 Xiaofei Gao1
Xiaofei Gao1 Ming Zhao
Ming Zhao- 1Hunan Key Laboratory of Medical Epigenomics, Department of Dermatology, The Second Xiangya Hospital of Central South University, Changsha, China
- 2Department of Pharmacy, The Third Xiangya Hospital, Central South University, Changsha, China
Non-coding RNAs (ncRNAs) are indispensable for CD4+ T cell differentiation and functions. By directly or indirectly regulating immune gene expression, ncRNAs give flexible instructions to guide the biological processes of CD4+ T cells and play a vital role in maintaining immune homeostasis. However, the dysfunction of ncRNAs alters the gene expression profiles, disturbs the normal biological processes of CD4+ T cells, and leads to the functional changes of CD4+ T cells, which is an underlying cause of systemic lupus erythematosus (SLE). In this review, we focus on the recent advances in the roles of ncRNAs in CD4+ T cell functions and differentiation, as well as their potential applications in the diagnosis and treatment of SLE.
Introduction
Systemic lupus erythematosus (SLE) is a highly heterogeneous autoimmune disease triggered by a large number of autoantibodies and the formation of immune complexes, which cause multiple organ damage and serious disease outcomes. Therefore, in order to develop earlier diagnosis and more effective treatment, it is necessary to better understand the pathogenesis of SLE.
SLE is characterized by overactive T cells that differentiate into different T helper (Th) subsets secreting inflammatory cytokines and supporting B cells to produce autoantibodies. Naive CD4+ T cells in lupus undergo the proinflammatory shifts in epigenetics that favor T cell activation and effector T cell immune responses, which trigger lupus flares (1). Our previous studies demonstrate that several hypomethylated genes, such as CD70, CD40LG, ITGAL, and PRF1, are associated with the hyperactivity of T cells in SLE (2–4). The imbalance of Th cell subsets, particularly the deficient proportion of regulatory T cells and the abundance of effector T cells, including Th1, Th2, Th17, and Tfh cells, contributes to the development of SLE and correlates closely with the disease activity (5). The ratio of Th1/Th2 significantly increases in patients with SLE, which is related to the severity of renal damage (6). In addition, Th1 cells promote the secretion of IgG2a and IgG3 antibodies by secreting IFN-γ, thus accelerating the development of immune complex glomerulonephritis (7). Th17 cells exert its effector function mainly by secreting proinflammatory cytokine IL-17 that is increased in SLE patients, indicating the important role of Th17 cells in SLE (8). The phenotypic and functional heterogeneity of Treg cells is also involved in SLE development. Interestingly, the percentage of circulating CD4+ CD25+ Foxp3+ Treg cells decreases significantly in SLE and negatively correlates with the SLE Disease Activity Index (SLEDAI) score (9). CD4+ CD25+ Treg cells freshly isolated from SLE patients secrete increased proinflammatory cytokines IFN-γ and IL-10 and decreased anti-inflammatory cytokine TGF-β (9). On the contrary, circulating CD4+ CD25− Foxp3+ Treg cells, with a pro-inflammatory feature and a deficiency in suppressive function, accumulate in SLE and correlate with the disease activity (10–12). In patients with SLE, peripheral CD4+ CXCR5+ PD-1+ Tfh cells (regarded as a GC-Tfh counterpart) are increased and are associated with a more serious disease phenotype, and CCR7− PD-1hi CXCR5+ CD4+ T cells (regarded as an activated Tfh cell phenotype) are associated with elevated autoantibody titers and the disease activity (13, 14). Imbalanced cytokine profiles in CD4+ T cell subsets, especially the predominance of inflammatory cytokines, are also involved in the pathogenesis of SLE (15). However, the mechanism that causes the aberrant activation, differentiation and function of CD4+ T cells in SLE remains largely unclear.
Non-coding RNAs (ncRNAs) are widely involved in the biological processes of immune cells by altering target gene expression. Mounting evidence has demonstrated that the abnormal expression of ncRNAs plays a key role in CD4+ T cell dysfunction and SLE development. Researches on the roles of ncRNAs in CD4+ T cell differentiation and functions provide new insights into the phenotypic and functional heterogeneity of CD4+ T cells, as well as the pathogenic mechanism of SLE. Furthermore, current studies focus on digging up available ncRNA biomarkers for the clinical management of SLE. Here, we review the current understanding of the roles of ncRNAs in CD4+ T cell differentiation and functions, as well as their potential applications in SLE.
Classification and Functions of ncRNAs
Generally, the most widely studied ncRNAs are microRNAs (miRNAs), long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs). Processed by the RNase III enzyme DROSHA and DICER, precursor miRNAs become mature miRNAs, a type of small (~21 nucleotides) endogenously expressed ncRNAs, which can post-transcriptionally suppress gene expression by binding to specific miRNA response elements (MREs) on their target transcripts (16). The canonical regulatory mechanism of miRNAs is binding to the site in 3′UTR of target mRNA through a form of RNA-induced silencing complex (RISC) containing specialized nuclear Argonaute (Ago) protein, resulting in mRNA degradation or translational inhibition (17). A single miRNA can tune the production of hundreds of proteins by directly or indirectly regulating thousands of genes, suggesting the profound and far-reaching implications of miRNAs on eukaryotic transcriptomes and proteomes (18). However, with a deeper understanding of the characteristics of miRNAs, researchers have uncovered the positive role of miRNAs in activating gene expression, thus making it more difficult to explain the regulatory mechanism of miRNAs. Previously, both Ago1 and Ago2 were considered to suppress gene expression (19). Nonetheless, a later research has reported that Ago2, but not Ago1, can also activate gene expression when its target mRNA lacks poly (A) tail, suggesting that the role of Ago2 is dependent on the length of mRNA poly(A) tail (20).
LncRNAs are a kind of ncRNAs more than 200 nucleotides and have various biological functions based on their structural characteristics and loci to the protein-coding genes (21). According to the location to protein-coding genes, lncRNAs can be classified into five categories, including long intergenic non-coding RNAs (lincRNAs), sense lncRNAs, anti-sense lncRNAs, bidirectional lncRNAs, intronic lncRNAs, each of them has potential in gene regulation. LncRNAs can regulate the expression of nearby protein-coding genes, also known as cis-regulatory action. For example, Wang et al. (22) found that chromosome looping brings lncRNA HOTAIR to the adjacent position of its target gene, and then HOTAIR targets WDR5/MLL complex by directly binding with WDR5, driving histone H3 lysine 4 trimethylation and gene transcription. Furthermore, lncRNAs are involved in adjusting the activity and localization of specific proteins, such as transcription factors. Specifically, lncRNA NRON can bind with the transcription factor NFAT and transcriptional regulators by targeting a scaffold protein to form an RNA-protein scaffold complex, which hinders the interaction between NFAT and phosphatases and prevents NFAT from dephosphorylation, thus inhibiting NFAT-mediated gene transcription (23). LncRNAs can act as miRNA sponges to regulate gene expression, which is also called competing endogenous RNA (ceRNA) mechanism (24, 25). For instance, linc-RoR acts as a miR-145 sponge to prevent Oct4, Sox2, and Nanog from miR-145-mediated degradation (26). In addition, when anti-sense lncRNA gene and protein-coding gene overlap, anti-sense lncRNA transcription inhibits the recruitment of RNA polymerase (Pol II) to the protein-coding gene promoter, leading to the transcriptional inhibition of the protein-coding gene (27).
CircRNAs, first identified in early 1991, are considered as a novel ncRNA subclass with a covalent-closed and stable loop structure (28). Pre-mRNAs undergo canonical splicing to form linear mRNAs, while circRNAs are produced when pre-mRNAs undergo backsplicing. Endogenous circRNAs are resistant to exonucleases-mediated degradation, which makes them get a long half-life and stably exist in mammals. Similar to lncRNAs, most circRNAs carry MREs and can serve as miRNA sponges to affect gene expression (25, 29). For instance, ciRS-7 acts as a miR-7 sponge to regulate Ago protein, and circular sex-determining region Y (Sry) acts as a miR-138 sponge to reserve miR-138-mediated inhibition of Ago2 (30). Besides, circRNAs can be miRNA “reservoirs” to stabilize the function of miRNAs. Hansen et al. (31) first reported this novel pattern of miRNA “reservoir” in the miR-7/miR-671/cisR-7 regulatory axis. In this signaling pathway, miR-671-mediated cleavage of cisR-7 results in the release of miR-7, resulting in the significant down-regulation of miR-7 targets. CircRNAs also control gene transcription through a special form of protein sponge. For example, circ-Foxo3 binds with functional proteins P21 and CDK1, forming a large circRNA-protein complex to regulate target gene transcription (32).
The Role of ncRNAs in CD4+ T Cell Differentiation and Functions
ncRNAs in Th1 Cells
Th1 cells are characterized as expressing the vital transcription factor T-bet and secreting effector cytokines IFN-γ and IL-10, which play a role in mediating chronic inflammatory responses against viruses, intracellular pathogens and tumors. Besides, STAT1 is crucial to IFN-γ signaling, and STAT4 is critical for IL-12 signaling, both of which are the key regulators for Th1 cell differentiation (33) (Table 1).
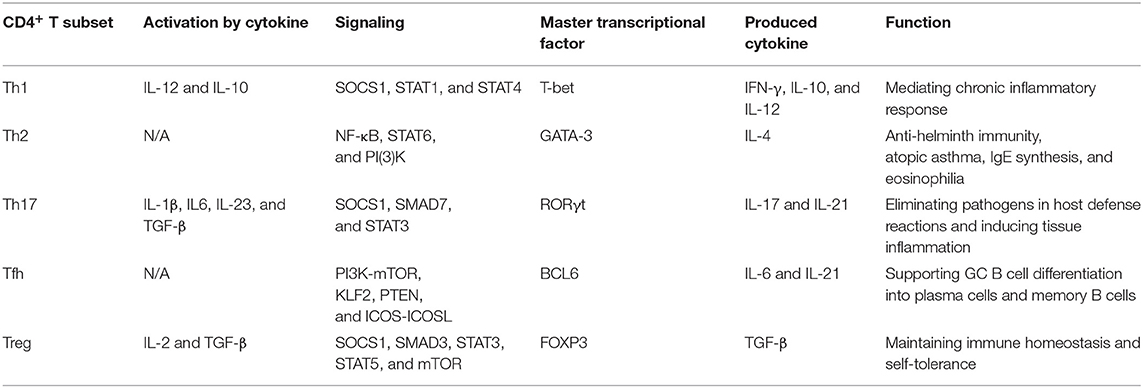
Table 1. The characteristics of CD4+ T subsets.
DICER-deficient T cells lose the ability to generate mature miRNAs and are inclined to differentiate into Th1 cells, suggesting the role of miRNAs in Th1 cell differentiation (34). Furthermore, several miRNAs, such as miR-21, and miR-29, are down-regulated in DICER-deficient CD4+ T cells (34). miR-29 limits the differentiation of Th1 cells and the production of IFN-γ by targeting T-bet and Eomes directly (35). Inhibiting miR-21 shifts the balance of Th1/Th2 toward Th1 cells by improving the secretion of IL-12 in dendritic cells (DCs) and NK cells (36). miR-148a controls Th1 cell survival by targeting the pro-apoptotic gene Bim, and the expression of miR-148a can be induced by Twist1 and T-bet, the critical transcription factors controlling Th1 cell fate (37, 38). Similarly, the overexpression of miR-142a-5p in activated lymphocytes contributes to T cell differentiation toward Th1 cells by targeting SOCS1 and TGFBR1 (39). miRNAs also play a part in regulating the migration and retention of Th1 cells. Deleting miR-31 promotes the expression of genes involved in T cell activation and chemotaxis, leading to the increased migratory ability of Th1 cells. Th1 transcription factor T-bet and FOXO1, respectively, act as positive and negative regulators for miR-31, indicating the interplay between miRNAs and cell signaling molecules (40). In addition, miRNAs can affect the propensity of cytokine production in Th1 cells. The differentiation of IL-10+ Th1 cells and IFN-γ+ Th1 cells are reciprocally restricted, as the increased IL-10 secreted by Th1 cells limits the differentiation of IFN-γ-secreting Th1 cells (41). miR-150 promotes IL-10-secreting Th1 cell differentiation by targeting SLC2A1 and modulating glucose uptake. However, the expression of miR-150 is decreased in IFN-γ-secreting Th1 cells, suggesting that miR-150 serves as a switch to promote IL-10+ Th1 cell differentiation and inhibit IFN-γ secretion (42). LncRNA-Ifng-AS1, also named NeST or Tmevpg1, is essential for the development of Th1 cells. Collier et al. (43) found that Ifng-AS1 and its human ortholog IFNG-AS1 are located near the IFN-γ encoding gene Ifng. LncRNA-Ifng-AS1 cooperates with T-bet or other critical factors to promote Ifng expression, but lncRNA-Ifng-AS1 alone is insufficient for regulating Ifng gene transcription. The abnormal expression of IFNG-AS1 in Th1 cells also correlates with several autoimmune disorders, such as multiple sclerosis (MS) and Hashimoto's Thyroiditis (HT) (44, 45) (Table 2).
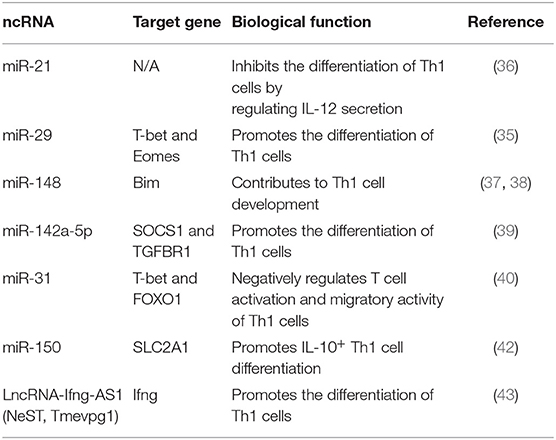
Table 2. ncRNAs involved in Th1 cells.
ncRNAs in Th2 Cells
Th2 cells secrete the master functional cytokine IL-4 and play a critical role in mediating IgE synthesis, eosinophilia, anti-helminth immunity, and atopic asthma. GATA-3, the central regulator of Th2 cells, is necessary and sufficient for the expression of IL-4 in CD4+ T cells, which further activates STAT6 to inhibit Th1 cell differentiation, thus determining the commitment to Th2 phenotype (46) (Table 1).
The miRNA expression profiling of human airway-infiltrating CD4+T cells reveals that miR-19, a member of the miR-17~92 clusters, is highly expressed in asthma, and cells lacking miR-17~92 clusters are compromised in terms of Th2 cell-mediated responses. Functionally, miR-19 facilitates Th2 cell-related cytokine production by targeting PTEN, SOCS1 and A20 to amplify NF-κB, JAK-STAT and PI(3)K signaling pathways (47). miR-23~27~24 clusters also play an important part in Th2-mediated immune responses. miR-24 and miR-27 collaboratively inhibit the differentiation of Th2 cells and the production of functional cytokine IL-4. miR-27 limits IL-4 production by directly repressing the transcription factor GATA-3. However, other direct targets of miR-24 and miR-27, including Cnot6, Clcn3, Ikzf1, Gpr174, and Galnt3, have few effects on IL-4, but they may alter the expression of molecules related to bypassing signaling pathways (48, 49). In addition, Th2 cells lacking miR-155 exhibit a significant deficiency in migrating ability. In-depth study finds that miR-155 can target S1pr1 to promote Th2-mediated airway allergy (50) (Table 3).

Table 3. ncRNAs involved in Th2 cells.
ncRNAs in Th17 Cells
Th17 cells are characterized by producing functional cytokines IL-17 and IL-21, as well as expressing the central transcription factor RORγt. TGF-β serves as a key regulator for the differentiation and function of Th17 cells by inducing RORγt expression (51). Th17 cell-mediated host defense plays a critical role in eliminating pathogens, however, the uncontrolled immune responses can also be a trigger for tissue inflammations and autoimmune diseases (52) (Table 1).
Mounting evidence has demonstrated that miRNAs are involved in Th17 cell-mediated inflammatory responses and autoimmunity. For instance, miR-155 facilitates the differentiation and function of Th17 cells by targeting SOCS1 (53). Moreover, miR-155 drives DCs to produce pro-inflammatory cytokines crucial for the differentiation of Th17 cells (54). Mice conditionally knocking out miR-223 in myeloid dendritic cells (mDCs) exhibit increased expression of PD-L1 and decreased levels of pro-inflammatory cytokines IL-1β, IL-6, and IL-23 necessary for driving pathogenic Th17 cell differentiation, thus slowing the progression of experimental autoimmune encephalomyelitis (EAE) (55). Similarly, mice lacking miR-21 are deficient in Th17 cell differentiation and are resistant to EAE. Functionally, miR-21 suppresses the expression of smad7, a negative regulator of TGF-β, which further activates the downstream SMAD-2/3 signaling pathway that induces TGF-β expression, thus contributing to Th17 cell differentiation (56). Dicer1-regulated microRNA-183-96-182 clusters (miR-183C) contribute to the pathogenicity of Th17 cells in autoimmunity. Deleting Dicer1, an encoding gene responsible for the biogenesis and processing of miRNAs, inhibits the production of inflammatory cytokines in Th17 cells, suggesting the critical role of miRNAs in regulating the pathogenic function of Th17 cells. Furthermore, the expression of miR-183C significantly increases in Th17 cells under the induction of IL-6-STAT3 signaling. Enhanced miR-183C further promotes the production of pathogenic cytokines in Th17 cells via suppressing Foxo1, a negative transcriptional factor of Th17 cells, resulting in serious autoimmune diseases (57). The increased expression of miR-210 induced by hypoxia plays a key role in Th17 cell differentiation. Under normoxic conditions, hypoxia-inducible factor 1α (HIF-1α) is proline hydroxylated by prolyl hydroxylases (PHDs), and then hydroxylated HIF-1α is recognized by the von Hippel Lindau protein (VHL) E3 ligase complex, resulting in HIF-1α ubiquitination and proteasomal degradation (58). On the contrary, hypoxic conditions protect HIF-1α from VHL-mediated ubiquitination, thus promoting its expression. HIF-1α expresses shortly after T cell activation, which initiates metabolic reprogramming and the transcription of Th17 lineage-defining transcription factor RORγt. The expression of miR-210 induced by HIF-1α occurs late in Th17 differentiation, which limits the function of HIF-1α via negative feedback. Under hypoxic conditions, inhibiting miR-210 promotes the differentiation of Th17 cells by enhancing the expression of HIF-1α (59). LincRNA-p21, a kind of hypoxia-induced lncRNAs, inhibits VHL-mediated HIF-1α ubiquitination by binding with HIF-1α and VHL and disturbing their interaction, thus contributing to HIF-1α accumulation. Besides, lincRNA-p21 and HIF-1α can form positive feedback on HIF-1α expression and HIF-1α-dependent glycolysis (60). Furthermore, lncRNA 1700040D17Rik is significantly decreased in EAE mouse model, while highly expressed 1700040D17Rik is observed in mice after treated by rhIL23R-CHR. Functional study demonstrates that 1700040D17Rik is involved in Th17 cell differentiation by regulating RORγt expression (61) (Table 4).
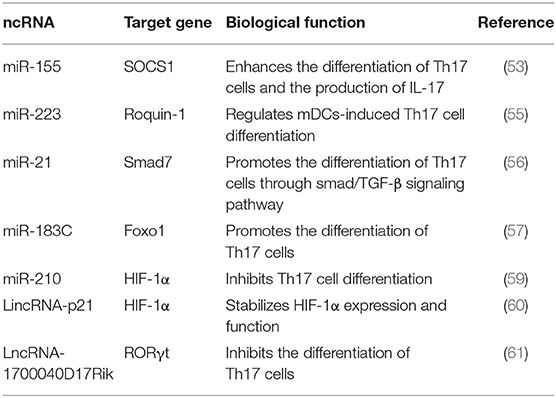
Table 4. ncRNAs involved in Th17 cells.
ncRNAs in Follicular Helper T Cell (Tfh) Cells
Tfh cell differentiation depends on the expression of lineage-defining transcription factor BCL6. Several characteristic molecules, such as CXCR5, PD-1, BTLA4, ICOS, IL-21, and SAP, also play important roles in the differentiation and function of Tfh cells, and they are indispensable for supporting the development of GC B cells (62). Generally, Tfh cell differentiation can be divided into two consecutive stages. The first stage is pre-GC Tfh cells, which is initialed by antigen-presenting cells (APCs). Under the stimulation from APCs, pre-GC Tfh cells migrate to the border of germinal centers (GCs), where the interaction of T/B cells occurs, and then highly express surface markers CXCR5, PD-1, ICOS, and SAP, which are necessary to determine the molecular basis for commitment to Tfh cell phenotype and promote GC formation (63, 64). The second stage is GC Tfh cells, which mainly participate in regulating GC responses and promote the differentiation of GC B cells into plasma cells and memory B cells. In GC Tfh cells, several important molecules, including CXCR5, PD-1, BCL6, BTLA4, ICOS, and SAP, reach a peak, while CD127, PSGL1, and EBI2 decline (64–66) (Table 1).
miRNAs are important for post-transcriptionally regulating Tfh cell differentiation in different stages. miR-17~92 plays a key role in the early stage differentiation of Tfh cells (67, 68). miR-17~92 facilitates robust Tfh cell differentiation after initial T cell activation by repressing Rora and other inappropriate genes (69). Interestingly, miR-17~92 induces the differentiation of CXCR5+ PD1hi Tfh cells without increasing BCL6 expression, which is consistent with the phenotype of pre-GC Tfh cells. Furthermore, miR-17~92 controls the migration of Tfh cells to B follicles by enhancing ICOS-PI(3)K signaling via repressing Akt phosphatase Phlpp2, an inhibitor for PI3K-Akt-mTOR signaling pathway (68). The RNA binding protein Roquin inhibits spontaneous T cell activation and Tfh cell differentiation via post-transcriptional regulation and plays a part in maintaining peripheral tolerance and preventing autoimmune diseases (70). Functionally, Roquin inhibits the PI3K-mTOR signaling pathway by preventing miR-17~92 binding with 3'UTR of PTEN, resulting in the upregulation of PTEN and the decreased differentiation of Tfh and Th17 cells (71). However, miR-92a, a member of miR-17~92 clusters, promotes Tfh cell development by inhibiting KLF2 and PTEN in human autoimmunity, indicating the variability of miRNA functions in different immune contexts (72). The transcription factor BCL6 can also affect miRNA expression to exert its regulatory role on Tfh cells. miR-31 limits the function of human Tfh cells by inhibiting the expression of molecules relevant to Tfh cell biology, such as SAP and CD40L. In human Tfh cells, miR-31 is specifically and strongly down-regulated by BCL6 (73). miR-155 is rapidly induced in activated T cells, which overlaps with the expression kinetics of miR-17~92 in activated CD4+ T cells. Consistently, deleting miR-155 in T cells compromises the early Tfh cell differentiation and expansion (74). Moreover, miR-155 promotes the proliferation of Tfh cells especially at the late stage of Tfh cell differentiation and positively controls CD40L expression in antigen-specific CD4+ T cells by suppressing the Peli1-c-Rel signaling pathway (75). Furthermore, the miR-155–Peli1–c-Rel pathway is important to Tfh cell differentiation but is dispensable for non-Tfh cell differentiation, highlighting its specific role for Tfh cell lineage (75). On the contrary, miR-146a plays a negative part in Tfh cell generation. miR-146a is induced by TCR-driven NF-κB activation and serves as a negative regulator in antigen-specific T cell response in vivo, for both primary and recall responses (76). The expression of miR-146a highly increases in Tfh and GC B cells, and peaks in the late stage of GC responses, coinciding with the decline of Tfh cell-mediated immune responses after immunization. miR-146a can repress multiple Tfh-related mRNAs, especially genes related to the ICOS-ICOSL signaling pathway in Tfh and GC B cells, thus inhibiting Tfh and GC B cell accumulation, which is important to maintain immunological tolerance and prevent autoimmune diseases (77). Besides, miR-146a-deficient T cells are hyperactive in chronic inflammation and are involved in T cell-mediated autoimmunity (76). Similarly, miR-146b, a paralog of miR-146a, also acts as an inhibitor of Tfh and Th17 cell differentiation (78). In addition, lncRNA Malat1 is highly expressed in T cell subsets and is widely involved in immune responses by regulating immune gene expression. Although the deficiency of Malat1 in T cells does not influence the differentiation and B cell helper function of Tfh cells, inhibiting Malat1 in monocytes can reduce the expression of proinflammatory cytokine IL-21 (79) (Table 5).
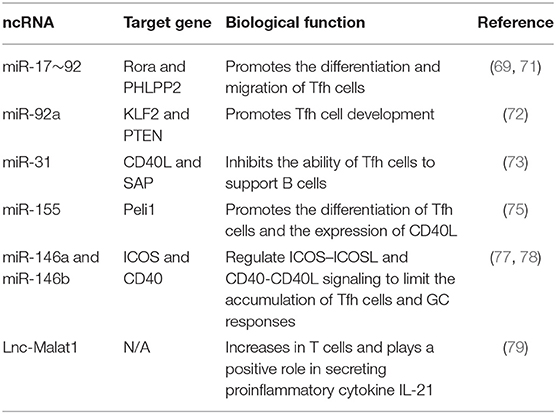
Table 5. ncRNAs involved in Tfh cells.
ncRNAs in Treg Cells
Treg cells characteristically express CD25 and the lineage-specific transcription factor Foxp3. CD25 is an original marker of Treg cells, which represents an activated state of CD4+ T cells, and Foxp3 determines the differentiation and suppressor function of Treg cells. Foxp3 expression is initialed by IL-2 signaling and influenced by particular TCR signaling and TGF-β (80). Furthermore, IL-2 receptor beta-dependent STAT5 activation is necessary for Foxp3 expression and Treg cell development (81). TGF-β is also responsible for balancing the reciprocal conversion between Th17 and Treg cells (82, 83). Foxp3+ Treg cells suppress overactive immune responses to maintain immune homeostasis and inhibit autoreactive T cells to preserve self-tolerance, whereas the dysfunction of Treg cells can cause autoimmune disorders and tumor immune escape (Table 1).
ncRNAs play a critical role in controlling the differentiation and function of Treg cells. The deficiency of miR-31 in CD4+ T cells promotes Treg cell development, leading to the alleviation of EAE and Ang II (Angiotensin II)-induced hypertension in mice, while the overexpression of miR-31 inhibits Treg cell differentiation by targeting Foxp3, Gprc5a, and Protein phosphatase 6c (Ppp6C) that are indispensable for Treg cell development (84, 85). On the contrary, miR-125a stabilizes the immunoregulatory capacity of Treg cells by suppressing several effector T lineage genes, including Stat3, Ifng, and Il13. miR-125a-deficient mice are inclined to develop more severe symptoms of EAE, which are alleviated after rescuing miR-125a (86). In the setting of a limited number of IL-2, miR-155 is required for maintaining the competitive fitness of Treg cells. Foxp3 controls the function of Treg cells by driving miR-155 expression, thus contributing to the reduced expression of SOCS1 protein and the increased level of high-affinity IL-2R in Treg cells (87). Treg cells contain a high level of cyclic AMP (cAMP) necessary for maintaining its suppressive function. However, upregulated miR-142-3p can restrict the level of cAMP in CD4+T cells, thus compromising the suppressive function of Treg cells (88). Interestingly, miR-142-5p, another mature isoform of miR-142, plays a positive role in regulating cAMP-mediated suppressor function of Treg cells. Deleting miR-142-5p decreases the concentration of intracellular cAMP by targeting AMP-hydrolyzing enzyme phosphodiesterase-3b (Pde3b), leading to the deficient function of Treg cells and immune disorder in mice despite preserving a number of Treg cells with a normal phenotype (89). miR-99a and miR-150 can target mTOR, which is responsible for cell growth, to promote the differentiation of Treg cells at the expense of inhibiting Th17 cells (90). miR-99a and miR-150 suppress the expression of mTOR by binding two adjacent target sites in it. Besides, an intact target site of miR-99a is necessary for miR-150-mediated regulation to mTOR, because miR-150 activation depends on miR-99a binding to its adjacent target site of mTOR (90).
Recently, scientists have uncovered a distinctive lncRNA transcriptome of Treg cells. And lncRNA Flatr, a core member of the lncRNA transcriptome of Treg cells, promotes Treg cell development by enhancing the expression of Foxp3 (91). LncRNA-smad3 also plays a role in controlling Treg cell fate and regulating T cell-mediated autoimmunity. Functionally, lnc-smad3 recruits histone deacetylase HDAC1 to the smad3 promoter, which silences smad3 transcription by altering smad3 locus accessibility, leading to the reduced expression of Foxp3 in Treg cells. Conversely, TGF-β activation inhibits lnc-smad3 expression in Treg cells, therefore promoting Foxp3 expression via restoring the accessibility of smad3 (92). LncRNA Flicr adjacent to Foxp3 in mouse and human genomes can affect the chromatin accessibility of Foxp3. Flicr-deficient mice show resistance to autoimmune diabetes, indicating that Flicr-deficient Treg cells may more effectively function as an immune suppressor than normal Treg cells (93) (Table 6).
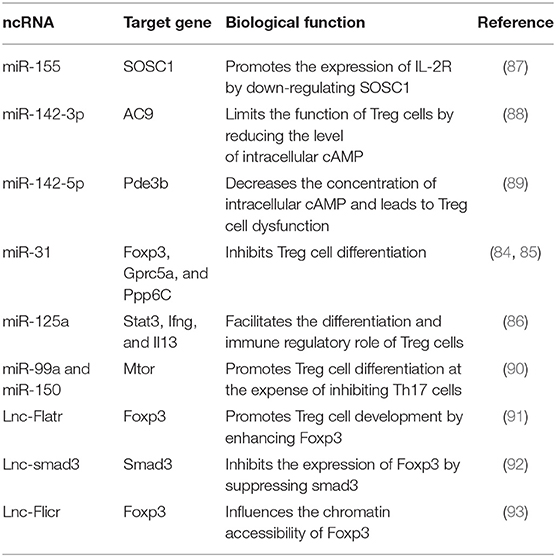
Table 6. ncRNAs involved in Treg cells.
The Roles of ncRNAs in the Aberrant Activation and Differentiation of CD4+ T Cells in SLE
The abnormal activation and differentiation of CD4+ T cells cause the pathogenic expansion of Th1/Th17/Tfh cells and Treg cell deficiency, which are implicated in SLE development. However, the regulatory mechanism leading to CD4+T cell dysfunction in SLE remains largely unknown. ncRNAs play an important role in CD4+ T cell activation and differentiation. Many studies have identified that the expression of above-mentioned ncRNAs, such as miR-21, miR-155, miR-31, miR-29a, miR-126, miR-142-3p/5p, and miR-183C, as well as several newly discovered lncRNAs and circRNAs, are markedly altered in the CD4+ T cells or peripheral blood mononuclear cells (PBMCs) of SLE, and are involved in SLE development by directly or indirectly regulating the aberrant activation and differentiation of CD4+ T cells.
ncRNAs Regulate the Over-Activation of CD4+ T Cells in SLE
ncRNAs are implicated in regulating lupus CD4+ T cell over-activation. miR-148a and miR-21 can target DNA methyltransferase 1 (DNMT1) to promote the DNA demethylation of CD70 and LFA-1 that are responsible for T cell hyperactivity (94). Likewise, miR-29a facilitates the expression of immune gene CD70 and CD11a in lupus CD4+ T cells by targeting sp1 to repress DNMT1 (95). Our previous studies identify several miRNAs, including miR-126 and miR-142-3p/5p that are involved in CD4+ T cell over-activation in SLE (96–98). We found that upregulated miR-126 promotes the expression of CD11a and CD70 in lupus CD4+ T cells via inhibiting DNMT1-mediated DNA methylation (96). In addition, we also reported that the decreased expression of miR-142-3p/5p in lupus CD4+ T cells is involved in T cell hyperactivity and B cell hyperstimulation by targeting SAP, CD84, and IL-10 (97). Enhancer of zeste homolog 2 (EZH2), an epigenetic modulator important to T cell activation and differentiation, is over-activated in lupus CD4+ T cells. Coit et al. (1) found that the reduced expression of miR-26a and miR-101 is responsible for the increased activity of EZH2 in lupus CD4+T cells, which causes the epigenetic reprogramming that favors T cell activation and effector T cell differentiation, leading to lupus flares.
Recently, Chen et al. have reported that circRNAs form small imperfect RNA duplexes to bind the protein kinase (PKR) and inhibit its activation, while circRNAs are mostly degraded by a PKR-dependent manner. Improving the level of dsRNA-containing circRNAs suppresses the aberrant activation of PKR in lupus T cells, indicating the role of circRNAs in controlling PKR activity in SLE (99). It was reported that RANTES is involved in the development of SLE, and the transcriptional factor KLF13 regulates the expression of RANTES in activated T cells (100). Zhang et al. (101) demonstrated that hsa_circ_0012919 is highly expressed in lupus CD4+ T cells and participates in regulating the expression of RANTES and KLF13 by serving as the miR-125a-3p sponge. Moreover, inhibiting hsa_circ_0012919 corrects the DNA hypomethylation of CD70 and CD11a in lupus CD4+T cells by promoting DNMT1 activity (101). The AKT signaling is over-activated in CD4+ T cells from SLE patients (102–104). And miR-29a regulates the AKT signaling pathway in lupus CD4+ T cells by indirectly targeting DNMT1 (95). Wang et al. (105) found that circIBTK is down-regulated in SLE and negatively correlates with the SLEDAI score and anti-double-stranded (ds)DNA antibodies but positively correlates with the level of complement component 3 (C3) in patients with SLE. CircIBTK serves as the miR-29b sponge, which may inhibit the activation of the AKT signaling pathway in lupus CD4+ T cells by sponging miR-29a. LncRNAs also play a role in CD4+ T cell over-activation in SLE. LncRNA UCA1 is highly expressed in the PBMCs of SLE patients and positively correlates with the SLEDAI score and the levels of C3 and anti-dsDNA antibodies. Furthermore, UCA1 promotes the expression of AKT and PI3K correlated with SLE progression (106) (Table 7).

Table 7. The roles of ncRNAs in the aberrant activation of CD4+ T cells in SLE.
ncRNAs Regulate the Aberrant Differentiation of CD4+ T Subsets in SLE
MiR-155 is dysregulated in CD4+ T cells from both patients with SLE and lupus-prone mice. Divekar et al. (107) found that in lupus-prone mice, the expression of miR-155 significantly increases in Treg cells with an abnormal phenotype (CD62L−CD69+) and a deficient suppressive capability. Functionally, miR-155 targets CD62L to alter the phenotype of Treg cells in lupus-prone mice, suggesting that miR-155 is involved in the phenotypic heterogeneity of Treg cells. Deleting miR-155 limits the proportion of activated T cells and the levels of inflammatory cytokines IFN-γ, IL-4 and IL-17 secreted by Th1, Th2, and Th17 cells, thus alleviating the disease severity of lupus-prone mice (108). Diffuse alveolar hemorrhage (DAH) is a severe complication of SLE. Zhou et al. (109) reported that down-regulating miR-155 corrects the abnormal activation of NF-κB signaling and ameliorates the lung inflammation of pristane-induced DAH. On the contrary, Lashine et al. (110) found that the expression of miR-155 decreases in PBMCs from juvenile SLE and negatively correlates with the disease activity and proteinuria but positively correlates with the white blood cell (WBC) count. Besides, correcting the expression of miR-155 represses the expression of phosphatase 2A (PP2A) and promotes the secretion of IL-2 in PBMCs from juvenile SLE patients. To sum up, as SLE is a highly heterogeneous autoimmune disease, the differences in patients and disease stages probably count for the contradiction in the expression and role of miR-155 in SLE.
miR-21 is upregulated in both PBMCs or CD4+T cells from SLE patients and splenic CD4+ T cells from lupus-prone mice (111, 112). miR-21 contributes to aberrant CD4+ T cell responses in human SLE by targeting PDCD4, while inhibiting miR-21 in vitro improves abnormal CD4+ T cell responses, including the production of IL-10, and the expression of CD40L, as well as the capability to support B cell development, which indicates the pathogenic role of miR-21 in SLE (113). Garchow et al. (114) reported that miR-21 deficiency protects mice from chronic graft-versus-host disease (cGVHD)-induced lupus by regulating Th17/Treg balance. They observed that cGVHD-affected mice with miR-21 deficiency have a reduced proportion of Th17 cells and an expanded compartment of Treg cells. Moreover, miR-21 inhibits the CD40: CD40L and CD28: CD80/86 co-stimulation pathways that are involved in establishing T cell tolerance and limiting adaptive immune response (114). Silencing miR-21 with a tiny seed-targeting LNA alleviates splenomegaly in B6.Sle123 mice and decreases the expression of PDCD4 and the population of Fas receptor-expressing lymphocytes (115). In SLE patients, the expression of miR-21 positively correlates with the disease activity and negatively correlates with the levels of C3 and IL-2, while the expression of miR-31 decreases and positively correlates with IL-2 production (116, 117). And IL-2 deficiency is responsible for the compromised suppressor ability of Treg cells in SLE (118). Furthermore, miR-21 and miR-155 are highly expressed in PBMCs from SLE patients with active nephritis and are the risk factors for lupus nephritis (LN), suggesting that miR-21 and miR-155 are potential biomarkers for assessing the severity of LN (119).
In addition to miR-155, miR-21, and miR-31, other miRNAs are also reported to be involved in aberrant differentiation of CD4+ T cell subsets in SLE, including miR-183C, miR-17-92 and miR-873. miR-183C is upregulated in both PBMCs from SLE patients and splenocytes from MRL/lpr mice (112). Interestingly, Li et al. (120) found that miR-183C is decreased in LN patients. Although it was reported that the increased expression of miR-183C plays a positive role in driving pathogenic Th17 cell differentiation and autoimmune diseases (57), their studies showed that overexpressing miR-183C in lupus mice reverses the ratio of Th17/Treg by regulating mTOR signaling, thus improving the severity of LN and the survival rate of lupus mice (120). miR-17-92 is critical for Tfh cell differentiation and is implicated in autoimmune diseases (68, 121). In addition, the members of miR-17-92 clusters (except miR-92a) are upregulated in CD4+ T cells from MRL/lpr mice and patients with SLE (122, 123). Upregulating miR-17-92 in mice facilitates the spontaneous activation and abnormal expansion of CD4+ T cells, and skews CD4+T cell differentiation toward Tfh cells, thus contributing to lymphoproliferative diseases and SLE-like symptoms (68, 124). In patients with SLE, the expression of miR-17, miR-19b and miR-20a is positively correlated with the SLEDAI score, and miR-17 and miR-20a are also positively associated with the titers of anti-dsDNA antibodies. Besides, miR-17 and miR-20a are negatively correlated with the level of C3, suggesting the different roles of miR-17-92 cluster members in indicating the disease activity of SLE (123). miR-873 is upregulated in SLE patients and positively correlates with the disease severity of SLE, and CD4+ T cells are the major source of miR-873 expression. Furthermore, miR-873 contributes to Th17 cell differentiation by targeting forkhead box O1 (Foxo1). Inhibiting miR-873 in vivo decreases the secretion of IL-17A and alleviates the progression of spontaneous SLE in MRL/lpr mice (125). Shao et al. reported that miR-34a limits the differentiation of Treg cells by inhibiting Foxp3 in CD4+ T cells. They found that the level of murine miR-34a decreases in TGF-β induced Treg cells but increases in Th17 cells induced in vitro. Moreover, the level of miR-34a positively correlates with the Th17 lineage-specific transcription factor RORγt but negatively correlates with the mRNA expression of FOXP3 in PBMCs of SLE, which indicates that miR-34a may play a part in the imbalance of Th17/Treg in SLE (126). In Treg cells of new-onset SLE, the expression of miR-326 is increased and negatively correlates with the mRNA expression of Ets-1 and positively correlates with the levels of CRP and anti-C1q antibodies from new-onset SLE patients, suggesting that miR-326 may regulate Treg cell dysfunction via targeting Est-1 in SLE (127).
LncRNA-NEAT1 is highly expressed in PBMCs from SLE patients and positively correlates with the SLEDAI score (128). Inhibiting NEAT1 in LN patients reduces the expression of a group of chemokines and cytokines, including IL-6, CXCL10, CCL8, etc., which are involved in glomerular Th1 cell predominance (129). IL-6 is also important to support CD4+ T cell expansion and Tfh cell differentiation and functions as a major proinflammatory cytokine in SLE (15, 130). Furthermore, NEAT1 regulates a set of genes, such as IFI27, OAS1, CXCL9, IFI35, CXCL10, and CXCL11, involved in abnormal type I interferon signaling pathway in SLE (128) (Table 8) (Figure 1).
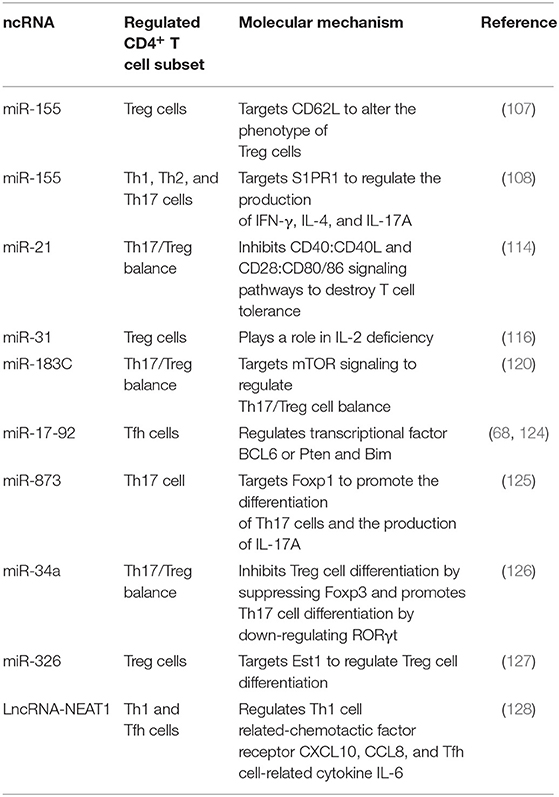
Table 8. The roles of ncRNAs in the aberrant differentiation of CD4+ T cells in SLE.
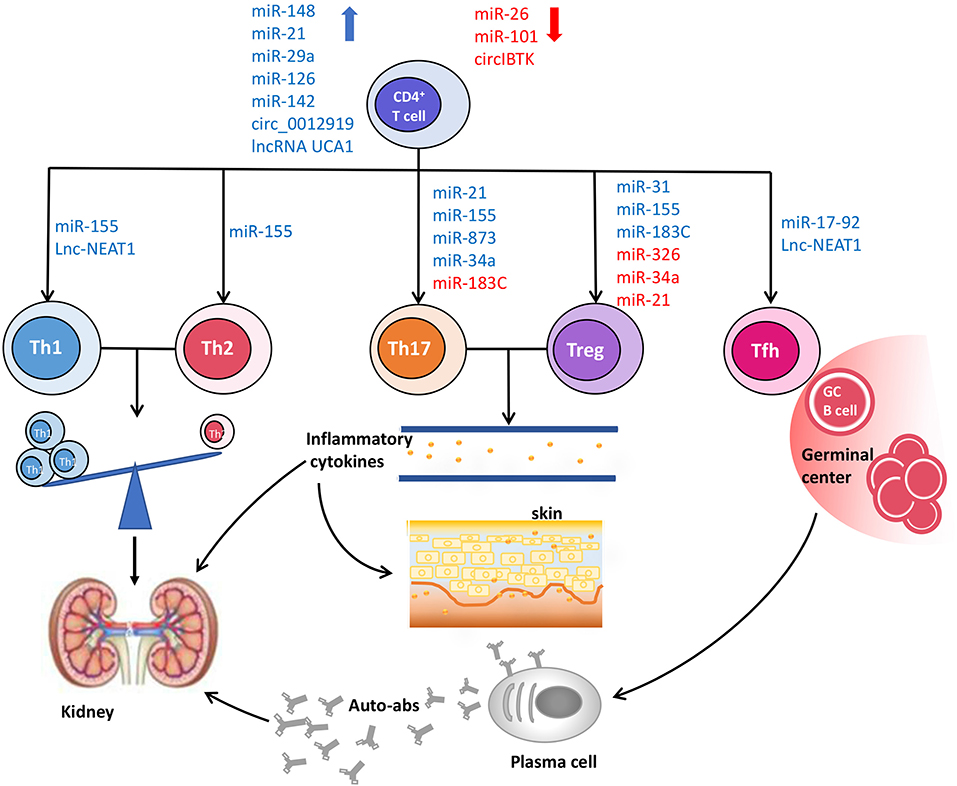
Figure 1. The roles of ncRNAs in CD4+ T cell dysfunction and pathogenesis of SLE. The aberrant expression of ncRNAs disturbs the normal biological processes of CD4+ T cells, and then leads to the phenotypic and functional changes of CD4+ T cells, including the imbalance of Th1/Th2, increased proportions of pro-inflammatory Th17 cells and Tfh cells, as well as deficient frequency and function of Treg cells, which contribute to the occurrence and development of SLE.
Conclusion and Outlook
In summary, the functions and roles of many miRNAs are demonstrated in regulating CD4+T cell activation and differentiation and the processes of SLE. However, the specific functions of lncRNAs and circRNAs in CD4+ T cell development and their roles in the pathogenesis of SLE are largely unknown. The functional studies about circRNAs and lncRNAs mainly arrive from cancer disease and anti-tumor immunity. Using RNA-seq, researchers have discovered distinctive expression profiles of circRNAs in different immune cells of SLE, but the functions and mechanisms of those circRNAs in SLE are unclear. The future of ncRNA researches in the immune system is both promising and challenging, since the reported ncRNAs are just the tip of the iceberg in the human genome, and most of their functions need to be investigated by experimental methods. Studying the roles of ncRNAs in CD4+ T cell differentiation and their applications in SLE will help us to understand how ncRNA disorders cause CD4+ T cell dysfunction and the pathogenesis of SLE, thus paving the way for the development of diagnostics and therapeutics of SLE.
Author Contributions
XG wrote the manuscript. LL, XM, and SJ edited the manuscript. MZ revised the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81874243 and No. 81602766).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Coit P, Dozmorov MG, Merrill JT, McCune WJ, Maksimowicz-McKinnon K, Wren JD, et al. Epigenetic reprogramming in naive CD4+ T cells favoring T cell activation and non-Th1 effector T cell immune response as an early event in lupus flares. Arthritis Rheumatol. (2016) 68:2200–9. doi: 10.1002/art.39720
2. Lu Q, Kaplan M, Ray D, Ray D, Zacharek S, Gutsch D, et al. Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis Rheum. (2002) 46:1282–91. doi: 10.1002/art.10234
3. Oelke K, Lu Q, Richardson D, Wu A, Deng C, Hanash S, et al. Overexpression of CD70 and overstimulation of IgG synthesis by lupus T cells and T cells treated with DNA methylation inhibitors. Arthritis Rheum. (2004) 50:1850–60. doi: 10.1002/art.20255
4. Lu Q, Wu A, Tesmer L, Ray D, Yousif N, Richardson B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J Immunol. (2007) 179:6352–8. doi: 10.4049/jimmunol.179.9.6352
5. He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med. (2016) 22:991–3. doi: 10.1038/nm.4148
6. Akahoshi M, Nakashima H, Tanaka Y, Kohsaka T, Nagano S, Ohgami E, et al. Th1/Th2 balance of peripheral T helper cells in systemic lupus erythematosus. Arthritis Rheum. (1999) 42:1644–8. doi: 10.1002/1529-0131(199908)42:8<1644::AID-ANR12>3.0.CO;2-L
7. Snapper CM, McIntyre TM, Mandler R, Pecanha LM, Finkelman FD, Lees A, et al. Induction of IgG3 secretion by interferon gamma: a model for T cell-independent class switching in response to T cell-independent type 2 antigens. J Exp Med. (1992) 175:1367–71. doi: 10.1084/jem.175.5.1367
8. Kwan BC, Tam LS, Lai KB, Lai FM, Li EK, Wang G, et al. The gene expression of type 17 T-helper cell-related cytokines in the urinary sediment of patients with systemic lupus erythematosus. Rheumatology. (2009) 48:1491–7. doi: 10.1093/rheumatology/kep255
9. Yang X, Wang W, Xu J, Zhang MS, Mei H, Shen Y, et al. Significant association of CD4+CD25+Foxp3+ regulatory T cells with clinical findings in patients with systemic lupus erythematosus. Ann Transl Med. (2019) 7:93. doi: 10.21037/atm.2019.01.38
10. Valencia X, Yarboro C, Illei G, Lipsky PE. Deficient CD4+CD25high T regulatory cell function in patients with active systemic lupus erythematosus. J Immunol. (2007) 178:2579–88. doi: 10.4049/jimmunol.178.4.2579
11. Bonelli M, Savitskaya A, von Dalwigk K, Steiner CW, Aletaha D, Smolen JS, et al. Quantitative and qualitative deficiencies of regulatory T cells in patients with systemic lupus erythematosus (SLE). Int Immunol. (2008) 20:861–8. doi: 10.1093/intimm/dxn044
12. Yin ZJ, Ju BM, Zhu L, Hu N, Luo J, He M, et al. Increased CD4(+)CD25(-)Foxp3(+) T cells in Chinese systemic lupus erythematosus: correlate with disease activity and organ involvement. Lupus. (2018) 27:2057–68. doi: 10.1177/0961203318804881
13. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity. (2013) 39:770–81. doi: 10.1016/j.immuni.2013.09.007
14. Choi JY, Ho JH, Pasoto SG, Bunin V, Kim ST, Carrasco S, et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol. (2015) 67:988–99. doi: 10.1002/art.39020
15. Talaat RM, Mohamed SF, Bassyouni IH, Raouf AA. Th1/Th2/Th17/Treg cytokine imbalance in systemic lupus erythematosus (SLE) patients: Correlation with disease activity. Cytokine. (2015) 72:146–53. doi: 10.1016/j.cyto.2014.12.027
16. Hutvágner G, McLachlan J, Pasquinelli AE, Bálint É, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme dicer in the maturation of the let-7 small temporal RNA. Science. (2001) 293:834–8. doi: 10.1126/science.1062961
18. Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. (2008) 455:58–63. doi: 10.1038/nature07228
19. Pillai RS, Artus CG, Filipowicz W. Tethering of human Ago proteins to mRNA mimics the miRNA-mediated repression of protein synthesis. RNA. (2004) 10:1518–25. doi: 10.1261/rna.7131604
20. Turchinovich A, Burwinkel B. Distinct AGO1 and AGO2 associated miRNA profiles in human cells and blood plasma. RNA Biol. (2012) 9:1066–75. doi: 10.4161/rna.21083
21. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. (2011) 43:904–14. doi: 10.1016/j.molcel.2011.08.018
22. Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. (2011) 472:120–4. doi: 10.1038/nature09819
23. Sharma S, Findlay GM, Bandukwala HS, Oberdoerffer S, Baust B, Li Z, et al. Dephosphorylation of the nuclear factor of activated T cells (NFAT) transcription factor is regulated by an RNA-protein scaffold complex. Proc Natl Acad Sci USA. (2011) 108:11381–6. doi: 10.1073/pnas.1019711108
24. Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. (2007) 4:721–6. doi: 10.1038/nmeth1079
25. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. (2014) 505:344–52. doi: 10.1038/nature12986
26. Wang Y, Xu Z, Jiang J, Xu C, Kang J, Xiao L, et al. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev Cell. (2013) 25:69–80. doi: 10.1016/j.devcel.2013.03.002
27. Latos PA, Pauler FM, Koerner MV, Senergin HB, Hudson QJ, Stocsits RR, et al. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science. (2012) 338:1469–72. doi: 10.1126/science.1228110
28. Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, et al. Scrambled exons. Cell. (1991) 64:607–13. doi: 10.1016/0092-8674(91)90244-S
29. Kristensen LS, Hansen TB, Veno MT, Kjems J. Circular RNAs in cancer: opportunities and challenges in the field. Oncogene. (2018) 37:555–65. doi: 10.1038/onc.2017.361
30. Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature. (2013) 495:384–8. doi: 10.1038/nature11993
31. Hansen TB, Kjems J, Damgaard CK. Circular RNA and miR-7 in cancer. Cancer Res. (2013) 73:5609–12. doi: 10.1158/0008-5472.CAN-13-1568
32. Du WW, Zhang C, Yang W, Yong T, Awan FM, Yang BB. Identifying and characterizing circRNA-protein interaction. Theranostics. (2017) 7:4183–91. doi: 10.7150/thno.21299
33. O'Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. (2008) 28:477–87. doi: 10.1016/j.immuni.2008.03.002
34. Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. J Exp Med. (2005) 202:261–9. doi: 10.1084/jem.20050678
35. Steiner DF, Thomas MF, Hu JK, Yang Z, Babiarz JE, Allen CD, et al. MicroRNA-29 regulates T-box transcription factors and interferon- γ production in helper T cells. Immunity. (2011) 35:169–81. doi: 10.1016/j.immuni.2011.07.009
36. Lu TX, Hartner J, Lim EJ, Fabry V, Mingler MK, Cole ET, et al. MicroRNA-21 limits in vivo immune response-mediated activation of the IL-12/IFN-gamma pathway, Th1 polarization, and the severity of delayed-type hypersensitivity. J Immunol. (2011) 187:3362–73. doi: 10.4049/jimmunol.1101235
37. Haftmann C, Stittrich AB, Zimmermann J, Fang Z, Hradilkova K, Bardua M, et al. miR-148a is upregulated by Twist1 and T-bet and promotes Th1-cell survival by regulating the proapoptotic gene Bim. Eur J Immunol. (2015) 45:1192–205. doi: 10.1002/eji.201444633
38. Maschmeyer P, Petkau G, Siracusa F, Zimmermann J, Zugel F, Kuhl AA, et al. Selective targeting of pro-inflammatory Th1 cells by microRNA-148a-specific antagomirs in vivo. J Autoimmun. (2018) 89:41–52. doi: 10.1016/j.jaut.2017.11.005
39. Talebi F, Ghorbani S, Chan WF, Boghozian R, Masoumi F, Ghasemi S, et al. MicroRNA-142 regulates inflammation and T cell differentiation in an animal model of multiple sclerosis. J Neuroinflammation. (2017) 14:55. doi: 10.1186/s12974-017-0832-7
40. Bardua M, Haftmann C, Durek P, Westendorf K, Buttgereit A, Tran CL, et al. MicroRNA-31 reduces the motility of proinflammatory T helper 1 lymphocytes. Front Immunol. (2018) 9:2813. doi: 10.3389/fimmu.2018.02813
41. Oleszycka E, McCluskey S, Sharp FA, Munoz-Wolf N, Hams E, Gorman AL, et al. The vaccine adjuvant alum promotes IL-10 production that suppresses Th1 responses. Eur J Immunol. (2018) 48:705–15. doi: 10.1002/eji.201747150
42. King BC, Esguerra JL, Golec E, Eliasson L, Kemper C, Blom AM. CD46 Activation regulates miR-150-mediated control of GLUT1 expression and cytokine secretion in human CD4+ T cells. J Immunol. (2016) 196:1636–45. doi: 10.4049/jimmunol.1500516
43. Collier SP, Collins PL, Williams CL, Boothby MR, Aune TM. Cutting edge: influence of Tmevpg1, a long intergenic noncoding RNA, on the expression of Ifng by Th1 cells. J Immunol. (2012) 189:2084–8. doi: 10.4049/jimmunol.1200774
44. Peng H, Liu Y, Tian J, Ma J, Tang X, Rui K, et al. The long noncoding RNA IFNG-AS1 promotes T helper type 1 cells response in patients with hashimoto's thyroiditis. Sci Rep. (2015) 5:17702. doi: 10.1038/srep17702
45. Hosseini A, Teimuri S, Ehsani M, Rasa SMM, Etemadifar M, Nasr Esfahani MH, et al. LncRNAs associated with multiple sclerosis expressed in the Th1 cell lineage. J Cell Physiol. (2019). doi: 10.1002/jcp.28779
46. O'Garra A, Arai N. The molecular basis of T helper 1 and T helper 2 cell differentiation. Trends Cell Biol. (2000) 10:542–50. doi: 10.1016/S0962-8924(00)01856-0
47. Simpson LJ, Patel S, Bhakta NR, Choy DF, Brightbill HD, Ren X, et al. A microRNA upregulated in asthma airway T cells promotes TH2 cytokine production. Nat Immunol. (2014) 15:1162–70. doi: 10.1038/ni.3026
48. Cho S, Wu CJ, Yasuda T, Cruz LO, Khan AA, Lin LL, et al. miR-23 approximately 27 approximately 24 clusters control effector T cell differentiation and function. J Exp Med. (2016) 213:235–49. doi: 10.1084/jem.20150990
49. Pua HH, Steiner DF, Patel S, Gonzalez JR, Ortiz-Carpena JF, Kageyama R, et al. MicroRNAs 24 and 27 suppress allergic inflammation and target a network of regulators of T helper 2 cell-associated cytokine production. Immunity. (2016) 44:821–32. doi: 10.1016/j.immuni.2016.01.003
50. Okoye IS, Czieso S, Ktistaki E, Roderick K, Coomes SM, Pelly VS, et al. Transcriptomics identified a critical role for Th2 cell-intrinsic miR-155 in mediating allergy and antihelminth immunity. Proc Natl Acad Sci USA. (2014) 111:E3081–90. doi: 10.1073/pnas.1406322111
51. Zhang S. The role of transforming growth factor β in T helper 17 differentiation. Immunology. (2018) 155:24–35. doi: 10.1111/imm.12938
52. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
53. Yao R, Ma YL, Liang W, Li HH, Ma ZJ, Yu X, et al. MicroRNA-155 modulates Treg and Th17 cells differentiation and Th17 cell function by targeting SOCS1. PLoS ONE. (2012) 7:e46082. doi: 10.1371/journal.pone.0046082
54. O'Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. (2010) 33:607–19. doi: 10.1016/j.immuni.2010.09.009
55. Ifergan I, Chen S, Zhang B, Miller SD. Cutting edge: MicroRNA-223 regulates myeloid dendritic cell-driven Th17 responses in experimental autoimmune encephalomyelitis. J Immunol. (2016) 196:1455–9. doi: 10.4049/jimmunol.1501965
56. Murugaiyan G, da Cunha AP, Ajay AK, Joller N, Garo LP, Kumaradevan S, et al. MicroRNA-21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J Clin Invest. (2015) 125:1069–80. doi: 10.1172/JCI74347
57. Ichiyama K, Gonzalez-Martin A, Kim BS, Jin HY, Jin W, Xu W, et al. The MicroRNA-183-96-182 cluster promotes T helper 17 cell pathogenicity by negatively regulating transcription factor foxo1 expression. Immunity. (2016) 44:1284–98. doi: 10.1016/j.immuni.2016.05.015
58. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. (2001) 292:468–72. doi: 10.1126/science.1059796
59. Wang H, Flach H, Onizawa M, Wei L, McManus MT, Weiss A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat Immunol. (2014) 15:393–401. doi: 10.1038/ni.2846
60. Yang F, Zhang H, Mei Y, Wu M. Reciprocal regulation of HIF-1α and lincRNA-p21 modulates the Warburg effect. Mol Cell. (2014) 53:88–100. doi: 10.1016/j.molcel.2013.11.004
61. Guo W, Lei W, Yu D, Ge Y, Chen Y, Xue W, et al. Involvement of lncRNA-1700040D17Rik in Th17 cell differentiation and the pathogenesis of EAE. Int Immunopharmacol. (2017) 47:141–9. doi: 10.1016/j.intimp.2017.03.014
62. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Annu Rev Immunol. (2016) 34:335–68. doi: 10.1146/annurev-immunol-041015-055605
63. Dorfman DM, Brown JA, Shahsafaei A, Freeman GJ. Programmed death-1 (PD-1) is a marker of germinal center-associated T cells and angioimmunoblastic T-cell lymphoma. Am J Surg Pathol. (2006) 30:802–10. doi: 10.1097/01.pas.0000209855.28282.ce
64. Yu D, Vinuesa CG. The elusive identity of T follicular helper cells. Trends Immunol. (2010) 31:377–83. doi: 10.1016/j.it.2010.07.001
65. Suan D, Nguyen A, Moran I, Bourne K, Hermes JR, Arshi M, et al. T follicular helper cells have distinct modes of migration and molecular signatures in naive and memory immune responses. Immunity. (2015) 42:704–18. doi: 10.1016/j.immuni.2015.03.002
66. Li J, Lu E, Yi T, Cyster JG. EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature. (2016) 533:110–4. doi: 10.1038/nature17947
67. Bronevetsky Y, Villarino AV, Eisley CJ, Barbeau R, Barczak AJ, Heinz GA, et al. T cell activation induces proteasomal degradation of Argonaute and rapid remodeling of the microRNA repertoire. J Exp Med. (2013) 210:417–32. doi: 10.1084/jem.20111717
68. Kang SG, Liu WH, Lu P, Jin HY, Lim HW, Shepherd J, et al. MicroRNAs of the miR-17 approximately 92 family are critical regulators of T(FH) differentiation. Nat Immunol. (2013) 14:849–57. doi: 10.1038/ni.2648
69. Baumjohann D, Kageyama R, Clingan JM, Morar MM, Patel S, de Kouchkovsky D, et al. The microRNA cluster miR-17 approximately 92 promotes TFH cell differentiation and represses subset-inappropriate gene expression. Nat Immunol. (2013) 14:840–8. doi: 10.1038/ni.2642
70. Vogel KU, Edelmann SL, Jeltsch KM, Bertossi A, Heger K, Heinz GA, et al. Roquin paralogs 1 and 2 redundantly repress the Icos and Ox40 costimulator mRNAs and control follicular helper T cell differentiation. Immunity. (2013) 38:655–68. doi: 10.1016/j.immuni.2012.12.004
71. Essig K, Hu D, Guimaraes JC, Alterauge D, Edelmann S, Raj T, et al. Roquin suppresses the PI3K-mTOR signaling pathway to inhibit T helper cell differentiation and conversion of Treg to Tfr cells. Immunity. (2017) 47:1067–82.e12. doi: 10.1016/j.immuni.2017.11.008
72. Serr I, Furst RW, Ott VB, Scherm MG, Nikolaev A, Gokmen F, et al. miRNA92a targets KLF2 and the phosphatase PTEN signaling to promote human T follicular helper precursors in T1D islet autoimmunity. Proc Natl Acad Sci USA. (2016) 113:E6659–68. doi: 10.1073/pnas.1606646113
73. Ripamonti A, Provasi E, Lorenzo M, De Simone M, Ranzani V, Vangelisti S, et al. Repression of miR-31 by BCL6 stabilizes the helper function of human follicular helper T cells. Proc Natl Acad Sci USA. (2017) 114:12797–802. doi: 10.1073/pnas.1705364114
74. Hu R, Kagele DA, Huffaker TB, Runtsch MC, Alexander M, Liu J, et al. miR-155 promotes T follicular helper cell accumulation during chronic, low-grade inflammation. Immunity. (2014) 41:605–19. doi: 10.1016/j.immuni.2014.09.015
75. Liu WH, Kang SG, Huang Z, Wu CJ, Jin HY, Maine CJ, et al. A miR-155-Peli1-c-Rel pathway controls the generation and function of T follicular helper cells. J Exp Med. (2016) 213:1901–19. doi: 10.1084/jem.20160204
76. Yang L, Boldin MP, Yu Y, Liu CS, Ea CK, Ramakrishnan P, et al. miR-146a controls the resolution of T cell responses in mice. J Exp Med. (2012) 209:1655–70. doi: 10.1084/jem.20112218
77. Pratama A, Srivastava M, Williams NJ, Papa I, Lee SK, Dinh XT, et al. MicroRNA-146a regulates ICOS-ICOSL signalling to limit accumulation of T follicular helper cells and germinal centres. Nat Commun. (2015) 6:6436. doi: 10.1038/ncomms7436
78. Cho S, Lee HM, Yu IS, Choi YS, Huang HY, Hashemifar SS, et al. Differential cell-intrinsic regulations of germinal center B and T cells by miR-146a and miR-146b. Nat Commun. (2018) 9:2757. doi: 10.1038/s41467-018-05196-3
79. Yao Y, Guo W, Chen J, Guo P, Yu G, Liu J, et al. Long noncoding RNA Malat1 is not essential for T cell development and response to LCMV infection. RNA Biol. (2018) 15:1477–86. doi: 10.1080/15476286.2018.1551705
80. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
81. Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. (2007) 178:280–90. doi: 10.4049/jimmunol.178.1.280
82. Konkel JE, Zhang D, Zanvit P, Chia C, Zangarle-Murray T, Jin W, et al. Transforming growth factor-β signaling in regulatory T cells controls T helper-17 cells and tissue-specific immune responses. Immunity. (2017) 46:660–74. doi: 10.1016/j.immuni.2017.03.015
83. Liu M, Li S, Li MO. TGF-β control of adaptive immune tolerance: a break from Treg cells. Bioessays. (2018) 40:e1800063. doi: 10.1002/bies.201800063
84. Rouas R, Fayyad-Kazan H, El Zein N, Lewalle P, Rothe F, Simion A, et al. Human natural Treg microRNA signature: role of microRNA-31 and microRNA-21 in FOXP3 expression. Eur J Immunol. (2009) 39:1608–18. doi: 10.1002/eji.200838509
85. Zhang L, Ke F, Liu Z, Bai J, Liu J, Yan S, et al. MicroRNA-31 negatively regulates peripherally derived regulatory T-cell generation by repressing retinoic acid-inducible protein 3. Nat Commun. (2015) 6:7639. doi: 10.1038/ncomms8639
86. Pan W, Zhu S, Dai D, Liu Z, Li D, Li B, et al. MiR-125a targets effector programs to stabilize Treg-mediated immune homeostasis. Nat Commun. (2015) 6:7096. doi: 10.1038/ncomms8096
87. Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. (2009) 30:80–91. doi: 10.1016/j.immuni.2008.11.010
88. Huang B, Zhao J, Lei Z, Shen S, Li D, Shen GX, et al. miR-142-3p restricts cAMP production in CD4+CD25− T cells and CD4+CD25+ TREG cells by targeting AC9 mRNA. EMBO Rep. (2009) 10:180–5. doi: 10.1038/embor.2008.224
89. Anandagoda N, Willis JC, Hertweck A, Roberts LB, Jackson I, Gokmen MR, et al. microRNA-142-mediated repression of phosphodiesterase 3B critically regulates peripheral immune tolerance. J Clin Invest. (2019) 129:1257–71. doi: 10.1172/JCI124725
90. Warth SC, Hoefig KP, Hiekel A, Schallenberg S, Jovanovic K, Klein L, et al. Induced miR-99a expression represses Mtor cooperatively with miR-150 to promote regulatory T-cell differentiation. EMBO J. (2015) 34:1195–213. doi: 10.15252/embj.201489589
91. Brajic A, Franckaert D, Burton O, Bornschein S, Calvanese AL, Demeyer S, et al. The long non-coding RNA Flatr anticipates Foxp3 expression in regulatory T cells. Front Immunol. (2018) 9:1989. doi: 10.3389/fimmu.2018.01989
92. Xia M, Liu J, Liu S, Chen K, Lin H, Jiang M, et al. Ash1l and lnc-Smad3 coordinate Smad3 locus accessibility to modulate iTreg polarization and T cell autoimmunity. Nat Commun. (2017) 8:15818. doi: 10.1038/ncomms15818
93. Zemmour D, Pratama A, Loughhead SM, Mathis D, Benoist C. Flicr, a long noncoding RNA, modulates Foxp3 expression and autoimmunity. Proc Natl Acad Sci USA. (2017) 114:E3472–80. doi: 10.1073/pnas.1700946114
94. Pan W, Zhu S, Yuan M, Cui H, Wang L, Luo X, et al. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol. (2010) 184:6773–81. doi: 10.4049/jimmunol.0904060
95. Qin H, Zhu X, Liang J, Wu J, Yang Y, Wang S, et al. MicroRNA-29b contributes to DNA hypomethylation of CD4+ T cells in systemic lupus erythematosus by indirectly targeting DNA methyltransferase 1. J Dermatol Sci. (2013) 69:61–7. doi: 10.1016/j.jdermsci.2012.10.011
96. Zhao S, Wang Y, Liang Y, Zhao M, Long H, Ding S, et al. MicroRNA-126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum. (2011) 63:1376–86. doi: 10.1002/art.30196
97. Ding S, Liang Y, Zhao M, Liang G, Long H, Zhao S, et al. Decreased microRNA-142-3p/5p expression causes CD4+ T cell activation and B cell hyperstimulation in systemic lupus erythematosus. Arthritis Rheum. (2012) 64:2953–63. doi: 10.1002/art.34505
98. Tang Q, Yang Y, Zhao M, Liang G, Wu H, Liu Q, et al. Mycophenolic acid upregulates miR-142-3P/5P and miR-146a in lupus CD4+T cells. Lupus. (2015) 24:935–42. doi: 10.1177/0961203315570685
99. Liu CX, Li X, Nan F, Jiang S, Gao X, Guo SK, et al. Structure and degradation of circular RNAs regulate PKR activation in innate immunity. Cell. (2019) 177:865–80.e21. doi: 10.1016/j.cell.2019.03.046
100. Eriksson C, Eneslatt K, Ivanoff J, Rantapaa-Dahlqvist S, Sundqvist KG. Abnormal expression of chemokine receptors on T-cells from patients with systemic lupus erythematosus. Lupus. (2003) 12:766–74. doi: 10.1191/0961203303lu467oa
101. Zhang C, Wang X, Chen Y, Wu Z, Zhang C, Shi W. The down-regulation of hsa_circ_0012919, the sponge for miR-125a-3p, contributes to DNA methylation of CD11a and CD70 in CD4(+) T cells of systemic lupus erythematous. Clin Sci. (2018) 132:2285–98. doi: 10.1042/CS20180403
102. Barber DF, Bartolome A, Hernandez C, Flores JM, Redondo C, Fernandez-Arias C, et al. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med. (2005) 11:933–5. doi: 10.1038/nm1291
103. Tang H, Tan G, Guo Q, Pang R, Zeng F. Abnormal activation of the Akt-GSK3beta signaling pathway in peripheral blood T cells from patients with systemic lupus erythematosus. Cell Cycle. (2009) 8:2789–93. doi: 10.4161/cc.8.17.9446
104. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. (2011) 12:295–303. doi: 10.1038/ni.2005
105. Wang X, Zhang C, Wu Z, Chen Y, Shi W. CircIBTK inhibits DNA demethylation and activation of AKT signaling pathway via miR-29b in peripheral blood mononuclear cells in systemic lupus erythematosus. Arthritis Res Ther. (2018) 20:118. doi: 10.1186/s13075-018-1618-8
106. Jiang CR, Li TH. Circulating UCA1 is highly expressed in patients with systemic lupus erythematosus and promotes the progression through the AKT pathway. Eur Rev Med Pharmacol Sci. (2018) 22:2364–71. doi: 10.26355/eurrev_201804_14828
107. Divekar AA, Dubey S, Gangalum PR, Singh RR. Dicer insufficiency and microRNA-155 overexpression in lupus regulatory T cells: an apparent paradox in the setting of an inflammatory milieu. J Immunol. (2011) 186:924–30. doi: 10.4049/jimmunol.1002218
108. Leiss H, Salzberger W, Jacobs B, Gessl I, Kozakowski N, Bluml S, et al. MicroRNA 155-deficiency leads to decreased autoantibody levels and reduced severity of nephritis and pneumonitis in pristane-induced lupus. PLoS ONE. (2017) 12:e0181015. doi: 10.1371/journal.pone.0181015
109. Zhou S, Wang Y, Meng Y, Xiao C, Liu Z, Brohawn P, et al. In vivo therapeutic success of MicroRNA-155 antagomir in a mouse model of lupus alveolar hemorrhage. Arthritis Rheumatol. (2016) 68:953–64. doi: 10.1002/art.39485
110. Lashine YA, Salah S, Aboelenein HR, Abdelaziz AI. Correcting the expression of miRNA-155 represses PP2Ac and enhances the release of IL-2 in PBMCs of juvenile SLE patients. Lupus. (2015) 24:240–7. doi: 10.1177/0961203314552117
111. Chen JQ, Papp G, Poliska S, Szabo K, Tarr T, Balint BL, et al. MicroRNA expression profiles identify disease-specific alterations in systemic lupus erythematosus and primary Sjogren's syndrome. PLoS ONE. (2017) 12:e0174585. doi: 10.1371/journal.pone.0174585
112. Wang Z, Heid B, Dai R, Ahmed SA. Similar dysregulation of lupus-associated miRNAs in peripheral blood mononuclear cells and splenic lymphocytes in MRL/lpr mice. Lupus Sci Med. (2018) 5:e000290. doi: 10.1136/lupus-2018-000290
113. Stagakis E, Bertsias G, Verginis P, Nakou M, Hatziapostolou M, Kritikos H, et al. Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: miR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann Rheum Dis. (2011) 70:1496–506. doi: 10.1136/ard.2010.139857
114. Garchow B, Kiriakidou M. MicroRNA-21 deficiency protects from lupus-like autoimmunity in the chronic graft-versus-host disease model of systemic lupus erythematosus. Clin Immunol. (2016) 162:100–6. doi: 10.1016/j.clim.2015.11.010
115. Garchow BG, Bartulos Encinas O, Leung YT, Tsao PY, Eisenberg RA, Caricchio R, et al. Silencing of microRNA-21 in vivo ameliorates autoimmune splenomegaly in lupus mice. EMBO Mol Med. (2011) 3:605–15. doi: 10.1002/emmm.201100171
116. Amr KS, Bayoumi FS, Elgengehy FT, Abdallah SO, Ahmed HH, Eissa E. The role of microRNA-31 and microRNA-21 as regulatory biomarkers in the activation of T lymphocytes of Egyptian lupus patients. Rheumatol Int. (2016) 36:1617–25. doi: 10.1007/s00296-016-3550-z
117. Suo QF, Sheng J, Qiang FY, Tang ZS, Yang YY. Association of long non-coding RNA GAS5 and miR-21 levels in CD4(+) T cells with clinical features of systemic lupus erythematosus. Exp Ther Med. (2018) 15:345–50. doi: 10.3892/etm.2017.5429
118. von Spee-Mayer C, Siegert E, Abdirama D, Rose A, Klaus A, Alexander T, et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheum Dis. (2016) 75:1407–15. doi: 10.1136/annrheumdis-2015-207776
119. Khoshmirsafa M, Kianmehr N, Falak R, Mowla SJ, Seif F, Mirzaei B, et al. Elevated expression of miR-21 and miR-155 in peripheral blood mononuclear cells as potential biomarkers for lupus nephritis. Int J Rheum Dis. (2019) 22:458–67. doi: 10.1111/1756-185X.13410
120. Li X, Luo F, Li J, Luo C. MiR-183 delivery attenuates murine lupus nephritis-related injuries via targeting mTOR. Scand J Immunol. (2019) 90:e12810. doi: 10.1111/sji.12810
121. Jiang SH, Shen N, Vinuesa CG. Posttranscriptional T cell gene regulation to limit Tfh cells and autoimmunity. Curr Opin Immunol. (2015) 37:21–7. doi: 10.1016/j.coi.2015.09.003
122. Dai R, Zhang Y, Khan D, Heid B, Caudell D, Crasta O, et al. Identification of a common lupus disease-associated microRNA expression pattern in three different murine models of lupus. PLoS ONE. (2010) 5:e14302. doi: 10.1371/journal.pone.0014302
123. Qin HH, Zhu XH, Liang J, Wu JF, Yang YS, Xu JH. The expression and significance of miR-17-92 cluster miRs in CD4+ T cells from patients with systemic lupus erythematosus. Clin Exp Rheumatol. (2013) 31:472–3.
124. Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell. (2009) 136:26–36. doi: 10.1016/j.cell.2008.12.027
125. Liu L, Liu Y, Yuan M, Xu L, Sun H. Elevated expression of microRNA-873 facilitates Th17 differentiation by targeting forkhead box O1 (Foxo1) in the pathogenesis of systemic lupus erythematosus. Biochem Biophys Res Commun. (2017) 492:453–60. doi: 10.1016/j.bbrc.2017.08.075
126. Xie M, Wang J, Gong W, Xu H, Pan X, Chen Y, et al. NF-κB-driven miR-34a impairs Treg/Th17 balance via targeting Foxp3. J Autoimmun. (2019) 102:96–113. doi: 10.1016/j.jaut.2019.04.018
127. Sun XG, Tao JH, Xiang N, Li XM, Wang GS, Fang X, et al. Negative correlation between miR-326 and Ets-1 in regulatory T cells from new-onset SLE patients. Inflammation. (2016) 39:822–9. doi: 10.1007/s10753-016-0312-8
128. Zhang F, Wu L, Qian J, Qu B, Xia S, La T, et al. Identification of the long noncoding RNA NEAT1 as a novel inflammatory regulator acting through MAPK pathway in human lupus. J Autoimmun. (2016) 75:96–104. doi: 10.1016/j.jaut.2016.07.012
129. Teramoto K, Negoro N, Kitamoto K, Iwai T, Iwao H, Okamura M, et al. Microarray analysis of glomerular gene expression in murine lupus nephritis. J Pharmacol Sci. (2008) 106:56–67. doi: 10.1254/jphs.FP0071337
Keywords: non-coding RNAs, CD4+ T cells, T helper cells, biomarkers, systemic lupus erythematosus
Citation: Gao X, Liu L, Min X, Jia S and Zhao M (2020) Non-Coding RNAs in CD4+ T Cells: New Insights Into the Pathogenesis of Systemic Lupus Erythematosus. Front. Immunol. 11:568. doi: 10.3389/fimmu.2020.00568
Received: 08 December 2019; Accepted: 12 March 2020;
Published: 03 April 2020.
Edited by:
Stefania Gallucci, Temple University, United StatesReviewed by:
Anne Cooke, University of Cambridge, United KingdomUma Sriram, Temple University, United States
Copyright © 2020 Gao, Liu, Min, Jia and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Zhao, emhhb21pbmczMDcmI3gwMDA0MDtjc3UuZWR1LmNu