
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 09 April 2020
Sec. Autoimmune and Autoinflammatory Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00578
 Sophie Steiner1
Sophie Steiner1 Sonya C. Becker1†
Sonya C. Becker1† Jelka Hartwig1
Jelka Hartwig1 Franziska Sotzny1
Franziska Sotzny1 Sebastian Lorenz1Sandra Bauer1Madlen Löbel2
Sebastian Lorenz1Sandra Bauer1Madlen Löbel2 Anna B. Stittrich3,4Patricia Grabowski1
Anna B. Stittrich3,4Patricia Grabowski1 Carmen Scheibenbogen1,3*
Carmen Scheibenbogen1,3*Single nucleotide polymorphisms (SNP) in various genes have been described to be associated with susceptibility to autoimmune disease. In this study, myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) patients and controls were genotyped for five immune gene SNPs in tyrosine phosphatase non-receptor type 22 (PTPN22, rs2476601), cytotoxic T-lymphocyte-associated protein 4 (CTLA4, rs3087243), tumor necrosis factor (TNF, rs1800629 and rs1799724), and interferon regulatory factor 5 (IRF5, rs3807306), which are among the most important risk variants for autoimmune diseases. Analysis of 305 ME/CFS patients and 201 healthy controls showed significant associations of the PTPN22 rs2476601 and CTLA4 rs3087243 autoimmunity-risk alleles with ME/CFS. The associations were only found in ME/CFS patients, who reported an acute onset of disease with an infection (PTPN22 rs2476601: OR 1.63, CI 1.04–2.55, p = 0.016; CTLA4 rs3087243: OR 1.53, CI 1.17–2.03, p = 0.001), but not in ME/CFS patients without infection-triggered onset (PTPN22 rs2476601: OR 1.09, CI 0.56–2.14, p = 0.398; CTLA4 rs3087243: OR 0.89, CI 0.61–1.30, p = 0.268). This finding provides evidence that autoimmunity might play a role in ME/CFS with an infection-triggered onset. Both genes play a key role in regulating B and T cell activation.
With an estimated prevalence of 0.2–0.3%, myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is a frequent and severe chronic multisystemic disease. Patients suffer from persistent exhaustion, cognitive dysfunctions, pain and flu-like symptoms, leading to a substantial reduction of life quality (1). The underlying pathomechanism is not well-understood, but there is convincing evidence that, at least in a subset of ME/CFS patients, autoimmunity contributes to disease etiology (2, 3). Autoantibodies against various antigens, including neurotransmitter receptors, were reported by several groups (3). Comorbidity with Hashimoto's thyroiditis, fibromyalgia, postural orthostatic tachycardia syndrome and a higher rate of autoimmune disease were reported for ME/CFS patients and their families (2, 3).
Enhanced immune activation and impairment of immunological tolerance are considered as important risk factors for autoimmunity. Autoimmune diseases are understood as being multifactorial with involvement of genetic and environmental factors. Disease onset is often triggered by infections, and the link between infections and autoimmune diseases is well-established (4). In recent years, genome-wide association studies have revealed single nucleotide polymorphisms (SNPs) in various genes to be associated with the risk to develop autoimmune diseases. For most of these SNPs, it was found that they confer gain- or loss-of-function in enzymes or transcription factors that play a role in B and T cell activation or cytokine production and cytokine signaling, which are crucial mechanisms for the development of autoimmune diseases (5–8). Among the most frequent variants associated with multiple autoimmune diseases are the tyrosine phosphatase non-receptor type 22 (PTPN22) SNP rs2476601, the cytotoxic T-lymphocyte-associated protein 4 (CTLA4) SNP rs3087243, the interferon regulatory factor 5 (IRF5) SNP rs3807306, and the SNP rs1800629 in the gene tumor necrosis factor (TNF).
The PTPN22 SNP rs2476601 is one of the most important susceptibility loci for autoimmunity and is associated with myasthenia gravis (MG), type 1 diabetes (T1D), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and other autoimmune disorders (9). This SNP was shown to play an important role in T and B cell receptor signaling (5). The SNP rs3087243 in CTLA4 is associated with several autoimmune diseases, including SLE, RA, T1D, and Graves' disease (8, 10–14). The CTLA4 SNP has a protective effect, and the major allele G is associated with autoimmunity. The G allele leads to an impaired negative regulation of T cell activation. Within multiple sclerosis (MS) patients, homozygous carriers of the G allele were shown to express significantly lower CTLA4 protein levels in CD4+ T cells (15). A SNP in IRF5, rs3807306, is another common autoimmune susceptibility locus associated with RA, SLE, MS, and Crohn's disease (CD) and is linked to higher serum interferon-α (IFNα) activity (measured by a reporter cell line) in SLE (7, 16–18). The SNP rs1800629 in TNF is linked to various autoimmune diseases, including CD, celiac disease, RA, and SLE (6, 19–21). Another SNP in the TNF gene rs1799724 is associated with CD and autoimmune hepatitis (AIH) (22, 23). Higher serum levels of TNFα were shown in carriers of the TNF rs1799724 risk genotype, while for the SNP rs1800629, data on an association with elevated TNFα serum levels are inconclusive (6, 24).
A recently published systematic review of Wang et al. summarizes the studies of genetic variants, which are associated with ME/CFS (25). Several SNPs in cytokines and human leukocyte antigen (HLA) associations were found in ME/CFS. Two SNPs in TNF, rs1800629 (TNF-308) and rs1799724 (TNF-857), were comparatively analyzed between a ME/CFS and control cohort (26). A higher frequency of the TNF-857 T allele was found in ME/CFS. Analysis of genetic variants in PTPN22, CTLA4, and IRF5 were not described in ME/CFS so far.
In the present study, we comparatively analyzed the prevalence of the five SNPs described above in ME/CFS patients and healthy controls. ME/CFS onset is triggered by an infection in approximately two thirds of patients (27). The link between infections as a trigger of autoimmune diseases is well-established (4). Therefore, we also studied a possible association of these SNPs with an infection-triggered disease onset (ITO).
ME/CFS patients were diagnosed at the outpatient clinic for immunodeficiencies at the Institute for Medical Immunology at the Charité-Universitätsmedizin Berlin, Germany. Diagnosis of ME/CFS is based on Canadian Consensus Criteria (CCC) (28) and exclusion of other medical or neurological diseases that may cause fatigue. Autoimmune diseases were an exclusion criterion, with the exception of Hashimoto's thyroiditis, which is classified as comorbidity in ME/CFS (1). Controls not suffering from fatigue were recruited from staff. Characteristics of patients are shown in Table 1. 232 of 305 patients stated that they had an acute onset of illness with an infection (ITO). We retrospectively collected the data of the types of infection from the patients records and could classify most patients into the categories “respiratory or gastrointestinal tract infection,” “primary EBV,” or “history of viral or bacterial infection.” All patients and controls were caucasian. The study was approved by the Ethics Committee of Charité-Universitätsmedizin Berlin in accordance with the 1964 Declaration of Helsinki and its later amendments (EA4-090-10). All patients and healthy controls recruited from staff gave written informed consent.
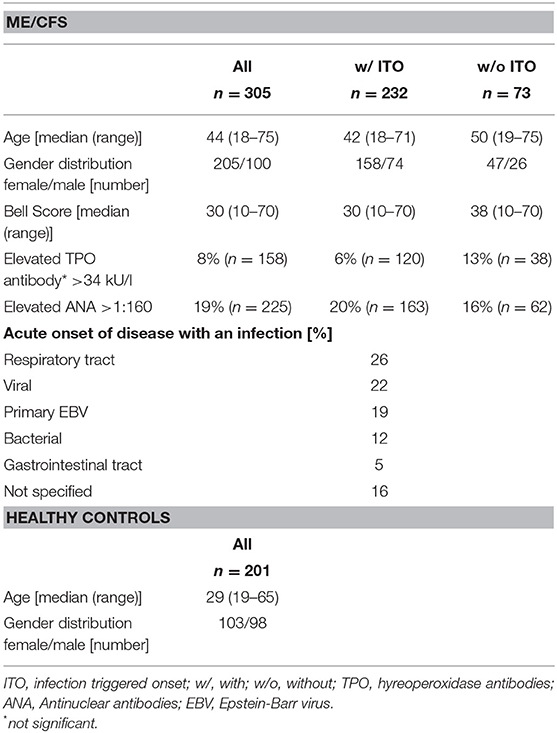
Table 1. Cohort characteristics.
We analyzed SNPs in genomic coding regions described as risk factors for autoimmunity (Table 2) by allelic discrimination (AD) polymerase chain reaction (PCR). Genomic DNA from patients and healthy controls was obtained from whole blood samples or peripheral blood mononuclear cells (PBMCs) using the QIAmp DNA Blood Mini Kit (QIAGEN) and stored at −20°C in nuclease-free water until use. DNA was quantified using NanoDrop™ spectrophotometer. Genotyping of all five SNPs (PTPN22 rs2476601, ID: C__16021387_20; CTLA4 rs3087243, ID: C___3296043_10; TNF rs1800629, ID: C___7514879_10; TNF rs1799724, ID: C__11918223_10; and IRF5 rs3807306, ID: C___2691231_10) was performed using pre-designed TaqMan allelic discrimination assay probes (Thermo Fisher). The 10 μl PCR reactions contained 2 μl of template DNA (5 ng/μl), 0.5 μl of the SNP genotyping Assay Mix, 5 μl of TaqMan Universal Mastermix II no UNG (Applied Biosystems), and 2 μl sterile distilled water. PCR run was performed in a 7500 Fast & 7500 Real-Time PCR System (Applied Biosystems). Background fluorescence was recorded performing a pre-read run for 1 min at 60°C. Amplification was achieved through activation of DNA polymerase for 10 min at 95°C, following 40 cycles of melting for 15 s at 92°C and annealing/extension for 1 min at 60°C. Afterwards, background fluorescence was automatically subtracted from the amplification results by running a post-read for 1 min at 60°C. AD PCR results were visualized in a cluster plot of allele X vs. allele Y to distinguish between homo- and heterozygosity.
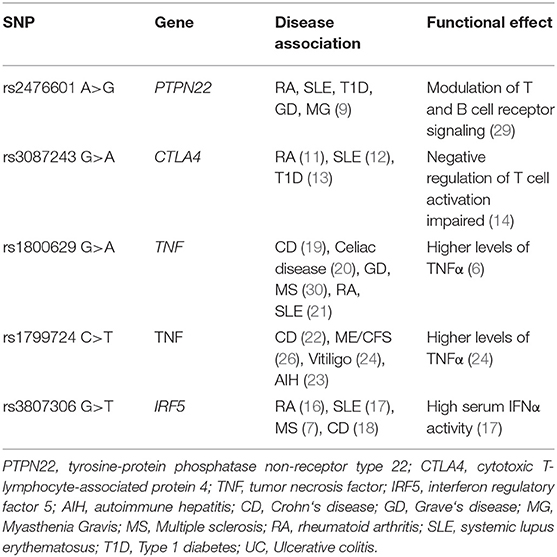
Table 2. SNPs associated with autoimmune diseases and their functional effect.
Lymphocyte phenotyping by flow cytometry including CD3+, CD4+, and CD8+ T cells and CD19+ B cells and serum protein analysis including C reactive protein (CrP), complement proteins C3 and C4, soluble IL-2 receptor (IL-2R), IgG, and TNFα in LPS-stimulated whole blood cells was carried out by Labor Berlin, Charité Vivantes GmbH, Berlin.
Allele distribution was analyzed using the additive model in a 2 × 2 and genotype distribution using a 2 × 3 contingency table and tested for significance by χ2-square test. One-tailed t-tests were performed to test for increased frequency of the known autoimmunity risk alleles in ME/CFS patients. Odds ratio (OR) and 95% confidence interval (CI) were calculated from the same contingency tables.
The Hardy-Weinberg equilibrium (HWE) was tested using a 2 × 3 χ2 table comparing the expected and observed genotypes of each SNP. No significant deviation from HWE was observed for any of the studied SNPs in patients or controls. The five SNP loci analyzed, reside on different chromosomes (PTPN22 rs2476601 on chr1, CTLA4 rs3087243 on chr2, TNF SNPs rs1799724 and rs1800629 on chr6, and IRF5 rs3807306 on chr7) therefore, a physical linkage can be excluded.
Analysis of association of serum proteins and immune cells with onset type or PTPN22 rs2476601 and CTLA4 rs3087243 risk variants were analyzed using the two-tailed Mann–Whitney U test.
Statistical tests were performed using GraphPad Prism software (Version 6.00) or R (31). A p ≤ 0.05 was considered statistically significant.
We analyzed a cohort of 305 ME/CFS patients and 201 healthy controls for potential disease association of the PTPN22, CTLA4, TNF, and IRF5 SNPs that were described to be risk loci for various autoimmune diseases (Table 2 and Supplementary Table 1).
We found a significantly increased frequency of the PTPN22 rs2476601 risk allele A, that confers susceptibility to autoimmunity, in ME/CFS patients (allele frequency (AF) 12%, odds ratio (OR) 1.50, p = 0.033) compared to healthy controls (AF 8%, n = 201; Table 3). When subgrouping patients according to the type of onset of disease, we observed the significant association of the PTPN22 rs2476601 risk allele A only in patients with ITO (AF 13%, OR 1.63, n = 232, p = 0.016), but not in those without ITO (AF 9%), compared to healthy controls (AF 8%).
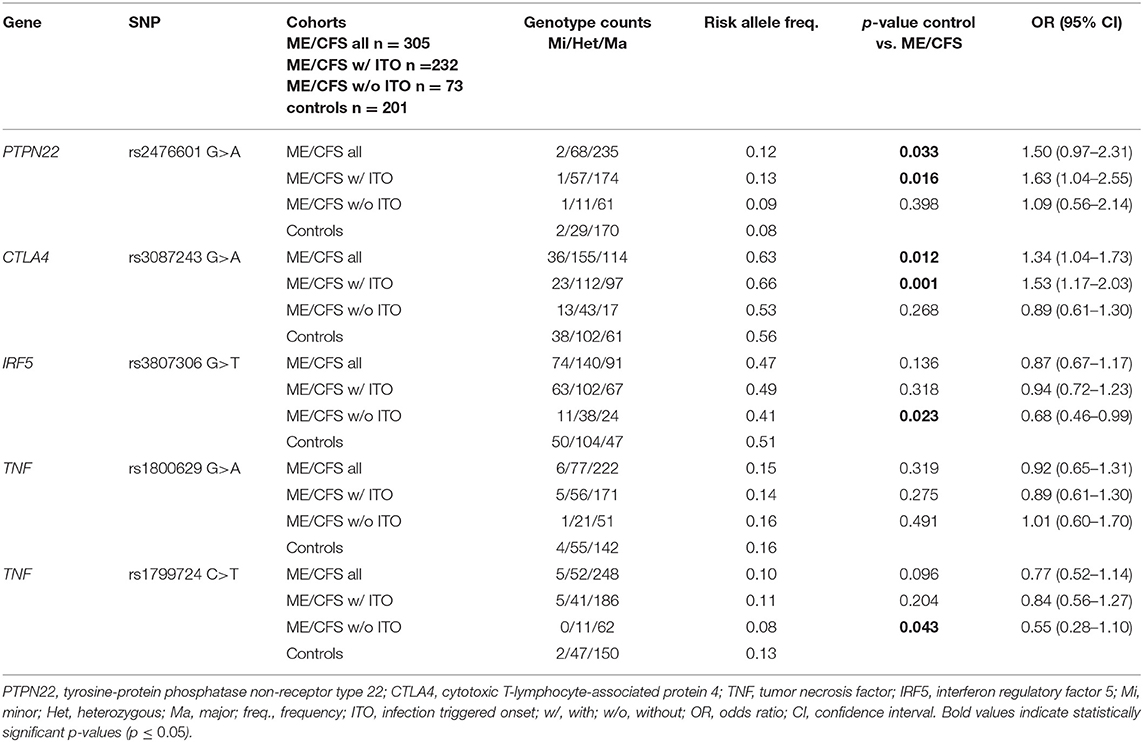
Table 3. Allele frequencies and genotypes.
Equally, the frequency of the CTLA4 rs3087243 risk allele G is significantly higher in ME/CFS patients (AF 63%, OR 1.34), compared to healthy controls (AF 56%; Table 3). Again, we observed a significant association of the CTLA4 rs3087243 risk allele G only in patients with ITO (AF = 66%, OR = 1.53, p = 0.001), but not in those wthout ITO (AF 53%) compared to healthy controls (AF 56%).
Oppositely, the AF of the IRF5 rs3807306 risk allele T was significantly lower in ME/CFS patients without ITO (AF 41%, OR 0.68, p = 0.023), compared to healthy controls (AF 51%; Table 3). No difference in the risk AF of the TNF rs1800629 SNP between patients and controls was observed, but the AF of the TNF rs1799724 risk allele T again was significantly lower in ME/CFS patients without ITO (AF 8%, OR 0.55, p = 0.043), compared to healthy controls (AF 13%, Table 3). No associations with IRF5 or TNF was found in patients with ITO.
Further, we analyzed the genotype distribution for CTLA4 rs3087243 and PTPN22 rs2476601 (Figure 1). We found a significant difference between the ME/CFS patients with an ITO, compared to healthy controls (CTLA4 rs3087243: p = 0.0061, PTPN22 rs2476601: p = 0.026). ME/CFS patients without ITO had a similar genotype distribution as healthy controls.
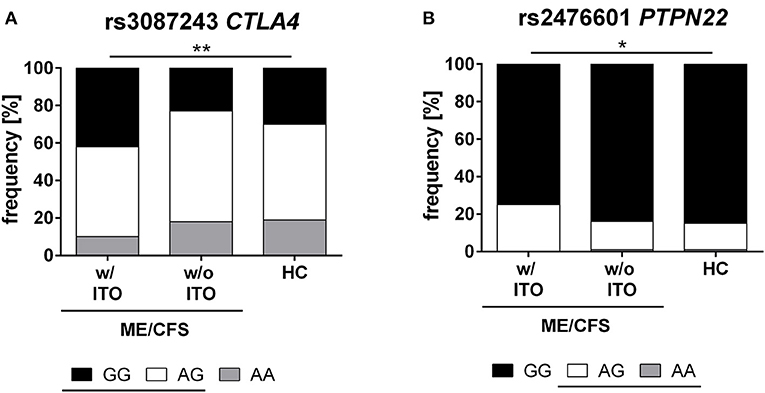
Figure 1. Genotype distribution of CTLA4 and PTPN22 SNPs. Distribution of major (black), hetero (white), and minor (gray) genotypes of CTLA4 rs3087243 (A) and PTPN22 rs2476601 (B) in ME/CFS patients with (n = 232) and without infection triggered onset (n = 73), and healthy controls (n = 201). The risk allele G in rs3087243 and A in rs2476601 are underlined. Statistical analyses were performed using 2 × 3 contingency tables and χ2- test. A p ≤ 0.05 was considered as statistically significant. HC, healthy control; w/ ITO, with infection-triggered onset; w/o ITO, without infection-triggered onset.
We comparatively analyzed numbers of CD3+, CD4+, and CD8+ T cells and CD19+ B cells, CrP, complement proteins C3 and C4, soluble IL-2R, IgG, and TNFα in ME/CFS patients with and without ITO. Patients with ITO had lower CD19+ B cell levels (p = 0.05) and a trend toward lower C3 levels (Figure 2A and Supplementary Table 2). In patients with both, the risk allele in PTPN22 rs2476601 (Figure 2B and Supplementary Table 3) or the homozygous risk allele for CTLA4 rs3087243 (Figure 2C and Supplementary Table 4), C4 levels (p = 0.06 and 0.004, respectively) but not C3 levels were decreased, while there was no difference in B cell numbers.
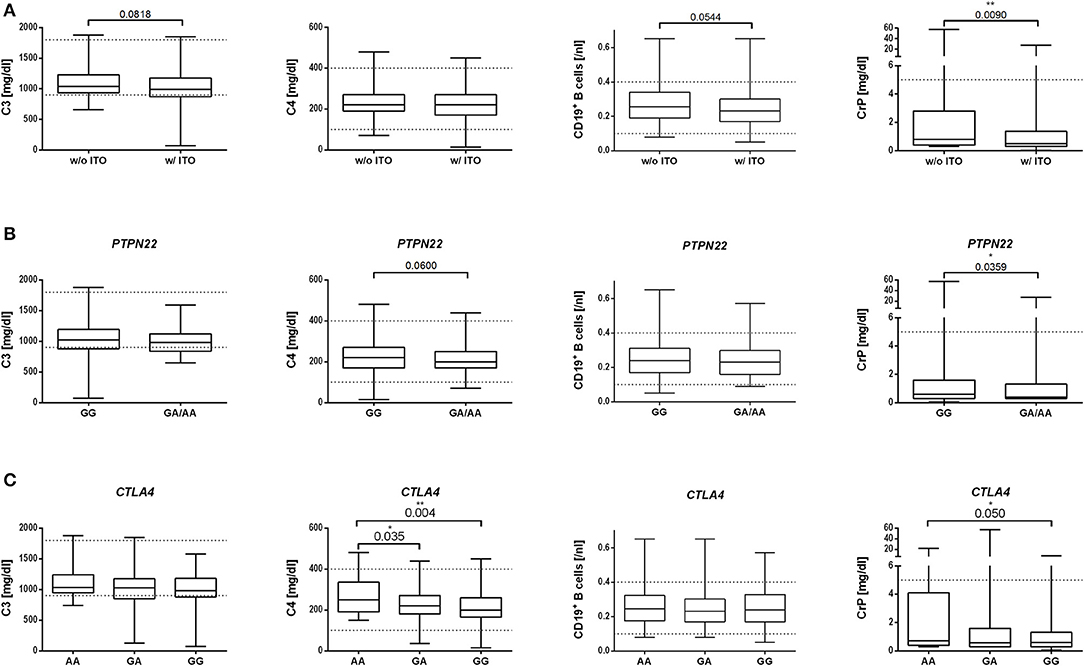
Figure 2. Association of disease onset type (A), PTPN22 SNP (B), or CTLA4 SNP (C) in ME/CFS patients with immune marker. Median with range of C3 and C4 complement levels [mg/dl], CD19+ B cells [/nl], and CrP [mg/dl] is shown for the ME/CFS subgroups (A) with (w/) or without (w/o) infection-triggered onset (ITO). (B) without or with PTPN22 rs2476601 risk allele A or (C) without or with CTLA4 rs3087243 risk allele G. Statistical analysis was performed using Mann–Whitney U test. A p ≤ 0.05 was considered as statistically significant.
CrP concentrations were significantly higher in ME/CFS patients without ITO (Figure 2A), and without the risk alleles of PTPN22 rs2476601-A (Figure 2B) and CTLA4 rs3087243-G (Figure 2C and Supplementary Tables 2–4).
CD3+, CD4+, and CD8+ T cells and soluble IL-2R, TNFα, and IgG concentrations did not differ between patient subgroups and risk variants (Supplementary Tables 2–4).
In this study, we evaluated five important SNPs that are associated with various autoimmune diseases in ME/CFS patients. Patients selected are representative of ME/CFS cohorts reported by other groups (with two-thirds of patients being female and reporting an ITO and an average disease severity of a Bell score of 30) (27). In accordance with this recent US study, ME/CFS onset in our patients was triggered most frequently by an acute respiratory infection or suspected viral infection (27). Also, a primary EBV infection in adolescence is a well-known trigger of ME/CFS and was reported in 19% of our patients. A primary EBV infection in adulthood is known as a risk factor for various autoimmune diseases (32).
We found a strong association of the autoimmunity risk alleles in CTLA4 rs3087243-G and PTPN22 rs2476601-A with ITO ME/CFS. Carrying the PTPN22 rs2476601 risk allele A increased the odds 1.6-fold for developing infection-triggered ME/CFS. Carriers of the risk allele G of CTLA4 rs3087243 had 1.5-fold-higher odds of developing ITO ME/CFS. Accordingly, the genotype distribution for both SNPs was significantly different in ME/CFS patients with ITO compared to healthy controls (Figure 1).
Both, the PTPN22 rs2476601 and CTLA4 rs3087243 SNPs were shown to be associated with numerous autoimmune diseases, including T1D, SLE, and RA (8–10, 33). PTPN22 encodes a lymphoid tyrosine phosphatase that acts as a strong negative regulator of T cell activation. A study by Vang et al. provides evidence that the autoimmune risk allele A of PTPN22 rs2476601 confers gain-of-function. This may lead to lower T cell receptor signaling and the inability to delete autoreactive T cells or diminished activity of Tregs (5). Further, the PTPN22 rs2476601 risk allele may contribute to the generation of autoreactive B cells via disturbances in clonal deletion and receptor editing thus leading to impaired central tolerance (29). This is supported by the finding that the strongest associations with this SNP are found in autoimmune diseases in which autoreactive T cells and autoantibodies have a major role in the pathogenesis of the disease (29). In contrast, autoimmune diseases in which Th17 cells play an important role, and those of mucosal sites, tend to have no association with PTPN22 (29).
CTLA4 is expressed on CD4+ and CD8+ T cells upon activation and has a crucial role as negative T cell regulator to switch-off activated T cells (34). The risk allele G of rs3087243 leads to reduced levels of soluble CTLA4 mRNA, which results in enhanced T cell activity (10, 15, 33).
Similar to our findings, both the PTPN22 rs2476601 and CTLA4 rs3087243 risk variants were shown to be susceptibility loci for autoimmune diseases including anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) and autoimmune polyglandular autoimmunity (35, 36). To our knowledge, the frequency of these SNPs has not been studied in other ME/CFS patients yet.
We further correlated our SNP data with immune markers. In patients with both the risk allele in PTPN22 rs2476601 or the homozygous risk alleles for CTLA4 rs3087243, C4 but not C3 levels were decreased. An association of these risk variants with C4 levels is not described in the literature. Low C4 levels are, however, frequent in SLE (37), a disease which is associated with both the risk allele in PTPN22 and CTLA4.
When comparing patient subgroups, only those with ITO had diminished CD19+ B cells. B cell lymphocytopenia is a frequent finding in SLE and RA and provides further evidence for an autoimmune mechanism in ME/CFS patients with ITO (38, 39). In contrast, levels of CrP were significantly higher in ME/CFS patients without ITO and in those without the risk alleles. This may suggest that, within the subgroup of ME/CFS patients without ITO, there is a tendency to a more pro-inflammatory response similar to patients with cancer-related fatigue (40).
For the IRF5 rs3807306 risk allele T we observed a significantly lower AF in ME/CFS patients without ITO compared to controls. This finding is unexpected and needs to be confirmed. We could not detect a difference in the frequency of the TNF rs1800629 AF between ME/CFS patients and healthy controls, but we again found a significantly lower AF of the TNF rs1799724 in ME/CFS patients without ITO compared to controls. This finding is in contrast to the previous study by Carlo-Stella et al., analyzing 80 ME/CFS patients, which found a higher frequency of the TNF-857 rs1799724 risk allele (26). We have no obvious explanation for this difference. The patient cohort in the 2006 study was diagnosed according to Fukuda criteria, which are less strict than the CCC criteria used in our study. Interestingly, these SNPs are associated with enhanced serum levels of TNFα and IFNα and also with autoimmune diseases of mucosal sites (19, 41). It is tempting to speculate that patients, who can produce higher levels of TNFα and IFNα, have a more effective mucosal immunity and are therefore less prone to develop ME/CFS. This is in line with our previous observation, that a deficiency of the complement factor mannose binding lectin (MBL), which results in susceptibility to infections, is associated with ME/CFS (42).
There are other variants of immune or immune-related genes described in ME/CFS as recently summarized (25). Several SNPs in HLA class II region were found to be associated with ME/CFS (43, 44). This association is another evidence for an autoimmune pathomechanism, as the link between certain HLA alleles and autoimmunity is well-established (45). Moreover, there are studies showing an association with several SNPs in the glucocorticoid receptor gene Nuclear Receptor Subfamily 3 Group C Member 1 (NR3C1). This receptor plays a role in the regulation of the inflammatory response by affecting the hypothalamic–pituitary–adrenal (HPA) axis activity trough cortisol release (30, 46). For ME/CFS, a deregulated HPA axis has been reported (47). In a similar manner, SNP variants in the catechol-O-methyltransferase (COMT) gene and the ß2 adrenergic receptor described in ME/CFS may modulate the immune response (48). Further studies found associations with SNPs not related to the immune system, like variants associated with muscle metabolism (49) and serotonergic system (50).
In conclusion, our study shows that the PTPN22 rs2476601 and CTLA4 rs3087243 autoimmunity risk variants are more frequent in patients with ITO ME/CFS. This finding provides further evidence that there is a genetic predisposition for ME/CFS. The associations with PTPN22 and CTLA4 SNPs point to a role of autoreactive T and B cells in the pathomechanism of ME/CFS. In contrast, the lack of association of CTLA4 and PTPN22 SNPs and the lower frequencies of IRF5 and TNF risk variants in ME/CFS patients without ITO suggest that the pathomechanism is distinct. These associations need to be confirmed in other ME/CFS patient cohorts. Our findings prompt us to intensify research into autoimmune mechanisms and perform clinical studies with drugs targeting autoreactive B cells.
The SNP data has been deposited to ClinVar - SCV001167677 and SCV001167678. Other raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
The studies involving human participants were reviewed and approved by Ethics Committee of Charité-Universitätsmedizin Berlin EA4-090-10. The patients/participants provided their written informed consent to participate in this study.
SS and SCB performed the research. SL, JH, FS, AS, and SB participated in data analysis. CS and PG contributed patient material. CS and ML designed the research project. SS and CS wrote the paper. All authors provided final approval of the version to be published.
This study was supported by the RAMSAY Research Grants in Basic, Preclinical, Clinical and Epidemiology Research 2016 from Solve ME/CFS Initiative Autoimmune signature in CFS/ME and a grant from the Weidenhammer-Zöbele Foundation. SS received a scholarship from the Lost Voices Foundation. We acknowledge support from the German Research Foundation (DFG) and the Open Access Publication Fund of Charité-Universitätsmedizin Berlin.
AS is employed by the company Labor Berlin—Charité Vivantes GmbH, and ML is employed by Carl-Thiem-Klinikum Cottbus GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00578/full#supplementary-material
Supplementary Table 1. Genotype Counts.xlx.
Supplementary Table 2. Soluble immune markers and lymphocytes for ME/CFS patients with or without ITO.
Supplementary Table 3. Soluble immune markers and lymphocytes for ME/CFS according to non-risk and risk genotype for PTPN22 rs2476601.
Supplementary Table 4. Soluble immune markers and lymphocytes for ME/CFS according to non-risk and risk alleles for CTLA4 rs3087243.
1. Carruthers BM, van de Sande MI, De Meirleir KL, Klimas NG, Broderick G, Mitchell T, et al. Myalgic encephalomyelitis: international consensus criteria. J Intern Med. (2011) 270:327–38. doi: 10.1111/j.1365-2796.2011.02428.x
2. Blomberg J, Gottfries C-G, Elfaitouri A, Rizwan M, Rosén A. Infection elicited autoimmunity and myalgic encephalomyelitis/chronic fatigue syndrome: an explanatory model. Front Immunol. (2018) 9:229. doi: 10.3389/fimmu.2018.00229
3. Sotzny F, Blanco J, Capelli E, Castro-Marrero J, Steiner S, Murovska M, et al. Myalgic encephalomyelitis/chronic fatigue syndrome – evidence for an autoimmune disease. Autoimmun Rev. (2018) 17:601–9. doi: 10.1016/j.autrev.2018.01.009
4. Ascherio A, Munger KL. EBV and Autoimmunity. In: C. Münz, editor. Epstein Barr Virus Volume 1: One Herpes Virus: Many Diseases. Cham: Springer International Publishing (2015). p. 365–85. doi: 10.1007/978-3-319-22822-8_15
5. Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. (2005) 37:1317–9. doi: 10.1038/ng1673
6. Lee YH, Harley JB, Nath SK. Meta-analysis of TNF-alpha promoter−308 A/G polymorphism and SLE susceptibility. Eur J Hum Genet. (2006) 14:364–71. doi: 10.1038/sj.ejhg.5201566
7. Kristjansdottir G, Sandling JK, Bonetti A, Roos IM, Milani L, Wang C, et al. Interferon regulatory factor 5 (IRF5) gene variants are associated with multiple sclerosis in three distinct populations. J Med Genet. (2008) 45:362–9. doi: 10.1136/jmg.2007.055012
8. Ting WH, Chien MN, Lo FS, Wang CH, Huang CY, Lin CL, et al. Association of cytotoxic T-lymphocyte-associated protein 4 (CTLA4) gene polymorphisms with autoimmune thyroid disease in children and adults: case-control study. PLoS ONE. (2016) 11:e0154394. doi: 10.1371/journal.pone.0154394
9. Mustelin T, Bottini N, Stanford SM. The contribution of PTPN22 to rheumatic disease. Arthr Rheumatol. (2019) 71:486–95. doi: 10.1002/art.40790
10. Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. (2003) 423:506–11. doi: 10.1038/nature01621
11. Li X, Zhang C, Zhang J, Zhang Y, Wu Z, Yang L, et al. Polymorphisms in the CTLA-4 gene and rheumatoid arthritis susceptibility: a meta-analysis. J Clin Immunol. (2012) 32:530–9. doi: 10.1007/s10875-012-9650-y
12. Liu J, Zhang H-X. CTLA-4 polymorphisms and systemic lupus erythematosus: a comprehensive meta-analysis. Genet Test Mol Biomark. (2013) 17:226–31. doi: 10.1089/gtmb.2012.0302
13. Wang J, Liu L, Ma J, Sun F, Zhao Z, Gu M. Common variants on cytotoxic T lymphocyte antigen-4 polymorphisms contributes to type 1 diabetes susceptibility: evidence based on 58 studies. PLoS ONE. (2014) 9:e85982. doi: 10.1371/journal.pone.0085982
14. Tu Y, Fan G, Dai Y, Zeng T, Xiao F, Chen L, et al. Association between rs3087243 and rs231775 polymorphism within the cytotoxic T-lymphocyte antigen 4 gene and Graves' disease: a case/control study combined with meta-analyses. Oncotarget. (2017) 8:110614–24. doi: 10.18632/oncotarget.22702
15. Karabon L, Kosmaczewska A, Bilinska M, Pawlak E, Ciszak L, Jedynak A, et al. The CTLA-4 gene polymorphisms are associated with CTLA-4 protein expression levels in multiple sclerosis patients and with susceptibility to disease. Immunology. (2009) 128:e787–96. doi: 10.1111/j.1365-2567.2009.03083.x
16. Sigurdsson S, Padyukov L, Kurreeman FAS, Liljedahl U, Wiman A-C, Alfredsson L, et al. Association of a haplotype in the promoter region of the interferon regulatory factor 5 gene with rheumatoid arthritis. Arthr Rheumat. (2007) 56:2202–10. doi: 10.1002/art.22704
17. Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthrit Rheum. (2008) 58:2481–7. doi: 10.1002/art.23613
18. Chua KH, Lian LH, Khor WC, Lee WS, Hilmi I, Goh KL, et al. Association between genetic polymorphisms in interferon regulatory factor 5 (IRF5) gene and Malaysian patients with Crohn's disease. J Dig Dis. (2015) 16:205–16. doi: 10.1111/1751-2980.12229
19. Ferreira AC, Almeida S, Tavares M, Canedo P, Pereira F, Regalo G, et al. NOD2/CARD15 and TNFA, but not IL1B and IL1RN, are associated with Crohn's disease. Inflamm Bowel Dis. (2005) 11:331–9. doi: 10.1097/01.MIB.0000158153.71579.b4
20. Khan S, Mandal RK, Jawed A, Dar SA, Wahid M, Panda AK, et al. TNF-α−308 G > A (rs1800629) polymorphism is associated with celiac disease: a meta-analysis of 11 case-control studies. Sci Rep. (2016) 6:32677. doi: 10.1038/srep32677
21. Chen L, Huang Z, Liao Y, Yang B, Zhang J. Association between tumor necrosis factor polymorphisms and rheumatoid arthritis as well as systemic lupus erythematosus: a meta-analysis. Brazil J Med Biol Res. (2019) 52:e7927. doi: 10.1590/1414-431x20187927
22. Mao YQ, Dong SQ, Gao M. Association between TNF-αrs1799724 and rs1800629 polymorphisms and the risk of Crohn's disease. Genet Mol Res. (2015) 14:15811–21. doi: 10.4238/2015.December.1.33
23. Motawi TK, El-Maraghy SA, Sharaf SA, Said SE. Association of CARD10 rs6000782 and TNF rs1799724 variants with paediatric-onset autoimmune hepatitis. J Adv Res. (2019) 15:103–10. doi: 10.1016/j.jare.2018.10.001
24. Laddha NC, Dwivedi M, Begum R. Increased tumor necrosis factor (TNF)-α and its promoter polymorphisms correlate with disease progression and higher susceptibility towards vitiligo. PLoS ONE. (2012) 7:e52298. doi: 10.1371/journal.pone.0052298
25. Wang T, Yin J, Miller AH, Xiao C. A systematic review of the association between fatigue and genetic polymorphisms. Brain Behav Immun. (2017) 62:230–44. doi: 10.1016/j.bbi.2017.01.007
26. Carlo-Stella N, Badulli C, De Silvestri A, Bazzichi L, Martinetti M, Lorusso L, et al. A first study of cytokine genomic polymorphisms in CFS: Positive association of TNF-857 and IFNgamma 874 rare alleles. Clin Exp Rheumatol. (2006) 24:179–82. Available online at: https://www.clinexprheumatol.org/abstract.asp?a=2817
27. Chu L, Valencia IJ, Garvert DW, Montoya JG. Onset patterns and course of myalgic encephalomyelitis/chronic fatigue syndrome. Front Pediatr. (2019) 7:e228546. doi: 10.3389/fped.2019.00012
28. Carruthers BM, De Meirleir KL. Myalgic encephalomyelitis/chronic fatigue syndrome: clinical working case definition, diagnostic and treatment protocols. J Chronic Fatigue Syndr. (2003) 11:7–116. doi: 10.1300/J092v11n01_02
29. Stanford SM, Bottini N. PTPN22: the archetypal non-HLA autoimmunity gene. Nature Rev Rheumatol. (2014) 10:602–11. doi: 10.1038/nrrheum.2014.109
30. Lee E, Cho S, Kim K, Park T. An integrated approach to infer causal associations among gene expression, genotype variation, and disease. Genomics. (2009) 94:269–77. doi: 10.1016/j.ygeno.2009.06.002
31. R Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing (2019). Available online at: https://www.R-project.org/
32. Niller HH, Wolf H, Ay E, Minarovits J. Epigenetic dysregulation of epstein-barr virus latency and development of autoimmune disease. Adv Exp Med Biol. (2011) 711:82–102. doi: 10.1007/978-1-4419-8216-2_7
33. Repnik K, Potocnik U. CTLA4 CT60 single-nucleotide polymorphism is associated with Slovenian inflammatory bowel disease patients and regulates expression of CTLA4 isoforms. DNA Cell Biol. (2010) 29:603–10. doi: 10.1089/dna.2010.1021
34. McCoy KD, Le Gros G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol Cell Biol. (1999) 77:1–10. doi: 10.1046/j.1440-1711.1999.00795.x
35. Carr EJ, Niederer HA, Williams J, Harper L, Watts RA, Lyons PA, et al. Confirmation of the genetic association of CTLA4 and PTPN22 with ANCA-associated vasculitis. BMC Med Genet. (2009) 10:121. doi: 10.1186/1471-2350-10-121
36. Houcken J, Degenhart C, Bender K, König J, Frommer L, Kahaly GJ. PTPN22 and CTLA-4 polymorphisms are associated with polyglandular autoimmunity. J Clin Endocrinol Metabol. (2018) 103:1977–84. doi: 10.1210/jc.2017-02577
37. Tsang-A-Sjoe MWP, Bultink IEM, Korswagen LA, van der Horst A, Rensink I, de Boer M, et al. Comprehensive approach to study complement C4 in systemic lupus erythematosus: gene polymorphisms, protein levels and functional activity. Mol Immunol. (2017) 92:125–31. doi: 10.1016/j.molimm.2017.10.004
38. Odendahl M, Jacobi A, Hansen A, Feist E, Hiepe F, Burmester GR, et al. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol. (2000) 165:5970. doi: 10.4049/jimmunol.165.10.5970
39. Wagner U, Kaltenhäuser S, Pierer M, Wilke B, Arnold S, Häntzschel H. B lymphocytopenia in rheumatoid arthritis is associated with the DRB1 shared epitope and increased acute phase response. Arthritis Res. (2002) 4:R1. doi: 10.1186/ar420
40. Collado-Hidalgo A, Bower JE, Ganz PA, Cole SW, Irwin MR. Inflammatory biomarkers for persistent fatigue in breast cancer survivors. Clin Cancer Res. (2006) 12:2759. doi: 10.1158/1078-0432.CCR-05-2398
41. Li P, Lv H, Yang H, Qian J-M. IRF5, but not TLR4, DEFEB1, or VDR, is associated with the risk of ulcerative colitis in a Han Chinese population. Scand J Gastroenterol. (2013) 48:1145–51. doi: 10.3109/00365521.2013.828775
42. Guenther S, Loebel M, Mooslechner AA, Knops M, Hanitsch LG, Grabowski P, et al. Frequent IgG subclass and mannose binding lectin deficiency in patients with chronic fatigue syndrome. Hum Immunol. (2015) 76:729–35. doi: 10.1016/j.humimm.2015.09.028
43. Smith J, Fritz EL, Kerr JR, Cleare AJ, Wessely S, Mattey DL. Association of chronic fatigue syndrome with human leucocyte antigen class II alleles. J Clin Pathol. (2005) 58:860–3. doi: 10.1136/jcp.2004.022681
44. Carlo-Stella N, Bozzini S, De Silvestri A, Sbarsi I, Pizzochero C, Lorusso L, et al. Molecular study of receptor for advanced glycation endproduct gene promoter and identification of specific HLA haplotypes possibly involved in chronic fatigue syndrome. Int J Immunopathol Pharmacol. (2009) 22:745–54. doi: 10.1177/039463200902200320
45. Bodis G, Toth V, Schwarting A. Role of human leukocyte antigens (HLA) in autoimmune diseases. Rheumatol Ther. (2018) 5:5–20. doi: 10.1007/s40744-018-0100-z
46. Rajeevan MS, Smith AK, Dimulescu I, Unger ER, Vernon SD, Heim C, et al. Glucocorticoid receptor polymorphisms and haplotypes associated with chronic fatigue syndrome. Genes Brain Behav. (2007) 6:167–76. doi: 10.1111/j.1601-183X.2006.00244.x
47. Cleare AJ. The HPA axis and the genesis of chronic fatigue syndrome. Trends Endocrinol Metabol. (2004) 15:55–9. doi: 10.1016/j.tem.2003.12.002
48. Sommerfeldt L, Portilla H, Jacobsen L, Gjerstad J, Wyller VB. Polymorphisms of adrenergic cardiovascular control genes are associated with adolescent chronic fatigue syndrome. Acta Paediatr. (2011) 100:293–8. doi: 10.1111/j.1651-2227.2010.02072.x
49. Vladutiu GD, Natelson BH. Association of medically unexplained fatigue with ACE insertion/deletion polymorphism in gulf war veterans. Muscle Nerve. (2004) 30:38–43. doi: 10.1002/mus.20055
Keywords: single nucleotide polymorphism (SNP), tyrosine phosphatase non-receptor type 22 (PTPN22), cytotoxic T-lymphocyte-associated protein 4 (CTLA4), interferon regulatory factor 5 (IRF5), tumor necrosis factor (TNF), myalgic encephalomyelitis (ME), chronic fatigue syndrome (CFS), autoimmunity
Citation: Steiner S, Becker SC, Hartwig J, Sotzny F, Lorenz S, Bauer S, Löbel M, Stittrich AB, Grabowski P and Scheibenbogen C (2020) Autoimmunity-Related Risk Variants in PTPN22 and CTLA4 Are Associated With ME/CFS With Infectious Onset. Front. Immunol. 11:578. doi: 10.3389/fimmu.2020.00578
Received: 11 October 2019; Accepted: 12 March 2020;
Published: 09 April 2020.
Edited by:
Zhiguang Zhou, Central South University, ChinaReviewed by:
Jillian M. Richmond, University of Massachusetts Medical School, United StatesCopyright © 2020 Steiner, Becker, Hartwig, Sotzny, Lorenz, Bauer, Löbel, Stittrich, Grabowski and Scheibenbogen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmen Scheibenbogen, Y2FybWVuLnNjaGVpYmVuYm9nZW5AY2hhcml0ZS5kZQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.