
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 05 June 2020
Sec. Microbial Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00786
This article is part of the Research TopicExploiting Novel Combined Host- and Pathogen-Directed Therapies for Combating Bacterial Multidrug ResistanceView all 12 articles
 Katie Watson1,2
Katie Watson1,2 Clark D. Russell1,2,3
Clark D. Russell1,2,3 J. Kenneth Baillie3
J. Kenneth Baillie3 Kev Dhaliwal2J. Ross Fitzgerald3
Kev Dhaliwal2J. Ross Fitzgerald3 Timothy J. Mitchell4A. John Simpson5
Timothy J. Mitchell4A. John Simpson5 Stephen A. Renshaw6
Stephen A. Renshaw6 David H. Dockrell1,2* on behalf of the SHIELD consortium
David H. Dockrell1,2* on behalf of the SHIELD consortiumAntimicrobial therapy has provided the main component of chemotherapy against bacterial pathogens. The effectiveness of this strategy has, however, been increasingly challenged by the emergence of antimicrobial resistance which now threatens the sustained utility of this approach. Humans and animals are constantly exposed to bacteria and have developed effective strategies to control pathogens involving innate and adaptive immune responses. Impaired pathogen handling by the innate immune system is a key determinant of susceptibility to bacterial infection. However, the essential components of this response, specifically those which are amenable to re-calibration to improve host defense, remain elusive despite extensive research. We provide a mini-review focusing on therapeutic targeting of microbicidal responses in macrophages and neutrophils to de-stress reliance on antimicrobial therapy. We highlight pre-clinical and clinical data pointing toward potential targets and therapies. We suggest that developing focused host-directed therapeutic strategies to enhance “pauci-inflammatory” microbial killing in myeloid phagocytes that maximizes pathogen clearance while minimizing the harmful consequences of the inflammatory response merits particular attention. We also suggest the importance of One Health approaches in developing host-based approaches through model development and comparative medicine in informing our understanding of how to deliver this strategy.
Antimicrobial chemotherapy has formed the cornerstone of our therapeutic strategy against bacterial disease since penicillin was first developed. Prior to this, developing host-based therapy was a major focus, including Fleming's original work on lysozyme, a humoral microbicide he isolated while seeking antimicrobial factors in pus (1). The first therapeutic use of penicillin in 1930 (treating eye infections in babies in Sheffield by Cecil Paine), and the pioneering work of Florey, Chain and colleagues in Oxford who developed innovations in penicillin synthesis to allow the first clinical trials in 1941, established antimicrobial chemotherapy as the pre-eminent therapeutic approach to bacterial disease (2). This has had a major impact on human health but arguably diverted focus away from host-based approaches other than vaccination.
Recent public health estimates suggest antimicrobial resistant bacteria cause 131 infections/100,000 population in Europe and that two-thirds are nosocomial (3). The disability adjusted life years of these infections approximates tuberculosis, influenza and HIV combined (3). In addition, development of new antimicrobials has been declining (4). There is thus a pressing need to develop new antimicrobials, improved antimicrobial stewardship, better diagnostics to identify the patients who truly need antimicrobials, and alternative approaches, for example those involving bacteriophage therapy, nanoparticle-based therapy, photodynamic light therapy and antimicrobial peptides (AMP) to manage infection with antimicrobial resistant ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.) pathogens (5). While vaccination remains a major focus, the concept of developing host-based therapy is gaining traction.
Pathogenic bacteria commonly colonize healthy individuals without causing disease. S. aureus is carried by >40% of infants after birth and ~50% of adults are permanent or intermittent carriers (6, 7). Uropathogenic Escherichia coli is typically part of an individual's fecal microbiota and healthy individuals carry a large number of potentially pathogenic strains (8). In other cases, pathogens are harmless microbiome constituents but cause opportunistic infections in patients whose immune system is impaired by medical co-morbidity, such as nosocomial enterococcal infections (9). This apparent paradox, between common carriage but uncommon disease, suggests most infections are readily controlled by the host yet the specific microbicidal responses that control infection when small numbers of colonizing bacteria translocate to new sites is incompletely defined. Broadly, the innate immune system ensures a rapid response, working in concert with any adaptive immune responses to the pathogen. There are many components to the innate immune system including mucosal barrier function, humoral factors released in mucosal secretions and a range of innate cellular responses that are not restricted to myeloid phagocytes but also include innate lymphoid cells. These responses are modified through adaptive immune responses, but the focus of this review is exclusively on myeloid phagocyte responses.
Professional phagocytes (macrophages and neutrophils) clear bacteria from mucosa associated with a low-density microbiome, for example the distal airway or bladder (10). Macrophages play a critical role in the initial response as the resident phagocytes in tissues, using pattern recognition receptors (PRRs) to detect pathogens and orchestrate the inflammatory response. They are efficient at phagocytosing bacteria and utilize a range of microbicidal strategies to kill ingested bacteria. Tissue macrophage function is tightly controlled by activation state which is regulated by a cell network including epithelial, endothelial, T- and B- lymphocytes, as well as tissue resident innate lymphoid cells. The resulting cytokine networks reflect the importance of environmental cues (11). Innate immune memory ensures previous pathogen exposure modulates macrophage function via epigenetic imprinting of monocytes to induce “training” (enhanced microbicidal responses to repeat challenge) and “tolerance” (reduced deleterious responses to repeat challenge) to pathogen-associated molecular patterns (12, 13). Lipopolysaccharide (LPS) engagement of Toll-like receptor (TLR) 4 is just one example amongst several of a microbial stimulus that can on repeat stimulation be associated with tolerance manifest as reduced generation of pro-inflammatory cytokines and reactive species (14). This has implications for monocyte-derived macrophage populations but the extent to which it also influences resident macrophage populations with distinct ontogeny remains to be established. Though capable of avid phagocytosis, tissue macrophages have a finite capacity to kill ingested bacteria (15). This capacity can be diminished by interactions with other microorganisms e.g., viruses, environmental factors or co-morbidity, resulting in increased susceptibility to bacterial disease. For example, both HIV-1 infection and chronic obstructive pulmonary disease (COPD) impair alveolar macrophage (AM) killing of pneumococci (16, 17). Furthermore, pathogenic bacteria have evolved mechanisms to withstand microbicides, such as antioxidant systems (18). Successful pathogens such as S. aureus inhibit phagosomal maturation contributing to intracellular survival (19), while others that are more readily killed may escape killing in subsets of macrophages, as exemplified by survival of pneumococci in permissive CD169+ splenic macrophages in murine and porcine models (20). Several potentially AMR pathogens such as K. pneumoniae and P. aeruginosa can subvert phagosomal maturation in macrophages (21, 22). Traditional paradigms of intracellular and extracellular bacteria are blurring and the intracellular fate of the so-called extracellular bacteria (including medically important ESKAPE pathogens, Haemophilus influenzae and Streptococcus pneumoniae) is likely a major determinant of infection outcome.
When the intracellular killing capacity of resident tissue macrophages is overwhelmed, they orchestrate recruitment of neutrophils and other inflammatory cells. Murine models of clodronate-mediated AM depletion illustrate how escalating bacterial challenge shifts the role of AM from primary effectors of bacterial clearance to regulators of the inflammatory response, with neutrophils required for pathogen clearance (15, 23). The exhaustion of macrophage clearance capacity is likely also a feature of systemic infections, as evidenced for Kupffer cells in the liver and is augmented by commensal bacteria (24). This represents the transition from sub-clinical infection to clinical disease, and signs of neutrophilic inflammation are used to establish a clinical diagnosis. The inflammatory response, however, contributes to tissue injury since potent microbicides, such as reactive oxygen species (ROS), can cause tissue injury and organ dysfunction (25). Nevertheless, this inflammatory response is essential and neutrophil deficiency results in severe bacterial infection (26). Neutrophil microbicidal responses have been extensively characterized and include ROS, AMP, divalent metal iron-sequestering proteins (e.g., lactoferrin), proteases such as the serine proteases contained in azurophilic granules (e.g., cathepsin G and neutrophil elastase) and acid hydrolases in lysosomes (26). The pre-eminence of ROS as a direct microbicidal mechanism has been challenged by observations that it is the associated ionic changes in the phagosome, activating granule-associated serine proteases, that actually mediate microbicidal killing (27). Neutrophils can also release granule contents and DNA extracellular traps to kill bacteria (28).
The challenge is therefore to generate an effective response that maximizes pathogen clearance and minimizes the inflammatory response, either by enhancing the macrophage response to raise the threshold for induction of neutrophilic inflammation or by ensuring the neutrophilic component achieves pathogen clearance yet limits bystander tissue injury. We term this desirable microbicidal profile a “pauci-inflammatory microbicidal response” recognizing that its characteristics include rapid induction, effective pathogen killing, and controlled recruitment of inflammatory cells when needed, but also early resolution and tightly regulated production of potentially damaging microbicidal species (Figure 1). It builds on concepts articulated by Sears and colleagues in chronic parasitic infections where the cost of the host response (immunopathology) is weighed against resistance to the pathogen (29). In the case of common “extracellular” bacterial disease, the primary cost becomes tissue injury/organ dysfunction due to the microbicidal response and chronic infection is a rare outcome. If initial microbicidal responses by phagocytes are sub-optimal, the inflammatory response is escalated with increased recruitment of neutrophils, macrophages and lymphocytes that have the potential to promote self-propagating waves of inflammation driven by release of damage-associated molecular patterns in response to tissue injury. Excessive production of cytokines, reactive species, proteases, phospholipids and eicosanoids mediate inflammatory tissue injury, induction of various cell death paradigms and ultimately loss of tissue homeostasis. These principles are well-exemplified by the development of acute respiratory distress syndrome (ARDS) (30). Organ specific injury is also associated with a systemic inflammatory response which can cause multiorgan failure (31). In addition, the generalized inflammatory response can lead to immunosuppression with impaired immune responses on subsequent pathogen challenge (32). It is therefore essential to limit these dysregulated inflammatory responses and induce a more limited inflammatory response with optimal pathogen clearance, by targeting microbicidal responses. To target potential bottlenecks in the host microbicidal response, we must identify optimal responses that promote resilience in the healthy population and patient groups in whom these fail. We need to develop assays to assess the host response and effect of therapy.
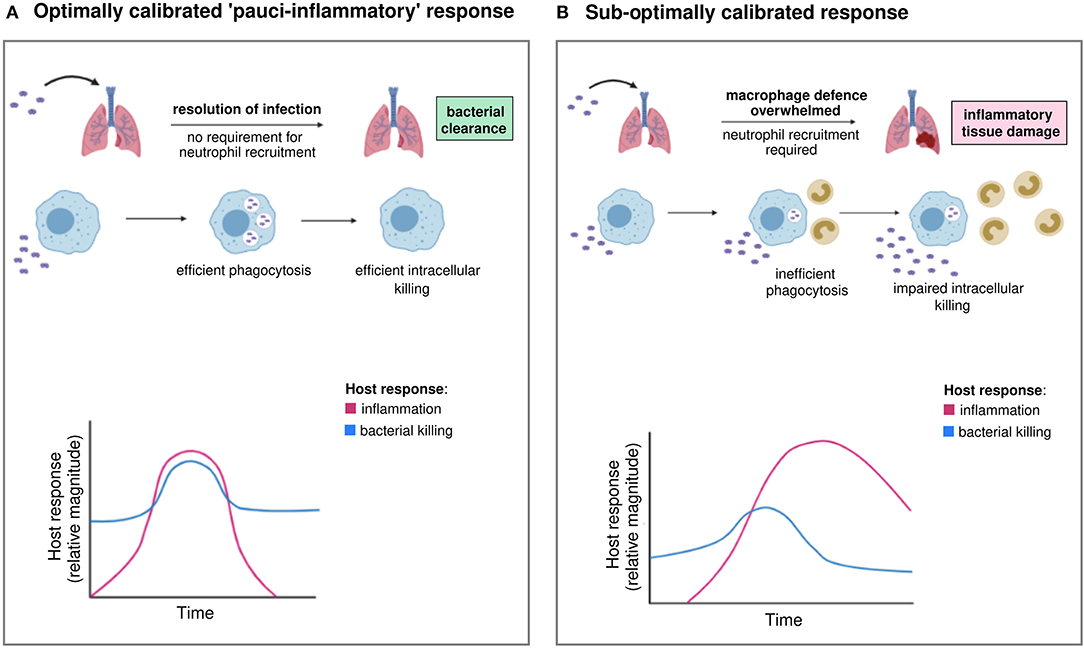
Figure 1. Optimal and sub-optimal inflammatory and bacterial killing trajectories during infection. (A) Invading extracellular bacteria are recognized and phagocytosed by macrophages, followed by intracellular killing. Pathogen clearance is optimal and achieved without the requirement for neutrophil recruitment. Inflammation is tightly controlled and resolves without causing tissue damage. We term this optimally calibrated response “pauci-inflammatory.” (B) In hosts with sub-optimally calibrated responses, there is inefficient phagocytosis and/or intracellular killing by macrophages, resulting in incomplete bacterial clearance. When macrophage defense is overwhelmed beyond a “tipping point,” neutrophil recruitment is required to control the invading pathogen. Inflammation is more prolonged and sustained by pathogen persistence and/or tissue damage. Inflammatory responses give rise to clinically recognizable features of disease, for example pneumonia. Images created using BioRender.com.
A critical bottleneck in host defense involves macrophage bacterial clearance (19, 33). However, therapeutic modulation of this is impeded by limitations in our understanding of microbicidal responses in tissue macrophages, which are often inferred from neutrophils, monocytes or monocytic cell lines. Well-established microbicidal mechanisms in other phagocytes may not operate in tissue macrophages, which (excluding those in atherosclerotic plaques) lack the ability to produce the more potent halogenated ROS like hypochlorous acid (34, 35). Some microbicidal responses are more convincingly demonstrated in mice than man, for example those involving nitric oxide (NO), which may be produced at lower levels in human macrophages, although several groups have detected it following bacterial challenge (36). Effective responses likely require combinations of microbicidals. Defining these has been limited by how well in vitro macrophage cultures mirror tissue macrophages in vivo. Many tissue macrophages with low-level homeostatic turnover arise from embryonic yolk sac or fetal liver hematopoietic stem cell progenitors and are maintained by division of resident cells, e.g., AM derived from fetal liver precursors (37). Monocyte-derived macrophages (MDM) give rise to macrophages in the gut and peritoneum, populations associated with a higher turnover, but we cannot assume their microbicidal responses are identical to macrophages derived from embryonic progenitors. In addition, tissue macrophage maturation is heavily influenced by environmental cues and their transcriptional profiles are as distinct as they are from monocytes (38).
Irrespective of these limitations there are many similarities between microbicidal mechanisms of different macrophage populations. A range of primary human macrophages (including MDM and AM) and murine models demonstrate an initial phase of extensive intracellular killing, activated in the phagosome. For pathogens such as pneumococci, this is followed by a delayed phase of bacterial killing, involving apoptosis-associated killing that clears residual viable bacteria (16, 19, 33). These responses often involve combinations of microbicidals (Figure 2), for example ROS and NO, which helps subvert pathogen resistance (33). Tissue macrophages modify the phagosomal environment to inhibit bacterial survival; phagolysosomal acidification and restriction of divalent metal cations inhibits bacterial enzymes, including manganese-containing superoxide dismutase. Nevertheless, the role of these responses is more established in killing intracellular bacteria, compared to internalized extracellular bacteria (39). These defenses are complemented by AMP and proteases. Matrix metalloproteinase 12 contributes to early killing of bacteria in macrophages (40). The cathelicidin LL-37 enhances killing of bacteria including S. aureus in macrophages and is taken up from exogenous sources to complement ROS generation and lysosome fusion (41). AMR in E. coli can increase the sensitivity to AMP, suggesting host-based strategies can synergize with antimicrobials or with antimicrobial selective pressure (42). Similarly, a synthetic peptide derived from human lactoferrin synergizes with antimicrobials against a carbapenemase-producing K. pneumoniae (43). However, there are also examples where mutations inducing AMR may also enable resistance to AMP; modification of K. pneumoniae lipid A not only enables resistance to polymyxins but also β-defensins and human neutrophil peptide-1 (44). Many other AMP and proteases contribute to microbicidal responses, but the mechanism may be indirect. For example cathepsin D enhances apoptosis-associated killing by increasing proteasomal degradation of the anti-apoptotic Bcl-2 family member Mcl-1 (45).
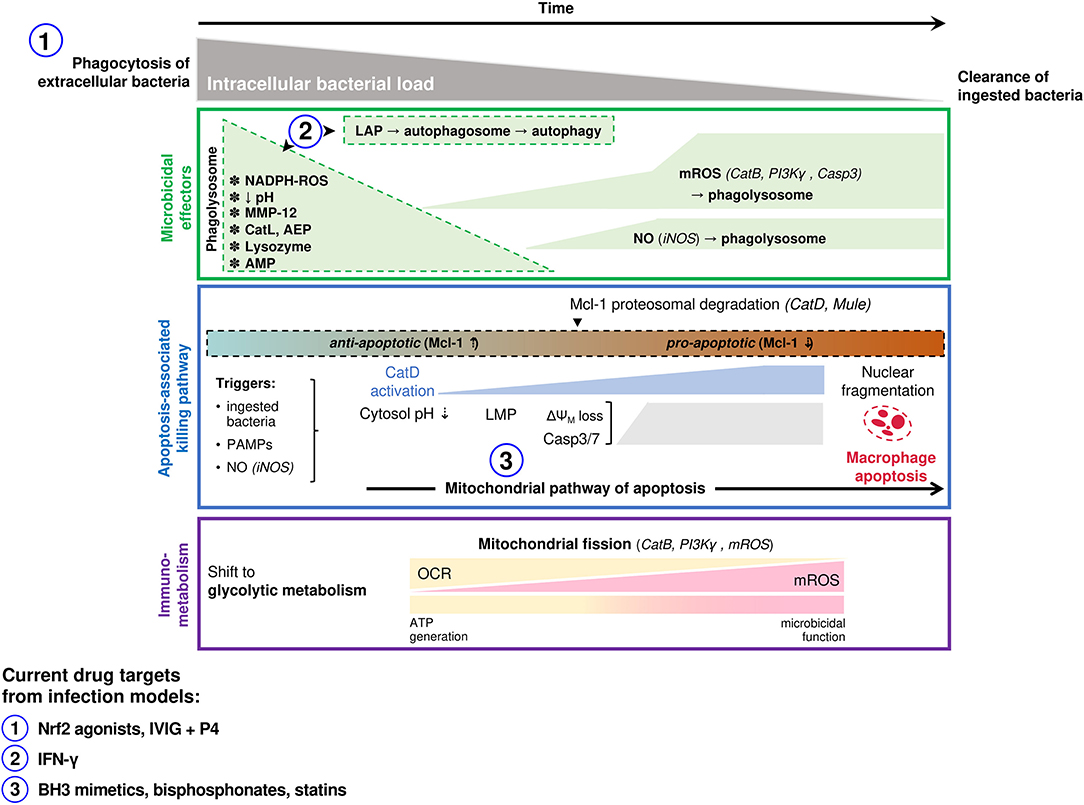
Figure 2. Macrophage microbicidal responses involved in successful clearance of extracellular bacterial pathogens. Macrophage responses to ingested extracellular bacteria (e.g., S. pneumoniae, S. aureus, P. aeruginosa) are summarized. Following phagocytosis of bacteria an initial microbicidal response occurs in the phagolysosome (top panel). Specific effectors with demonstrated microbicidal roles differs based on the ingested organism, and include NADPH derived ROS, MMP-12 (S. aureus), cathepsin L (S. aureus), asparagine endopeptidase (P. aeruginosa), lysozyme and antimicrobial peptides. Microbicidal species produced later that co-localize to bacteria-containing phagolysosomes include NO and mROS which have demonstrated roles in killing ingested pneumococci. A mitochondrial pathway of host-directed apoptosis is engaged in response to live ingested pneumococci, involving recognition of pneumolysin and accumulation of NO (middle panel). This has been best studied in pneumococcal models, where it allows pauci-inflammatory clearance of bacteria that have survived initial phagolysosomal killing, but may occur for other extracellular bacteria also. Immuno-metabolic changes that underpin the microbicidal function of macrophages have also been characterized well in pneumococcal models and also in some other extracellular bacterial infections (bottom panel). This involves an early shift to glycolysis and a progressive transition of mitochondrial function from ATP generation (oxidative phosphorylation) to become microbicidal organelles (mROS generation). Targets of host-directed therapeutics that have been investigated in infection studies (clinical or pre-clinical) are indicated. The number corresponding to each indicates the stage in the killing process where it acts, as indicated on the panels above. LAP, LC-3 associated phagocytosis; MMP, matrix metalloproteinase; Cat, cathepsin; AEP, asparagine endopeptidase; AMP, antimicrobial peptide; ROS, reactive oxygen species; mROS, mitochondrial ROS; NO, nitric oxide; PI3K, phosphoinositide 3-kinase; Casp, caspase; iNOS, inducible nitric oxide synthase; Mcl-1, myeloid cell leukemia-1; PAMP, pathogen-associated molecular pattern; LMP, lysosomal membrane permeabilization; ΔΨM, mitochondrial membrane potential; OCR, oxygen consumption rate; IVIG, intravenous immunoglobulin; IFN, interferon.
The ability to perform lentiviral delivery of genome-scale clustered regularly interspaced short palindromic repeats (CRISPR)-associated nuclease Cas9 knock-out (GeCKO) pooled libraries to human cells allows whole genome screening with the potential to shed new light on microbicidal mechanisms (46, 47). A further potential approach is to harness comparative biology and aims to use convergent evolution of pathogens as they shift species tropism (48) or divergent evolution within species as they rapidly evolve under a host-selective pressure (49), to probe microbicidal mechanisms. Nevertheless, identifying microbicidal mechanisms as targets for immunomodulation will also require evidence that these are sub-optimally calibrated in patient groups with increased susceptibility to bacterial disease. For example, AM from patients with COPD fail to enhance mitochondrial ROS (mROS) production following bacterial challenge (16). This is important since mROS has recently emerged as a key microbicide affecting bacterial killing in the macrophage phagolysosome (33, 50). Evaluation of potential microbicidal targets will also require application of super-resolution microscopy and other advanced imaging modalities, combined with advances in probes, optics and analytics to provide temporal and spatial resolution of microbicidal generation. In the past, generation at a population level using automated systems such as flow cytometry has been assumed to be a surrogate for this but may be insufficient to allow optimal characterization. In vivo imaging is also a valuable adjunct and comparative medicine using large animals such as pigs, whose immune system is similar to humans, and studies in humans will aid translation in models of infection (51, 52).
Only a few strategies to modulate the host response to bacteria have progressed to clinical trials, and specific assessment of target microbicidal responses is often lacking (Table 1). Interferon (IFN)-γ is established in the treatment of chronic granulomatous disease (CGD), a genetic disorder in which deficiency in one of the components of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase leads to increased susceptibility to a range of infections. While this is an extreme case of adjusting an immune response, it shows immunomodulation can be used to enhance microbicidal responses. Clinical trial data shows IFN-γ reduces the frequency of severe infections in CGD and it has also been investigated for multi-drug resistant tuberculosis, Mycobacterium avium complex and Cryptococcus neoformans infections (53, 68). IFN-γ enhances several microbicidal mechanisms and has been shown to correct defective ex vivo killing of the intracellular pathogen Burkholderia cenocepacia in cystic fibrosis (CF) MDM by enhancing autophagy, a regulated cellular process that enables removal and recycling of macromolecules and organelles to promote cellular homeostasis and a related cell process using autophagy machinery that leads to killing of ingested bacteria termed xenophagy (59). However, nebulized IFN-γ did not reduce bacterial density or inflammation in a clinical trial in CF (69). In critically ill adults, clinical trial data demonstrates that IFN-γ is associated with clearance of persistent bacteremia and improved cytokine profiles in the setting of sepsis-induced immunosuppression. Further investigation in clinical trials in sepsis is ongoing (70). It has also been shown to correct HLA-DR expression on monocytes in patients with sepsis which provides a useful marker of response (56). In a case report, IFN-γ enabled clearance of persistent S. aureus bacteremia in association with transcriptional profiles associated with a shift toward Th1/Th17 responses and antigen-specific T-regs, though the specific consequences for microbicidal responses were not examined (58). In patients with septic shock and lymphopenia, IL-7 has been shown to reverse sepsis-induced lymphopenia (55).
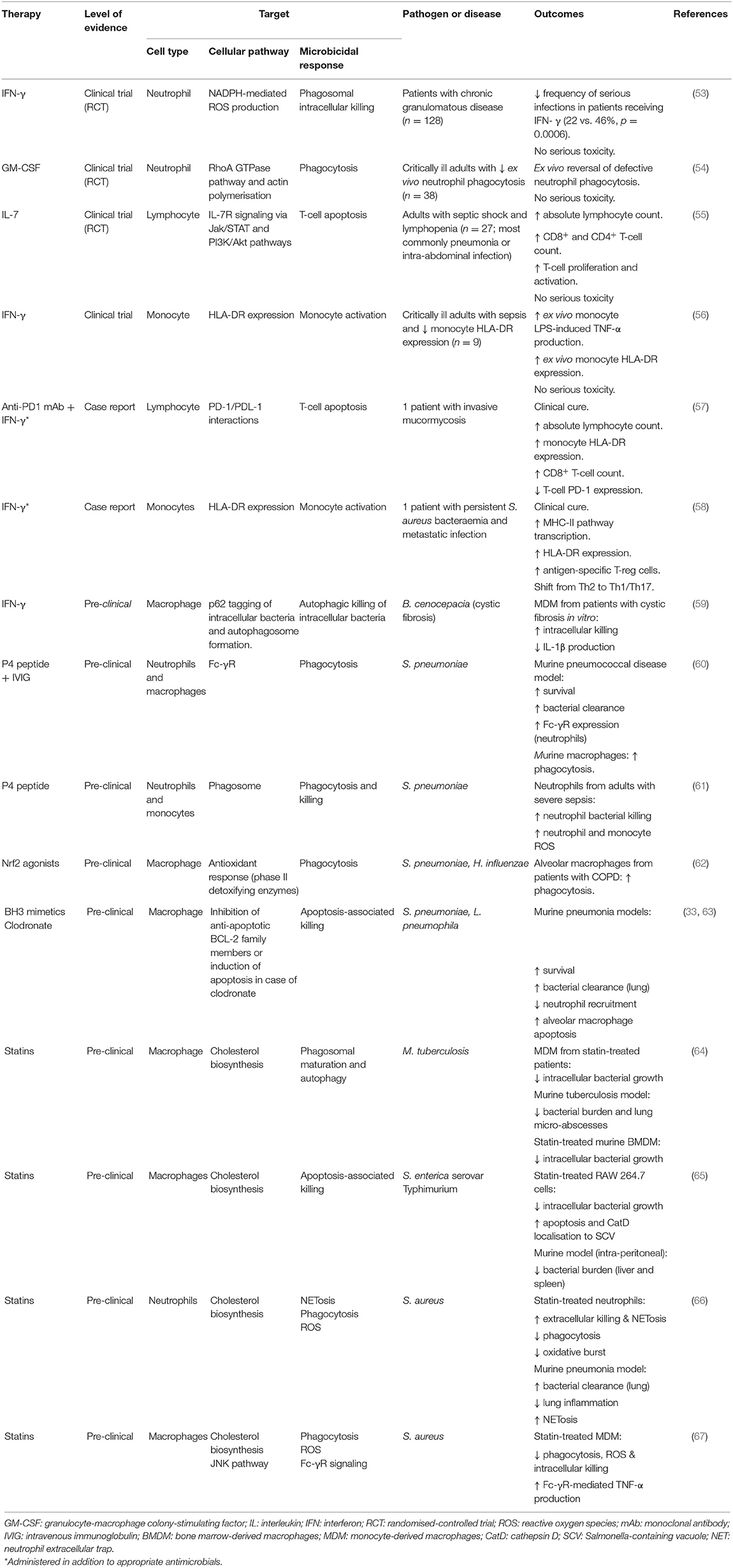
Table 1. Examples of host-directed therapies in infectious diseases from clinical and pre-clinical studies.
GM-CSF and G-CSF enhance macrophage and neutrophil phagocytosis and microbicidal responses in vitro and are used to restore functional phagocyte numbers in patients receiving bone marrow-suppressive chemotherapy. GM-/G-CSF have also been investigated in patients with sepsis, with a meta-analysis suggesting a trend toward benefit (71, 72). Timing may be important with GM-CSF and it may have most efficacy when targeted to patients with low monocyte HLA-DR (73). Whilst the impact on microbicidal responses is often not studied, a recent clinical trial showed GM-CSF targeted to critically ill patients with defects in ex vivo neutrophil phagocytosis could ameliorate this defect and increase monocyte HLA-DR (54). Both GM-CSF and IFN-γ will, with subtle differences, contribute to macrophage activation phenotypes that promote microbicidal responses, particularly against pathogens with significant intracellular survival. Other cytokines will have similar effects (74). As with many other approaches listed, each can impact more than one cellular process directly or indirectly, affecting microbicidal responses (Table 2). For example, IFN-γ can also enhance myeloid cell recruitment in clinical trials (68).
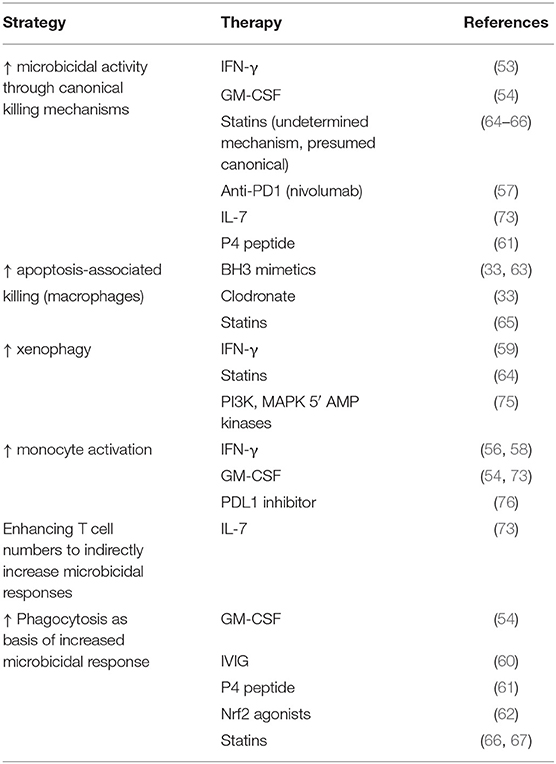
Table 2. Summary of strategies of host-directed therapy.
Other investigational approaches include the use of check-point inhibitors, such as anti-programmed cell death protein-1 (anti-PD-1) or anti-cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) monoclonal antibodies (73). These inhibitors aim to reverse suppression of T-cell inflammatory responses. Nivolumab, an anti-PD-1 monoclonal antibody, is being tested in a clinical trial in sepsis, and while such therapies are anticipated to modulate the inflammatory response, they may also target microbicidal responses. For example, there is a case report of Nivolumab being used in combination with IFN-γ to successfully treat an intractable fungal infection (57). A PD-1 ligand inhibitor has also been shown to increase monocyte HLA-DR expression (76). Other immune modulating strategies that can be expected to modulate microbicidal responses include recombinant IL-7, which corrects lymphopenia and will enhance IFN-γ, and intravenous immunoglobulin (IVIG), which in addition to immunomodulation enhances pathogen clearance through phagocytosis (73). Immunomodulatory peptides have also been combined with IVIG, specifically the P4 peptide (derived from the immunomodulatory pneumococcal lipopeptide Pneumococcal surface adhesin A), resulting in increased pneumococcal clearance in mice and enhanced neutrophil and monocyte bacterial killing (60, 61).
Studies in relevant in vitro and animal models, and human patient groups, can identify host microbicidal targets. But there is then a need to develop therapeutic approaches to modulate these targets. This will inevitably be constrained by cost, but this can potentially be reduced by re-purposing existing agents that are found to modify the host response of interest (75).
Critical illness can be associated with the compensatory anti-inflammatory response syndrome and temporary immunoparesis, after the initial stages of innate immune activation. This is characterized by reduced Th1 and monocyte responses, which increase the risk of nosocomial infection (77). Reducing PRR engagement and subsequent immune activation, such as through reduction in TLR activation in the early stages of illness, could potentially reverse this phenomenon and the turmeric constituent curcumin appears to down-regulate signaling through a range of TLRs (78, 79).
Phagocytosis of bacteria activates phagosomal microbicidal responses in myeloid cells (80). Although phagocytosis is not usually a rate limiting process, in conditions such as COPD macrophage phagocytosis may be reduced. This is associated with increased airway bacterial burden (62). This defect is related to cellular oxidative stress (62, 81). Nrf2 agonists are in development, which enhance the host cell's anti-oxidant host defenses, and in COPD AM can enhance phagocytosis as well as clearance of P. aeruginosa in mice exposed to cigarette smoke (62, 82).
Xenophagy is selective autophagy that aids clearance of intracellular pathogens such as Mycobacterium tuberculosis (83) and some extracellular bacteria. Of note, Streptococcus pyogenes subverts this process in endothelial cells (84). Activation of autophagy via inhibition of inhibitory pathways, such as class I phosphoinositide-3-kinase, mitogen-activated protein kinases or 5'-AMP-activated protein kinases, could be a tractable microbicidal strategy and drugs already under development for other indications could be re-purposed (75).
Another novel microbicidal response in macrophages and potentially other myeloid cells involves apoptosis-associated killing. BH3 mimetics enhance killing of S. pneumoniae and Legionella pneumophila in murine models through augmentation/restoration of this pathway (33, 63). Bisphosphonates also enhance macrophage apoptosis-associated killing of bacteria (33), while fluoroquinolones cause lysosomal permeabilization, sensitizing cells to this pathway (45, 85).
3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase inhibitors, termed statins, are used as cholesterol lowering medicines. Statins enhance bacterial clearance in a murine sickle cell model of pneumococcal disease. The impact was limited to the sickle cell mice with no response seen in wild type (86). One potential mechanism was downregulation of platelet-activating factor receptor required for bacterial translocation from the lung in the sickle cell mice. However, the microbicidal basis for the enhanced clearance was not established beyond the association of increased clearance with reduced sickle cell-associated inflammation. In the case of M. tuberculosis, statins enhance phagosomal maturation and xenophagy (64), while for Salmonella enterica serovar Typhimurium they enhance cathepsin D localization to phagosomes and apoptosis induction (65). Whether they also enhance these processes for extracellular pathogens is not established. They can enhance neutrophil and monocyte killing by extracellular traps (66). However, they inhibit phagocytosis and microbicidal responses in other models such as Fcγ-receptor mediated uptake of opsonized S. aureus (67) and reduce bacterial killing by neutrophils in a murine pneumonia model (87). Therefore, how they would be best used requires further elucidation, as reflected in contradictory findings from clinical studies. For example, a reduced risk of community-acquired S. aureus bacteremia (88) and reduced mortality in pneumonia were reported (89, 90) yet no reduction in mortality was observed in another pneumonia study (91) or in a study of ventilator-associated pneumonia (92).
Recalibrating responses will likely require a personalized medicine approach. Individual pathogens would need varying degrees of engagement of a given response. S. aureus inhibits apoptosis-associated killing in macrophages so might need a greater degree of enhancement, or might require an alternative approach, while for S. pneumoniae in which apoptosis-associated killing is already engaged, the adjustment might only need to be of a more modest extent in a subset of individuals (33). Certain responses might need engagement in select patient groups such as those with medical comorbidities that adjust the response. Alternatively these responses might not be suitable for enhancement in certain groups. For example, patients with COPD might not be amenable to enhancement of mROS production or might require reduction in high baseline levels of antioxidants to enhance this microbicidal response (16). Such personalized approaches would require validated tests to help calibrate an individual response.
Another challenge is that where responses need to be recalibrated it will be important that responses do not over shoot and result in overproduction of factors that could lead to tissue injury if there is excessive production of microbicidals or inflammatory cells (30). This is most likely to be prevented where the responses enhanced are intracellular, generated at high levels adjacent to bacteria and transient. Responses will require application of techniques to measure the individuals response through use of appropriate biomarkers or imaging modalities and would benefit from approaches that combine these measures with microdosing experiments and endomicroscopy (the application of in vivo microscopy applied through endoscopy to allow optical biopsy) to test the efficacy of recalibration (93).
The ineluctable progression of AMR necessitates investigation of novel strategies for treating bacterial disease. Based on the observation that exposure to potentially pathogenic bacteria infrequently leads to disease, we contend that identification and exploitation of specific determinants of host defense represents a tractable alternative to antimicrobials (host-based therapy). While there are many potential aspects of the host response that represent tractable targets, including humoral factors (e.g., AMP), epithelial barrier function, and lymphoid populations, we suggest approaches that promote pauci-inflammatory macrophage and neutrophil microbicidal responses can improve outcomes. We have highlighted a number of promising in vitro, animal model, human and pre-clinical observations that support this viewpoint and provide a roadmap for future research.
KW, CR, and DD wrote the initial drafts of the article. JB, KD, JF, TM, AS, and SR provided critical comment and revised the document.
The authors are supported by the MRC SHIELD consortium investigating novel host based antimicrobial responses to antimicrobial resistant bacteria (MRNO2995X/1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Hare R. The scientific activities of Alexander Fleming, other than the discovery of penicillin. Med Hist. (1983) 27:347–72. doi: 10.1017/s0025727300043386
2. Arseculeratne SN, Arseculeratne G. A re-appraisal of the conventional history of antibiosis and Penicillin. Mycoses. (2017) 60:343–7. doi: 10.1111/myc.12599
3. Cassini A, Hogberg LD, Plachouras D, Quattrocchi A, Hoxha A, Simonsen GS, et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis. Lancet Infect Dis. (2019) 19:56–66. doi: 10.1016/S1473-3099(18)30605-4
4. Czaplewski L, Bax R, Clokie M, Dawson M, Fairhead H, Fischetti V, et al. Alternatives to antibiotics-a pipeline portfolio review. Lancet Infect Dis. (2016) 16:239–51. doi: 10.1016/S1473-3099(15)00466-1
5. Mulani MS, Kamble EE, Kumkar SN, Tawre MS, Pardesi KR. Emerging Strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: a review. Front Microbiol. (2019) 10:539. doi: 10.3389/fmicb.2019.00539
6. Peacock SJ, Justice A, Griffiths D, de Silva GDI, Kantzanou MN, Crook D, et al. Determinants of acquisition and carriage of Staphylococcus aureus in infancy. J Clin Microbiol. (2003) 41:5718–25. doi: 10.1128/jcm.41.12.5718-5725.2003
7. Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, et al. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis. (2005) 5:751–62. doi: 10.1016/S1473-3099(05)70295-4
8. Nielsen KL, Dynesen P, Larsen PN. Frimodt-Moller. Faecal Escherichia coli from patients with E. coli urinary tract infection and healthy controls who have never had a urinary tract infection. J Med Microbiol. (2014) 63:582–9. doi: 10.1099/jmm.0.068783-0
9. Gao W, Howden BP, Stinear TP. Evolution of virulence in Enterococcus faecium, a hospital-adapted opportunistic pathogen. Curr Opin Microbiol. (2018) 41:76–82. doi: 10.1016/j.mib.2017.11.030
10. Turvey SE, Broide DH. Innate immunity. J Allergy Clin Immunol. (2010) 125(Suppl. 2):S24–32. doi: 10.1016/j.jaci.2009.07.016
11. Mortha A, Burrows K. Cytokine networks between innate lymphoid cells and myeloid cells. Front Immunol. (2018) 9:191. doi: 10.3389/fimmu.2018.00191
12. Ifrim DC, Quintin J, Joosten LAB, Jacobs C, Jansen T, Jacobs L, Gow NAR, et al. Trained immunity or tolerance: opposing functional programs induced in human monocytes after engagement of various pattern recognition receptors. Clin Vaccine Immunol. (2014) 21:534–45. doi: 10.1128/CVI.00688-13
13. Saeed S, Quintin J, Kerstens HH, Rao NA, Sharifi N, Janssen-Megens EM, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. (2014) 345:1251086. doi: 10.1126/science.1251086
14. Medvedev AE, Sabroe I, Hasday JD, Vogel SN. Tolerance to microbial TLR ligands: molecular mechanisms and relevance to disease. J Endotoxin Res. (2006) 12:133–50. doi: 10.1179/096805106X102255
15. Dockrell DH, Marriott HM, Prince LR, Ridger VC, Ince PG, Hellewell PG, et al. Alveolar macrophage apoptosis contributes to pneumococcal clearance in a resolving model of pulmonary infection. J Immunol. (2003) 171:5380–8. doi: 10.4049/jimmunol.171.10.5380
16. Bewley MA, Preston JA, Mohasin M, Marriott HM, Budd RC, Swales J, et al. Impaired mitochondrial microbicidal responses in chronic obstructive pulmonary disease macrophages. Am J Respir Crit Care Med. (2017) 196:845–55. doi: 10.1164/rccm.201608-1714OC
17. Collini PJ, Bewley MA, Mohasin M, Marriott H, Miller RF, Geretti A-M, et al. HIV gp120 in the lungs of antiretroviral therapy-treated individuals impairs alveolar macrophage responses to pneumococci. Am J Respir Crit Care Med. (2018) 197:1604–15. doi: 10.1164/rccm.201708-1755OC
18. Aberdein JD, Cole J, Bewley MA, Marriott H, Dockrell DH. Alveolar macrophages in pulmonary host defence the unrecognized role of apoptosis as a mechanism of intracellular bacterial killing. Clin Exp Immunol. (2013) 174:193–202. doi: 10.1111/cei.12170
19. Jubrail J, Morris P, Bewley MA, Stoneham S, Johnston SA, Foster SJ, et al. Inability to sustain intraphagolysosomal killing of Staphylococcus aureus predisposes to bacterial persistence in macrophages. Cell Microbiol. (2016) 18:80–96. doi: 10.1111/cmi.12485
20. Ercoli G, Fernandes VE, Chung WY, Wanford J, Thomson S, Bayliss CD, et al. Intracellular replication of Streptococcus pneumoniae inside splenic macrophages serves as a reservoir for septicaemia. Nat Microbiol. (2018) 3:600–10. doi: 10.1038/s41564-018-0147-1
21. Cano V, March C, Insua JL, Aguilo N, Llobet E, Llobet E, Moranta D, et al. Klebsiella pneumoniae survives within macrophages by avoiding delivery to lysosomes. Cell Microbiol. (2015) 17:1537–60. doi: 10.1111/cmi.12466
22. Mukherjee K, Khatua B, Mandal C. Sialic acid-siglec-e interactions during pseudomonas aeruginosa infection of macrophages interferes with phagosome maturation by altering intracellular calcium concentrations. Front Immunol. (2020) 11:332. doi: 10.3389/fimmu.2020.00332
23. Knapp S, Leemans JC, Florquin S, Branger J, Maris NA, Pater J, et al. Alveolar macrophages have a protective antiinflammatory role during murine pneumococcal pneumonia. Am J Respir Crit Care Med. (2003) 167:171–9. doi: 10.1164/rccm.200207-698OC
24. Boldock E, Surewaard BGJ, Shamarina D, Na M, Fei Y, Ali A, et al. Human skin commensals augment Staphylococcus aureus pathogenesis. Nat Microbiol. (2018) 3:881–90. doi: 10.1038/s41564-018-0198-3
25. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. (2014) 20:1126–67. doi: 10.1089/ars.2012.5149
26. Segal W. How neutrophils kill microbes. Annu Rev Immunol. (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
27. Reeves EP, Lu H, Jacobs HL, Messina CGM, Bolsover S, Gabella G, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. (2002) 416:291–7. doi: 10.1038/416291a
28. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss D, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
29. Sears BF, Rohr JR, Allen JE, Martin LB. The economy of inflammation: when is less more? Trends Parasitol. (2011) 27:382–7. doi: 10.1016/j.pt.2011.05.004
30. Han S, Mallampalli RK. The acute respiratory distress syndrome: from mechanism to translation. J Immunol. (2015) 194:855–60. doi: 10.4049/jimmunol.1402513
31. Meduri GU, Annane D, Chrousos GP, Marik PE, Sinclair SE, et al. Activation and regulation of systemic inflammation in ARDS: rationale for prolonged glucocorticoid therapy. Chest. (2009) 136:1631–43. doi: 10.1378/chest.08-2408
32. Buttenschoen K, Kornmann M, Berger D, Leder G, Beger H, Vasilescu GC. Endotoxemia and endotoxin tolerance in patients with ARDS. Langenbecks Arch Surg. (2008) 393:473–8. doi: 10.1007/s00423-008-0317-3
33. Preston JA, Bewley MA, Marriott HM, McGarry Houghton A, Mohasin M, Jubrail J, et al. Alveolar macrophage apoptosis-associated bacterial killing helps prevent murine pneumonia. Am J Respir Crit Care Med. (2019) 200:84–97. doi: 10.1164/rccm.201804-0646OC
34. Koeffler HP, Ranyard J, Pertcheck M. Myeloperoxidase: its structure and expression during myeloid differentiation. Blood. (1985) 65:484–91
35. Sugiyama S, Okada Y, Sukhova GK, Virmani R, Heinecke JW, Libby P, et al. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. (2001) 158:879–91. doi: 10.1016/S0002-9440(10)64036-9
36. Thomas AC, Mattila JT. “Of mice and men”: arginine metabolism in macrophages. Front Immunol. (2014) 5:479. doi: 10.3389/fimmu.2014.00479
37. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. (2016) 44:439–49. doi: 10.1016/j.immuni.2016.02.024
38. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. (2012) 13:1118–28. doi: 10.1038/ni.2419
39. Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. (2009) 7:355–66. doi: 10.1038/nrmicro2128
40. Houghton AM, Hartzell WO, Robbins CS, Gomis-Rüth FX, Shapiro SD, et al. Macrophage elastase kills bacteria within murine macrophages. Nature. (2009) 460:637–41. doi: 10.1038/nature08181
41. Tang X, Basavarajappa D, Haeggstrom JZ, Wan M. P2X7 receptor regulates internalization of antimicrobial peptide LL-37 by human macrophages that promotes intracellular pathogen clearance. J Immunol. (2015) 195:1191–201. doi: 10.4049/jimmunol.1402845
42. Lazar V, Martins A, Spohn R, Daruka L, Grezal G, Fekete G, et al. Antibiotic-resistant bacteria show widespread collateral sensitivity to antimicrobial peptides. Nat Microbiol. (2018) 3:718–31. doi: 10.1038/s41564-018-0164-0
43. Morici P, Florio W, Rizzato C, Ghelardi E, Tavanti A, Rossolini G, et al. Synergistic activity of synthetic N-terminal peptide of human lactoferrin in combination with various antibiotics against carbapenem-resistant Klebsiella pneumoniae strains. Eur J Clin Microbiol Infect Dis. (2017) 36:1739–48. doi: 10.1007/s10096-017-2987-7
44. Kidd TJ, Mills G, Sa-Pessoa J, Dumigan A, Frank CG, Insua JL, et al. A Klebsiella pneumoniae antibiotic resistance mechanism that subdues host defences and promotes virulence. EMBO Mol Med. (2017) 9:430–47. doi: 10.15252/emmm.201607336
45. Bewley MA, Marriott HM, Tulone C, Francis SE, Mitchell TJ, et al. A cardinal role for cathepsin d in co-ordinating the host-mediated apoptosis of macrophages and killing of pneumococci. PLoS Pathog. (2011) 7:e1001262. doi: 10.1371/journal.ppat.1001262
46. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. (2014) 343:84–7. doi: 10.1126/science.1247005
47. Li B, Clohisey SM, Chia BS, Wang B, Cui A, Eisenhaure T, et al. Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection. Nat Commun. (2020) 11:164. doi: 10.1038/s41467-019-13965-x
48. Viana D, Comos M, McAdam PR, Ward MJ, Selva L, Guinane CM, et al. A single natural nucleotide mutation alters bacterial pathogen host tropism. Nat Genet. (2015) 47:361–6. doi: 10.1038/ng.3219
49. Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol. (2011) 187:434–40. doi: 10.4049/jimmunol.1003143
50. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. (2011) 472:476–80. doi: 10.1038/nature09973
51. Akram AR, Chankeshwara SV, Scholefield E, Aslam T, McDonald N, Megia-Fernandez A, et al. In situ identification of gram-negative bacteria in human lungs using a topical fluorescent peptide targeting lipid A. Sci Transl Med. (2018) 10:eaal0033. doi: 10.1126/scitranslmed.aal0033
52. Meurens F, Summerfield A, Nauwynck H, Saif L, Gerdts V. The pig: a model for human infectious diseases. Trends Microbiol. (2012) 20:50–7. doi: 10.1016/j.tim.2011.11.002
53. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. The international chronic granulomatous disease cooperative study group. N Engl J Med. (1991) 324:509–16. doi: 10.1056/NEJM199102213240801
54. Pinder EM, Rostron AJ, Hellyer TP, Ruchaud-Sparagano MH, Scott J, Macfarlane JG, et al. Randomised controlled trial of GM-CSF in critically ill patients with impaired neutrophil phagocytosis. Thorax. (2018) 73:918–25. doi: 10.1136/thoraxjnl-2017-211323
55. Francois B, Jeannet R, Daix T, Walton AH, Shotwell MS, Unsinger J, et al. Interleukin-7 restores lymphocytes in septic shock: the IRIS-7 randomized clinical trial. JCI Insight. (2018) 3:98960. doi: 10.1172/jci.insight.98960
56. Döcke WD, Randow F, Syrbe U, Krausch D, Asadullah K, Reinke P, et al. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med. (1997) 3:678–81. doi: 10.1038/nm0697-678
57. Grimaldi D, Pradier O, Hotchkiss RS, Vincent JL. Nivolumab plus interferon-gamma in the treatment of intractable mucormycosis. Lancet Infect Dis. (2017) 17:18. doi: 10.1016/S1473-3099(16)30541-2
58. Nalos M, Santner-Nanan B, Parnell G, Tang B, McLean AS, Nanan R. Immune effects of interferon gamma in persistent staphylococcal sepsis. Am J Respir Crit Care Med. (2012) 185:110–2. doi: 10.1164/ajrccm.185.1.110
59. Assani K, Tazi MF, Amer AO Kopp BT. IFN-gamma stimulates autophagy-mediated clearance of Burkholderia cenocepacia in human cystic fibrosis macrophages. PLoS ONE. (2014) 9:e96681. doi: 10.1371/journal.pone.0096681
60. Bangert M, Bricio-Moreno L, Gore S, Rajam G, Ades EW, Gordon SB, et al. P4-mediated antibody therapy in an acute model of invasive pneumococcal disease. J Infect Dis. (2012) 205:1399–407. doi: 10.1093/infdis/jis223
61. Morton B, Mitsi E, Pennington SH, Reiné J, Wright AD, Parker R, et al. Augmented passive immunotherapy with P4 peptide improves phagocyte activity in severe sepsis. Shock. (2016) 46:635–41. doi: 10.1097/SHK.0000000000000715
62. Bewley MA, Budd RC, Ryan E, Cole J, Collini P, Marshall J, et al. Opsonic phagocytosis in chronic obstructive pulmonary disease is enhanced by Nrf2 agonists. Am J Respir Crit Care Med. (2018) 198:739–50. doi: 10.1164/rccm.201705-0903OC
63. Speir M, Lawlor KE, Glaser SP, Abraham G, Chow S, Vogrin A, et al. Eliminating Legionella by inhibiting BCL-XL to induce macrophage apoptosis. Nat Microbiol. (2016) 1:15034. doi: 10.1038/nmicrobiol.2015.34
64. Parihar SP, Guler R, Khutlang R, Lang DM, Hurdayal R, Mhlanga MM, et al. Statin therapy reduces the Mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis. (2014) 209:754–63. doi: 10.1093/infdis/jit550
65. Catron DM, Lange Y, Borensztajn J, Sylvester MD, Jones BD, Haldar K. Salmonella enterica serovar Typhimurium requires nonsterol precursors of the cholesterol biosynthetic pathway for intracellular proliferation. Infect Immun. (2004) 72:1036–42. doi: 10.1128/iai.72.2.1036-1042.2004
66. Chow OA, von Kockritz-Blickwede M, Bright AT, Hensler ME, Zinkernagel AS, Cogen AL, et al. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe. (2010) 8:445–54. doi: 10.1016/j.chom.2010.10.005
67. Benati D, Ferro M, Savino MT, Ulivieri C, Schiavo E, Nuccitelli A, et al. Opposite effects of simvastatin on the bactericidal and inflammatory response of macrophages to opsonized S. aureus. J Leukoc Biol. (2010) 87:433–42. doi: 10.1189/jlb.0409273
68. Miller CH, Maher SG, Young HA. Clinical use of interferon-gamma. Ann N Y Acad Sci. (2009) 1182:69–79. doi: 10.1111/j.1749-6632.2009.05069.x
69. Moss RB, Mayer-Hamblett N, Wagener J, Daines C, Hale K, Ahrens R, et al. Randomized, double-blind, placebo-controlled, dose-escalating study of aerosolized interferon gamma-1b in patients with mild to moderate cystic fibrosis lung disease. Pediatr Pulmonol. (2005) 39:209–18. doi: 10.1002/ppul.20152
70. Payen D, Faivre V, Miatello J, Leentjens J, Brumpt C, Tissieres P, et al. Multicentric experience with interferon gamma therapy in sepsis induced immunosuppression. A case series. BMC Infect Dis. (2019) 19:931. doi: 10.1186/s12879-019-4526-x
71. Bo L, Wang F, Zhu J, Li J, Deng X. Granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) for sepsis: a meta-analysis. Crit Care. (2011) 15:R58. doi: 10.1186/cc10031
72. Mathias B, Szpila BE, Moore FA, Efron PA, Moldawer LL. A review of GM-CSF therapy in sepsis. Medicine. (2015) 94:e2044. doi: 10.1097/MD.0000000000002044
73. Davies R, O'Dea K, Gordon A. Immune therapy in sepsis: Are we ready to try again? J Intensive Care Soc. (2018) 19:326–44. doi: 10.1177/1751143718765407
74. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
75. Brown D. Antibiotic resistance breakers: can repurposed drugs fill the antibiotic discovery void? Nat Rev Drug Discov. (2015) 14:821–32. doi: 10.1038/nrd4675
76. Hotchkiss RS, Colston E, Yende S, Angus DC, Moldawer LL, Crouser ED, et al. Immune checkpoint inhibition in sepsis: a phase 1b randomized, placebo-controlled, single ascending dose study of antiprogrammed cell death-ligand 1 antibody (BMS-936559) Crit Care Med. (2019) 47:632–42. doi: 10.1097/CCM.0000000000003685
77. Ward NS, Casserly B, Ayala A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med. (2008) 29:617–25. doi: 10.1016/j.ccm.2008.06.010
78. Gradisar H, Keber MM, Pristovsek P, Jerala R. MD-2 as the target of curcumin in the inhibition of response to LPS. J Leukoc Biol. (2007) 82:968–74. doi: 10.1189/jlb.1206727
79. Shuto T, Ono T, Ohira Y, Shimasaki S, Mizunoe S, Watanabe K, et al. Curcumin decreases toll-like receptor-2 gene expression and function in human monocytes and neutrophils. Biochem Biophys Res Commun. (2010) 398:647–52. doi: 10.1016/j.bbrc.2010.06.126
80. DeLeo FR, Allen LA, Apicella M, Nauseef WM. NADPH oxidase activation and assembly during phagocytosis. J Immunol. (1999) 163:6732–40
81. Belchamber KBR, Singh R, Batista CM, Whyte MK, Dockrell DH, Kilty I, et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur Respir J. (2019) 54:1802244. doi: 10.1183/13993003.02244-2018
82. Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH, et al. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med. (2011) 3:78ra32. doi: 10.1126/scitranslmed.3002042
83. Sharma V, Verma S, Seranova E, Sarkar S, Kumar D. Selective autophagy and xenophagy in infection and disease. Front Cell Dev Biol. (2018) 6:147. doi: 10.3389/fcell.2018.00147
84. Cheng YL, Kuo CF, Lu SL, Hiroko O, Wu YN, Hsieh CL, et al. Group A Streptococcus induces laposomes via SLO/beta1 integrin/NOX2/ROS pathway in endothelial cells that are ineffective in bacterial killing and suppress xenophagy. mBio. (2019) 10:e02148–19. doi: 10.1128/mBio.02148-19
85. Bewley MA, Naughton M, Preston J, Mitchell A, Holmes A, Marriott HM, et al. Pneumolysin activates macrophage lysosomal membrane permeabilization and executes apoptosis by distinct mechanisms without membrane pore formation. mBio. (2014) 5:e01710–14. doi: 10.1128/mBio.01710-14
86. Rosch JW, Boyd AR, Hinojosa E, Pestina T, Hu Y, Persons DA, et al. RStatins protect against fulminant pneumococcal infection and cytolysin toxicity in a mouse model of sickle cell disease. J Clin Invest. (2010) 120:627–35. doi: 10.1172/JCI39843
87. Fessler MB, Young SK, Jeyaseelan S, Lieber JG, Arndt PG, Nick JA, et al. A role for hydroxy-methylglutaryl coenzyme a reductase in pulmonary inflammation and host defense. Am J Respir Crit Care Med. (2005) 171:606–15. doi: 10.1164/rccm.200406-729OC
88. Smit J, López-Cortés LE, Thomsen RW, Schønheyder HC, Nielsen H, Frøslev T, et al. Statin use and risk of community-acquired Staphylococcus aureus bacteremia: a population-based case-control study. Mayo Clin Proc. (2017) 92:1469–1478. doi: 10.1016/j.mayocp.2017.07.008
89. Schlienger RG, Fedson DS, Jick SS, Jick H, Meier CR. Statins and the risk of pneumonia: a population-based, nested case-control study. Pharmacotherapy. (2007) 27:325–32. doi: 10.1592/phco.27.3.325
90. Thomsen RW, Riis A, Kornum JB, Christensen S, Johnsen SP, Srensen HT. Preadmission use of statins and outcomes after hospitalization with pneumonia: population-based cohort study of 29,900 patients. Arch Intern Med. (2008) 168:2081–7. doi: 10.1001/archinte.168.19.2081
91. Majumdar SR, McAlister FA, Eurich DT, Padwal RS, Marrie TJ. Statins and outcomes in patients admitted to hospital with community acquired pneumonia: population based prospective cohort study. BMJ. (2006) 333:999. doi: 10.1136/bmj.38992.565972.7C
92. Papazian L, Roch A, Charles PE, Penot-Ragon C, Perrin G, Roulier P, et al. Effect of statin therapy on mortality in patients with ventilator-associated pneumonia: a randomized clinical trial. JAMA. (2013) 310:1692–700. doi: 10.1001/jama.2013.280031
Keywords: antimicrobial resistance, macrophage, neutrophil, host-based therapies, innate immunity
Citation: Watson K, Russell CD, Baillie JK, Dhaliwal K, Fitzgerald JR, Mitchell TJ, Simpson AJ, Renshaw SA and Dockrell DH (2020) Developing Novel Host-Based Therapies Targeting Microbicidal Responses in Macrophages and Neutrophils to Combat Bacterial Antimicrobial Resistance. Front. Immunol. 11:786. doi: 10.3389/fimmu.2020.00786
Received: 14 February 2020; Accepted: 07 April 2020;
Published: 05 June 2020.
Edited by:
Marco Rinaldo Oggioni, University of Leicester, United KingdomReviewed by:
Arshad Khan, University of Texas Health Science Center at Houston, United StatesCopyright © 2020 Watson, Russell, Baillie, Dhaliwal, Fitzgerald, Mitchell, Simpson, Renshaw and Dockrell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David H. Dockrell, ZGF2aWQuZG9ja3JlbGxAZWQuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.