 Mengxiao Xie1,2†
Mengxiao Xie1,2† Jian Xu
Jian Xu- 1Department of Laboratory Medicine, The First Affiliated Hospital of Nanjing Medical University, Nanjing, China
- 2National Key Clinical Department of Laboratory Medicine, Nanjing, China
Lung cancer is the leading cause of cancer-associated deaths worldwide, with non-small cell-lung cancer (NSCLC) accounting for approximately 80% of cases. Immune escape has been demonstrated to play a key role in the initiation and progression of NSCLC, although the underlying mechanisms are diverse and their puzzling nature is far from being understood. As a critical participant in immune escape, the CD4+ T cell subset of regulatory T (Treg) cells, with their immunosuppressive functions, has been implicated in the occurrence of many types of cancers. Additionally, therapies based on Treg blockade have benefited a portion of cancer patients, including those with NSCLC. Accumulating literature has noted high Treg infiltration in NSCLC tumor tissues, bone marrow, lymph nodes and/or blood; moreover, the tumor milieu is involved in regulating the proliferation, differentiation, recruitment and suppressive functions of Treg cells. Multifarious mechanisms by which CD4+ Treg cells are generated, attracted and modulated in the NSCLC milieu will be discussed in this review.
Introduction
Lung cancer is continuously evaluated and is considered the leading cause of cancer-related mortality worldwide, with a dismal 5-year survival rate of approximately 19% in the United States (1, 2). According to histopathological features, lung cancer can be further classified into small-cell lung cancer (SCLC), which accounts for approximately 10% ∼ 15% of lung cancer cases, and non-small-cell lung cancer (NSCLC), comprising the remaining 85% ∼ 90% (3, 4). NSCLC has three histological subtypes: squamous cell carcinoma, adenocarcinoma, and large cell carcinoma (5). Although genetic susceptibility and environmental hazards (i.e., cigarette smoke) that trigger chronic pulmonary inflammation have been confirmed as high-risk factors for NSCLC (6); their precise regulatory mechanisms are still puzzling.
Cancer immune escape, involving (a) loss of antigenicity, (b) loss of immunogenicity, and (c) the presence of a complicated immunosuppressive tumor microenvironment, is a major mechanism antagonizing antitumor immune responses (7). Published literature has emphasized that tumor milieu-induced activation of immunosuppressive cells enhances cancer occurrence, growth, invasion, and metastasis by diminishing tumor-killing immune responses (8–11). More importantly, therapeutics targeting the contributing factors in the tumor microenvironment have achieved an unexpected breakthrough in some patients, including those suffering from NSCLC (12–14), implying the non-redundant role of immunosuppression in modulating tumor biology.
CD4+ regulatory T (Treg) cells, which express the X chromosome-linked, linage specific transcription factor Foxp3, are potent immunosuppressive cells and can serve as brakes during immune responses. Numerous studies on autoimmune diseases have emphasized the protective role of Treg cells in remitting inflammation and the requirement for Treg supplementation therapy in these inflammatory diseases (15). However, Treg cells play an opposite role in cancer immunity as Treg cells recruited in tumor tissues become accomplices that help cancer cells escape from immunological surveillance. Heterogenetic Tregs with high frequencies in tumor tissues, bone marrow, lymph nodes or peripheral blood from NSCLC patients are considered predictors of disease outcome (16–18). Further, accumulating evidence suggests that cytokines or other agents derived from NSCLC tissues, as well as phenotype modulators, might be the key factors leading to the differentiation, trafficking and immunosuppressive effects of Tregs. In the following sections of this review, we will discuss the underlying mechanisms in detail.
Generation or Expansion of Treg Cells
CD4+ Treg cells are currently classified into two main subtypes, natural Tregs (nTregs) and inducible Tregs (iTregs), according to their origins, determinant markers and distributions (19). Despite the existence of delicate differences, both subtypes express the linage specific transcription factor Foxp3 and can exert immunosuppressive effects. Considering that conventional T cells undergo reprogramming, proliferation and differentiation in the NSCLC milieu, it is worthwhile summarizing the mechanisms of Treg cell generation induced by factors in NSCLC microenvironment.
TCR Signaling
Recognition of antigen peptide-MHC complexes by T cell receptors (TCRs) is the first step in T cell proliferation and differentiation. An updated published work revealed that the TCR repertoires of intratumoral Tregs from patients with metastatic melanoma, gastrointestinal, and ovarian cancer have a unique TCR repertoire different from that of other intratumoral CD4+ T cells (20). These results emphasize the possibility that ex vivo generation or expansion of NSCLC-infiltrating Tregs, like other T cell subsets, also requires distinct TCR signaling in response to neoantigens, which determines their heterogeneity. This section will be discussed in the following part.
Coinhibitory Ligands and Receptors
Following TCR stimulation, T cells undergo further proliferation and lineage fate determination subsequent to CD28-CD80/CD86 costimulatory interaction (21). Additionally, coinhibitory crosslinking, including cytotoxic T lymphocyte associated antigen-4 (CTLA-4)-CD80/86 and programmed cell death protein-1 (PD-1)-programmed death-ligand-1 (PD-L1) binding, both of which serve as brakes in the process for T cell activation, can occur.
CTLA-4, a CD28 family receptor, is not expressed by resting T cells but can be induced by de novo transcription and accumulates on membranes upon T cell stimulation (22). On the one hand, CTLA-4 induced by activated T cells can compete with CD28 to interact with CD80/86 with high affinity, causing T cell anergy (23); on the other hand, it has a positive effect on iTreg cell differentiation (24). Although the current mechanisms by which CTLA-4 promotes Treg generation in vitro remain unelucidated, this activity could be ascribed to an emulative CTLA-4 mediated reduction in CD28-CD80/86-interaction-induced NF-κB activity, which is specially required for iTreg, but not nTreg differentiation, potentially in an miR-34a-dependent manner (25–27). Alternatively, Treg generation can be achieved via indoleamine 2,3-dioxygenase (IDO) production by dendritic cells (DCs) upon CTLA-4-CD80/86 interaction, which favors in vitro differentiation of iTregs (28–30). Emerging evidence has indicated that CTLA-4 expression level is markedly elevated in tumor-infiltrating T cells of NSCLC patients (31), which might contribute to their conversion into iTreg cells (Figure 1A). So far, two CTLA-4 monoclonal antibodies, namely ipilimumab and tremelimumab, have been developed to enhance antitumor immune responses by recovering T cell activation status (32, 33). Ipilimumab has been evaluated in advanced NSCLC in combination with chemotherapy in a Phase II study and the results showed that phased ipilimumab plus chemotherapy significantly improved progression-free survival (PFS) compared with chemotherapy alone (34). Notably, anti-CTLA-4 therapy has shown a promising outcome for decreasing Treg cell numbers, which has been mentioned and suggested for NSCLC treatment (35–37); however, the definite effect of CTLA-4-based therapies on Treg cell numbers needs further investigation.
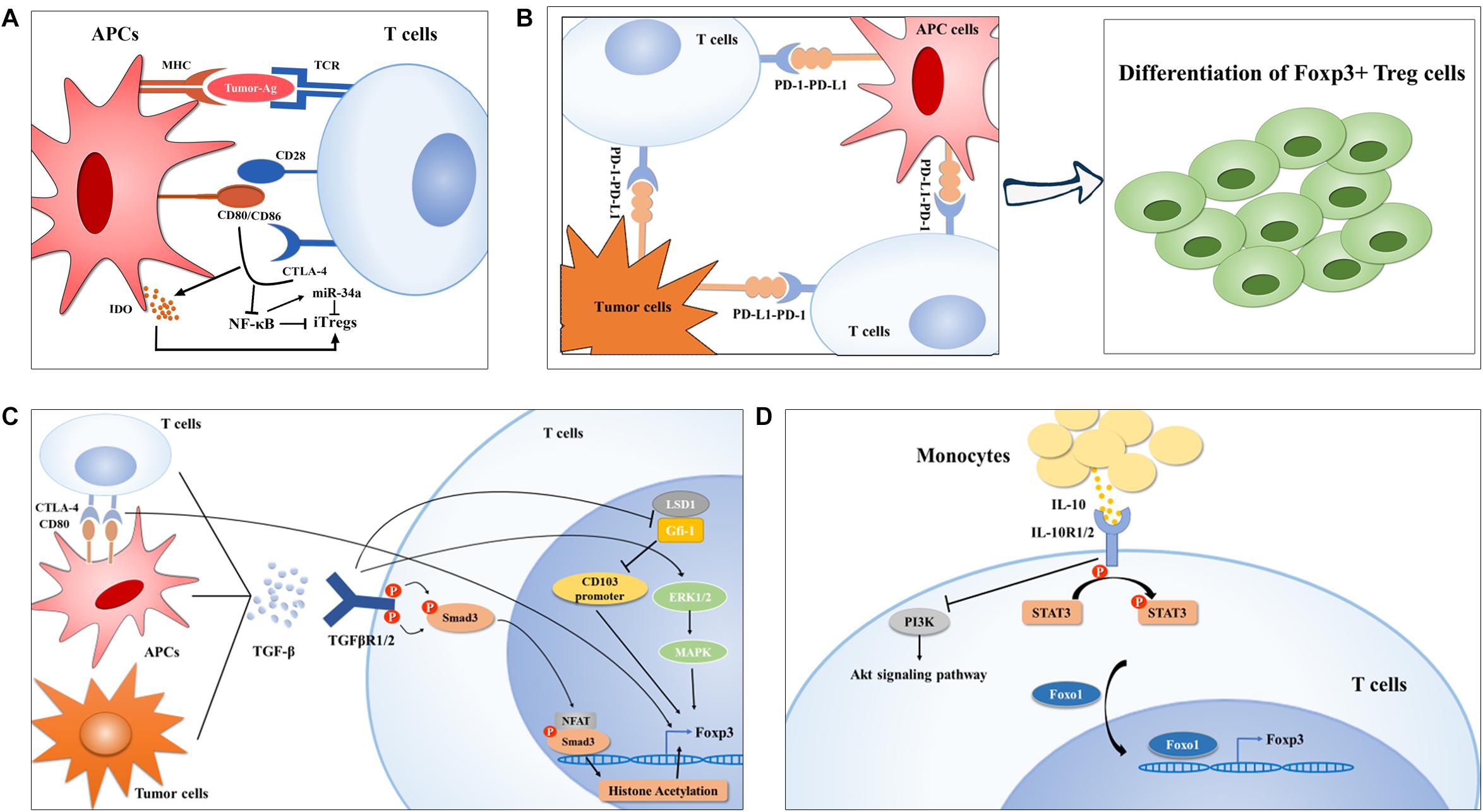
Figure 1. Treg cell generation in lung cancer. (A) Ex vivo generation of Tregs is modulated by the first and second signaling of T cell activation in lung cancer. In brief, neoantigens determines the TCR repertoire of Tregs (left) and CTLA-4-CD80/CD86 crosslink downregulates NF-κB activity, which was reported to inhibit Foxp3 expression by upregulating miR-34a, finally promoting Treg cell polarization. (B-C) APC- or tumor cell-derived PD-L1 or TGF-β can also induce Treg cell generation by interaction their corresponding receptors, respectively, on TILs via diverse mechanisms. On the one hand, TGF-β induces CTLA-4 expression on TILs, on the other hand, TGF-mediated activation of Smad and ERK1/2 can enhance Foxp3 expression in Treg cells. Moreover, TGF-β inhibits LSD1-Gfi-1 axis via an unknown mechanism to enhance immunosuppressive CD103+ Treg differentiation. (D) IL-10 induced Foxo1 translocation into nucleus facilities its occupation in Foxp3 promoter upon STAT3 activation and PI3K-Akt inactivation.
PD-1, also called CD279, is an immune checkpoint receptor that is a CD28 family receptor and is expressed on diverse types of immune cells including Tregs (38–41). PD-L1, also termed CD274 or B7-H1, is a transmembrane protein that transmits an inhibitory signal promoting T cells to undergo apoptosis and anergy by binding to its receptor—PD-1 (42–44). Numerous studies in human NSCLC patients or a mouse model of EGFR-driven adenocarcinomas have implicated hyperactivation of the PD-1-PD-L1 axis in tumor immune escape and malignant progression (45–47), and manipulation of Treg generation driven by this axis constitutes one of the most predominant mechanisms of NSCLC occurrence (Figure 1B). Using TCR transgenic CD4+ OT-II T cells, Wang et al. (48) found that the conversion of OT-II T cells into iTreg cells was notably diminished after PD-L1 blockade iv vivo, and that the loss of PD-L1 in DCs failed to induce iTreg differentiation ex vivo, preliminarily suggesting a PD-L1-dependent mechanism for Treg cell generation. Chen et al. (49) showed that conventional CD4+ T cells with PD-1 knockout displayed a strikingly diminished tendency toward differentiation into iTreg cells, arguing that the PD-1-PD-L1 pathway, in addition to limiting conventional T cell activation and proliferation, can also provoke peripheral tolerance by supporting iTreg generation. Reportedly, genomic alterations including gene amplification and translocation at chromosome 9p24.1 (50) and genomic structural alteration in the PD-L1 3′-untranslated region (3′-UTR) (51), several epigenetic modifiers, comprising the histone acetylation modifier bromodomain and extra-terminal (BET) proteins (52, 53) and mixed-lineage leukemia 1 (MLL1) with histone methyltransferase activities (54), as well as oncogenic signaling pathways, involving JAK-STAT (55), NF-κB (56), PI3K-Akt-mTOR (57), MAPKs (58, 59), and Hippo (60), have been implicated in PD-L1 expression at different levels. Further investigations are needed to disclose the precise mechanisms underlying the hyper-induction of infiltrating Treg cells in NSCLC driven by PD-1-PD-L1 activation.
TGF-β Cascade
Transforming growth factor beta (TGF-β) is a polypeptide cytokine of the transforming growth factor superfamily that includes three different isoforms (TGF-β1, 2, and 3) and regulates multiple biological processes involved in embryonic development, stem cell differentiation, immune regulation, wound healing, and inflammation (61–63). This pathway is initiated by TGF-β signaling through the cell-surface receptors, TGF-βR1 and TGF-βR2, which are dual specificity kinases and intracellular signal transducer proteins. Upon activation of these receptors, Smad proteins undergo phosphorylation by TGF-βR1 kinase at the two carboxy-terminal serine residues and then translocate into the nucleus to modulate target gene expression (64). In addition, non-redundant pathways through which TGF-β can also activate MAPK, Rho-like GTPase and PI3K pathways independent of Smad have been recorded (65).
Both in vivo and ex vivo investigations suggested that TGF-β signaling is required for the induction of Foxp3 in peripheral CD4+ T cells through different mechanisms (66–68). For instance, Smad3 can induce Foxp3 expression by binding the conserved non-coding sequence 1 (CNS1) region of Foxp3 enhancer or facilitating binding of the transcription factor nuclear factor of activated T cells (NFAT) to Foxp3 enhancer, further triggering histone acetylation at this locus (69, 70). Zheng and colleagues (24) revealed that TGF-β can accelerate the expression of CTLA-4, whose binding to CD80 shortly after T cell activation enables Foxp3 induction in conventional CD4+ cells and to endows them with suppressive activity, implying that TGF-β participates in the coinhibitory crosslinking between CTLA-4 and CD80. In addition, Lu and colleagues (71) documented that TGF-β boosts iTreg cell differentiation through Smad2/3 and MAPK-ERK1/2 signaling, suggesting the involvement of non-redundant Smad and non-Smad signaling in the development of Treg cells. Moreover, in the subset of CD103+ iTreg cells which were defined as tumor-infiltrating Tregs (72), TGF-β reportedly inhibited the expression of growth factor independent 1 (Gfi-1), a transcriptional repressor that can form a repressive complex with lysine-specific demethylase 1 (LSD1) in the intergenic region of CD103 intron 1, resulting in epigenetic upregulation of CD103 and differentiation of CD103+ iTreg cells (73) (Figure 1C). It has been demonstrated that the concentration of TGF-β in the plasma of NSCLC patients positively correlated with the frequency of circulating Treg cells and that TGF-β and Foxp3 were co-expressed in serial sections from tumor tissues of lung cancer (74), implying a contributing role of TGF-β in driving Treg generation in the process of NSCLC development. More importantly, TGF-β signaling blockade has been shown to decrease Treg cell numbers in a lewis lung carcinoma (LLC) mouse model (75), indicating a potential strategy targeting Treg cells for NSCLC treatment. Currently, the TGF-βR1 inhibitor galunisertib is under clinical development in combination with the PD-1 inhibitor, including nivolumab or durvalumab, in NSCLC patients (76).
IL-10 Signaling
IL-10, an anti-inflammatory cytokine reportedly found at high levels in the cancer microenvironment including in NSCLC (77), is not only an effector, but also a strong inducer of Treg cells (78, 79). IL-10 signals through a receptor complex consisting of two IL-10R1 and two IL-10R2 subunits, which then triggers STAT3 phosphorylation at tyrosine 705 and serine 727 via the phosphorylation of the cytoplasmic tails of IL-10R1 and IL-10R2 driven by JAK1 and Tyk2, respectively (80–82). Accumulating evidence indicates that massive production of IL-10 is indispensable for iTreg generation in NSCLC. Hsu and his colleagues (79) found that human monocyte derived-IL-10 potentiated TGF-β-induced iTreg generation, which was abolished in patients with IL-10R gene deficiency and dominant-negative STAT3 mutation. Mechanistic studies revealed that IL-10 not only triggered STAT3 phosphorylation, but also prevented PI3K-Akt activation, both of which induce the transcription factor Foxo1 to translocate into the nucleus via a direct protein-protein interaction or phosphorylation (79, 83). Foxo1 then functions as an upstream transcription factor regulating Foxp3 expression during IL-10 stimulated iTreg cell differentiation (84) (Figure 1D).
Migration of Treg Cells
CCL22/CCL17-CCR4 Axis
Recruitment of Tregs into NSCLC tumor tissues relies on chemokines overproduced by innate immune cells, tumor cells, as well as stromal cells that act through their corresponding receptors (Figure 2). It has been proposed that chemokine (C-C motif) ligand 22 (CCL22), expressed by different immune cells including DCs, macrophages and so on, can induce Treg cell migration by interacting with chemokine receptors such as C-C chemokine receptor type 4 (CCR4) on Treg cells (8, 85, 86). The CCL22-CCR4 axis is currently the most reported mechanism underlying Treg trafficking. TGF-β, the potent inducer of tumor progression and metastasis (87, 88), can augment CCL22 production by reducing the level of its upstream antagonist miR-34a in hepatocellular carcinoma (HCC) (89). Similarly, Wiedemann et al. (90) confirmed that IL-1α-induced Treg migration in HCC was also CCL22 dependent. This precise mechanism may also explain why Treg cells are recruited into NSCLC tumor tissues, as TGF-β and CCL22 are also enriched in these areas (91, 92). Zaynagetdinov et al. (93) found that IL-5, a cytokine involved in allergic and infectious diseases, promotes lung metastasis of tumor cells. Further mechanistic studies revealed that IL-5 contributes to CCL22 production by eosinophils in lung tissues, causing Treg cell recruitment and immunosuppression in the lung cancer milieu.
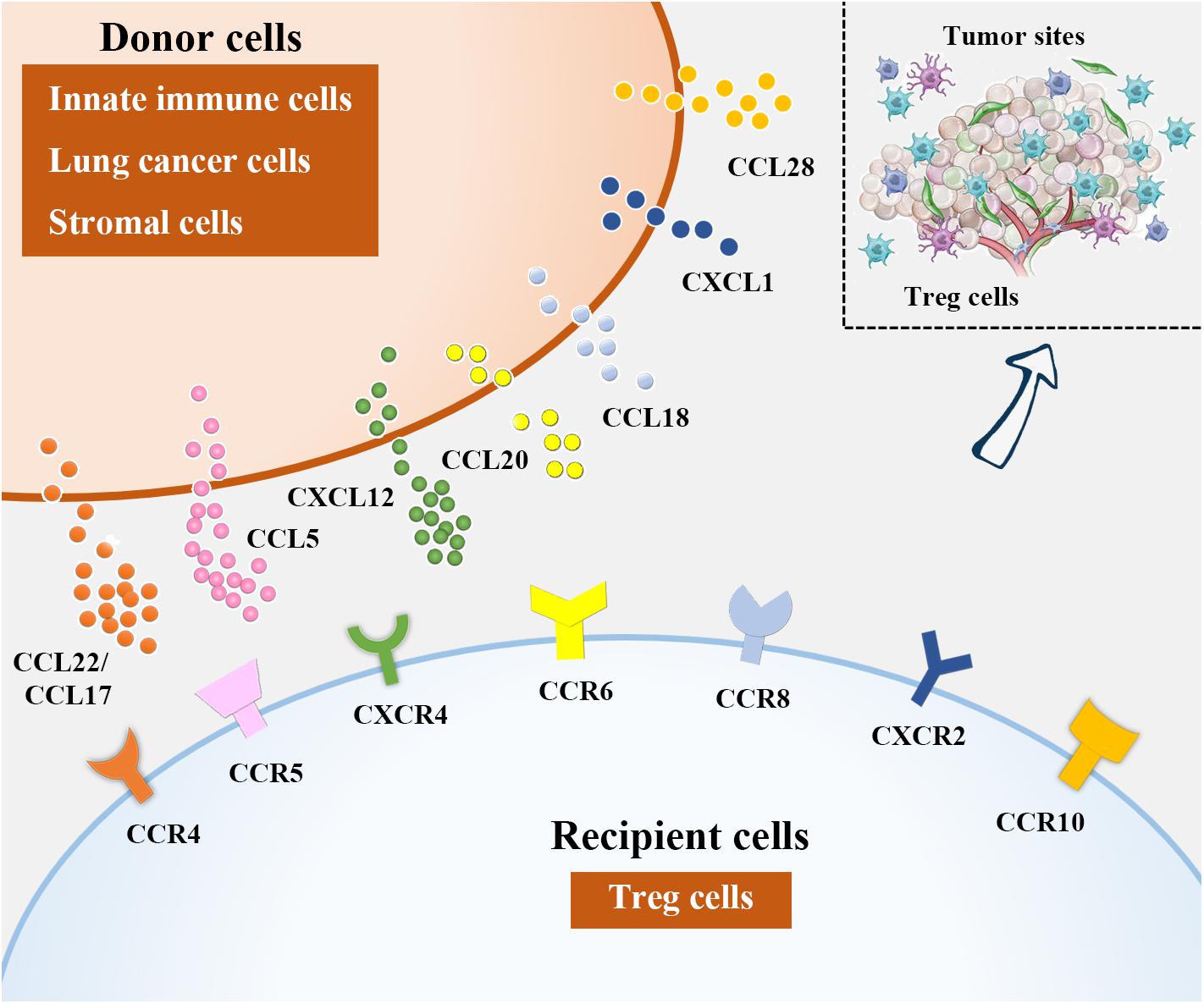
Figure 2. Treg cell recruitment into lung cancer. In NSCLC microenvironment, chemokines attract Tregs by binding to their corresponding receptors. Among them, DCs, macrophages and eosinophils are sources of CCL22, CCL17 is derived from DCs and lung cancer cells, CCL15 can be produced from lung cancer cells and MDCSs, lung cancer cells and stromal cells in NSCLC microenvironment are the major producers of CXCL12 and CCL20, respectively. Besides, CCL18 is generated from DCs and macrophages, lung cancer cells are responsible for CXCL1 and CCL28 production.
CCL17, derived from DCs and tumor cells including lung cancer cells, is another reported ligand for CCR4 during Treg cell recruitment (8, 94–96). It has been indicated that the abundance of CCR4+ Treg cells attracted by CCL17 was correlated with the clinical stage in NSCLC patients (97). To date, approaches based on Treg depletion in NSCLC patients via CCR4 neutralization have shown promise (98, 99).
CCL5-CCR5 Axis
CCL5 (also called RANTES) is a member of the CC family of chemokines. The roles of CCL5 in cancer biology are versatile as this ligand not only triggers antitumor immune responses but also is implicated in tumor progression and metastasis formation (100, 101). CCL5 has been implicated as a pathogenic factor in NSCLC. By establishing a murine intratibial model of cancer-associated bone resorption, Kim et al. (102) showed that Runt-related transcription factor 3 (RUNX3) silencing in NSCLC cells triggered cell proliferation, migration and invasion, accompanied by osteolytic lesions, by enhancing CCL5 levels, suggesting that CCL5 may be a potent promoter of NSCLC progression. Additionally, Schlecker et al. (103) revealed that MDSC-derived CCL5 infiltration into tumor tissues shared a mechanism similar to CCL5-CCR5 crosslinking to induce Treg cell accumulation.
FOXP3 expression has been reported in several tumors such as melanoma, pancreatic cancer, breast cancer and lung cancer (and is referred to as cancer-FOXP3) despite its non-redundant role in the regulation of Treg cell differentiation, development and functions (104, 105). A positive link between cancer-FOXP3 expression levels and Treg cell accumulation in pancreatic ductal adenocarcinoma (PDAC)-derived tumor tissues was recently shown. Mechanistically, cancer-FOXP3 directly transactivates CCL5, and the CCL5-CCR5 axis promotes Treg cell accumulation in tumor lesions from peripheral blood in vitro and in vivo (106). Considering that cancer-FOXP3 has been identified as a biomarker for poor prognosis in NSCLC (107, 108), it is rational to propose that FOXP3-initiated CCL5-CCR5 interactions may also participate in Treg cell migration in NSCLC.
The mechanisms underlying CCL5 elevation in the tumor microenvironment are increasingly clear. Nuclear focal adhesion kinase (FAK) was reported to induce Treg cell accumulation in the tumor milieu predominantly by modifying chemokine/cytokine and ligand-receptor crosstalk, including initiation of CCL5 transcription. Mechanistic investigations revealed that FAK integrated with chromatin in the nucleus could form a complex with transcription factors and their upstream regulators that control CCL5 expression (109).
Notably, the migration of Treg cells into tumor areas was reduced when the CCL5-CCR5 signaling was disrupted via the reduction of CCL5 production or systemic administration of the CCR5 inhibitor TAK-779 (110), suggesting that selective interventions with CCL5-CCR5 axis might represent a novel immunomodulatory strategy for NSCLC treatment.
Other Chemokine-Receptor Crosslinks
Reportedly, interactions between CXCL12 and CXCR4 (111), CCL20 and CCR6 (112), CCL18 and CCR8 (113, 114), CXCL1 and CXCR2 (115), and CCL28 and CCR10 (116, 117) are also important contributors to Treg accumulation in NSCLC.
Heterogeneity of Tregs
Cancer immunotherapies based on Treg blockade have shown sustained clinical responses in NSCLC treatment (118, 119), but their therapeutic efficacy varies and depends partially on the cell phenotype, gene expression, and the functional activities of Treg cells, which endow them with high heterogeneity. In other words, the heterogeneous characteristics of Treg cells make immunotherapy more complicated than ever expected and emphasize a better definition of the cells in lung cancer milieu.
As mentioned above, TCR signaling in response to neoantigens, might determine the heterogeneity of Tregs. By characterizing the TCR profiles of Treg cells from patients with metastatic melanoma, gastrointestinal, and ovarian cancers, Ahmadzadeh et al. (20) showed that the TCR repertoire of intratumoral Treg cells was distinct from that of intratumoral conventional T cells and that tumor antigens can induce the clonal expansion of intratumoral Tregs. Their findings also suggested a dynamic migration of tumor-antigen specific Treg cells from circulation into tumor sites due to the overlap between the TCRB clonotypes of intratumoral Treg cells and those of circulating Treg cells. Thus, it is reasonable that heterogeneous Tregs might receive TCR signaling from tumor antigens to proliferate during lung cancer progression. More importantly, the subtypes and abundances of functional characteristics of activated Treg cells are also heterogeneous in tumor-resident Treg cells, which might be correlated with their immunosuppressive activities.
Treg-Locking Transcription Factors
By studying the phenotypic, functional, as well as the transcriptional features of Treg cells in 92 NSCLC patients at the single-cell level, Akimova et al. (17) documented that the number and suppressive functions of intratumoral Tregs were dramatically elevated versus those of Tregs in the blood, lungs, and lymph nodes. This group also demonstrated that tumor Tregs exhibited a phenotype of increased abundance of Foxp3, as well as other transcription factors including Eos, IRF-4, Satb1, and Gata-1. Furthermore, the expression of these “Treg-locking” transcription factors was positively correlated with Foxp3 mRNA level, with the highest correlations for Eos and Satb1. Eos had an additional, Foxp3 mRNA-independent, positive correlation with Foxp3 protein level in NSCLC tumor Treg cells (17). Mechanistically, Eos limits Treg plasticity by preventing their reprogramming (120) and IRF-4 and Satb1 promote Treg cell differentiation via regulating IL-10 and Foxp3, respectively (121, 122).
GARP
Glycoprotein A repetitions predominant (GARP), encoded by leucine-rich repeat containing 32 (LRRC32), is expressed on the surface of activated human Treg cells and endows them with TGF-β-dependent bioavailability (123). Exposure to soluble GARP (sGARP) induces Foxp3, represses interleukin IL-2 and IFN-γ production and reduces T cell proliferation, promoting naïve T cells to skew toward iTreg cells. This process is attributed to Smad2/3 phosphorylation (124). Jin et al. (125) found that GARP expression was enhanced in Tregs from the tumor tissues of lung cancer patients and was associated with lymph node metastasis, distant metastasis, and clinical stage, which was partially consistent with Akimova’s findings (17). Furthermore, infiltrating Tregs from patients with early stage lung cancer displayed higher GARP expression than those from patients with advanced cancer, indicating a role for GARP in early diagnosis. Ex vivo studies demonstrated that human lung cancer cells might induce the expression of GARP in Tregs by cell contact-independent mechanisms.
TNFRSF9
To shed light on the baseline landscape of the composition, lineage and functional states of tumor-infiltrating lymphocytes (TILs) in NSCLC, deep single-cell RNA sequencing of 12,346 T cells from 14 treatment-naïve NSCLC patients was performed by Zhang’s group (126). Their findings showed that tumor Treg clonal expansion was mostly cluster-specific and occurred within tumors. Closer examination of the tumor-resident Treg cluster with suppressive functions revealed heterogeneity based on genes correlated with Treg functions. Notably, tumor necrosis factor receptor superfamily member 9 (TNFRSF9), a known activation marker for antigen-specific Tregs, exhibited a striking bimodal expression distribution within tumor-infiltrating Treg cells. Compared to the TNFRSF9– Treg population, the TNFRSF9+ population exhibited high levels of a set of 260 genes, including those performing immunosuppressive functions, further supporting the finding that TNFRSF9+ cells not only are antigen-experienced but also form the major population of functional tumor Tregs. Survival analysis showed that the 260-gene signature was predictive of worse patient prognosis.
CD45RA
Treg cells have been proposed to exist in multiple differentiation states with naive, effector and memory phenotypes (127). Two functionally distinct phenotypes of Tregs with suppressive activities, including CD45RA+ Foxp3lo naive Treg and CD45RA– Foxp3hi effector Treg (eTreg) populations, have been identified (128). The tumor milieu is fulfilled with Treg cells with an effector phenotype (129). It has been demonstrated in lung cancer patients that the number of CD45RA– Foxp3hi effector Tregs, but not the populations with the naïve CD45RA+ Foxp3lo phenotype, was increased in TILs compared to the number of those cells in peripheral blood monocyte cells (PBMCs). However, further studies are needed to explore the mechanisms of CD45RA in modulating the suppressive activities of eTreg cells and to evaluate the role of these subpopulations of Tregs in the modulation of NSCLC progression.
Perspective
Many cancers, including NSCLC, are not easily diagnosed by early laboratorial examination. Immunosuppression is not only a result of cancer progression but also a critical contributing factor for cancer initiation. With the introduction and development of precision medicine together with several developmental sequencing techniques, particularly single-cell sequencing (130), recent advances in examining the mechanisms underlying Treg migration, differentiation and phenotypes have opened new avenues for identifying Treg-based therapeutic strategies for NSCLC. Immune checkpoint inhibitors targeting CTLA-4 and PD-1, as well as neutralizing antibodies against chemokines or the corresponding receptors on Tregs, are showing promise in many cancers such as NSCLC (98, 131). Currently, the burgeoning immunometabolism field has revealed unexpected links to cancer immunology (132, 133). We consider identification of novel mechanisms within this field to be highly valuable, because metabolism disorder may be a mediator between the NSCLC milieu and infiltrating Treg cells. Overall, identification of the precise mechanisms by which Treg cells are recruited, generated or activated in NSCLC tissues not only is essential for improving the understandings of NSCLC pathogenesis but also can provide new insights into other types of cancers with analogous etiologies.
Author Contributions
MX and JW wrote the manuscript. JX modified this manuscript.
Funding
This work was supported by a grant from the National Natural Science Foundation of China (81772269).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. (2019) 69:7–34. doi: 10.3322/caac.21551
3. Marshall EA, Ng KW, Kung SH, Conway EM, Martinez VD, Halvorsen EC, et al. Emerging roles of T helper 17 and regulatory T cells in lung cancer progression and metastasis. Mol Cancer. (2016) 15:67.
4. Pikor LA, Ramnarine VR, Lam S, Lam WL. Genetic alterations defining NSCLC subtypes and their therapeutic implications. Lung Cancer. (2013) 82:179–89. doi: 10.1016/j.lungcan.2013.07.025
5. Thomas A, Liu SV, Subramaniam DS, Giaccone G. Refining the treatment of NSCLC according to histological and molecular subtypes. Nat Rev Clin Oncol. (2015) 12:511–26. doi: 10.1038/nrclinonc.2015.90
6. Malhotra J, Malvezzi M, Negri E, La Vecchia C, Boffetta P. Risk factors for lung cancer worldwide. Eur Respir J. (2016) 48:889–902. doi: 10.1183/13993003.00359-2016
7. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. (2015) 21:687–92. doi: 10.1158/1078-0432.CCR-14-1860
8. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. (2017) 17:559–72. doi: 10.1038/nri.2017.49
9. Zhang H, Maric I, DiPrima MJ, Khan J, Orentas RJ, Kaplan RN, et al. Fibrocytes represent a novel MDSC subset circulating in patients with metastatic cancer. Blood. (2013) 122:1105–13.
10. Dijkgraaf EM, Heusinkveld M, Tummers B, Vogelpoel LT, Goedemans R, Jha V, et al. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res. (2013) 73:2480–92. doi: 10.1158/0008-5472.CAN-12-3542
11. Kalathil S, Lugade AA, Miller A, Iyer R, Thanavala Y. Higher frequencies of GARP(+)CTLA-4(+)Foxp3(+) T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T-cell functionality. Cancer Res. (2013) 73:2435–44. doi: 10.1158/0008-5472.CAN-12-3381
12. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348:56–61. doi: 10.1126/science.aaa8172
13. Swartz MA, Iida N, Roberts EW, Sangaletti S, Wong MH, Yull FE, et al. Tumor microenvironment complexity: emerging roles in cancer therapy. Cancer Res. (2012) 72:2473–80. doi: 10.1158/0008-5472.CAN-12-0122
14. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
15. Miyara M, Ito Y, Sakaguchi S. TREG-cell therapies for autoimmune rheumatic diseases. Nat Rev Rheumatol. (2014) 10:543–51. doi: 10.1038/nrrheum.2014.105
16. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity. (2016) 45:1135–47. doi: 10.1016/j.immuni.2016.10.021
17. Akimova T, Zhang T, Negorev D, Singhal S, Stadanlick J, Rao A, et al. Human lung tumor FOXP3+ Tregs upregulate four “Treg-locking” transcription factors. JCI Insight. (2017) 2:e94075.
18. O’Callaghan DS, Rexhepaj E, Gately K, Coate L, Delaney D, O’Donnell DM, et al. Tumour islet Foxp3+ T-cell infiltration predicts poor outcome in nonsmall cell lung cancer. Eur Respir J. (2015) 46:1762–72. doi: 10.1183/13993003.00176-2014
19. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. (2013) 14:307–8.
20. Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanovic S, Robbins PF, et al. Tumor-infiltrating human CD4(+) regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci Immunol. (2019) 4:eaao4310. doi: 10.1126/sciimmunol.aao4310
21. Williams JA, Zhang J, Jeon H, Nitta T, Ohigashi I, Klug D, et al. Thymic medullary epithelium and thymocyte self-tolerance require cooperation between CD28-CD80/86 and CD40-CD40L costimulatory pathways. J Immunol. (2014) 192:630–40.
22. Qureshi OS, Kaur S, Hou TZ, Jeffery LE, Poulter NS, Briggs Z, et al. Constitutive clathrin-mediated endocytosis of CTLA-4 persists during T cell activation. J Biol Chem. (2012) 287:9429–40. doi: 10.1074/jbc.M111.304329
23. Linch SN, Kasiewicz MJ, McNamara MJ, Hilgart-Martiszus IF, Farhad M, Redmond WL. Combination OX40 agonism/CTLA-4 blockade with HER2 vaccination reverses T-cell anergy and promotes survival in tumor-bearing mice. Proc Natl Acad Sci USA. (2016) 113:E319–27.
24. Zheng SG, Wang JH, Stohl W, Kim KS, Gray JD, Horwitz DA. TGF-beta requires CTLA-4 early after T cell activation to induce FoxP3 and generate adaptive CD4+CD25+ regulatory cells. J Immunol. (2006) 176:3321–9.
25. Valk E, Rudd CE, Schneider H. CTLA-4 trafficking and surface expression. Trends Immunol. (2008) 29:272–9.
26. Molinero LL, Miller ML, Evaristo C, Alegre ML. High TCR stimuli prevent induced regulatory T cell differentiation in a NF-kappaB-dependent manner. J Immunol. (2011) 186:4609–17.
27. Xie M, Wang J, Gong W, Xu H, Pan X, Chen Y, et al. NF-kappaB-driven miR-34a impairs Treg/Th17 balance via targeting Foxp3. J Autoimmun. (2019) 102:96–113. doi: 10.1016/j.jaut.2019.04.018
28. Han Y, Chen Z, Yang Y, Jiang Z, Gu Y, Liu Y, et al. Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology. (2014) 59:567–79. doi: 10.1002/hep.26694
29. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep. (2015) 13:412–24. doi: 10.1016/j.celrep.2015.08.077
30. Manches O, Munn D, Fallahi A, Lifson J, Chaperot L, Plumas J, et al. HIV-activated human plasmacytoid DCs induce Tregs through an indoleamine 2,3-dioxygenase-dependent mechanism. J Clin Invest. (2008) 118:3431–9. doi: 10.1172/JCI34823
31. Erfani N, Mehrabadi SM, Ghayumi MA, Haghshenas MR, Mojtahedi Z, Ghaderi A, et al. Increase of regulatory T cells in metastatic stage and CTLA-4 over expression in lymphocytes of patients with non-small cell lung cancer (NSCLC). Lung Cancer. (2012) 77:306–11. doi: 10.1016/j.lungcan.2012.04.011
32. Mastracci L, Fontana V, Queirolo P, Carosio R, Grillo F, Morabito A, et al. Response to ipilimumab therapy in metastatic melanoma patients: potential relevance of CTLA-4(+) tumor infiltrating lymphocytes and their in situ localization. Cancer Immunol Immunother. (2020) 69:653–62.
33. Calabro L, Morra A, Giannarelli D, Amato G, D’Incecco A, Covre A, et al. Tremelimumab combined with durvalumab in patients with mesothelioma (NIBIT-MESO-1): an open-label, non-randomised, phase 2 study. Lancet Respir Med. (2018) 6:451–60. doi: 10.1016/S2213-2600(18)30151-6
34. Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. (2012) 30:2046–54.
35. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. (2013) 210:1695–710. doi: 10.1084/jem.20130579
36. Pai CS, Simons DM, Lu X, Evans M, Wei J, Wang YH, et al. Tumor-conditional anti-CTLA4 uncouples antitumor efficacy from immunotherapy-related toxicity. J Clin Invest. (2019) 129:349–63.
37. Aerts JG, Hegmans JP. Tumor-specific cytotoxic T cells are crucial for efficacy of immunomodulatory antibodies in patients with lung cancer. Cancer Res. (2013) 73:2381–8. doi: 10.1158/0008-5472.CAN-12-3932
38. Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y, Zang X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol Med (2015) 21:24–33. doi: 10.1016/j.molmed.2014.10.009
39. Lim TS, Chew V, Sieow JL, Goh S, Yeong JP, Soon AL, et al. PD-1 expression on dendritic cells suppresses CD8(+) T cell function and antitumor immunity. Oncoimmunology. (2016) 5:e1085146. doi: 10.1080/2162402x.2015.1085146
40. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. (2017) 545:495–9. doi: 10.1038/nature22396
41. Quatrini L, Wieduwild E, Escaliere B, Filtjens J, Chasson L, Laprie C, et al. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat Immunol. (2018) 19:954–62. doi: 10.1038/s41590-018-0185-0
42. Sun C, Mezzadra R, Schumacher TN. Regulation and Function of the PD-L1 Checkpoint. Immunity. (2018) 48:434–52. doi: 10.1016/j.immuni.2018.03.014
43. Sahin IH, Askan G, Hu ZI, O’Reilly EM. Immunotherapy in pancreatic ductal adenocarcinoma: an emerging entity? Ann Oncol. (2017) 28:2950–61. doi: 10.1093/annonc/mdx503
44. Baas M, Besancon A, Goncalves T, Valette F, Yagita H, Sawitzki B, et al. TGFbeta-dependent expression of PD-1 and PD-L1 controls CD8(+) T cell anergy in transplant tolerance. eLife. (2016) 5:e08133. doi: 10.7554/eLife.08133
45. Sacher AG, Gandhi L. Biomarkers for the clinical Use of PD-1/PD-L1 Inhibitors in non-small-cell lung cancer: a review. JAMA Oncol. (2016) 2:1217–22. doi: 10.1001/jamaoncol.2016.0639
46. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. (2013) 3:1355–63. doi: 10.1158/2159-8290.CD-13-0310
47. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med. (2018) 24:994–1004. doi: 10.1038/s41591-018-0057-z
48. Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci USA. (2008) 105:9331–6.
49. Chen X, Fosco D, Kline DE, Meng L, Nishi S, Savage PA, et al. PD-1 regulates extrathymic regulatory T-cell differentiation. Eur J Immunol. (2014) 44:2603–16.
50. Roemer MG, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J Clin Oncol. (2016) 34:2690–7.
51. Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature. (2016) 534:402–6. doi: 10.1038/nature18294
52. Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep. (2016) 16:2829–37.
53. Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, et al. BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. (2017) 18:2162–74. doi: 10.1016/j.celrep.2017.02.011
54. Lu C, Paschall AV, Shi H, Savage N, Waller JL, Sabbatini ME, et al. The MLL1-H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion. J Natl Cancer Inst. (2017) 109:djw283. doi: 10.1093/jnci/djw283
55. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. (2017) 19:1189–201. doi: 10.1016/j.celrep.2017.04.031
56. Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell. (2016) 30:925–39. doi: 10.1016/j.ccell.2016.10.010
57. Lastwika KJ, Wilson W III, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. (2016) 76:227–38. doi: 10.1158/0008-5472.CAN-14-3362
58. Coelho MA, de Carne Trecesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity. (2017) 47:1083–99.e86. doi: 10.1016/j.immuni.2017.11.016
59. Atefi M, Avramis E, Lassen A, Wong DJ, Robert L, Foulad D, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. (2014) 20:3446–57. doi: 10.1158/1078-0432.CCR-13-2797
60. Janse HJ, van Rensburg Azad T, Ling M, Hao Y, Snetsinger B, Khanal P, et al. The hippo pathway component TAZ promotes immune evasion in human cancer through PD-L1. Cancer Res. (2018) 78:1457–70.
61. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. (2016) 12:325–38.
62. Zhang H, Kozono DE, O’Connor KW, Vidal-Cardenas S, Rousseau A, Hamilton A, et al. TGF-beta inhibition rescues hematopoietic stem cell defects and bone marrow failure in fanconi anemia. Cell Stem Cell. (2016) 18:668–81. doi: 10.1016/j.stem.2016.03.002
63. Hart KM, Fabre T, Sciurba JC, Gieseck RL III, Borthwick LA, Vannella KM, et al. Type 2 immunity is protective in metabolic disease but exacerbates NAFLD collaboratively with TGF-beta. Sci Transl Med. (2017) 9:eaal3694. doi: 10.1126/scitranslmed.aal3694
64. Hata A, Chen YG. TGF-beta Signaling from Receptors to Smads. Cold Spring Harb Perspect Biol. (2016) 8:a022061. doi: 10.1101/cshperspect.a022061
65. Zhang YE. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb Perspect Biol. (2017) 9:a022129. doi: 10.1101/cshperspect.a022129
66. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
67. Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu Rev Immunol. (2012) 30:733–58. doi: 10.1146/annurev-immunol-020711-075043
68. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. (2003) 198:1875–86.
69. Schlenner SM, Weigmann B, Ruan Q, Chen Y, von Boehmer H. Smad3 binding to the foxp3 enhancer is dispensable for the development of regulatory T cells with the exception of the gut. J Exp Med. (2012) 209:1529–35. doi: 10.1084/jem.20112646
70. Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. (2008) 9:194–202. doi: 10.1038/ni1549
71. Lu L, Wang J, Zhang F, Chai Y, Brand D, Wang X, et al. Role of SMAD and non-SMAD signals in the development of Th17 and regulatory T cells. J Immunol. (2010) 184:4295–306. doi: 10.4049/jimmunol.0903418
72. Anz D, Mueller W, Golic M, Kunz WG, Rapp M, Koelzer VH, et al. CD103 is a hallmark of tumor-infiltrating regulatory T cells. Int J Cancer. (2011) 129:2417–26. doi: 10.1002/ijc.25902
73. Zhu J, Davidson TS, Wei G, Jankovic D, Cui K, Schones DE, et al. Down-regulation of Gfi-1 expression by TGF-beta is important for differentiation of Th17 and CD103+ inducible regulatory T cells. J Exp Med. (2009) 206:329–41. doi: 10.1084/jem.20081666
74. Baratelli F, Lee JM, Hazra S, Lin Y, Walser TC, Schaue D, et al. PGE(2) contributes to TGF-beta induced T regulatory cell function in human non-small cell lung cancer. Am J Transl Res. (2010) 2:356–67.
75. Takasaka N, Seed RI, Cormier A, Bondesson AJ, Lou J, Elattma A, et al. Integrin alphavbeta8-expressing tumor cells evade host immunity by regulating TGF-beta activation in immune cells. JCI Insight. (2018) 3:e122591.
76. Akhurst RJ. Targeting TGF-beta Signaling for Therapeutic Gain. Cold Spring Harb Perspect Biol. (2017) 9:a022301. doi: 10.1101/cshperspect.a022301
77. Sato T, Terai M, Tamura Y, Alexeev V, Mastrangelo MJ, Selvan SR. Interleukin 10 in the tumor microenvironment: a target for anticancer immunotherapy. Immunol Res. (2011) 51:170–82. doi: 10.1007/s12026-011-8262-6
78. Akdis CA, Akdis M. Mechanisms of immune tolerance to allergens: role of IL-10 and Tregs. J Clin Invest. (2014) 124:4678–80. doi: 10.1172/JCI78891
79. Hsu P, Santner-Nanan B, Hu M, Skarratt K, Lee CH, Stormon M, et al. IL-10 Potentiates differentiation of human induced regulatory T cells via STAT3 and Foxo1. J Immunol. (2015) 195:3665–74. doi: 10.4049/jimmunol.1402898
80. Zhu L, Shi T, Zhong C, Wang Y, Chang M, Liu X. IL-10 and IL-10 receptor mutations in very early onset inflammatory bowel disease. Gastroenterology Res. (2017) 10:65–9. doi: 10.14740/gr740w
81. Degboe Y, Rauwel B, Baron M, Boyer JF, Ruyssen-Witrand A, Constantin A, et al. Polarization of rheumatoid macrophages by TNF targeting through an IL-10/STAT3 mechanism. Front Immunol. (2019) 10:3. doi: 10.3389/fimmu.2019.00003
82. Boyle DL, Soma K, Hodge J, Kavanaugh A, Mandel D, Mease P, et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann Rheum Dis. (2015) 74:1311–6. doi: 10.1136/annrheumdis-2014-206028
83. Oh HM, Yu CR, Dambuza I, Marrero B, Egwuagu CE. STAT3 protein interacts with Class O Forkhead transcription factors in the cytoplasm and regulates nuclear/cytoplasmic localization of FoxO1 and FoxO3a proteins in CD4(+) T cells. J Biol Chem. (2012) 287:30436–43.
84. Harada Y, Harada Y, Elly C, Ying G, Paik JH, DePinho RA, et al. Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J Exp Med. (2010) 207:1381–91. doi: 10.1084/jem.20100004
85. Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. (2009) 69:2000–9.
86. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. (2013) 5:200ra116. doi: 10.1126/scitranslmed.3006504
87. Seoane J, Gomis RR. TGF-beta Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb Perspect Biol. (2017) 9:a022277. doi: 10.1101/cshperspect.a022277
88. Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S, et al. Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat Med. (2015) 21:1262–71. doi: 10.1038/nm.3961
89. Yang P, Li QJ, Feng Y, Zhang Y, Markowitz GJ, Ning S, et al. TGF-beta-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell. (2012) 22:291–303. doi: 10.1016/j.ccr.2012.07.023
90. Wiedemann GM, Knott MM, Vetter VK, Rapp M, Haubner S, Fesseler J, et al. Cancer cell-derived IL-1alpha induces CCL22 and the recruitment of regulatory T cells. Oncoimmunology (2016) 5:e1175794. doi: 10.1080/2162402x.2016.1175794
91. Li C, Wan L, Liu Z, Xu G, Wang S, Su Z, et al. Long non-coding RNA XIST promotes TGF-beta-induced epithelial-mesenchymal transition by regulating miR-367/141-ZEB2 axis in non-small-cell lung cancer. Cancer Lett. (2018) 418:185–95. doi: 10.1016/j.canlet.2018.01.036
92. Ma H, Shu Y, Pan S, Chen J, Dai J, Jin G, et al. Polymorphisms of key chemokine genes and survival of non-small cell lung cancer in Chinese. Lung Cancer. (2011) 74:164–9. doi: 10.1016/j.lungcan.2011.03.005
93. Zaynagetdinov R, Sherrill TP, Gleaves LA, McLoed AG, Saxon JA, Habermann AC, et al. Interleukin-5 facilitates lung metastasis by modulating the immune microenvironment. Cancer Res. (2015) 75:1624–34. doi: 10.1158/0008-5472.CAN-14-2379
94. Pere H, Montier Y, Bayry J, Quintin-Colonna F, Merillon N, Dransart E, et al. A CCR4 antagonist combined with vaccines induces antigen-specific CD8+ T cells and tumor immunity against self antigens. Blood. (2011) 118:4853–62. doi: 10.1182/blood-2011-01-329656
95. Weber C, Meiler S, Doring Y, Koch M, Drechsler M, Megens RT, et al. CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J Clin Invest. (2011) 121:2898–910. doi: 10.1172/JCI44925
96. Monick MM, Powers LS, Hassan I, Groskreutz D, Yarovinsky TO, Barrett CW, et al. Respiratory syncytial virus synergizes with Th2 cytokines to induce optimal levels of TARC/CCL17. J Immunol. (2007) 179:1648–58.
97. Liu W, Wei X, Li L, Wu X, Yan J, Yang H, et al. CCR4 mediated chemotaxis of regulatory T cells suppress the activation of T cells and NK cells via TGF-beta pathway in human non-small cell lung cancer. Biochem Biophys Res Commun. (2017) 488:196–203. doi: 10.1016/j.bbrc.2017.05.034
98. Kurose K, Ohue Y, Sato E, Yamauchi A, Eikawa S, Isobe M, et al. Increase in activated Treg in TIL in lung cancer and in vitro depletion of Treg by ADCC using an antihuman CCR4 mAb (KM2760). J Thorac Oncol. (2015) 10:74–83. doi: 10.1097/JTO.0000000000000364
99. Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, et al. Phase ia study of FoxP3+ CD4 treg depletion by infusion of a humanized anti-CCR4 antibody. KW-0761, in cancer patients. Clin Cancer Res. (2015) 21:4327–36. doi: 10.1158/1078-0432.CCR-15-0357
100. Bronte V, Bria E. Interfering with CCL5/CCR5 at the Tumor-Stroma Interface. Cancer Cell. (2016) 29:437–9. doi: 10.1016/j.ccell.2016.03.019
101. Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, et al. Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti-CCR5 therapy in cancer patients. Cancer Cell. (2016) 29:587–601. doi: 10.1016/j.ccell.2016.03.005
102. Kim HJ, Park J, Lee SK, Kim KR, Park KK, Chung WY. Loss of RUNX3 expression promotes cancer-associated bone destruction by regulating CCL5, CCL19 and CXCL11 in non-small cell lung cancer. J Pathol. (2015) 237:520–31. doi: 10.1002/path.4597
103. Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol. (2012) 189:5602–11. doi: 10.4049/jimmunol.1201018
104. Yang S, Liu Y, Li MY, Ng CSH, Yang SL, Wang S, et al. FOXP3 promotes tumor growth and metastasis by activating Wnt/beta-catenin signaling pathway and EMT in non-small cell lung cancer. Mol Cancer. (2017) 16:124. doi: 10.1186/s12943-017-0700-1
105. Martin F, Ladoire S, Mignot G, Apetoh L, Ghiringhelli F. Human FOXP3 and cancer. Oncogene. (2010) 29:4121–9. doi: 10.1038/onc.2010.174
106. Wang X, Lang M, Zhao T, Feng X, Zheng C, Huang C, et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3(+)Treg cells in pancreatic ductal adenocarcinoma. Oncogene. (2017) 36:3048–58. doi: 10.1038/onc.2016.458
107. Triulzi T, Tagliabue E, Balsari A, Casalini P. FOXP3 expression in tumor cells and implications for cancer progression. J Cell Physiol. (2013) 228:30–5. doi: 10.1002/jcp.24125
108. Tao H, Mimura Y, Aoe K, Kobayashi S, Yamamoto H, Matsuda E, et al. Prognostic potential of FOXP3 expression in non-small cell lung cancer cells combined with tumor-infiltrating regulatory T cells. Lung Cancer. (2012) 75:95–101. doi: 10.1016/j.lungcan.2011.06.002
109. Serrels A, Lund T, Serrels B, Byron A, McPherson RC, von Kriegsheim A, et al. Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell. (2015) 163:160–73. doi: 10.1016/j.cell.2015.09.001
110. Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. (2009) 182:1746–55.
111. Scala S. Molecular pathways: targeting the CXCR4-CXCL12 axis–untapped potential in the tumor microenvironment. Clin Cancer Res. (2015) 21:4278–85. doi: 10.1158/1078-0432.CCR-14-0914
112. Zhang CY, Qi Y, Li XN, Yang Y, Liu DL, Zhao J, et al. The role of CCL20/CCR6 axis in recruiting Treg cells to tumor sites of NSCLC patients. Biomed Pharmacother. (2015) 69:242–8. doi: 10.1016/j.biopha.2014.12.008
113. Bremnes RM, Al-Shibli K, Donnem T, Sirera R, Al-Saad S, Andersen S, et al. The role of tumor-infiltrating immune cells and chronic inflammation at the tumor site on cancer development, progression, and prognosis: emphasis on non-small cell lung cancer. J Thorac Oncol. (2011) 6:824–33. doi: 10.1097/JTO.0b013e3182037b76
114. Chenivesse C, Chang Y, Azzaoui I, Yahia SA, Morales O, Ple C, et al. Pulmonary CCL18 recruits human regulatory T cells. J Immunol. (2012) 189:128–37. doi: 10.4049/jimmunol.1003616
115. Lv M, Xu Y, Tang R, Ren J, Shen S, Chen Y, et al. miR141-CXCL1-CXCR2 signaling-induced Treg recruitment regulates metastases and survival of non-small cell lung cancer. Mol Cancer Ther. (2014) 13:3152–62. doi: 10.1158/1535-7163.MCT-14-0448
116. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. (2011) 475:226–30.
117. Yang D, Zhou J, Zeng T, Yang Z, Wang X, Hu J, et al. Serum chemokine network correlates with chemotherapy in non-small cell lung cancer. Cancer Lett. (2015) 365:57–67. doi: 10.1016/j.canlet.2015.05.001
118. Pircher A, Gamerith G, Amann A, Reinold S, Popper H, Gachter A, et al. Neoadjuvant chemo-immunotherapy modifies CD4(+)CD25(+) regulatory T cells (Treg) in non-small cell lung cancer (NSCLC) patients. Lung Cancer. (2014) 85:81–7. doi: 10.1016/j.lungcan.2014.04.001
119. Roselli M, Cereda V, di Bari MG, Formica V, Spila A, Jochems C, et al. Effects of conventional therapeutic interventions on the number and function of regulatory T cells. Oncoimmunology. (2013) 2:e27025. doi: 10.4161/onci.27025
120. Sharma MD, Huang L, Choi JH, Lee EJ, Wilson JM, Lemos H, et al. An inherently bifunctional subset of Foxp3+ T helper cells is controlled by the transcription factor eos. Immunity. (2013) 38:998–1012. doi: 10.1016/j.immuni.2013.01.013
121. Cretney E, Xin A, Shi W, Minnich M, Masson F, Miasari M, et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat Immunol. (2011) 12:304–11. doi: 10.1038/ni.2006
122. Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, et al. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat Immunol. (2017) 18:173–83. doi: 10.1038/ni.3646
123. Wu BX, Li A, Lei L, Kaneko S, Wallace C, Li X, et al. Glycoprotein A repetitions predominant (GARP) positively regulates transforming growth factor (TGF) beta3 and is essential for mouse palatogenesis. J Biol Chem. (2017) 292:18091–7. doi: 10.1074/jbc.M117.797613
124. Hahn SA, Stahl HF, Becker C, Correll A, Schneider FJ, Tuettenberg A, et al. Soluble GARP has potent antiinflammatory and immunomodulatory impact on human CD4(+) T cells. Blood. (2013) 122:1182–91. doi: 10.1182/blood-2012-12-474478
125. Jin H, Sun L, Tang L, Yu W, Li H. Expression of GARP Is Increased in tumor-infiltrating regulatory t cells and is correlated to clinicopathology of lung cancer patients. Front Immunol. (2017) 8:138. doi: 10.3389/fimmu.2017.00138
126. Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. (2018) 24:978–85. doi: 10.1038/s41591-018-0045-3
127. Lin YC, Chang LY, Huang CT, Peng HM, Dutta A, Chen TC, et al. Effector/memory but not naive regulatory T cells are responsible for the loss of concomitant tumor immunity. J Immunol. (2009) 182: 6095–104.
128. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. (2009) 30:899–911. doi: 10.1016/j.immuni.2009.03.019
129. Ward-Hartstonge KA, Kemp RA. Regulatory T-cell heterogeneity and the cancer immune response. Clin Transl Immunol. (2017) 6:e154. doi: 10.1038/cti.2017.43
130. Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet. (2013) 14:618–30. doi: 10.1038/nrg3542
131. Brahmer JR, Pardoll DM. Immune checkpoint inhibitors: making immunotherapy a reality for the treatment of lung cancer. Cancer Immunol Res. (2013) 1:85–91. doi: 10.1158/2326-6066.CIR-13-0078
132. Newton R, Priyadharshini B, Turka LA. Immunometabolism of regulatory T cells. Nat Immunol. (2016) 17:618–25. doi: 10.1038/ni.3466
Keywords: non-small cell-lung cancer, tumor microenvironment, CD4+ Tregs, differentiation, proliferation, chemotaxis, phenotype
Citation: Xie M, Wei J and Xu J (2020) Inducers, Attractors and Modulators of CD4+ Treg Cells in Non-Small-Cell Lung Cancer. Front. Immunol. 11:676. doi: 10.3389/fimmu.2020.00676
Received: 23 December 2019; Accepted: 26 March 2020;
Published: 28 April 2020.
Edited by:
Peter Brossart, University of Bonn, GermanyReviewed by:
Ying Ma, The University of Texas MD Anderson Cancer Center, United StatesDennis O. Adeegbe, Moffitt Cancer Center, United States
Copyright © 2020 Xie, Wei and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Xu, eHVfamlhbjc5QDE2My5jb20=
†These authors have contributed equally to this work