 Takuya Takeichi
Takuya Takeichi Masashi Akiyama
Masashi Akiyama- Department of Dermatology, Nagoya University Graduate School of Medicine, Nagoya, Japan
Keratosis linearis with ichthyosis congenita and sclerosing keratoderma (KLICK) syndrome is a rare autosomal recessive skin disorder characterized by palmoplantar keratoderma, linear hyperkeratotic plaques, ichthyosiform scaling, circular constrictions around the fingers, and numerous papules distributed linearly in the arm folds and on the wrists. Histologically, the affected skin shows hypertrophy and hyperplasia of the spinous, granular, and horny epidermal layers with mild infiltration of inflammatory cells in the upper dermis. There are 14 patients with KLICK syndrome described in the literature, and they all carry the same nucleotide deletion. Proteasome maturation protein (POMP), encoded by POMP, is an ubiquitously expressed protein that functions as a chaperone for proteasome maturation. KLICK syndrome is caused by a reduction in POMP levels that leads to proteasome insufficiency in differentiating keratinocytes. It is noteworthy that POMP is also known to be the causative gene for proteasome-associated autoinflammatory syndrome-2 (PRAAS2). It is considered that the disrupted proteasome assembly caused by the POMP mutation might lead to both skin inflammation and then hyperkeratosis in KLICK syndrome. Inflammation caused by the hyperactivation of innate immunity occasionally leads to inflammatory diseases of the skin, recently denoted as autoinflammatory keratinization diseases (AiKDs). We propose that KLICK syndrome caused by the specific 1-bp nucleotide deletion mutation in the regulatory region of POMP might be in a spectrum of proteasome-associated phenotypes.
Introduction
Keratosis linearis with ichthyosis congenita and sclerosing keratoderma (KLICK) syndrome (MIM 601952) is an autosomal recessive skin disorder characterized by palmoplantar keratoderma, linear hyperkeratotic plaques, ichthyosiform scaling, circular constrictions around the fingers, and numerous papules distributed linearly in the arm folds and on the wrists (1, 2). KLICK syndrome is a rare disease, with only several pedigrees having been described. In 2010, a single nucleotide deletion in the 5′ untranslated region (UTR) of POMP (rs112368783) was identified in 12 KLICK patients (3). All described patients with KLICK syndrome harbored the same homozygous 1-bp deletion in the 5′ UTR of the POMP gene (3–5).
Autoinflammatory keratinization disease (AiKD) is an umbrella term recently introduced to describe inflammatory keratinization diseases caused by mutations in single genes associated with autoinflammatory diseases (6, 7). AiKDs are genetically heterogeneous, and their different subtypes show various clinical features, complications, and prognoses (8–11). We propose that KLICK syndrome associated with the POMP mutation be categorized as an AiKD.
What is Klick Syndrome?
In 1989, Pujol RM et al. reported four members of a consanguineous family presenting a disorder similar to KLICK syndrome (12). They described a congenital syndrome consisting of (i) generalized ichthyosiform dermatosis, (ii) diffuse palmoplantar keratoderma with sclerosis, deformities, pseudoainhum, and functional impairment, (iii) multiple keratotic papules in a symmetrical linear cordlike arrangement involving the flexures and exhibiting peculiar acrosyringial keratoses, (iv) a possible autosomal recessive pattern of inheritance, (v) inconsistent dental abnormalities, and (vi) the absence of systemic involvement (e.g., neurological or ophthalmological) (12). Their peculiar clinical pictures were described as “congenital ichthyosiform dermatosis with linear keratotic flexural papules and sclerosing palmoplantar keratoderma” (12). A biopsy specimen from an area with ichthyosiform dermatosis showed irregular hyperplasia, hypergranulosis, hyperkeratosis, and parakeratosis (12). In addition, the dermis showed mild superficial perivascular lymphohistiocytic infiltrates. In 1997, Vahlquist et al. reported an additional case and proposed the acronym KLICK to define this uncommon disorder (2, 13). Using a combination of homozygosity mapping and candidate gene screening, Dahlqvist J et al. identified a single-nucleotide deletion in the 5′ UTR of POMP that was identified in 12 KLICK patients (2, 12, 14, 15). The families were nonrelated and originated from Spain, Italy, Netherlands, Sweden, and Norway (3). Haplotype analysis using microsatellite markers flanking POMP in the eight affected probands found at least five different haplotypes, suggesting that the c.-95delC variant is a recurrent, rather than a founder, mutation (3).
Recently, an unusual case of KLICK syndrome was reported whose initial clinical diagnosis was erythrokeratoderma or loricrin keratoderma (5). The patient had diffuse thin white scaling skin and well-demarcated nonmigratory symmetrical erythematous and hyperkeratotic plaques on the limbs and extremities (5). A skin biopsy revealed irregular acanthosis and hypergranulosis associated with numerous enlarged keratohyaline granules (5). The presence of well-demarcated erythematous and hyperkeratotic plaques, as seen in erythrokeratoderma, is not a clinical feature that has been commonly reported for KLICK syndrome (5). To date, ~20 cases of KLICK syndrome associated with the recurrent hotspot mutation in the 5′ UTR of POMP have been reported. Some cases of KLICK syndrome show significant improvement of the skin eruptions with etretinate therapy (4, 5, 13).
Klick Syndrome and Proteasome Insufficiency
POMP, encoded by POMP, is an ubiquitously expressed protein that functions as a chaperone for proteasome maturation of the standard proteasome and the immunoproteasome (3, 16). Constitutive proteasomes and immunoproteasomes shape the peptide repertoire presented by major histocompatibility complex class I (MHC-I) molecules by harboring different sets of catalytically active subunits and plays a critical role in homeostasis and immunity (17). The ubiquitin–proteasome system (UPS) is a selective proteolytic system in which substrates are recognized and tagged with ubiquitin for processive degradation by the proteasome (18). Cells rapidly shift to immunoproteasome formation in response to proinflammatory cytokines produced by the innate immune system early upon infection (19). POMP is strongly and consistently expressed from the basal to the granular layer of the epidermis in sections from healthy subjects, whereas in KLICK patients, the staining is strong in the basal layer with a gradual decrease toward the granular layer (3). Thus, KLICK syndrome is caused by a reduction in POMP levels that leads to proteasome insufficiency in differentiating keratinocytes (20). POMP functions as a chaperone for proteasome assembly and interacts with an initially formed α ring for subsequent sequential incorporation of β subunits into both the standard multiprotein complex 20S proteasome and the immunoproteasome (16). Proteasome inhibition is known to cause increased endoplasmic reticulum (ER) stress (21). CCAAT/enhancer-binding protein homologous protein (CHOP) is a transcription factor induced by persistently elevated ER stress and by the unfolded protein response (UPR). Immunostaining of CHOP shows a gradual, but moderate, increase from the spinous to the granular layer in the normal skin. In contrast, the staining of CHOP is clearly increased in the granular layer, and weak staining is also seen in the horny layer in KLICK skin (3). This staining pattern is consistent with the abnormal distribution of POMP and the proteasome subunits, α7 and β5, in the granular layer of individuals with KLICK (3).
Dahlqvist et al. revealed that the knockdown of POMP expression in cell cultures results in decreased amounts of proteasome subunits (20). Additionally, POMP knockdown causes a slight increase in the ER chaperone BiP in keratinocyte-derived HaCaT, an immortalized human keratinocyte cell line, cells but not in HeLa cells, supporting the idea of tissue-specific sensitivity to ER stress (20). ER stress is activated by impairment in the degradation of misfolded proteins due to dysfunctional proteasomes (22). Importantly, physiological ER stress is required for the maintenance of normal biological functions in skin, including keratinocyte differentiation, a vital process in competent skin barrier formation (23). Activation of the UPR likely leads to the disturbance in terminal differentiation of keratinocytes (22). The ER stress that leads to UPR in differentiating cells is very mild and occurs at levels that do not cause keratinocytes apoptosis, whereas severe ER stress, i.e., that which exceeds mild ER stress, does cause epidermal apoptosis (22). Excessive ER stress has been reported to be involved in the pathogenesis of certain skin disorders, including inflammatory skin diseases (i.e., psoriasis, rosacea, vitiligo, etc.) (23).
Pomp-Related Systemic Autoinflammatory Diseases
It is noteworthy that POMP is also known to be a causative gene for proteasome-associated autoinflammatory syndrome (PRAAS2) (24), also known as POMP-related autoinflammation and immune dysregulation disease (PRAID) (16). In fact, a homozygous single-nucleotide deletion in the 5′ UTR of POMP causes KLICK syndrome, whereas heterozygous frameshift variants in the penultimate exon of POMP result in systemic autoinflammatory diseases (Table 1) (16, 24). Autosomal recessive homozygous or compound heterozygous loss-of-function mutations in PSMB8, which encodes the inducible proteasome component β5i, cause a syndrome that has historically been referred to as joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced childhood-onset lipodystrophy syndrome (25), Nakajo–Nishimura syndrome (26, 27), or chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) (28). These conditions form a single disease spectrum of PRAAS (24). Brehm et al. reported additional disease-causing variants in three proteasome genes, PSMA3 encoding α7, PSMB4 encoding β7, PSMB9 encoding β1i, and POMP, and also established the digenic inheritance of PRAAS (24). Additive depletion of two proteasome subunits by small-interfering RNA (siRNA) was shown to cause more severe assembly defects and decreases in proteolytic function than monogenic inherited PRAAS (24). Recently, Sarrabay et al. described a patient exhibiting a PRAAS phenotype due to a homozygous mutation in PSMB10, which led to interferon (IFN) type I dysregulation, PSMB10 maturation defect, and enzymatic impairment (29). Notably, among these mutations, the 5′ UTR of PSMB4 variant (c.-9G>A) reduced the expression levels of the mutant transcripts (24). Patients with PRAID show a unique constellation of early-onset combined immunodeficiency, inflammatory neutrophilic dermatosis, and autoimmunity (16). Although the proposed mechanism of diseases is different (haploinsufficiency vs. dominant-negative effect), the clinical inflammatory phenotype of PRAID is similar to that of PRAAS. PRAID has been considered as the proteasome-associated autoinflammatory syndrome-2 (PRAAS2, MIM 618048).
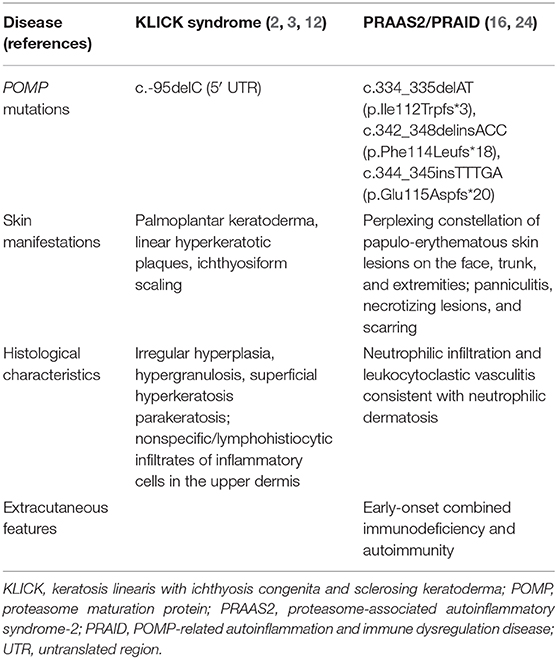
Table 1. Comparison of two syndromes associated with POMP mutations.
The first reported patient with PRAAS2 had a heterozygous frameshift mutation c.344_345insTTTGA (p.Glu115Aspfs*20) in exon 5 of POMP (24). This POMP mutation likely causes haploinsufficiency, as supported by previous findings in which an ~50% reduction in POMP levels is sufficient to cause impaired proteasome activity and cell death in vitro (30, 31). Although the detailed cutaneous features of PRAAS2 were lacking, this patient with POMP mutation presented with periorbital erythema, annular plaques, and acanthosis nigricans (24). Subsequently, Poli et al. detected two de novo frameshift POMP mutations, c.334_335delAT (p.Ile112Trpfs*3) and c.342_348delinsACC (p.Phe114Leufs*18), in two unrelated affected individuals with PRAID. Both heterozygous frameshift mutations in the penultimate exon of POMP escape nonsense-mediated messenger RNA (mRNA) decay and result in a truncated protein that perturbs proteasome assembly by a dominant-negative mechanism (16). The aggregation of ubiquitin-modified proteins that resulted from the proteasome dysfunction was observed in fibroblast cell lines from these two patients (16). The cutaneous manifestations of PRAID are a perplexing constellation of papulo-erythematous skin lesions on the face, trunk, and extremities, and these eruptions can progress to necrotizing lesions and subsequent scarring (16). Skin biopsies from the affected lesions in both PRAID patients revealed neutrophilic infiltration and leukocytoclastic vasculitis consistent with neutrophilic dermatosis (16). These clinical and histological findings suggest more severe inflammation in the skin eruptions of PRAID than in those of KLICK syndrome. HEK293T cells transfected with the mutant POMP constructs detected in the patients with PRAID showed increased expression of genes that are induced by type 1 interferon, indicating a disease-promoting toxic dominant-negative effect of the truncated protein and resembling the findings in patients with PRAID (16).
Autoinflammatory Diseases Caused by Inflammasome Hyperactivation
The KLICK syndrome phenotype, which is limited to the epidermis, is reminiscent of another group of diseases that are caused by mutations in the pyrin/leucine-rich repeat (LRR) domains of NLRP1, mutations that lead to multiple self-healing palmoplantar carcinoma (MSPC)/familial keratosis lichenoides chronica (FKLC); also, the PRAID phenotype is reminiscent of diseases that are caused by mutations in the NLRP1 gene causative of NLRP1-associated autoinflammation with arthritis and dyskeratosis (NAIAD)/juvenile-onset recurrent respiratory papillomatosis (JRRP) (32). Characteristic clinical features of FKLC include tiny papules on the trunk and extremities that become confluent, resulting in linear and reticulate patterns, and seborrheic dermatitis-like eruptions on the face (32). The lesions have a chronic and often progressive course. Excessive activation of inflammasomes has been demonstrated in patient keratinocytes, and inflammasome-dependent interleukin (IL)-1 cytokines have been shown to cause inflammatory FKLC (32). We have classified FKLC as an original member of the AiKDs (6). Thereafter, one family with an NLRP1 mutation between the NACHT and LRR domains and one sporadic patient with an NLRP1 mutation in the FIIND domain were reported to have autoinflammation symptoms, including follicular keratosis in the skin, as well as polyarthritis (NAIAD), and those symptoms were responsive to IL-1 inhibition (33) (Supplementary Table 1). Serum IL-18 and caspase-1 levels were elevated in the patients with NAIAD, which suggests the hyperactivation of the NLRP1 inflammasome (33). Moreover, very recently, a homozygous NLRP1 gain-of-function mutation located between the NACHT and LRR domains in siblings with a syndromic form of JRRP was reported (34). JRRP is a rare and debilitating childhood disease that presents with recurrent growth of papillomas in the upper airway (34). Drutman et al. revealed that patient-derived keratinocytes secreted elevated levels of IL-1β at baseline, and both patients displayed elevated levels of inflammasome-induced cytokines in the serum (34). Notably, these patients had unique cutaneous eruptions: keratosis pilaris on the legs and lower trunk, palmoplantar wart-like hyperkeratotic papules, and atrophoderma vermiculata on the cheeks (34). Thus, several distinct phenotypes resulting from the different mutations in NLRP1 have been reported. Although phenotype/genotype correlations are still unclear, the pathogenesis of these skin inflammatory diseases is linked to gain of function in the NLRP1 inflammasome.
Other Cutaneous Phenotypes Associated With Autoinflammation in the Skin
Recently, we proposed that porokeratosis be categorized as an AiKD (8). Porokeratosis is a genetically heterogeneous disorder that can be caused by mutations in any of the four genes involved in the mevalonate pathway (MVK, MVD, PMVK, and FDPS) or by mutations in SLC17A9 (8, 35). Interestingly, one of the major causative genes of porokeratosis, MVK, is also known to be causative of a conventional autoinflammatory disease, hyperimmunoglobulinemia D, and periodic fever syndrome (MIM 260920) (8). In 2010, mutations in genes encoding γ-secretase subunits (NCSTN, PSENEN, and PSEN1) were reported in patients presenting with hidradenitis suppurativa (HS) (36). The essential subunit of the γ-secretase complex, an endoprotease complex, catalyzes the intramembrane cleavage of integral membrane proteins, such as Notch receptors, and amyloid-beta precursor protein (37). γ-Secretase deficiency could also regulate inflammation by processing important cytokine receptors such as IL-1β R1/R2 and IL-6R (37). HS caused by mutations in genes encoding γ-secretase subunits was suggested to have features characteristic of an AiKD (9, 38). Mutations involving different autoinflammatory genes (MEFV, NLRP3, NLRP12, NOD2, LPIN2, and PSTPIP1) have been reported in syndromic HS [pyoderma gangrenosum, acne, and hidradenitis suppurativa (PASH) syndrome] as well as in pyoderma gangrenosum (PG), a prototypic neutrophilic dermatosis (39). Marzano et al. reported that increase in skin expression of IL-1β and IL-17 and the presence of mutations in these genes involved in autoinflammation indicate that PG is a polygenic autoinflammatory condition, as previously demonstrated in PASH (39, 40).
Some studies of mouse models of autoinflammatory diseases mimicking neutrophilic dermatosis in humans were also reported. Neutrophilic dermatosis encompasses disorders that are characterized by neutrophilic infiltration with ulceration in the upper dermis not associated with infection, such as Sweet's syndrome and PG (41). Mutations in the PTPN6 gene that encodes the protein tyrosine phosphatase Src homology region 2 (SH2) domain-containing phosphatase 1 (SHP-1) have been linked with autoinflammatory and autoimmune diseases in humans (41). Hypomorphic Ptpn6 mutant mice with a homozygous Tyr208Asn amino acid alteration mutation (exhibiting spontaneous inflammation or spin) develop persistent footpad swelling and suppurative inflammation that are very similar to neutrophilic dermatosis in humans (42–44).
Klick Syndrome Has Features Characteristic of an Aikd
A number of unique features support KLICK syndrome as an AiKD (6, 7). First, the skin of affected individuals histologically shows hypertrophy and hyperplasia of the spinous, granular, and horny epidermal layers (2, 5, 12). In addition, mild, spare nonspecific/lymphohistiocytic infiltrates of inflammatory cells are seen in the upper dermis (2, 12). These histological features suggest that the primary and main inflammation sites of KLICK are the epidermis and the upper dermis. Second, the ichthyosis, the palmoplantar keratoderma with constricting bands, and the keratotic papules that are seen in KLICK syndrome are usual and common eruptions caused by hyperkeratosis, which is the main characteristic phenotype of AiKDs. Finally, the causal genetic variant of KLICK is consistent with proteasome dysfunction (3, 20). Although the molecular mechanism of the disease is unclear, KLICK patients exhibit increased expression of the ER stress markers BiP and/or CHOP, and prolonged ER stress is known to induce inflammation as well as to be responsible for the pathogenesis of numerous chronic inflammatory diseases (16, 20). Although there are no reports of hyperactivation of innate immunity or of the induction of a type 1 interferon response in KLICK patients, the chronic ER stress present in the epidermis of KLICK patients may eventually trigger autoinflammation (16).
Like porokeratosis and hidradenitis suppurativa, as novel genetic causes and predisposing factors for inflammatory keratinization disorders have been revealed, we assume that more skin inflammatory diseases will be categorized as AiKDs. In KLICK syndrome, the disrupted proteasome assembly caused by the POMP mutation leads to hyperkeratosis (22) and might also lead to autoinflammation by increased type 1 interferon signaling (Figure 1). Recently, significant upregulation of IL-17/tumor necrosis factor alpha (TNF-α)-related genes has been reported in autosomal recessive congenital ichthyosis patients (45, 46). Furthermore, clinically, anti-inflammatory therapies (e.g., anti-TNF-α antibodies, anti-IL-17 antibodies) have been reported as useful treatments for inherited ichthyoses and in other AiKDs (7, 47, 48). Therefore, because KLICK syndrome is also a form of inherited ichthyosis (49), anti-inflammatory therapies might be useful for KLICK patients (48).
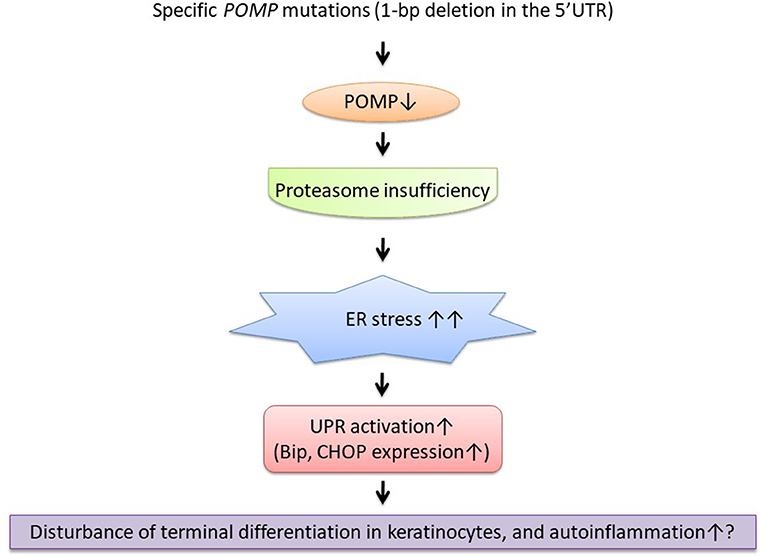
Figure 1. The suggestive pathogenesis of keratosis linearis with ichthyosis congenita and sclerosing keratoderma (KLICK) syndrome as an autoinflammatory keratinization disease (AiKD). ER, endoplasmic reticulum; POMP, proteasome maturation protein; UPR, unfolded protein response.
Discussion
This review summarized the pathogenesis and clinical features of KLICK syndrome associated with a specific POMP mutation. Some KLICK cases show improvement of the skin eruptions with etretinate, one of the systemic retinoids, therapy. Although oral retinoids have been described as efficient therapeutic agents for severe ichthyotic disorders, those treatments have various severe side effects. Moderate to severe ichthyosis is known to have a significant impact on quality of life (50). Thus, we strongly hope that safe, effective therapies will be established for patients with KLICK syndrome in the near future based on an understanding of the molecular pathogenesis of KLICK syndrome.
Both the clinical eruptions and the histological findings described in the reported KLICK patients clearly show mild to moderate inflammation in the affected skin (2, 5, 12). As we have cited that prolonged ER stress is known to induce inflammation in numerous chronic inflammatory skin diseases (23), the inflammation seen in KLICK syndrome could be considered to result from continuous abnormal ER stress in keratinocytes. Hetz et al. notes that under “irremediable ER stress,” the UPR actively promotes proteotoxicity, sterile inflammation, and apoptosis (51). Agyemang et al. report that Mendelian defects in the proteasome cause protein accumulation, which can trigger interferon-dependent autoinflammatory disease (52). However, in the literature, there are little data revealing the detailed inflammatory pathways from ER stress and UPR activation in KLICK syndrome. Although there are no therapeutic interventions available that target proteasome assembly, improving the proteostatic potential of cells by intervention with UPR (51) might be a therapeutic strategy for KLICK syndrome. To reduce patient distress, further clinical and laboratory investigations for the diagnosis and treatment of KLICK are needed.
Author Contributions
TT and MA contributed conception and design of the study and read and approved the submitted version. TT wrote the first draft of the manuscript. MA contributed to manuscript revision.
Funding
This work was supported by Grant-in-Aid for Scientific Research (B) 18H02832 to MA and by Grant-in-Aid for Young Scientists 18K16058 to TT from the Japan Society for the Promotion of Science (JSPS). This work was also supported by funding from Advanced Research and Development Programs for Medical Innovation (AMED-CREST) 19gm0910002h0105 to MA. This research was supported by AMED under Grant Numbers JP19ek0109281 and JP19ek0109295. The authors thank JSID's Fellowship Shiseido Research Grant 2019.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00641/full#supplementary-material
Abbreviations
AiKDs, autoinflammatory keratinization diseases; ER, endoplasmic reticulum; HS, hidradenitis suppurativa; KLICK, keratosis linearis with ichthyosis congenita and sclerosing keratoderma; PG, pyoderma gangrenosum; POMP, proteasome maturation protein; PRAAS2, proteasome-associated autoinflammatory syndrome; PRAID, POMP-related autoinflammation and immune dysregulation disease; UPR, unfolded protein response; UTR, untranslated region.
References
1. Takeichi T, Akiyama M. Inherited ichthyosis: non-syndromic forms. J Dermatol. (2016) 43:242–51. doi: 10.1111/1346-8138.13243
2. Vahlquist A, Ponten F, Pettersson A. Keratosis linearis with ichthyosis congenita and sclerosing keratoderma (KLICK-syndrome): a rare, autosomal recessive disorder of keratohyaline formation? Acta Derm Venereol. (1997) 77:225–7. doi: 10.2340/0001555577225227
3. Dahlqvist J, Klar J, Tiwari N, Schuster J, Torma H, Badhai J, et al. A single-nucleotide deletion in the POMP 5′ UTR causes a transcriptional switch and altered epidermal proteasome distribution in KLICK genodermatosis. Am J Hum Genet. (2010) 86:596–603. doi: 10.1016/j.ajhg.2010.02.018
4. Morice-Picard F, Jonca N, Pichery M, Mermin D, Leaute-Labreze C, Taieb A, et al. KLICK syndrome: recognizable phenotype and hot-spot POMP mutation. J Eur Acad Dermatol Venereol. (2017) 31:e154–e6. doi: 10.1111/jdv.13898
5. Onnis G, Bourrat E, Jonca N, Dreyfus I, Severino-Freire M, Pichery M, et al. KLICK syndrome: an unusual phenotype. Br J Dermatol. (2018) 178:1445–6. doi: 10.1111/bjd.16318
6. Akiyama M, Takeichi T, McGrath JA, Sugiura K. Autoinflammatory keratinization diseases. J Allergy Clin Immunol. (2017) 140:1545–7. doi: 10.1016/j.jaci.2017.05.019
7. Akiyama M, Takeichi T, McGrath JA, Sugiura K. Autoinflammatory keratinization diseases: An emerging concept encompassing various inflammatory keratinization disorders of the skin. J Dermatol Sci. (2018) 90:105–11. doi: 10.1016/j.jdermsci.2018.01.012
8. Takeichi T, Akiyama M. Familial or sporadic porokeratosis as an autoinflammatory keratinization disease. J Dermatol. (2019) 46:e125–e6. doi: 10.1111/1346-8138.14666
9. Takeichi T, Matsumoto T, Nomura T, Takeda M, Niwa H, Kono M, et al. A novel NCSTN missense mutation in the signal peptide domain causes hidradenitis suppurativa, which has features characteristic of an autoinflammatory keratinization disease. Br J Dermatol. (2019) doi: 10.1111/bjd.18445
10. Murase Y, Takeichi T, Akiyama M. Aberrant CARD14 function might cause defective barrier formation. J Allergy Clin Immunol. (2019) 143:1656–7. doi: 10.1016/j.jaci.2018.11.044
11. Takeichi T, Sugiura K, Nomura T, Sakamoto T, Ogawa Y, Oiso N, et al. Pityriasis rubra pilaris type V as an autoinflammatory disease by CARD14 mutations. JAMA Dermatol. (2017) 153:66–70. doi: 10.1001/jamadermatol.2016.3601
12. Pujol RM, Moreno A, Alomar A, de Moragas JM. Congenital ichthyosiform dermatosis with linear keratotic flexural papules and sclerosing palmoplantar keratoderma. Arch Dermatol. (1989) 125:103–6.
13. Pujol RM, Alomar A, De Moragas JM. A new type of erythrokeratoderma, or KLICK syndrome? Br J Dermatol. (2005) 153:461; author reply 462. doi: 10.1111/j.1365-2133.2005.06790.x
14. van Steensel MA, van Geel M, Steijlen PM. A new type of erythrokeratoderma. Br J Dermatol. (2005) 152:155–158. doi: 10.1111/j.1365-2133.2005.06319.x
15. Chaves AJ, Merchan-Garcia R, Fernandez-Recio JM, Rodriguez-Nevado I, de Argila D. [Keratosis linearis with ichthyosis congenita and sclerosing keratoderma (KLICK syndrome)]. Actas Dermosifiliogr. (2006) 97:342–4. doi: 10.1016/s0001-7310(06)73415-9
16. Poli MC, Ebstein F, Nicholas SK, de Guzman MM, Forbes LR, Chinn IK, et al. Heterozygous truncating variants in POMP escape nonsense-mediated decay and cause a unique immune dysregulatory syndrome. Am J Hum Genet. (2018) 102:1126–42. doi: 10.1016/j.ajhg.2018.04.010
17. Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M, et al. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell. (2012) 148:727–38. doi: 10.1016/j.cell.2011.12.030
18. Ji CH, Kwon YT. Crosstalk and interplay between the ubiquitin-proteasome system and autophagy. Mol Cells. (2017) 40:441–9. doi: 10.14348/molcells.2017.0115
19. Seifert U, Bialy LP, Ebstein F, Bech-Otschir D, Voigt A, Schroter F, et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell. (2010) 142:613–24. doi: 10.1016/j.cell.2010.07.036
20. Dahlqvist J, Torma H, Badhai J, Dahl N. siRNA silencing of proteasome maturation protein (POMP) activates the unfolded protein response and constitutes a model for KLICK genodermatosis. PLoS ONE. (2012) 7:e29471. doi: 10.1371/journal.pone.0029471
21. Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. (2008) 4:141–150. doi: 10.4161/auto.5190
22. Sugiura K. Unfolded protein response in keratinocytes: impact on normal and abnormal keratinization. J Dermatol Sci. (2013) 69:181–6. doi: 10.1016/j.jdermsci.2012.12.002
23. Park K, Lee SE, Shin KO, Uchida Y. Insights into the role of endoplasmic reticulum stress in skin function and associated diseases. FEBS J. (2019) 286:413–25. doi: 10.1111/febs.14739
24. Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest. (2015) 125:4196–211. doi: 10.1172/JCI81260
25. Agarwal AK, Xing C, DeMartino GN, Mizrachi D, Hernandez MD, Sousa AB, et al. PSMB8 encoding the beta5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet. (2010) 87:866–72. doi: 10.1016/j.ajhg.2010.10.031
26. Kanazawa N. Nakajo-Nishimura syndrome: an autoinflammatory disorder showing pernio-like rashes and progressive partial lipodystrophy. Allergol Int. (2012) 61:197–206. doi: 10.2332/allergolint.11-RAI-0416
27. Arima K, Kinoshita A, Mishima H, Kanazawa N, Kaneko T, Mizushima T, et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc Natl Acad Sci USA. (2011) 108:14914–14919. doi: 10.1073/pnas.1106015108
28. Torrelo A. CANDLE Syndrome As a Paradigm of Proteasome-Related Autoinflammation. Front Immunol. (2017) 8:927. doi: 10.3389/fimmu.2017.00927
29. Sarrabay G, Mechin D, Salhi A, Boursier G, Rittore C, Crow Y, et al. PSMB10, the last immunoproteasome gene missing for PRAAS. J Allergy Clin Immunol. (2019) doi: 10.1016/j.jaci.2019.11.024
30. Heink S, Ludwig D, Kloetzel PM, Kruger E. IFN-gamma-induced immune adaptation of the proteasome system is an accelerated and transient response. Proc Natl Acad Sci USA. (2005) 102:9241–46. doi: 10.1073/pnas.0501711102
31. Glynne R, Powis SH, Beck S, Kelly A, Kerr LA, Trowsdale J. A proteasome-related gene between the two ABC transporter loci in the class II region of the human MHC. Nature. (1991) 353:357–60. doi: 10.1038/353357a0
32. Zhong FL, Mamai O, Sborgi L, Boussofara L, Hopkins R, Robinson K, et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell. (2016) 167:187–202 e117. doi: 10.1016/j.cell.2016.09.001
33. Grandemange S, Sanchez E, Louis-Plence P, Tran Mau-Them F, Bessis D, Coubes C, et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann Rheum Dis. (2017) 76:1191–8. doi: 10.1136/annrheumdis-2016-210021
34. Drutman SB, Haerynck F, Zhong FL, Hum D, Hernandez NJ, Belkaya S, et al. Homozygous NLRP1 gain-of-function mutation in siblings with a syndromic form of recurrent respiratory papillomatosis. Proc Natl Acad Sci U S A. (2019) 116:19055–63. doi: 10.1073/pnas.1906184116
35. Zhang SQ, Jiang T, Li M, Zhang X, Ren YQ, Wei SC, et al. Exome sequencing identifies MVK mutations in disseminated superficial actinic porokeratosis. Nat Genet. (2012) 44:1156–60. doi: 10.1038/ng.2409
36. Wang B, Yang W, Wen W, Sun J, Su B, Liu B, et al. Gamma-secretase gene mutations in familial acne inversa. Science. (2010) 330:1065. doi: 10.1126/science.1196284
37. Tricarico PM, Boniotto M, Genovese G, Zouboulis CC, Marzano AV, Crovella S. An integrated approach to unravel hidradenitis suppurativa etiopathogenesis. Front Immunol. (2019) 10:892. doi: 10.3389/fimmu.2019.00892
38. De Vita V, McGonagle D. Hidradenitis suppurativa as an autoinflammatory keratinization disease. J Allergy Clin Immunol. (2018) 141:1953. doi: 10.1016/j.jaci.2018.01.010
39. Marzano AV, Damiani G, Ceccherini I, Berti E, Gattorno M, Cugno M. Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis). Br J Dermatol. (2017) 176:1588–98. doi: 10.1111/bjd.15226
40. Marzano AV, Ceccherini I, Gattorno M, Fanoni D, Caroli F, Rusmini M, et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine. (2014) 93:e187. doi: 10.1097/MD.0000000000000187
41. Tartey S, Gurung P, Dasari TK, Burton A, Kanneganti TD. ASK1/2 signaling promotes inflammation in a mouse model of neutrophilic dermatosis. J Clin Invest. (2018) 128:2042–2047. doi: 10.1172/JCI98446
42. Nesterovitch AB, Gyorfy Z, Hoffman MD, Moore EC, Elbuluk N, Tryniszewska B, et al. Alteration in the gene encoding protein tyrosine phosphatase nonreceptor type 6 (PTPN6/SHP1) may contribute to neutrophilic dermatoses. Am J Pathol. (2011) 178:1434–41. doi: 10.1016/j.ajpath.2010.12.035
43. Nesterovitch AB, Szanto S, Gonda A, Bardos T, Kis-Toth K, Adarichev VA, et al. Spontaneous insertion of a b2 element in the ptpn6 gene drives a systemic autoinflammatory disease in mice resembling neutrophilic dermatosis in humans. Am J Pathol. (2011) 178:1701–1714. doi: 10.1016/j.ajpath.2010.12.053
44. Lukens JR, Vogel P, Johnson GR, Kelliher MA, Iwakura Y, Lamkanfi M, et al. RIP1-driven autoinflammation targets IL-1alpha independently of inflammasomes and RIP3. Nature. (2013) 498:224–7. doi: 10.1038/nature12174
45. Paller AS, Renert-Yuval Y, Suprun M, Esaki H, Oliva M, Huynh TN, et al. An IL-17-dominant immune profile is shared across the major orphan forms of ichthyosis. J Allergy Clin Immunol. (2017) 139:152–65. doi: 10.1016/j.jaci.2016.07.019
46. Malik K, He H, Huynh TN, Tran G, Mueller K, Doytcheva K, et al. Ichthyosis molecular fingerprinting shows profound TH17 skewing and a unique barrier genomic signature. J Allergy Clin Immunol. (2019) 143:604–18. doi: 10.1016/j.jaci.2018.03.021
47. Roda A, Mendonca-Sanches M, Travassos AR, Soares-de-Almeida L, Metze D. Infliximab therapy for Netherton syndrome: A case report. JAAD Case Rep. (2017) 3:550–2. doi: 10.1016/j.jdcr.2017.07.019
48. Takeichi T, Akiyama M. Generalized pustular psoriasis: clinical management and update on autoinflammatory aspects. Am J Clin Dermatol. (2020) 21:227–36. doi: 10.1007/s40257-019-00492-0
49. Oji V, Tadini G, Akiyama M, Blanchet Bardon C, Bodemer C, Bourrat E, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Soreze 2009. J Am Acad Dermatol. (2010) 63:607–41. doi: 10.1016/j.jaad.2009.11.020
50. Murase C, Takeichi T, Shibata A, Nakatochi M, Kinoshita F, Kubo A, et al. Cross-sectional survey on disease severity in Japanese patients with harlequin ichthyosis/ichthyosis: Syndromic forms and quality-of-life analysis in a subgroup. J Dermatol Sci. (2018) 92:127–33. doi: 10.1016/j.jdermsci.2018.08.008
51. Hetz C, Chevet E, Oakes SA. Proteostasis control by the unfolded protein response. Nat Cell Biol. (2015) 17:829–38. doi: 10.1038/ncb3184
Keywords: autoinflammatory keratinization diseases, keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome, inflammation, proteasome maturation protein, unfolded protein response
Citation: Takeichi T and Akiyama M (2020) KLICK Syndrome Linked to a POMP Mutation Has Features Suggestive of an Autoinflammatory Keratinization Disease. Front. Immunol. 11:641. doi: 10.3389/fimmu.2020.00641
Received: 22 August 2019; Accepted: 20 March 2020;
Published: 30 April 2020.
Edited by:
Ivona Aksentijevich, National Human Genome Research Institute (NHGRI), United StatesReviewed by:
Elke Krüger, Greifswald University Hospital, GermanyAngelo Valerio Marzano, University of Milan, Italy
Copyright © 2020 Takeichi and Akiyama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takuya Takeichi, takeichi@med.nagoya-u.ac.jp; Masashi Akiyama, makiyama@med.nagoya-u.ac.jp