) Estimated relative risks and 95% confidence intervals for the durable control of pVL during MAP obtained from univariate log-binomial regression models.
To assess re-seeding of the viral reservoir during the MAP, total HIV-1 DNA was measured at MAP0 (n = 13), on the day of ART resumption (n = 10), and 6 months after for the 10 MAP-NC (n = 8 available), and at MAP32 for the three MAP-C. We did not observe any significant change in total HIV-1 DNA during MAP in participants with early ART resumption (Wilcoxon signed-rank, p = 0.5759 for MAP-NC) and, noteworthy, nor in the three participants with sustained low level viremia for 32 weeks. Moreover, one of the three MAP-C showed a 2-fold reduction in the HIV-1 DNA (from 34 at MAP0 to 16 copies/106 CD4+ cells at MAP32, Figure 5C). This was the only participant undergoing MAP carrying HLA alleles associated with natural HIV control (HLA-B*27:01/HLA-B*51:01) (45, 46).
Supplementary Table 1 shows the summary of variables explored to explain the binary outcome defined as MAP-NC vs. MAP-C. The estimated relative risks obtained from the log-binomial models for different covariates analyzed are shown in Figure 5D.
Univariate log-binomial regression models used to detect factors associated with virologic control during MAP revealed that pVL before ART initiation (pre-ART pVL) was the only factor statistically significantly associated with control of viral rebound after ART interruption. For each log increase on the pre-ART pVL, the probability of becoming a MAP-C decreased by 66% (RR 0.34; 95% CI 0.14, 0.79). Interestingly, albeit not statistically significant in the univariate models, the 3 MAP-C had among the lowest reservoir levels at treatment interruption time point (16, 54 and 122 copies/106 CD4+ T cells) –consistent with lower pre-ART pVL– and showed the highest shift in CTL immunodominance pattern after vaccination (>700 HIVconsv-specific SFC/106 PBMCs and >75% of HIVconsv dominance at peak immunogenicity time point, Supplementary Figure 6).
In this single-arm, open-label, phase I, proof-of-concept trial performed in HIV-1-infected ART-suppressed individuals treated during acute/recent HIV-1 infection, we show that combination of the HIVconsv vaccines with RMD as a latency reversing agent was safe, highly immunogenic, and induced bursts of viral transcription. The combined intervention resulted in a tendency toward a reduction in proviral DNA levels and was followed by a prolonged viremic control in 23% of participants after ART interruption without evidence of reservoir re-seeding.
Early-treated individuals typically show less immune exhaustion (47) and reduced frequency of immune-escaped viral variants compared to individuals initiating ART at later stages of HIV-1 infection (48), offering a potentially favorable setting to induce a protective immune response upon therapeutic vaccinations. In the parental BCN01 trial, a prime/boost vaccine regimen with ChAdV63.HIVconsv and MVA.HIVconsv induced high frequencies of T cells with high in vitro suppressive capacity that markedly shifted the focus of the CD8+ CTL response toward HIVconsv sequences that are subdominant during natural infection (20). In the BCN02 trial, after 3 years of viral suppression and 2 years since the last vaccination in BCN01, booster MVA.HIVconsv vaccinations were still immunogenic and further increased breadth, magnitude and immunodominance of CTL responses toward HIVconsv sequences.
Along with a strong vaccine-elicited CTL activity (“kill”), the ability to simultaneously induce reactivation of the viral reservoir (“kick”) is a critical feature for the success of the kick&kill strategy (24). A 3-dose regimen of weekly RMD at 5 mg/m2 BSA was selected based on results from previous trials (33, 34). Consistently, we observed a direct in vivo effect of RMD in histone 3 acetylation upon each RMD dose, which was followed by changes in cell-associated HIV-1 RNA levels. Conversely, a placebo-controlled dose-escalating trial (ACTG 5315) testing 3 RMD doses (5 mg/m2 BSA) administered every two weeks in chronically suppressed individuals, did not show changes in viral transcription (49). The weekly administration regimen and the intensive sampling after each RMD dose in our study allowed us to detect increases in CA HIV-1 RNA above 2-fold in 80% of individuals at any time point after RMD administrations. These changes were followed by consistent increases in T cell activation markers, which remarkably, were maintained one week after RMD3, likely reflecting a direct and cumulative effect of weekly RMD dosing and an effective induction of viral transcription. Noteworthy, changes above 2.1-fold in CA-RNA have been estimated to occur in <5% of repeated measurements in an individual (50).
Despite the induction of viral transcription, the kinetics of plasma HIV-1 RNA followed an unclear pattern, similar to previous studies showing variable changes in plasma viremia following LRA administration (34, 51, 52). This variability might reflect suboptimal potency of the agents tested so far and/or the reactivation of predominantly defective proviruses. In the context of the current study, elimination of reactivated cells by vaccine-elicited T cells may have additionally blunted quantification of plasma viremia.
A critical objective of the use of LRA in a kick&kill strategy is to mobilize and ultimately reduce the viral reservoir. Our findings showed that, despite robust immunogenicity of HIVconsv vaccines and at least partial reactivation of the viral reservoir induced by RMD, the net effect on the proviral DNA levels was modest. All participants had detectable levels of HIV-1 DNA at the time of treatment interruption although a tendency toward a decrease by 19.3% from baseline to week 17 (Wilcoxon signed-rank, p = 0.0599) was detected. Conversely, a mean 39.7% decrease in reservoir size was observed in the REDUC trial (34). This discrepancy between REDUC and BCN02 results may be explained by the inclusion of early-treated individuals in our study, in which already baseline levels of proviral DNA were substantially lower, challenging the quantification of the effects of the intervention on HIV-1 DNA levels. We acknowledge that the translation of HIV-1 protein expression into antigen presentation—even in case of defective proviruses (1)—upon LRA reactivation is poorly understood. Likewise, the ability of LRA-induced HIV-1 protein expression to effectively induce recognition and killing by CD8+ T cells remains to be fully elucidated (53) and therefore, the potential effects of further RMD administrations on the viral reservoir are to be determined. A potential significant toxicity—suggested in in vitro assays (54)—on vaccine-induced T cells might also have limited our capability to observe a further reduction in the reservoir size in our study. This hypothesis is consistent with the fact that HIVconsv vaccines induced higher levels of activated T cells compared to Vacc-4x vaccination in the REDUC trial. Nevertheless, the potential toxicity of RMD in vivo, its relationship with RMD exposure and, ultimately, whether vaccine-induced T cells were able to sensor and remove infected cells in response to HIV-1 reactivation remains to be determined.
To attain a functional cure, a persistent immune-mediated control of residual HIV-1 might be as relevant as achieving an absolute reduction on the proviral DNA levels. In this regard, the three BCN02 MAP-C, were among the subjects with both lower viral reservoir levels at MAP and higher vaccine-induced responses. In our study, having lower pre-ART pVL was the only outstanding marker associated with viral control during MAP, which correlates with the size of the viral reservoir after ART suppression. These relationships are consistent with previous studies suggesting a role of a low viral reservoir on analytical treatment interruptions (ATI) outcomes (55, 56). Furthermore, after ART discontinuation, MAP-C did not show an initial burst in pVL followed by a fast post-peak control as described in several post treatment controllers (PTC) (8, 57). Collectively, the findings from this and other studies suggest that a small reservoir size, resulting from early ART or another intervention, may be essential to achieve sustained post-intervention control (55) but also, that a potent vaccine-induced immune pressure might contribute to prevent a peak burst of viremia and maintain suppressed viremia for a substantial period of time. This control, mediated by immune pressure, is supported by the absence of re-seeding of the viral reservoir in the BCN02 MAP-C, in contrast to reports from previous ATI trials (58, 59).
The interpretation of the outcome of kick&kill studies may be confounded by individuals controlling HIV rebound after treatment interruption without the need for a prior therapeutic intervention. The prevalence and mechanisms driving such PTC in natural HIV infection are not well-understood. A recent metanalysis (CHAMP study) including 14 interruption trials estimated a 13% rate of PTC among early-treated individuals (57). Importantly, and in contrast to the three BCN02 MAP-C who did not show a transient high burst of viremia, 32% of the 61 PTCs analyzed in the CHAMP metanalysis had peaks of viremia ranging from 1,000 to over 10,000 copies/ml within the first 24 weeks after treatment interruption. Thus, in addition to the different behavior of the 3 MAP-C (23%) with respect to the PTC, according to this metanalysis, the BCN02 trial may have missed additional MAP-C due to the conservative ART resumption criteria used (two consecutive pVL over 2,000 copies/ml).
The safety and tolerability profiles of MVA.HIVconsv and RMD were similar to those reported in previous studies (19, 20, 34). However, there was a SAE in one participant. This case highlights the need for planning intensive monitoring in this kind of pilot trials, even if not powered to detect low-frequency AE, and points toward the need for a trade-off between the number of participants potentially put at risk in well-powered controlled trials and for caution with the use of uncompletely characterized agents in large numbers of individuals. Given that natural PTC rates are considered to be up to 13% in early-treated individuals, powering trials to show viral control efficacy after an ATI becomes challenging (60). Despite frequent clinical monitoring for pVL and access to psychological support, protocol violation during MAP occurred in one individual, probably due to anxiety secondary to the antiretroviral interruption. This case warrants close psychological management in longer term ATIs.
We fully acknowledge the limitations of the small sample size and lack of a control arm in the present study. Therefore, we interpret these results with caution and regard this study only as hypothesis-generating trial for future interventions. BCN02 eligibility was restricted to vaccinated participants in the parental open-label BCN01 trial. This intrinsic restriction limited the sample size to a small number of previously vaccinated individuals and precluded the inclusion of a control arm. At the time of trial design, interventional trials including an ATI were typically small and included very conservative ART resumption criteria (58, 61). Furthermore, ATI acceptability by participants, risks of HIV-1 transmission to others in the absence of available PrEP, and potential viral re-seeding upon treatment interruption were of special concern in early-treated individuals, who had limited viral reservoirs both in size and diversity (48).
Altogether, the results from this pilot study suggest a potential role for kick&kill strategies in inducing durable immune-mediated HIV-1 control in a proportion of early-treated individuals. In view of these results, future controlled studies to identify the mechanisms underlying sustained HIV-1 suppression are warranted.
All datasets generated for this study are included in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by Hospital Germans Trias i Pujol and Hospital Clínic (AC-15-108-R). The patients/participants provided their written informed consent to participate in this study.
BM, JM, BC, and CB conceived and designed the study. CMa, TH, JMM, JM-P, AB, PC, MM, and MV contributed to the study design. CMi, CMa, IR, JT, ACa, MR-U, and MP contributed contributed to data management. BM, MR-U, MP, ML, SM-L, AL, JM, PC, ACa, CMi, SC, AB, and CMa performed the experiments. JM, BM, MP, MV, YA-S, GM, and KL undertook the statistical analysis. ACr and TH contributed with reagents, materials, analysis tools. BM, JM, and CB drafted the manuscript. TH, MR-U, CMa, MP, SM-L, JM-P, JMM, MV, and BC participated in study analyses and revised the manuscript critically for important intellectual content. All authors reviewed and approved the final version of the manuscript.
IrsiCaixa AIDS Research Institute-HIVACAT, Hospital Universitari Germans Trias i Pujol, Badalona, Spain: Susana Benet, Christian Brander, Samandhy Cedeño, Bonaventura Clotet, Pep Coll, Anuska Llano, Javier Martinez-Picado, Marta Marszalek, Sara Morón-López, Beatriz Mothe, Roger Paredes, Maria C. Puertas, Miriam Rosás-Umbert, Marta Ruiz-Riol. Fundació Lluita contra la Sida, Infectious Diseases Department, Hospital Universitari Germans Trias i Pujol, Badalona, Spain: Roser Escrig, Silvia Gel, Miriam López, Cristina Miranda, José Moltó, Jose Muñoz, Nuria Perez-Alvarez, Jordi Puig, Boris Revollo, Jessica Toro. Germans Trias i Pujol Research Institute, Badalona, Spain: Ana María Barriocanal, Cristina Perez-Reche. Clinical Pharmacology Unit, Hospital Universitari Germans Trias i Pujol, Badalona, Spain: Magí Farré. Pharmacokinetic/pharmacodynamic modeling and simultation, Institut de Recerca de l'Hospital de la Santa Creu i Sant Pau-IIB Sant Pau, Barcelona, Spain: Marta Valle. Hospital Clinic-HIVACAT, IDIBAPS, University of Barcelona, Barcelona, Spain: Christian Manzardo, Juan Ambrosioni, Irene Ruiz, Cristina Rovira, Carmen Ligero, Emma Fernández, and Jose M. Miró. Projecte dels NOMS-Hispanosida, BCN Checkpoint, Barcelona, Spain: Antonio Carrillo, Michael Meulbroek, Ferran Pujol and Jorge Saz. The Jenner Institute, The Nuffield Department of Medicine, University of Oxford, UK: Nicola Borthwick, Alison Crook, Edmund G. Wee and Tomáš Hanke.
BCN02 study was an investigator-initiated study funded by the ISCIII PI15/01188 grant, the HIVACAT Catalan research program for an HIV vaccine and the Fundació Gloria Soler. Some sub-analyses were partly funded by the European Union's Horizon 2020 research and innovation program under grant agreement 681137-EAVI2020 and by NIH grant P01-AI131568. The GMP manufacture of the MVA.HIVconsv vaccines was jointly funded by the Medical Research Council (MRC) UK and the UK Department for International Development (DFID) under the MRC/DFID Concordat agreements (G0701669) and kindly provided through an IIS agreement. Romidepsin was kindly provided by Celgene Ltd through an IIS agreement. BM held a post-doctoral fellowship grant from ISCIII (JR 13/00024) from 2014–16. CMa held a post-doctoral research grant (PERIS- 2016/2020) from the Departament de Salut de la Generalitat de Catalunya, Barcelona, Spain during the conduct of the study. SM-L held a Ph.D. grant from DGR (2013FI_B 00275) from 2013–16. JM received a 80:20 research grant from IDIBAPS, Barcelona, Spain, during 2017–21. GM, KL, and YS have been partially supported by Grant MTM2015-64465-C2-1-R from Ministerio de Economía y Competitividad.
JMM reports grants and personal fees from Abbvie, Angelini, Contrafect, Genentech, Gilead, Jansen, Medtronic, MSD, Pfizer, ViiV Healthcare, outside the submitted work. TH reports grants from Medical Research Council UK, during the conduct of the study, and has a patent US 7981430B2 issued. CB was founder, CSO and shareholder of AELIX THERAPEUTICS, S.L. BM was a consultant for AELIX THERAPEUTICS, S.L., outside the submitted work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank i2e3 for providing medical writing support. We especially thank Agueda Fernandez, Rafaela Ayen, Lucía Gómez and Carmen Ligero for their technical assistance. Special thanks to all the volunteers participating in this study for their perseverance and dedication.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00823/full#supplementary-material
1. Bruner KM, Murray AJ, Pollack RA, Soliman MG, Laskey SB, Capoferri AA, et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat Med. (2016) 22:1043–9. doi: 10.1038/nm.4156
2. Davey RT Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar RN, et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci USA. (1999) 96:15109–14. doi: 10.1073/pnas.96.26.15109
3. Mothe B, Brander C. HIV T-cell vaccines. In: editors. Advances in Experimental Medicine and Biology, Vol. 1075. Springer: New York LLC (2018). p. 31–51. doi: 10.1007/978-981-13-0484-2_2
4. Autran B, Murphy RL, Costagliola D, Tubiana R, Clotet B, Gatell J, et al. Greater viral rebound and reduced time to resume antiretroviral therapy after therapeutic immunization with the ALVAC-HIV vaccine (vCP1452). AIDS. (2008) 22:1313–22. doi: 10.1097/QAD.0b013e3282fdce94
5. Schooley RT, Spritzler J, Wang H, Lederman MM, Havlir D, Kuritzkes DR, et al. AIDS clinical trials group 5197: a placebo-controlled trial of immunization of HIV-1–infected persons with a replication-deficient adenovirus type 5 vaccine expressing the HIV-1 core protein. J Infect Dis. (2010) 202:705–16. doi: 10.1086/655468
6. Harrer T, Plettenberg A, Arastéh K, Van Lunzen J, Fätkenheuer G, Jaeger H, et al. Safety and immunogenicity of an adjuvanted protein therapeutic HIV-1 vaccine in subjects with HIV-1 infection: a randomised placebo-controlled study. Vaccine. (2014) 32:2657–65. doi: 10.1016/j.vaccine.2013.10.030
7. Evans TG, Keefer MC, Weinhold KJ, Wolff M, Montefiori D, Gorse GJ, et al. A canarypox vaccine expressing multiple human immunodeficiency virus type 1 genes given alone or with Rgp120 elicits broad and durable CD8+ cytotoxic T lymphocyte responses in seronegative volunteers. J Infect Dis. (1999) 180:290–8. doi: 10.1086/314895
8. Sneller MC, Justement JS, Gittens KR, Petrone ME, Clarridge KE, Proschan MA, et al. A randomized controlled safety/efficacy trial of therapeutic vaccination in HIV-infected individuals who initiated antiretroviral therapy early in infection. Sci Transl Med. (2017) 9:eaan8848. doi: 10.1126/scitranslmed.aan8848
9. Fischer W, Perkins S, Theiler J, Bhattacharya T, Yusim K, Funkhouser R, et al. Polyvalent vaccines for optimal coverage of potential T-cell epitopes in global HIV-1 variants. Nat Med. (2007) 13:100–6. doi: 10.1038/nm1461
10. Theiler J, Yoon H, Yusim K, Picker LJ, Fruh K, Korber B. Epigraph: a vaccine design tool applied to an HIV therapeutic vaccine and a pan-filovirus vaccine. Sci Rep. (2016) 6:33987. doi: 10.1038/srep33987
11. Mothe B, Hu X, Llano A, Rosati M, Olvera A, Kulkarni V, et al. A human immune data-informed vaccine concept elicits strong and broad T-cell specificities associated with HIV-1 control in mice and macaques. J Transl Med. (2015) 13:60. doi: 10.1186/s12967-015-0392-5
12. Létourneau S, Im EJ, Mashishi T, Brereton C, Bridgeman A, Yang H, et al. Design and pre-clinical evaluation of a universal HIV-1 vaccine. PLoS ONE. (2007) 2:e984. doi: 10.1371/journal.pone.0000984
13. Rolland M, Nickle DC, Mullins JI. HIV-1 group M conserved elements vaccine. PLoS Pathog. (2007) 3:e157. doi: 10.1371/journal.ppat.0030157
14. Ondondo B, Murakoshi H, Clutton G, Abdul-Jawad S, Wee EG, Gatanaga H, et al. Novel conserved-region T-cell mosaic vaccine with high global HIV-1 coverage is recognized by protective responses in untreated infection. Mol Ther. (2016) 24:832–42. doi: 10.1038/mt.2016.3
15. Gaiha GD, Rossin EJ, Urbach J, Landeros C, Collins DR, Nwonu C, et al. Structural topology defines protective CD8 + T cell epitopes in the HIV proteome. Science. (2019) 364:480–4. doi: 10.1126/science.aav5095
16. Borthwick N, Ahmed T, Ondondo B, Hayes P, Rose A, Ebrahimsa U, et al. Vaccine-elicited human T cells recognizing conserved protein regions inhibit HIV-1. Mol Ther. (2014) 22:464–75. doi: 10.1038/mt.2013.248
17. Hayton EJ, Rose A, Ibrahimsa U, Del Sorbo M, Capone S, Crook A, et al. Safety and tolerability of conserved region vaccines vectored by plasmid DNA, simian adenovirus and modified vaccinia virus ankara administered to human immunodeficiency virus type 1-uninfected adults in a randomized, single-blind phase I trial. PLoS ONE. (2014) 9:e101591. doi: 10.1371/journal.pone.0101591
18. Mutua G, Farah B, Langat R, Indangasi J, Ogola S, Onsembe B, et al. Broad HIV-1 inhibition in vitro by vaccine-elicited CD8+ T cells in African adults. Mol Ther Methods Clin Dev. (2016) 3:16061. doi: 10.1038/mtm.2016.61
19. Hancock G, Morón-López S, Kopycinski J, Puertas MC, Giannoulatou E, Rose A, et al. Evaluation of the immunogenicity and impact on the latent HIV-1 reservoir of a conserved region vaccine, MVA.HIVconsv, in antiretroviral therapy-treated subjects. J Int AIDS Soc. (2017) 20:1–11. doi: 10.7448/IAS.20.1.21171
20. Mothe B, Manzardo C, Sanchez-Bernabeu A, Coll P, Morón-López S, Puertas MC, et al. Therapeutic vaccination refocuses T-cell responses towards conserved regions of HIV-1 in early treated individuals (BCN 01 study). Clin Med. (2019) 11:65–80. doi: 10.1016/j.eclinm.2019.05.009
21. Hartnell F, Brown A, Capone S, Kopycinski J, Bliss C, Makvandi-Nejad S, et al. A novel vaccine strategy employing serologically different chimpanzee adenoviral vectors for the prevention of HIV-1 and HCV coinfection. Front Immunol. (2018) 9:3175. doi: 10.3389/fimmu.2018.03175
22. Borthwick NJ, Lane T, Moyo N, Crook A, Shim JM, Baines I, et al. Randomized phase I trial HIV-CORE 003: depletion of serum amyloid P component and immunogenicity of DNA vaccination against HIV-1. Borrow R, ed. PLoS ONE. (2018) 13:e0197299. doi: 10.1371/journal.pone.0197299
23. Fidler S, Stöhr W, Pace M, Dorrell L, Lever A, Pett S, et al. Antiretroviral therapy alone versus antiretroviral therapy with a kick and kill approach, on measures of the HIV reservoir in participants with recent HIV infection (the RIVER trial): a phase 2, randomised trial. The Lancet. (2020) 395:888–98. doi: 10.1016/S0140-6736(19)32990-3
24. Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. (2012) 36:491–501. doi: 10.1016/j.immuni.2012.01.014
25. Sengupta S, Siliciano RF. Targeting the latent reservoir for HIV-1. Immunity. (2018) 48:872–95. doi: 10.1016/j.immuni.2018.04.030
26. Pankrac J, Klein K, Mann JFS. Eradication of HIV-1 latent reservoirs through therapeutic vaccination. AIDS Res Ther. (2017) 14:45. doi: 10.1186/s12981-017-0177-4
27. Rasmussen TA, Schmeltz Søgaard O, Brinkmann C, Wightman F, Lewin SR, Melchjorsen J, et al. Comparison of HDAC inhibitors in clinical development: effect on HIV production in latently infected cells and T-cell activation. Hum Vaccin Immunother. (2013) 9:993–1001. doi: 10.4161/hv.23800
28. Bullen CK, Laird GM, Durand CM, Siliciano JD, Siliciano RF. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat Med. (2014) 20:425–9. doi: 10.1038/nm.3489
29. Shan L, Xing S, Yang H-C, Zhang H, Margolick JB, Siliciano RF. Unique characteristics of histone deacetylase inhibitors in reactivation of latent HIV-1 in Bcl-2-transduced primary resting CD4+ T cells. J Antimicrob Chemother. (2014) 69:28–33. doi: 10.1093/jac/dkt338
30. Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. (2012) 487:482–5. doi: 10.1038/nature11286
31. Lehrman G, Hogue IB, Palmer S, Jennings C, Spina CA, Wiegand A, et al. Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet. (2005) 366:549–55. doi: 10.1016/S0140-6736(05)67098-5
32. Wei DG, Chiang V, Fyne E, Balakrishnan M, Barnes T, Graupe M, et al. Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathog. (2014) 10:e1004071. doi: 10.1371/journal.ppat.1004071
33. Søgaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, et al. The depsipeptide romidepsin reverses HIV-1 latency in vivo. Siliciano RF, ed. PLoS Pathog. (2015) 11:e1005142. doi: 10.1371/journal.ppat.1005142
34. Leth S, Schleimann MH, Nissen SK, Højen JF, Olesen R, Graversen ME, et al. Combined effect of Vacc-4x, recombinant human granulocyte macrophage colony-stimulating factor vaccination, and romidepsin on the HIV-1 reservoir (REDUC): a single-arm, phase 1B/2A trial. Lancet HIV. (2016) 3:e463–72. doi: 10.1016/S2352-3018(16)30055-8
35. Moron-Lopez S, Kim P, Søgaard OS, Tolstrup M, Wong JK, Yukl SA. Characterization of the HIV-1 transcription profile after romidepsin administration in ART-suppressed individuals. AIDS. (2019) 33:425–31. doi: 10.1097/QAD.0000000000002083
36. Moltó J, Perez C, Valle M, Mothe B, Miranda C, Farré M, et al. Population pharmacokinetics of romidepsin as a latency reactivating agent in HIV-infected adults. In: 18th International Workshop on Clinical Pharmacologyof Antiviral Therapy. Chicago: IL (2017).
37. Morón-López S, Puertas MC, Gálvez C, Navarro J, Carrasco A, Esteve M, et al. Sensitive quantification of the HIV-1 reservoir in gut-associated lymphoid tissue. Roques P, ed. PLoS ONE. (2017) 12:e0175899. doi: 10.1371/journal.pone.0175899
38. Gupta RK, Abdul-Jawad S, McCoy LE, Mok HP, Peppa D, Salgado M, et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature. (2019) 568:244–8. doi: 10.1038/s41586-019-1027-4
39. Kawana-Tachikawa A, Llibre JM, Bravo I, Escrig R, Mothe B, Puig JP, et al. Effect of maraviroc intensification on HIV-1-specific T cell immunity in recently HIV-1-infected individuals. PLoS ONE. (2014) 9:e87334. doi: 10.1371/journal.pone.0087334
40. Sáez-Cirión A, Bacchus C, Hocqueloux L, Avettand-Fenoel V, Girault I, Lecuroux C, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI study. PLoS Pathog. (2013) 9:e1003211. doi: 10.1371/journal.ppat.1003211
41. Wacholder S. Binomial regression in GLIM: estimating risk ratios and risk differences. Am J Epidemiol. (1986) 123:174–84. doi: 10.1093/oxfordjournals.aje.a114212
42. R Core Team. R: A Language and Environment for Statistical Computing. (2017). Available online at: https://www.r-project.org/ (accessed February 1, 2017).
43. Woo S, Gardner ER, Chen X, Ockers SB, Baum CE, Sissung TM, et al. Population pharmacokinetics of romidepsin in patients with cutaneous T-cell lymphoma and relapsed peripheral T-cell lymphoma. Clin Cancer Res. (2009) 15:1496–503. doi: 10.1158/1078-0432.CCR-08-1215
44. Mogal A, Abdulkadir SA. Effects of histone deacetylase inhibitor (HDACi) trichostatin-A (TSA) on the expression of housekeeping genes. Mol Cell Probes. (2006) 20:81–6. doi: 10.1016/j.mcp.2005.09.008
45. Gao X, Bashirova A, Iversen AK, Phair J, Goedert JJ, Buchbinder S, et al. AIDS restriction HLA allotypes target distinct intervals of HIV-1 pathogenesis. Nat Med. (2005) 11:1290–92. doi: 10.1038/nm1333
46. Carrington M, O'Brien SJ. The influence of HLA genotype on AIDS. Annu Rev Med. (2003) 54:535–51. doi: 10.1146/annurev.med.54.101601.152346
47. Robb ML, Ananworanich J. Lessons from acute HIV infection. Curr Opin HIV AIDS. (2016) 11:555–60. doi: 10.1097/COH.0000000000000316
48. Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature. (2015) 517:381–5. doi: 10.1038/nature14053
49. McMahon D, Zheng L, Cyktor JC, Aga E, Macatangay BJ, Godfrey C, et al. Multidose IV romidepsin: no increased HIV-1 expression in persons on ART, ACTG A5315. Conference Retrovirus and Opportunistic Infections, March 4–7 2019, Seattle (#26). Seattle: Washington (2019).
50. Leth S, Nymann R, Jørgensen S, Olesen R, Rasmussen TA, Østergaard L, et al. HIV-1 transcriptional activity during frequent longitudinal sampling in aviremic patients on antiretroviral therapy. AIDS. (2016) 30:713–21. doi: 10.1097/QAD.0000000000000974
51. Elliott JH, McMahon JH, Chang CC, Lee SA, Hartogensis W, Bumpus N, et al. Short-term administration of disulfiram for reversal of latent HIV infection: a phase 2 dose-escalation study. Lancet HIV. (2015) 2:e520–29. doi: 10.1016/S2352-3018(15)00226-X
52. Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, et al. Panobinostat, a histone deacetylase inhibitor, for latentvirus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV. (2014) 1:e13–21. doi: 10.1016/S2352-3018(14)70014-1
53. Ruiz A, Blanch-Lombarte O, Jimenez-Moyano E, Ouchi D, Mothe B, Peña R, et al. Antigen production after latency reversal and expression of inhibitory receptors in CD8+ T cells limit the killing of HIV-1 reactivated cells. Front Immunol. (2019) 9:3162. doi: 10.3389/fimmu.2018.03162
54. Jones RB, O'Connor R, Mueller S, Foley M, Szeto GL, Karel D, et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog. (2014) 10:e1004287. doi: 10.1371/journal.ppat.1004287
55. Li JZ, Etemad B, Ahmed H, Aga E, Bosch RJ, Mellors JW, et al. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS. (2016) 30:343–53. doi: 10.1097/01.aids.0000499516.66930.89
56. Williams JP, Hurst J, Stöhr W, Robinson N, Brown H, Fisher M, et al. HIV-1 DNA predicts disease progression and post-treatment virological control. Elife. (2014) 3:e03821. doi: 10.7554/eLife.03821
57. Namazi G, Fajnzylber JM, Aga E, Bosch RJ, Acosta EP, Sharaf R, et al. The control of HIV after antiretroviral medication pause (CHAMP) study: posttreatment controllers identified from 14 clinical studies. J Infect Dis. (2018) 218:1954–63. doi: 10.1093/infdis/jiy479
58. Colby DJ, Trautmann L, Pinyakorn S, Leyre L, Pagliuzza A, Kroon E, et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in fiebig I acute HIV infection. Nat Med. (2018) 24:923–6. doi: 10.1038/s41591-018-0026-6
59. Huiting ED, Gittens K, Justement JS, Shi V, Blazkova J, Benko E, et al. Impact of treatment interruption on HIV reservoirs and lymphocyte subsets in individuals who initiated antiretroviral therapy during the early phase of infection. J Infect Dis. (2019) 220:270–74. doi: 10.1093/infdis/jiz100
60. Julg B, Dee L, Ananworanich J, Barouch DH, Bar K, Caskey M, et al. Recommendations for analytical antiretroviral treatment interruptions in HIV research trials—report of a consensus meeting. Lancet HIV. (2019) 6:e259–68. doi: 10.1016/S2352-3018(19)30052-9
Keywords: romidepsin, HDAC inhibitor, kick&kill strategy, HIVconsv, early-treatment
Citation: Mothe B, Rosás-Umbert M, Coll P, Manzardo C, Puertas MC, Morón-López S, Llano A, Miranda C, Cedeño S, López M, Alarcón-Soto Y, Melis GG, Langohr K, Barriocanal AM, Toro J, Ruiz I, Rovira C, Carrillo A, Meulbroek M, Crook A, Wee EG, Miró JM, Clotet B, Valle M, Martinez-Picado J, Hanke T, Brander C, Moltó J and the BCN02 Study Investigators (2020) HIVconsv Vaccines and Romidepsin in Early-Treated HIV-1-Infected Individuals: Safety, Immunogenicity and Effect on the Viral Reservoir (Study BCN02). Front. Immunol. 11:823. doi: 10.3389/fimmu.2020.00823
Received: 13 February 2020; Accepted: 09 April 2020;
Published: 06 May 2020.
Edited by:
Carolina Garrido, University of North Carolina at Chapel Hill, United StatesReviewed by:
Paul W. Denton, University of Nebraska Omaha, United StatesCopyright © 2020 Mothe, Rosás-Umbert, Coll, Manzardo, Puertas, Morón-López, Llano, Miranda, Cedeño, López, Alarcón-Soto, Melis, Langohr, Barriocanal, Toro, Ruiz, Rovira, Carrillo, Meulbroek, Crook, Wee, Miró, Clotet, Valle, Martinez-Picado, Hanke, Brander, Moltó and the BCN02 Study Investigators. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beatriz Mothe, Ym1vdGhlQGlyc2ljYWl4YS5lcw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.